 James P. Higham1†
James P. Higham1† Bilal R. Malik1†
Bilal R. Malik1† Edgar Buhl1
Edgar Buhl1 Jennifer M. Dawson1
Jennifer M. Dawson1 Anna S. Ogier1
Anna S. Ogier1 Katie Lunnon2
Katie Lunnon2 James J. L. Hodge1*
James J. L. Hodge1*- 1School of Physiology, Pharmacology and Neuroscience, University of Bristol, Bristol, United Kingdom
- 2University of Exeter Medical School, University of Exeter, Exeter, United Kingdom
Alzheimer’s disease (AD) is the most common form of dementia and is characterized by intracellular neurofibrillary tangles of hyperphosphorylated Tau, including the 0N4R isoform and accumulation of extracellular amyloid beta (Aβ) plaques. However, less than 5% of AD cases are familial, with many additional risk factors contributing to AD including aging, lifestyle, the environment and epigenetics. Recent epigenome-wide association studies (EWAS) of AD have identified a number of loci that are differentially methylated in the AD cortex. Indeed, hypermethylation and reduced expression of the Ankyrin 1 (ANK1) gene in AD has been reported in the cortex in numerous different post-mortem brain cohorts. Little is known about the normal function of ANK1 in the healthy brain, nor the role it may play in AD. We have generated Drosophila models to allow us to functionally characterize Drosophila Ank2, the ortholog of human ANK1 and to determine its interaction with human Tau and Aβ. We show expression of human Tau 0N4R or the oligomerizing Aβ 42 amino acid peptide caused shortened lifespan, degeneration, disrupted movement, memory loss, and decreased excitability of memory neurons with co-expression tending to make the pathology worse. We find that Drosophila with reduced neuronal Ank2 expression have shortened lifespan, reduced locomotion, reduced memory and reduced neuronal excitability similar to flies overexpressing either human Tau 0N4R or Aβ42. Therefore, we show that the mis-expression of Ank2 can drive disease relevant processes and phenocopy some features of AD. Therefore, we propose targeting human ANK1 may have therapeutic potential. This represents the first study to characterize an AD-relevant gene nominated from EWAS.
Introduction
Alzheimer’s disease (AD) is the most common form of dementia, with patients suffering from premature death and accelerated cognitive decline including memory loss. Post-mortem examination of AD brain samples reveals the accumulation of extracellular amyloid-beta (Aβ) plaques and intracellular neurofibrillary tangles (NFTs) of hyperphosphorylated microtubule associated protein Tau (MAPT), which is accompanied by gliosis, neuronal cell loss, and brain atrophy. Aβ is produced by the amyloidogenic cleavage of the amyloid precursor protein (APP) by β and γ secretases resulting in the formation of neurotoxic aggregating Aβ peptides with the 42 amino acid (aa) peptide (Aβ42) being more toxic than Aβ40 (Selkoe and Hardy, 2016). Tau exists in six different isoforms, which vary with the number of N-terminal domains (0N, 1N or 2N) and the number of C-terminal aggregating tubulin binding repeats (3R or 4R) (Arendt et al., 2016). The 4R isoforms are upregulated in AD brain and show stronger tubulin binding and aggregation than the 3R isoforms (Arendt et al., 2016). Another contributing factor to Tau aggregation is its hyperphosphorylation by a number of different kinases including glycogen synthase kinase-3β, cyclin-dependent kinase 5, JNK, microtubule-associated regulatory kinase, DYRK1a and CaMKII (Hanger et al., 1992; Ferrer et al., 2005; Plattner et al., 2006; Wang et al., 2007; Dolan and Johnson, 2010; Ghosh and Giese, 2015). It is not currently known how Aβ and Tau pathology are linked, however, the amyloid cascade hypothesis suggests that Aβ pathology leads to the other hallmarks of AD, including the spread of NFTs (Selkoe and Hardy, 2016) and recent work suggests changes in neuronal excitability and calcium (Ca2+) signaling may be important for their connection and disease progression (Spires-Jones and Hyman, 2014; Wu et al., 2016), but exactly how remains unknown.
Less than 5% of AD cases are due to autosomal dominant mutations in the APP, presenilin 1 (PSEN1) or PSEN2 genes. The remainder of AD cases are sporadic, with incidence attributed to both genetic and environmental risk factors. Genome-wide association studies (GWAS) have identified a number of genes where common genetic variation is associated with increased risk of sporadic AD, including APOE (𝜀4 allele), BIN1 and PICALM genes amongst others (Lambert et al., 2013) with many additional risk factors for AD being associated with lifestyle and/or the environment.
Epigenetics refers to the mitotically and meiotically heritable changes in gene expression without alterations in the underlying DNA sequence for instance by DNA methylation and downregulation of genes (Gräff and Sanchez-Mut, 2015). This also potentially allows for alterations in gene expression in response to environmental variation, such as stress, diet or exposure to environmental chemicals. In order to characterize the contribution of epigenetic mechanisms to AD etiology, recent epigenome-wide association studies (EWAS) of AD have been performed and have identified a number of genetic loci that are associated with increased risk of AD (De Jager et al., 2014; Lunnon et al., 2014; Smith et al., 2018). One locus that showed consistent cortical AD-associated hypermethylation in five independent cohorts resided in the Ankyrin 1 (ANK1) gene (De Jager et al., 2014; Lunnon et al., 2014; Smith and Lunnon, 2017). A recent publication has demonstrated that ANK1 DNA methylation in the entorhinal cortex is observed in only certain neurodegenerative diseases (Smith et al., 2019). This study reported disease-associated hypermethylation in AD, Huntington’s disease and to a lesser extent Parkinson’s disease (PD). The authors showed that disease-associated hypermethylation was only seen in donors with vascular dementia (VaD) and dementia with Lewy bodies (DLB) when individuals had co-existing AD pathology; in individuals with “pure” VaD or DLB, no ANK1 hypermethylation was observed. ANK1 is an integral membrane and adaptor protein, that mediates the attachment of membrane proteins such as ion channels, cell adhesion proteins and receptors with the spectrin-actin cytoskeleton and is important for cell proliferation, mobility, activation, and maintenance of specialized membrane domains (Smith and Penzes, 2018).
Most of our understanding of the molecular changes that cause AD pathology comes largely from experiments using rodents to model genetic variation; however, these are models of familial AD, and do not recapitulate sporadic disease. As such, new drugs effective in these rodents have not translated to any new successful treatments for AD highlighting the need to generate and characterize new models of sporadic AD (McGowan et al., 2006; Crews and Masliah, 2010; Van Dam and De Deyn, 2011; Gama Sosa et al., 2012; Guo et al., 2012; De Jager et al., 2014). In Drosophila, neuronal overexpression of different human APP products (including Aβ42) and mutants has been reported to cause degeneration of the photoreceptor neurons of the fly eye, shortened lifespan, change in neuronal excitability as well as movement, circadian, sleep, and learning defects in a number of different studies (Iijima et al., 2004; Chiang et al., 2010; Speretta et al., 2012; Chen et al., 2014; Blake et al., 2015; Ping et al., 2015; Tabuchi et al., 2015). Likewise, neuronal overexpression of human Tau isoforms associated with AD have been shown to result in degeneration of the photoreceptor neurons of the fly eye, shortened lifespan, movement and learning defects in many different studies (Wittmann et al., 2001; Folwell et al., 2010; Iijima-Ando and Iijima, 2010; Kosmidis et al., 2010; Beharry et al., 2013; Papanikolopoulou and Skoulakis, 2015; Sealey et al., 2017). Fewer animal models have determined the effect of expression of human Aβ and Tau quantifying the effect on lifespan, axonal transport, synaptic morphology, degeneration of the eye and climbing (Folwell et al., 2010; Iijima et al., 2010; Lee et al., 2012; Shulman et al., 2014). Co-expression more accurately reflects the progression of AD in humans (Guerreiro and Hardy, 2011; Selkoe, 2012; Spires-Jones and Hyman, 2014; Bouleau and Tricoire, 2015; Arendt et al., 2016; Selkoe and Hardy, 2016). Furthermore, fewer studies have compared the effect of common variants nominated from GWAS for AD (Shulman et al., 2011, 2014; Gotz et al., 2012; Chapuis et al., 2013; Dourlen et al., 2016) and to our knowledge none from EWAS for AD. Thus, many genomic and epigenomic loci nominated in these studies remain uncharacterized in any living organism, with many of these genes or loci having a completely unknown function in the brain (Guerreiro and Hardy, 2011; Shulman et al., 2011; Gotz et al., 2012; De Jager et al., 2014; Del-Aguila et al., 2015; Devall et al., 2016).
In order to address these issues, we have characterized the first animal model to investigate the function of a locus nominated from EWAS in AD. We have investigated ANK mis-expression and compared its AD relevant phenotypes to Drosophila models expressing either (a) human mutant APP (which results in an aggregating form of oligomerized Aβ42), (b) MAPT (resulting in 0N4R Tau), (c) APP (Aβ42) with MAPT (0N4R Tau), and (d) APP (Aβ42) or MAPT (0N4R Tau) with Ank mis-expression, finding that mis-expression of these AD associated genes cause similar reduction in lifespan, movement, memory and neuronal excitability. We also report for the first time the effect of human Aβ42, 0N4R Tau and co-expression on 1 h memory and determine how Aβ42 and 0N4R Tau change mushroom body (MB) memory neuron excitability prior to neurodegeneration.
Materials and Methods
Drosophila Genetics
Flies were raised at a standard density with a 12 h:12 h light dark (LD) cycle with lights on at ZT 0 (Zeitgeber time) on standard Drosophila medium (0.7% agar, 1.0% soya flour, 8.0% polenta/maize, 1.8% yeast, 8.0% malt extract, 4.0% molasses, 0.8% propionic acid, and 2.3% nipagen) at 25°C. The following flies used in this study were previously described or obtained from the Bloomington and Vienna fly stock centers: wild type control was Canton S w- (CSw-) (gift from Dr. Scott Waddell, University of Oxford). Experimental genotypes were elav-Gal4 (Bloomington stock center line number BL8760), OK107-Gal4 (BL854), GMR-Gal4 (BL9146), uas-human MAPT (TAU 0N4R) wild type (gift from Dr. Linda Partridge, University College London) (Wittmann et al., 2001; Kerr et al., 2011), uas-human tandem Aβ42-22 amino acid linker-Aβ42 (gift from Dr. Damian Crowther, University of Cambridge) (Speretta et al., 2012), uas-GFP (gift from Dr. Mark Wu, Johns Hopkins University), uas-GCaMP6f [BL42747 (Shaw et al., 2018)], uas-Ank2-RNAi line A (BL29438), uas-Ank2 RNAi (Vienna Drosophila RNAi Center stock number VDRC107238 and KK10497) and uas-Ank2 (gift from Dr. Ron R. Dubreuil, University of Illinois) (Mazock et al., 2010).
Survival Assay
Approximately 2 days after eclosion 10 mated females were transferred to a vial containing standard food and maintained at 25°C throughout. Deaths were scored every 2 days and then transferred to a fresh food vial (Kerr et al., 2011). Data was presented as Kaplan–Meier survival curves with statistical analysis performed using log-rank tests to compare survival between genotypes. All statistical tests were performed using Prism (GraphPad Software Inc., La Jolla, CA, United States).
Eye Degeneration Assay
Two to five day old adult flies were anesthetized by CO2 prior to immersion in ethanol in order to euthanize the fly to prevent any further movement during image capture (Folwell et al., 2010). The eyes were imaged with a Zeiss AxioCam MRm camera attached to a stereomicroscope (Zeiss SteREO Discovery.V8, up to 8× magnification). Surface area was quantified using Zeiss Zen software and normalized to the mean size of the wild type control. One-way ANOVA with Dunnett’s multiple comparisons was used to analyze data.
Climbing Assay
Ten 2–5 day old flies were collected and given 1 h to acclimatize to the test vial in an environmentally controlled room at 25°C and 70% humidity. Using the negative geotaxis reflex of Drosophila, flies were gently tapped to the bottom of a 7.5 cm plastic vial and the number of flies that crossed a line drawn 2 cm from the top of the tube in 10 s was counted, and then expressed as a % which was referred to as the climbing performance or climbing index (Iijima et al., 2004; Sun et al., 2018). One-way ANOVA with Dunnett’s multiple comparisons was used to analyze data.
Memory
One hour memory and sensory controls were performed as previously described using the olfactory-shock aversive conditioning assay (Cavaliere et al., 2013; Malik et al., 2013; Malik and Hodge, 2014). Experiments were performed with groups of 30–50 flies aged between 2 and 5 days old of a given genotype, in a T-maze apparatus housed in an environmentally controlled room at 25°C and 70% humidity under dim red light. Flies were exposed to either 4-methylcyclohexanol (MCH, Sigma, ∼1:200) or 3-octanol (OCT, Sigma, ∼1:100) diluted in mineral oil (Sigma) paired with 1.5 s pulses of 60 V electric shock interspersed with 3.5 s pauses from shock for a minute. After 30 s of fresh air the flies were exposed to the reciprocal order without shock for another minute. After a 1 h rest, memory was assessed by transferring the flies to a choice point of the T maze, with one arm containing the shock paired odor and the other the non-shock paired odor, flies showed learning by avoiding the shock paired odor (i.e., correct flies).
To eliminate odor bias, the assay was performed with two groups of flies (30–50 flies in each), one shocked with MCH and then the other shocked with OCT. The average of the two groups was taken to give an n = 1 PI value. Control experiments were performed to show that the different genotypes of flies could respond to MCH, OCT and shock alone. One-way ANOVA with Dunnett’s multiple comparisons was used to analyze data.
Calcium Imaging and Microscopy
Calcium imaging and imaging of the MB structure was performed using a genetically encoded GCaMP Ca2+ reporter and adapting previously published protocols (Cavaliere et al., 2012; Gillespie and Hodge, 2013; Malik et al., 2013; Schlichting et al., 2016; Shaw et al., 2018). Adult flies of the indicated genotypes using OK107-Gal4 and uas-GCaMP6f were collected between 2 and 5 days post-eclosion, decapitated and the brain dissected in extracellular saline solution containing (in mM): 101 NaCl, 1 CaCl2, 4 MgCl2, 3 KCl, 5 glucose, 1.25 NaH2PO4, 20.7 NaHCO3, pH adjusted to 7.2. Brains were placed ventral side up in the recording chamber, secured with a custom-made anchor and continuously perfused with aerated saline. Images of the whole MB of each genotype was captured in the resting state. To activate neurons, high concentration KCl (100 mM in saline) was bath applied through the perfusion system for 4 min and then washed out. The calcium fluorescence signal was acquired using a CCD camera (Zeiss Axiocam) and a 470 nm LED light source (Colibri, Zeiss) on an upright Zeiss Examiner microscope with a 20× water immersion lens, recorded with ZEN (Zeiss, 4 frames/sec) and plotted with Microsoft Excel. One-way ANOVA with Dunnett’s multiple comparisons was used to analyze data.
Results
Mis-Expression of Human Mutant APP, MAPT or Ank2 Shortens Lifespan
Increased neuronal levels of Aβ or Tau lead to AD pathology and early death (Selkoe, 2012; Selkoe and Hardy, 2016). We therefore overexpressed in all neurons either an aggregating form of human mutant APP that encodes oligomerized human Aβ42 [tandem Aβ42 (Speretta et al., 2012)] or human MAPT [0N4R Tau (Papanikolopoulou and Skoulakis, 2015; Sealey et al., 2017)] both of which caused premature death reducing the flies’ lifespan by about 25% (Figures 1A,C). In order to test for a genetic interaction between the two neurotoxic genes, we overexpressed both mutant APP (Aβ42) and MAPT (0N4R) together in all neurons and got a further reduction to exactly half the lifespan of a normal fly. The reduction in median lifespan due to co-expression was equivalent to an additive effect of the shortening of life due to mutant APP (Aβ42) and MAPT (0N4R) alone, this suggests that Aβ42 and Tau pathology may act in separate pathways to cause neurotoxicity and early death.
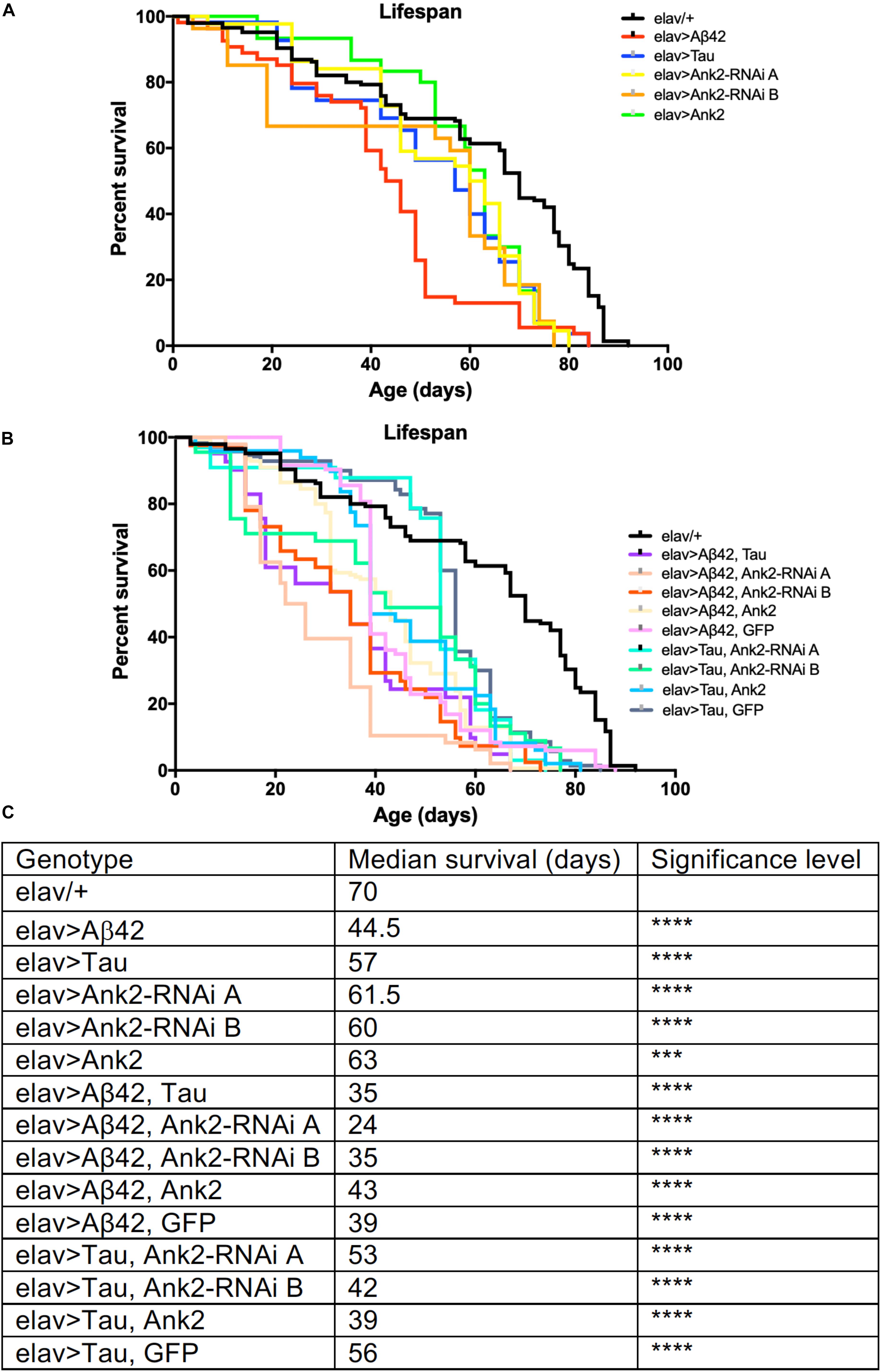
Figure 1. The effect of expression of human mutant APP (Aβ42), MAPT (Tau 0N4R) and Drosophila Ank2 on Drosophila lifespan. (A) Survival curves of pan-neuronal expressing Aβ42 (elav > Aβ42), Tau (elav > Tau), Ank2-RNAi (elav > Ank2-RNAi line A or B), Ank2 (elav > Ank2), (B) Tau with Aβ42 (elav > Tau, Aβ42), Aβ42 with Ank2-RNAi (elav > Aβ42, Ank2-RNAi line A or B), Aβ42 with Ank2 (elav > Aβ42, Ank2), Aβ42 with Ank2 (elav > Aβ42, Ank2), Aβ42 with Ank2 (elav > Aβ42, GFP), Tau with Ank2-RNAi (elav > Tau, Ank2-RNAi line A or B), Tau with Ank2 (elav > Tau, Ank2), Tau with GFP (elav > Aβ42, GFP) compared to wild type control (elav/+) flies kept at 25°C. Misexpression of all Alzheimer’s disease (AD) genes caused a significant reduction in lifespan compared to control using the Kaplan–Meier and log rank test (n > 100 per genotype of flies). (C) Table listing all genotypes characterized with median lifespan (days) and significant reductions in lifespan as determined by Kaplan–Meier and log rank test and indicated as ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001 and used in all subsequent figures.
Recently, human ANK1 gene has been shown to be differentially methylated in AD (Lunnon et al., 2014). There are two other ANK genes in the human genome, ANK2, and ANK3. In Drosophila, there are two Ank genes, the ubiquitously expressed Ank1, and neuron specific Ank2 (Koch et al., 2008; Pielage et al., 2008; Mazock et al., 2010; Keller et al., 2011; Siegenthaler et al., 2015). The closest ortholog to Drosophila Ank1 is human ANK3 with 51% total amino acid identity while the closest ortholog of Drosophila Ank2 is human ANK1 with 43% amino acid identity. Human ANK2 is more similar to Drosophila Ank1 (49% identity) than Drosophila Ank2 (33% identity). Because hypermethylation of human ANK1 has been reported in AD cortex (Lunnon et al., 2014), we used RNAi to knock-down expression of Ank1 and Ank2 comparing the effect of reducing these genes in all Drosophila neurons using two different RNAi transgenes designed to non-overlapping regions of each gene. Pan-neural reduction in expression of Drosophila Ank1 did not cause any AD relevant behavioral deficits (Figures 2A,B), while reduction in Ank2 using the same promoter and assay did cause a reduction in 1 h memory (Figure 2B). Therefore, we conclude, that Drosophila Ank2 is the closest functional ortholog of human ANK1, which we characterized in subsequent experiments.
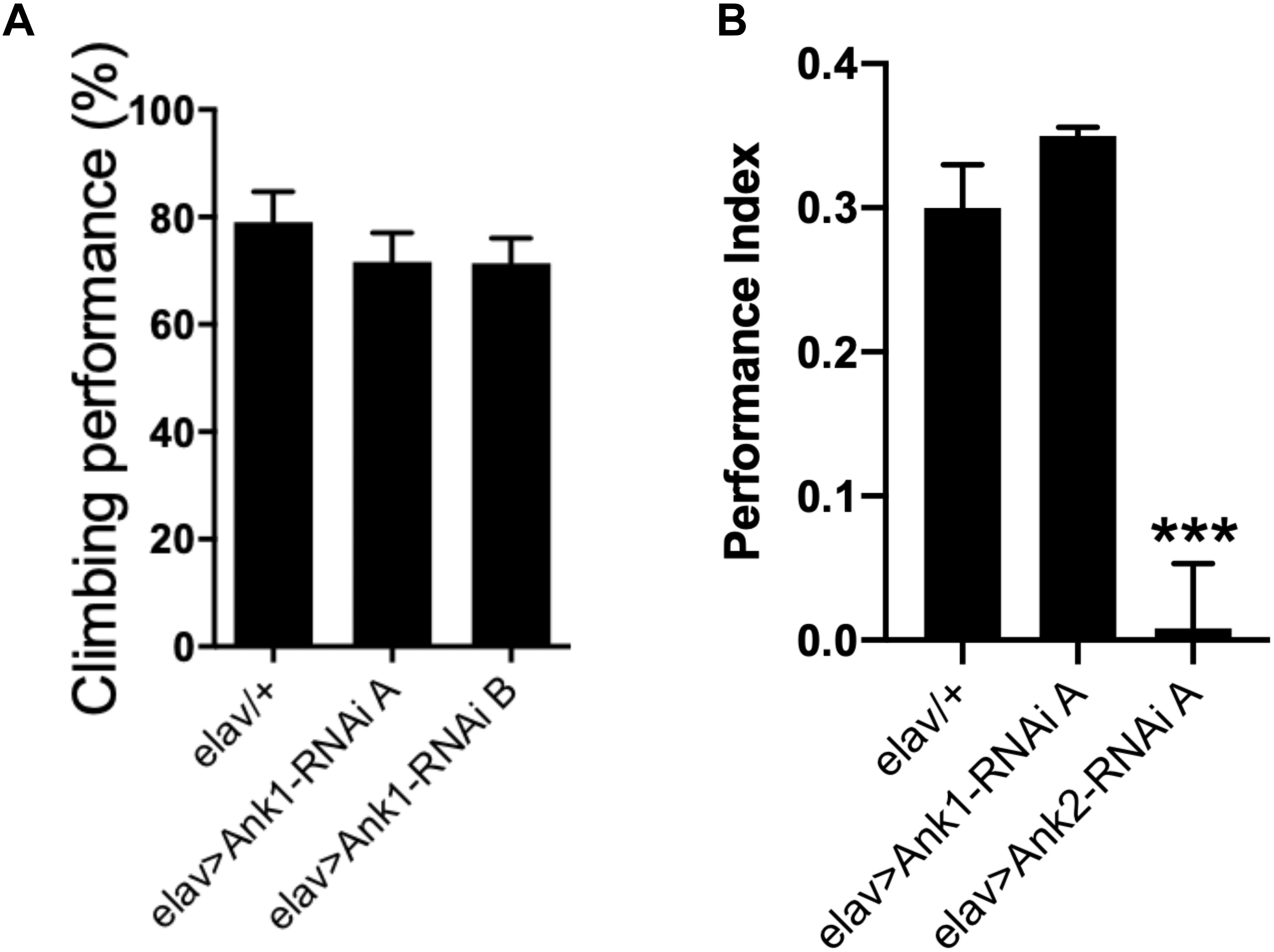
Figure 2. The effect of pan-neuronal expression of Drosophila Ank1 and Ank2 on behavior. (A) The negative geotaxis-climbing reflex was used to quantify motor deficits of 2–5 day old flies. Pan-neuronal reduction of Ank1 (elav-Gal4 > Ank1-RNAi line A or B) was compared to control using one-way ANOVA with Dunnett’s multiple comparison and showed no significant difference. n ≥ 6 groups of 10 flies. (B) One hour memory of 2–5 day old flies was assessed using the olfactory shock-conditioning assay. Pan-neuronal reduction in Ank2 (elav > Ank2-RNAi line A) as opposed to Ank1 (elav > Ank1-RNAi line A) caused a significant reduction in memory compared to control using one-way ANOVA with Dunnett’s multiple comparisons. Error bars are standard error of the mean (SEM). n ≥ 4 groups of ∼50 flies.
We found that pan-neural reduction of Ank2 resulted in a shortening of lifespan by 15% compared to control (Figures 1B,C). We verified our results by using two independent RNAi lines (A and B) to non-overlapping sequences in Ank2, both lines gave similar results in all assays. Depending on the genomic location of DNA methylation, it can result in either increased or decreased expression of the target gene (Suzuki and Bird, 2008); we therefore tested if neuronal Ank2 overexpression affected longevity and found that it also shortened lifespan (∼10%), but at a lower significance level to all other genotypes. In order to test if there was a genetic interaction between mutant APP (Aβ42) or MAPT (0N4R) and Ank2 we generated flies that co-expressed mutant APP (Aβ42) or MAPT (0N4R) with the Ank2 transgenes. We found that the detrimental effect of each gene on lifespan remained (Figures 1B,C), suggesting changing the level of fly Ank2 could not rescue the shortened lifespan caused by overexpression of human mutant APP (Aβ42) or MAPT (0N4R) to a level similar to wild type control (elav/+). One potential cause of a reduction or suppression of toxicity of a gene product is dilution of Gal4. This is due to a single Gal4 transcription factor driving expression by binding to a single UAS transgene [e.g., mutant APP (Aβ42) or MAPT (0N4R)] which may get diluted when adding a second UAS site of a gene (e.g., Ank2). This results in a reduction in the amount of the AD toxic gene product being made, giving a false positive of a suppression phenotype. To control for such a dilution effect, we generated flies that co-expressed a second unrelated neutral gene product (GFP) with either human mutant APP (Aβ42) or MAPT (0N4R Tau). There was no significant change in neurotoxicity of the mutant APP (Aβ42) or MAPT (0N4R Tau) when expressed alone or with GFP.
Overexpression of Human Mutant APP or Mutant MAPT but Not Mis-Expression of Ank2 Caused Degeneration of Photoreceptor Neurons
Increased levels of aggregating toxic Aβ and Tau lead to neurodegeneration and AD. In order to model this neurotoxicity in Drosophila, human mutant APP causing Aβ42 production (Figure 3B) or mutant MAPT (0N4R Tau) (Figure 3C) was expressed throughout development and adulthood in the photoreceptor neurons of the eye, resulting in a so called “rough eye” phenotype, where the degeneration and loss of the normally regularly arrayed ommatidia of the compound eye of wild type flies (Figure 3A) gives rise to a disorganized and smaller eye. The loss of photoreceptors could be quantified by measuring the total surface area of the eye, which showed that expression of human mutant APP (Aβ42) and MAPT (0N4R Tau) reduced the size of the eye by 37% and 40%, respectively (Figure 3H). Co-expression of human mutant APP (Aβ42) and MAPT (0N4R Tau) (Figures 3D,H), resulted in a 53% reduction in eye size, suggesting that Aβ42 and Tau act in partially overlapping pathways to cause neurotoxicity in the fly eye. Reduction of Ank2 (Figures 3E,F), overexpression of Ank2 (Figure 3G) and co-expression of Ank2 and Tau did not cause degeneration or reduction in size of the eye (Figure 3H), although the latter cannot be considered a rescue as although the eye was significantly larger (p < 0.01) than eyes expressing Tau it was not significantly bigger than those expressing Tau and GFP suggesting any suppression maybe due to a Gal4 dilution effect.
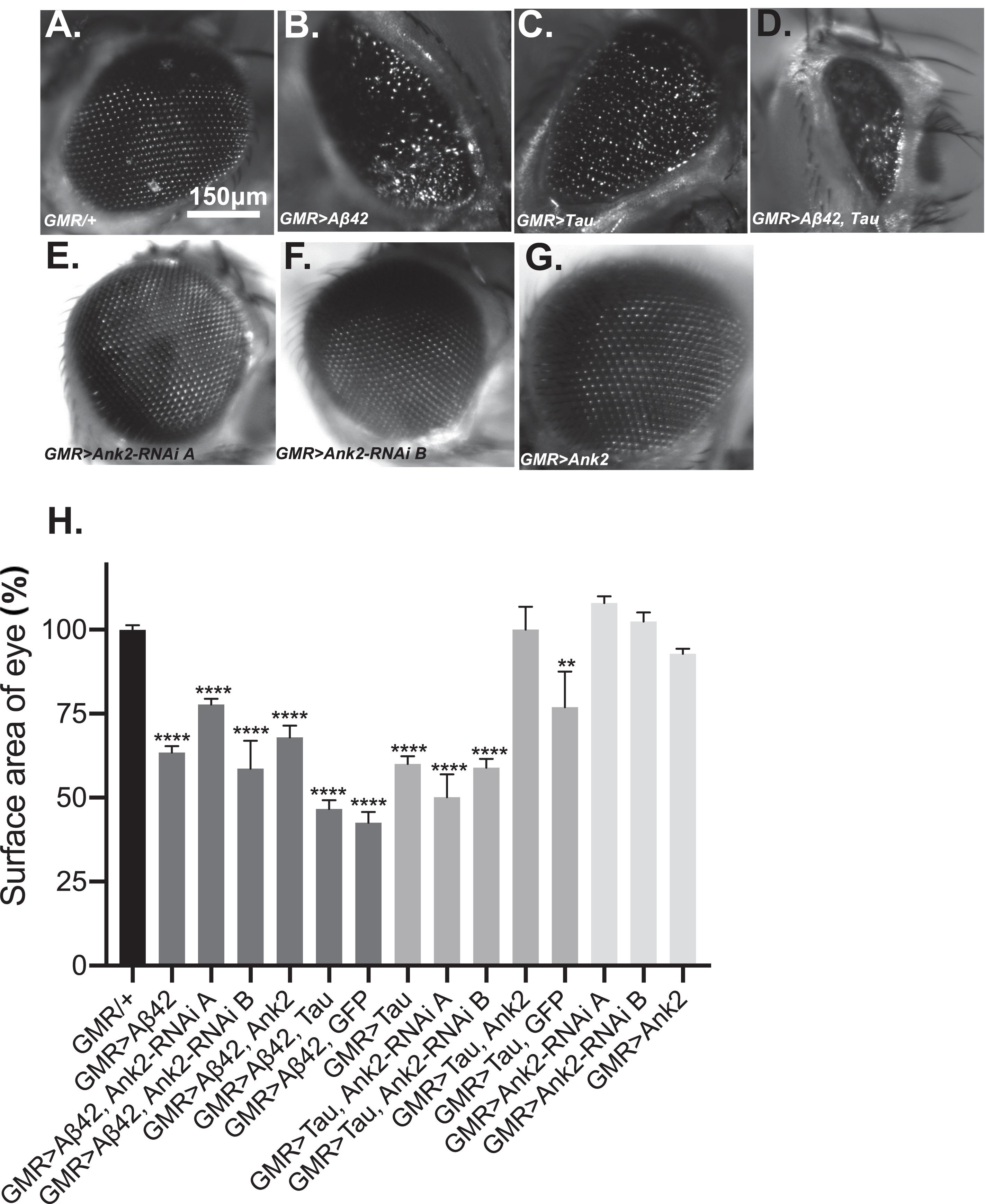
Figure 3. The effect of expression of human mutant APP (Aβ42), MAPT (Tau 0N4R), and Drosophila Ank2 on degeneration of photoreceptor neurons. Images of 2–5 day old compound eyes of (A) control fly (GMR-Gal4/+) showing the regular alignment of ommatidia compared to photoreceptors neurons overexpressing, (B) Aβ42 (GMR-Gal4 > Aβ42), (C) Tau (GMR-Gal4 > Tau), and (D) Tau with Aβ42 (GMR-Gal4 > Tau, Aβ42) which were smaller and displayed a “rough eye” phenotype. (E,F) photoreceptors expressing Ank2-RNAi (GMR > Ank2-RNAi line A or B) or (G) Ank2 (GMR > Ank2) appeared normal. (H) Degeneration of photoreceptor neurons was quantified as normalized percentage surface area of the eye of genotypes compared to the mean of the control (GMR/+) which was set at 100%, comparisons were made between to mutant genotypes compared to control using one-way ANOVA with Dunnett’s multiple comparisons. Genotypes that included expression of APP (Aβ42) or MAPT (Tau 0N4R) in the eye showed a significantly reduced size of eye, while those mis-expressing Drosophila Ank2 did not. Co-expression of MAPT (Tau 0N4R) and Ank2 rescued eye size to a level indistinguishable from wild type. Error bars are SEM. n ≥ 7 eyes per genotype.
Overexpression of Human Mutant APP and MAPT Genes and Reduction in Ank2 Expression Caused Locomotor Deficits
Flies exhibit a negative geotaxis reflex, such that after tapping on a surface ∼80% of 2–5 day old wild type flies will climb to the top of a tube in 10 s. This startle-induced reflex requires the co-ordinated activity of dopaminergic neurons afferent to the MB, and of MB Kenyon cells, in addition to MB efferent neurons which communicate with central motor centers (Sun et al., 2018). In order to quantify any detrimental effect of human mutant APP (Aβ42) and MAPT (0N4R Tau) on this behavior, we pan-neuronally (elav-Gal4 expressed human mutant APP (Aβ42) and MAPT (0N4R Tau), which resulted in a 20% and 40% reduction in climbing ability, respectively. Likewise, reduction in Ank2 expression caused a 15% reduction in climbing, while Ank2 overexpression had no effect. All other gene combinations were found to significantly reduce gross climbing performance (Figure 4).
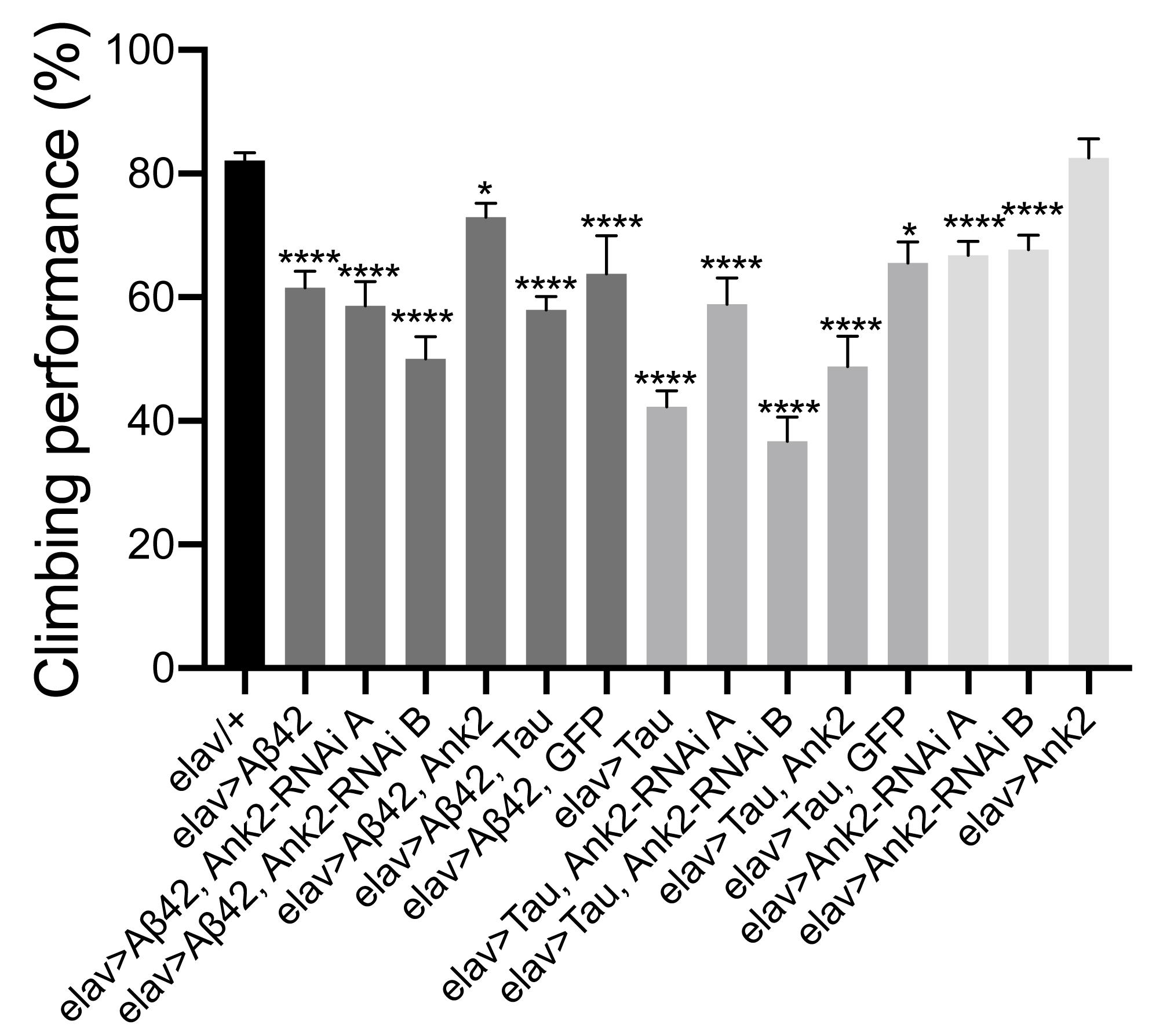
Figure 4. The effect of expression of human mutant APP (Aβ42), MAPT (Tau 0N4R) or Drosophila Ank2 on locomotor behavior. The negative geotaxis-climbing reflex was used to quantify locomotor behavior of 2–5 day old flies. About 80% of control (elav-Gal4/+) flies were able to climb to the top of test vial within 10 s; this compared to experimental genotypes expressing AD transgenes throughout their nervous system. Mis-expression of all AD associated genes except for overexpression of Ank2 alone, resulted in a significant reduction in climbing performance using one-way ANOVA with Dunnett’s multiple comparisons. Error bars are SEM. n ≥ 15 groups of 10 flies.
Overexpression of Human Mutant APP and MAPT Genes and Reduced Ank2 Causes Memory Deficits
As multiple forms and phases of memory, including anterograde memory, are affected by AD (Walsh and Selkoe, 2004), therefore we measured the effect of the human mutant APP (Aβ42), MAPT (0N4R Tau) and Drosophila Ank2 on memory. We performed the olfactory shock assay on 2–5 day old flies (Cavaliere et al., 2013; Malik et al., 2013; Malik and Hodge, 2014) assessing associative memory at the 1 h time point, which is considered to be intermediate memory in Drosophila. Olfactory shock memory is mediated by MB neurons and we therefore drove expression of the genes using a promoter (OK107-Gal4) with broad expression in these neurons (Cavaliere et al., 2013; Malik et al., 2013). We found MB expression of human mutant APP (Aβ42) or MAPT (0N4R Tau) caused a large reduction in memory (Figure 5). Ank2 is highly expressed in the adult MB as shown by Ank2 reporter line (R54H11-Gal4) expression (Pfeiffer et al., 2008) and previous studies (Siegenthaler et al., 2015). Reduction, as opposed to overexpression, of Ank2 also caused a reduction in memory (Figure 5). Co-expression of human mutant APP (Aβ42) and MAPT (0N4R Tau; Figure 5) or either AD gene with Ank2-RNAi caused a similar reduction in memory, suggesting that all three genes may act in the same pathway in MB neurons. Overexpression of Ank2, or co-expression of Ank2 with either human mutant APP (Aβ42) or MAPT (0N4R Tau) did not cause a significant reduction in memory. The latter cannot be considered rescue of the memory deficit caused by expression of Aβ42 or Tau alone, as the memory of Ank2 co-expressed with either human mutant APP (Aβ42) or MAPT (0N4R Tau) was not significantly greater than that of human mutant APP (Aβ42) or MAPT (0N4R Tau) co-expressed with GFP, again suggesting a Gal4 dilution effect was contributing to the rescue of the mutant phenotype.
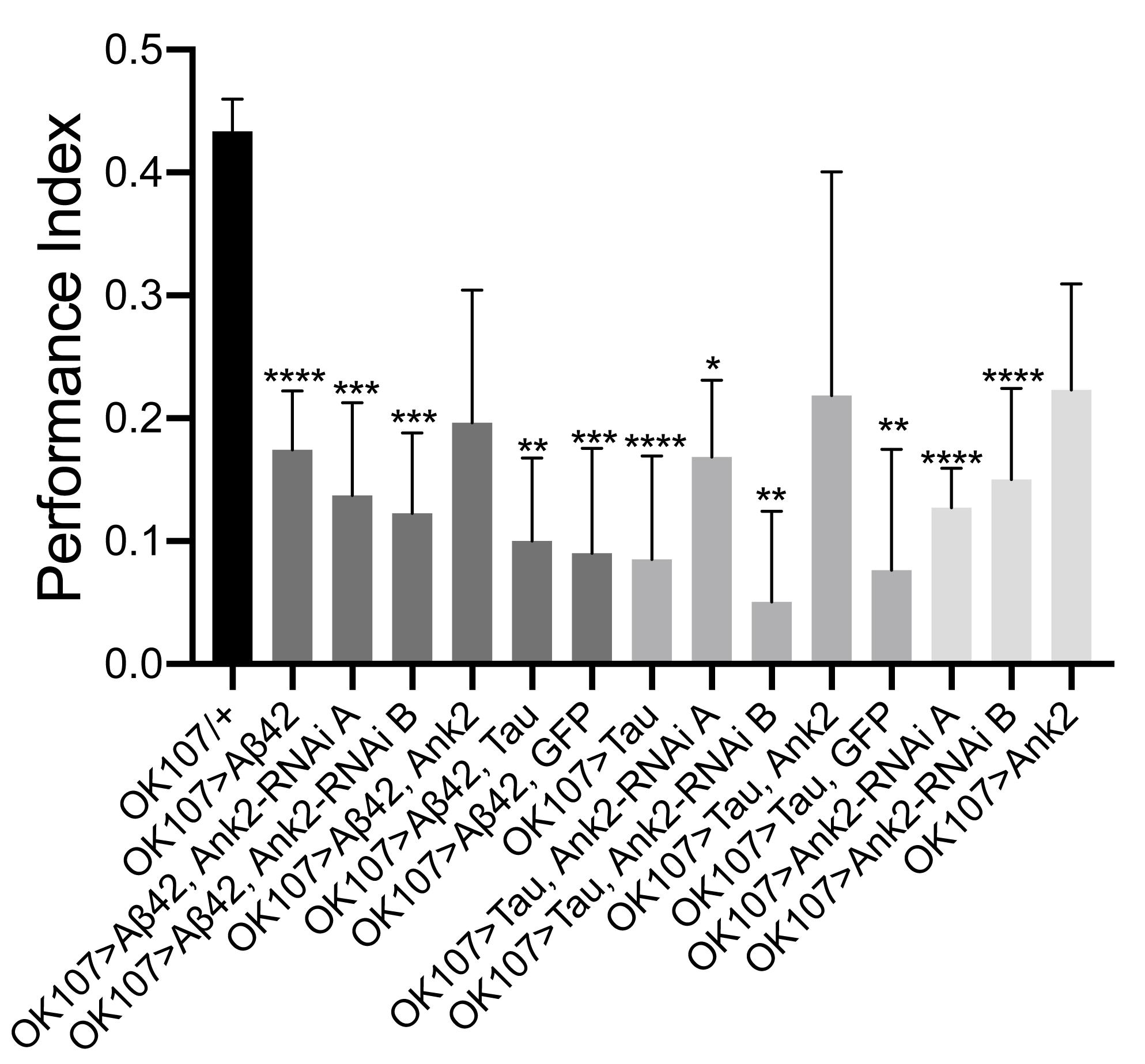
Figure 5. The effect of expression of human mutant APP (Aβ42), MAPT (Tau 0N4R) and Drosophila Ank2 on memory. One hour memory of 2–5 day old flies was assessed using the olfactory shock-conditioning assay. The performance index of flies expressing AD transgenes throughout their mushroom body (MB) was compared to control (OK107-Gal4/+) and were found to show a significant reduction in memory using one-way ANOVA with Dunnett’s multiple comparisons, except genotypes overexpressing Ank2. Error bars are SEM. n ≥ 4 groups of ∼100 flies.
For flies to be able to perform the olfactory shock assay, the fly must be able to respond normally to shock and the odors used in the memory task. Therefore, we performed behavioral controls on 2–5 day old flies that showed that there was no significant difference between mutant genotypes and wild type in terms of avoidance of electric shock (Figure 6A), octanol (Figure 6B) and methylcyclohexanol (MCH; Figure 6C) odors (Cavaliere et al., 2013; Malik et al., 2013; Malik and Hodge, 2014). Therefore, any mutant genotype showing a significant reduction in memory in Figure 5 was a bona fide memory mutant.
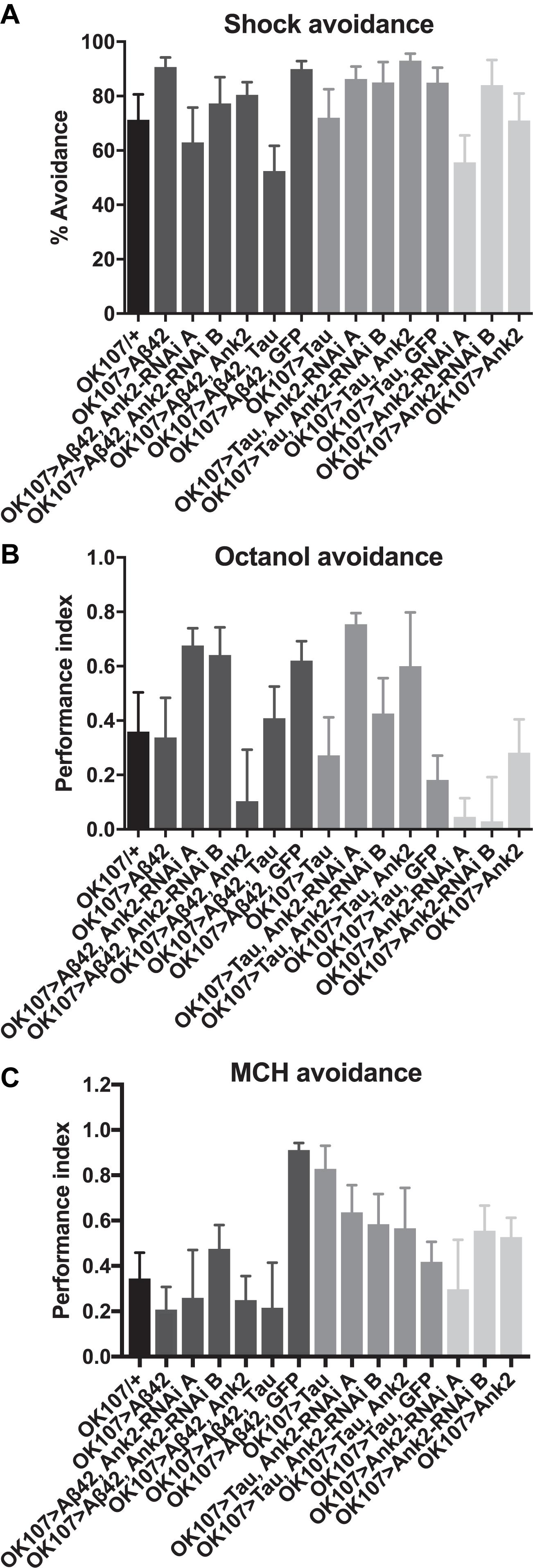
Figure 6. The effect of expression of human mutant APP (Aβ42), MAPT (Tau 0N4R) and Drosophila Ank2 on response to negative reinforcement (shock) and olfaction. (A) The response of 2–5 day old flies to negative reinforcement was quantified as % avoidance of shock, with experimental genotypes with AD genes expressed throughout their MB compared to control (OK107/+). The avoidance of these genotypes of flies to 3-octanol (B) and methylcyclohexanol (MCH) (C) odors was expressed as a performance index. All genotypes responded to odors and shock in a manner not significantly different from wild type using one-way ANOVA with Dunnett’s multiple comparisons. n ≥ 4 groups of ∼50 flies.
Mis-Expression of Human Mutant APP (Aβ42), MAPT (0N4R Tau), and Ank2 Decreased the Neural Excitability of Memory Neurons Prior to Any Observable Neurodegeneration
Changes in neuronal excitability and Ca2+ signaling are thought to occur early in disease progression prior to neurodegeneration and are proposed to mediate early changes in behavior in AD, such as memory loss (Spires-Jones and Hyman, 2014; Wu et al., 2016). Therefore, in order to determine how mis-expression of human mutant APP (Aβ42), MAPT (0N4R Tau), and Ank2 may lead to changes in neuronal function and AD relevant phenotypes, we expressed the genetically encoded Ca2+ reporter, GCaMP6f using the same OK107-Gal4 MB promoter as for the memory experiments and then measuring peak intracellular Ca2+ in response to high [K+] solution that non-specifically depolarizes neurons (Malik et al., 2013). The axons and synaptic terminals of MB neurons form a pair of bilaterally arranged and symmetrically lobed structures which show low basal Ca2+ fluorescence levels (Figure 7A left panel) which, in response to depolarizing high [K+] saline, show a large peak in Ca2+ fluorescence levels (Figure 7A right panel). The increase in neuronal activity can be expressed as the relative fluorescence change (ΔF/F0) over time (Figure 7B) showing that high K+ causes a rapid peak Ca2+ influx that returns to baseline after washing. MB wide overexpression of human mutant APP (Aβ42), MAPT (0N4R Tau) or mis-expression of Ank2 all caused a similar decrease in peak Ca2+ influx of memory neurons (Figure 7C), suggesting a reduction in neuronal excitability. A previous study showed human 0N4R tau expression in γ MB neurons reduced learning and 1.5 h memory in 3–5 day old young flies which were demonstrated to still have their γ neurons intact. However, by day 45, the 0N4R expressing γ neurons had started to neurodegenerate (Mershin et al., 2004). In order to verify the MB were intact in our 2–5 day old flies overexpressing human mutant APP (Aβ42), MAPT (0N4R Tau) or mis-expressing fly Ank2 throughout the MB we imaged the MB with GCaMP6f and saw that all the genotypes had intact MB α, β, γ, α′, and β′ lobes (Figure 8).
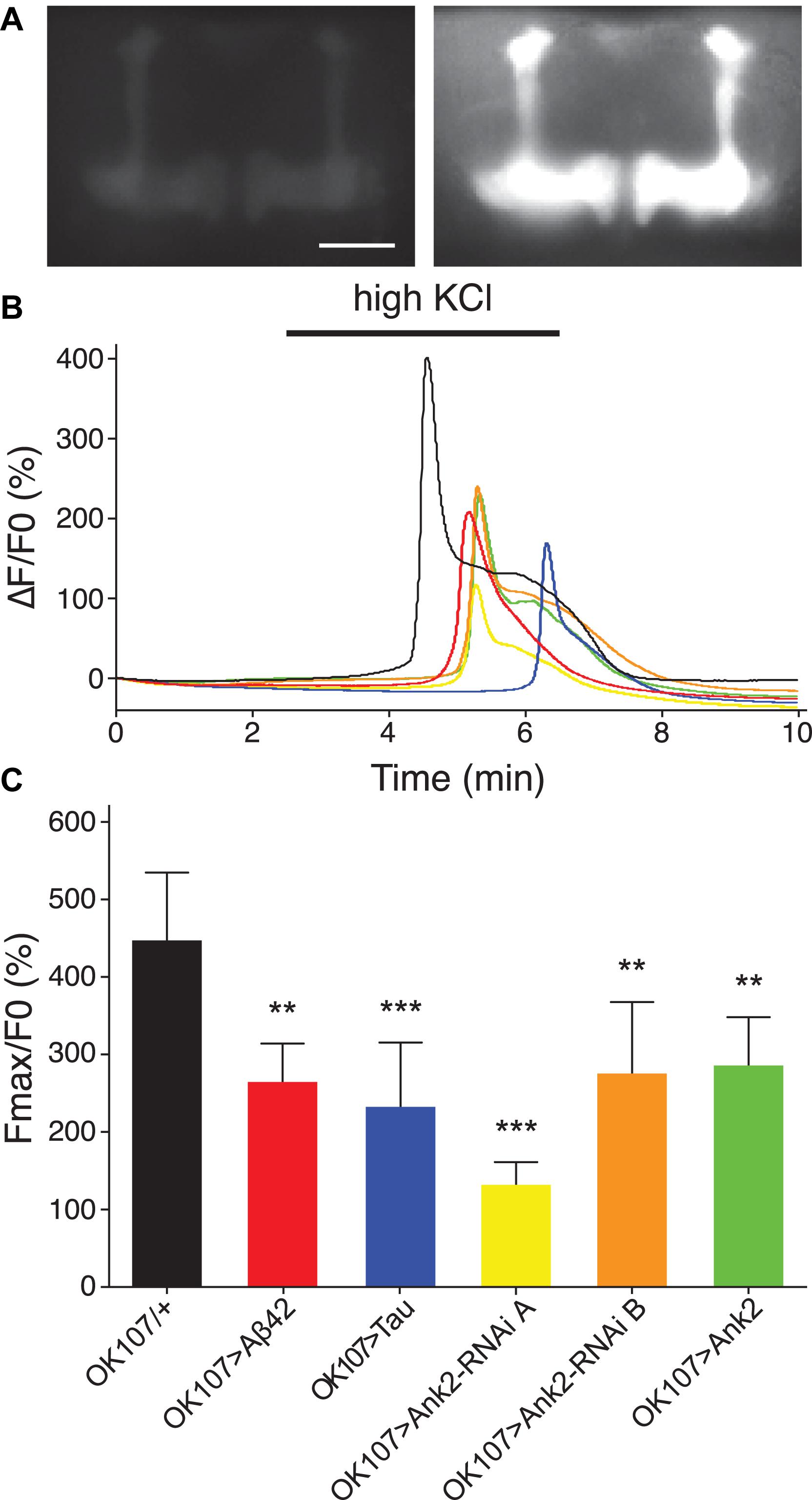
Figure 7. The effect of expression of human mutant APP (Aβ42), MAPT (Tau 0N4R), and Drosophila Ank2 on activation of mushroom body neurons. (A) Exemplary control 2–5 day old fly MB expressing the calcium reporter GCaMP6f shows a big increase in relative fluorescence in response to elevated KCl (100 mM, bath-applied). Images show the same brain before (left) and during (right) KCl application; scale bar 50 μm. (B) The increase in neuronal activity is expressed as the relative change in fluorescence (ΔF/F0) over time showing that high KCl (indicated by bar) causes peak Ca2+ influx that returns to baseline after washing. (C) Quantitative analysis of the maximal response for the indicated genotypes shows a reduced responsiveness of all mutants. Data was analyzed with one-way ANOVA with Tukey’s post hoc and error bars are standard deviation, n ≥ 5 brains.
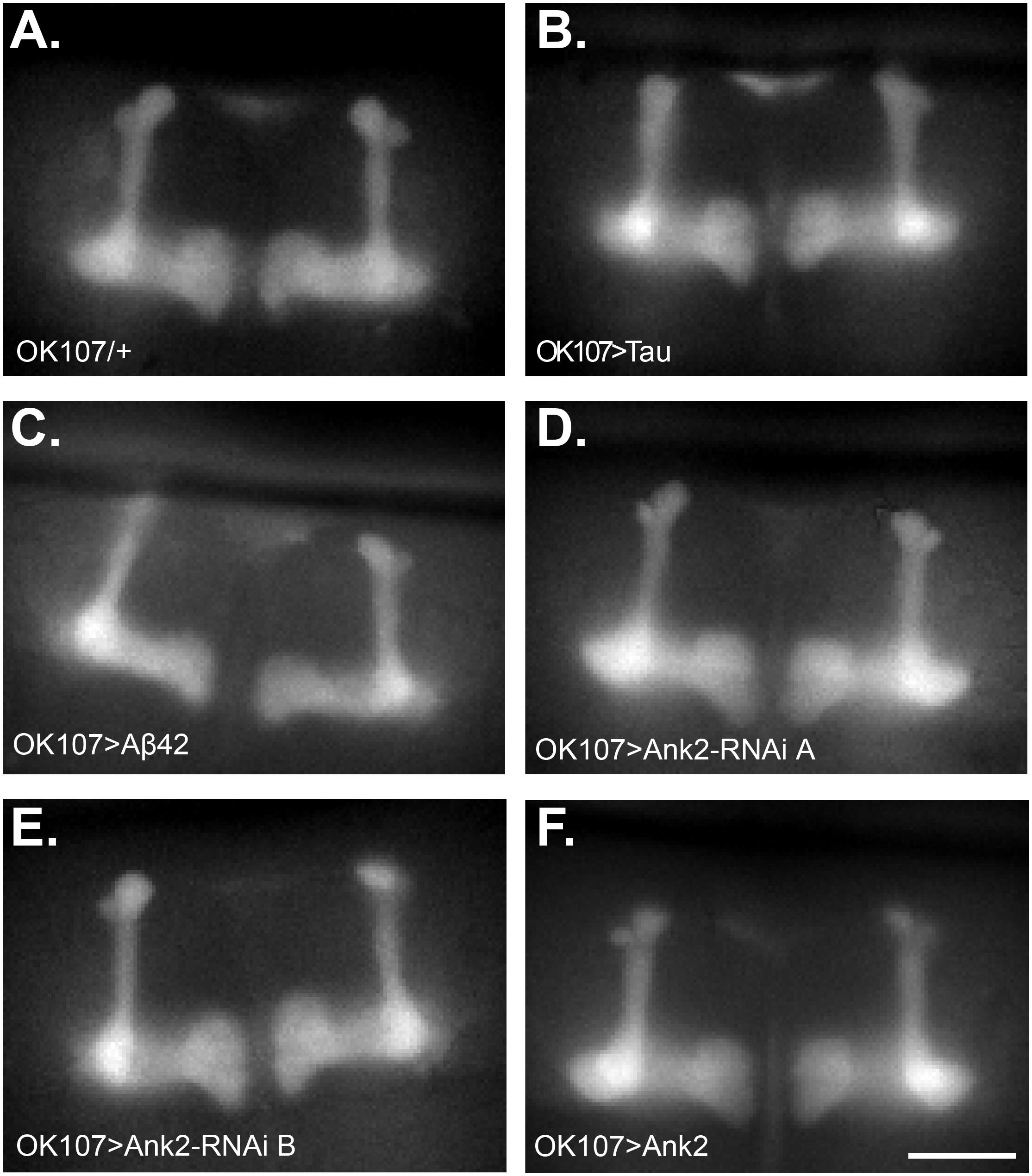
Figure 8. Mushroom body morphology is not affected by expression of human mutant APP (Aβ42), MAPT (Tau 0N4R), and Drosophila Ank2 in young flies. Images show representative mushroom bodies of 2–5 day old flies visualized by expression of the Ca2+ reporter GCaMP6f of (A) a control fly (OK107/+), (B) OK107 > Tau, (C) OK107 > Aβ42, (D) OK107 > Ank2-RNAi line A, (E) OK107 > Ank2-RNAi line B, and (F) OK107 > Ank2 which all show the regular organization of α, β, γ, α′ and β′ lobes. Scale bar is 50 μm.
Discussion
In this study, we have characterized the first animal model based on mis-expression of the AD EWAS nominated ANK1 gene ortholog in Drosophila. Altered DNA methylation of human ANK1 occurs in AD brains, especially in regions that show gross AD pathology, such as the entorhinal cortex (De Jager et al., 2014; Lunnon et al., 2014). Here, we found reduction in expression of the fly ortholog of human ANK1, which is Drosophila Ank2, in areas of the brain responsible for memory (the MB), caused memory impairment similar in magnitude to that caused by overexpression of human mutant APP resulting in an oligomerizing form of Aβ42 or MAPT producing the 0N4R isoform of Tau, both of which are particularly associated with the disease. In flies, overexpression of fly Ank2, did not result in degeneration of the eye, locomotor or memory defects but did lead to mild shortening of lifespan (by ∼10%) and reduced MB excitability.
In addition to memory loss, animals with reduced neuronal Ank2 also recapitulated the shortening of lifespan seen in those with AD, again a phenotype seen in flies overexpressing human mutant APP (Aβ42) or MAPT (0N4R Tau). Co-expression of human mutant APP (Aβ42) and MAPT (0N4R Tau) caused a further reduction in lifespan, suggesting that the two molecules may act in partially non-overlapping and therefore additive pathways that lead to the pathology causing the flies to die early. A similar effect on lifespan has been reported previously for another form of APP that resulted in a non-oligomerizing version of Aβ42 and another form of MAPT, resulting in the 1N4R isoform of Tau, however, the longevity assays were only run for roughly half the lifespan of the flies (Folwell et al., 2010). We also found a reduction in locomotion in flies expressing human mutant APP (Aβ42), MAPT (0N4R Tau) and reduced Ank2. Interestingly, a recent study has shown that ANK1 hypermethylation is observed in PD, a movement disorder, that can also be associated with dementia (Smith et al., 2019). In the eye, overexpression of human mutant APP (Aβ42) or MAPT (0N4R Tau) were both neurotoxic causing degeneration of the fly eye photoreceptor neurons. Co-expression caused a further reduction implying the two act in separate and additive pathways to cause neurotoxicity and neuronal death. Reduction in Ank2 did not cause degeneration of the eye suggesting that Ank2 may affect neuronal function independent of degeneration. This is likely to be via changes in neuronal excitability, as we saw when the gene was misexpressed in MB neurons (Smith and Penzes, 2018). We found misexpression of human mutant APP (Aβ42), MAPT (0N4R Tau) and Ank2 all reduced the peak Ca2+ response of MB neurons, a decrease in excitability likely to contribute to the memory deficits of these flies.
In Drosophila, Ank2 has been shown to be important for synaptic plasticity and stability (Koch et al., 2008; Pielage et al., 2008; Massaro et al., 2009; Keller et al., 2011; Bulat et al., 2014) and is involved in a glia mediated pathway that causes degeneration of motor neurons (Keller et al., 2011). Therefore, it is possible that reduction of Ank2 may only cause degeneration in certain types of neurons. Furthermore, because human ANK1 has also been shown to be misexpressed in glia in the AD brain (Mastroeni et al., 2017), it is also possible that neurodegeneration results from misexpression of Ank2 in glia.
We do not know how Ankyrin interacts with Tau and what changes in Tau protein level, location or phosphorylation result from Ankyrin misexpression. We know that Ankyrins are adaptor proteins that attach to integral membrane proteins such as the Na+/K+ ATPase, voltage-gated Na+ (Nav1), voltage-gated K+ (KCNQ) and TrpA1 channels, and then link them to the actin-spectrin based membrane cytoskeleton, with some of these molecules having been associated with AD (Bennett and Baines, 2001; Yamamoto et al., 2007; Smith and Penzes, 2018). Therefore the loss of Ankyrin might be predicted to disrupt the proper clustering of these ion channels, some of which are involved in MB-memory and calcium signaling (Cavaliere et al., 2013), and hence may result in the decreased neuronal excitability detected by our MB calcium imaging experiments and which might be required for proper memory formation. The changes in calcium influx may also cause dysregulation of calcium dependent phosphorylation of Tau, also known to disrupt MB dependent memory and shorten lifespan (Papanikolopoulou and Skoulakis, 2015). Finally, the loss of Ankyrin may distrupt the actin-spectrin cytoskeleton, which may result in pathological changes in microtubules exacerbated by their association with Tau. More experiments are required to investigate the nature of the interaction between Ankyrin and Tau in AD.
In summary, we have characterized the first animal model of a gene implicated in AD that was nominated from EWAS. We found that mis-expression of the fly ortholog of this gene, Ank2, in central neurons caused a range of AD-relevant phenotypes such as shortened lifespan, memory loss, and changes in neuronal excitability similar to those resulting from human Aβ42 and 0N4R Tau. In alignment with previous AD studies, we conclude that Ank2 – or, in humans, ANK1 – likely plays a role in AD neuropathology.
Author Contributions
JH, BM, EB, JD, AO, and JJLH performed the experiments. JH devised the experiments and wrote the manuscript. JJLH and KL secured the funding. JH, BM, EB, and KL edited the drafts of the manuscript.
Funding
This work was supported by a GW4 accelerator (GW4-AF2-002) award to Drs. KL, Jonathan Mill, Jon Brown, Nick Allen, Vasanta Subramanian, and JJLH, as well as by Alzheimer’s Society undergraduate and a Leverhulme Trust project grant (RPG-2016-31) grants to JJLH. This manuscript has been released as a Pre-Print at BioRxiv (Higham et al., 2018).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Drs. Damian Crowther, Ronald Dubreuil, Linda Partridge, Scott Waddell, and Mark Wu for Drosophila stocks and Dr. Owen Peters for helpful comments on the manuscript.
References
Arendt, T., Stieler, J. T., and Holzer, M. (2016). Tau and tauopathies. Brain Res. Bull. 126, 238–292. doi: 10.1016/j.brainresbull.2016.08.018
Beharry, C., Alaniz, M. E., and Alonso Adel, C. (2013). Expression of Alzheimer-like pathological human tau induces a behavioral motor and olfactory learning deficit in Drosophila melanogaster. J. Alzheimer’s Dis. 37, 539–550. doi: 10.3233/JAD-130617
Bennett, V., and Baines, A. J. (2001). Spectrin and ankyrin-based pathways: metazoan inventions for integrating cells into tissues. Physiol. Rev. 81, 1353–1392. doi: 10.1152/physrev.2001.81.3.1353
Blake, M. R., Holbrook, S. D., Kotwica-Rolinska, J., Chow, E. S., Kretzschmar, D., and Giebultowicz, J. M. (2015). Manipulations of amyloid precursor protein cleavage disrupt the circadian clock in aging Drosophila. Neurobiol. Dis. 77, 117–126. doi: 10.1016/j.nbd.2015.02.012
Bouleau, S., and Tricoire, H. (2015). Drosophila models of Alzheimer’s disease: advances, limits, and perspectives. J. Alzheimer’s Dis. 45, 1015–1038. doi: 10.3233/JAD-142802
Bulat, V., Rast, M., and Pielage, J. (2014). Presynaptic CK2 promotes synapse organization and stability by targeting Ankyrin2. J Cell Biol 204, 77–94. doi: 10.1083/jcb.201305134
Cavaliere, S., Gillespie, J. M., and Hodge, J. J. (2012). KCNQ channels show conserved ethanol block and function in ethanol behaviour. PLoS One 7:e50279. doi: 10.1371/journal.pone.0050279
Cavaliere, S., Malik, B. R., and Hodge, J. J. (2013). KCNQ channels regulate age-related memory impairment. PLoS One 8:e62445. doi: 10.1371/journal.pone.0062445
Chapuis, J., Hansmannel, F., Gistelinck, M., Mounier, A., Van Cauwenberghe, C., Kolen, K. V., et al. (2013). Increased expression of BIN1 mediates Alzheimer genetic risk by modulating tau pathology. Mol. Psychiatry 18, 1225–1234. doi: 10.1038/mp.2013.1
Chen, K. F., Possidente, B., Lomas, D. A., and Crowther, D. C. (2014). The central molecular clock is robust in the face of behavioural arrhythmia in a Drosophila model of Alzheimer’s disease. Dis. Models Mech. 7, 445–458. doi: 10.1242/dmm.014134
Chiang, H. C., Wang, L., Xie, Z., Yau, A., and Zhong, Y. (2010). PI3 kinase signaling is involved in Abeta-induced memory loss in Drosophila. Proc. Natl. Acad. Sci. U.S.A. 107, 7060–7065. doi: 10.1073/pnas.0909314107
Crews, L., and Masliah, E. (2010). Molecular mechanisms of neurodegeneration in Alzheimer’s disease. Hum. Mol. Genet. 19:R12–R20. doi: 10.1093/hmg/ddq160
De Jager, P. L., Srivastava, G., Lunnon, K., Burgess, J., Schalkwyk, L. C., Yu, L., et al. (2014). Alzheimer’s disease: early alterations in brain DNA methylation at ANK1. BIN1, RHBDF2 and other loci. Nat. Neurosci. 17, 1156–1163. doi: 10.1038/nn.3786
Del-Aguila, J. L., Koboldt, D. C., Black, K., Chasse, R., Norton, J., Wilson, R. K., et al. (2015). Alzheimer’s disease: rare variants with large effect sizes. Curr. Opin. Genet. Dev. 33, 49–55. doi: 10.1016/j.gde.2015.07.008
Devall, M., Roubroeks, J., Mill, J., Weedon, M., and Lunnon, K. (2016). Epigenetic regulation of mitochondrial function in neurodegenerative disease: new insights from advances in genomic technologies. Neurosci. Lett. 625, 47–55. doi: 10.1016/j.neulet.2016.02.013
Dolan, P. J., and Johnson, G. V. (2010). The role of tau kinases in Alzheimer’s disease. Curr. Opin. Drug Dis. Dev. 13, 595–603.
Dourlen, P., Fernandez-Gomez, F. J., Dupont, C., Grenier-Boley, B., Bellenguez, C., Obriot, H., et al. (2016). Functional screening of Alzheimer risk loci identifies PTK2B as an in vivo modulator and early marker of tau pathology. Mol. Psychiatry 22, 874–883. doi: 10.1038/mp.2016.59
Ferrer, I., Barrachina, M., Puig, B., Martinez de Lagran, M., Marti, E., Avila, J., et al. (2005). Constitutive Dyrk1A is abnormally expressed in Alzheimer disease. Down syndrome, Pick disease, and related transgenic models. Neurobiol. Dis. 20, 392–400. doi: 10.1016/j.nbd.2005.03.020
Folwell, J., Cowan, C. M., Ubhi, K. K., Shiabh, H., Newman, T. A., Shepherd, D., et al. (2010). Abeta exacerbates the neuronal dysfunction caused by human tau expression in a Drosophila model of Alzheimer’s disease. Exp. Neurol. 223, 401–409. doi: 10.1016/j.expneurol.2009.09.014
Gama Sosa, M. A., De Gasperi, R., and Elder, G. A. (2012). Modeling human neurodegenerative diseases in transgenic systems. Hum. Genet. 131, 535–563. doi: 10.1007/s00439-011-1119-1
Ghosh, A., and Giese, K. P. (2015). Calcium/calmodulin-dependent kinase II and Alzheimer’s disease. Mol. Brain 8:78. doi: 10.1016/j.yjmcc.2013.03.005
Gillespie, J. M., and Hodge, J. J. (2013). CASK regulates CaMKII autophosphorylation in neuronal growth, calcium signaling, and learning. Front. Mol. Neurosci. 6:27. doi: 10.3389/fnmol.2013.00027
Gotz, J., Matamales, M., Gotz, N. N., Ittner, L. M., and Eckert, A. (2012). Alzheimer’s disease models and functional genomics-How many needles are there in the haystack? Front. Physiol. 3:320. doi: 10.3389/fphys.2012.00320
Gräff, J., and Sanchez-Mut, J. V. (2015). Epigenetic alterations in Alzheimer’s Disease. Front. Behav. Neurosci. 9:347. doi: 10.3389/fnbeh.2015.00347
Guerreiro, R. J., and Hardy, J. (2011). Alzheimer’s disease genetics: lessons to improve disease modelling. Biochem. Soc. Trans. 39, 910–916. doi: 10.1042/BST0390910
Guo, Q., Wang, Z., Li, H., Wiese, M., and Zheng, H. (2012). APP physiological and pathophysiological functions: insights from animal models. Cell Res. 22, 78–89. doi: 10.1038/cr.2011.116
Hanger, D. P., Hughes, K., Woodgett, J. R., Brion, J. P., and Anderton, B. H. (1992). Glycogen synthase kinase-3 induces Alzheimer’s disease-like phosphorylation of tau: generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci. Lett. 147, 58–62. doi: 10.1016/0304-3940(92)90774-2
Higham, J. P., Malik, B. R., Buhl, E., Dawson, J., Ogier, A. S., Lunnon, K., et al. (2018). Mis-expression of the Alzheimer’s disease associated gene Ankyrin causes memory loss and shortened lifespan in Drosophila. bioRxiv
Iijima, K., Gatt, A., and Iijima-Ando, K. (2010). Tau Ser262 phosphorylation is critical for Abeta42-induced tau toxicity in a transgenic Drosophila model of Alzheimer’s disease. Hum. Mol. Genet. 19, 2947–2957. doi: 10.1093/hmg/ddq200
Iijima, K., Liu, H. P., Chiang, A. S., Hearn, S. A., Konsolaki, M., and Zhong, Y. (2004). Dissecting the pathological effects of human Abeta40 and Abeta42 in Drosophila: a potential model for Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 101, 6623–6628. doi: 10.1073/pnas.0400895101
Iijima-Ando, K., and Iijima, K. (2010). Transgenic Drosophila models of Alzheimer’s disease and tauopathies. Brain Struc. Func. 214, 245–262. doi: 10.1007/s00429-009-0234-4
Keller, L. C., Cheng, L., Locke, C. J., Muller, M., Fetter, R. D., and Davis, G. W. (2011). Glial-derived prodegenerative signaling in the Drosophila neuromuscular system. Neuron 72, 760–775. doi: 10.1016/j.neuron.2011.09.031
Kerr, F., Augustin, H., Piper, M. D., Gandy, C., Allen, M. J., Lovestone, S., et al. (2011). Dietary restriction delays aging, but not neuronal dysfunction, in Drosophila models of Alzheimer’s disease. Neurobiol. Aging 32, 1977–1989. doi: 10.1016/j.neurobiolaging.2009.10.015
Koch, I., Schwarz, H., Beuchle, D., Goellner, B., Langegger, M., and Aberle, H. (2008). Drosophila ankyrin 2 is required for synaptic stability. Neuron 58, 210–222. doi: 10.1016/j.neuron.2008.03.019
Kosmidis, S., Grammenoudi, S., Papanikolopoulou, K., and Skoulakis, E. M. (2010). Differential effects of Tau on the integrity and function of neurons essential for learning in Drosophila. J. Neurosci. 30, 464–477. doi: 10.1523/JNEUROSCI.1490-09.2010
Lambert, J. C., Ibrahim-Verbaas, C. A., Harold, D., Naj, A. C., Sims, R., Bellenguez, C., et al. (2013). Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 45, 1452–1458. doi: 10.1038/ng.2802
Lee, S., Wang, J. W., Yu, W., and Lu, B. (2012). Phospho-dependent ubiquitination and degradation of PAR-1 regulates synaptic morphology and tau-mediated Abeta toxicity in Drosophila. Nat. Commun. 3:1312. doi: 10.1038/ncomms2278
Lunnon, K., Smith, R., Hannon, E., De Jager, P. L., Srivastava, G., Volta, M., et al. (2014). Methylomic profiling implicates cortical deregulation of ANK1 in Alzheimer’s disease. Nat. Neurosci. 17, 1164–1170. doi: 10.1038/nn.3782
Malik, B. R., Gillespie, J. M., and Hodge, J. J. (2013). CASK and CaMKII function in the mushroom body α′/β′ neurons during Drosophila memory formation. Front. Neural Cir. 7:52. doi: 10.3389/fncir.2013.00052
Malik, B. R., and Hodge, J. J. (2014). Drosophila adult olfactory shock learning. J. Visual. Exp. 90:e50107.
Massaro, C. M., Pielage, J., and Davis, G. W. (2009). Molecular mechanisms that enhance synapse stability despite persistent disruption of the spectrin/ankyrin/microtubule cytoskeleton. J. Cell Biol. 187, 101–117. doi: 10.1083/jcb.200903166
Mastroeni, D., Sekar, S., Nolz, J., Delvaux, E., Lunnon, K., Mill, J., et al. (2017). ANK1 is up-regulated in laser captured microglia in Alzheimer’s brain; the importance of addressing cellular heterogeneity. PLoS One 12:e0177814. doi: 10.1371/journal.pone.0177814
Mazock, G. H., Das, A., Base, C., and Dubreuil, R. R. (2010). Transgene rescue identifies an essential function for Drosophila beta spectrin in the nervous system and a selective requirement for ankyrin-2-binding activity. Mol. Biol. Cell 21, 2860–2868. doi: 10.1091/mbc.E10-03-0180
McGowan, E., Eriksen, J., and Hutton, M. (2006). A decade of modeling Alzheimer’s disease in transgenic mice. Trends Genet. 22, 281–289. doi: 10.1016/j.tig.2006.03.007
Mershin, A., Pavlopoulos, E., Fitch, O., Braden, B. C., Nanopoulos, D. V., and Skoulakis, E. M. (2004). Learning and memory deficits upon TAU accumulation in Drosophila mushroom body neurons. Learn. Mem. 11, 277–287. doi: 10.1101/lm.70804
Papanikolopoulou, K., and Skoulakis, E. M. (2015). Temporally distinct phosphorylations differentiate Tau-dependent learning deficits and premature mortality in Drosophila. Hum. Mol. Genet. 24, 2065–2077. doi: 10.1093/hmg/ddu726
Pfeiffer, B. D., Jenett, A., Hammonds, A. S., Ngo, T. T., Misra, S., Murphy, C., et al. (2008). Tools for neuroanatomy and neurogenetics in Drosophila. Proc. Natl. Acad. Sci. U.S.A. 105, 9715–9720. doi: 10.1073/pnas.0803697105
Pielage, J., Cheng, L., Fetter, R. D., Carlton, P. M., Sedat, J. W., and Davis, G. W. (2008). A Presynaptic giant ankyrin stabilizes the NMJ through regulation of presynaptic microtubules and transsynaptic cell adhesion. Neuron 58, 195–209. doi: 10.1016/j.neuron.2008.02.017
Ping, Y., Hahm, E. T., Waro, G., Song, Q., Vo-Ba, D. A., Licursi, A., et al. (2015). Linking abeta42-induced hyperexcitability to neurodegeneration, learning and motor deficits, and a shorter lifespan in an Alzheimer’s model. PLoS Genet. 11:e1005025. doi: 10.1371/journal.pgen.1005025
Plattner, F., Angelo, M., and Giese, K. P. (2006). The roles of cyclin-dependent kinase 5 and glycogen synthase kinase 3 in tau hyperphosphorylation. J. Biol. Chem. 281, 25457–25465. doi: 10.1074/jbc.m603469200
Schlichting, M., Menegazzi, P., Lelito, K. R., Yao, Z., Buhl, E., Dalla Benetta, E., et al. (2016). A neural network underlying circadian entrainment and photoperiodic adjustment of sleep and activity in Drosophila. J. Neurosci. 36, 9084–9096. doi: 10.1523/JNEUROSCI.0992-16.2016
Sealey, M. A., Vourkou, E., Cowan, C. M., Bossing, T., Quraishe, S., Grammenoudi, S., et al. (2017). Distinct phenotypes of three-repeat and four-repeat human tau in a transgenic model of tauopathy. Neurobiol. Dis. 105, 74–83. doi: 10.1016/j.nbd.2017.05.003
Selkoe, D. J., and Hardy, J. (2016). The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 8, 595–608. doi: 10.15252/emmm.201606210
Shaw, R. E., Kottler, B., Ludlow, Z. N., Buhl, E., Kim, D., Morais da Silva, S., et al. (2018). In vivo expansion of functionally integrated GABAergic interneurons by targeted increase in neural progenitors. EMBO J. 37:e98163. doi: 10.15252/embj.201798163
Shulman, J. M., Chipendo, P., Chibnik, L. B., Aubin, C., Tran, D., Keenan, B. T., et al. (2011). Functional screening of Alzheimer pathology genome-wide association signals in Drosophila. Am. J. Hum. Genet. 88, 232–238. doi: 10.1016/j.ajhg.2011.01.006
Shulman, J. M., Imboywa, S., Giagtzoglou, N., Powers, M. P., Hu, Y., Devenport, D., et al. (2014). Functional screening in Drosophila identifies Alzheimer’s disease susceptibility genes and implicates Tau-mediated mechanisms. Hum. Mol. Genet. 23, 870–877. doi: 10.1093/hmg/ddt478
Siegenthaler, D., Enneking, E. M., Moreno, E., and Pielage, J. (2015). L1CAM/Neuroglian controls the axon-axon interactions establishing layered and lobular mushroom body architecture. J. Cell Biol. 208, 1003–1018. doi: 10.1083/jcb.201407131
Smith, A. R., Smith, R. G., Burrage, J., Troakes, C., Al-Sarraj, S., Kalaria, R. N., et al. (2019). A cross-brain regions study of ANK1 DNA methylation in different neurodegenerative diseases. Neurobiol. Aging 74, 70–76. doi: 10.1016/j.neurobiolaging.2018.09.024
Smith, K. R., and Penzes, P. (2018). Ankyrins: roles in synaptic biology and pathology. Mol. Cell Neurosci. 91, 131–139. doi: 10.1016/j.mcn.2018.04.010
Smith, R. G., Hannon, E., De Jager, P. L., Chibnik, L., Lott, S. J., Condliffe, D., et al. (2018). Elevated DNA methylation across a 48-kb region spanning the HOXA gene cluster is associated with Alzheimer’s disease neuropathology. Alzheimer’s Dementia 14, 1580–1588. doi: 10.1016/j.jalz.2018.01.017
Smith, R. G., and Lunnon, K. (2017). DNA Modifications and Alzheimer’s Disease. Adv. Exp. Med. Biol. 978, 303–319.
Speretta, E., Jahn, T. R., Tartaglia, G. G., Favrin, G., Barros, T. P., Imarisio, S., et al. (2012). Expression in Drosophila of tandem amyloid beta peptides provides insights into links between aggregation and neurotoxicity. J. Biol. Chem. 287, 20748–20754. doi: 10.1074/jbc.M112.350124
Spires-Jones, T. L., and Hyman, B. T. (2014). The intersection of amyloid beta and tau at synapses in Alzheimer’s disease. Neuron 82, 756–771. doi: 10.1016/j.neuron.2014.05.004
Sun, J., Xu, A. Q., Giraud, J., Poppinga, H., Riemensperger, T., Fiala, A., et al. (2018). Neural control of startle-induced locomotion by the mushroom bodies and associated neurons in Drosophila. Front. Sys. Neurosci. 12:6. doi: 10.3389/fnsys.2018.00006
Suzuki, M. M., and Bird, A. (2008). DNA methylation landscapes: provocative insights from epigenomics. Nat. Rev. Genet. 9, 465–476. doi: 10.1038/nrg2341
Tabuchi, M., Lone, S. R., Liu, S., Liu, Q., Zhang, J., Spira, A. P., et al. (2015). Sleep interacts with abeta to modulate intrinsic neuronal excitability. Curr. Biol. 25, 702–712. doi: 10.1016/j.cub.2015.01.016
Van Dam, D., and De Deyn, P. P. (2011). Animal models in the drug discovery pipeline for Alzheimer’s disease. Br. J. Pharmacol. 164, 1285–1300. doi: 10.1111/j.1476-5381.2011.01299.x
Walsh, D. M., and Selkoe, D. J. (2004). Deciphering the molecular basis of memory failure in Alzheimer’s disease. Neuron 44, 181–193. doi: 10.1016/j.neuron.2004.09.010
Wang, J. Z., Grundke-Iqbal, I., and Iqbal, K. (2007). Kinases and phosphatases and tau sites involved in Alzheimer neurofibrillary degeneration. Eur. J. Neurosci. 25, 59–68. doi: 10.1111/j.1460-9568.2006.05226.x
Wittmann, C. W., Wszolek, M. F., Shulman, J. M., Salvaterra, P. M., Lewis, J., Hutton, M., et al. (2001). Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science 293, 711–714. doi: 10.1126/science.1062382
Wu, J. W., Hussaini, S. A., Bastille, I. M., Rodriguez, G. A., Mrejeru, A., Rilett, K., et al. (2016). Neuronal activity enhances tau propagation and tau pathology in vivo. Nat. Neurosci. 19, 1085–1092. doi: 10.1038/nn.4328
Keywords: Alzheimer’s disease, Drosophila, memory, lifespan, locomotion, neurodegeneration, Tau, Ankyrin
Citation: Higham JP, Malik BR, Buhl E, Dawson JM, Ogier AS, Lunnon K and Hodge JJL (2019) Alzheimer’s Disease Associated Genes Ankyrin and Tau Cause Shortened Lifespan and Memory Loss in Drosophila. Front. Cell. Neurosci. 13:260. doi: 10.3389/fncel.2019.00260
Received: 01 February 2019; Accepted: 23 May 2019;
Published: 11 June 2019.
Edited by:
Miguel Medina, Network Biomedical Research Center on Neurodegenerative Diseases (CIBERNED), SpainReviewed by:
Kunie Ando, Free University of Brussels, BelgiumEfthimios M. C. Skoulakis, Alexander Fleming Biomedical Sciences Research Center, Greece
Copyright © 2019 Higham, Malik, Buhl, Dawson, Ogier, Lunnon and Hodge. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: James J. L. Hodge, amFtZXMuaG9kZ2VAYnJpc3RvbC5hYy51aw==
†These authors have contributed equally to this work