 Bruno A. Cisterna
Bruno A. Cisterna Pablo Arroyo
Pablo Arroyo Carlos Puebla
Carlos Puebla- 1Escuela de Medicina, Universidad de Talca, Talca, Chile
- 2Centro para el Desarrollo de la Nanociencia y Nanotecnología (CEDENNA), Universidad de Santiago de Chile, Santiago, Chile
- 3Facultad de Medicina, Pontificia Universidad Católica de Chile, Santiago, Chile
- 4Instituto de Ciencias Biomédicas, Facultad de Ciencias de la Salud, Universidad Autónoma de Chile, Santiago, Chile
Peripheral nerves have the capacity to conduct action potentials along great distances and quickly recover following damage which is mainly due to Schwann cells (SCs), the most abundant glial cells of the peripheral nervous system (PNS). SCs wrap around an axonal segment multiple times, forming a myelin sheath, allowing for a significant increase in action potential conduction by insulating the axons. Mature myelin consists of compact and non-compact (or cytoplasmic) myelin zones. Non-compact myelin is found in paranodal loops bordering the nodes of Ranvier, and in the inner and outermost cytoplasmic tongues and is the region in which Schmidt-Lanterman incisures (SLI; continuous spirals of overlapping cytoplasmic expansions within areas of compact myelin) are located. Using different technologies, it was shown that the layers of non-compact myelin could be connected to each other by gap junction channels (GJCs), formed by connexin 32 (Cx32), and their relative abundance allows for the transfer of ions and different small molecules. Likewise, Cx29 is expressed in the innermost layer of the myelin sheath. Here it does not form GJCs but colocalizes with Kv1, which implies that the SCs play an active role in the electrical condition in mammals. The critical role of GJCs in the functioning of myelinating SCs is evident in Charcot-Marie-Tooth disease (CMT), X-linked form 1 (CMTX1), which is caused by mutations in the gap junction protein beta 1 (GJB1) gene that codes for Cx32. Although the management of CMT symptoms is currently supportive, there is a recent method for targeted gene delivery to myelinating cells, which rescues the phenotype in KO-Cx32 mice, a model of CMTX1. In this mini-review article, we discuss the current knowledge on the role of Cxs in myelin-forming SCs and summarize recent discoveries that may become a real treatment possibility for patients with disorders such as CMT.
Myelinating Schwann Cells
The ability of peripheral nerves to recover quickly after damage is mostly due to the plasticity of Schwann cells (SCs; Boerboom et al., 2017). SCs are the principal glia of the peripheral nervous system (PNS; Fehmi et al., 2018). In the development of vertebrate PNS, SCs are derived from neural crest cells that differentiate into SC precursors and then into immature SCs (Jessen and Mirsky, 2005; Boerboom et al., 2017). The process of myelination begins around birth, where SCs cover axons that are larger than ~1 μm in diameter (Fehmi et al., 2018).
Myelinating SCs allow for the fast conduction of action potentials by the axons (Jessen and Mirsky, 2005; Boerboom et al., 2017). Each myelin internode is flanked by the nodes of Ranvier, which are bare patches of the axon plasma membrane that are enriched in voltage-gated Na+ channels (essential for saltatory conduction). In turn, K+ channels are clustered at the juxtaparanodal region of the axon. Thus, the two primary types of ion channels are separated by the paranode (Poliak and Peles, 2003).
An essential process in a functional interaction between axons and SCs, is myelin formation. The molecules that mediate axonal–glial interactions along the internode have been described (Trapp, 1990; Martini and Carenini, 1998; Maurel et al., 2007; Hayashi et al., 2008; Chen et al., 2016). The myelin-associated glycoprotein (MAG), a cell adhesion molecule member of the Ig-superfamily—expressed by myelinating SCs and oligodendrocytes [equivalent to the SCs but located in the central nervous system (CNS)]—has been localized in the internode (Trapp, 1990; Maurel et al., 2007). This protein interacts with several axonal components (Hannila et al., 2007), and it is crucial for the maintenance of myelin, evidenced by the degeneration of myelin and axons in MAG-deficient mice (Martini and Carenini, 1998). In the later stages of myelination, MAG is also found in Schmidt-Lanterman incisures (SLI; Maurel et al., 2007). Additionally, connexin 32 (Cx32) is another component type involved in the maintenance of myelination, which does not belong to the Ig-superfamily (Martini and Carenini, 1998).
Recently, it was reported that nectins and nectin-like molecules (Necl) are required for the axon–glial interactions along the internode through heterophilic interactions. In particular, for cell-cell context, both neurons and SCs express different sets of Necl proteins: axons express Necl-1 and Necl-2, and SCs express Necl-4 and some Necl-2. The main binding protein detected between axon and SCs was Necl-1 and Necl-4 (Maurel et al., 2007). Necl-1 activity involves the PI3 kinase/Akt signaling cascade (Chen et al., 2016). Moreover, Necl-1, Necl-2, and Necl-4 are also expressed at high levels in the SLI of myelinating SCs (Maurel et al., 2007).
The Issue of Intracellular Communication in Myelinated Schwann Cells
It has been proposed that the number of myelin lamellae in the sheath is directly proportional to the axonal circumference, for example, over the range of 10–80 lamellae in the sciatic and vagus nerves of mice (Friede and Samorajski, 1967), or 18–24 in the optic nerve in humans (Friede and Hu, 1967). A significant amount of myelin membrane has been proposed to increase the axonal membrane resistance and decrease the capacitance, increasing the conduction velocity. According to some estimates, myelinating SCs may produce up to 20 mm2 of compacted membrane around the largest axons, about 2,000 times more than typical epithelial cells (Kidd et al., 2013). This massive amount of membrane, concentrated in a small area is a challenge for the communication of the cell itself. The myelinating SCs manages this issue by separating the myelin in two distinct domains, compact myelin, and non-compact (or cytoplasmic) myelin (Kidd et al., 2013).
Compact myelin is a compact membrane spiral, with a periodicity of 13–18 nm per turn (Fernandez-Moran and Finean, 1957; Kirschner and Sidman, 1976), which begins and ends with the apposition of the SC plasma membrane against itself to form the inner mesaxon and the outer mesaxon. Compact myelin has a high (~70%) lipid content and is enriched in galactosphingolipids, saturated long-chain fatty acids and cholesterol (Saher and Simons, 2010), while it has a low protein content with a very low protein diversity; mainly P0 protein, maltose-binding protein (MBP), and peripheral myelin protein 22 (PMP22). Non-compact myelin is in paranodal loops (cytoplasmic extensions located in the lateral margins of the SCs, bordering nodes of Ranvier), and in SLI (continuous spirals of overlapping cytoplasmic expansions within areas of compact myelin), and in the inner and outermost cytoplasmic tongues. While, compaction of myelin removes the aqueous components of the cytoplasm, favoring electric conduction but hindering cellular processes such as intracellular communication, non-compact compartments concentrate the organelles and the necessary components for the cellular operation.
A pioneering observation of the myelin sheath, made using freeze-fracture electron microscopy, reported gap junction channels (GJCs)-like structures in the membrane of the mesaxons, paranodal loops and SLI (Mugnaini et al., 1977; Sandri et al., 1977; Bertaud, 1978; Tetzlaff, 1982). Likewise, the diffusion of fluorescent dyes across the cytoplasmic layers, primarily at SLI, suggest that GJCs provides a direct radial pathway for the transport of ions and small metabolites across the myelin sheath (Balice-Gordon et al., 1998). Then, considering that the unrolled myelin sheath is more than 4 mm long, while the compact myelin sheath is less than 4 μm thick (Friede and Bischhausen, 1980), this potential radial pathway would be more than 1,000-fold shorter than the circumferential pathway (Scherer et al., 1995; Kleopa et al., 2010). For diffusion time, it was proposed that the radial pathway is one million times faster than the circumferential pathway, because, in theory, the diffusion time in a plane is proportional to the square of the distance (Balice-Gordon et al., 1998). However, this investigation has some limitations, for example, the authors assumed that the ions showed a very slow velocity in circumferential diffusion, but they did not measure this pathway nor did they measure the rate of diffusion within the incisures. It is therefore difficult to determine that the radial pathway will be x-fold faster than the circumferential pathway, in fact, the most important real contribution is providing functional evidence, i.e., injection of sciatic nerve fibers, that GJCs mediate a radial pathway.
Gap Junction Channels in Myelinating Schwann Cells
GJCs connect adjacent cells (heterocellular) and isolated segments of the same cell (autocellular) and allow the diffusion of ions and small molecules across apposed cell membranes expressed in vertebrates (Sáez et al., 2003a). The single channel is formed by the interaction of two apposed hemichannels (HCs) or connexons, each of which is composed of six connexins (Cxs) arranged radially around a central pore (Sosinsky, 1996; Unger et al., 1997). It is worth mentioning that, Cx HCs are semipermeable channels that may allow the passage of small molecules such as ATP (Wang et al., 2013), IP3 and cAMP (Hernandez et al., 2007), NAD+ (Bruzzone et al., 2001), and glutamate (Sáez et al., 2003b; Ye et al., 2003), and the passage of ions driven by their electrochemical gradients (Sáez et al., 2005). Cxs are transmembrane proteins that belong to a multigene family with 20 and 21 members in the mouse and human genome, respectively (Söhl and Willecke, 2003). Each Cx HCs can be formed by a single type of Cx (homomeric) or possibly by two or even three Cxs (heteromeric). The GJCs formation can then give rise to homotypic or heterotypic intercellular channels, when they are the same or different Cxs in apposed cells (Jiang and Goodenough, 1996; Koval et al., 2014). Every one of these channel combinations displays different permeability properties and are regulated differently (Weber et al., 2004). Thus, Cxs diversity provides essential variations in cell-to-cell communication.
The presence of the Cx32 is well documented by immunocytochemistry in inner mesaxons, at paranodal loops and SLI of the myelin sheath (Bergoffen et al., 1993a; Scherer et al., 1995; Spray and Dermietzel, 1995). Likewise, these findings were confirmed by freeze-fracture replica immunogold labeling (FRIL). Cx32 is also present at the two outer layers of myelin, suggesting that Cx32 forms GJCs between the non-compact layers of the SCs myelin sheath (Meier et al., 2004).
The Cxs play a critical role in the functioning of the myelinating SCs. Indeed, mutations in the human gene gap junction protein beta 1 (GJB1), which encodes Cx32, lead to the pathological phenotype of the X-chromosomal form of Charcot-Marie-Tooth (CMT1X or CMTX1), where inflammatory processes in peripheral nerves decrease conduction velocity of action potentials, leading to muscle atrophy (Bergoffen et al., 1993a; Fischbeck et al., 1996; Abrams et al., 2002). This disease is the second most common form of hereditary motor and sensory neuropathy, and there is no cure (Kleopa et al., 2012). Despite the abundant evidence that relates the absence or altered function of Cx directly to CMT1X, the role of Cxs in myelin physiology remains poorly understood.
Cx32 has multifaceted functions described in glial cells in the CNS (Abrams, 2017) and PNS (Bortolozzi, 2018). Recent evidence indicated the expression of functional Cx32 HCs that release ATP during electrical stimulation on mice sciatic nerves, which from cultured SCs, by depolarization evoked through a high extracellular potassium concentration (Nualart-Marti et al., 2013). ATP release was then significantly decreased after the sciatic nerve was treated with HCs inhibitors (octanol or carbenoxolone) or after silencing Cx32 from cultured SCs, suggesting that purinergic mediated signaling could contribute to intracellular communication (Nualart-Marti et al., 2013). On the other hand, Ca2+ waves do not seem to utilize GJCs in glial cells of the brain. Instead, Ca2+ waves are propagated by an alternative extracellular mechanism, involving ATP release and possibly requiring Cx HC activity (Bennett et al., 2003; Nedergaard et al., 2003), suggesting that Ca2+ waves may be propagated by an ATP-induced ATP release mechanism. However, the functions of HCs in myelinating SC still needs to be demonstrated. The Cx32 is even involved in the proliferation of SCs, related to neuregulin-1, which does not involve Cx32-mediated intercellular communication (Freidin et al., 2009). Likewise, mice lacking Cx32 display features of CMTX, such as onion bulb formation and slow nerve conduction in myelinating SC (Anzini et al., 1997; Scherer et al., 1998). Moreover, cultured SCs, from Cx32-null mice, are still electrically coupled and even allow diffusion of fluorescent dye (Zhao et al., 1999), implying the presence of other Cxs forming gap junctions (Balice-Gordon et al., 1998).
Connexin 29 (Cx29), a 29-kDa protein (and equivalent to human ortholog Cx31.3; Söhl and Willecke, 2003) was also found in the myelin sheath, by freeze-FRIL, but only in the innermost layer, in close association with the hexagonally arranged intramembrane particle (IMP) “rosettes” in axolemma of large myelinated axons (Li et al., 2002). Similarly, through immunofluorescence (IF), Cx29 was localized in the innermost layer of the myelin sheath, in the paranode and juxtaparanode, tightly colocalized with Kv1.2 channels in the axolemma (Altevogt et al., 2002). Recently, through IF and FRIL, Kv1.1/Kv1.2 channels were identified within the axonal rosette in rodent sciatic nerve and the molecular coalignment 1:1 with Cx29 was proposed to form a xenotypic channel (Rash et al., 2016). On the other hand, K+ conductance occurs neither in the perinodal, nor in the submyelinic extracellular spaces, as previously envisioned; it is now proposed to occur in the innermost myoplasm of SCs, which is electrically isolated by the myelin plasma membrane (Rash, 2010). Likewise, the proposed K+ recycling from axoplasm to myoplasm to axoplasm could be energetically cheaper than using Na+-K+ ATPase to axoplasmic replacement. This axon-to-glia linkage implies that myelin may play an electrically active role that underlies both the faster axonal repolarization and the faster conduction velocity of mammalian myelinated axon. However, Cx29 does not prevent the development of demyelinating neuropathy in Cx32-null mice (Scherer et al., 1998), or in Cx32 mutant mice (Jeng et al., 2006; Sargiannidou et al., 2009). Although Cx29 may form HCs (Ahn et al., 2008), these Cx proteins do not form GJCs (in Xenopus oocytes or N2A cells), but at least in vitro, it may modify the properties of Cx32 GJCs (Altevogt et al., 2002).
Other Cx proteins, such as Cx26, Cx43 and Cx46, have also been reported to be expressed in SCs (Chandross et al., 1996a,b; Yoshimura et al., 1996; Mambetisaeva et al., 1999; Li et al., 2007), but there are no published data that currently establishes any of these Cxs in SCs. In particular Cx43, together with Cx29 and Cx32 are present during SCs development in mice and displayed a consecutive onset of expression (Li et al., 2007). Similarly, it has been suggested that Cx43 and Cx46 could coordinate cellular responses of SCs after a peripheral nerve’s injury (Chandross et al., 1996a,b).
In summary, Cx32 and Cx29 are the main Cxs detectable in myelinating SCs in an asymmetric subcellular distribution, with a partial overlapping distribution. Cx32 was found to be concentrated in GJCs on the nuclear end of the myelin (abaxonal), whereas Cx29 form HCs was found to be concentrated closer to the paranodal loop and axon (abaxonal). This suggests that they could both contribute to reflexive junctions across the myelin sheath, speeding up communication through the myelin layers that separate the adaxonal and the abaxonal cytoplasm (Figure 1).
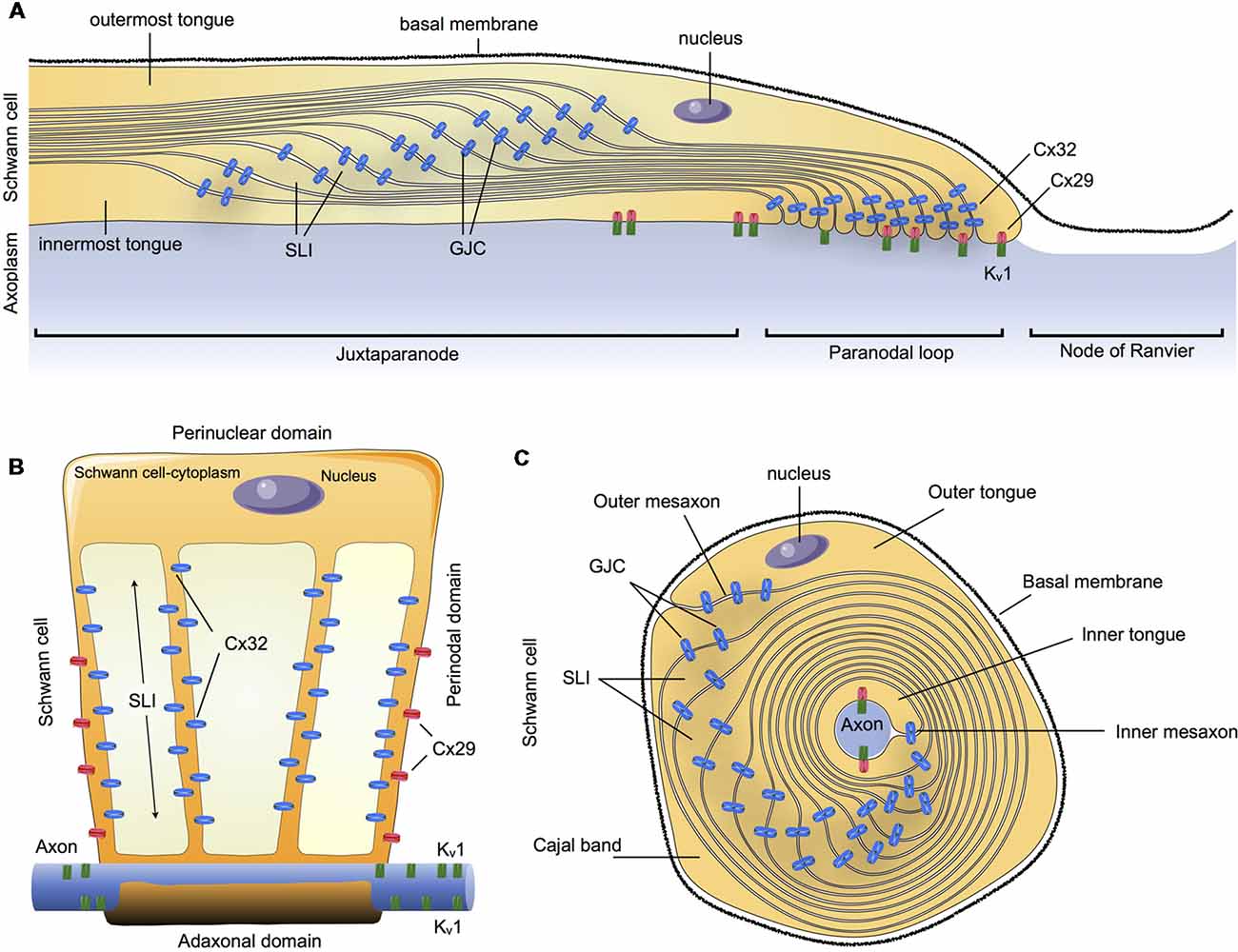
Figure 1. Schematic summary of connexins (Cxs) in the communication of myelin sheath in Schwann cells (SCs). (A) Longitudinal view of myelinated SCs showing the different compartments: outermost tongue, innermost tongue, Schmidt-Lanterman incisures (SLI) and the distribution of the connexins forming gap junction channels (GJCs) or hemichannels (HCs) formed by Cxs (Cx HCs) along the juxtaparanode, paranodal loop or the node of Ranvier. (B) Myelinated SCs unwrapped from the axon it invests. (C) Transversal view. Cx29, connexin29; Cx32, connexin 32; Kv1, Potassium voltage-gated channel subfamily 1.
Diseases Associated With Dysfunction of Connexins in Schwann Cells
Charcot-Marie-Tooth disease (CMT) is a large group of disorders caused by different types of mutations in several genes whose protein are expressed in myelin and/or axonal structures within PNS (Lupski, 1999; Kamholz et al., 2000). Usually, the initial symptoms of CMT are distal weakness and muscle atrophy, manifesting with foot drop and pes cavus. Also, sensory symptoms are often present but tend to be less prominent. Later, these patients present foot deformities, such as hammertoes, together with hand weakness and atrophy (Saporta, 2014).
The categories of CMT are CMT types 1 through 7, as well as an X-linked category, CMTX. Then, there are X-linked dominant and recessive forms of CMT involving different loci. Together, the X-linked forms account for 10% to 15% of all CMT cases (Ouvrier et al., 2007). CMTX1, the X-linked dominant form of CMT, is the second most common form of CMT and the first X-linked form of CMT, with 7% to 12%, and 50% of all CMT cases and X-linked cases, respectively (Boerkoel et al., 2002; Huttner et al., 2006; Ouvrier et al., 2007; Pareyson and Marchesi, 2009; Fridman et al., 2015).
CMTX1 symptoms are more prominent in boys, who present gait problems in infancy or later in childhood (e.g., toe walking, flat-footed walking, falls, difficulty running; Yiu et al., 2011). Less common features include tremors, hand weakness, and sensorineural deafness. Reflexes are lost at the ankles in all cases, whereas patellar reflexes are retained in half of the cases in girls. The neuropathy may be asymmetric and therefore mimic an acquired immune-mediated neuropathy (Saporta, 2014). Typically, the pathophysiology of CMTX1 includes features of both demyelination and axon loss, and this is reflected in neurophysiologic studies. Nerve conduction velocities are moderately slowed (Nicholson and Nash, 1993). Demyelination and axonal loss are observed histologically, but onion bulb formation is minimal.
CMTX1 is caused by mutations in the GJB1 gene, i.e., Cx32 gene, on chromosome Xq13.1 (Hahn et al., 1990; Bergoffen et al., 1993a,b; Keller and Chance, 1999). Most GJB1 mutations cause disability through the loss of function of Cx32 (Bruzzone et al., 1994; Kleopa et al., 2012). In particular, Cx32 mutations related to CMTX1 could be organized into five classes: (1) Cx32 protein is not synthesized; (2) mutant Cx32 protein has a reduced expression (with a normal transcription), (3) mutant Cx32 protein has a normal expression but is not transported to the plasma membrane; (4) mutant Cx32 protein forms HCs but not functional GJCs; and (5) mutant Cx32 protein forms GJCs and HCs with altered electrical, gating or permeability properties (Bortolozzi, 2018).
It is worth mentioning that some mutants that appear as “functional,” with respect to the WT model, cause severe phenotypes of CMTX1. This issue may be explained by the fact that in most of the studies only the activity of GJCs was evaluated, without considering the gating/permeability dysfunction of mutant Cx32 HCs. Recently, an investigation uncovered that the loss of the C-terminus in Cx32 (R220X mutation), inhibits Cx32 HCs gating. However, it does not significantly alter the intercellular diffusion mediated by GJCs: the unitary permeabilities to ions, signaling molecules (cAMP) or larger solutes (Lucifer yellow) concerning the wild-type (Carrer et al., 2018). These findings support the hypothesis that paracrine signaling alteration, due to Cx32 HCs dysfunction, underlies CMTX1 pathogenesis.
Although it is described that two linked connexons, i.e., a GJCs, may contain the same connexin proteins, or different ones, an interesting case has been described, in myelinated axons, where a Cx29-based connexon is linked to a K+ channel (Rash et al., 2016; Traub et al., 2018). This xenotypic channel could be helpful to understand why Cx29 KO mice do not show important neuropathic symptoms, such as Cx32 KO. First, the formation of axolemmal rosettes is an inherent property of KV1 channels, does not depend on a linkage with Cx29 (Rash et al., 2016), so it is possible to suggest that in the absence of Cx29 it is still present in “half” of the activity of this xenotypic channel. Second, the absence of the Cx32 protein necessarily involves a total loss of Cx32-based channel activity in myelinated cells, with no compensation from other Cx proteins (Sargiannidou et al., 2009), so the absence of Cx32 would lead to worse results than the absence of Cx29. It was also recently proposed that Cx32 HCs could underlie CMTX1 by altering the purinergic signaling controlling SCs myelination (Nualart-Marti et al., 2013; Carrer et al., 2018). Additionally, if we consider that a heteromeric mixing of Cx29 and Cx32 (i.e., Cx32-Cx29/Cx32-Cx32) produced channels with properties different from those of Cx32 alone (Altevogt et al., 2002), in cases of Cx32 mutations, an unexplored (and speculative) possibility is that these mutations can change the affinity of Cx32 for Cx29 and these “new” connexons could be involved in different failures of Cx32 GJCs activity. The Cx32 protein then becomes an essential protein in the physiology of PNS (Bortolozzi, 2018).
Finally, considering that the current “treatments” include only palliative care, it is necessary to advance new therapies. A recent study obtained a cell-specific expression of Cx32 in up to 50% of SCs, in multiple lumbar spinal roots and peripheral nerves, after a single intrathecal gene delivery into adult GJB1-KO mice. Thus, treated mice showed a reduced amount of demyelinated fibers and inflammatory cells with motor performance, quadriceps muscle contractility, and sciatic nerve conduction velocities improved (Kagiava et al., 2016). The same gene therapy was performed in CMT1X mice expressing different Cx32 mutants, which showed different results, as CMT1X mutants may interfere with gene addition therapy (Kagiava et al., 2018). A better approach may be one that does not incorporate the genome but leads to permanent modification of the targeted genetic defect. In this regard, recent reports of treating mouse models of Duchenne muscular dystrophy, with adeno-associated viral vectors, which are not integrated into the genome, is promising. This method used the clustered regularly interspaced short palindromic repeats system (CRISPR)-Cas9 to target and eliminate mutant exons of dystrophin (Nelson et al., 2016; Tabebordbar et al., 2016). In summary, the actual data suggest that gene therapy might become a realistic possibility for patients with these disorders.
Conclusions
Myelinating SCs and peripheral axons present an interesting example of two specialized cells, where each is indispensable for the functioning of the other. In this sense, the communication through GJCs is a significant evolutionary advance which can solve the issue of the reflexive GJs in the myelinating SCs. This cellular specialization has also led to the differential expression and specific roles of Cx29 and Cx32. In particular, CMTX1 is a clear example of diseases that are caused by mutations in only one GJ gene (Cx32), and despite this broad expression pattern, peripheral neuropathy is the main clinical manifestation. Since several mutations of the Cx32 gene in SCs lead to lasting and severe motor and neurological problems, the search for curative treatment points towards gene therapies in an attempt to replace the defective gene. These approaches however, have their limitations because of toxicity from random integrations in the genome.
Author Contributions
CP and BC conceived, researched and wrote this review article. PA assisted in writing and editing.
Funding
This work was supported by FONDECYT no. 11160536 (CP) and no. 3170938 (BC).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the Universidad Autónoma de Chile.
References
Abrams, C. K. (2017). Diseases of connexins expressed in myelinating glia. Neurosci. Lett. doi: 10.1016/j.neulet.2017.05.037 [Epub ahead of print].
Abrams, C. K., Bennett, M. V., Verselis, V. K., and Bargiello, T. A. (2002). Voltage opens unopposed gap junction hemichannels formed by a connexin 32 mutant associated with X-linked Charcot-Marie-Tooth disease. Proc. Natl. Acad. Sci. U S A 99, 3980–3984. doi: 10.1073/pnas.261713499
Ahn, M., Lee, J., Gustafsson, A., Enriquez, A., Lancaster, E., Sul, J. Y., et al. (2008). Cx29 and Cx32, two connexins expressed by myelinating glia, do not interact and are functionally distinct. J. Neurosci. Res. 86, 992–1006. doi: 10.1002/jnr.21561
Altevogt, B. M., Kleopa, K. A., Postma, F. R., Scherer, S. S., and Paul, D. L. (2002). Connexin29 is uniquely distributed within myelinating glial cells of the central and peripheral nervous systems. J. Neurosci. 22, 6458–6470. doi: 10.1523/JNEUROSCI.22-15-06458.2002
Anzini, P., Neuberg, D. H., Schachner, M., Nelles, E., Willecke, K., Zielasek, J., et al. (1997). Structural abnormalities and deficient maintenance of peripheral nerve myelin in mice lacking the gap junction protein connexin 32. J. Neurosci. 17, 4545–4551. doi: 10.1523/JNEUROSCI.17-12-04545.1997
Balice-Gordon, R. J., Bone, L. J., and Scherer, S. S. (1998). Functional gap junctions in the schwann cell myelin sheath. J. Cell Biol. 142, 1095–1104. doi: 10.1083/jcb.142.4.1095
Bennett, M. V., Contreras, J. E., Bukauskas, F. F., and Sáez, J. C. (2003). New roles for astrocytes: gap junction hemichannels have something to communicate. Trends Neurosci. 26, 610–617. doi: 10.1016/j.tins.2003.09.008
Bergoffen, J., Scherer, S. S., Wang, S., Scott, M. O., Bone, L. J., Paul, D. L., et al. (1993a). Connexin mutations in X-linked Charcot-Marie-Tooth disease. Science 262, 2039–2042. doi: 10.1126/science.8266101
Bergoffen, J., Trofatter, J., Pericak-Vance, M. A., Haines, J. L., Chance, P. F., and Fischbeck, K. H. (1993b). Linkage localization of X-linked Charcot-Marie-Tooth disease. Am. J. Hum. Genet. 52, 312–318.
Bertaud, W. S. (1978). Membrane junctions in the myelin sheath of goldfish lateral nerve. J. Cell Sci. 30, 77–85.
Boerboom, A., Dion, V., Chariot, A., and Franzen, R. (2017). Molecular Mechanisms involved in schwann cell plasticity. Front. Mol. Neurosci. 10:38. doi: 10.3389/fnmol.2017.00038
Boerkoel, C. F., Takashima, H., and Lupski, J. R. (2002). The genetic convergence of Charcot-Marie-Tooth disease types 1 and 2 and the role of genetics in sporadic neuropathy. Curr. Neurol. Neurosci. Rep. 2, 70–77. doi: 10.1007/s11910-002-0056-8
Bortolozzi, M. (2018). What’s the function of connexin 32 in the peripheral nervous system? Front. Mol. Neurosci. 11:227. doi: 10.3389/fnmol.2018.00227
Bruzzone, S., Guida, L., Zocchi, E., Franco, L., and De Flora, A. (2001). Connexin 43 hemi channels mediate Ca2+-regulated transmembrane NAD+ fluxes in intact cells. FASEB J. 15, 10–12. doi: 10.1096/fj.00-0566fje
Bruzzone, R., White, T. W., Scherer, S. S., Fischbeck, K. H., and Paul, D. L. (1994). Null mutations of connexin32 in patients with X-linked Charcot-Marie-Tooth disease. Neuron 13, 1253–1260. doi: 10.1016/0896-6273(94)90063-9
Carrer, A., Leparulo, A., Crispino, G., Ciubotaru, C. D., Marin, O., Zonta, F., et al. (2018). Cx32 hemichannel opening by cytosolic Ca2+ is inhibited by the R220X mutation that causes Charcot-Marie-Tooth disease. Hum. Mol. Genet. 27, 80–94. doi: 10.1093/hmg/ddx386
Chandross, K. J., Kessler, J. A., Cohen, R. I., Simburger, E., Spray, D. C., Bieri, P., et al. (1996a). Altered connexin expression after peripheral-nerve injury. Mol. Cell. Neurosci. 7, 501–518. doi: 10.1006/mcne.1996.0036
Chandross, K. J., Spray, D. C., Cohen, R. I., Kumar, N. M., Kremer, M., Dermietzel, R., et al. (1996b). TNF α inhibits Schwann cell proliferation, connexin46 expression, and gap junctional communication. Mol. Cell. Neurosci. 7, 479–500. doi: 10.1006/mcne.1996.0035
Chen, M. S., Kim, H., Jagot-Lacoussiere, L., and Maurel, P. (2016). Cadm3 (Necl-1) interferes with the activation of the PI3 kinase/Akt signaling cascade and inhibits Schwann cell myelination in vitro. Glia 64, 2247–2262. doi: 10.1002/glia.23072
Fehmi, J., Scherer, S. S., Willison, H. J., and Rinaldi, S. (2018). Nodes, paranodes and neuropathies. J. Neurol. Neurosurg. Psychiatry 89, 61–71. doi: 10.1136/jnnp-2016-315480
Fernandez-Moran, H., and Finean, J. B. (1957). Electron microscope and low-angle x-ray diffraction studies of the nerve myelin sheath. J. Biophys. Biochem. Cytol. 3, 725–748. doi: 10.1083/jcb.3.5.725
Fischbeck, K. H., Deschênes, S. M., Bone, L. J., and Scherer, S. S. (1996). Connexin32 and X-linked Charcot-Marie-Tooth disease. Cold Spring Harb. Symp. Quant. Biol. 61, 673–677. doi: 10.1101/SQB.1996.061.01.067
Freidin, M., Asche, S., Bargiello, T. A., Bennett, M. V., and Abrams, C. K. (2009). Connexin 32 increases the proliferative response of Schwann cells to neuregulin-1 (Nrg1). Proc. Natl. Acad. Sci. U S A 106, 3567–3572. doi: 10.1073/pnas.0813413106
Fridman, V., Bundy, B., Reilly, M. M., Pareyson, D., Bacon, C., Burns, J., et al. (2015). CMT subtypes and disease burden in patients enrolled in the Inherited Neuropathies Consortium natural history study: a cross-sectional analysis. J. Neurol. Neurosurg. Psychiatry 86, 873–878. doi: 10.1136/jnnp-2014-308826
Friede, R. L., and Bischhausen, R. (1980). The precise geometry of large internodes. J. Neurol. Sci. 48, 367–381. doi: 10.1016/0022-510X(80)90109-4
Friede, R. L., and Hu, K. H. (1967). Increase in cholesterol along human optic nerve. J. Neurochem. 14, 307–315. doi: 10.1111/j.1471-4159.1967.tb09528.x
Friede, R. L., and Samorajski, T. (1967). Relation between the number of myelin lamellae and axon circumference in fibers of vagus and sciatic nerves of mice. J. Comp. Neurol. 130, 223–231. doi: 10.1002/cne.901300304
Hahn, A. F., Brown, W. F., Koopman, W. J., and Feasby, T. E. (1990). X-linked dominant hereditary motor and sensory neuropathy. Brain 113, 1511–1525. doi: 10.1093/brain/113.5.1511
Hannila, S. S., Siddiq, M. M., and Filbin, M. T. (2007). Therapeutic approaches to promoting axonal regeneration in the adult mammalian spinal cord. Int. Rev. Neurobiol. 77, 57–105. doi: 10.1016/s0074-7742(06)77003-9
Hayashi, A., Moradzadeh, A., Tong, A., Wei, C., Tuffaha, S. H., Hunter, D. A., et al. (2008). Treatment modality affects allograft-derived Schwann cell phenotype and myelinating capacity. Exp. Neurol. 212, 324–336. doi: 10.1016/j.expneurol.2008.04.018
Hernandez, V. H., Bortolozzi, M., Pertegato, V., Beltramello, M., Giarin, M., Zaccolo, M., et al. (2007). Unitary permeability of gap junction channels to second messengers measured by FRET microscopy. Nat. Methods 4, 353–358. doi: 10.1038/nmeth1031
Huttner, I. G., Kennerson, M. L., Reddel, S. W., Radovanovic, D., and Nicholson, G. A. (2006). Proof of genetic heterogeneity in X-linked Charcot-Marie-Tooth disease. Neurology 67, 2016–2021. doi: 10.1212/01.wnl.0000247271.40782.b7
Jeng, L. J., Balice-Gordon, R. J., Messing, A., Fischbeck, K. H., and Scherer, S. S. (2006). The effects of a dominant connexin32 mutant in myelinating Schwann cells. Mol. Cell. Neurosci. 32, 283–298. doi: 10.1016/j.mcn.2006.05.001
Jessen, K. R., and Mirsky, R. (2005). The origin and development of glial cells in peripheral nerves. Nat. Rev. Neurosci. 6, 671–682. doi: 10.1038/nrn1746
Jiang, J. X., and Goodenough, D. A. (1996). Heteromeric connexons in lens gap junction channels. Proc. Natl. Acad. Sci. U S A 93, 1287–1291. doi: 10.1073/pnas.93.3.1287
Kagiava, A., Karaiskos, C., Richter, J., Tryfonos, C., Lapathitis, G., Sargiannidou, I., et al. (2018). Intrathecal gene therapy in mouse models expressing CMT1X mutations. Hum. Mol. Genet. 27, 1460–1473. doi: 10.1093/hmg/ddy056
Kagiava, A., Sargiannidou, I., Theophilidis, G., Karaiskos, C., Richter, J., Bashiardes, S., et al. (2016). Intrathecal gene therapy rescues a model of demyelinating peripheral neuropathy. Proc. Natl. Acad. Sci. U S A 113, E2421–E2429. doi: 10.1073/pnas.1522202113
Kamholz, J., Menichella, D., Jani, A., Garbern, J., Lewis, R. A., Krajewski, K. M., et al. (2000). Charcot-Marie-Tooth disease type 1: molecular pathogenesis to gene therapy. Brain 123, 222–233. doi: 10.1093/brain/123.2.222
Keller, M. P., and Chance, P. F. (1999). Inherited peripheral neuropathy. Semin. Neurol. 19, 353–362. doi: 10.1055/s-2008-1040850
Kidd, G. J., Ohno, N., and Trapp, B. D. (2013). Biology of Schwann cells. Handb. Clin. Neurol. 115, 55–79. doi: 10.1016/B978-0-444-52902-2.00005-9
Kirschner, D. A., and Sidman, R. L. (1976). X-ray diffraction study of myelin structure in immature and mutant mice. Biochim. Biophys. Acta 448, 73–87. doi: 10.1016/0005-2736(76)90077-8
Kleopa, K. A., Abrams, C. K., and Scherer, S. S. (2012). How do mutations in GJB1 cause X-linked Charcot-Marie-Tooth disease? Brain Res. 1487, 198–205. doi: 10.1016/j.brainres.2012.03.068
Kleopa, K. A., Orthmann-Murphy, J., and Sargiannidou, I. (2010). Gap junction disorders of myelinating cells. Rev. Neurosci. 21, 397–419. doi: 10.1515/revneuro.2010.21.5.397
Koval, M., Molina, S. A., and Burt, J. M. (2014). Mix and match: investigating heteromeric and heterotypic gap junction channels in model systems and native tissues. FEBS Lett. 588, 1193–1204. doi: 10.1016/j.febslet.2014.02.025
Li, J., Habbes, H. W., Eiberger, J., Willecke, K., Dermietzel, R., and Meier, C. (2007). Analysis of connexin expression during mouse Schwann cell development identifies connexin29 as a novel marker for the transition of neural crest to precursor cells. Glia 55, 93–103. doi: 10.1002/glia.20427
Li, X., Lynn, B. D., Olson, C., Meier, C., Davidson, K. G., Yasumura, T., et al. (2002). Connexin29 expression, immunocytochemistry and freeze-fracture replica immunogold labelling (FRIL) in sciatic nerve. Eur. J. Neurosci. 16, 795–806. doi: 10.1046/j.1460-9568.2002.02149.x
Lupski, J. R. (1999). Charcot-Marie-Tooth polyneuropathy: duplication, gene dosage and genetic heterogeneity. Pediatr. Res. 45, 159–165. doi: 10.1203/00006450-199902000-00001
Mambetisaeva, E. T., Gire, V., and Evans, W. H. (1999). Multiple connexin expression in peripheral nerve, Schwann cells, and Schwannoma cells. J. Neurosci. Res. 57, 166–175. doi: 10.1002/(sici)1097-4547(19990715)57:2<166::aid-jnr2>3.3.co;2-p
Martini, R., and Carenini, S. (1998). Formation and maintenance of the myelin sheath in the peripheral nerve: roles of cell adhesion molecules and the gap junction protein connexin 32. Microsc. Res. Tech. 41, 403–415. doi: 10.1002/(sici)1097-0029(19980601)41:5<403::aid-jemt7>3.3.co;2-#
Maurel, P., Einheber, S., Galinska, J., Thaker, P., Lam, I., Rubin, M. B., et al. (2007). Nectin-like proteins mediate axon Schwann cell interactions along the internode and are essential for myelination. J. Cell Biol. 178, 861–874. doi: 10.1083/jcb.200705132
Meier, C., Dermietzel, R., Davidson, K. G., Yasumura, T., and Rash, J. E. (2004). Connexin32-containing gap junctions in Schwann cells at the internodal zone of partial myelin compaction and in Schmidt-Lanterman incisures. J. Neurosci. 24, 3186–3198. doi: 10.1523/JNEUROSCI.5146-03.2004
Mugnaini, E., Osen, K. K., Schnapp, B., and Friedrich, V. L. Jr. (1977). Distribution of Schwann cell cytoplasm and plasmalemmal vesicles (caveolae) in peripheral myelin sheaths. An electron microscopic study with thin sections and freeze-fracturing. J. Neurocytol. 6, 647–668. doi: 10.1007/bf01176378
Nedergaard, M., Ransom, B., and Goldman, S. A. (2003). New roles for astrocytes: redefining the functional architecture of the brain. Trends Neurosci. 26, 523–530. doi: 10.1016/j.tins.2003.08.008
Nelson, C. E., Hakim, C. H., Ousterout, D. G., Thakore, P. I., Moreb, E. A., Castellanos Rivera, R. M., et al. (2016). In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science 351, 403–407. doi: 10.1126/science.aad5143
Nicholson, G., and Nash, J. (1993). Intermediate nerve conduction velocities define X-linked Charcot-Marie-Tooth neuropathy families. Neurology 43, 2558–2564. doi: 10.1212/wnl.43.12.2558
Nualart-Marti, A., del Molino, E. M., Grandes, X., Bahima, L., Martin-Satué, M., Puchal, R., et al. (2013). Role of connexin 32 hemichannels in the release of ATP from peripheral nerves. Glia 61, 1976–1989. doi: 10.1002/glia.22568
Ouvrier, R., Geevasingha, N., and Ryan, M. M. (2007). Autosomal-recessive and X-linked forms of hereditary motor and sensory neuropathy in childhood. Muscle Nerve 36, 131–143. doi: 10.1002/mus.20776
Pareyson, D., and Marchesi, C. (2009). Diagnosis, natural history, and management of Charcot-Marie-Tooth disease. Lancet Neurol. 8, 654–667. doi: 10.1016/s1474-4422(09)70110-3
Poliak, S., and Peles, E. (2003). The local differentiation of myelinated axons at nodes of Ranvier. Nat. Rev. Neurosci. 4, 968–980. doi: 10.1038/nrn1253
Rash, J. E. (2010). Molecular disruptions of the panglial syncytium block potassium siphoning and axonal saltatory conduction: pertinence to neuromyelitis optica and other demyelinating diseases of the central nervous system. Neuroscience 168, 982–1008. doi: 10.1016/j.neuroscience.2009.10.028
Rash, J. E., Vanderpool, K. G., Yasumura, T., Hickman, J., Beatty, J. T., and Nagy, J. I. (2016). KV1 channels identified in rodent myelinated axons, linked to Cx29 in innermost myelin: support for electrically active myelin in mammalian saltatory conduction. J. Neurophysiol. 115, 1836–1859. doi: 10.1152/jn.01077.2015
Sáez, J. C., Berthoud, V. M., Branes, M. C., Martinez, A. D., and Beyer, E. C. (2003a). Plasma membrane channels formed by connexins: their regulation and functions. Physiol. Rev. 83, 1359–1400. doi: 10.1152/physrev.00007.2003
Sáez, J. C., Contreras, J. E., Bukauskas, F. F., Retamal, M. A., and Bennett, M. V. (2003b). Gap junction hemichannels in astrocytes of the CNS. Acta Physiol Scand. 179, 9–22. doi: 10.1046/j.1365-201x.2003.01196.x
Sáez, J. C., Retamal, M. A., Basilio, D., Bukauskas, F. F., and Bennett, M. V. (2005). Connexin-based gap junction hemichannels: gating mechanisms. Biochim. Biophys. Acta 1711, 215–224. doi: 10.1016/j.bbamem.2005.01.014
Saher, G., and Simons, M. (2010). Cholesterol and myelin biogenesis. Subcell. Biochem. 51, 489–508. doi: 10.1007/978-90-481-8622-8_18
Sandri, C., Van Buren, J. M., and Akert, K. (1977). Membrane morphology of the vertebrate nervous system. A study with freeze-etch technique. Prog. Brain Res. 46, 1–384.
Saporta, M. A. (2014). Charcot-Marie-Tooth disease and other inherited neuropathies. Continuum 20, 1208–1225. doi: 10.1212/01.con.0000455885.37169.4c
Sargiannidou, I., Vavlitou, N., Aristodemou, S., Hadjisavvas, A., Kyriacou, K., Scherer, S. S., et al. (2009). Connexin32 mutations cause loss of function in Schwann cells and oligodendrocytes leading to PNS and CNS myelination defects. J. Neurosci. 29, 4736–4749. doi: 10.1523/JNEUROSCI.0325-09.2009
Scherer, S. S., Deschenes, S. M., Xu, Y. T., Grinspan, J. B., Fischbeck, K. H., and Paul, D. L. (1995). Connexin32 is a myelin-related protein in the PNS and CNS. J. Neurosci. 15, 8281–8294. doi: 10.1523/JNEUROSCI.15-12-08281.1995
Scherer, S. S., Xu, Y. T., Nelles, E., Fischbeck, K., Willecke, K., and Bone, L. J. (1998). Connexin32-null mice develop demyelinating peripheral neuropathy. Glia 24, 8–20. doi: 10.1002/(sici)1098-1136(199809)24:1<8::aid-glia2>3.0.co;2-3
Söhl, G., and Willecke, K. (2003). An update on connexin genes and their nomenclature in mouse and man. Cell Commun Adhes. 10, 173–180. doi: 10.1080/714040423
Sosinsky, G. E. (1996). Molecular organization of gap junction membrane channels. J. Bioenerg. Biomembr. 28, 297–309. doi: 10.1007/bf02110106
Spray, D. C., and Dermietzel, R. (1995). X-linked dominant Charcot-Marie-Tooth disease and other potential gap-junction diseases of the nervous system. Trends Neurosci. 18, 256–262. doi: 10.1016/0166-2236(95)93911-g
Tabebordbar, M., Zhu, K., Cheng, J. K. W., Chew, W. L., Widrick, J. J., Yan, W. X., et al. (2016). In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science 351, 407–411. doi: 10.1126/science.aad5177
Tetzlaff, W. (1982). Tight junction contact events and temporary gap junctions in the sciatic nerve fibres of the chicken during Wallerian degeneration and subsequent regeneration. J. Neurocytol. 11, 839–858. doi: 10.1007/bf01153522
Trapp, B. D. (1990). Myelin-associated glycoprotein. Location and potential functions. Ann. N Y Acad. Sci. 605, 29–43. doi: 10.1111/j.1749-6632.1990.tb42378.x
Traub, R. D., Whittington, M. A., Gutiérrez, R., and Draguhn, A. (2018). Electrical coupling between hippocampal neurons: contrasting roles of principal cell gap junctions and interneuron gap junctions. Cell Tissue Res. 373, 671–691. doi: 10.1007/s00441-018-2881-3
Unger, V. M., Kumar, N. M., Gilula, N. B., and Yeager, M. (1997). Projection structure of a gap junction membrane channel at 7 A resolution. Nat. Struct. Biol. 4, 39–43. doi: 10.1038/nsb0197-39
Wang, N., De Bock, M., Decrock, E., Bol, M., Gadicherla, A., Vinken, M., et al. (2013). Paracrine signaling through plasma membrane hemichannels. Biochim. Biophys. Acta 1828, 35–50. doi: 10.1016/j.bbamem.2012.07.002
Weber, P. A., Chang, H. C., Spaeth, K. E., Nitsche, J. M., and Nicholson, B. J. (2004). The permeability of gap junction channels to probes of different size is dependent on connexin composition and permeant-pore affinities. Biophys. J. 87, 958–973. doi: 10.1529/biophysj.103.036350
Ye, Z. C., Wyeth, M. S., Baltan-Tekkok, S., and Ransom, B. R. (2003). Functional hemichannels in astrocytes: a novel mechanism of glutamate release. J. Neurosci. 23, 3588–3596. doi: 10.1523/JNEUROSCI.23-09-03588.2003
Yiu, E. M., Geevasinga, N., Nicholson, G. A., Fagan, E. R., Ryan, M. M., and Ouvrier, R. A. (2011). A retrospective review of X-linked Charcot-Marie-Tooth disease in childhood. Neurology 76, 461–466. doi: 10.1212/WNL.0b013e31820a0ceb
Yoshimura, T., Satake, M., and Kobayashi, T. (1996). Connexin43 is another gap junction protein in the peripheral nervous system. J. Neurochem. 67, 1252–1258. doi: 10.1046/j.1471-4159.1996.67031252.x
Keywords: connexins, gap junction channels, myelin sheath, Schmidt-Lanterman incisure, Charcot-Marie-Tooth disease, CMTX1
Citation: Cisterna BA, Arroyo P and Puebla C (2019) Role of Connexin-Based Gap Junction Channels in Communication of Myelin Sheath in Schwann Cells. Front. Cell. Neurosci. 13:69. doi: 10.3389/fncel.2019.00069
Received: 15 September 2018; Accepted: 12 February 2019;
Published: 01 March 2019.
Edited by:
Marion Baraban, University of Edinburgh, United KingdomReviewed by:
Mario Bortolozzi, University of Padua, ItalyJohn E. Rash, Colorado State University, United States
Georg Zoidl, York University, Canada
Copyright © 2019 Cisterna, Arroyo and Puebla. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bruno A. Cisterna, YmNpc3Rlcm5hQHVjLmNs
Carlos Puebla, Y2FybG9zLnB1ZWJsYUB1YXV0b25vbWEuY2w=