 Shin-ya Kawaguchi1,2,3*
Shin-ya Kawaguchi1,2,3*- 1Society-Academia Collaboration for Innovation, Kyoto University, Kyoto, Japan
- 2Department of Biophysics, Graduate School of Science, Kyoto University, Kyoto, Japan
- 3Institute for Advanced Study, Kyoto University, Kyoto, Japan
Small size of an axon and presynaptic structures have hindered direct functional analysis of axonal signaling and transmitter release at presynaptic boutons in the central nervous system. However, recent technical advances in subcellular patch-clamp recordings and in fluorescent imagings are shedding light on the dynamic nature of axonal and presynaptic mechanisms. Here I summarize the functional design of an axon and presynaptic boutons, such as diversity and activity-dependent changes of action potential (AP) waveforms, Ca2+ influx, and kinetics of transmitter release, revealed by the technical tour de force of direct patch-clamp recordings and the leading-edge fluorescent imagings. I highlight the critical factors for dynamic modulation of transmitter release and presynaptic short-term plasticity.
Introduction
Direct patch-clamp recording from an axonal compartment tells a lot about the axon physiology and mechanisms of synaptic transmission. Unfortunately an axon and presynaptic terminals are usually too small to apply patch-clamp technique, except for unusually large structures, such as synapses of calyx of Held in the auditory brainstem and presynaptic boutons of hippocampal mossy fiber (Forsythe, 1994; Borst et al., 1995; Geiger and Jonas, 2000). Most conventional synapses in the central nervous system have much smaller presynaptic boutons (∼1 μm) containing only 1–2 active zones, from which direct recording has been almost impossible to perform. However, in this decade remarkable technical advances have been accomplished in recording even from conventional small presynaptic structures in primary culture system, such as boutons of hippocampal pyramidal cells (∼1 μm, Novak et al., 2013), cerebellar Purkinje cell (PC) axon terminals (∼2–3 μm, Kawaguchi and Sakaba, 2015), and cerebellar granule cell (GC) axon varicosities (∼1 μm, Kawaguchi and Sakaba, 2017). These advancements have provided detailed information about the mechanisms of presynaptic function, such as dynamics of presynaptic membrane excitability, Ca2+ influx and buffering, pool size of readily releasable synaptic vesicles, and functional coupling between Ca2+ and release machinery. In this article, I like to overview these recently clarified presynaptic mechanisms for transmitter release, particularly focusing on small synapses.
Factors to Determine Presynaptic Transmitter Release
Fluorescent labeling of axons and presynaptic boutons makes it possible to precisely position a thin tip of glass pipette at a small structure for patch-clamp recordings. Voltage-clamp of an axon terminal allows to study quantitative relationship between the Ca2+ influx to a presynaptic bouton and the amount of exocytosis of synaptic vesicles (Figure 1). For example, a long-duration of depolarization pulse applied to a bouton triggers fusion of almost all release-ready synaptic vesicles to the cytoplasmic membrane, resulting in an increase of surface membrane area size which can be recorded as a sudden jump of membrane capacitance (Cm) (Lindau and Neher, 1988; Figure 1). The amount of Cm increase, that is, the number of synaptic vesicles in readily releasable pool (RRP) changes depending on the type of synapses. For example, calyx of Held synapse (diameter, ∼10 μm) shows about 400 fF of Cm increase upon a large Ca2+ influx as ∼2 nA upon a presynaptic depolarization pulse to 0 mV (Sun and Wu, 2001). On the other hand, ∼100 fF of Cm increase is caused by ∼200 pA of Ca2+ currents at mossy fiber boutons in hippocampus (∼5 μm) and those in the cerebellum (∼6 μm) (Hallermann et al., 2003; Delvendahl et al., 2013). Recently, an inhibitory neuronal presynaptic terminal, a PC bouton with a size of 2∼3 μm was patched, and a Cm increase of 80 fF was observed upon depolarization-induced Ca2+ influx of ∼300 pA (Kawaguchi and Sakaba, 2015; Figure 1). Thus, given that a single synaptic vesicle has a Cm of ∼ 0.1 fF, about 1,000 synaptic vesicles are exocytosed from a single presynaptic bouton of a mossy fiber or a PC within several hundred milliseconds. On the other hand, a much smaller conventional presynaptic structure (∼1 μm), an axon varicosity of a cerebellar GC, exhibited a ∼20 fF of Cm increase with ∼60 pA of Ca2+ currents upon the identical depolarization pulse (Kawaguchi and Sakaba, 2017; Figure 1). Thus, even at a small axon terminal about 200 synaptic vesicles rapidly undergo fusion, which surprisingly corresponds to one third of total synaptic vesicles in a single bouton, based on the estimation by an electron microscopic analysis (Xu-Friedman et al., 2001). It should be noted that the quality of patch-clamp recording (i.e., physical access to the cytosol reflected by the series resistance) is critical for precise activation and measurement (in the extent and time course) of Ca2+ currents and vesicular release. These measurements of presynaptic Cm increase upon strong Ca2+ influx suggests that substantial number of synaptic vesicles rapidly undergo exocytosis at most synapses irrespective of their being excitatory or inhibitory.
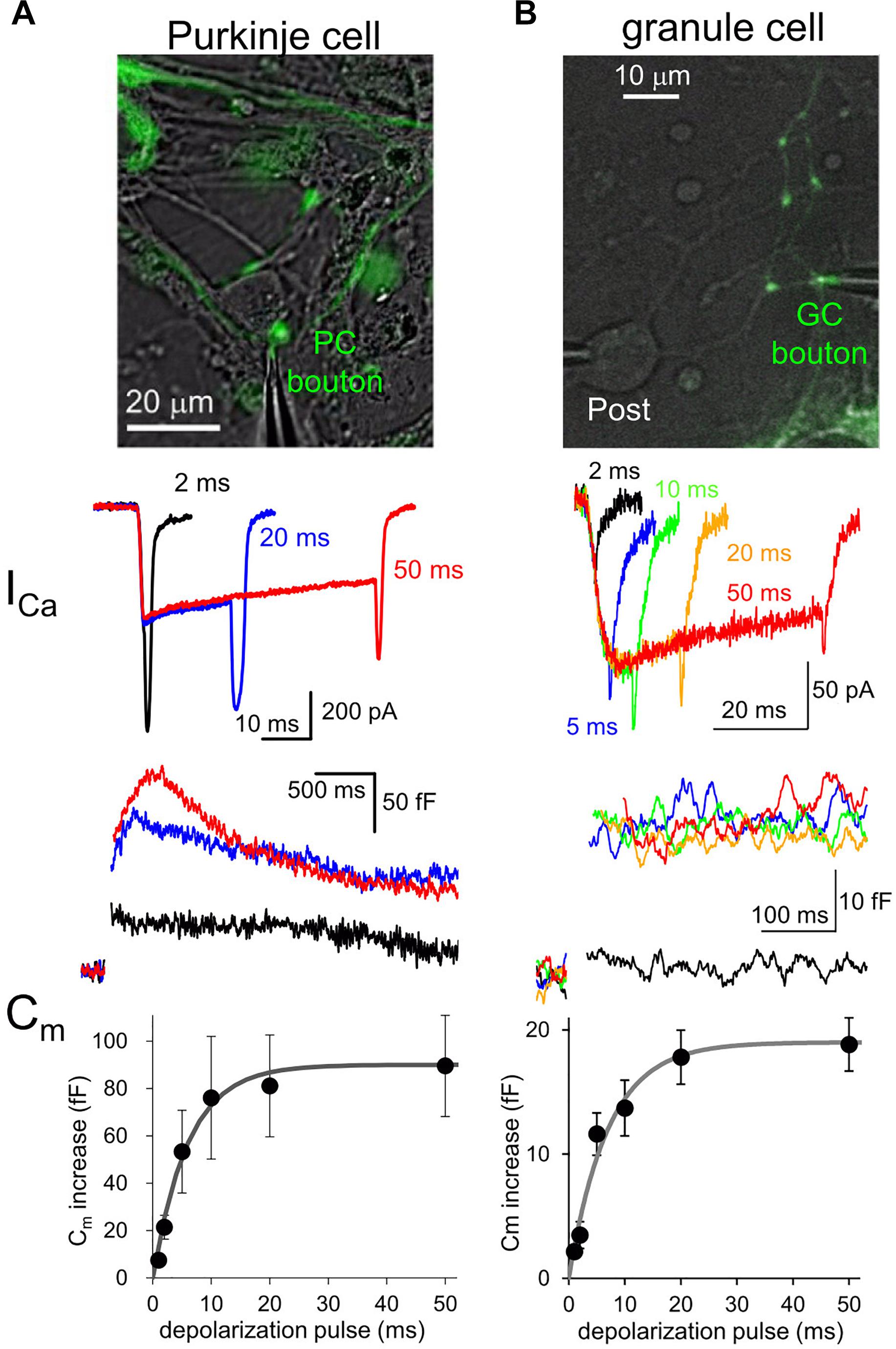
Figure 1. Presynaptic transmitter releases studied by direct patch clamp recordings. (A,B) Patch-clamp recordings from a EGFP-labeled axon varicosity of a cerebellar PC (A) or of GC (B), adapted from Kawaguchi and Sakaba (2015, 2017), respectively, with permission from Elsevier. Top, images of EGFP-labeled axon terminals of each neuron. Middle, presynaptic Ca2+ currents (ICa) upon different durations of depolarization pulses to 0 mV. Bottom, presynaptic Cm increase triggered by Ca2+ currents indicated in the middle. Average Cm increases plotted against duration of depolarization pulses are also shown.
Interestingly, comparison of various neuronal terminals implies a relationship that the maximal Cm increase and presynaptic Ca2+ current amplitude likely depend on the first to second power of presynaptic diameter. Thus, we are tempted to assume that the RRP size estimated by Cm measurement simply reflects the surface area size of presynaptic structure, implying that the release machinery downstream of activation by Ca2+ may operate in a similar manner at various neuronal boutons. However, it should be noted that the vesicular release estimated by Cm increases upon depolarization for tens of ms sometimes reflects the fusion of multiple states of synaptic vesicles, including those already primed at and those loosely coupled to the release sites (Sakaba and Neher, 2001), and even newly recruited ones at an extremely rapid rate (∼ several ms) (Saviane and Silver, 2006; Kawaguchi and Sakaba, 2017). Recent elegant glutamate imaging coupled with super-resolution analysis of active zone proteins and electron microscopic analysis suggested that the exact number of release sites, which corresponds to synaptic vesicles maximally ready for immediate exocytosis upon Ca2+ influx, is defined by the number of protein complex clusters consisting of Ca2+ channels, and some active zone protein like Munc-13 (Miki et al., 2017; Sakamoto et al., 2018). Thus, the molecular organizations of presynaptic active zone and the vesicular recruitment mechanisms toward empty release sites determine the strength and sustainability of presynaptic transmitter release.
Different neuronal terminals show diversity in the process from Ca2+ channel activation to triggering of vesicular release. First, Ca2+ current is rapidly activated at a PC bouton (Kawaguchi and Sakaba, 2015) like a calyx of Held synapse (Borst et al., 1995), whereas a GC bouton exhibits slow activation of Ca2+ current upon the identical depolarization pulses (Kawaguchi and Sakaba, 2017; Figure 1). In addition, the Ca2+-release coupling estimated by the sensitivity to Ca2+ chelators like EGTA varies in different neuronal boutons: a PC bouton shows tight coupling with low sensitivity to EGTA (Díaz-Rojas et al., 2015; Kawaguchi and Sakaba, 2015), as in other inhibitory interneurons (Bucurenciu et al., 2008; Eggermann et al., 2012); a GC bouton shows loose coupling with high EGTA sensitivity (Kawaguchi and Sakaba, 2017), in a similar manner to hippocampal mossy fiber boutons (Vyleta and Jonas, 2014). Furthermore, even action potential (AP) waveforms are quite different in axon terminals of PCs and GCs (see Figure 2).
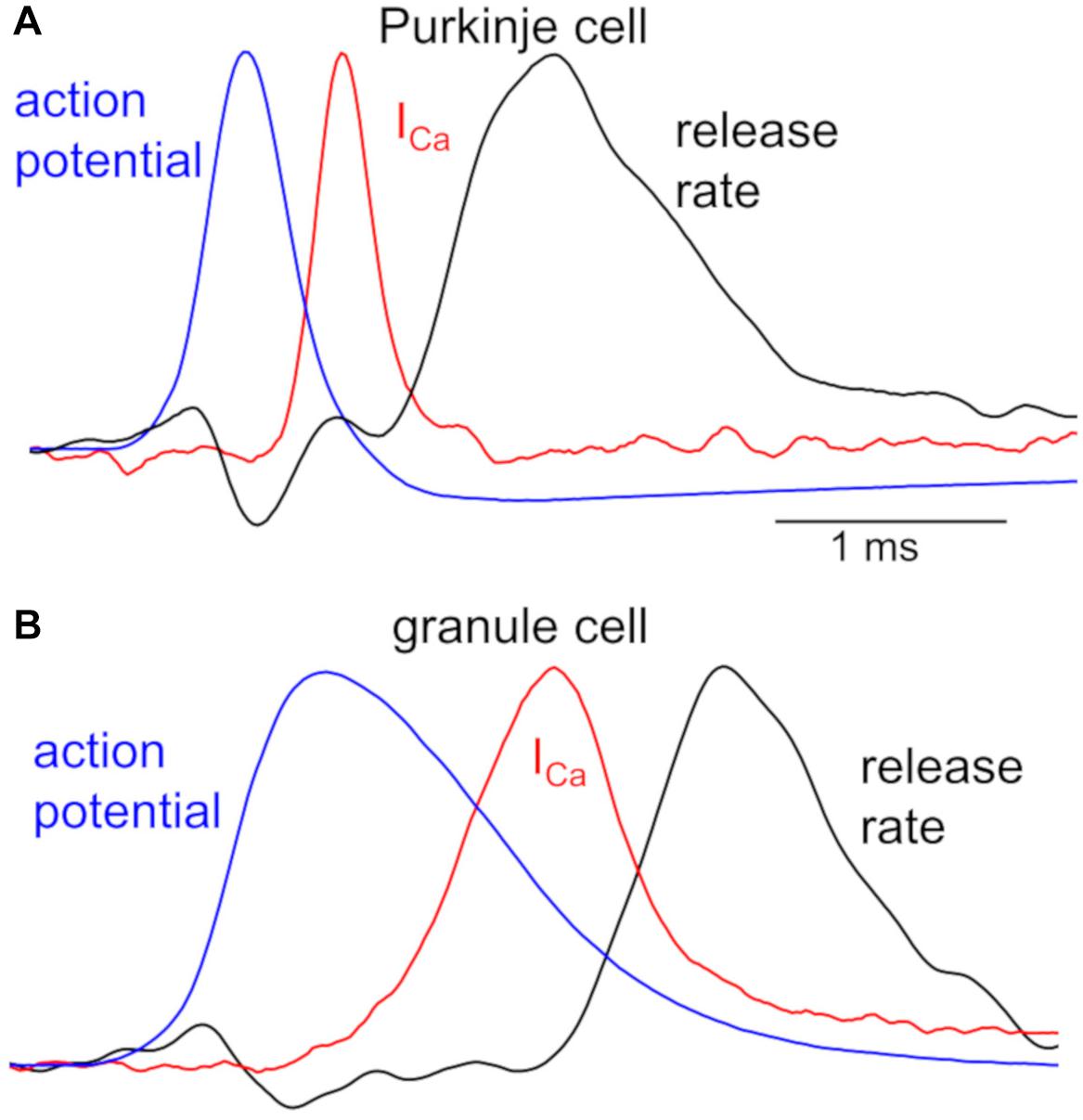
Figure 2. Kinetics of APs, Ca2+ influx, and transmitter release in a PC and GC bouton. Peak-scaled representative waveforms of APs, presynaptic Ca2+ currents (ICa), and transmitter release rate obtained by simultaneous recordings from a representative pair of presynaptic bouton (A, PC; B, GC) and postsynaptic cell. Spontaneous APs (blue), recorded from an EGFP-labeled axon varicosity of a PC (A, peak of +30 mV) and of a GC (B, peak of +37 mV), were used as voltage commands for stimulation to the voltage-clamped bouton, and presynaptic ICa (red, average of 5 traces from a pair) and postsynaptic currents were simultaneously recorded. Time courses of transmitter release rate (black, average of 5 traces recorded from a pair) were calculated by the deconvolution of the postsynaptic responses evoked by the presynaptic ICa, based on the miniature postsynaptic responses. Presynaptic recordings from a bouton of PC axon and that of GC axon had similar recording conditions, precluding the possibility of the distinct kinetics being due to the quality of patch-clamp recordings. All traces are adapted from Kawaguchi and Sakaba (2015, 2017) with permission from Elsevier.
Simultaneous recordings both from a presynaptic bouton and a postsynaptic cell provide more quantitative information about synaptic transmission (Sakaba and Neher, 2001; Sun and Wu, 2001). Figure 2 illustrates time courses of presynaptic APs and Ca2+ influx recorded from a PC or GC bouton, together with the kinetics of vesicular release estimated from postsynaptic responses (Kawaguchi and Sakaba, 2015, 2017). The remarkably different AP waveforms, when applied to the voltage-clamped bouton as voltage commands, cause clearly distinct kinetics of Ca2+ influxes. This difference in Ca2+ influx might be partly due to the slower activation of Ca2+ channels in a GC than in a PC bouton (see Figure 1), in addition to the distinct AP kinetics (Figure 2). Furthermore, the initiation of transmitter release delays more in a GC axon varicosity after the Ca2+ influx (∼500 μs after the time of half-maximal ICa) than in a PC bouton (∼300 μs), in spite that once started the release kinetics are similar (Figure 2). This distinct delay of release onset may partially reflect the different Ca2+-release coupling: tight coupling at a PC bouton, whereas loose coupling in a GC bouton. On the other hand, similar time course of release rate by itself implies that release machinery may operate in a similar manner at these neurons. Thus, AP waveforms, the resultant activation of Ca2+ channels, and the Ca2+-driven activation of release machinery, seem to change depending on the neuronal type, and likely become as key factors to impact the transmitter release. Interestingly, recent imaging studies demonstrated that Ca2+-release coupling becomes tighter after the chemical induction of presynaptic long-term potentiation (LTP) in hippocampal mossy fiber boutons (Midorikawa and Sakaba, 2017).
Digital and Analog Signaling in an Axon
Traditionally AP has been regarded as an all-or-none type of digital signal, reliably conveying information toward terminals in the central nervous system of vertebrates. However, direct recordings of APs by axonal patch-clamp methods and fluorescent imagings of membrane potential with a genetically encoded or chemical voltage indicator, have demonstrated activity-and/or location-dependent dynamic changes of AP waveforms in axon terminals (Kole et al., 2007; Hoppa et al., 2014; Kawaguchi and Sakaba, 2015; Rowan et al., 2016). In addition, sub-threshold electrical signals affect AP waveforms by modulating axonal K+ channels and/or by passively propagating into an axon over hundreds μm even around distal regions (Pouzat and Marty, 1999; Alle and Geiger, 2006; Shu et al., 2006; Kole et al., 2007; Paradiso and Wu, 2009; Trigo et al., 2010; Pugh and Jahr, 2013; Zbili et al., 2016), which results in the modulation of transmitter release from presynaptic terminals (Rama et al., 2015; Rowan and Christie, 2017; Zorrilla de San Martin et al., 2017). Thus, the axonal AP signaling is modified in a manner like a hybrid of analog and digital transmission (Clark and Häusser, 2006; Debanne et al., 2011).
Because all Ca2+ channels at a presynaptic structure are not necessarily activated by individual APs, change of an AP waveform potentially impacts the Ca2+ influx into presynaptic cytoplasm, leading to modulation of transmitter release. In a cerebellar PC, high frequency AP firing results in attenuation of AP amplitude at a bouton, decreasing peak Ca2+ influx, and hence synaptic outputs (Kawaguchi and Sakaba, 2015). On the other hand, at hippocampal mossy fiber synapses, activity-dependent attenuation of AP is accompanied with the slower decay and hence causes larger net influx of Ca2+, leading to augmented transmission (Geiger and Jonas, 2000). Distinct Ca2+-release coupling in PC boutons and mossy fiber boutons might be partially responsible for the apparently opposite effects of activity-dependent AP changes on transmitter release (Vyleta and Jonas, 2014; Kawaguchi and Sakaba, 2015). The synapses with tighter Ca2+-release coupling like PC boutons would show steeper dependence of transmitter release on the number of activated Ca2+ channels rather than the total amount of Ca2+ influx. Then, the AP amplitude controlling the maximal number of activated Ca2+ channels could be more influential for the release in such tightly coupled synapses than the decay time course affecting the duration of Ca2+ influx. In contrast, the total Ca2+ influx would more preferentially control transmitter release in loosely coupled synapses. Thus, the impact of AP waveform modulation is determined how the synapse is functionally designed at the molecular level.
The presence of receptors for neurotransmitters in an axon has been demonstrated at various brain region and influences axonal excitability and neurotransmitter release (Debanne et al., 2011). Because of the substantial passive traveling of electrical signals in an axon, even a limited number of receptors in individual boutons could cooperatively contribute to changing the local membrane potential in an axon. For example, the axonal GABAARs have been reported at various excitatory and inhibitory neurons, such as those in calyx of Held, posterior pituitary, cerebral cortex, hippocampus, and cerebellum (MacDermott et al., 1999; Trigo et al., 2008; Zorrilla de San Martin et al., 2017). Axonal GABAARs may monitor the level of GABAergic inhibition around the postsynaptic target, locally adjusting the amount GABA release. Perforated patch-clamp technique using gramicidin clarified the reversal potential of GABA-mediated current (EGABA) as ∼-50 mV in a terminal of the calyx of Held synapse (Price and Trussell, 2006) and -45 mV in a PC bouton (Zorrilla de San Martin et al., 2017). Because of the depolarized EGABA, presynaptic GABAAR activation depolarizes presynaptic boutons, facilitating Ca2+ influx upon the subsequent AP arrival and hence synaptic transmission. In contrast, axonal GABAARs also exert inhibitory effects in different neurons (Xia et al., 2014).
Short-Term Plasticity
Activity-dependent short-term plasticity of synaptic transmission lasting for milliseconds to minutes is an important element in the neuronal computation (Abbott and Regehr, 2004; Regehr, 2012). A lot of central synapses exhibit increase or decrease of strength, termed short-term synaptic facilitation, or depression, respectively, upon repetitive activity at short time intervals (Zucker and Regehr, 2002; Fioravante and Regehr, 2011). Dynamic changes of presynaptic Ca2+ concentration and downstream processes of transmitter release mediate short-term plasticity. Residual Ca2+ hypothesis is the simplest candidate mechanism for facilitation (Katz and Miledi, 1968). Temporal summation of residual Ca2+ remaining in the cytoplasm after the first AP was suggested to facilitate the following AP-triggered transmitter release based on the ∼third power Ca2+-dependence of release (Neher and Sakaba, 2008). However, the residual Ca2+ increase is too small compared with the local Ca2+ to explain synaptic facilitation in most cases.
P/Q-type of Ca2+ channels, which predominantly mediates transmitter release at many synapses, exhibit remarkable facilitation of its current in a Ca2+-dependent manner, resulting in facilitation of transmitter release (Catterall and Few, 2008; Ben-Johny and Yue, 2014). The association of Ca2+ to C-lobe of calmodulin, which is basically bound with the P/Q-type channels, is thought to rapidly augment the Ca2+ current by increasing the probability of channel opening. Exogenous expression of mutant Ca2+ channels in superior cervical ganglion neurons in culture demonstrated a tight correlation between facilitation of Ca2+ current and that of synaptic transmission (Mochida et al., 2008). Paired recordings from pre- and postsynaptic structures of a PC-PC pair showed that facilitation is almost exclusively mediated by the Ca2+ current facilitation (Díaz-Rojas et al., 2015). Similar Ca2+-dependent facilitation of presynaptic Ca2+ influx takes place at glutamatergic boutons in the calyx of Held, which partly contributes to facilitation of synaptic transmission (Felmy et al., 2003; Müller et al., 2008; Hori and Takahashi, 2009). However, recently it was reported that a mutant mice lacking Ca2+-facilitation of P-type channels still exhibit short-term facilitation at many synapses in a physiological condition, suggesting just a minor contribution of Ca2+-current facilitation to short-term synaptic facilitation (Weyrer et al., 2019).
Another candidate mechanism for synaptic facilitation is the Ca2+ buffer saturation hypothesis. If Ca2+ buffer molecules remain occupied by Ca2+ that entered into a bouton upon the first AP, cytoplasmic free Ca2+ becomes more abundant upon the following APs, and leading to larger transmitter release. Indeed, calbindin contributes to the facilitation at hippocampal mossy fiber-CA3 excitatory synapses and inhibitory synapses in the cerebral cortex (Blatow et al., 2003). The buffer saturation model relies on the loose coupling between Ca2+ and release machinery (Neher, 1998; Rozov et al., 2001; Vyleta and Jonas, 2014). Recently, direct recordings from a cerebellar GC axon varicosity demonstrated that Ca2+-release coupling is indeed loose there, and facilitation depends on the presence of low concentration of fast Ca2+ buffer in the terminal (Kawaguchi and Sakaba, 2017), in line with the Ca2+ buffer saturation model.
The next hypothesis for short-term facilitation is the Ca2+-dependent facilitation of release sensors (Regehr, 2012). Recent accumulating results indicate the critical role of Syt7 as such a facilitation sensor at various synapses (Jackman et al., 2016; Turecek and Regehr, 2018). The mechanism how Syt7 contributes to facilitation of transmitter release remains to be clarified in the future. However, it should be noted that some facilitation remains even with the genetic ablation of Syt7, implying that different combination of several facilitation mechanisms described above would operate together at distinct synapses.
In addition to facilitation, short-term depression has also been studied, and the RRP depletion is thought to be the major mechanism (Zucker and Regehr, 2002; Fioravante and Regehr, 2011). Other mechanisms, such as Ca2+ current inactivation and AP conduction failure at axonal branches (Brody and Yue, 2000; Xu and Wu, 2005) and inactivation of release sites (Neher and Sakaba, 2008; Pan and Zucker, 2009), could also contribute. GABAergic synapses on deep cerebellar nuclei neurons innervated from PCs also exhibit short-term depression upon high frequency activation (Telgkamp and Raman, 2002; Pedroarena and Schwarz, 2003). Recently, direct axonal patch-clamp recordings together with fluorescent imaging of synapto-pHluorin demonstrated frequency-dependent attenuation of AP propagation from axonal tract toward the terminals in PCs (Kawaguchi and Sakaba, 2015). Smaller number of Ca2+ channels activated by a smaller AP is responsible for the depression of transmitter release at high-frequency activation in a PC bouton. Importantly, the AP conduction faithfulness seems to increase in PC axons along with the development, resulting in smaller frequency-dependent depression at this synapse in mature animals (Turecek et al., 2016).
Conclusion and Future Direction
As overviewed here, recent technical advances to study axon physiology are shedding light on rich computational ability of an axon affecting synaptic outputs, rather than the classical view of an axon as a simple reliable digital signal conductor. Transmitter release from a presynaptic bouton is dynamically controlled by a variety of activity-dependent modulations of critical factors, such as AP waveforms, Ca2+ channel activation, and Ca2+-triggered release processes based on fine molecular organizations of release sites and vesicular replenishment systems. Different combinations of these dynamic elements would define the neuronal type-specific functional design of synapses and their short- and/or long-term plasticity. Rapid expansion of techniques for direct recording of axonal signaling with subcellular patch-clamp methods and/or fluorescent imagings is going to clarify neuron-specific and common functional designs of a variety of CNS synapses in the near future, which enriches our understanding of neuronal circuitry.
Author Contributions
S-yK wrote the manuscript and prepared the figures.
Funding
This work was supported by the KAKENHI grants from the JSPS/MEXT, Japan (15KT0082, 18H02527, and the Grant-in Scientific Research on Innovation Areas 19H04750), the Naito Science Foundation, the Takeda Science Foundation, and by the JSPS Core-to-Core Program A Advanced Research Networks.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Alle, H., and Geiger, J. R. P. (2006). Combined analog and action potential coding in hippocampal mossy fibers. Science 311, 1290–1293. doi: 10.1126/science.1119055
Ben-Johny, M., and Yue, D. T. (2014). Calmodulin regulation (calmodulation) of voltage-gated calcium channels. J. Gen. Physiol. 143, 679–692. doi: 10.1085/jgp.201311153
Blatow, M., Caputi, A., Burnashev, N., Monyer, H., and Rozov, A. (2003). Ca2+ buffer saturation underlies paired pulse facilitation in calbindin-D28k-containing terminals. Neuron 38, 79–88. doi: 10.1016/s0896-6273(03)00196-x
Borst, J. G., Helmchen, F., and Sakmann, B. (1995). Pre- and postsynaptic whole-cell recordings in the medial nucleus of the trapezoid body of the rat. J. Physiol. 489, 825–840. doi: 10.1113/jphysiol.1995.sp021095
Brody, D. L., and Yue, D. T. (2000). Release-independent short-term synaptic depression in cultured hippocampal neurons. J. Neurosci. 20, 2480–2494. doi: 10.1523/jneurosci.20-07-02480.2000
Bucurenciu, I., Kulik, A., Schwaller, B., Frotscher, M., and Jonas, P. (2008). Nanodomain coupling between Ca2+ channels and Ca2+ sensors promotes fast and efficient transmitter release at a cortical GABAergic synapse. Neuron 57, 536–545. doi: 10.1016/j.neuron.2007.12.026
Catterall, W. A., and Few, A. P. (2008). Calcium channel regulation and presynaptic plasticity. Neuron 59, 882–901. doi: 10.1016/j.neuron.2008.09.005
Clark, B., and Häusser, M. (2006). Neural coding: hybrid analog and digital signalling in axons. Curr. Biol. 16, R585–R588.
Debanne, D., Campanac, E., Bialowas, A., Carlier, E., and Alcaraz, G. (2011). Axon physiology. Physiol. Rev. 91, 555–602. doi: 10.1152/physrev.00048.2009
Delvendahl, I., Weyhersmüller, A., Ritzau-Jost, A., and Hallermann, S. (2013). Hippocampal and cerebellar mossy fibre boutons - same name, different function. J. Physiol. 591, 3179–3188. doi: 10.1113/jphysiol.2012.248294
Díaz-Rojas, F., Sakaba, T., and Kawaguchi, S. Y. (2015). Ca2+ current facilitation determines short-term facilitation at inhibitory synapses between cerebellar Purkinje cells. J. Physiol. 593, 4889–4904. doi: 10.1113/JP270704
Eggermann, E., Bucurenciu, I., Goswami, S. P., and Jonas, P. (2012). Nanodomain coupling between Ca2+ channels and sensors of exocytosis at fast mammalian synapses. Nature Rev. Neurosci. 13, 7–21. doi: 10.1038/nrn3125
Felmy, F., Neher, E., and Schneggenburger, R. (2003). Probing the intracellular calcium sensitivity of transmitter release during synaptic facilitation. Neuron 37, 801–811. doi: 10.1016/s0896-6273(03)00085-0
Fioravante, D., and Regehr, W. G. (2011). Short-term forms of presynaptic plasticity. Curr. Opin. Neurobiol. 21, 269–274. doi: 10.1016/j.conb.2011.02.003
Forsythe, I. D. (1994). Direct patch recording from identified presynaptic terminals mediating glutamatergic EPSCs in the rat CNS, in vitro. J. Physiol. 479, 381–387. doi: 10.1113/jphysiol.1994.sp020303
Geiger, J. R., and Jonas, P. (2000). Dynamic control of presynaptic Ca(2+) inflow by fast-inactivating k(+) channels in hippocampal mossy fiber boutons. Neuron 28, 927–939. doi: 10.1016/s0896-6273(00)00164-1
Hallermann, S., Pawlu, C., Jonas, P., and Heckmann, N. (2003). A large pool of releasable vesicles in a cortical glutamatergic synapse. Proc. Natl. Acad. Sci. U.S.A. 100, 8975–8980. doi: 10.1073/pnas.1432836100
Hoppa, M. B., Gouzer, G., Armbruster, M., and Ryan, T. A. (2014). Control and plasticity of the presynaptic action potential waveform at small CNS nerve terminals. Neuron 84, 778–789. doi: 10.1016/j.neuron.2014.09.038
Hori, T., and Takahashi, T. (2009). Mechanisms underlying short-term modulation of transmitter release by presynaptic depolarization. J. Physiol. 587, 2987–3000. doi: 10.1113/jphysiol.2009
Jackman, S. L., Turecek, J., Belinsky, J. E., and Regehr, W. G. (2016). The calcium sensor synaptotagmin 7 is required for synaptic facilitation. Nature 529, 88–91. doi: 10.1038/nature16507
Katz, B., and Miledi, R. (1968). The role of calcium in neuromuscular facilitation. J. Physiol. 195, 481–492. doi: 10.1113/jphysiol.1968.sp008469
Kawaguchi, S. Y., and Sakaba, T. (2015). Control of inhibitory synaptic outputs by low excitability of axon terminals revealed by direct recording. Neuron 85, 1273–1288. doi: 10.1016/j.neuron.2015.02.013
Kawaguchi, S. Y., and Sakaba, T. (2017). Fast Ca2+ buffer-dependent reliable but plastic transmission at small CNS synapses revealed by direct bouton recording. Cell Rep. 21, 3338–3345. doi: 10.1016/j.celrep.2017.11.072
Kole, M. H., Letzkus, J. J., and Stuart, G. J. (2007). Axon initial segment Kv1 channels control axonal action potential waveform and synaptic efficacy. Neuron 55, 633–647. doi: 10.1016/j.neuron.2007.07.031
Lindau, M., and Neher, E. (1988). Patch-clamp techniques for time-resolved capacitance measurements in single cells. Pflugers. Arch. 411, 137–146. doi: 10.1007/bf00582306
MacDermott, A. B., Role, L. W., and Siegelbaum, S. A. (1999). Presynaptic ionotropic receptors and the control of transmitter release. Annu. Rev. Neurosci. 22, 443–485. doi: 10.1146/annurev.neuro.22.1.443
Midorikawa, M., and Sakaba, T. (2017). Kinetics of releasable synaptic vesicles and their plastic changes at hippocampal mossy fiber synapses. Neuron 96, 1033–1040. doi: 10.1016/j.neuron.2017.10.016
Miki, T., Kaufmann, W. A., Malagon, G., Gomez, L., Tabuchi, K., Watanabe, M., et al. (2017). Numbers of presynaptic Ca2+ channel clusters match those of functionally defined vesicular docking sites in single central synapses. Proc. Natl. Acad. Sci. U.S.A. 114, E5246–E5255. doi: 10.1073/pnas.1704470114
Mochida, S., Few, A. P., Scheuer, T., and Catterall, W. A. (2008). Regulation of presynaptic Ca(V)2.1 channels by Ca2+ sensor proteins mediates short-term synaptic plasticity. Neuron 57, 210–216. doi: 10.1016/j.neuron.2007.11.036
Müller, M., Felmy, F., and Schneggenburger, R. (2008). A limited contribution of Ca2+ current facilitation to paired-pulse facilitation of transmitter release at the rat calyx of Held. J. Physiol. 586, 5503–5520. doi: 10.1113/jphysiol.2008.155838
Neher, E. (1998). Vesicle pools and Ca2+ microdomains: new tools for understanding their roles in neurotransmitter release. Neuron 20, 389–399. doi: 10.1016/s0896-6273(00)80983-6
Neher, E., and Sakaba, T. (2008). Multiple roles of calcium ions in the regulation of transmitter release. Neuron 59, 861–872. doi: 10.1016/j.neuron.2008.08.019
Novak, P., Gorelik, J., Vivekananda, U., Shevchuk, A. I., Ermolyuk, Y. S., Bailey, R. J., et al. (2013). Nanoscale-targeted patch-clamp recordings of functional presynaptic ion channels. Neuron 79, 1067–1077. doi: 10.1016/j.neuron.2013.07.012
Pan, B., and Zucker, R. S. (2009). A general model of synaptic transmission and short-term plasticity. Neuron 62, 539–554. doi: 10.1016/j.neuron.2009.03.025
Paradiso, K., and Wu, L.-G. (2009). Small voltage changes at nerve terminals travel up axons to affect action potential initiation. Nat. Neurosci. 12, 541–543. doi: 10.1038/nn.2301
Pedroarena, C. M., and Schwarz, C. (2003). Efficacy and short-term plasticity at GABAergic synapses between Purkinje and deep cerebellar nuclei neurons. J. Neurophysiol. 89, 704–716.
Pouzat, C., and Marty, A. (1999). Somatic recording of gabaergic autoreceptor current in cerebellar stellate and basket cells. J. Neurosci. 19, 1675–1690. doi: 10.1523/jneurosci.19-05-01675.1999
Price, G. D., and Trussell, L. O. (2006). Estimate of the chloride concentration in a central glutamatergic terminal: a gramicidin perforated-patch study on the calyx of held. J. Neurosci. 26, 11432–11436. doi: 10.1523/jneurosci.1660-06.2006
Pugh, J. R., and Jahr, C. E. (2013). Activation of axonal receptors by gaba spillover increases somatic firing. J. Neurosci. 33, 16924–16929. doi: 10.1523/JNEUROSCI.2796-13.2013
Rama, S., Zbili, M., Bialowas, A., Fronzaroli-Molinieres, L., Ankri, N., Carlier, E., et al. (2015). Presynaptic hyperpolarization induces a fast analogue modulation of spike-evoked transmission mediated by axonal sodium channels. Nat. Commun. 6:10163. doi: 10.1038/ncomms10163
Regehr, W. G. (2012). Short-term presynaptic plasticity. Cold Spring Harb. Perspet. Biol. 4:a005072. doi: 10.1101/cshperspect.a005702
Rowan, M. J., DelCanto, G., Yu, J. J., Kamasawa, N., and Christie, J. M. (2016). Synapse-Level determination of action potential duration by k(+) channel clustering in axons. Neuron 91, 370–383. doi: 10.1016/j.neuron.2016.05.035
Rowan, M. J. M., and Christie, J. M. (2017). Rapid state-dependent alteration in kv3 channel availability drives flexible synaptic signaling dependent on somatic subthreshold depolarization. Cell Rep. 18, 2018–2029. doi: 10.1016/j.celrep.2017.01.068
Rozov, A., Burnashev, N., Sakmann, B., and Neher, E. (2001). Transmitter release modulation by intracellular Ca2+ buffers in facilitating and depressing nerve terminals of pyramidal cells in layer 2/3 of the rat neocortex indicates a target cell-specific difference in presynaptic calcium dynamics. J. Physiol. 531, 807–826. doi: 10.1111/j.1469-7793.2001.0807h.x
Sakaba, T., and Neher, E. (2001). Calmodulin mediates rapid recruitment of fast-releasing synaptic vesicles at a calyx-type synapse. Neuron 32, 1119–1131. doi: 10.1016/s0896-6273(01)00543-8
Sakamoto, H., Ariyoshi, T., Kimpara, N., Sugao, K., Taiko, I., Takikawa, K., et al. (2018). Synaptic weight set by Munc13-1 supramolecular assemblies. Nat. Neurosci. 21, 41–49. doi: 10.1038/s41593-017-0041-9
Saviane, C., and Silver, R. A. (2006). Fast vesicle reloading and a large pool sustain high bandwidth transmission at a central synapse. Nature 439, 983–987. doi: 10.1038/nature04509
Shu, Y., Hasenstaub, A., Duque, A., Yu, Y., and McCormick, D. A. (2006). Modulation of intracortical synaptic potentials by presynaptic somatic membrane potential. Nature 441, 761–765. doi: 10.1038/nature04720
Sun, J. Y., and Wu, L. G. (2001). Fast kinetics of exocytosis revealed by simultaneous measurements of presynaptic capacitance and postsynaptic currents a central synapse. Neuron 30, 171–182. doi: 10.1016/s0896-6273(01)00271-9
Telgkamp, P., and Raman, I. M. (2002). Depression of inhibitory synaptic transmission between Purkinje cells and neurons of the cerebellar nuclei. J. Neurosci. 22, 8447–8457. doi: 10.1523/jneurosci.22-19-08447.2002
Trigo, F. F., Bouhours, B., Rostaing, P., Papageorgiou, G., Corrie, J. E. T., Triller, A., et al. (2010). Presynaptic miniature GABAergic currents in developing interneurons. Neuron 66, 235–247. doi: 10.1016/j.neuron.2010.03.030
Trigo, F. F., Marty, A., and Stell, B. M. (2008). Axonal GABAA receptors. Eur. J. Neurosci. 28, 841–848. doi: 10.1111/j.1460-9568.2008.06404.x
Turecek, J., Jackman, S. L., and Regehr, W. G. (2016). Synaptic specializations support frequency-independent purkinje cell output from the cerebellar cortex. Cell Rep. 17, 3256–3268. doi: 10.1016/j.celrep.2016.11.081
Turecek, J., and Regehr, W. G. (2018). Synaptotagmin 7 mediates both facilitation and asynchronous release at granule cell synapses. J. Neurosci. 38, 3240–3251. doi: 10.1523/JNEUROSCI.3207-17.2018
Vyleta, N. P., and Jonas, P. (2014). Loose coupling between Ca2+ channels and release sensors at a plastic hippocampal synapse. Science 343, 665–670. doi: 10.1126/science.1244811
Weyrer, C., Turecek, J., Niday, Z., Liu, P. W., Nanou, E., Catterall, W. A., et al. (2019). The role of cav2.1 channel facilitation in synaptic facilitation. Cell Rep. 26:2289-2297.e3. doi: 10.1016/j.celrep.2019.01.114
Xia, Y., Zhao, Y., Yang, M., Zeng, S., and Shu, Y. (2014). Regulation of action potential waveforms by axonal GABAA receptors in cortical pyramidal neurons. PLoS One 9:e100968. doi: 10.1371/journal.pone.0100968
Xu, J., and Wu, L. G. (2005). The decrease in the presynaptic calcium current is a major cause of short-term depression at a calyx synapse. Neuron 46, 633–645. doi: 10.1016/j.neuron.2005.03.024
Xu-Friedman, M. A., Harris, K. M., and Regehr, W. G. (2001). Three-dimensional comparison of ultrastructural characteristics at depressing and facilitating synapses onto cerebellar Purkinje cells. J. Neurosci. 21, 6666–6672. doi: 10.1523/jneurosci.21-17-06666.2001
Zbili, M., Rama, S., and Debanne, D. (2016). Dynamic control of neurotransmitter release by presynaptic potential. Front. Cell. Neurosci. 10:278. doi: 10.3389/fncel.2016.00278
Zorrilla de San Martin, J., Trigo, F. F., and Kawaguchi, S. Y. (2017). Axonal GABAA receptors depolarize presynaptic terminals and facilitate transmitter release in cerebellar Purkinje cells. J. Physiol. 595, 7477–7493. doi: 10.1113/JP275369
Keywords: axon, patch-clamp, transmitter release, presynaptic terminal, action potential, short-term plasticity
Citation: Kawaguchi S-y (2019) Dynamic Factors for Transmitter Release at Small Presynaptic Boutons Revealed by Direct Patch-Clamp Recordings. Front. Cell. Neurosci. 13:269. doi: 10.3389/fncel.2019.00269
Received: 26 April 2019; Accepted: 29 May 2019;
Published: 12 June 2019.
Edited by:
Josef Bischofberger, University of Basel, SwitzerlandReviewed by:
Stefan Hallermann, Leipzig University, GermanyDominique Debanne, INSERM U1072 Neurobiologie des Canaux Ioniques et de la Synapse, France
Copyright © 2019 Kawaguchi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shin-ya Kawaguchi, a2F3YWd1Y2hpLnNoaW55YS43bUBreW90by11LmFjLmpw