 Jong M. Rho
Jong M. Rho Li-Rong Shao
Li-Rong Shao Carl E. Stafstrom
Carl E. Stafstrom- 1Section of Pediatric Neurology, Department of Pediatrics, Alberta Children’s Hospital Research Institute, Hotchkiss Brain Institute, Cumming School of Medicine, University of Calgary, Calgary, AB, Canada
- 2Department of Clinical Neurosciences, Alberta Children’s Hospital Research Institute, Hotchkiss Brain Institute, Cumming School of Medicine, University of Calgary, Calgary, AB, Canada
- 3Department of Physiology and Pharmacology, Alberta Children’s Hospital Research Institute, Hotchkiss Brain Institute, Cumming School of Medicine, University of Calgary, Calgary, AB, Canada
- 4Division of Pediatric Neurology, Department of Neurology, Johns Hopkins University School of Medicine, Baltimore, MD, United States
Current anti-seizure drugs (ASDs) are believed to reduce neuronal excitability through modulation of ion channels and transporters that regulate excitability at the synaptic level. While most patients with epilepsy respond to ASDs, many remain refractory to medical treatment but respond favorably to a high-fat, low-carbohydrate metabolism-based therapy known as the ketogenic diet (KD). The clinical effectiveness of the KD has increasingly underscored the thesis that metabolic factors also play a crucial role in the dampening neuronal hyperexcitability that is a hallmark feature of epilepsy. This notion is further amplified by the clinical utility of other related metabolism-based diets such as the modified Atkins diet and the low-glycemic index treatment (LGIT). Traditional high-fat diets are characterized by enhanced fatty acid oxidation (which produces ketone bodies such as beta-hydroxybutyrate) and a reduction in glycolytic flux, whereas the LGIT is predicated mainly on the latter observation of reduced blood glucose levels. As dietary implementation is not without challenges regarding clinical administration and patient compliance, there is an inherent desire and need to determine whether specific metabolic substrates and/or enzymes might afford similar clinical benefits, hence validating the concept of a “diet in a pill.” Here, we discuss the evidence for one glycolytic inhibitor, 2-deoxyglucose (2DG) and one metabolic substrate, β-hydroxybutyrate (BHB) exerting direct effects on neuronal excitability, highlight their mechanistic differences, and provide the strengthening scientific rationale for their individual or possibly combined use in the clinical arena of seizure management.
Key Points
• Cellular metabolism plays a key role in the modulation of neuronal excitability.
• 2-Deoxyglucose (2DG), an inhibitor of glycolysis, abrogates seizure activity and retards epilepsy progression both in vitro and in vivo.
• Beta-hydroxybutyrate (BHB), a by-product of fatty acid oxidation, exerts both anti-seizure and neuroprotective effects.
• 2DG and BHB offer potential novel avenues for suppressing seizure activity and possibly epileptogenesis.
Introduction
For decades, the mechanistic focus in the field of epilepsy research has been on ion channels and transporters that regulate neuronal excitability at the synapse. Indeed, virtually all anti-seizure drugs (ASDs) are believed to exert their clinical effects on synaptic targets, whether at the pre- or post-synaptic terminals (Rogawski et al., 2016). However, it has become increasingly clear that metabolic factors can potently affect neuronal excitability and even influence the activity of key cellular membrane-bound ion channels such as inhibitory adenosine and GABAA receptors and ATP-sensitive potassium (KATP) channels (Reid et al., 2014; Bazzigaluppi et al., 2017). The steadily increasing scientific arena of metabolic control of neuronal excitability has been catalyzed by the clinical success and evidence-based validation of the high-fat, low-carbohydrate ketogenic diet (KD) and its variants [i.e., medium-chain triglyceride (MCT), modified Atkins diet (MAD), and low-glycemic index treatment (LGIT)] (Neal et al., 2008; Rho and Stafstrom, 2012; Lutas and Yellen, 2013; Gano et al., 2014). While the fundamental mechanisms underlying the efficacy of these metabolic treatments across numerous epileptic conditions remain unclear, there is growing evidence that specific metabolic substrates and enzymes may act directly or indirectly to induce anti-seizure (and even neuroprotective) effects. Moreover, substrate or pharmacological modulation of key biochemical pathways has shown similar benefits (Rogawski et al., 2016).
Inhibition of Glycolysis
The KD originated from the centuries-old observation that fasting led to seizure control and was thus formulated to mimic the physiological effects of fasting (Gano et al., 2014). Clinicians also noted that seizure control while on the KD could be abruptly lost when carbohydrates, which would break the ketosis, were ingested (Huttenlocher, 1976). This observation led to the general hypothesis that carbohydrate restriction alone might protect against seizure activity (Greene et al., 2003). Further amplifying this idea was the growing notion that reducing total calorie intake per se could also suppress seizures and provide neuroprotection (Greene et al., 2003; Ingram and Roth, 2011; Yuen and Sander, 2014; Pani, 2015).
Glucose is an obligate energy source for the brain, which is a highly energy-dependent organ, consuming approximately 20% of the body’s total caloric requirements at rest (Magistretti and Allaman, 2015). Seizure activity places further demands on the overall brain metabolic milieu due to excessive neuronal activity – reflected by the aberrant high-voltage activity seen from single neurons to brain networks using microelectrodes and extracellular field and surface scalp electrodes. Neurometabolic coupling during seizure activity not only depends on energy metabolism of neurons, but may also involve astrocytes as they may provide neurons with fuel (i.e., lactate) through the lactate shuttle (Cloix and Hévor, 2009; Magistretti and Allaman, 2015; Boison and Steinhäuser, 2018, but see Dienel, 2017). In addition, brain microvasculature integrity is of paramount importance in supporting the neurometabolic fluctuations required to enable neuronal excitability (Librizzi et al., 2018). Not surprisingly then, deficits in glucose availability and usage have been linked to several neurological disorders (Mergenthaler et al., 2013). By contrast, enhanced neuronal activity, such as during epileptic seizures, significantly increases regional blood glucose utilization, as shown by human positron emission tomography (PET) studies (Cendes et al., 2016), thus suggesting a rationale for potential seizure control through metabolic interventions.
2-Deoxyglucose, A Glycolysis Inhibitor
As mentioned above, the KD mimics fasting in restricting the intake of the main source of brain energy (i.e., carbohydrates) while supplying fat and protein to generate ketone bodies as an alternative energy source. While the mechanisms of seizure control by the KD are likely to be multi-faceted (Kawamura et al., 2016), it is important to note that the KD bypasses glycolysis, and an intake of even a small amount of sugar quickly reverses its otherwise seizure-stabilizing effects (Huttenlocher, 1976). This suggests that energy production by glycolysis may be important for seizure activity and bypassing or suppressing glycolysis may represent a key mechanism involved in KD treatment. Collectively, these observations provide the rationale for the notion that inhibitors of glycolysis may mimic in part the therapeutic effects of the KD. It is also well known that ketolysis itself decreases glycolytic flux, and it has been proposed that ketone bodies attenuate neuronal cellular excitability through this mechanism (Lutas and Yellen, 2013). As there are known agents that restrict glycolytic flux, this overarching hypothesis is eminently testable.
One promising glycolysis inhibitor for seizure protection is the glucose analog 2-deoxyglucose (2DG) which differs from glucose by the substitution of oxygen from the 2′ position (Figure 1). Similar to glucose, 2DG is transported into cells and is phosphorylated to 2DG-6-phosphate at the 6′ position by hexokinase (HK), but this phosphorylated substrate cannot be converted to fructose-6-phosphate by phosphoglucose isomerase (PGI), and is thus trapped in the cell. The accumulation of 2DG-6-phosphate competitively inhibits the rate-limiting enzymes, primarily PGI (Wick et al., 1957) but also HK (Pelicano et al., 2006), hence partially blocking glycolysis. In addition, inhibition of PGI would divert glycolysis to the pentose phosphate pathway (PPP), producing ribulose and glutathione. It should be kept in mind that 2DG, like glucose, is not only taken up by neurons (via glucose transporter 3) but is also taken up by glial cells (via glucose transporter 1), inhibiting astrocytic glycolysis. Recent studies hypothesize that astrocytes may transport their glycolytic end-product, lactate, as an alternative fuel source to neurons through the “astrocyte-neuron lactate shuttle” (ANLS) (Pellerin and Magistretti, 1994, but see Dienel, 2017). Therefore, 2DG may potentially affect neuronal activity indirectly by suppressing astrocytic glycolysis. This biochemical feature has been successfully exploited to identify energetically active cells, notably hyperexcitable brain cells or rapidly dividing cancer cells (Pelicano et al., 2006; Cheong et al., 2011). Cancer cells, even in aerobic conditions, tend to use glycolysis for energy production over oxidative phosphorylation (Warburg effect); 2DG enhances oxidative phosphorylation, countering the Warburg effect (de Padua et al., 2017). For decades, fluorinated 2DG (18F-2DG) has been safely used as a tracer to reveal regional glucose utilization by PET (Wree, 1990; Cendes et al., 2016).
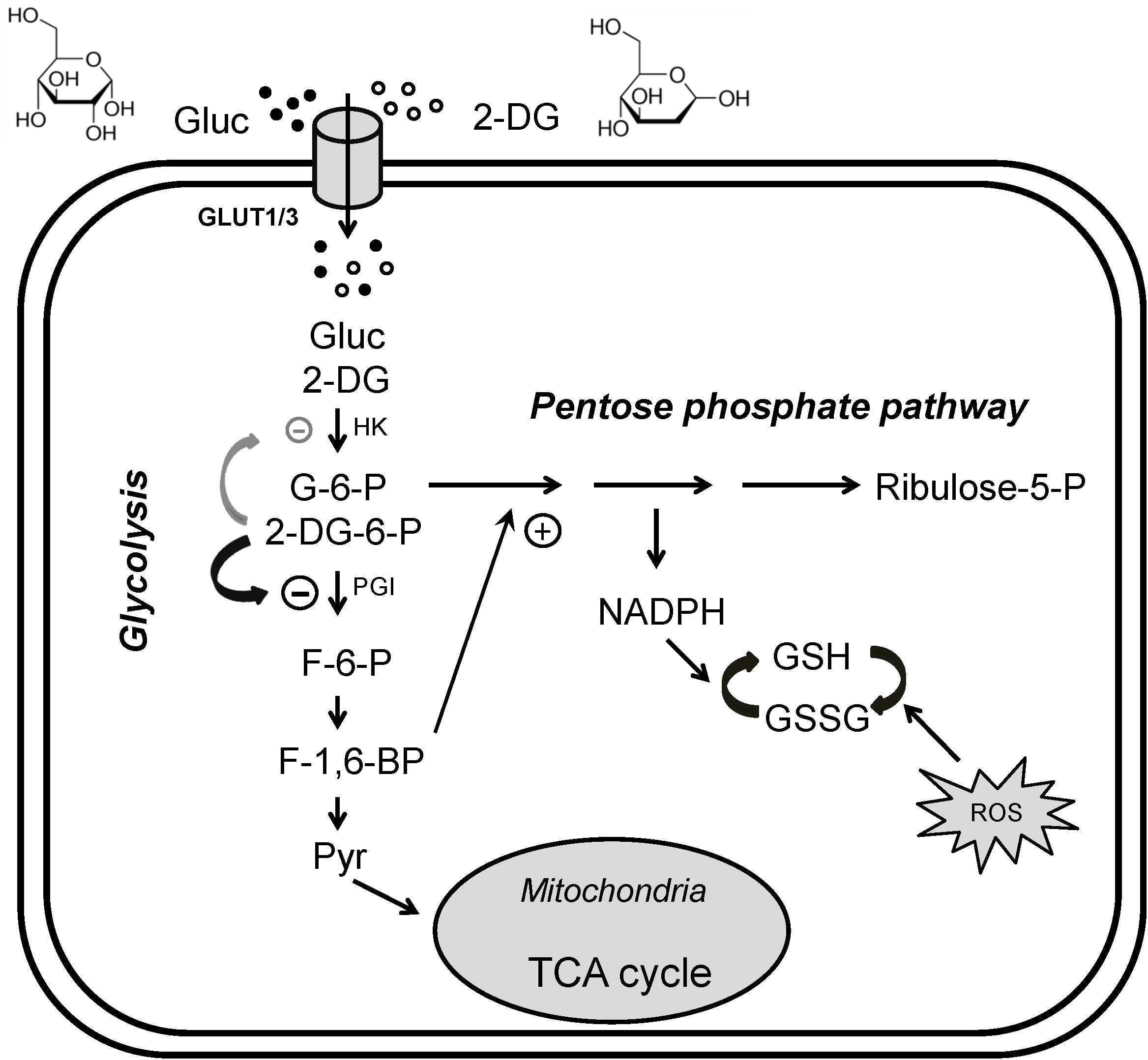
Figure 1. Glycolytic inhibition via 2-deoxyglucose (2DG). 2DG decreases glycolytic flux, reduces cell energy production and produces anti-seizure effects. Glucose (Gluc) and 2DG enter the cytoplasm via glucose transporters (GLUT3 in neurons or GLUT1 in glia). During glycolysis, glucose is first phosphorylated to glucose-6-phosphate (G-6-P), which is then converted into fructose-6-phosphate (F-6-P). F-6-P is converted into fructose-1,6-bisphosphate (FBP) and ultimately to pyruvate via multiple steps. Pyruvate is the final product of glycolysis; it enters mitochondria to participate in the tricarboxylic acid (TCA) cycle for oxidative ATP production. G-6-P also enters the pentose phosphate pathway (PPP), generating reduced nicotinamide adenine dinucleotide phosphate (NADPH) and glutathione (GSH), which attenuates cell damage caused by reactive oxygen species (ROS). In addition, GSH may have antiseizure properties. Once inside the cell, 2DG is phosphorylated to 2DG-6-P, which cannot be further metabolized. 2DG-6-P is trapped and accumulates in the cell, competitively inhibiting phosphoglucose isomerase (PGI, primary effect) and hexokinase (HK), limiting the conversion of G-6-P to F-6-P and glucose to G-6-P, two of the rate-limiting steps in glycolysis. Increased FBP (e.g., by exogenous administration) may also enhance PPP activity and produce an antiseizure effect (plus sign). The chemical structures of glucose and 2DG are indicated at the top of the figure. Modified from Shao et al. (2018), with permission from Epilepsia Open.
With respect to epilepsy, 2DG has been shown to exert broad activity in several in vitro and in vivo models of acute seizures (Stafstrom et al., 2009). In vitro, 2DG attenuates high-frequency epileptiform bursting in the high extracellular potassium model, and in response to two chemoconvulsants – 4-aminopyridine (4-AP, a non-selective potassium channel blocker) and bicuculline [a γ-aminobutyric acid (GABAA) receptor antagonist] (Stafstrom et al., 2009). 2DG was also effective in hippocampal brain slices whether synaptic activity in CA3 pyramidal neurons was left intact or blocked by AMPA, NMDA, and GABAA receptor antagonists (DNQX, APV, and bicuculline, respectively) (Shao and Stafstrom, 2017). Interestingly, CA3 neuronal firing persisted even after intracellular administration of 2DG, suggesting that glycolytic inhibition of individual neurons is not sufficient to stop neuronal firing. Indeed, bath application of 2DG in hippocampal slices abolished epileptiform bursts induced by a Mg2+-free medium containing 4-AP in a dose-dependent manner (Shao and Stafstrom, 2017). Collectively, these results indicate that 2DG can markedly block epileptiform activity in both interictal and ictal states. Recently, however, a study reported that 2DG reduced Cs+- and bicuculline-induced interictal-like activity while increasing ictal-like activity in rat hippocampal slices (Nedergaard and Andreasen, 2018). The precise reasons for this discrepancy are unclear. Possibly, in most of the aforementioned in vitro seizure models, either K+ channel or GABAergic inhibition is unaffected, whereas in the model used by Nedergaard and Andreasen (2018), both inhibitory mechanisms are blocked, which may hamper seizure termination.
The anti-seizure effects of 2DG were also demonstrated in different types of animal models of seizures. 2DG increased the mean after-discharge threshold in the rat perforant path kindling model (Garriga-Canut et al., 2006; Stafstrom et al., 2009). Further, 2DG blocked seizure activity in the 6-Hz corneal stimulation model (Barton et al., 2001; Stafstrom et al., 2009; Gasior et al., 2010) and in Frings audiogenic seizure-susceptible mice (Stafstrom et al., 2009). In addition, 2DG significantly delayed seizure onset and diminished seizure severity and duration in the pilocarpine model, and delayed seizure onset in the kainate (KA) model (Lian et al., 2007). In contrast, 2DG was unable to protect against seizures induced by pentylenetetrazol (PTZ) or maximal electroshock (MES) (Gasior et al., 2010). While 2DG’s spectrum of activity was not universal in some standard experimental models, its profile has certainly been shown to be unique compared to existing anti-seizure medications. Another important aspect of 2DG function deserves mention – 2DG is mostly taken up by cells that are metabolically active. This use-dependence affords a distinct advantage for any therapeutic strategy aimed at targets in brain areas exhibiting hyperexcitable and hypersynchronous discharge, as seen with epileptic seizures.
The acute and chronic effects of 2DG in different model systems are likely due to distinct cellular and molecular mechanisms. With respect to anti-epileptogenesis as reflected in the rat kindling model, the most intriguing mechanism reported is 2DG’s actions in decreasing brain-derived neurotrophic factor (BDNF) and its receptor trkB (Garriga-Canut et al., 2006), both of which are necessary for kindling to occur (He et al., 2004). Specifically, these changes are mediated by the transcriptional repressor Neuron Restrictive Silencing Factor (NRSF) and its co-repressor, the nicotinamide adenine dinucleotide hydride (NADH)-sensitive carboxy-terminal binding protein (CtBP), which together target the promoter regions of both Bdnf and TrkB genes (Garriga-Canut et al., 2006). There is growing evidence that repression of BDNF and trkB retards kindling progression, and this may be the key mechanism underlying the anti-epileptogenic effects of 2DG (Garriga-Canut et al., 2006; Huang and McNamara, 2006; Hu et al., 2011). An additional mechanism implicated in 2DG action is upregulation of mRNAs of the ATP-sensitive potassium (KATP) channel subunits Kir6.1 and Kir6.2 (Yang et al., 2013). Plasmalemmal KATP channels, when open and thus allowing potassium efflux, hyperpolarize the membrane. 2DG may also modulate GABAergic signaling, specifically by potentiating extra-synaptic tonic GABAergic currents through activation of neurosteroidogenesis (Forte et al., 2016).
Other than its anti-seizure activity, 2DG has also been shown to be neuroprotective. Specifically, 2DG protected against the deleterious oxidative and metabolic effects of kainic acid in rat hippocampal cell cultures by enhancing stress responses (Lee et al., 1999) and reduced seizure occurrence in the mouse bilateral carotid artery occlusion model (Redjak et al., 2001). These neuroprotective actions may result from possible enhancement of PPP flux which provides endogenous antioxidant glutathione, but also appear to involve mechanisms distinct from direct glycolytic restriction, including reduced oxidative stress through ketogenesis and sustaining mitochondrial function (Yao et al., 2011) and activation of adenosine monophosphate (AMP)-activated protein kinase which confer resistance to oxidative stress (Park et al., 2009).
Notably, other researchers reported that 2DG might exert pro-seizure/pro-epileptogenic effects under certain conditions, which adds to the complexity of 2DG action. One study observed that the latency to seizure onset was decreased after 2DG administration in the PTZ and kainate models (but prolonged in the 6 Hz model) (Gasior et al., 2010). While the mechanism(s) underlying these findings remain(s) unclear, 2DG is known to increase cerebral blood flow (Elman et al., 1999), so it is possible that seizure activity might be promoted acutely as initial neuronal and network excitability is fueled, which may confound the interpretation of these findings. More recently, it was reported that daily intracerebroventricular administration of 2DG for 4 weeks resulted in spontaneous seizures in two of ten rats and a depolarized GABA equilibrium potential (EGABA) suggesting a pro-epileptogenic effect, although the acute effects of 2DG were more complex (i.e., decreased synaptic excitability but depolarized membrane potential and EGABA) (Samokhina et al., 2017). It is not known what underlies the discrepancies seen with 2DG in different seizure models, and between acute and chronic treatment protocols with 2DG. Clearly, more studies are needed to resolve these differences and to further understand the effects of 2DG, and more broadly metabolic interventions for seizures/epilepsy.
Ketone Bodies as Anti-Seizure Agents
While the efficacy of the KD in patients with medically intractable epilepsy is no longer debatable (Freeman et al., 1998; Neal et al., 2008, 2009; Lambrechts et al., 2017), the mechanisms underlying its clinical effects remain unclear. Many hypotheses have been advanced, including but not limited to favorable changes in neurotransmitter systems (e.g., GABA, glutamate, and adenosine), glycolytic restriction/diversion, enhancement of tricarboxylic acid (TCA) cycle function, improved cellular bioenergetics and mitochondrial function (with decreased oxidative stress), and direct inhibitory effects of fatty acids (Rogawski et al., 2016).
The hallmark feature of the KD is the production of ketone bodies (KB) [i.e., β-hydroxybutyrate (BHB), acetoactate (ACA) and acetone] through fatty acid oxidation primarily in the liver, and possibly the brain (Aalling et al., 2018). Ordinarily, fatty acids are converted to acetyl-CoA which then enters the TCA cycle. However, when fatty acid levels rise and exceed maximal TCA cycle capacity – as in the case with KD treatment – acetyl-CoA is shunted to ketogenesis. Acetoacetyl-CoA is produced from two acetyl-CoA molecules through a thiolase enzyme and is then is used for the synthesis of ACA which is interconverted with BHB through a dehydrogenase enzyme. Acetone is a spontaneous decarboxylation product of ACA and is eliminated primarily through the lungs and kidneys. BHB and ACA are transported to the brain interstitial space via monocarboxylic acid transporters (MCTs) and enter mitochondria which convert these ketone bodies to acetyl-CoA through several enzymatic steps. The reconstituted acetyl-CoA species are then utilized by the TCA cycle for ATP production. Increased ATP production can then lead to further biochemical and physiological effects that can contribute to seizure control and neuroprotection (see below).
In this regard, a persistent question has been whether ketone bodies are direct mediators; that is, do ketone bodies exert biological effects that attenuate neuronal hyperexcitability and/or synchrony, beyond their classic role as fuel for ATP production? The common belief is that they do not, based on clinical studies demonstrating that blood levels of ketone bodies do not correlate well with seizure control (Gilbert et al., 2000; van Delft et al., 2010). Additionally, the mechanistic role for KB is most seriously challenged when considering the low-glycemic index therapy (LGIT) which is an effective treatment for intractable forms of epilepsy but is not associated with significant elevations in blood ketone levels (Pfeifer and Thiele, 2005). Thus, the LGIT must work through distinct mechanisms that have yet to be fully elucidated but may involve KATP channels and adenosine receptors (Ma et al., 2007; Kawamura et al., 2010).
At least in experimental models, ketosis does not appear to be necessary for seizure control (Thavendiranathan et al., 2000; Dallérac et al., 2017). For example, a MCT KD was unable to block seizures induced by MES, threshold electroconvulsive shock, threshold PTZ and maximal PTZ, despite significant elevations in blood ketone levels (Thavendiranathan et al., 2000). Moreover, a nutritional strategy targeting specific amino acids, fatty acids and carbohydrates constituting a low ketogenic ratio (i.e., the ratio of fat to carbohydrate plus protein by weight) without significant ketosis still resulted in seizure protection in the kainic acid mouse model of epilepsy (Dallérac et al., 2017). It should be noted that the Dallérac et al. (2017) study utilized MCTs (i.e., decanoate) that are known to directly block α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors (Chang et al., 2016). Thus, while various dietary approaches may all work to control epileptic seizures, the underlying mechanisms are likely distinct, making it somewhat difficult to compare these with the classic or even modified KD.
Notwithstanding these clinical and laboratory observations, more recent clinical data suggest otherwise (Kim et al., 2015; Buchhalter et al., 2017) and the growing mechanistic properties of ketone bodies strengthen the biological plausibility that these substrates afford both structural and functional neuroprotection in the epileptic brain. In a prospective pilot clinical study of children with medically intractable epilepsy who were treated with the KD, blood BHB levels determined by tandem mass spectrometry were inversely related to seizure frequency (Buchhalter et al., 2017). This is consistent with an earlier report that blood ketone levels (i.e., BHB) correlated better with seizure control than urine ketone levels (i.e., ACA) (Gilbert et al., 2000).
The earliest laboratory reports of ketone bodies exerting anti-seizure effects came in the early 1930s, shortly after the introduction of the KD in clinical practice (Keith, 1931, 1932, 1933, 1935). Specifically, ACA was shown to protect against seizures induced by thujone, a constituent of wormwood oil later shown to be an antagonist of GABAA receptors (Höld et al., 2000). Decades later, acetone was shown to provide dose-dependent beneficial effects in the MES, subcutaneous PTZ, amygdala kindling, and the AY-9944 (an inhibitor of cholesterol biosynthesis, which evokes atypical absence seizures) models of induced seizures or epileptogenesis (Likhodii et al., 2003). Further, in support of Keith’s original studies, ACA was effective in blocking seizures in a model of sensory-evoked reflex seizures, the Frings audiogenic seizure-susceptible mice (Rho et al., 2002). More recently, several research teams demonstrated that BHB, when administered acutely or chronically, conferred seizure protection in immature rat pups against flurothyl-induced (Minlebaev and Khazipov, 2011) and in the rat betamethasone-NMDA model of infantile spasms (Yum et al., 2015). In another study, using the clinically relevant Kcna1-null mouse model of developmental limbic epilepsy (that lacks the delayed rectifier voltage-gated potassium channel α subunit), Kim et al. (2015) found that BHB afforded the same degree of seizure control as the full KD. Taken together, these and other studies support the hypothesis that KB can themselves induce anti-seizure effects separate from their role in ATP production. Hence, the possibility that these compounds contribute directly to the clinical effects of the KD cannot be readily dismissed.
So, what are the key molecular targets of ketone body action? As KB were historically viewed as metabolic substrates and not direct modulators of synaptic function, it was not surprising that low millimolar concentrations of BHB and ACA did not influence the activity of GABAA receptors, ionotropic glutamate receptors, or voltage-gated sodium channels in normal rodent hippocampus (Thio et al., 2000; Yang et al., 2007), in contrast to our current understanding of how most of clinically approved ASDs work (Rogawski et al., 2016). However, in a more elegant study, it was shown that neuronal excitability could be suppressed by ketone bodies through a pre-synaptic mechanism (Juge et al., 2010). Specifically, BHB and ACA inhibited the presynaptic release of glutamate by directly competing with Cl- at the allosteric modulation site on vesicular glutamate transporters (VGLUTs). These investigators also demonstrated that the proconvulsant effects of 4-AP, accompanied by presynaptic release of glutamate, could be attenuated by ACA (Juge et al., 2010).
The first mechanistic insights into KB action, however, came from neurochemical experiments in the 1990s. Erecinska et al. (1996) and Daikhin and Yudkoff (1998) demonstrated that ketone bodies (i.e., BHB) altered glutamate metabolism within the tripartite neurovascular unit, comprising neurons, astrocytes and the capillary microvasculature. In the ketotic state, the aspartate-to-glutamate ratio is reduced based on a change in the equilibrium of the aspartate aminotransferase reaction. As the transamination of glutamate to aspartate is decreased, glutamate decarboxylation to GABA is increased (Erecinska et al., 1996; Daikhin and Yudkoff, 1998). Thus, increased synaptic levels of GABA would enable increased inhibitory neurotransmission and dampen seizure activity. While this has long stood as a rational and intuitive mechanistic argument, supported by predicted neurochemical changes in certain brain regions but not the key seizure-prone areas such as the neocortex or hippocampus, more direct evidence has not been forthcoming (Yudkoff et al., 2001; Lund et al., 2009, 2011; Valente-Silva et al., 2015; Zhang et al., 2015). Further, the GABAergic hypothesis of ketone body action doesn’t explain why the KD would be effective in blocking seizure activity in patients with epilepsy who were refractory to GABAergic medications (Freeman et al., 2006).
A generation ago, it was speculated that KATP channels might be an important link between cellular energy metabolism and plasmalemmal membrane excitability (Schwartzkroin, 1999). Indeed, it has long been known that KATP channels are activated when the intracellular ATP-to-ADP ratio falls, resulting in efflux of potassium and hence causing membrane hyperpolarization. However, it was not until much later that Ma et al. (2007) demonstrated that ketone bodies influence the activity of KATP channels. Using brain slices from normal and genetically engineered mice that lack KATP channels, they showed that BHB and ACA decreased the spontaneous firing of GABAergic interneurons in the substantia nigra pars reticulata (a subcortical modulator of seizure propagation), and that this effect was dependent on KATP channels and GABAB receptors (Ma et al., 2007). The central thesis has been that ATP levels subjacent to cell membrane-bound KATP channels are depleted due to heightened neuronal excitability during seizure activity which requires energy, and this would lead to activation of inhibitory KATP channels.
Despite its intrinsic appeal, there is yet no direct confirmation of this mechanistic hypothesis. Moreover, the KD and ketone bodies have been shown to increase cellular levels of ATP via enhanced mitochondrial biogenesis and respiration (DeVivo et al., 1978; Bough et al., 2006; Kim et al., 2010). Another explanation for ketone body action on KATP channels was provided by Kawamura et al. (2010) who showed that low-glucose conditions lead to opening of pannexin channels and efflux of ATP through these channels on CA3 pyramidal neurons, subsequent conversion of ATP to adenosine via extracellular ectonucleotidases, and activation of adenosine A1 inhibitory receptors, which are coupled to KATP channels (Kawamura et al., 2010). Thus, BHB might induce a form of autocrine regulation by enhancing ATP synthesis, with one downstream consequence being cellular membrane inhibition.
Other novel targets for ketone body action have been described in recent years. In the study by Kim et al. (2015) mentioned above, BHB-induced block of spontaneous recurrent seizures in Kcna1-null mice was shown to be mediated by an interaction with the mitochondrial permeability transition (mPT) pore, which when activated triggers the release of calcium and pro-apoptotic factors and eventual cell death (Izzo et al., 2016). Previous reports indicated that BHB exerts multiple effects on mitochondria, including increased mitochondrial respiration, enhanced NADH oxidation and ATP production, and reduced reactive oxygen species (ROS) production, all of which either singly or in combination would raise the threshold for mPT (Maalouf et al., 2007; Maalouf and Rho, 2008; Kim et al., 2010). Intriguingly, this was the first demonstration that inhibition of mPT could result in anti-seizure effects.
There have been a few additional seminal studies showing the pleiotropic activity of ketone bodies, and while these novel targets were revealed in experimental systems outside the context of epileptic tissues, they nevertheless highlight important molecular actions that have become increasingly relevant in epilepsy research. First, BHB was shown to inhibit histone deacetylases (HDACs) in renal tissue, which resulted in increased resistance to oxidative stress via transcription of antioxidant genes regulated by FOXO3A (Shimazu et al., 2013). The relevance of HDACs in epilepsy is underscored by the fact that the broad-spectrum ASD valproic acid is in part an HDAC inhibitor (Monti et al., 2009). Further, BHB’s neuroprotective properties stem from (among other actions) an interaction with the hydroxycarboxylic acid 2 (HCA2) receptor (or GPR109A, also known as NIACR1 or the niacin receptor 1, and encoded by the HCAR2 gene), a G protein-coupled receptor found on adipocytes, neutrophils, tissue macrophages, and in the anterior cingulate cortex (Rahman et al., 2014). The net effect of BHB binding to HCA2 is a profound anti-inflammatory effect. BHB also targets other key components of the innate immune system. Specifically, it has been shown that BHB prevents the assembly of the innate immune sensor NOD-like receptor protein 3 (NLRP3) inflammasome, a multiprotein complex that induces caspase-1 activation and release of the pro-inflammatory cytokines IL-1β and IL-18 in macrophages (Youm et al., 2015). As neuroinflammation is now unquestionably linked to seizure genesis and epileptogenesis (Vezzani et al., 2015), strategies to mitigate aberrant brain inflammation would be expected to be both anticonvulsant and neuroprotective. Indeed, BHB possesses anti-inflammatory properties, consistent with the fact the KD is highly effective against neuroinflammation-induced epilepsy such as FIRES (febrile infection-related epilepsy syndrome) (Nabbout et al., 2010). A mechanistic summary of ketone body action illustrated in Figure 2.
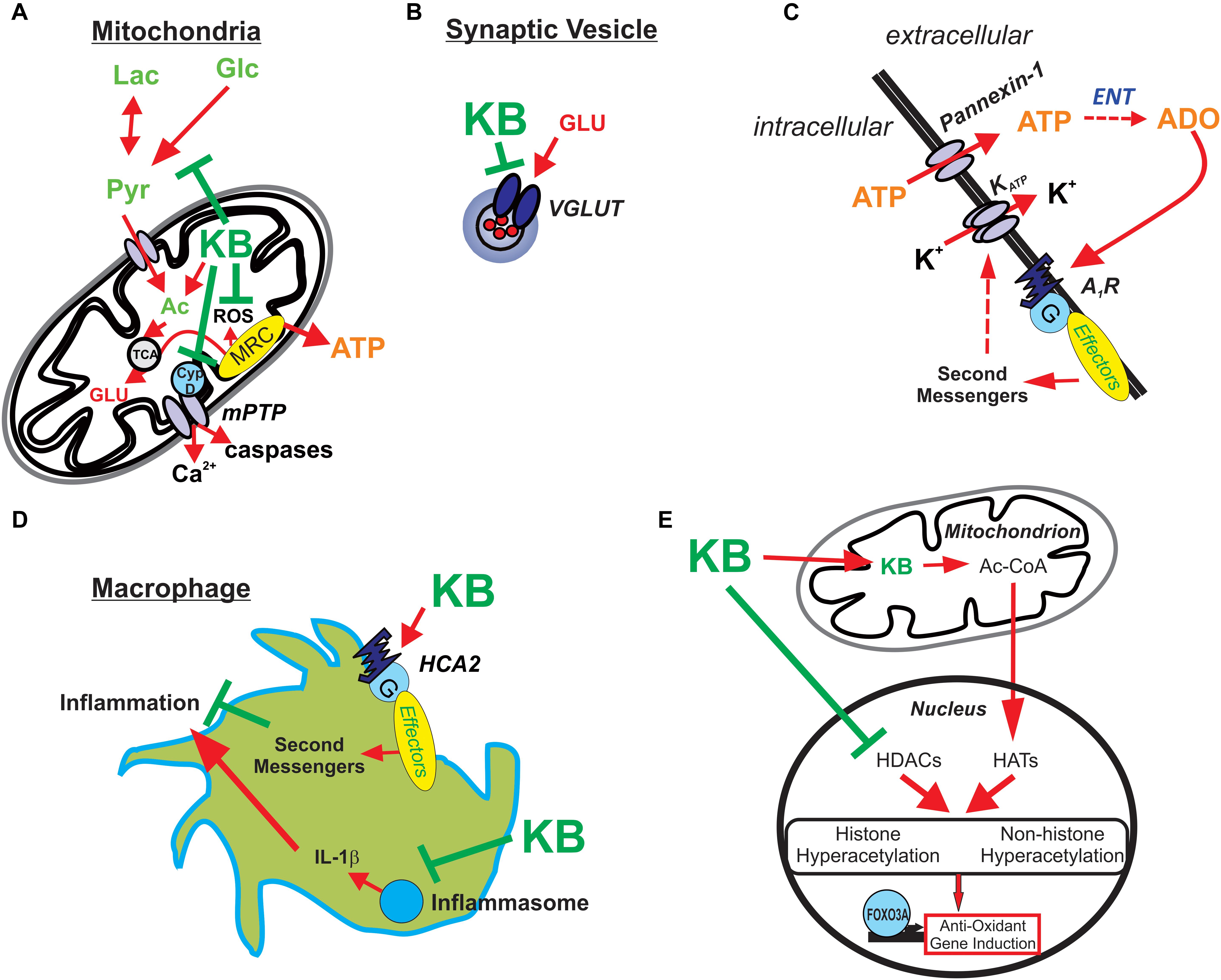
Figure 2. Potential mechanisms underlying ketone body-mediated attenuation of CNS hyperexcitability and neuroprotection. (A) Ketone bodies (KB) may enhance ATP production by providing acetyl-CoA (Ac) and inhibiting production of reactive oxygen species (ROS) and the mitochondrial permeability transition (mPT) pore, thereby protecting the cell against oxidative injury and preventing excessive release of calcium. (B) KB may inhibit vesicular glutamate transporters (VGLUT), decreasing the amount of glutamate loaded in vesicles and reducing the size of glutamate quanta released during synaptic transmission. (C) KB-mediated increases in intracellular ATP and subsequent release through pannexin channels lead to adenosine (ADO) synthesis via ectonucleotidases (ENT) in the extracellular space. ADO in turn binds to inhibitory adenosine type 1 receptors (A1Rs) which are coupled to the indirect opening of KATP channels. (D) KB activates HCA2 receptors and inhibit the assembly of the NRLP3 inflammasome; thus, KB attenuate inflammatory mediators produced by infiltrating macrophages. (E) KB also promote histone and non-histone hyperacetylation by increasing acetyl-CoA, a substrate for histone acetyltransferases (HATs), and directly inhibiting histone deacetylases (HDACs) – with the net result of increasing endogenous anti-oxidants (among other actions). Reprinted with permission from Simeone et al. (2018).
2-Deoxyglucose Versus/Plus Ketone Bodies
From the foregoing discussion, it is important to note both 2DG and the KD can suppress seizure activity but likely exert their effects through distinct yet overlapping mechanisms (Table 1). Ketone bodies have also been shown to possess anti-seizure properties, albeit in preclinical models (Simeone et al., 2018). One major distinction between 2DG and the KD is the fact that 2DG does not induce ketosis. And while 2DG and the KD both block seizures in the 6-Hz corneal stimulation and Frings audiogenic seizure susceptible models, 2DG is ineffective in the MES test which models focal-onset seizures with secondary generalization (Hartman et al., 2008). Further, despite different induction protocols, the progression of kindling is delayed by both the KD and 2DG (Hori et al., 1997; Garriga-Canut et al., 2006; Stafstrom et al., 2009). Thus, while seizure susceptibility is modified by both 2DG and the KD (also ketone bodies) through modulation of biochemical pathways involved in energy metabolism, these interventions do not possess the same anti-seizure profile and cannot be directly compared. That said, the fact that the mechanistic profiles of 2DG and ketone bodies suggest that a combined approach may yield either additive or synergistic effects, especially since a broader spectrum of mechanistic targets and pathways would be influenced. Also, it will be interesting to assess the combined efficacy of 2DG and BHB on seizure control in other chronic epilepsy models with spontaneous seizures such as the pilocarpine and kainate models.

Table 1. Mechanistic comparison between 2-deoxyglucose (2DG) and β-hydroxybutyrate.
Before formulations of 2DG or BHB are approved for clinical use in patients with epilepsy, efficacy and safety need to be clearly demonstrated. In this regard, the relative safety profile of 2DG has already been established in cancer trials (Pelicano et al., 2006), and epilepsy clinical trials are in progress (Bialer et al., 2017). The half-life of 2DG in humans is estimated to be 5–7 h, so this therapy would be potentially useful with twice-daily dosing (Gounder et al., 2012). With respect to BHB, ketone esters have been investigated preclinically as potential therapeutic agents, primarily due to their ability to produce prolonged elevations in blood ketone bodies (D’Agostino et al., 2013). However, the pharmacokinetics of BHB have not been carefully studied in humans. While ketone esters have yet to be studied in humans with epilepsy, there is already a medical formulation of MCTs that induces significant ketosis in humans (Henderson et al., 2009), and various ketone salts, esters and oils are already widely available through commercial sources and through the Internet. In addition, several metabolites or metabolic strategies have been reported to exhibit potent anticonvulsant actions via alternative mechanisms, including fructose-1,6-bisphosphate (Lian et al., 2007; Shao et al., 2018), decanoic acid (Chang et al., 2016), inhibition of lactate dehydrogenase (Sada et al., 2015), D-leucine (Hartman et al., 2015), and polyunsaturated fatty acids (Dustin and Stafstrom, 2016), all of which are worthy of further exploration for epilepsy therapeutics.
Conclusion
Metabolic control of neuronal excitability has increasingly become a worthy scientific paradigm for therapeutic development in the field of epilepsy, impacting key mechanisms in seizure genesis and possibly epileptogenesis. The origin and evolution of this approach have been inspired by the clinical success of the KD and its variants. Within this framework, 2DG and BHB represent two promising approaches for seizure control, the former primarily through partial inhibition of glycolysis and downstream transcriptional regulation, and the latter via pleiotropic actions at mitochondrial, epigenetic and cellular levels. While 2DG and BHB each possesses unique preclinical profiles, both have been shown to reduce seizure susceptibility in several animal models and in multiple in vitro cellular systems.
However, clinical implementation of metabolism-based approaches such as the KD is not without pragmatic challenges regarding administration and patient compliance. Thus, there is a strong need to identify specific metabolic substrates and/or enzymes that might induce clinical benefits comparable to the KD as a necessary condition for rational experimental therapeutics and later clinical validation. 2DG and BHB may only represent component pieces of diet-based treatments for epilepsy, but together, since they recapitulate two core features of the KD (i.e., glycolytic restriction and products of increased fatty acid oxidation), they could in time help validate the concept of a “diet in a pill” (Rho and Sankar, 2008), or polypharmacy in a pill. Of course, the KD induces far broader mechanistic effects than either 2DG or BHB alone (Rogawski et al., 2016), but the current major advantage of these substrates is that they can already be delivered in palatable forms to patients with epilepsy.
Author Contributions
JR, CS, and L-RS wrote the manuscript.
Conflict of Interest Statement
We confirm that we have read the journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines. JR has served as a paid consultant for Accera Pharma, Xenon Pharmaceuticals, Danone Nutricia, and Ajinomoto USA. CS has served as a paid consultant for Mallinckrodt and has received speaker fees from Nutricia. JR and CS have received royalties from UpToDate for a chapter about the mechanisms of seizures and epilepsy. CS is an inventor on a patent application for the use of 2DG as a clinical antiseizure medication through theWisconsin Alumni Research Foundation.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
JR’s research has been supported by the Canadian Institutes of Health Research. CS’s research has been supported by The Charlie Foundation, the Mathias Koch Memorial Fund of the Community Foundation of Southern Wisconsin, and the Sandra and Malcolm Berman Foundation.
References
Aalling, N. N., Nedergaard, M., and DiNuzzo, M. (2018). Cerebral metabolic changes during sleep. Curr. Neurol. Neurosci. Rep. 18:57. doi: 10.1007/s11910-018-0868-9
Barton, M. E., Klein, B. D., Wolf, H. H., and White, H. S. (2001). Pharmacological characterization of the 6 Hz psychomotor seizure model of partial epilepsy. Epilepsy Res. 47, 217–227. doi: 10.1016/s0920-1211(01)00302-3
Bazzigaluppi, P., EbrahimAmini, A., Weisspapir, I., Stefanovic, B., and Carlen, P. L. (2017). Hungry neurons: metabolic insights on seizure dynamics. Int. J. Mol. Sci. 18:E2269. doi: 10.3390/ijms18112269
Bialer, M., Johannessen, S. I., Levy, R. H., Perucca, E., Tomson, T., and White, H. S. (2017). Progress report on new antiepileptic drugs: a summary of the thirteenth Eilat conference on new antiepileptic drugs and devices (EILAT XIII). Epilepsia 58, 181–221. doi: 10.1111/epi.13634
Boison, D., and Steinhäuser, C. (2018). Epilepsy and astrocyte energy metabolism. Glia 66, 1235–1243. doi: 10.1002/glia.23247
Bough, K. J., Wetherington, J., Hassel, B., Pare, J. F., Gawryluk, J. W., Greene, J. G., et al. (2006). Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenic diet. Ann. Neurol. 60, 223–235. doi: 10.1002/ana.20899
Buchhalter, J. R., D’Alfonso, S., Connolly, M., Fung, E., Michoulas, A., Sinasac, D., et al. (2017). The relationship between D-beta-hydroxybutyrate blood concentrations and seizure control in children treated with the ketogenic diet for medically intractable epilepsy. Epilepsia Open 2, 317–321. doi: 10.1002/epi4.12058
Cendes, F., Theodore, W. H., Brinkmann, B. H., Sulc, V., and Cascino, G. D. (2016). Neuroimaging of epilepsy. Handb. Clin. Neurol. 136, 985–1014.
Chang, P., Augustin, K., Boddum, K., Williams, S., Sun, M., Terschak, J. A., et al. (2016). Seizure control by decanoic acid through direct AMPA receptor inhibition. Brain 139, 431–443. doi: 10.1093/brain/awv325
Cheong, J. H., Park, E. S., Liang, J., Dennison, J. B., Tsavachidou, D., Nguyen-Charles, C., et al. (2011). Dual inhibition of tumor energy pathway by 2-deoxyglucose and metformin is effective against a broad spectrum of preclinical cancer models. Mol. Cancer Ther. 10, 2350–2362. doi: 10.1158/1535-7163.MCT-11-0497
Cloix, J. F., and Hévor, T. (2009). Epilepsy, regulation of brain energy metabolism and neurotransmission. Curr. Med. Chem. 16, 841–853. doi: 10.2174/092986709787549316
D’Agostino, D. P., Pilla, R., Held, H. E., Landon, C. S., Puchowicz, M., Brunengraber, H., et al. (2013). Therapeutic ketosis with ketone ester delays central nervous system oxygen toxicity seizures in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 304, R829–R836. doi: 10.1152/ajpregu.00506.2012
Daikhin, Y., and Yudkoff, M. (1998). Ketone bodies and brain glutamate and GABA metabolism. Dev. Neurosci. 20, 358–364. doi: 10.1159/000017331
Dallérac, G., Moulard, J., Benoist, J. F., Rouach, S., Auvin, S., Guilbot, A., et al. (2017). Non-ketogenic combination of nutritional strategies provides robust protection against seizures. Sci. Rep. 7:5496. doi: 10.1038/s41598-017-05542-3
de Padua, M. C., Delodi, G., Vuèetiæ, M., Durivault, J., Vial, V., Bayer, P., et al. (2017). Disrupting glucose-6-phosphate isomerase fully suppresses the “Warburg effect” and activates OXPHOS with minimal impact on tumor growth except in hypoxia. Oncotarget 8, 87623–87637. doi: 10.18632/oncotarget.21007
DeVivo, D. C., Leckie, M. P., Ferrendelli, J. S., and McDougal, D. B. Jr. (1978). Chronic ketosis and cerebral metabolism. Ann. Neurol. 3, 331–337. doi: 10.1002/ana.410030410
Dienel, G. A. (2017). Lack of appropriate stoichiometry: strong evidence against an energetically important astrocyte-neuron lactate shuttle in brain. J. Neurosci. Res. 95, 2103–2125. doi: 10.1002/jnr.24015
Dustin, S. M., and Stafstrom, C. E. (2016). Ketogenic diet, but not polyunsaturated fatty acid diet, reduces spontaneous seizures in juvenile rats with kainic acid-induced epilepsy. J. Epilepsy Res. 6, 1–7. doi: 10.14581/jer.16001
Elman, I., Sokoloff, L., Adler, C. M., Weisenfeld, N., and Breier, A. (1999). The effects of pharmacological doses of 2-deoxyglucose on cerebral blood flow in healthy volunteers. Brain Res. 815, 243–249. doi: 10.1016/s0006-8993(98)01137-8
Erecinska, M., Nelson, D., Daikhin, Y., and Yudkoff, M. (1996). Regulation of GABA level in rat brain synaptosomes: fluxes through enzymes of the GABA shunt and effects of glutamate, calcium, and ketone bodies. J. Neurochem. 67, 2325–2334. doi: 10.1046/j.1471-4159.1996.67062325.x
Forte, N., Medrihan, L., Cappetti, B., Baldelli, P., and Benfenati, F. (2016). 2-Deoxy-D-glucose enhances tonic inhibition through the neurosteroid-mediated activation of extrasynaptic GABAA receptors. Epilepsia 57, 1987–2000. doi: 10.1111/epi.13578
Freeman, J., Veggiotti, P., Lanzi, G., Tagliabue, A., and Perucca, E. (2006). The ketogenic diet: from molecular mechanisms to clinical effects. Epilepsy Res. 68, 145–180. doi: 10.1016/j.eplepsyres.2005.10.003
Freeman, J. M., Vining, E. P., Pillas, D. J., Pyzik, P. L., Casey, J. C., and Kelly, L. M. (1998). The efficacy of the ketogenic diet -: a prospective evaluation of intervention in 150 children. Pediatrics 1998, 1358–1363. doi: 10.1542/peds.102.6.1358
Gano, L. B., Patel, M., and Rho, J. M. (2014). Ketogenic diets, mitochondria, and neurological diseases. J. Lipid Res. 55, 2211–2228. doi: 10.1194/jlr.R048975
Garriga-Canut, M., Schoenike, B., Qazi, R., Bergendahl, K., Daley, T. J., Pfender, R. M., et al. (2006). 2-Deoxy-D-glucose reduces epilepsy progression by NRSF-CtBP-dependent metabolic regulation of chromatin structure. Nat. Neurosci. 9, 1382–1387. doi: 10.1038/nn1791
Gasior, M., Yankura, J., Hartman, A. L., French, A., and Rogawski, M. A. (2010). Anticonvulsant and proconvulsant actions of 2-deoxy-D-glucose. Epilepsia 5, 1385–1394. doi: 10.1111/j.1528-1167.2010.02593.x
Gilbert, D. L., Pyzik, P. L., and Freeman, J. M. (2000). The ketogenic diet: seizure control correlates better with serum beta-hydroxybutyrate than with urine ketones. J. Child Neurol. 15, 787–790. doi: 10.1177/088307380001501203
Gounder, M. K., Lin, H., Stein, M., Goodin, S., Bertino, J. R., Kong, A. N., et al. (2012). A validated bioanalytical HPLC method for pharmacokinetic evaluation of 2-deoxyglucose in human plasma. Biomed. Chromatogr. 26, 650–654. doi: 10.1002/bmc.1710
Greene, A. E., Todorova, M. T., and Seyfried, T. N. (2003). Perspectives on the metabolic management of epilepsy through dietary reduction of glucose and elevation of ketone bodies. J. Neurochem. 86, 529–537. doi: 10.1046/j.1471-4159.2003.01862.x
Hartman, A. L., Lyle, M., Rogawski, M. A., and Gasior, M. (2008). Efficacy of the ketogenic diet in the 6-Hz seizure test. Epilepsia 49, 334–339. doi: 10.1111/j.1528-1167.2007.01430.x
Hartman, A. L., Santos, P., O’Riordan, K. J., Stafstrom, C. E., and Hardwick, J. M. (2015). Potent anti-seizure effects of D-leucine. Neurobiol. Dis. 82, 46–53. doi: 10.1016/j.nbd.2015.05.013
He, X. P., Kotloski, R., Nef, S., Luikart, B. W., Parada, L. F., and McNamara, J. O. (2004). Conditional deletion of TrkB but not BDNF prevents epileptogenesis in the kindling model. Neuron 43, 31–42. doi: 10.1016/j.neuron.2004.06.019
Henderson, S. T., Vogel, J. L., Barr, L. J., Garvin, F., Jones, J. J., and Costantini, L. C. (2009). Study of the ketogenic agent AC-1202 in mild to moderate Alzheimer’s disease: a randomized, double-blind, placebo-controlled, multicenter trial. Nutr. Metab. 6:31. doi: 10.1186/1743-7075-6-31
Höld, K. M., Sirisoma, N. S., Ikeda, T., Narahashi, T., and Casida, J. E. (2000). Alpha-thujone (the active component of absinthe): gamma-aminobutyric acid type A receptor modulation and metabolic detoxification. Proc. Natl. Acad. Sci. U.S.A. 97, 3826–3831. doi: 10.1073/pnas.070042397
Hori, A., Tandon, P., Holmes, G. L., and Stafstrom, C. E. (1997). Ketogenic diet: effects on expression of kindled seizures and behavior in adult rats. Epilepsia 38, 750–758. doi: 10.1111/j.1528-1157.1997.tb01461.x
Hu, X. L., Cheng, X., Fei, J., and Xiong, Z. Q. (2011). Neuron-restrictive silencer factor is not required for the antiepileptic effect of the ketogenic diet. Epilepsia 52, 1609–1616. doi: 10.1111/j.1528-1167.2011.03171.x
Huang, Y. Z., and McNamara, J. O. (2006). Inhibiting glycolysis to reduce seizures: how it might work. Nat. Neurosci. 9, 1351–1352. doi: 10.1038/nn1106-1351
Huttenlocher, P. R. (1976). Ketonemia and seizures: metabolic and anticonvulsant effects of two ketogenic diets in childhood epilepsy. Pediatr. Res. 10, 536–540. doi: 10.1203/00006450-197605000-00006
Ingram, D. K., and Roth, G. S. (2011). Glycolytic inhibition as a strategy for developing calorie restriction mimetics. Exp. Gerontol. 46, 148–154. doi: 10.1016/j.exger.2010.12.001
Izzo, V., Bravo-San Pedro, J. M., Sica, V., Kroemer, G., and Galluzzi, L. (2016). Mitochondrial permeability transition: new findings and persisting uncertainties. Trends Cell Biol. 26, 655–667. doi: 10.1016/j.tcb.2016.04.006
Juge, N., Gray, J. A., Omote, H., Miyaji, T., Inoue, T., Hara, C., et al. (2010). Metabolic control of vesicular glutamate transport and release. Neuron 68, 99–112. doi: 10.1016/j.neuron.2010.09.002
Kawamura, M. Jr., Ruskin, D. N., and Masino, S. A. (2010). Metabolic autocrine regulation of neurons involves cooperation among pannexin hemichannels, adenosine receptors, and KATP channels. J. Neurosci. 30, 3886–3895. doi: 10.1523/JNEUROSCI.0055-10.2010
Kawamura, M. Jr., Ruskin, D. N., and Masino, S. A. (2016). Metabolic therapy for temporal lobe epilepsy in a dish: investigating mechanisms of ketogenic diet using electrophysiological recordings in hippocampal slices. Front. Mol. Neurosci. 9:112. doi: 10.3389/fnmol.2016.00112
Keith, H. M. (1931). The effect of various factors on experimentally produced convulsions. Am. J. Dis. Child 41, 532–543.
Keith, H. M. (1932). Further studies of the control of experimentally produced convulsions. Pharmacol. Exp. Ther. 44, 449–455.
Keith, H. M. (1933). Factors influencing experimentally produced convulsions. Arch. Neurol. Psychiatry 29, 148–154.
Keith, H. M. (1935). Experimental convulsions induced by administration of thujone. Arch. Neurol. Psychiatry 34, 1022–1040.
Kim, D. Y., Simeone, K. A., Simeone, T. A., Pandya, J. D., Wilke, J. C., Ahn, Y., et al. (2015). Ketone bodies mediate antiseizure effects through mitochondrial permeability transition. Ann. Neurol. 78, 77–87. doi: 10.1002/ana.24424
Kim, D. Y., Vallejo, J., and Rho, J. M. (2010). Ketones prevent synaptic dysfunction induced by mitochondrial respiratory complex inhibitors. J. Neurochem. 114, 130–141. doi: 10.1111/j.1471-4159.2010.06728.x
Lambrechts, D. A., de Kinderen, R. J., Vles, J. S., de Louw, A. J., Aldenkamp, A. P., and Majoie, H. J. (2017). A randomized controlled trial of the ketogenic diet in refractory childhood epilepsy. Acta Neurol. Scand. 135, 231–239. doi: 10.1111/ane.12592
Lee, J., Bruce-Keller, A. J., Kruman, Y., Chan, S. L., and Mattson, M. P. (1999). 2-Deoxy-D-glucose protects hippocampal neurons against excitotoxic and oxidative injury: evidence for the involvement of stress proteins. J. Neurosci. Res. 57, 48–61. doi: 10.1002/(sici)1097-4547(19990701)57:1<48::aid-jnr6>3.3.co;2-c
Lian, X. Y., Khan, F. A., and Stringer, J. L. (2007). Fructose-1,6-bisphosphate has anticonvulsant activity in models of acute seizures in adult rats. J. Neurosci. 27, 12007–12011. doi: 10.1523/jneurosci.3163-07.2007
Librizzi, L., de Curtis, M., Janigro, D., Runtz, L., de Bock, F., Barbier, E. L., et al. (2018). Cerebrovascular heterogeneity and neuronal excitability. Neurosci. Lett. 667, 75–83. doi: 10.1016/j.neulet.2017.01.013
Likhodii, S. S., Serbanescu, I., Cortez, M. A., Murphy, P., Snead, O. C. III, and Burnham, W. M. (2003). Anticonvulsant properties of acetone, a brain ketone elevated by the ketogenic diet. Ann. Neurol. 54, 219–226. doi: 10.1002/ana.10634
Lund, T. M., Obel, L. F., Risa, O., and Sonnewald, U. (2011). Beta-Hydroxybutyrate is the preferred substrate for GABA and glutamate synthesis while glucose is indispensable during depolarization in cultured GABAergic neurons. Neurochem. Int. 59, 309–318. doi: 10.1016/j.neuint.2011.06.002
Lund, T. M., Risa, O., Sonnewald, U., Schousboe, A., and Waagepetersen, H. S. (2009). Availability of neurotransmitter glutamate is diminished when beta-hydroxybutyrate replaces glucose in cultured neurons. J. Neurochem. 110, 80–91. doi: 10.1111/j.1471-4159.2009.06115.x
Lutas, A., and Yellen, G. (2013). The ketogenic diet: metabolic influences on brain excitability and epilepsy. Trends Neurosci. 36, 32–40. doi: 10.1016/j.tins.2012.11.005
Ma, W., Berg, J., and Yellen, G. (2007). Ketogenic diet metabolites reduce firing in central neurons by opening KATP channels. J. Neurosci. 27, 3618–3625. doi: 10.1523/jneurosci.0132-07.2007
Maalouf, M., and Rho, J. M. (2008). Oxidative impairment of hippocampal long-term potentiation involves activation of protein phosphatase 2A and is prevented by ketone bodies. J. Neurosci. Res. 86, 3322–3330. doi: 10.1002/jnr.21782
Maalouf, M., Sullivan, P. G., Davis, L., Kim, D. Y., and Rho, J. M. (2007). Ketones inhibit mitochondrial production of reactive oxygen species production following glutamate excitotoxicity by increasing NADH oxidation. Neuroscience 145, 256–264. doi: 10.1016/j.neuroscience.2006.11.065
Magistretti, P. J., and Allaman, I. (2015). A cellular perspective on brain energy metabolism and functional imaging. Neuron 86, 883–901. doi: 10.1016/j.neuron.2015.03.035
Mergenthaler, P., Lindauer, U., Dienel, G. A., and Meisel, A. (2013). Sugar for the brain: the role of glucose in physiological and pathological brain function. Trends Neurosci. 36, 587–597. doi: 10.1016/j.tins.2013.07.001
Minlebaev, M., and Khazipov, R. (2011). Antiepileptic effects of endogenous beta-hydroxybutyrate in suckling infant rats. Epilepsy Res. 95, 100–109. doi: 10.1016/j.eplepsyres.2011.03.003
Monti, B., Polazzi, E., and Contestabile, A. (2009). Biochemical, molecular and epigenetic mechanisms of valproic acid neuroprotection. Curr. Mol. Pharmacol. 2, 95–109. doi: 10.2174/1874467210902010095
Nabbout, R., Mazzuca, M., Hubert, P., Peudennier, S., Allaire, C., Flurin, V., et al. (2010). Efficacy of ketogenic diet in severe refractory status epilepticus initiating fever induced refractory epileptic encephalopathy in school age children (FIRES). Epilepsia 51, 2033–2037. doi: 10.1111/j.1528-1167.2010.02703.x
Neal, E. G., Chaffe, H., Schwartz, R. H., Lawson, M. S., Edwards, N., Fitzsimmons, G., et al. (2008). The ketogenic diet for the treatment of childhood epilepsy: a randomised controlled trial. Lancet Neurol. 7, 500–506. doi: 10.1016/S1474-4422(08)70092-9
Neal, E. G., Chaffe, H., Schwartz, R. H., Lawson, M. S., Edwards, N., Fitzsimmons, G., et al. (2009). A randomized trial of classical and medium-chain triglyceride ketogenic diets in the treatment of childhood epilepsy. Epilepsia 50, 1109–1117. doi: 10.1111/j.1528-1167.2008.01870.x
Nedergaard, S., and Andreasen, M. (2018). Opposing effects of 2-deoxy-D-glucose on interictal- and ictal-like activity when K+ currents and GABAA receptors are blocked in rat hippocampus in vitro. J. Neurophysiol. 119, 1912–1923. doi: 10.1152/jn.00732.2017
Pani, G. (2015). Neuroprotective effects of dietary restriction: evidence and mechanisms. Semin. Cell Dev. Biol. 40, 106–114. doi: 10.1016/j.semcdb.2015.03.004
Park, M., Song, K. S., Kim, H. K., Park, Y. J., Kim, H. S., Bae, M. I., et al. (2009). 2-Deoxy-D-glucose protects neural progenitor cells against oxidative stress through the activation of AMP-activated protein kinase. Neurosci. Lett. 449, 201–206. doi: 10.1016/j.neulet.2008.11.007
Pelicano, H., Martin, D. S., Xu, R.-H., and Huang, P. (2006). Glycolysis inhibition for anticancer treatment. Oncogene 25, 4633–4646. doi: 10.1038/sj.onc.1209597
Pellerin, L., and Magistretti, P. J. (1994). Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc. Natl. Acad. Sci. U.S.A. 91, 10625–10629. doi: 10.1073/pnas.91.22.10625
Pfeifer, H. H., and Thiele, E. A. (2005). Low-glycemic-index treatment: a liberalized ketogenic diet for treatment of intractable epilepsy. Neurology 65, 1810–1812. doi: 10.1212/01.wnl.0000187071.24292.9e
Rahman, M., Muhammad, S., Khan, M. A., Chen, H., Ridder, D. A., Muller-Fielitz, H., et al. (2014). The beta-hydroxybutyrate receptor HCA2 activates a neuroprotective subset of macrophages. Nat. Commun. 5:3944. doi: 10.1038/ncomms4944
Redjak, K., Redjak, R., Sieklucka-Dziuba, M., Stelmasiak, Z., and Grieb, P. (2001). 2-Deoxyglucose enhances epileptic tolerance evoked by transient incomplete brain ischemia in mice. Epilepsy Res. 43, 271–278. doi: 10.1016/s0920-1211(01)00184-x
Reid, C. A., Mullen, S., Kim, T. H., and Petrou, S. (2014). Epilepsy, energy deficiency and new therapeutic approaches including diet. Pharmacol. Ther. 144, 192–201. doi: 10.1016/j.pharmthera.2014.06.001
Rho, J. M., Anderson, G. D., Donevan, S. D., and White, H. S. (2002). Acetoacetate, acetone, and dibenzylamine (a contaminant in l-(+)-beta-hydroxybutyrate) exhibit direct anticonvulsant actions in vivo. Epilepsia 43, 358–361. doi: 10.1046/j.1528-1157.2002.47901.x
Rho, J. M., and Sankar, R. (2008). The ketogenic diet in a pill: is this possible? Epilepsia 49(Suppl. 8), 127–133. doi: 10.1111/j.1528-1167.2008.01857.x
Rho, J. M., and Stafstrom, C. E. (2012). The ketogenic diet: what has science taught us? Epilepsy Res. 100, 210–217. doi: 10.1016/j.eplepsyres.2011.05.021
Rogawski, M. A., Loscher, W., and Rho, J. M. (2016). Mechanisms of action of antiseizure drugs and the ketogenic diet. Cold Spring Harb. Perspect. Med. 6:a022780. doi: 10.1101/cshperspect.a022780
Sada, N., Lee, S., Katsu, T., Otsuki, T., and Inoue, T. (2015). Epilepsy treatment. Targeting LDH enzymes with a stiripentol analog to treat epilepsy. Science 347, 1362–1367. doi: 10.1126/science.aaa1299
Samokhina, E., Popova, I., Malkov, A., Ivanov, A. I., Papadia, D., Osypov, A., et al. (2017). Chronic inhibition of brain glycolysis initiates epileptogenesis. J. Neurosci. Res. 95, 2195–2206. doi: 10.1002/jnr.24019
Schwartzkroin, P. A. (1999). Mechanisms underlying the anti-epileptic efficacy of the ketogenic diet. Epilepsy Res. 37, 171–180. doi: 10.1016/s0920-1211(99)00069-8
Shao, L. R., and Stafstrom, C. E. (2017). Glycolytic inhibition by 2-deoxy-D-glucose abolishes both neuronal and network bursts in an in vitro seizure model. J. Neurophysiol. 118, 103–113. doi: 10.1152/jn.00100.2017
Shao, L. R., Wang, G., and Stafstrom, C. E. (2018). The glycolytic metabolite, fructose-1,6-bisphosphate, blocks epileptiform bursts by attenuating voltage-activated calcium currents in hippocampal slices. Front. Cell. Neurosci. 12:168. doi: 10.3389/fncel.2018.00168
Shimazu, T., Hirschey, M. D., Newman, J., He, W., Shirakawa, K., Le Moan, N., et al. (2013). Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 339, 211–214. doi: 10.1126/science.1227166
Simeone, T. A., Simeone, K. A., Stafstrom, C. E., and Rho, J. M. (2018). Do ketone bodies mediate the anti-seizure effects of the ketogenic diet? Neuropharmacology 133, 233–241. doi: 10.1016/j.neuropharm.2018.01.011
Stafstrom, C. E., Ockuly, J. C., Murphree, L., Valley, M. T., Roopra, A., and Sutula, T. P. (2009). Anticonvulsant and antiepileptic actions of 2-deoxy-D-glucose in epilepsy models. Ann. Neurol. 65, 435–447. doi: 10.1002/ana.21603
Thavendiranathan, P., Mendonca, A., Dell, C., Likhodii, S. S., Musa, K., Iracleous, C., et al. (2000). The MCT ketogenic diet: effects on animal seizure models. Exp. Neurol. 161, 696–703. doi: 10.1006/exnr.1999.7298
Thio, L. L., Wong, M., and Yamada, K. A. (2000). Ketone bodies do not directly alter excitatory or inhibitory hippocampal synaptic transmission. Neurology 54, 325–331.
Valente-Silva, P., Lemos, C., Kofalvi, A., Cunha, R. A., and Jones, J. G. (2015). Ketone bodies effectively compete with glucose for neuronal acetyl-CoA generation in rat hippocampal slices. NMR Biomed. 28, 1111–1116. doi: 10.1002/nbm.3355
van Delft, R., Lambrechts, D., Verschuure, P., Hulsman, J., and Majoie, M. (2010). Blood beta-hydroxybutyrate correlates better with seizure reduction due to ketogenic diet than do ketones in the urine. Seizure 19, 36–39. doi: 10.1016/j.seizure.2009.10.009
Vezzani, A., Lang, B., and Aronica, E. (2015). Immunity and inflammation in epilepsy. Cold Spring Harb. Perspect. Med. 6:a022699. doi: 10.1101/cshperspect.a022699
Wick, A. N., Drury, D. R., Nakada, H. I., and Wolf, J. B. (1957). Localization of the primary metabolic block produced by 2-deoxyglucose. J. Biol. Chem. 224, 963–969.
Wree, A. (1990). Principles of the 2-deoxyglucose method for the determination of the local cerebral glucose utilization. Eur. J. Morphol. 28, 132–138.
Yang, H., Guo, R., Wu, J., Peng, Y., Xie, D., Zheng, W., et al. (2013). The antiepileptic effect of the glycolytic inhibitor 2-deoxy-D-glucose is mediated by upregulation of KATP channel subunits Kir6.1 and Kir6.2. Neurochem. Res. 38, 677–685. doi: 10.1007/s11064-012-0958-z
Yang, L., Zhao, J., Milutinovic, P. S., Brosnan, R. J., Eger, E. I. II, and Sonner, J. M. (2007). Anesthetic properties of the ketone bodies beta-hydroxybutyric acid and acetone. Anesth. Analg. 105, 673–679. doi: 10.1213/01.ane.0000278127.68312.dc
Yao, J., Chen, S., Mao, Z., Cadenas, E., and Brinton, R. D. (2011). 2-Deoxy-D-glucose treatment induces ketogenesis, sustains mitochondrial function, and reduces pathology in female mouse model of Alzheimer’s disease. PLoS One 6:e21788. doi: 10.1371/journal.pone.0021788
Youm, Y. H., Nguyen, K. Y., Grant, R. W., Goldberg, E. L., Bodogai, M., Kim, D., et al. (2015). The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med. 21, 263–269. doi: 10.1038/nm.3804
Yudkoff, M., Daikhin, Y., Nissim, I., Lazarow, A., and Nissim, I. (2001). Brain amino acid metabolism and ketosis. J. Neurosci. Res. 66, 272–281. doi: 10.1002/jnr.1221
Yuen, A. W. C., and Sander, J. W. (2014). Rationale for using intermittent calorie restriction as a dietary treatment for drug resistant epilepsy. Epilepsy Behav. 33, 110–114. doi: 10.1016/j.yebeh.2014.02.026
Yum, M. S., Lee, M., Woo, D. C., Kim, D. W., Ko, T. S., and Velisek, L. (2015). Beta-Hydroxybutyrate attenuates NMDA-induced spasms in rats with evidence of neuronal stabilization on MR spectroscopy. Epilepsy Res. 117, 125–132. doi: 10.1016/j.eplepsyres.2015.08.005
Zhang, D., Li, J., Wang, F., Hu, J., Wang, S., and Sun, Y. (2014). 2-Deoxy-D-glucose targeting of glucose metabolism in cancer cells as a potential therapy. Cancer Lett. 355, 176–183. doi: 10.1016/j.canlet.2014.09.003
Keywords: epilepsy, 2-deoxyglucose (2DG), glycolysis, ketosis, ketone body, beta-hydroxybutyrate, ketogenic diet, metabolic therapy
Citation: Rho JM, Shao L-R and Stafstrom CE (2019) 2-Deoxyglucose and Beta-Hydroxybutyrate: Metabolic Agents for Seizure Control. Front. Cell. Neurosci. 13:172. doi: 10.3389/fncel.2019.00172
Received: 20 July 2018; Accepted: 11 April 2019;
Published: 30 April 2019.
Edited by:
Antonio Gambardella, Università degli Studi “Magna Græcia” di Catanzaro, ItalyReviewed by:
Ursula Susan Sandau, Oregon Health & Science University, United StatesMauro DiNuzzo, Independent Researcher, Rome, Italy
Copyright © 2019 Rho, Shao and Stafstrom. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jong M. Rho, am9uZy5yaG9AYWhzLmNh