 Kenneth J. Burke Jr.
Kenneth J. Burke Jr. Kevin J. Bender
Kevin J. Bender- Neuroscience Graduate Program and Department of Neurology, Kavli Institute for Fundamental Neuroscience, Weill Institute for Neurosciences, University of California, San Francisco, San Francisco, CA, United States
The axon is responsible for integrating synaptic signals, generating action potentials (APs), propagating those APs to downstream synapses and converting them into patterns of neurotransmitter vesicle release. This process is mediated by a rich assortment of voltage-gated ion channels whose function can be affected on short and long time scales by activity. Moreover, neuromodulators control the activity of these proteins through G-protein coupled receptor signaling cascades. Here, we review cellular mechanisms and signaling pathways involved in axonal ion channel modulation and examine how changes to ion channel function affect AP initiation, AP propagation, and the release of neurotransmitter. We then examine how these mechanisms could modulate synaptic function by focusing on three key features of synaptic information transmission: synaptic strength, synaptic variability, and short-term plasticity. Viewing these cellular mechanisms of neuromodulation from a functional perspective may assist in extending these findings to theories of neural circuit function and its neuromodulation.
Introduction
Neuromodulators exert powerful control over both neuronal circuit activity and animal behavior throughout the brain (Marder, 2012; Nadim and Bucher, 2014). Neuromodulatory transmitters engage G-protein coupled receptors (GPCRs), activating intracellular signaling cascades that then can directly activate or modify the properties of ion channels. Neuromodulatory transmitters can bind GPCRs many microns from the site of release, regulating activity within a volume of neuropil (Agnati et al., 1995; Rice and Cragg, 2008; Liu et al., 2018), though cases of more direct synapse-like transmission are also found throughout the brain (Kia et al., 1996; Sesack et al., 2003; Gantz et al., 2013; Courtney and Ford, 2016). Neuromodulatory regulation of ion channels affects how ion channels respond to voltage deflections on short and long time scales, thus affecting how certain features of synaptic input are transformed into neuronal output. This process occurs throughout neuronal arbors, including dendritic and axonal arbors (Athilingam et al., 2017; Labarrera et al., 2018; Yu et al., 2018). Here, we focus on neuromodulation of ion channels in the axon. Recent advances, including the ability to more directly interrogate ion channel function in small axonal compartments, has improved our understanding of how channel function is regulated in these compartments. These modulatory events dramatically affect how synaptic information is integrated to generate patters of action potentials (APs) as well as how those APs are transformed into transmitter release at axon terminals (Figure 1A).
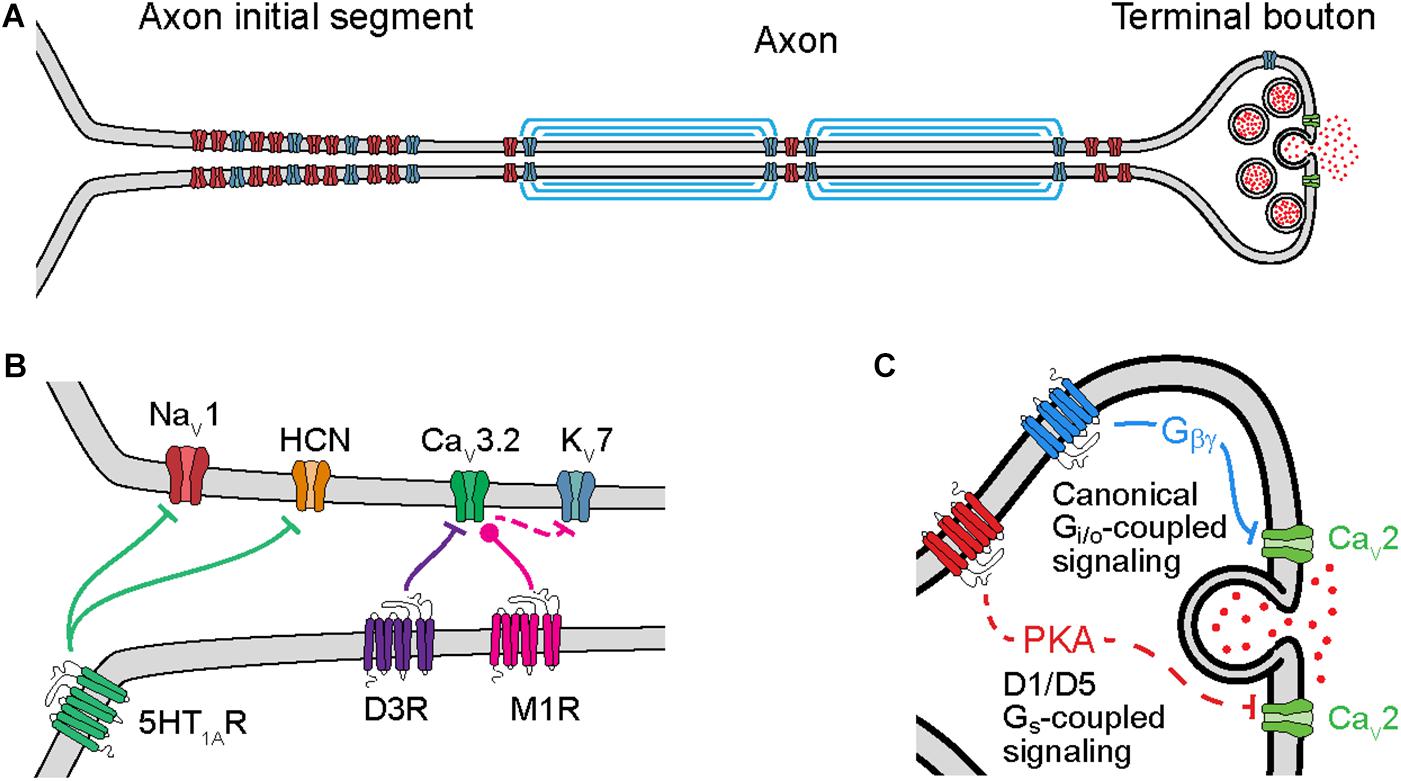
Figure 1. Cellular mechanisms of neuromodulation of axonal ion channels. (A) Schematic of axon subcompartments. Sodium (NaV), potassium (KV), and calcium (CaV) permeable voltage-gated ion channels are shown in red, blue, and green, respectively. (B) Schematic of GPCR neuromodulation of voltage-gated ion channels in the axon initial segment. Bars or circles at end of lines indicate a net reduction or increase, respectively, in target channel ion flux as a result of neuromodulation. Note that inhibition of KV7 channels is a downstream consequence of CaV3.2 modulation due to changes in intracellular calcium concentration. (C) Schematic of GPCR neuromodulation of CaV in the terminal bouton. Solid lines indicate direct binding of Gβγ subunits to CaVs, dashed lines indicate intermediate steps.
Axonal ion channels are important for many aspects of neuronal function, from the initiation and propagation of APs to the release of neurotransmitter (Figure 1A). APs are initiated in the axon initial segment (AIS), a cellular compartment enriched with voltage-gated ion channels and GPCRs (Figure 1B). At this location, synaptic currents are converted from a graded voltage signal into a train of APs. Due in part to the AP initiation threshold, this transformation is fundamentally non-linear; as a result, the output spike pattern of a neuron is highly sensitive to the neuromodulation of the small fraction of ion channels localized to the AIS.
Neuromodulation of ion channels further down the axon also significantly affects how information is propagated to downstream synapses. Like in the AIS, modulation of ion channels at axonal boutons, the site of neurotransmitter release, also occurs through activation of GPCRs (Figure 1C). Known targets of neuromodulation at the axonal bouton include voltage-gated sodium (NaV), potassium (KV), and calcium (CaV) channels. In particular, modulation of CaVs can strongly impact the release of neurotransmitters because vesicle release is a direct (but probabilistic and non-linear) function of calcium concentration. Intracellular calcium also influences how the release of neurotransmitter changes with different patterns of presynaptic activity, a critical component of short-term plasticity (STP). As a result, subtle changes to ion channel function at the axonal bouton can result in large effects on synaptic strength, STP, and synaptic information transmission.
In this review, we will provide brief overviews of the biophysical processes involved in AP initiation, propagation, and neurotransmitter release, with an emphasis on how various neuromodulatory mechanisms that target axonal ion channels alter the dynamics of vesicle release and STP at the synapse. We focus primarily on studies performed in vertebrate central circuits, drawing from neuromodulatory systems like dopaminergic and GABAB receptor systems in which significant mechanistic detail has been elucidated. For a recent review of membrane excitability across axonal compartments (see Alpizar et al., 2019). We intend to provide the reader with an intuitive understanding of the connection between neuromodulatory mechanism and function; for more mathematical descriptions of presynaptic release, STP and information transfer, see these references (Tsodyks and Markram, 1997; Dayan and Abbott, 2001; Silver, 2010; Hennig, 2013). By focusing on these biophysical intervention points for neuromodulation and their functional impact, we hope to frame presynaptic neuromodulation in terms that are more easily translated to the study of neuronal circuit dynamics.
Regulation of Action Potential Initiation
Action potential initiation and propagation are regulated by neuromodulators at several sites. APs are first generated in the AIS, a specialized axonal compartment adjacent to the somatodendritic compartment (Bender and Trussell, 2012; Kole and Stuart, 2012; Huang and Rasband, 2018). The AIS is enriched with sodium and potassium channels that underlie to the rising and falling phases of the AP, as well as other ion channel classes that can augment the AP initiation process, including CaVs and hyperpolarization-activated cyclic nucleotide-gated (HCN) cation channels (Bender and Trussell, 2009; Yu et al., 2010; Martinello et al., 2015; Ko et al., 2016). The precise site of AP initiation can be regulated by activity on short time scales (Scott et al., 2014), and can be structurally modified over longer time scales (Grubb and Burrone, 2010; Kuba et al., 2010). Furthermore, activation of neuromodulatory GPCRs can either enhance or weaken the function of many of these ion channel classes.
CaV3.2 channels, which are expressed in the AIS of many neuronal classes (Bender and Trussell, 2009; Martinello et al., 2015; Clarkson et al., 2017; Dumenieu et al., 2018), are the target of neuromodulation by both dopamine and acetylcholine. These CaVs contribute to subthreshold depolarization and high-frequency AP bursts in many systems (Cain and Snutch, 2013). Dopamine, acting through D3 receptors, hyperpolarizes CaV3.2 voltage-dependent steady state inactivation (Yang et al., 2016). Because the steady-state inactivation level of CaV3.2 channels varies markedly near typical resting membrane potentials (−60 to −90 mV, Serrano et al., 1999; Yang et al., 2016), changes in voltage-dependent inactivation can affect the number of channels available during AP generation. Ultimately, the net effect of dopaminergic modulation is to reduce burstiness, which has been observed in both auditory brainstem and neocortical neurons (Bender et al., 2010, 2012; Clarkson et al., 2017). Cholinergic modulation, by contrast, hyperpolarizes voltage-dependent activation properties of AIS-localized CaV3.2 channels in hippocampal granule cells, increasing AIS calcium concentration near resting membrane potentials (Martinello et al., 2015). The predominant effect of increased basal calcium levels is to reduce calcium-sensitive KV7 potassium current, which in turn lowers the threshold for AP initiation. Thus, neuromodulation that affects different biophysical aspects of CaV3.2 function can have bidirectional effects on AP generation.
Axon initial segment HCN channels and sodium channels can also be regulated by neuromodulators, specifically serotonin 5-HT1A receptors. In auditory brainstem, serotonin suppresses HCN channels via Gi/o-mediated inhibition of cyclic AMP, leading to a hyperpolarization of resting membrane potential and AP threshold (Ko et al., 2016). In mouse cortex and frog spinal cord, serotonin instead has inhibitory effects on axonal sodium channels, reducing sodium channel current density (Cotel et al., 2013; Yin et al., 2015). In cortex, this has a preferential effect on the NaV1.2 channel subtype specifically but appears to have a more ubiquitous effect on NaVs in the spinal cord. Serotonin’s effects in the spinal cord, however, appear to be due to 5-HT1A receptors expressed in the soma rather than the AIS itself (Cotel et al., 2013). This insight may help explain why physiological effects of serotonin can be observed in the AIS despite uncertainty over the precise subcellular localization of 5-HT1A receptors (Petersen et al., 2017).
Regulation of Action Potential Propagation and Waveform
Once APs are initiated, they propagate down the axon to sites of vesicle release known as active zones (AZs). This AP propagation itself is not perfectly reliable. For example, APs may fail at axonal branch points (reviewed in Kress and Mennerick, 2009), or near nerve terminals (Kawaguchi and Sakaba, 2015). Furthermore, some spikes generated during high frequency bursts of APs can fail to propagate to release sites (Khaliq and Raman, 2005; Monsivais et al., 2005; Roberts et al., 2008), or exhibit variable conduction velocities that can be augmented in part by dopaminergic regulation of axonal HCN channels (Ballo et al., 2010, 2012). Additionally, astrocytes can regulate AP waveform through ionotropic receptor activation or buffering of extracellular potassium (Menichella et al., 2006; Perea et al., 2007; Bay and Butt, 2012; Bellot-Saez et al., 2017).
Those APs that do arrive at axonal terminals produce a local depolarization that activates CaVs. The resulting calcium influx drives transmitter release via a non-linear reaction dependent on the third- to fourth-order of the local calcium concentration (Katz and Miledi, 1970; Reid et al., 1998; Schneggenburger and Neher, 2000; Scimemi and Diamond, 2012). This non-linear relationship between release and calcium is dependent on several factors, including the physical distance between CaVs and calcium sensors, calcium buffer dynamics and the geometry of the bouton, and the binding affinity and cooperativity requirements of these sensors for calcium (Augustine, 2001; Stanley, 2016; Brunger et al., 2018). Because of this non-linearity, small changes in AP waveform that affect calcium influx can have large effects on release.
Interestingly, however, neuromodulators that regulate AP waveform tend to also have other, more direct mechanisms for influencing Pr. For example, dopamine can broaden or narrow APs via D1- or D2-family receptors, respectively, as measured in the primary axon of cortical pyramidal cells (Yang et al., 2013). But the dominant effect of D1 receptors at release sites appears to be direct regulation of CaV function, rather than AP waveform modulation (Burke et al., 2018). Similarly, while Gi/o-coupled receptors including the CB1 cannabinoid receptor have been shown to activate G-protein coupled inward rectifier potassium channels (GIRKs) in the axon (Alger et al., 1996; Daniel and Crepel, 2001; Diana and Marty, 2003), the dominant functional effect of Gi/o-coupled receptor activation at boutons is the direct regulation of CaVs through Gβγ-dependent signaling (Lüscher et al., 1997; Brown et al., 2004, but see Zurawski et al., 2018), as discussed below.
Action potential waveform adaptation during high-frequency activity has also been repeatedly shown to either suppress or enhance vesicle release, depending on the mechanism of adaptation. In cultured Purkinje cells, attenuation of AP height near terminals strongly suppresses transmitter release (Kawaguchi and Sakaba, 2015). Surprisingly, at hippocampal mossy fiber boutons a similar activity-dependent shortening and widening of axonal APs at high frequency leads to an increase in vesicle release probability (Geiger and Jonas, 2000). The difference between these two synapses may lie in the mechanisms that underlie spike adaptation. AP height is reduced during ongoing activity in both synapses, largely due to reductions in sodium or potassium currents in Purkinje and mossy fiber boutons, respectively. This would be predicted on its own to decrease the activation of CaVs and reduce calcium influx. However, reductions in potassium currents lead to a larger broadening of the AP waveform in mossy fiber boutons than in Purkinje cell axons, increasing the total time the membrane is depolarized. This increased duration of membrane depolarization appears to be sufficient to counteract the reduction in AP height and ultimately increase the overall calcium influx. Indeed, in cerebellar stellate interneurons, a similar widening of presynaptic AP waveforms in response to prolonged somatic depolarization also leads to enhanced transmitter release due to the inactivation of KV3 potassium channels (Rowan et al., 2016; Rowan and Christie, 2017). Thus, a subtle mechanistic difference in spike waveform adaptation can lead to the opposite outcome for vesicle release probability.
Axon terminal voltage can also be affected by several mechanisms beyond manipulations of AP waveform, leading to alterations in transmitter release. Terminal CaVs can be sensitive to subthreshold depolarization (Awatramani et al., 2005). This subthreshold activity can be mediated by the local activity of ionotropic receptors, including GABAA receptors which may depolarize axon terminals (Price and Trussell, 2006; Christie et al., 2011), nicotinic acetylcholine receptors (McKay et al., 2007), or glutamatergic receptors (Pinheiro and Mulle, 2008). Alternatively, subthreshold activity at the soma, filtered by axonal cable properties, can propagate to and affect the terminal membrane (Alle and Geiger, 2006; Shu et al., 2006), especially in cells with high input resistance (Christie et al., 2011). This so-called “analog signaling” interacts with APs at the terminal to alter release (Rama et al., 2018). Thus, multiple mechanisms exist to regulate the excitability of the presynaptic bouton and vesicle release, including neuromodulatory GPCR signaling, activity-dependent AP waveform adaptation and subthreshold voltage fluctuations.
Regulation of Active Zone Voltage-Gated Calcium Channels
Calcium channels localized to active zones are among the most extensively studied effector targets of neuromodulation (reviewed in: Catterall and Few, 2008; Zamponi and Currie, 2013). Typically, presynaptic terminals are enriched with the CaV2 class of calcium channels. This group is comprised of three different isoforms: CaV2.1 (P/Q-type), CaV2.2 (N-type), and CaV2.3 (R-type). Axonal expression of each of these isoforms varies by cell type, and, at times, postsynaptic target (Éltes et al., 2017). For example, some GABAergic synapses express CaV2.1 or CaV2.2 channels exclusively (Poncer et al., 1997; Li et al., 2007; Bender and Trussell, 2009; Cao and Tsien, 2010; Szabo et al., 2014). By contrast, many glutamatergic synapses express a mix of CaV2.1, CaV2.2, and CaV2.3 channels (Brown et al., 2004; Ritzau-Jost et al., 2014). Calcium influx via CaV2.1 and 2.2 channels tends to dominate AP-evoked release (Turner et al., 1992; Wheeler et al., 1994; Bucurenciu et al., 2010; Ritzau-Jost et al., 2014; Burke et al., 2018), whereas 2.3 channels have been shown to be more critical for AP-independent spontaneous release (Ermolyuk et al., 2013). These channels have different kinetics (Colecraft et al., 2001) and exhibit differential activation depending on AP duration. For example, at mossy fiber boutons, CaV2.1 channels are best recruited by fast AP waveforms whereas longer duration waveforms recruit CaV2.1, 2.2, and 2.3 channels to comparable levels (Li et al., 2007). Thus, mechanisms of AP waveform neuromodulation or adaptation discussed earlier may impact synapses differentially depending on the expression levels of different CaV isoforms. Moreover, isoform-specific mechanisms of neuromodulation could result in synapse-specific changes to short term plasticity (Colecraft et al., 2001). Such changes have been observed with presynaptic forms of long-term potentiation where CaV2.2 channels are preferentially incorporated into synapses following plasticity induction (Ahmed and Siegelbaum, 2009).
Direct inhibition of CaV2 channels by Gβγ subunits is perhaps the most common and best understood form of G-protein-mediated neuromodulation of presynaptic calcium channels (reviewed in: Currie, 2010; Padgett and Slesinger, 2010). This form of modulation is common across presynaptically expressed Gi/o-coupled receptors, including GABAB receptors (Bean, 1989; Mintz and Bean, 1993; Otis and Trussell, 1996; Park and Dunlap, 1998; Takahashi et al., 1998; Chalifoux and Carter, 2011), CB1 cannabinoid receptors (Hoffman and Lupica, 2000; Huang et al., 2001; Kreitzer and Regehr, 2001; Wilson et al., 2001; Zhang and Linden, 2009), type 2 muscarinic acetylcholine receptors (Qin et al., 1997), D2 dopamine receptors (Pisani et al., 2000; Momiyama and Koga, 2001), opioid receptors (Endo and Yawo, 2000; Hjelmstad and Fields, 2003), metabotropic glutamate receptors (Faas et al., 2002), and adenosine receptors (Yawo and Chuhma, 1993; Lüscher et al., 1997). Direct binding of Gβγ subunits to CaV2 channels affects channel biophysics by both slowing activation kinetics and by depolarizing the voltage dependence of activation (Bean, 1989; Colecraft et al., 2000). Interestingly, Gβγ-bound CaV2.2 exhibits a greater number of “reluctant” openings than CaV2.1 channels in which channel open duration is reduced or delayed. Thus, in response to AP depolarizations, the net effect of Gβγ-mediated inhibition is to reduce calcium influx. However, these calcium currents are also modestly shorter in duration for Gβγ-bound CaV2.2 channels than CaV2.1 channels (Colecraft et al., 2001). All of these mechanisms converge on reduced calcium influx per AP, which subsequently reduces the probability of neurotransmitter release.
While Gβγ-dependent signaling is a well-understood form of GPCR-mediated CaV2 channel regulation, GPCRs can also regulate CaV2 channels through other pathways. Dopamine and nociceptin receptors have both been shown to promote channel internalization through GPCR-channel complexes (Altier et al., 2006; Kisilevsky and Zamponi, 2008; Kisilevsky et al., 2008), though whether such mechanisms occur at presynaptic terminals remains unclear (Kisilevsky et al., 2008; Murali et al., 2012). If present at presynaptic terminals, channel internalization could lead to a functional silencing of release sites, essentially reducing synapse number.
D1/D5 dopamine receptors have also been shown to regulate calcium influx through high-voltage activated (presumably CaV2) calcium channels in dissociated striatal neurons via PKA-dependent signaling (Surmeier et al., 1995; Zhang et al., 2002). Similarly, D1/D5 receptors regulate CaV2.1 and CaV2.2 calcium influx in select glutamatergic inputs to prefrontal pyramidal neurons, also via a PKA-dependent pathway (Burke et al., 2018). The precise mechanism of calcium channel modulation remains unclear, but it is clearly distinct from Gβγ-mediated effects because dopaminergic modulation did not occlude further modulation via GABAB receptors. Moreover, in contrast to Gβγ-dependent modulation, dopamine did not reduce a single channel’s calcium influx per AP, but rather the probability individual channels would open in response to an AP. This small difference in mechanism resulted in differential functional effects on release; while both dopamine and GABAB reduced Pr at this synapse, only GABAB altered STP (further discussed below). This form of presynaptic regulation without marked changes in STP has also been observed for noradrenergic (Delaney et al., 2007) and kappa opioid receptor-dependent modulation (Li et al., 2012; Tejeda et al., 2017). In these cases, the mechanism of action appeared to be either Gβγ-dependent effects at sites downstream of CaVs (e.g., SNAP25, see Delaney et al., 2007; Zurawski et al., 2018), or via other signaling cascades (e.g., ERK, see Li et al., 2012).
Overall, even when focusing only on ion channel function in axons, there exist many different cellular mechanisms to modulate axon excitability and synaptic transmission. While many of these mechanisms were initially identified due to their functional consequences of regulating vesicle release, we continue to gain insight into how modulation of ion channel biophysical properties affects information transmission across synapses. Further characterization of synaptic transmission before and after neuromodulation will help in identifying the functional role that these mechanisms play in vivo.
Functional Consequences
Neuromodulation of synaptic transmission at the presynaptic axon results in marked changes in information transfer from presynaptic to postsynaptic target. Neuromodulation can affect this process on multiple timescales, affecting three key aspects of transmission at the terminal: (1) synaptic strength, (2) synaptic variability, and (3) STP. By regulating presynaptic calcium influx and vesicle release, axonal neuromodulation can control the strength of specific subsets of synaptic inputs relative to others. But because transmitter release is probabilistic, it also regulates the extent to which individual presynaptic APs are reliably transmitted to the postsynaptic cell. Finally, regulation of STP controls a release site’s strength as a dynamic function of its past activity, which can create a frequency-sensitive postsynaptic signal from a sequence of all-or-none presynaptic APs. Considering synaptic neuromodulation from this functional lens can lead to insight into how cellular mechanisms in the axon might be employed in neural information processing in vivo.
Synaptic Strength and Variability
Neuromodulatory GPCRs can exert fast and reversible control of synaptic strength by altering the probability of neurotransmitter release, Pr. In order to focus on broader functional impacts, we will define Pr at the level of the synapse, i.e., the probability that the active zone successfully releases a vesicle of neurotransmitter in response to an AP. This quantity is the product of many complex underlying variables, including the number of vesicles available for release, their distance to sources of calcium influx, and the probability of release of that each of these vesicles as a function of intracellular calcium. Within this framework, synaptic strength is therefore defined as the product of Pr, the number of release sites N, and the quantal size q (i.e., the amount of postsynaptic current generated from one vesicle).
Changes to both presynaptic release through modification of Pr and postsynaptic sensitivity to neurotransmitter q will change synaptic strength linearly. Importantly, however, this is only true on average, because presynaptic neuromodulation also changes the variability of the synapse, whereas postsynaptic modifications of q do not (Figure 2A; for comprehensive review of Pr and synaptic variability, see Branco and Staras, 2009). This variability in vesicle release is a major source of overall variability in synaptic transmission, and due to the non-linear relationship between calcium influx and Pr, neuromodulators that regulate presynaptic calcium influx will strongly influence both synaptic strength and variance.
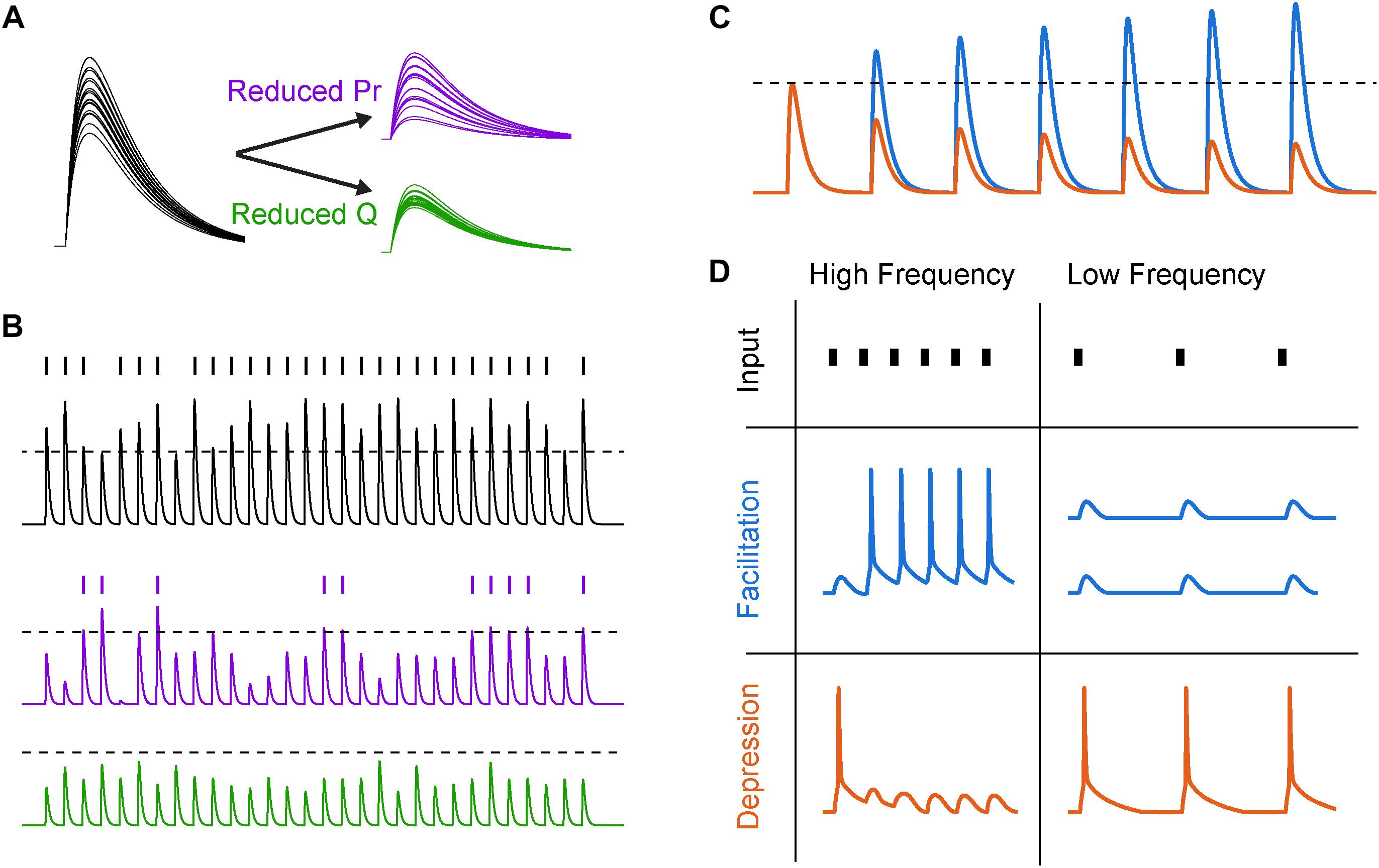
Figure 2. Synaptic variability and short-term plasticity (STP). (A) Synaptic variability is differentially affected by presynaptic and postsynaptic neuromodulation. Twenty synaptic potential amplitudes were simulated as a result of a binomial process with Gaussian noise under different conditions. Left, under baseline conditions with several release sites and high release probability (N = 5, Pr = 0.8), synaptic responses are large and reliable. Right, top, after reducing release probability by 60%, the trial-to-trial variability increases significantly. Right, bottom, reducing postsynaptic charge transfer per vesicle q by the same fraction does not lead to an increase in synaptic variability. (B) Increased synaptic variability counteracts mean amplitude suppression for simulated presynaptic, but not postsynaptic, mechanisms of neuromodulation. Top, a similar binomial process as in (A) was used to generate a sequence of simulated synaptic potentials. An arbitrary AP threshold is shown as a dotted line, with the raster plot above indicating crossings of that threshold. Middle, after reducing release probability, the arbitrary threshold is still crossed but at a lower rate. Bottom, after reducing postsynaptic charge transfer per vesicle, the synapse can no longer cross threshold. (C) Schematized subthreshold synaptic potentials in response to a series of presynaptic APs for a facilitating (blue) and depressing (orange) synapse. Dotted line indicates expected synaptic potential amplitude in the absence of STP. (D) Responses to high- and low-frequency synaptic input under different STP dynamics. In first row, tick marks indicate timing of presynaptic APs. In second and third row, traces indicate postsynaptic AP generation in response to a strong synapse with facilitating and depressing STP dynamics, respectively.
While the importance of average synaptic strength in information transmission may be intuitively clear, the role of synaptic variability may be less obvious. Naively, an increase in synaptic variability through reductions in Pr might be predicted to make the synaptic signals noisier, degrading the ability of the synapse to convey information about presynaptic AP trains to postsynaptic targets (Zador, 1998; but see Goldman, 2004). However, this may depend on how those synaptic signals are integrated in the postsynaptic cell. For example, in a cell where very few strong input synapses drive APs, reducing q below AP threshold could completely abolish postsynaptic spiking, whereas reducing Pr would simply reduce the likelihood of AP generation (Figure 2B). If synapses are modeled as simple binomial processes, then the result of reducing q is a sharp reduction of spiking near the AP threshold non-linearity, contrasted with a more gradual reduction in postsynaptic AP generation when Pr is reduced. In the case of strong q suppression (Figure 2B, bottom), the postsynaptic spike sequence then carries no information about the sequence of synaptic inputs, whereas in the latter case of modulating Pr this information transmission is merely diminished.
This generalization does not, however, extend to all circumstances. First, it ignores non-linearities in dendritic integration, where synchronized excitatory inputs can generate dendritic calcium or NMDA spikes that are either independent of or coordinated with spikes generated in the AIS (Larkum et al., 1999; Schiller et al., 2000; Branco and Häusser, 2011). Whether the neuromodulation of synaptic variability impacts dendritic spikes similarly to how it impacts AIS spikes remains unstudied. Second, for cells that receive many weak synaptic inputs with low Pr (e.g., some synapses between neocortical excitatory neurons; Markram et al., 1997), the effects of synaptic variability are more complex and depend on the voltage of the postsynaptic membrane. If the postsynaptic membrane voltage is close to AP threshold, an increase in synaptic variability can push weaker signals over the AP threshold and increase postsynaptic output that is correlated with the weak signal; conversely, if the postsynaptic cell is well above threshold, synaptic noise and increased membrane conductance can lead to reduced output (Shu et al., 2003; Azouz, 2005). In this way, a neuromodulatory weakening of Pr and increase in synaptic variability can surprisingly improve the encoding capability of a postsynaptic neuron by broadening its dynamic range (Silver, 2010). Thus, modulation of synaptic strength through modification of release probability can have many indirect effects on neuronal information processing beyond linear changes in synaptic strength.
Short-Term Plasticity
At many synapses throughout the brain, the strength of a single synapse strength is a function of its previous activity (Figures 2C,D). This process of use-dependent regulation of vesicle release, termed STP, can be altered markedly by neuromodulatory GPCRs. STP depends on many factors, including variable Pr, availability of readily releasable vesicles, and AP waveform adaptation with repeated activity. For example, short-term facilitation is mediated through specific isoforms of calcium sensing proteins including synaptotagmin 7, which increase Pr in response to relatively low levels of calcium (Jackman et al., 2016). Because the concentration of free calcium in the bouton decays on the timescale of tens of milliseconds (Brenowitz and Regehr, 2007), these proteins allow the calcium from recent APs to non-linearly boost vesicle release over physiologically relevant frequencies of APs. However, the pool of readily releasable neurotransmitter vesicles is finite and depletion of this pool can lead to activity-dependent short-term depression of transmitter release when presynaptic AP rates are high (Betz, 1970; for review see Zucker and Regehr, 2002).
The interplay between these mechanisms of facilitation and depression (and many others) can lead to frequency-dependent filtering of synaptic strength (Tsodyks and Markram, 1997; Dittman et al., 2000). This transformation of presynaptic spike trains into graded, frequency-dependent postsynaptic signals has many functional use cases. For example, facilitating synapses can effectively propagate bursts and minimize low-frequency single APs, which may serve to enhance the signal-to-noise ratio of bursts of activity that encode behaviorally relevant information (Laviolette et al., 2005; Burgos-Robles et al., 2007; Holmes et al., 2012). Conversely, depressing synapses can subtract these bursts from the sequence transmitted to the postsynaptic cell, for example as in adaptation to sensory stimuli (Chance et al., 1998; Chung et al., 2002). Depression has also been shown to implement gain control over inputs with different baseline activity levels, allowing for the postsynaptic cell to respond to relative, rather than absolute, changes in input firing rate (Abbott et al., 1997). Finally, combinations of facilitation and depression can implement band-pass filtering, where activity would be weakened both above and below a characteristic frequency band. Temporal filtering of this sort can enhance responses to transient sensory stimuli (Chance et al., 1998) and may match frequency bands of behaviorally relevant circuit oscillations (Pietersen et al., 2009).
Canonical Presynaptic Neuromodulation
Short-term plasticity is often regulated in parallel with synaptic strength when a neuromodulator modifies presynaptic calcium influx. For example, changes in calcium influx will affect the calcium sensors that mediate short-term facilitation, but changes in Pr will also affect the extent of depletion-mediated short-term depression. In fact, these effects can both occur simultaneously to conceal each other. For example, at high-Pr synapses short-term facilitation can be limited by vesicle depletion. At such a synapse, reductions in calcium influx can both reduce Pr and unveil underlying facilitation (Zucker and Regehr, 2002; Sakaba, 2006). Indeed, reductions in Pr with parallel increases in short-term facilitation as measured by the “paired-pulse ratio” (PPR) are so common that they are considered a hallmark of presynaptic neuromodulation of release (Dittman and Regehr, 1998).
A common example of synaptic suppression with changes in STP is activation of presynaptic GABAB receptors. As described above, GABAB receptor activation suppresses presynaptic calcium currents via Gβγ-dependent signaling (Mintz and Bean, 1993; Wu and Saggau, 1995). Alongside other calcium-independent mechanisms (Dittman and Regehr, 1996; Sakaba and Neher, 2003), this leads to a potent suppression of release probability while increasing short-term facilitation of subsequent events (Chalifoux and Carter, 2011; Burke et al., 2018). This combination of effects has been hypothesized to support faithful transmission at high frequencies in auditory brainstem (Brenowitz et al., 1998). The behavioral role that this shift in STP plays is unclear; while genetic ablation of the GABAB receptor appears to reduce both anxiety and depressive phenotypes in mice (Mombereau et al., 2004), these mutations likely silence GABAB receptor expression at all subcellular locations. Better tools to disentangle the many cellular functions of the GABAB receptor (including increasing postsynaptic conductance, hyperpolarizing postsynaptic voltage, reducing Pr, and increasing facilitation) will be required to attribute behavioral effects to these transformations of synaptic function. Furthermore, tools to precisely separate the two presynaptic effects of GABAB (reducing Pr and increasing facilitation) would allow us to better understand the function of other presynaptic neuromodulators that also employ Gβγ-dependent signaling pathways, such as the CB1 receptor. Alternatively, experiments that compare activation of presynaptic neuromodulators with different effects on synaptic transmission (e.g., two presynaptic inhibitors of Pr that differ in effects on facilitation) could more clearly tie these effects on STP to circuit activity and behavior.
Non-canonical Presynaptic Neuromodulation
While Gβγ-dependent signaling canonically regulates both synaptic strength and STP, which we will term temporal modulation, some presynaptic neuromodulators appear to regulate Pr in isolation, which we will term gain modulation. These latter cases violate the dogma that presynaptic modulation also regulates PPR and demonstrate that temporal and gain modulation do not map cleanly onto presynaptic and postsynaptic mechanisms, respectively. Indeed, any form of modulation that essentially silences synapses (permanently or stochastically) can functionally be characterized as gain modulation. This includes both presynaptic mechanisms (e.g., axonal AP propagation failures, removal of synapses or active zones, slowly dissociating CaV antagonists) as well as postsynaptic mechanisms (e.g., changes in postsynaptic receptor density or ion channel amplification of synaptic potentials). Similarly, while presynaptic mechanisms to modulate facilitation and depression have been extensively documented, some postsynaptic mechanisms have also been implicated (e.g., local reduction in synaptic driving force; Abrahamsson et al., 2012). Experimental identification of presynaptic or postsynaptic mechanisms therefore require more than simply measuring changes in levels of facilitation or depression (e.g., PPR) and should include other more direct measurements of release probability, such as measurements of synaptic variance (Sigworth, 1980; Saviane and Silver, 2006; Delaney et al., 2007) or optical measurements of transmission at individual synapses (e.g., optical quantal analysis, Oertner et al., 2002; Higley et al., 2009; Little and Carter, 2012; Sylantyev et al., 2013; Boddum et al., 2016; Burke et al., 2018).
One common mechanism for observing presynaptic gain modulation is direct blockade of calcium channels using slowly dissociating antagonists such as cadmium or conotoxin-MVIIC (Hefft et al., 2002; Hjelmstad, 2004; Scimemi and Diamond, 2012). Several neuromodulatory systems have also been found to exert similar effects, including D1/D5 in prefrontal cortex (Gao et al., 2001; Seamans et al., 2001; Burke et al., 2018), D1/D5 at the perforant pathway (Behr et al., 2000), kappa-opioid receptors at amygdalar inputs to the nucleus accumbens (Tejeda et al., 2017), kappa-opioid receptors in the bed nucleus of the stria terminalis (Li et al., 2012), and noradrenergic receptors in central amygdala (Delaney et al., 2007). While these neuromodulators likely employ different signaling pathways, they share in common the regulation of Pr apparently without changing STP. Importantly, this presynaptic neuromodulation only of synaptic strength is different from temporal modulation in how it transforms information as it is transmitted across the synapse. While canonical Gβγ-dependent signaling suppresses transmission from low-frequency presynaptic APs while preserving or enhancing high-frequency transmission, these non-canonical gain neuromodulators that only regulate Pr serve to control the average strength of the synapse while preserving the relative strength of transmitted frequencies.
Mechanisms of presynaptic gain modulation can be grouped into two major categories: first, changes in the number of vesicles available for release, and second, changes in release probability per vesicle. An example of this first category is found at the neuromuscular junction of Drosophila, where homeostatic adaptation to postsynaptic glutamate receptor blockade is achieved through an enhancement in the size of the readily releasable pool of vesicles (Weyhersmuller et al., 2011). This adaptation is best described as gain modulation because the change in short-term facilitation was significantly smaller than the change in synaptic strength. However, as noted earlier, a finite RRP can lead to vesicle depletion with activity, which leads to frequency-dependent short-term depression (Zucker and Regehr, 2002; Sakaba, 2006). Increasing RRP size could hypothetically remove this depression and unmask facilitation, thus acting as temporal modulation; therefore, increases in RRP size may only implement gain modulation if depletion is not a significant factor under baseline conditions.
An example of the second category, presynaptic gain modulation through changes in Pr, was recently observed at excitatory synapses in prefrontal cortex (Burke et al., 2018), and may underlie similar observations made with kappa opioid receptor-dependent modulation in subcortical regions (Li et al., 2012; Tejeda et al., 2017). Here, presynaptic release probability was suppressed by activation of dopaminergic D1 receptors. Unlike some other forms of presynaptic gain modulation, however, the underlying mechanism was a reduction in calcium influx. Interestingly, typical mechanisms that reduce presynaptic calcium, such as a reduction in extracellular calcium concentration, often appear as temporal modulation with strongly correlated with changes in facilitation (Zucker and Regehr, 2002). The gain modulation by the D1 receptor represents an important caveat to this common pattern; because D1 receptor activation suppressed calcium channel open probability, the functional consequence of this modulation was to reduce release probability in a nearly “all-or-none” fashion. Thus, the effect of this modulation bears more resemblance to an AP conduction failure than a “canonical” reduction in AP-evoked calcium channel currents.
Function Following Form at Nanodomain Synapses
Why do some neuromodulators that regulate Pr also regulate STP, whereas others do not? As mentioned earlier, the size of the RRP and the extent of vesicle depletion could be one explanation. Another is the functional coupling of CaVs and vesicles at active zones (Figure 3). The presynaptic configuration where release of each vesicle is driven by calcium influx through many CaVs is termed a calcium microdomain; when release is driven by very few CaVs per vesicle, this is termed a calcium nanodomain (for review, see Stanley, 2016). Regulation of the probability of individual CaV channel opening has been shown to lead to gain and temporal modulation in nanodomain and microdomain configurations, respectively; by contrast, CaV calcium influx per AP can lead to temporal modulation under both configurations (Figures 3B,C; Otis and Trussell, 1996; Hefft et al., 2002; Hjelmstad, 2004; Eggermann et al., 2012; Scimemi and Diamond, 2012; Burke et al., 2018). Thus, at synapses with nanodomain coupling between CaVs and vesicles, the precise mechanism by which CaVs are modulated can dictate whether a neuromodulator causes gain or temporal modulation. In other words, any observations made where a neuromodulator employs a mix of apparent pre- and postsynaptic mechanisms (e.g., an increase in CV without an increase in PPR) should be couched aside experiments designed to assess whether the same synapses utilize nano- or microdomain release mechanisms.
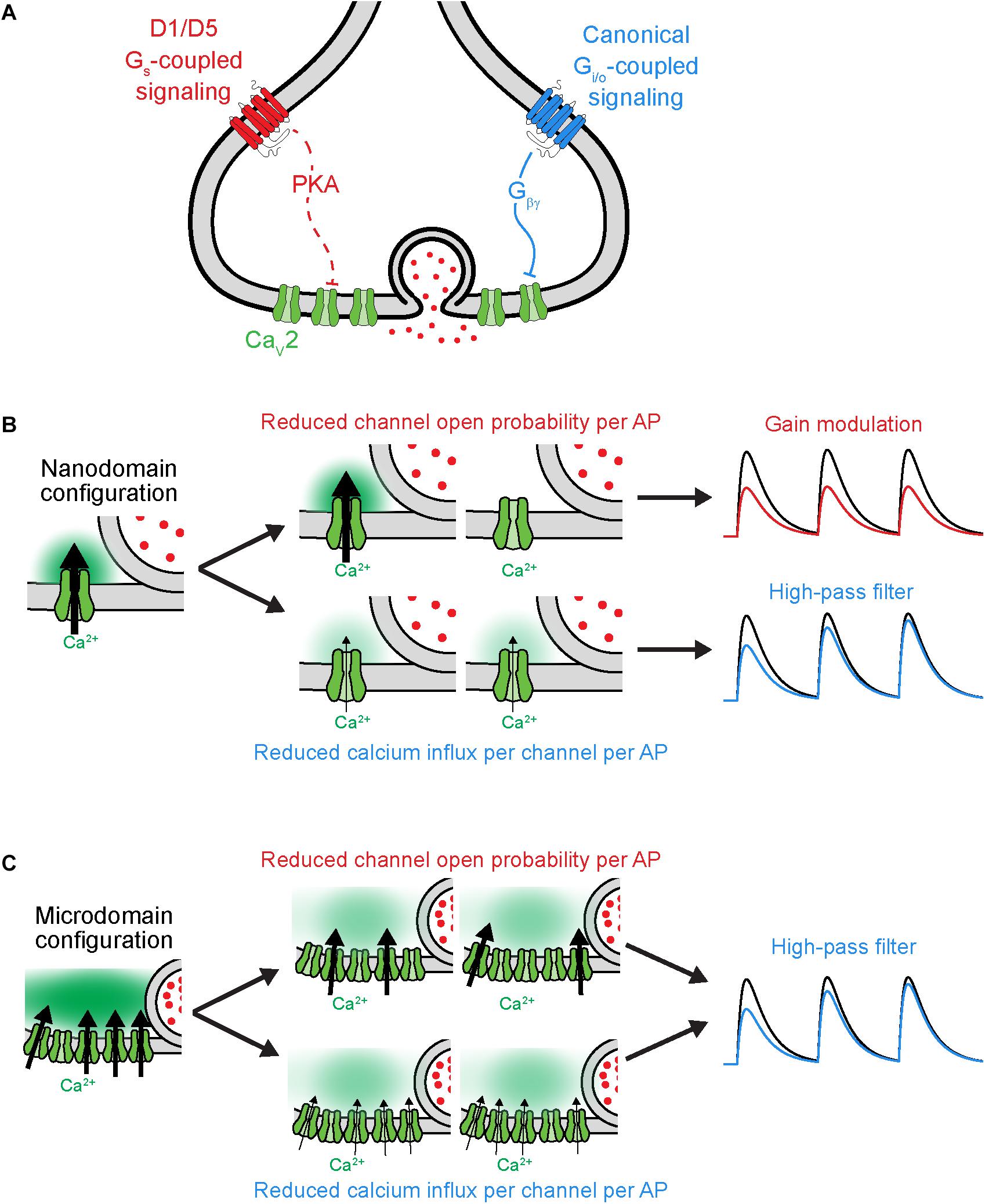
Figure 3. Distinct functional consequences for CaV2 neuromodulation at nanodomain synapses. (A) Schematic of GPCR neuromodulation of CaV2 at axonal bouton. (B) Functional consequences of different forms of CaV2 regulation under nanodomain coupling between CaV2 and vesicles. Left, schematic of nanodomain coupling indicating calcium influx through one channel influencing release. Center, top, two possible outcomes resulting from reduced open probability of channel. Center, bottom, two outcomes resulting from reducing the current influx through each channel. Right, result of two different forms of CaV2 regulation on rate of vesicle release (schematized as postsynaptic potential). Note that the two mechanisms of CaV2 neuromodulation lead to distinct effects under nanodomain coupling. Black and colorized traces indicate baseline transmission and post-CaV2 regulation, respectively. (C) As in (B), but under microdomain coupling between CaV2 and vesicles. Note that the two mechanisms of CaV2 neuromodulation lead to similar effects on vesicle release under microdomain coupling. Schematics adapted from Burke et al. (2018).
The neuromodulation of synaptic strength has been understudied in the context of functional coupling between CaVs and vesicles. The aforementioned experiments examining D1 receptor modulation in PFC suggested a critical role of CaV-vesicle functional coupling in its effects on synaptic transmission (Burke et al., 2018). These synapses appear to employ a nanodomain release configuration in which differential CaV recruitment per AP affects STP (Scimemi and Diamond, 2012). At these synapses in PFC, D1 receptor activation reduced individual CaV channel open probability at the presynaptic axon, leading to gain modulation of synaptic transmission, whereas activation of the GABAB receptor reduced CaV current per AP and led to temporal modulation. Taken together, these data led to the hypothesis that D1 activation would modulate postsynaptic responses to all input frequencies similarly, whereas GABAB activation would preferentially suppress responses to low-frequency inputs. Experiments quantifying postsynaptic AP generation confirmed this hypothesis. Whether this functional distinction between gain and temporal modulation extends to naturalistic patterns of activity seen in vivo, and what functional role it may play in circuit neuromodulation, has yet to be investigated.
Future Directions
While the use of fixed frequency synaptic stimulation is both common and important in investigations of cellular mechanisms of synaptic transmission, it also likely obscures how STP and its neuromodulation affect the noisy and irregular patterns of active neurons in vivo. Similarly, many effects of neuromodulation observed in ex vivo acute slice preparations have yet to be linked to specific behavioral outcomes in vivo. Recent work has begun to investigate how different synaptic mechanisms contribute to neuronal computations in vivo (Bolding and Franks, 2018; Evans et al., 2018; Lien and Scanziani, 2018). As the experimental technology to measure neuronal activity in vivo continues to improve, it is becoming increasingly possible to bridge the gap between mechanistic studies in vitro and functional studies in vivo and better incorporate synaptic neuromodulation into theories of neuronal circuit function. The highly non-linear behavior of synapses, including their variability, short-term dynamics and complex neuromodulation, are likely to be a critical component of future research into how cellular processes impact neural circuits and behavior. It will be critical to move from mere identification of synaptic connectivity to the identification of the neural codes employed by these connections, as well as identification of when, where, and how neuromodulators are engaged in vivo. To achieve this, new tools that can provide real-time measurements of neuromodulatory activity, including sensors for neurotransmitters (Jing et al., 2018; Marvin et al., 2018; Patriarchi et al., 2018; Sun et al., 2018) and intracellular signaling molecules (Violin et al., 2003; Jiang et al., 2017; Ma et al., 2018), will be critical. These tools can be complemented by methods to measure and manipulate activity patterns at individual neurons and even individual synapses (Lee et al., 2019), including both genetically and functionally defined neural circuits (Guenthner et al., 2013; Grewe et al., 2017; Wang et al., 2017; Parker et al., 2018). Together, these technological advances will allow us to quantify how neuromodulators regulate complex activity patterns as they propagate throughout synaptic networks. Moreover, more precise tools are needed to dissect the relative contributions of pre- and post-synaptic effects of neuromodulatory GPCRs, for example distinguishing the functional roles of GABAB activation in changing postsynaptic conductance versus presynaptic Pr and STP (Zurawski et al., 2018). The ability to disentangle these many distinct effects will be critical in developing a quantifiable and falsifiable theory of the role neuromodulation plays in synaptic computation.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by grants from the NIH (DA035913, MH112117, and MH112729).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Anna Lipkin and other members of the Bender Lab for comments and feedback.
References
Abbott, L. F., Varela, J. A., Sen, K., and Nelson, S. B. (1997). Synaptic depression and cortical gain control. Science 275, 221–224 doi: 10.1126/science.275.5297.221
Abrahamsson, T., Cathala, L., Matsui, K., Shigemoto, R., and DiGregorio, D. A. (2012). Thin dendrites of cerebellar interneurons confer sublinear synaptic integration and a gradient of short-term plasticity. Neuron 73, 1159–1172. doi: 10.1016/j.neuron.2012.01.027
Agnati, L. F., Zoli, M., Strömberg, I., and Fuxe, K. (1995). Intercellular communication in the brain: wiring versus volume transmission. Neuroscience 69, 711–726. doi: 10.1016/0306-4522(95)00308-6
Ahmed, M. S., and Siegelbaum, S. A. (2009). Recruitment of N-Type Ca2+ channels during LTP enhances low release efficacy of hippocampal CA1 perforant path synapses. Neuron 63, 372–385. doi: 10.1016/j.neuron.2009.07.013
Alger, B. E., Pitler, T. A., Wagner, J. J., Martin, L. A., Morishita, W., Kirov, S. A., et al. (1996). Retrograde signalling in depolarization-induced suppression of inhibition in rat hippocampal CA1 cells. J. Physiol. 496( Pt 1), 197–209. doi: 10.1113/jphysiol.1996.sp021677
Alle, H., and Geiger, J. R. P. (2006). Combined analog and action potential coding in hippocampal mossy fibers. Science 311, 1290–1293. doi: 10.1126/science.1119055
Alpizar, S. A., Cho, I. H., and Hoppa, M. B. (2019). Subcellular control of membrane excitability in the axon. Curr. Opin. Neurobiol. 57, 117–125. doi: 10.1016/j.conb.2019.01.020
Altier, C., Khosravani, H., Evans, R. M., Hameed, S., Peloquin, J. B., Vartian, B. A., et al. (2006). ORL1 receptor–mediated internalization of N-type calcium channels. Nat. Neurosci. 9, 31–40. doi: 10.1038/nn1605
Athilingam, J. C., Ben-Shalom, R., Keeshen, C. M., Sohal, V. S., and Bender, K. J. (2017). Serotonin enhances excitability and gamma frequency temporal integration in mouse prefrontal fast-spiking interneurons. eLife 6:e31991. doi: 10.7554/eLife.31991
Augustine, G. J. (2001). How does calcium trigger neurotransmitter release? Curr. Opin. Neurobiol. 11, 320–326. doi: 10.1016/s0959-4388(00)00214-2
Awatramani, G. B., Price, G. D., and Trussell, L. O. (2005). Modulation of transmitter release by presynaptic resting potential and background calcium levels. Neuron 48, 109–121. doi: 10.1016/j.neuron.2005.08.038
Azouz, R. (2005). Dynamic spatiotemporal synaptic integration in cortical neurons: neuronal gain, revisited. J. Neurophysiol. 94, 2785–2796. doi: 10.1152/jn.00542.2005
Ballo, A. W., Keene, J. C., Troy, P. J., Goeritz, M. L., Nadim, F., and Bucher, D. (2010). Dopamine modulates Ih in a motor axon. J. Neurosci. 30, 8425–8434. doi: 10.1523/JNEUROSCI.0405-10.2010
Ballo, A. W., Nadim, F., and Bucher, D. (2012). Dopamine modulation of ih improves temporal fidelity of spike propagation in an unmyelinated axon. J. Neurosci. 32, 5106–5119. doi: 10.1523/JNEUROSCI.6320-11.2012
Bay, V., and Butt, A. M. (2012). Relationship between glial potassium regulation and axon excitability: a role for glial Kir4.1 channels. Glia 60, 651–660. doi: 10.1002/glia.22299
Bean, B. P. (1989). Neurotransmitter inhibition of neuronal calcium currents by changes in channel voltage dependence. Nature 340, 153–156. doi: 10.1038/340153a0
Behr, J., Gloveli, T., Schmitz, D., and Heinemann, U. (2000). Dopamine depresses excitatory synaptic transmission onto rat subicular neurons via presynaptic D1-like dopamine receptors. J. Neurophysiol. 84, 112–119. doi: 10.1152/jn.2000.84.1.112
Bellot-Saez, A., Kékesi, O., Morley, J. W., and Buskila, Y. (2017). Astrocytic modulation of neuronal excitability through K+ spatial buffering. Neurosci. Biobehav. Rev. 77, 87–97. doi: 10.1016/j.neubiorev.2017.03.002
Bender, K. J., Ford, C. P., and Trussell, L. O. (2010). Dopaminergic modulation of axon initial segment calcium channels regulates action potential initiation. Neuron 68, 500–511. doi: 10.1016/j.neuron.2010.09.026
Bender, K. J., and Trussell, L. O. (2009). Axon initial segment Ca2+ channels influence action potential generation and timing. Neuron 61, 259–271. doi: 10.1016/j.neuron.2008.12.004
Bender, K. J., and Trussell, L. O. (2012). The physiology of the axon initial segment. Annu. Rev. Neurosci. 35, 249–265. doi: 10.1146/annurev-neuro-062111-150339
Bender, K. J., Uebele, V. N., Renger, J. J., and Trussell, L. O. (2012). Control of firing patterns through modulation of axon initial segment T-type calcium channels. J. Physiol. 590, 109–118. doi: 10.1113/jphysiol.2011.218768
Betz, W. J. (1970). Depression of transmitter release at the neuromuscular junction of the frog. J. Physiol. 206, 629–644. doi: 10.1113/jphysiol.1970.sp009034
Boddum, K., Jensen, T. P., Magloire, V., Kristiansen, U., Rusakov, D. A., Pavlov, I., et al. (2016). Astrocytic GABA transporter activity modulates excitatory neurotransmission. Nat. Commun. 7:13572. doi: 10.1038/ncomms13572
Bolding, K. A., and Franks, K. M. (2018). Recurrent cortical circuits implement concentration-invariant odor coding. Science 361:eaat6904. doi: 10.1126/science.aat6904
Branco, T., and Häusser, M. (2011). Synaptic integration gradients in single cortical pyramidal cell dendrites. Neuron 69, 885–892. doi: 10.1016/j.neuron.2011.02.006
Branco, T., and Staras, K. (2009). The probability of neurotransmitter release: variability and feedback control at single synapses. Nat. Rev. Neurosci. 10, 373–383. doi: 10.1038/nrn2634
Brenowitz, S., David, J., and Trussell, L. (1998). Enhancement of synaptic efficacy by presynaptic GABA(B) receptors. Neuron 20, 135–141. doi: 10.1016/s0896-6273(00)80441-9
Brenowitz, S. D., and Regehr, W. G. (2007). Reliability and heterogeneity of calcium signaling at single presynaptic boutons of cerebellar granule cells. J. Neurosci. 27, 7888–7898. doi: 10.1523/jneurosci.1064-07.2007
Brown, S. P., Safo, P. K., and Regehr, W. G. (2004). Endocannabinoids inhibit transmission at granule cell to purkinje cell synapses by modulating three types of presynaptic calcium channels. J. Neurosci. 24, 5623–5631. doi: 10.1523/jneurosci.0918-04.2004
Brunger, A. T., Choi, U. B., Lai, Y., Leitz, J., and Zhou, Q. (2018). Molecular mechanisms of fast neurotransmitter release. Annu. Rev. Biophys. 47, 469–497. doi: 10.1146/annurev-biophys-070816-034117
Bucurenciu, I., Bischofberger, J., and Jonas, P. (2010). A small number of open Ca2+ channels trigger transmitter release at a central GABAergic synapse. Nat. Neurosci. 13, 19–21. doi: 10.1038/nn.2461
Burgos-Robles, A., Vidal-Gonzalez, I., Santini, E., and Quirk, G. J. (2007). Consolidation of fear extinction requires NMDA receptor-dependent bursting in the ventromedial prefrontal cortex. Neuron 53, 871–880. doi: 10.1016/j.neuron.2007.02.021
Burke, K. J., Keeshen, C. M., and Bender, K. J. (2018). Two forms of synaptic depression produced by differential neuromodulation of presynaptic calcium channels. Neuron 99, 969–984.e7. doi: 10.1016/j.neuron.2018.07.030
Cain, S. M., and Snutch, T. P. (2013). T-type calcium channels in burst-firing, network synchrony, and epilepsy. Biochim. Biophys. Acta 1828, 1572–1578. doi: 10.1016/j.bbamem.2012.07.028
Cao, Y.-Q., and Tsien, R. W. (2010). Different relationship of N- and P/Q-type Ca2+ channels to channel-interacting slots in controlling neurotransmission at cultured hippocampal synapses. J. Neurosci. 30, 4536–4546. doi: 10.1523/JNEUROSCI.5161-09.2010
Catterall, W. A., and Few, A. P. (2008). Calcium channel regulation and presynaptic plasticity. Neuron 59, 882–901. doi: 10.1016/j.neuron.2008.09.005
Chalifoux, J. R., and Carter, A. G. (2011). GABAB receptor modulation of synaptic function. Curr. Opin. Neurobiol. 21, 339–344. doi: 10.1016/j.conb.2011.02.004
Chance, F. S., Nelson, S. B., and Abbott, L. F. (1998). Synaptic depression and the temporal response characteristics of V1 cells. J. Neurosci. 18, 4785–4799. doi: 10.1523/jneurosci.18-12-04785.1998
Christie, J. M., Chiu, D. N., and Jahr, C. E. (2011). Ca2+-dependent enhancement of release by subthreshold somatic depolarization. Nat. Neurosci. 14, 62–68. doi: 10.1038/nn.2718
Chung, S., Li, X., and Nelson, S. B. (2002). Short-term depression at thalamocortical synapses contributes to rapid adaptation of cortical sensory responses in vivo. Neuron 34, 437–446. doi: 10.1016/s0896-6273(02)00659-1
Clarkson, R. L., Liptak, A. T., Gee, S. M., Sohal, V. S., and Bender, K. J. (2017). D3 receptors regulate excitability in a unique class of prefrontal pyramidal cells. J. Neurosci. 37, 5846–5860. doi: 10.1523/JNEUROSCI.0310-17.2017
Colecraft, H. M., Brody, D. L., and Yue, D. T. (2001). G-protein inhibition of N- and P/Q-type calcium channels: distinctive elementary mechanisms and their functional impact. J. Neurosci. 21, 1137–1147. doi: 10.1523/jneurosci.21-04-01137.2001
Colecraft, H. M., Patil, P. G., and Yue, D. T. (2000). Differential occurrence of reluctant openings in G-protein-inhibited N- and P/Q-type calcium channels. J. Gen. Physiol. 115, 175–192. doi: 10.1085/jgp.115.2.175
Cotel, F., Exley, R., Cragg, S. J., and Perrier, J.-F. (2013). Serotonin spillover onto the axon initial segment of motoneurons induces central fatigue by inhibiting action potential initiation. Proc. Natl. Acad. Sci. U.S.A. 110, 4774–4779. doi: 10.1073/pnas.1216150110
Courtney, N. A., and Ford, C. P. (2016). Mechanisms of 5-HT1A receptor-mediated transmission in dorsal raphe serotonin neurons. J. Physiol. 594, 953–965. doi: 10.1113/JP271716
Currie, K. P. M. (2010). G protein inhibition of CaV2 calcium channels. Channels 4, 497–509. doi: 10.4161/chan.4.6.12871
Daniel, H., and Crepel, F. (2001). Control of Ca(2+) influx by cannabinoid and metabotropic glutamate receptors in rat cerebellar cortex requires K(+) channels. J. Physiol. 537, 793–800. doi: 10.1113/jphysiol.2001.013221
Delaney, A. J., Crane, J. W., and Sah, P. (2007). Noradrenaline modulates transmission at a central synapse by a presynaptic mechanism. Neuron 56, 880–892. doi: 10.1016/j.neuron.2007.10.022
Diana, M. A., and Marty, A. (2003). Characterization of depolarization-induced suppression of inhibition using paired interneuron–Purkinje cell recordings. J. Neurosci. 23, 5906–5918. doi: 10.1523/jneurosci.23-13-05906.2003
Dittman, J. S., Kreitzer, A. C., and Regehr, W. G. (2000). Interplay between facilitation, depression, and residual calcium at three presynaptic terminals. J. Neurosci. 20, 1374–1385. doi: 10.1523/jneurosci.20-04-01374.2000
Dittman, J. S., and Regehr, W. G. (1996). Contributions of calcium-dependent and calcium-independent mechanisms to presynaptic inhibition at a cerebellar synapse. J. Neurosci. 16, 1623–1633. doi: 10.1523/jneurosci.16-05-01623.1996
Dittman, J. S., and Regehr, W. G. (1998). Calcium dependence and recovery kinetics of presynaptic depression at the climbing fiber to Purkinje cell synapse. J. Neurosci. 18, 6147–6162. doi: 10.1523/jneurosci.18-16-06147.1998
Dumenieu, M., Senkov, O., Mironov, A., Bourinet, E., Kreutz, M. R., Dityatev, A., et al. (2018). The low-threshold calcium channel Cav3.2 mediates burst firing of mature dentate granule cells. Cereb. Cortex 28, 2594–2609. doi: 10.1093/cercor/bhy084
Eggermann, E., Bucurenciu, I., Goswami, S. P., and Jonas, P. (2012). Nanodomain coupling between Ca2+ channels and sensors of exocytosis at fast mammalian synapses. Nat. Rev. Neurosci. 13, 7–21. doi: 10.1038/nrn3125
Éltes, T., Kirizs, T., Nusser, Z., and Holderith, N. (2017). Target cell type-dependent differences in Ca2+ channel function underlie distinct release probabilities at hippocampal glutamatergic terminals. J. Neurosci. 37, 1910–1924. doi: 10.1523/JNEUROSCI.2024-16.2017
Endo, K., and Yawo, H. (2000). mu-Opioid receptor inhibits N-type Ca2+ channels in the calyx presynaptic terminal of the embryonic chick ciliary ganglion. J. Physiol. 524(Pt 3), 769–781. doi: 10.1111/j.1469-7793.2000.00769.x
Ermolyuk, Y. S., Alder, F. G., Surges, R., Pavlov, I. Y., Timofeeva, Y., Kullmann, D. M., et al. (2013). Differential triggering of spontaneous glutamate release by P/Q-, N- and R-type Ca2+ channels. Nat. Neurosci. 16, 1754–1763. doi: 10.1038/nn.3563
Evans, D. A., Stempel, A. V., Vale, R., Ruehle, S., Lefler, Y., and Branco, T. (2018). A synaptic threshold mechanism for computing escape decisions. Nature 558, 590–594. doi: 10.1038/s41586-018-0244-6
Faas, G. C., Adwanikar, H., Gereau, R. W., and Saggau, P. (2002). Modulation of presynaptic calcium transients by metabotropic glutamate receptor activation: a differential role in acute depression of synaptic transmission and long-term depression. J. Neurosci. 22, 6885–6890. doi: 10.1523/jneurosci.22-16-06885.2002
Gantz, S. C., Bunzow, J. R., and Williams, J. T. (2013). Spontaneous inhibitory synaptic currents mediated by a G Protein-coupled receptor. Neuron 78, 807–812. doi: 10.1016/j.neuron.2013.04.013
Gao, W.-J., Krimer, L. S., and Goldman-Rakic, P. S. (2001). Presynaptic regulation of recurrent excitation by D1 receptors in prefrontal circuits. Proc. Natl. Acad. Sci. U.S.A. 98, 295–300. doi: 10.1073/pnas.011524298
Geiger, J. R. P., and Jonas, P. (2000). Dynamic control of presynaptic Ca2+ inflow by fast-inactivating K+ channels in hippocampal mossy fiber boutons. Neuron 28, 927–939. doi: 10.1016/s0896-6273(00)00164-1
Goldman, M. S. (2004). Enhancement of information transmission efficiency by synaptic failures. Neural Comput. 16, 1137–1162. doi: 10.1162/089976604773717568
Grewe, B. F., Gründemann, J., Kitch, L. J., Lecoq, J. A., Parker, J. G., Marshall, J. D., et al. (2017). Neural ensemble dynamics underlying a long-term associative memory. Nature 543, 670–675. doi: 10.1038/nature21682
Grubb, M. S., and Burrone, J. (2010). Activity-dependent relocation of the axon initial segment fine-tunes neuronal excitability. Nature 465, 1070–1074. doi: 10.1038/nature09160
Guenthner, C. J., Miyamichi, K., Yang, H. H., Heller, H. C., and Luo, L. (2013). Permanent genetic access to transiently active neurons via TRAP: targeted recombination in active populations. Neuron 78, 773–784. doi: 10.1016/j.neuron.2013.03.025
Hefft, S., Kraushaar, U., Geiger, R. P. J., and Jonas, P. (2002). Presynaptic short-term depression is maintained during regulation of transmitter release at a GABAergic synapse in rat hippocampus. J. Physiol. 539, 201–208. doi: 10.1113/jphysiol.2001.013455
Hennig, M. H. (2013). Theoretical models of synaptic short term plasticity. Front. Comput. Neurosci. 7:45. doi: 10.3389/fncom.2013.00045
Higley, M. J., Soler-Llavina, G. J., and Sabatini, B. L. (2009). Cholinergic modulation of multivesicular release regulates striatal synaptic potency and integration. Nat. Neurosci. 12, 1121–1128. doi: 10.1038/nn.2368
Hjelmstad, G. O. (2004). Dopamine excites nucleus accumbens neurons through the differential modulation of glutamate and GABA release. J. Neurosci. 24, 8621–8628. doi: 10.1523/jneurosci.3280-04.2004
Hjelmstad, G. O., and Fields, H. L. (2003). Kappa opioid receptor activation in the nucleus accumbens inhibits glutamate and GABA release through different mechanisms. J. Neurophysiol. 89, 2389–2395. doi: 10.1152/jn.01115.2002
Hoffman, A. F., and Lupica, C. R. (2000). Mechanisms of cannabinoid inhibition of GABA(A) synaptic transmission in the hippocampus. J. Neurosci. 20, 2470–2479. doi: 10.1523/jneurosci.20-07-02470.2000
Holmes, A., Fitzgerald, P. J., MacPherson, K. P., DeBrouse, L., Colacicco, G., Flynn, S. M., et al. (2012). Chronic alcohol remodels prefrontal neurons and disrupts NMDAR-mediated fear extinction encoding. Nat. Neurosci. 15, 1359–1361. doi: 10.1038/nn.3204
Huang, C. C., Lo, S. W., and Hsu, K. S. (2001). Presynaptic mechanisms underlying cannabinoid inhibition of excitatory synaptic transmission in rat striatal neurons. J. Physiol. 532, 731–748. doi: 10.1111/j.1469-7793.2001.0731e.x
Huang, C. Y.-M., and Rasband, M. N. (2018). Axon initial segments: structure, function, and disease. Ann. N. Y. Acad. Sci. 1420, 46–61. doi: 10.1111/nyas.13718
Jackman, S. L., Turecek, J., Belinsky, J. E., and Regehr, W. G. (2016). The calcium sensor synaptotagmin 7 is required for synaptic facilitation. Nature 529, 88–91. doi: 10.1038/nature16507
Jiang, J. Y., Falcone, J. L., Curci, S., and Hofer, A. M. (2017). Interrogating cyclic AMP signaling using optical approaches. Cell Calcium 64, 47–56. doi: 10.1016/j.ceca.2017.02.010
Jing, M., Zhang, P., Wang, G., Feng, J., Mesik, L., Zeng, J., et al. (2018). A genetically encoded fluorescent acetylcholine indicator for in vitro and in vivo studies. Nat. Biotechnol. 36, 726–737. doi: 10.1038/nbt.4184
Katz, B., and Miledi, R. (1970). Further study of the role of calcium in synaptic transmission. J. Physiol. 207, 789–801. doi: 10.1113/jphysiol.1970.sp009095
Kawaguchi, S., and Sakaba, T. (2015). Control of inhibitory synaptic outputs by low excitability of axon terminals revealed by direct recording. Neuron 85, 1273–1288. doi: 10.1016/j.neuron.2015.02.013
Khaliq, Z. M., and Raman, I. M. (2005). Axonal propagation of simple and complex spikes in cerebellar purkinje neurons. J. Neurosci. 25, 454–463. doi: 10.1523/jneurosci.3045-04.2005
Kia, H. K., Brisorgueil, M.-J., Hamon, M., Calas, A., and Vergé, D. (1996). Ultrastructural localization of 5-hydroxytryptamine1A receptors in the rat brain. J. Neurosci. Res. 46, 697–708. doi: 10.1002/(sici)1097-4547(19961215)46:6<697::aid-jnr7>3.3.co;2-n
Kisilevsky, A. E., Mulligan, S. J., Altier, C., Iftinca, M. C., Varela, D., Tai, C., et al. (2008). D1 receptors physically interact with N-type calcium channels to regulate channel distribution and dendritic calcium entry. Neuron 58, 557–570. doi: 10.1016/j.neuron.2008.03.002
Kisilevsky, A. E., and Zamponi, G. W. (2008). D2 dopamine receptors interact directly with N-type calcium channels and regulate channel surface expression levels. Channels 2, 269–277. doi: 10.4161/chan.2.4.6402
Ko, K. W., Rasband, M. N., Meseguer, V., Kramer, R. H., and Golding, N. L. (2016). Serotonin modulates spike probability in the axon initial segment through HCN channels. Nat. Neurosci. 19, 826–834. doi: 10.1038/nn.4293
Kole, M. H. P., and Stuart, G. J. (2012). Signal processing in the axon initial segment. Neuron 73, 235–247. doi: 10.1016/j.neuron.2012.01.007
Kreitzer, A. C., and Regehr, W. G. (2001). Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron 29, 717–727. doi: 10.1016/s0896-6273(01)00246-x
Kress, G. J., and Mennerick, S. (2009). Action potential initiation and propagation: upstream influences on neurotransmission. Neuroscience 158, 211–222. doi: 10.1016/j.neuroscience.2008.03.021
Kuba, H., Oichi, Y., and Ohmori, H. (2010). Presynaptic activity regulates Na+ channel distribution at the axon initial segment. Nature 465, 1075–1078. doi: 10.1038/nature09087
Labarrera, C., Deitcher, Y., Dudai, A., Weiner, B., Kaduri Amichai, A., Zylbermann, N., et al. (2018). Adrenergic modulation regulates the dendritic excitability of layer 5 pyramidal neurons In Vivo. Cell Rep. 23, 1034–1044. doi: 10.1016/j.celrep.2018.03.103
Larkum, M. E., Zhu, J. J., and Sakmann, B. (1999). A new cellular mechanism for coupling inputs arriving at different cortical layers. Nature 398, 338–341. doi: 10.1038/18686
Laviolette, S. R., Lipski, W. J., and Grace, A. A. (2005). A subpopulation of neurons in the medial prefrontal cortex encodes emotional learning with burst and frequency codes through a dopamine D4 receptor-dependent basolateral amygdala input. J. Neurosci. 25, 6066–6075. doi: 10.1523/jneurosci.1168-05.2005
Lee, K.-S., Vandemark, K., Mezey, D., Shultz, N., and Fitzpatrick, D. (2019). Functional synaptic architecture of callosal inputs in mouse primary visual cortex. Neuron 101, 421–428.e5. doi: 10.1016/j.neuron.2018.12.005
Li, C., Pleil, K. E., Stamatakis, A. M., Busan, S., Vong, L., Lowell, B. B., et al. (2012). Presynaptic inhibition of gamma-aminobutyric acid release in the bed nucleus of the stria terminalis by kappa opioid receptor signaling. Biol. Psychiatry 71, 725–732. doi: 10.1016/j.biopsych.2011.11.015
Li, L., Bischofberger, J., and Jonas, P. (2007). Differential gating and recruitment of P/Q-, N-, and R-Type Ca2+ channels in hippocampal mossy fiber boutons. J. Neurosci. 27, 13420–13429. doi: 10.1523/jneurosci.1709-07.2007
Lien, A. D., and Scanziani, M. (2018). Cortical direction selectivity emerges at convergence of thalamic synapses. Nature 558, 80–86. doi: 10.1038/s41586-018-0148-5
Little, J. P., and Carter, A. G. (2012). Subcellular synaptic connectivity of layer 2 pyramidal neurons in the medial prefrontal cortex. J. Neurosci. 32, 12808–12819. doi: 10.1523/jneurosci.1616-12.2012
Liu, C., Kershberg, L., Wang, J., Schneeberger, S., and Kaeser, P. S. (2018). Dopamine secretion is mediated by sparse active zone-like release sites. Cell 172, 706–718.e15. doi: 10.1016/j.cell.2018.01.008
Lüscher, C., Jan, L. Y., Stoffel, M., Malenka, R. C., and Nicoll, R. A. (1997). G Protein-coupled inwardly rectifying K+ channels (GIRKs) mediate postsynaptic but not presynaptic transmitter actions in hippocampal neurons. Neuron 19, 687–695. doi: 10.1016/s0896-6273(00)80381-5
Ma, L., Jongbloets, B. C., Xiong, W.-H., Melander, J. B., Qin, M., Lameyer, T. J., et al. (2018). A highly sensitive a-kinase activity reporter for imaging neuromodulatory events in awake mice. Neuron 99, 665–679.e5. doi: 10.1016/j.neuron.2018.07.020
Marder, E. (2012). Neuromodulation of neuronal circuits: back to the future. Neuron 76, 1–11. doi: 10.1016/j.neuron.2012.09.010
Markram, H., Lübke, J., Frotscher, M., Roth, A., and Sakmann, B. (1997). Physiology and anatomy of synaptic connections between thick tufted pyramidal neurones in the developing rat neocortex. J. Physiol. 500( Pt 2), 409–440. doi: 10.1113/jphysiol.1997.sp022031
Martinello, K., Huang, Z., Lujan, R., Tran, B., Watanabe, M., Cooper, E. C., et al. (2015). Cholinergic afferent stimulation induces axonal function plasticity in adult hippocampal granule cells. Neuron 85, 346–363. doi: 10.1016/j.neuron.2014.12.030
Marvin, J. S., Scholl, B., Wilson, D. E., Podgorski, K., Kazemipour, A., Müller, J. A., et al. (2018). Stability, affinity, and chromatic variants of the glutamate sensor iGluSnFR. Nat. Methods 15, 936–939. doi: 10.1038/s41592-018-0171-3
McKay, B. E., Placzek, A. N., and Dani, J. A. (2007). Regulation of synaptic transmission and plasticity by neuronal nicotinic acetylcholine receptors. Biochem. Pharmacol. 74, 1120–1133. doi: 10.1016/j.bcp.2007.07.001
Menichella, D. M., Majdan, M., Awatramani, R., Goodenough, D. A., Sirkowski, E., Scherer, S. S., et al. (2006). Genetic and physiological evidence that oligodendrocyte gap junctions contribute to spatial buffering of potassium released during neuronal activity. J. Neurosci. 26, 10984–10991. doi: 10.1523/jneurosci.0304-06.2006
Mintz, I. M., and Bean, B. P. (1993). GABAB receptor inhibition of P-type Ca2+ channels in central neurons. Neuron 10, 889–898. doi: 10.1016/0896-6273(93)90204-5
Mombereau, C., Kaupmann, K., Froestl, W., Sansig, G., van der Putten, H., and Cryan, J. F. (2004). Genetic and pharmacological evidence of a role for GABAB receptors in the modulation of anxiety- and antidepressant-like behavior. Neuropsychopharmacology 29, 1050–1062. doi: 10.1038/sj.npp.1300413
Momiyama, T., and Koga, E. (2001). Dopamine D(2)-like receptors selectively block N-type Ca(2+) channels to reduce GABA release onto rat striatal cholinergic interneurones. J. Physiol. 533, 479–492. doi: 10.1111/j.1469-7793.2001.0479a.x
Monsivais, P., Clark, B. A., Roth, A., and Häusser, M. (2005). Determinants of action potential propagation in cerebellar purkinje cell axons. J. Neurosci. 25, 464–472. doi: 10.1523/jneurosci.3871-04.2005
Murali, S. S., Napier, I. A., Rycroft, B. K., and Christie, M. J. (2012). Opioid-related (ORL1) receptors are enriched in a subpopulation of sensory neurons and prolonged activation produces no functional loss of surface N-type calcium channels. J. Physiol. 590, 1655–1667. doi: 10.1113/jphysiol.2012.228429
Nadim, F., and Bucher, D. (2014). Neuromodulation of neurons and synapses. Curr. Opin. Neurobiol. 29, 48–56. doi: 10.1016/j.conb.2014.05.003
Oertner, T. G., Sabatini, B. L., Nimchinsky, E. A., and Svoboda, K. (2002). Facilitation at single synapses probed with optical quantal analysis. Nat. Neurosci. 5, 657–664. doi: 10.1038/nn867
Otis, T. S., and Trussell, L. O. (1996). Inhibition of transmitter release shortens the duration of the excitatory synaptic current at a calyceal synapse. J. Neurophysiol. 76, 3584–3588. doi: 10.1152/jn.1996.76.5.3584
Padgett, C. L., and Slesinger, P. A. (2010). GABAB receptor coupling to G-proteins and ion channels. Adv. Pharmacol. 58, 123–147. doi: 10.1016/S1054-3589(10)58006-2
Park, D., and Dunlap, K. (1998). Dynamic regulation of calcium influx by G-proteins, action potential waveform, and neuronal firing frequency. J. Neurosci. 18, 6757–6766. doi: 10.1523/jneurosci.18-17-06757.1998
Parker, J. G., Marshall, J. D., Ahanonu, B., Wu, Y.-W., Kim, T. H., Grewe, B. F., et al. (2018). Diametric neural ensemble dynamics in parkinsonian and dyskinetic states. Nature 557, 177–182. doi: 10.1038/s41586-018-0090-6
Patriarchi, T., Cho, J. R., Merten, K., Howe, M. W., Marley, A., Xiong, W.-H., et al. (2018). Ultrafast neuronal imaging of dopamine dynamics with designed genetically encoded sensors. Science 360:eaat4422. doi: 10.1126/science.aat4422
Perea, G., Araque, A., and Ikegaya, Y. (2007). Astrocytes potentiate transmitter release at single hippocampal synapses. Science 317, 1083–1086. doi: 10.1126/science.1144640
Petersen, A. V., Cotel, F., and Perrier, J.-F. (2017). Plasticity of the axon initial segment: fast and slow processes with multiple functional roles. Neuroscience 23, 364–373. doi: 10.1177/1073858416648311
Pietersen, A. N., Lancaster, D. M., Patel, N., Hamilton, J. B., and Vreugdenhil, M. (2009). Modulation of gamma oscillations by endogenous adenosine through A1 and A2A receptors in the mouse hippocampus. Neuropharmacology 56, 481–492. doi: 10.1016/j.neuropharm.2008.10.001
Pinheiro, P. S., and Mulle, C. (2008). Presynaptic glutamate receptors: physiological functions and mechanisms of action. Nat. Rev. Neurosci. 9, 423–436. doi: 10.1038/nrn2379
Pisani, A., Bonsi, P., Centonze, D., Calabresi, P., and Bernardi, G. (2000). Activation of D2-like dopamine receptors reduces synaptic inputs to striatal cholinergic interneurons. J. Neurosci. 20:RC69.
Poncer, J. C., McKinney, R. A., Gähwiler, B. H., and Thompson, S. M. (1997). Either N- or P-type calcium channels mediate GABA release at distinct hippocampal inhibitory synapses. Neuron 18, 463–472. doi: 10.1016/s0896-6273(00)81246-5
Price, G. D., and Trussell, L. O. (2006). Estimate of the chloride concentration in a central glutamatergic terminal: a gramicidin perforated-patch study on the calyx of held. J. Neurosci. 26, 11432–11436. doi: 10.1523/jneurosci.1660-06.2006
Qin, N., Platano, D., Olcese, R., Stefani, E., and Birnbaumer, L. (1997). Direct interaction of gbetagamma with a C-terminal gbetagamma-binding domain of the Ca2+ channel alpha1 subunit is responsible for channel inhibition by G protein-coupled receptors. Proc. Natl. Acad. Sci. U.S.A. 94, 8866–8871. doi: 10.1073/pnas.94.16.8866
Rama, S., Zbili, M., and Debanne, D. (2018). Signal propagation along the axon. Curr. Opin. Neurobiol. 51, 37–44. doi: 10.1016/j.conb.2018.02.017
Reid, C. A., Bekkers, J. M., and Clements, J. D. (1998). N- and P/Q-type Ca2+ channels mediate transmitter release with a similar cooperativity at rat hippocampal autapses. J. Neurosci. 18, 2849–2855. doi: 10.1523/jneurosci.18-08-02849.1998
Rice, M. E., and Cragg, S. J. (2008). Dopamine spillover after quantal release: rethinking dopamine transmission in the nigrostriatal pathway. Brain Res. Rev. 58, 303–313. doi: 10.1016/j.brainresrev.2008.02.004
Ritzau-Jost, A., Delvendahl, I., Rings, A., Byczkowicz, N., Harada, H., Shigemoto, R., et al. (2014). Ultrafast action potentials mediate kilohertz signaling at a central synapse. Neuron 84, 152–163. doi: 10.1016/j.neuron.2014.08.036
Roberts, M. T., Bender, K. J., and Trussell, L. O. (2008). Fidelity of complex spike-mediated synaptic transmission between inhibitory interneurons. J. Neurosci. 28, 9440–9450. doi: 10.1523/JNEUROSCI.2226-08.2008
Rowan, M. J. M., and Christie, J, M. (2017). Rapid state-dependent alteration in Kv3 channel availability drives flexible synaptic signaling dependent on somatic subthreshold depolarization. Cell Rep. 18, 2018–2029. doi: 10.1016/j.celrep.2017.01.068
Rowan, M. J. M., DelCanto, G., Yu, J. J., Kamasawa, N., and Christie, J. M. (2016). Synapse-level determination of action potential duration by K+ channel clustering in axons. Neuron 91, 370–383. doi: 10.1016/j.neuron.2016.05.035
Sakaba, T. (2006). Roles of the fast-releasing and the slowly releasing vesicles in synaptic transmission at the calyx of held. J. Neurosci. 26, 5863–5871. doi: 10.1523/jneurosci.0182-06.2006
Sakaba, T., and Neher, E. (2003). Direct modulation of synaptic vesicle priming by GABAB receptor activation at a glutamatergic synapse. Nature 424, 775–778. doi: 10.1038/nature01859
Saviane, C., and Silver, R. A. (2006). Errors in the estimation of the variance: implications for multiple-probability fluctuation analysis. J. Neurosci. Methods 153, 250–260. doi: 10.1016/j.jneumeth.2005.11.003
Schiller, J., Major, G., Koester, H. J., and Schiller, Y. (2000). NMDA spikes in basal dendrites of cortical pyramidal neurons. Nature 404, 285–289. doi: 10.1038/35005094
Schneggenburger, R., and Neher, E. (2000). Intracellular calcium dependence of transmitter release rates at a fast central synapse. Nature 406, 889–893. doi: 10.1038/35022702
Scimemi, A., and Diamond, J. S. (2012). The number and organization of Ca2+ channels in the active zone shapes neurotransmitter release from schaffer collateral synapses. J. Neurosci. 32, 18157–18176. doi: 10.1523/JNEUROSCI.3827-12.2012
Scott, R. S., Henneberger, C., Padmashri, R., Anders, S., Jensen, T. P., and Rusakov, D. A. (2014). Neuronal adaptation involves rapid expansion of the action potential initiation site. Nat. Commun. 5:3817. doi: 10.1038/ncomms4817
Seamans, J. K., Durstewitz, D., Christie, B. R., Stevens, C. F., and Sejnowski, T. J. (2001). Dopamine D1/D5 receptor modulation of excitatory synaptic inputs to layer V prefrontal cortex neurons. Proc. Natl. Acad. Sci. U.S.A. 98, 301–306. doi: 10.1073/pnas.011518798
Serrano, J. R., Perez-Reyes, E., and Jones, S. W. (1999). State-dependent inactivation of the alpha1G T-type calcium channel. J. Gen. Physiol. 114, 185–201.
Sesack, S. R., Carr, D. B., Omlchenko, N., and Pinto, A. (2003). Anatomical substrates for glutamate-dopamine interactions. Ann. N. Y. Acad. Sci. 1003, 36–52. doi: 10.1196/annals.1300.066
Shu, Y., Hasenstaub, A., Badoual, M., Bal, T., and McCormick, D. A. (2003). Barrages of synaptic activity control the gain and sensitivity of cortical neurons. J. Neurosci. 23, 10388–10401. doi: 10.1523/jneurosci.23-32-10388.2003
Shu, Y., Hasenstaub, A., Duque, A., Yu, Y., and McCormick, D. A. (2006). Modulation of intracortical synaptic potentials by presynaptic somatic membrane potential. Nature 441, 761–765. doi: 10.1038/nature04720
Sigworth, F. J. (1980). The variance of sodium current fluctuations at the node of Ranvier. J. Physiol. 307, 97–129. doi: 10.1113/jphysiol.1980.sp013426
Stanley, E. F. (2016). The nanophysiology of fast transmitter release. Trends Neurosci. 39, 183–197. doi: 10.1016/j.tins.2016.01.005
Sun, F., Zeng, J., Jing, M., Zhou, J., Feng, J., Owen, S. F., et al. (2018). A genetically encoded fluorescent sensor enables rapid and specific detection of dopamine in flies, fish, and mice. Cell 174, 481–496.e19. doi: 10.1016/j.cell.2018.06.042
Surmeier, D. J., Bargas, J., Hemmings, H. C., Nairn, A. C., and Greengard, P. (1995). Modulation of calcium currents by a D1 dopaminergic protein kinase/phosphatase cascade in rat neostriatal neurons. Neuron 14, 385–397. doi: 10.1016/0896-6273(95)90294-5
Sylantyev, S., Jensen, T. P., Ross, R. A., and Rusakov, D. A. (2013). Cannabinoid- and lysophosphatidylinositol-sensitive receptor GPR55 boosts neurotransmitter release at central synapses. Proc. Natl. Acad. Sci. U.S.A. 110, 5193–5198. doi: 10.1073/pnas.1211204110
Szabo, G. G., Lenkey, N., Holderith, N., Andrasi, T., Nusser, Z., and Hajos, N. (2014). Presynaptic calcium channel inhibition underlies CB1 cannabinoid receptor-mediated suppression of GABA release. J. Neurosci. 34, 7958–7963. doi: 10.1523/JNEUROSCI.0247-14.2014
Takahashi, T., Kajikawa, Y., and Tsujimoto, T. (1998). G-protein-coupled modulation of presynaptic calcium currents and transmitter release by a GABAB receptor. J. Neurosci. 18, 3138–3146. doi: 10.1523/jneurosci.18-09-03138.1998
Tejeda, H. A., Wu, J., Kornspun, A. R., Pignatelli, M., Kashtelyan, V., Krashes, M. J., et al. (2017). Pathway- and cell-specific kappa-opioid receptor modulation of excitation-inhibition balance differentially gates D1 and D2 accumbens neuron activity. Neuron 93, 147–163. doi: 10.1016/j.neuron.2016.12.005
Tsodyks, M. V., and Markram, H. (1997). The neural code between neocortical pyramidal neurons depends on neurotransmitter release probability. Proc. Natl. Acad. Sci. U.S.A. 94, 719–723. doi: 10.1073/pnas.94.2.719
Turner, T., Adams, M., and Dunlap, K. (1992). Calcium channels coupled to glutamate release identified by omega-Aga-IVA. Science 258, 310–313. doi: 10.1126/science.1357749
Violin, J. D., Zhang, J., Tsien, R. Y., and Newton, A. C. (2003). A genetically encoded fluorescent reporter reveals oscillatory phosphorylation by protein kinase C. J. Cell Biol. 161, 899–909. doi: 10.1083/jcb.200302125
Wang, W., Wildes, C. P., Pattarabanjird, T., Sanchez, M. I., Glober, G. F., Matthews, G. A., et al. (2017). A light- and calcium-gated transcription factor for imaging and manipulating activated neurons. Nat. Biotechnol. 35, 864–871. doi: 10.1038/nbt.3909
Weyhersmuller, A., Hallermann, S., Wagner, N., and Eilers, J. (2011). Rapid active zone remodeling during synaptic plasticity. J. Neurosci. 31, 6041–6052. doi: 10.1523/JNEUROSCI.6698-10.2011
Wheeler, D. B., Randall, A., and Tsien, R. W. (1994). Roles of N-type and Q-type Ca2+ channels in supporting hippocampal synaptic transmission. Science 264, 107–111. doi: 10.1126/science.7832825
Wilson, R. I., Kunos, G., and Nicoll, R. A. (2001). Presynaptic specificity of endocannabinoid signaling in the hippocampus. Neuron 31, 453–462. doi: 10.1016/s0896-6273(01)00372-5
Wu, L. G., and Saggau, P. (1995). GABAB receptor-mediated presynaptic inhibition in guinea-pig hippocampus is caused by reduction of presynaptic Ca2+ influx. J. Physiol. 485( Pt 3), 649–657. doi: 10.1113/jphysiol.1995.sp020759
Yang, J., Ye, M., Tian, C., Yang, M., Wang, Y., and Shu, Y. (2013). Dopaminergic modulation of axonal potassium channels and action potential waveform in pyramidal neurons of prefrontal cortex. J. Physiol. 591, 3233–3251. doi: 10.1113/jphysiol.2013.251058
Yang, S., Ben-Shalom, R., Ahn, M., Liptak, A. T., van Rijn, R. M., Whistler, J. L., et al. (2016). β-arrestin-dependent dopaminergic regulation of calcium channel activity in the axon initial segment. Cell Rep. 16, 1518–1526. doi: 10.1016/j.celrep.2016.06.098
Yawo, H., and Chuhma, N. (1993). Preferential inhibition of oω-conotoxin-sensitive presynaptic Ca2+ channels by adenosine autoreceptors. Nature 365, 256–258. doi: 10.1038/365256a0
Yin, L., Rasch, M. J., He, Q., Wu, S., Dou, F., and Shu, Y. (2015). Selective modulation of axonal sodium channel subtypes by 5-HT1A receptor in cortical pyramidal neuron. Cereb. Cortex 27, 509–521. doi: 10.1093/cercor/bhv245
Yu, W., Kwon, J., Sohn, J.-W., Lee, S. H., Kim, S., and Ho, W.-K. (2018). mGluR5-dependent modulation of dendritic excitability in CA1 pyramidal neurons mediated by enhancement of persistent Na + currents. J. Physiol. 596, 4141–4156. doi: 10.1113/JP275999
Yu, Y., Maureira, C., Liu, X., and McCormick, D. (2010). P/Q and N channels control baseline and spike-triggered calcium levels in neocortical axons and synaptic boutons. J. Neurosci. 30, 11858–11869. doi: 10.1523/JNEUROSCI.2651-10.2010
Zador, A. (1998). Impact of synaptic unreliability on the information transmitted by spiking neurons. J. Neurophysiol. 79, 1219–1229. doi: 10.1152/jn.1998.79.3.1219
Zamponi, G. W., and Currie, K. P. M. (2013). Regulation of CaV2 calcium channels by G protein coupled receptors. Biochim. Biophys. Acta 1828, 1629–1643. doi: 10.1016/j.bbamem.2012.10.004
Zhang, W., and Linden, D. J. (2009). Neuromodulation at single presynaptic boutons of cerebellar parallel fibers is determined by bouton size and basal action potential-evoked Ca transient amplitude. J. Neurosci. 29, 15586–15594. doi: 10.1523/JNEUROSCI.3793-09.2009
Zhang, X.-F., Cooper, D. C., and White, F. J. (2002). Repeated cocaine treatment decreases whole-cell calcium current in rat nucleus accumbens neurons. J. Pharmacol. Exp. Ther. 301, 1119–1125. doi: 10.1124/jpet.301.3.1119
Zucker, R. S., and Regehr, W. G. (2002). Short-term synaptic plasticity. Annu. Rev. Physiol. 64, 355–405.
Keywords: presynaptic, action potential, GPCR, modulation, neurotransmission
Citation: Burke KJ Jr and Bender KJ (2019) Modulation of Ion Channels in the Axon: Mechanisms and Function. Front. Cell. Neurosci. 13:221. doi: 10.3389/fncel.2019.00221
Received: 01 March 2019; Accepted: 01 May 2019;
Published: 17 May 2019.
Edited by:
Dominique Debanne, INSERM U1072 Unité de Neurobiologie des Canaux Ioniques et de la Synapse, FranceReviewed by:
Maarten H. P. Kole, Netherlands Institute for Neuroscience (KNAW), NetherlandsDmitri A. Rusakov, University College London, United Kingdom
Copyright © 2019 Burke and Bender. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kevin J. Bender, a2V2aW4uYmVuZGVyQHVjc2YuZWR1