 Xuewen Wu
Xuewen Wu Wenjuan Zhang
Wenjuan Zhang Yihui Li4
Yihui Li4 Xi Lin
Xi Lin- 1Department of Otolaryngology, Head-Neck and Surgery, Xiangya Hospital of Central South University, Changsha, China
- 2Department of Otolaryngology, Emory University School of Medicine, Atlanta, GA, United States
- 3Department of Otolaryngology, Wuhan Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 4Department of Pharmacy, Changsha Hospital of Traditional Medicine, Changsha, China
Connexins (Cxs) are ubiquitous membrane proteins that are found throughout vertebrate organs, acting as building blocks of the gap junctions (GJs) known to play vital roles in the normal function of many organs. Mutations in Cx genes (particularly GJB2, which encodes Cx26) cause approximately half of all cases of congenital hearing loss in newborns. Great progress has been made in understanding GJ function and the molecular mechanisms for the role of Cxs in the cochlea. Data reveal that multiple types of Cxs work together to ensure normal development and function of the cochlea. These findings include many aspects not proposed in the classic K+ recycling theory, such as the formation of normal cochlear morphology (e.g., the opening of the tunnel of Corti), the fine-tuning of the innervation of nerve fibers to the hair cells (HCs), the maturation of the ribbon synapses, and the initiation of the endocochlear potential (EP). New data, especially those collected from targeted modification of major Cx genes in the mouse cochlea, have demonstrated that Cx26 plays an essential role in the postnatal maturation of the cochlea. Studies also show that Cx26 and Cx30 assume very different roles in the EP generation, given that only Cx26 is required for normal hearing. This article will review our current understanding of the molecular structure, cellular distribution, and major functions of cochlear GJs. Potential implications of the knowledge of cochlear GJs on the design and implementation of translational studies of cochlear gene therapies for Cx mutations are also discussed.
Introduction
Connexins (Cxs) are ubiquitous membrane proteins that are present throughout the vertebrate organs. Six Cx subunits are assembled into a connexon, a hexameric structure in the cell membrane (also called a hemichannel; Dermietzel et al., 1990; Liu et al., 2006; Laird and Lampe, 2018). Undocked hemichannels provide conduits to connect intracellular and extracellular spaces when opened upon different stimuli and allow movement of molecules such as ions, ATP and fluorescent dyes (Alstrøm et al., 2015). Two hemichannels align to form one gap junction (GJ) channel that spans the plasma membrane and provides a conduit connecting the intracellular spaces of two adjacent cells (Dermietzel et al., 1990). These intercellular channels facilitate the movement of ions and biochemically-active molecules [e.g., glucose (Chang et al., 2008), ATP (Bevans and Harris, 1999; Anselmi et al., 2008), miRNA (Zong et al., 2016), and cell signaling molecules such as second messengers (Laird and Lampe, 2018)] that are essential for nutritional and signaling needs of cells. Invertebrates have similar proteins called innexins (Phelan et al., 1998; Güiza et al., 2018; Slivko-Koltchik et al., 2019). Another family of proteins shared between lower chordates and vertebrates are termed pannexins (Baranova et al., 2004; Tang et al., 2008), which generally form membrane channels that connect the intracellular and extracellular spaces, but rarely form GJs (Beyer and Berthoud, 2018). Currently there are 21 and 20 subtypes of Cxs found in humans and mice, respectively. Nineteen of these Cxs are orthologs that are shared between the two species (Söhl et al., 2003; Bedner et al., 2012). Compatible Cx subtypes co-assemble to form GJs in order to perform specific functions appropriate for local microenvironment (White, 2002; Ahmad et al., 2003). The most common GJs found in the adult rodent cochlea are co-assembled from Cx26 and Cx30, both of which are present in all types of non-sensory cells in the cochlea (Ahmad et al., 2003; Sun et al., 2005; Hoang Dinh et al., 2009).
GJs play vital roles in maintaining homeostasis of the microenvironment of cell-cell interactions in tissues (Zhao et al., 2006; Meşe et al., 2007). These intercellular channels are essential in the normal development and function of many organs, especially where microcirculation is poor, such as lens in the eyes (White, 2002) and the organ of Corti in the cochlea (Wang Y. et al., 2009) where physiology requirements demand special arrangements. The formation of morphogen gradients during development is critically dependent on GJs (Pietak and Levin, 2018). During cardiovascular and uterine muscle development, synchronization of myocyte contraction depends on the transmission of action potentials through GJ-mediated “electrical synapses” (Delorme et al., 1997). Mutations in various types of Cxs directly cause a large spectrum of human diseases (Srinivas et al., 2018). Most relevant to this review, mutations in Cxs cause more than half of all congenital cases of both syndromic and non-syndromic deafness (Hoang Dinh et al., 2009; Beheshtian et al., 2016).
The idea of GJs in the organ of Corti was first suggested in the 1970s, when ultrastructural examination of cochlear morphology led to the proposal of a “functional syncytium” among supporting cells in the organ of Corti (Jahnke, 1975; Iurato et al., 1977). The essential role of GJs in hearing is revealed through numerous genetic linkage analyses and functional studies associating mutations in Cx genes with congenital hearing impairment in humans (White et al., 1998; Bruzzone et al., 2003; Chang et al., 2003; Del Castillo et al., 2003). Meanwhile, various roles for hemichannels in cochlear supporting cells and lateral wall cells have been proposed as well, such as releasing of K+, ATP or IP3 into extracellular space by cochlear cells (Verselis, 2019). Abnormal opening of hemichannels caused by GJB2 mutations has serious consequences on the ability of cells to maintain homeostasis and are suspected to cause syndromic hearing loss that may lead to death in early childhood (Stong et al., 2006). The accumulation of data demonstrates that mutations in GJB2, which encodes Cx26, account for approximately half of all inherited prelingual non-syndromic deafness cases in both European and East Asian populations (Maw et al., 1995; Morell et al., 1998; Dai et al., 2009, 2015; Liu X. Z. et al., 2009). It is now clear that mutations in Cx genes are one of the most common genetic causes of hearing loss. Recently, a role for Cx26 in noise- and age-dependent hearing loss has also been proposed (Wang et al., 2014; Wu et al., 2014; Zhou et al., 2016; Zong et al., 2017). In addition, hundreds of mutations associated with human deafness have been identified in Cxs other than GJB2, such as GJB6 (encoding Cx30; Grifa et al., 1999), GJB3 (encoding Cx31; Xia et al., 1998; López-Bigas et al., 2001), GJE1 (encoding Cx29; Yang et al., 2007), and GJB1 (encoding Cx32; Rabionet et al., 20001). The deafness-linked GJB2 mutations include at least 93 truncation mutations, 239 point mutations that are known to cause either non-syndromic or syndromic deafness. Most of these mutations are inherited in the autosomal recessive manner, but there are at least 14 reported autosomal dominant point-mutations as well (Figure 1).
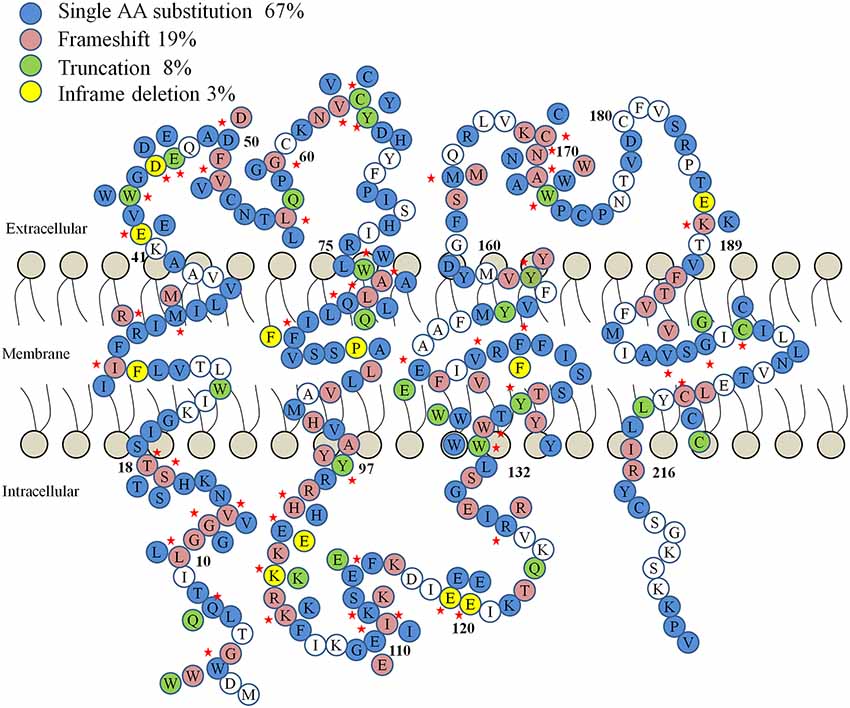
Figure 1. A diagram of human Connexin 26 (Cx26) protein mutations linked to hearing loss. The four types of mutations (frame shift, truncation, single amino acid substitution and in-frame deletion) are color-coded. Red stars denote residues with more than one type of mutation.
A complex set of Cxs forming homotypic/heterotypic/heteromeric GJs are expressed in the cochlea in developmentally-regulated and cell type-specific manners. Recent data suggest that Cx26 and other Cxs play essential roles in the development and maintenance of the cochlear function (Ahmad et al., 2003; Qu et al., 2012; Chang et al., 2015a; Mammano, 2018), such as the formation of the normal cochlear morphology, development of the endocochlear potential (EP; Qu et al., 2012; Wingard and Zhao, 2015; Mammano, 2018). In contrast to the assumptions made by the K+ recycling theory, Cx mutations clearly affect GJ function long before the establishment of the high concentration (~150 mM) of K+, the EP, and the onset of hearing (Kamiya et al., 2014). Many studies have suggested plausible theories (e.g., expression profile, functional maturation of the cochlea, hair cells (HCs) synaptic formation, etc.; Chang et al., 2015a; Jagger and Forge, 2015; Zhao, 2017). This review article will provide a brief review of our current understanding of the molecular structure, cellular distribution and function of cochlear GJs, as well as the translational relevance of knowledge of cochlear GJs to the success of cochlear gene therapies for Cx mutations.
Molecular Structure and Pattern of Expression of Cochlear GJs
General Protein Structure of Cochlear GJs
Most mammalian Cx genes consist of two exons, with exon 1 encoding the 5′-untranslated region (5′-UTR), while exon two contains the complete coding sequence (CDS) and the 3′-UTR. However, GJB1 and GJC1 (encoding Cx32 and Cx45, respectively) have more than two exons, with exon 3 encoding the CDS (Teunissen et al., 2007). Hydropathy plots of Cxs predict a common membrane topology consisting of four hydrophobic membrane-spanning domains (M1–M4), connected by two extracellular loops (El and E2) and one intracellular loop, with C- and N-termini both in the cytoplasm. The N-terminus and extracellular loops appear to be relatively conserved across paralogous Cxs, while the C-terminus shows the most variability. The C-terminus is not essential for surface expression of Cxs, as truncating mutations do not prevent trafficking to the membrane or GJ assembly (Martin et al., 2000). However, the C-terminus is the major site for phosphorylation and contains sites for interacting with non-Cx proteins (e.g., cell junction or cytoskeleton proteins) such as vinculin, calmodulin, ZO-1, and spectrin (Lampe and Lau, 2000; Batissoco et al., 2018). These findings suggest that variability in the C-terminus may affect the trafficking of Cxs. In contrast, the N-terminus of Cxs has been found to form part of the pore entrance (Purnick et al., 2000; Musa et al., 2004) together with the transmembrane M1 domain and extracellular E1 loop. The three domains are key components in GJ pore formation, as well as in voltage- and chemical-gating [e.g., by (Ca2+) changes] of GJs (Purnick et al., 2000; Sanchez and Verselis, 2014). The M1 domain also contributes to GJ voltage-gating polarity (Verselis et al., 1994), size selectivity, and unitary conductance (Kronengold et al., 2003). Importantly, the E1 domain constitutes the bulk of the aqueous pore and the E2 loop determines Cx compatibility in the assembly of heterotypic GJs, which are essential for GJ functions in cochlea and other organs (Sanchez and Verselis, 2014; Bai et al., 2018).
Cellular Expression Patterns of Various Subtypes of GJs in the Cochlea
Multiple subtypes of Cxs are found in the cochlea, and many exhibit temporal regulation of expression during development, starting from early embryonic stages. For instance, Cx31 is expressed starting at E12, Cx26 at E14.5, Cx30 at E15, Cx43 at E15.5, and Cx45 at E17.5 (Lautermann et al., 1998; Xia et al., 2000; López-Bigas et al., 2002; Ahmad et al., 2003; Cohen-Salmon et al., 2004; Sun et al., 2005). Given GJs play a vital role in embryonic stages, it is necessary to investigate the cellular pattern and developmental time course of Cx expression in order to better understand the molecular basis of hereditary deafness. One of the earliest studies utilized a cDNA dot-blot analysis method, to quantitatively investigate the expression levels of 15 Cx genes in the mouse cochlea (Ahmad et al., 2003). The expression patterns and relative abundance of four major cochlear Cxs were determined by this method (from highest to lowest expression: Gjb2>Gjc3>Gjb6≈Gja1). Multiple studies have demonstrated that major subtypes of Cxs expressed in the cochlea are Cx26 (Kikuchi et al., 1995; Lautermann et al., 1998; Forge et al., 1999; Chang et al., 2008) and Cx30 (Xia et al., 2001; Ahmad et al., 2003; Teubner et al., 2003) in non-sensory cells of cochlea [e.g., supporting cells, lateral wall fibrocytes and basal cells of stria vascularis (SV)]. The study by Tang et al. (2006) was the first to show the expression of Cx29 in the cochlear Schwann cells. Other studies have shown that Cx43 is expressed in the SV and mature bony capsule (Suzuki et al., 2003; Cohen-Salmon et al., 2004), and Cx45 is found in the cochlear blood vessels (Cohen-Salmon et al., 2004). Other reported but more controversial Cxs appear to be minor cochlear Cxs, including Cx23 (Locher et al., 2015), Cx31 (Xia et al., 2000), Cx30.2 (Buniello et al., 2004), Cx31.1 (Buniello et al., 2004), Cx32 (López-Bigas et al., 2002), Cx36 (Liu W. et al., 2009), Cx40 (Buniello et al., 2004), and Cx59 (Buniello et al., 2004). Expressions of pannexin subtypes 1, 2, and 3 (Wang X. H. et al., 2009) have also been reported, but their ability to form GJ channels is controversial (Sahu et al., 2014). Data support that it is unlikely that they are assembled into cochlear GJs, but may function as hemichannels (Penuela et al., 2007).
The compatible pairs of major GJ-forming Cxs in the cochlea are Cx26–Cx30 (Ahmad et al., 2003), and Cx26–Cx31 (Liu X. Z. et al., 2009). Depending on the Cx composition, heteromerically and heterotypically assembled GJs demonstrate characteristic unitary conductance, permeability, voltage- and chemical-gating properties that are suited for their specific physiological roles (White and Bruzzone, 1996). It has been shown that heteromeric GJ channels assembled from Cx26 and Cx30 facilitate intracellular Ca2+ signaling twice as fast as their homomeric counterparts (Sun et al., 2005). In addition, the permeability of hetero- or homomeric GJs constituting Cx26 and Cx30 are found to display different charge- and size-selective properties (Sun et al., 2005). The following sections provide more detailed information about molecular structure of cochlear GJs and their constituting Cxs.
Cx26 and Cx30
GJs constituting Cx26 and Cx30 are the predominant GJs in the cochlea. These GJs connect all types of non-sensory cells in the organ of Corti, the connective tissue fibrocytes in the lateral wall and at least the basal cells in the SV (the expression of Cxs in the intermediate cells in the SV is controversial; Forge et al., 2003; Sun et al., 2005; Zhao and Yu, 2006; Liu W. et al., 2009; Kamiya et al., 2014). Given that Cx26 and Cx30 are not expressed in cochlear HCs (Ahmad et al., 2003; Zhao and Yu, 2006; Liu W. et al., 2009; Kamiya et al., 2014), it is generally believed that there are no direct intercellular conduits linking HCs and supporting cells. Cx26 expression and formation of Cx26-containing GJs are detected in the mouse cochlea as early as E14.5 (Sun et al., 2005; Kamiya et al., 2014). mRNA levels of both Cx26 and Cx30 peaks in the cochlea around P10, just before the onset of hearing in mice (Ahmad et al., 2003). In contrast, Western blots demonstrate that protein levels of both Cxs saturate around P15, and stay at the adult patterns and levels afterward. Cx26 and Cx30 exhibit gradient expression in the basilar membrane, with three-fold greater expression in the apex than the base (Zhao and Yu, 2006). In animals other than mice, immunofluorescent staining of guinea pig and rat cochleae shows an expression pattern of Cx26 and Cx30 consistent with that found in mice (Zhao and Yu, 2006; Liu and Zhao, 2008), namely that Cx26 and Cx30 are not expressed in the IHCs or OHC but in all types of cochlear non-sensory cells. However, largely due to limited availability of materials, the expression profiles of Cx26 and Cx30 in the human cochlea are still unclear. One preliminary study using human adult cochlea (Liu W. et al., 2009) reported that Cx26 and Cx30 are widely expressed in the lateral wall fibrocytes and supporting cells of Organ of Corti. A subsequent study by the same group showed that Cx26 and Cx30 proteins may not necessarily be co-assembled in the lateral wall of adult human cochlea, and homomeric GJs consisting of either Cx26 or Cx30 may be more prevalently assembled than previously thought (Liu et al., 2016). In human embryonic cochlea, expression of Cx26 has been detected as early as 11 weeks of gestation (W11; Kammen-Jolly et al., 2001; Locher et al., 2015). At W18, Cx26 is consistently detected in the supporting cells of the organ of Corti and the Kolliker’s organ, the outer sulcus cells, and Claudius cells (Locher et al., 2015). Cx30 is also detected in human embryologic cochlea in the Kolliker’s organ and the cells lining the outer sulcus cells (Locher et al., 2015).
Quantitative comparison of Cx26 and Cx30 protein levels indicates that Cx26 is expressed earlier than Cx30 in the cochlea (Sun et al., 2005), suggesting that loss of Cx26 expression may transiently result in a near elimination of GJs in the developing cochlea. Likewise, mice with a conditional knockout of Cx26 in the cochlea exhibit more severe and rapid cellular degeneration than mice lacking Cx30 (Sun et al., 2009). More importantly, hearing could be normal in the absence of Cx30 as long as Cx26 protein level is maintained at the WT level (Ahmad et al., 2007; Qu et al., 2012; Boulay et al., 2013; Chang et al., 2015a; Jagger and Forge, 2015). These observations suggest that the role of Cx26 in the developing cochlea is not replaceable by Cx30 (Ahmad et al., 2003; Qu et al., 2012; Boulay et al., 2013).
Cx29
In the mouse cochlea, Cx29 expression was first detected by the cDNA dot-blot hybridization and immunolabeling methods, and it is localized to the cochlear Schwann cells after birth, but not in the embryologic cochlea (Ahmad et al., 2003; Tang et al., 2006). These findings were supported by additional studies using both in vitro and in vivo studies (Eiberger et al., 2006; Li et al., 2007). Approximately 50% of Cx29−/− mice exhibit early loss of high-frequency hearing and elevated sensitivity to noise damage (Tang et al., 2006), suggesting that Cx29 is required for normal cochlear function. Data from deaf patients have also linked mutations in Cx29 to hearing loss in humans, suggesting that Cx29 is a new candidate for studying auditory neuropathy (Yang et al., 2007).
Cx31
The expression of Cx31 is detected by in situ hybridization as early as E12 in Reissner’s membrane, SV and spiral limbus, the supporting cells, and fibrocytes in the spiral ligament (Xia et al., 2000; López-Bigas et al., 2002). In contrast, Cx31 expression disappears postnatally in most cells. After P12, only the type II and IV fibrocytes are positive for immunolabeling of Cx31 (Xia et al., 2000). However, no labeling in the sensory epithelial cells has been observed at any developmental stage (Xia et al., 2000; López-Bigas et al., 2002). Thus, the precise pattern of Cx31 expression in the cochlea is still unclear. Mutations in the GJB3 gene encoding Cx31 (e.g., in-frame 3 bp deletion: 423-425delATT) have been linked to hearing loss in Chinese families with recessive deafness (Xia et al., 1998; Liu X. Z. et al., 2009).
Cx32
Cx32 is reported to be expressed in the SV, spiral limbus, Reissner’s and basilar membranes, and spiral ganglion neurons (SGNs) as detected by in situ hybridization (López-Bigas et al., 2002). However, these results are inconsistent with other studies utilizing cDNA microarrays, immunolocalization, and western blotting (Forge et al., 1999; Ahmad et al., 2003). In particular, Forge et al. (2003) failed to detect Cx32 using both RT-PCR and Western blots. Overall, Cx32 appears to be a minor player among cochlear GJs, as no severe hearing loss is observed in Cx32 null mice (Scherer et al., 1998). Cx32 mutations (e.g., Va163Ile and Glu186Lys) have been linked to X-linked peripheral neuropathy (e.g., X-linked Charcot-Marie-Tooth disease), and deafness may indirectly manifest as one of many phenotypes in humans (Matsuyama et al., 2001).
Cx43
Cohen-Salmon et al. (2004) were the first to find Cx43 expression in the developing cochleae of mice. They reported that Cx43 expression starts as early as E15.5. Fibrocytes and the mesenchymal cells below the basilar membrane are labeled at E16.5. By P5, Cx43 expression is detected in fibrocytes of the spiral ligament, in the SV capillaries and mesenchymal cells lining the basilar membrane. From P8 onwards, the expression pattern of Cx43 changes to the bony layer of the otic capsule, and the expression level increases with maturation, which is consistent with findings obtained by cDNA microarrays (Ahmad et al., 2003). However, details of Cx43 cellular localization are still unclear (Lautermann et al., 1998; Liu et al., 2001; Suzuki et al., 2003). One consistent finding appears to locate the Cx43 to the capillaries of the SV in mice. In human fetal cochlea, Cx43 expression is detected by immunostaining in a subgroup of spiral ligament fibrocytes at W14. By W18, these cells are more clearly defined as type I fibrocytes (Locher et al., 2015). A missense mutation (e.g., 976C→T, Thr326Ser) in Cx43 has been linked to hereditary hearing loss in humans (Yang et al., 2007).
Cx45
Expression of Cx45 is detected in the mouse cochlea by E17.5, in fibrocytes of the spiral limbus and ligament, mesenchymal cells under the basilar membrane and lining the scala vestibule, and in capillary cells (Cohen-Salmon et al., 2004). At P1, SGNs are more intensely labeled compared to embryologic stage. After P8, Cx45 is detected mainly in the capillaries and mesenchymal cells lining the basilar membrane (Cohen-Salmon et al., 2004). However, other groups reported that Cx45 was not detectable by RT-PCR or by western-blot in mature mouse cochlea (Forge et al., 2003). To date, no pathogenic mutations have been identified in Cx45 (Ouyang et al., 2011).
Pannexins (Panxs)
Panx1 and 2 expressions were first reported by Tang et al. (2008) as early as E16.5 in the mouse cochlea. Western-blot and immunolabeling show that Panx1 is expressed in the inner and outer sulcus cell, the Claudius cells and the SGNs. In contrast, Panx2 is detected only in the soma and nerve fibers of SGNs (Tang et al., 2008). Other studies (Wang X. H. et al., 2009; Zhao et al., 2015; Zhao, 2016) reported labeling of Panx1 in the supporting cells of the organ of Corti, and the Panx2 in SV and SGNs, Panx3 was detected in cochlear bone. Only one case report suggested that a homozygous PANX1 variant (c.650G→A) may be associated with sensorineural hearing loss (Shao et al., 2016).
Properties and Proposed Functions of GJs in the Cochlea
GJ intercellular channels possess a relatively large pore size (10–15 Å; Wingard and Zhao, 2015; Zhu et al., 2015b) that allow the passage of ions, nucleotides, miRNA, second messengers and other small molecules up to 1.8 kDa (Neijssen et al., 2005). Gating of GJs may regulate permeability to both larger (e.g., via gating to stay at various incompletely closed sub-states; Hesketh et al., 2009) and small molecules (e.g., by completely closed GJs; Bukauskas et al., 2002). Based on the studies that electrical coupling was not significantly affected in many mutant GJs known to affect hearing in humans (Kameritsch et al., 2005; Zhang et al., 2005), regulation of GJ permeability for cell-signaling and biochemically-important molecules (e.g., glucose, cAMP, nitric oxide) has been considered as the major function of cochlear GJs. The importance of GJ-mediated biochemical coupling is further supported by studies demonstrating that Cx26 mutations linked to human deafness (e.g., V84L, V95M, A88S) specifically affect GJ-mediated biochemical coupling (Beltramello et al., 2005; Zhang et al., 2005). Based on current understanding about the molecular properties of gating, developmental and spatial expression patterns, and molecular composition of cochlear GJs, multiple theories/hypotheses have been proposed for their functions.
K+ Recycling Theory: GJs Facilitate the Diffusion of K+ Away From the Base of Hair Cells
The scala media is filled with endolymph which contains a high concentration of K+ of ~150 mM extracellularly. Acoustic stimulation induces K+ ions to flow into HCs through mechanotransduction channels, causing K+ ions to accumulate at the base of HCs if not quickly removed. The K+ recycling theory proposes that K+ accumulated during auditory transduction is removed by passive diffusion through intercellular GJs in supporting cells surrounding the HCs. These K+ ions are then recycled back into the endolymph through the GJs in the spiral limbus as well as the GJs in the lateral wall. One fault of this theory is that it does not identify the nature of the driving force for making the upward turn along the proposed K+ recycling pathway. Furthermore, new results obtained by multiple groups (Sun et al., 2009; Mammano, 2011; Zhao, 2017) from conditional Cx26 and Cx30 knockout mice are inconsistent with the predictions of classical K+ recycling theory:
(a) OHC degeneration starts days before the degeneration of IHCs (Wang Y. et al., 2009). As IHCs are the true mechanotransducer cells that are innervated by 90–95% of type I afferent fibers, and would be expected to accumulate large amount of the extracellular K+ that needs to be removed, this observation is not compatible with that expected by the K+ recycling theory.
(b) Data clearly show that the major cochlear GJs constituted by Cx26 play an essential role in the structural development (e.g., the opening of the tunnel of Corti) and functional maturation (e.g., cessation of spontaneous depolarization activities) of the cochlea (Wang Y. et al., 2009). Abnormal GJ formation is also observed in embryonic cochlea (Kamiya et al., 2014) weeks before the establishment of high K+, EP, and onset of hearing in mice (Wang Y. et al., 2009; Qu et al., 2012; Chang et al., 2015a; Jagger and Forge, 2015; Zhao, 2017). The timing of these observed events would make the proposed K+ recycling unnecessary.
(c) Conditional knockout of Cx26 before early postnatal stages (e.g., before P4) results in severe hearing loss in mice (Wang Y. et al., 2009; Chang et al., 2015a). Many cellular defects in the developing cochleae are observed, such as abnormal ribbon synapses, spontaneous depolarizing activities, pruning of type I and type II fibers of SGNs (Chang et al., 2015a). In contrast, conditional null of Cx26 expression in the cochlea after P16 does not significantly affect normal hearing (Chang et al., 2015a). These observations are not compatible with K+ recycling theory.
(d) There are other GJ systems in the cochlea, but not in the proposed K+ recycling pathway (e.g., GJs assembled from Cx29, Cx43), that play important cochlear functions that are clearly not linked to K+ recycling at all.
Initiation of the EP
The EP is generated by a complex regulation of K+ fluxes by ion channels, membrane transporters/co-transporters and GJs in all types of cells in the SV (Prazma, 1975; Nin et al., 2008). The EP starts to develop around P5 in mice and reaches maturity at P17–18 (Sadanaga and Morimitsu, 1995). Importantly, EP is never developed in Gjb6−/− mice, which lack Cx30, presumably due to disruption of the endothelial barrier in the SV (Teubner et al., 2003; Cohen-Salmon et al., 2007). Alternatively, the reduction of EP has been related to the absence of Cx30 and reduced Cx26 expression in the basal cells of the SV (Boulay et al., 2013; Chen et al., 2014; Mei et al., 2017). In contrast, conditional knockout of Cx26 in the mouse cochlea reduces but does not eliminate, positive EP (Chen et al., 2014; Mei et al., 2017; Mammano, 2018). However, this phenotype results may be due to incomplete elimination of Cx26 expression in basal cells and fibrocytes in the lateral wall in the conditional Cx26-null mouse model. These studies indicate that Cx30 is needed for the initiation of the EP, but may indirectly act by reducing Cx26 expression (Ahmad et al., 2007).
Intercellular Biochemical Signaling That Plays Vital Roles in Development and Survival of Cochlear Cells
Cochlear development requires well-established intercellular communication to coordinate cellular proliferation and differentiation. Given that the sensory epithelium in the cochlea is an avascular organ, GJs are proposed to act as an extension of the microvasculature to facilitate the transfer of metabolically-important molecules in cochlear supporting cells (Zhang et al., 2005; Chang et al., 2008). GJ-facilitated intercellular transfer of nutrient and signaling molecules may, therefore, play essential roles in cellular homeostasis during development and maintenance of cochlear functions. At the early postnatal stage, multiple phenotypes including abnormal ribbon synapse development, spontaneous depolarizing activities, fine-tuning of the innervation of the HCs are observed (Chang et al., 2015a). Missense or null mutations in Cx26 disrupt GJ formation as early as E14.5 in mice (Kamiya et al., 2014). In Cx30-null mice, double-electrode patch clamp recordings show that the absence of Cx30 does not significantly change GJ conductance among cochlear supporting cells (Chang et al., 2008). Dye diffusion assays, however, show that the rate and extent of intercellular transfer of multiple fluorescent dyes including a non-metabolizable D-glucose analog (2-NBDG) are severely reduced. In addition to glucose transport, a lack of Cx26 or Cx30 expression disrupts intercellular transfer of miRNAs (e.g., microRNA-96), inositol 1,4,5-trisphosphate, cAMP/cGMP, and ATP (Beltramello et al., 2005; Wang Y. et al., 2009; Conte et al., 2013; Forge et al., 2013; Zhu et al., 2015b; Mammano, 2018), all of which are likely to play critical roles in the cochlear development.
GJs Contribute to Active Cochlear Amplification
Disruption of GJ expression in supporting cells has been shown to change active cochlear mechanics (Zhu et al., 2013, 2015a). Targeted deletion of Cx26 specifically in the Deiter and outer pillar cells alters nonlinear capacitance of OHCs and reduces high frequency DPOAEs. Reduction in DPOAEs progressively extends to the middle and low frequency regions with aging of mice (Zong et al., 2017). The results suggest that Cx26-containing GJs in the supporting cells play important roles in active cochlear amplification (Zhu et al., 2013; Lukashkina et al., 2017).
Functions of GJs Constituted by Cxs Other Than Cx26 and Cx30
Cx29 is found to be exclusively expressed in Schwann cells surrounding SGNs in the cochlea (Tang et al., 2006). Cx29 does not form GJ plaques or functional GJ channels (Ahn et al., 2008), it is therefore proposed to play a major function as a hemichannel (Ahn et al., 2008) or by its novel association with the voltage-dependent K+ channel (Kv1; Altevogt et al., 2002; Cisterna et al., 2019) in the innermost layer of myelin-facing membranes of SGNs, allowing glial uptake of K+ from the extracellular space between axon and the myelin. The Gjc3−/−(Cx29 knockout) mice show a delay in the maturation of hearing thresholds and an early loss of high-frequency sensitivities. In addition, a prolongation in latency and distortion in the wave I of the auditory brainstem responses and elevated sensitivity to noise damages are found. However, the morphology of sensory HCs and DPOAE that depend on the integrity of outer HCs are normal in Gjc3−/− mice, indicating that the organ of Corti is not directly affected. The phenotypes in Gjc3−/− mice are explained by proposing that Cx29 hemichannels provide a pathway for removing accumulated K+ in peri-axonal space during high-frequency firings of the auditory nerve (Tang et al., 2006; Kagiava et al., 2015). Cx43 is found mostly in the bony capsule of the mature inner ear and is believed to play a critical role in the maturation of the otic capsule (Cohen-Salmon et al., 2004). Cx45 has been proposed to be involved in the inner ear vascular functions (Cohen-Salmon et al., 2004). Panx1 deficiency has been reported to activate the Caspase-3 cell apoptotic pathway and cause cochlear cell degeneration (particularly HCs; Zhao et al., 2015), thereby suggesting that Panx1 deficiency may lead to hearing loss. However, other studies have found that Panx1 is dispensable for normal hearing functions in mice (Zorzi et al., 2017; Abitbol et al., 2019).
Knowledge About Cochlear Cxs Affects Design and Implementation of Translational Studies of Cochlear Gene Therapy
According to the World Health Organization (WHO)2, hundreds of millions of people are affected by hearing-impairment (defined as >40 dB hearing loss in at least one ear). Population-based studies in Europe and North America have identified a prevalence of approximately 1/1,000 of children affected by hearing loss3. Early interventions have been found to be a key factor to improve speech and language acquisition skills in affected children (Chen and Oghalai, 2016; Abdurehim et al., 2017). For patients with severe to profound hearing loss, cochlear implantation is currently the best available treatment option. Meanwhile, biology-based treatments are explored by many labs around the world for their intrinsic advantages in terms of improved sound perception quality and costs of long-term usage. More than half of congenital hearing loss cases arise from genetic causes, most commonly due to mutations in GJB2 and GJB6. The most common GJB2 mutations are: 35delG for European populations, 167delT for the Ashkenazi Jewish and 235delC for Eastern Asian populations. All three mutations generate functional null of Cx26, theoretically give rise to effects similar to those observed in functional null mouse models (Denoyelle et al., 1998; Cohen-Salmon et al., 2002; Dere et al., 2003; Wang Y. et al., 2009). In addition to Cx26, mutations in Cx30, Cx31, and Cx29 are also reported to cause heritable hearing loss. In most cases, deafness is caused by recessive mutations in a single gene, though some special cases of digenic inheritance have been reported (Riazuddin et al., 2000; del Castillo et al., 2002; Liu X. Z. et al., 2009). Monogenic deafness is potentially amenable to treatment by gene replacement or supplementation therapies, typically by utilizing a viral vector to express a WT gene (Zhang et al., 2018).
However, there are many challenging hurdles that must be overcome before cochlear gene therapy can be applied to humans (Zhang et al., 2018), including therapeutic time window, safe vector delivery route, transfection efficacy, and the specificity of the target cells. To date, many viral vectors have been used for cochlear gene therapy in animal models, such as AAV, adenovirus, herpes simplex virus, lentivirus, Sendai virus (Kurioka et al., 2016). However, most viral vectors have low transfection efficiency in cochlear supporting cells (Kilpatrick et al., 2011; Kurioka et al., 2016; Shu et al., 2016). It is interesting that a new synthetic viral vector (AAV-ie) shows promising high transfection efficiency in all types of cochlear supporting cells even injected at the adult stage (Tan et al., 2019). The majority of studies have used AAVs and results suggest it is the most promising viral subtype to be used in the next translational stage. Optimization of viral subtypes and their modifications, surgery delivery method and delivery routes have been reviewed in many published articles (Sacheli et al., 2013; Zhang et al., 2018; Wang et al., 2019) and they are not the focus here. Mutations in a single gene can lead to both syndromic and non-syndromic hearing loss [e.g., in cases of GJB2 (MIM 121011), COL11A2 (MIM 120290), MYH9 (MIM 160775), MY07A (MIM 601317), PDS (MIM 274600), CDH23 (MIM 605516), and WFS1 (MIM 606201)]. Different mutations in the same gene may cause either dominant or recessive forms of non-syndromic hearing loss [e.g., GJB2 mutations may cause either DFNA3 (MIM 601544) or DFNB1A (MIM 220290)]. Treatment designs will need to be considerably different (Gao et al., 2018). The phenotypic diversity demonstrates how the type of mutation, the position of the mutation within the gene, and allelic combinations (i.e., compound heterozygosity) can affect the overall clinical presentation.
Studies conducted in mouse models show that null mutations in Cx26 predominantly affect the normal development of the sensory epithelium in the cochlea before the onset of hearing (Wang Y. et al., 2009; Chang et al., 2015a). Relatively few studies on the cochlear pathological changes induced by Cx26 or Cx30 mutations have been performed in human samples. In one case-report of a 41-year-old female with congenital severe hearing loss associated with the Cx26 35delG mutation, examination of the inner ear found near-total degeneration of cochlear HCs (Jun et al., 2000). The patient’s tectorial membrane was also abnormal, and there was mild vestibular hydrops. Importantly, there was no obvious neural degeneration in the cochlea. It is unclear whether these pathological changes in the inner ear started from birth or gradually developed. These findings from human samples are consistent with the massive cellular degeneration observed in the cochlear sensory epithelium of conditional Gjb2−/− mice after the onset of hearing, suggesting that any gene therapy attempts to correct Cx26 defects in adults is unlikely to be successful (Yu et al., 2014). Some studies have reported success in preventing hearing loss when treatment is performed at embryonic stages (e.g., at E11.5) in Cx30 KO mice (Miwa et al., 2013; Minoda et al., 2015). These findings suggest that the time window of cochlear gene therapy for GJB2 mutations need to be carried out early (perhaps embryonically) in humans to restore normal hearing (Zhang et al., 2018). Any treatment after malformation of the cochlea and HC death would have a significantly diminished chance of success.
Compared to Cx26 mutations, Cx30-null mutations show a slower time course of cellular degeneration in the mouse cochlea (Sun et al., 2009). The time window of treating patients with Cx30-null mutations may extend to a postnatal stage. Interestingly, over-expression of Cx26 can completely restore hearing sensitivity and prevent hair cell death in Cx30-null mice (Ahmad et al., 2007; Boulay et al., 2013), indicating that up-regulation of Cx26 might be a novel therapeutic strategy to prevent and treat deafness caused by Cx30 mutations. Another interesting finding is that mice homozygous for the A88V mutation in Cx30 show an improved high-frequency hearing threshold, while low frequency hearing is moderately impaired (Bosen et al., 2014; Kelly et al., 2019). This unusual finding may suggest a novel approach in clinical intervention using genome editing for treating high frequency hearing loss. Table 1 lists examples of Cx26 and Cx30 mutations that may demand different treatment time window. The expected outcomes of cochlear gene therapy based on our current knowledge learned from mouse models are also given.

Table 1. Types of Connexin26 (Cx26) and Cx30 human mutations and their possible treatment windows and outcomes by cochlear gene therapy.
Other factors that need to be considered in designing cochlear gene therapy for Cx-linked deafness should include the following: (1) Turnover rate of Cxs. Many Cxs, including Cx26, exhibit limited stability and degrade within 1–5 h (Fallon and Goodenough, 1981; Kelly et al., 2015). Cx30 exhibits a longer half-life of about 12 h (Kelly et al., 2015). It is unclear how over-expression of Cxs from viral vectors will affect this turnover rate, though such a strategy could still potentially alter GJ function and offer long-term treatment benefits; (2) Efficient delivery to the targeted cochlear cells. Studies have shown that ectopic Cx26 expression usually does not result in the formation of GJs in the cell membrane (Yu et al., 2014). Although it is unclear whether these intracellular or hemichannel Cx proteins are functional, the observation that the ectopically-expressed Cx26 in the cochlea of WT mice did not affect normal hearing (Yu et al., 2014) suggests that ectopically-expressed Cx26 does not seem to have harmful effects for the organ of Corti; and (3) A clinically-important question is how to maintain a long-term treatment efficacy of cochlear gene therapy. Many studies have shown that AAV1-mediated improvement of hearing is not necessarily long-lasting (Chang et al., 2015b; Kim et al., 2016; Isgrig et al., 2017). Therefore, more studies are needed before cochlear gene therapy can become clinically appealing.
Our current knowledge about GJ structure and functions in the cochlea indicates that Cxs play essential roles in the structure and functional maturation of the cochlea. Both Cx26 and Cx30 play vital roles in EP generation and maintenance. Reduced Cx26 expression is the likely cause of deafness even in cases of Cx30-null mutations since Cx26 expression at WT levels is sufficient for normal GJ function in the cochlea and the preservation of hearing. Before we can make a conclusion, one needs to keep in mind that studies have also shown that results obtained from mouse models may not necessarily be extrapolated to humans (and vice versa). Thus, larger animal models such as pig or non-human primates may be needed for further studies. Many human GJB2 point mutations (e.g., M34T, V37I, L90P) give only mild-to-moderate hearing loss (Norris et al., 2006; Minami et al., 2013), which is very different from hearing phenotypes found in knockout mice. These studies warn us of the danger of oversimplification. We, therefore, need to be cautious in reaching any conclusion before we can get more accurate genotype and phenotype information from human samples (e.g., high-quality cochlear histopathology from genotype-confirmed patient inner ear samples) and non-human primate studies. Bear these in mind, studies thus far from the mouse models (mainly based on results obtained from conditional Gjb2-null mice) have indicated that the appropriate time window of gene therapy of Cx26 seems to be in early development, possibly necessitating surgery in human embryos. The ramifications of such aggressive intervention will need to be carefully analyzed.
Author Contributions
XW and XL designed this article and generated Figure 1 and Table 1. WZ and YL made substantial contributions to the generation of Table 1.
Funding
This study was supported by grants to XL from the National Institute on Deafness and Other Communication Disorders (RO1 DC006483 and RO1 DC014496). XW is supported by grant from National Natural Science Foundation of China (81300819) and Natural Science Foundation of Hunan Province (2018JJ3842).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank two anonymous reviewers for carefully reading the manuscript. Their comments and suggestions have greatly helped in improving the quality of the review. We also thank Dr. George AndrewInglis for proofreading the manuscript carefully.
Footnotes
- ^ http://davinci.crg.es/deafness/
- ^ https://www.who.int/news-room/fact-sheets/detail/deafness-and-hearing-loss
- ^ https://www.cdc.gov/ncbddd/hearingloss/data.html
References
Abdurehim, Y., Lehmann, A., and Zeitouni, A. G. (2017). Predictive value of GJB2 mutation status for hearing outcomes of pediatric cochlear implantation. Otolaryngol. Head Neck Surg. 157, 16–24. doi: 10.1177/0194599817697054
Abitbol, J. M., O’Donnell, B. L., Wakefield, C. B., Jewlal, E., Kelly, J. J., Barr, K., et al. (2019). Double deletion of Panx1 and Panx3 affects skin and bone but not hearing. J. Mol. Med. 97, 723–736. doi: 10.1007/s00109-019-01779-9
Ahmad, S., Chen, S., Sun, J., and Lin, X. (2003). Connexins 26 and 30 are co-assembled to form gap junctions in the cochlea of mice. Biochem. Biophys. Res. Commun. 307, 362–368. doi: 10.1016/s0006-291x(03)01166-5
Ahmad, S., Tang, W., Chang, Q., Qu, Y., Hibshman, J., Li, Y., et al. (2007). Restoration of connexin26 protein level in the cochlea completely rescues hearing in a mouse model of human connexin30-linked deafness. Proc. Natl. Acad. Sci. U S A 104, 1337–1341. doi: 10.1073/pnas.0606855104
Ahn, M., Lee, J., Gustafsson, A., Enriquez, A., Lancaster, E., Sul, J. Y., et al. (2008). Cx29 and Cx32, two connexins expressed by myelinating glia, do not interact and are functionally distinct. J. Neurosci. Res. 86, 992–1006. doi: 10.1002/jnr.21561
Alstrøm, J. S., Hansen, D. B., Nielsen, M. S., and MacAulay, N. (2015). Isoform-specific phosphorylation-dependent regulation of connexin hemichannels. J. Neurophysiol. 114, 3014–3022. doi: 10.1152/jn.00575.2015
Altevogt, B. M., Kleopa, K. A., Postma, F. R., Scherer, S. S., and Paul, D. L. (2002). Connexin29 is uniquely distributed within myelinating glial cells of the central and peripheral nervous systems. J. Neurosci. 22, 6458–6470. doi: 10.1523/jneurosci.22-15-06458.2002
Anselmi, F., Hernandez, V. H., Crispino, G., Seydel, A., Ortolano, S., Roper, S. D., et al. (2008). ATP release through connexin hemichannels and gap junction transfer of second messengers propagate Ca2+ signals across the inner ear. Proc. Natl. Acad. Sci. U S A 105, 18770–18775. doi: 10.1073/pnas.0800793105
Bai, D., Yue, B., and Aoyama, H. (2018). Crucial motifs and residues in the extracellular loops influence the formation and specificity of connexin docking. Biochim. Biophys. Acta Biomembr. 1860, 9–21. doi: 10.1016/j.bbamem.2017.07.003
Baranova, A., Ivanov, D., Petrash, N., Pestova, A., Skoblov, M., Kelmanson, I., et al. (2004). The mammalian pannexin family is homologous to the invertebrate innexin gap junction proteins. Genomics 83, 706–716. doi: 10.1016/j.ygeno.2003.09.025
Batissoco, A. C., Salazar-Silva, R., Oiticica, J., Bento, R. F., Mingroni-Netto, R. C., and Haddad, L. A. (2018). A cell junctional protein network associated with connexin-26. Int. J. Mol. Sci. 19:E2535. doi: 10.3390/ijms19092535
Bedner, P., Steinhäuser, C., and Theis, M. (2012). Functional redundancy and compensation among members of gap junction protein families? Biochim. Biophys. Acta 1818, 1971–1984. doi: 10.1016/j.bbamem.2011.10.016
Beheshtian, M., Babanejad, M., Azaiez, H., Bazazzadegan, N., Kolbe, D., Sloan-Heggen, C., et al. (2016). Heterogeneity of hereditary hearing loss in iran: a comprehensive review. Arch. Iran. Med. 19, 720–728.
Beltramello, M., Piazza, V., Bukauskas, F. F., Pozzan, T., and Mammano, F. (2005). Impaired permeability to Ins(1,4,5)P3 in a mutant connexin underlies recessive hereditary deafness. Nat. Cell Biol. 7, 63–69. doi: 10.1038/ncb1205
Bevans, C. G., and Harris, A. L. (1999). Direct high affinity modulation of connexin channel activity by cyclic nucleotides. J. Biol. Chem. 274, 3720–3725. doi: 10.1074/jbc.274.6.3720
Beyer, E. C., and Berthoud, V. M. (2018). Gap junction gene and protein families: connexins, innexins and pannexins. Biochim. Biophys. Acta Biomembr. 1860, 5–8. doi: 10.1016/j.bbamem.2017.05.016
Bosen, F., Schütz, M., Beinhauer, A., Strenzke, N., Franz, T., and Willecke, K. (2014). The clouston syndrome mutation connexin30 A88V leads to hyperproliferation of sebaceous glands and hearing impairments in mice. FEBS Lett. 588, 1795–1801. doi: 10.1016/j.febslet.2014.03.040
Boulay, A. C., del Castillo, F. J., Giraudet, F., Hamard, G., Giaume, C., Petit, C., et al. (2013). Hearing is normal without connexin30. J. Neurosci. 33, 430–434. doi: 10.1523/jneurosci.4240-12.2013
Bruzzone, R., Veronesi, V., Gomes, D., Bicego, M., Duval, N., Marlin, S., et al. (2003). Loss-of-function and residual channel activity of connexin26 mutations associated with non-syndromic deafness. FEBS Lett. 533, 79–88. doi: 10.1016/s0014-5793(02)03755-9
Bukauskas, F. F., Bukauskiene, A., and Verselis, V. K. (2002). Conductance and permeability of the residual state of connexin43 gap junction channels. J. Gen. Physiol. 119, 171–185. doi: 10.1085/jgp.119.2.171
Buniello, A., Montanaro, D., Volinia, S., Gasparini, P., and Marigo, V. (2004). An expression atlas of connexin genes in the mouse. Genomics 83, 812–820. doi: 10.1016/j.ygeno.2003.10.011
Chang, E. H., Van Camp, G., and Smith, R. J. (2003). The role of connexins in human disease. Ear Hear. 24, 314–323. doi: 10.1097/01.AUD.0000079801.55588.13
Chang, Q., Tang, W., Ahmad, S., Zhou, B., and Lin, X. (2008). Gap junction mediated intercellular metabolite transfer in the cochlea is compromised in connexin30 null mice. PLoS One 3:e4088. doi: 10.1371/journal.pone.0004088
Chang, Q., Tang, W., Kim, Y., and Lin, X. (2015a). Timed conditional null of connexin26 in mice reveals temporary requirements of connexin26 in key cochlear developmental events before the onset of hearing. Neurobiol. Dis. 73, 418–427. doi: 10.1016/j.nbd.2014.09.005
Chang, Q., Wang, J., Li, Q., Kim, Y., Zhou, B., Wang, Y., et al. (2015b). Virally mediated Kcnq1 gene replacement therapy in the immature scala media restores hearing in a mouse model of human Jervell and Lange-Nielsen deafness syndrome. EMBO Mol. Med. 7, 1077–1086. doi: 10.15252/emmm.201404929
Chen, J., Zhu, Y., Liang, C., and Zhao, H. B. (2014). Deafness induced by connexin 26 (GJB2) deficiency is not determined by endocochlear potential (EP) reduction but is associated with cochlear developmental disorders. Biochem. Biophys. Res. Commun. 448, 28–32. doi: 10.1016/j.bbrc.2014.04.016
Chen, M. M., and Oghalai, J. S. (2016). Diagnosis and management of congenital sensorineural hearing loss. Curr. Treat. Options Pediatr. 2, 256–265. doi: 10.1007/s40746-016-0056-6
Cisterna, B. A., Arroyo, P., and Puebla, C. (2019). Role of connexin-based gap junction channels in communication of myelin sheath in schwann cells. Front. Cell. Neurosci. 13:69. doi: 10.3389/fncel.2019.00069
Cohen-Salmon, M., Maxeiner, S., Kruger, O., Theis, M., Willecke, K., and Petit, C. (2004). Expression of the connexin43- and connexin45-encoding genes in the developing and mature mouse inner ear. Cell Tissue Res. 316, 15–22. doi: 10.1007/s00441-004-0861-2
Cohen-Salmon, M., Ott, T., Michel, V., Hardelin, J. P., Perfettini, I., Eybalin, M., et al. (2002). Targeted ablation of connexin26 in the inner ear epithelial gap junction network causes hearing impairment and cell death. Curr. Biol. 12, 1106–1111. doi: 10.1016/s0960-9822(02)00904-1
Cohen-Salmon, M., Regnault, B., Cayet, N., Caille, D., Demuth, K., Hardelin, J. P., et al. (2007). Connexin30 deficiency causes instrastrial fluid-blood barrier disruption within the cochlear stria vascularis. Proc. Natl. Acad. Sci. U S A 104, 6229–6234. doi: 10.1073/pnas.0605108104
Conte, I., Banfi, S., and Bovolenta, P. (2013). Non-coding RNAs in the development of sensory organs and related diseases. Cell. Mol. Life Sci. 70, 4141–4155. doi: 10.1007/s00018-013-1335-z
Dai, P., Yu, F., Han, B., Liu, X., Wang, G., Li, Q., et al. (2009). GJB2 mutation spectrum in 2,063 Chinese patients with nonsyndromic hearing impairment. J. Transl. Med. 7:26. doi: 10.1186/1479-5876-7-26
Dai, Z. Y., Sun, B. C., Huang, S. S., Yuan, Y. Y., Zhu, Y. H., Su, Y., et al. (2015). Correlation analysis of phenotype and genotype of GJB2 in patients with non-syndromic hearing loss in China. Gene 570, 272–276. doi: 10.1016/j.gene.2015.06.038
Del Castillo, I., Moreno-Pelayo, M. A., Del Castillo, F. J., Brownstein, Z., Marlin, S., Adina, Q., et al. (2003). Prevalence and evolutionary origins of the del(GJB6–D13S1830) mutation in the DFNB1 locus in hearing-impaired subjects: a multicenter study. Am. J. Hum. Genet. 73, 1452–1458. doi: 10.1086/380205
del Castillo, I., Villamar, M., Moreno-Pelayo, M. A., del Castillo, F. J., Alvarez, A., Telleria, D., et al. (2002). A deletion involving the connexin 30 gene in nonsyndromic hearing impairment. N. Engl. J. Med. 346, 243–249. doi: 10.1056/NEJMoa012052
Delorme, B., Dahl, E., Jarry-Guichard, T., Briand, J. P., Willecke, K., Gros, D., et al. (1997). Expression pattern of connexin gene products at the early developmental stages of the mouse cardiovascular system. Circ. Res. 81, 423–437. doi: 10.1161/01.res.81.3.423
Denoyelle, F., Lina-Granade, G., Plauchu, H., Bruzzone, R., Chaib, H., Levi-Acobas, F., et al. (1998). Connexin 26 gene linked to a dominant deafness. Nature 393, 319–320. doi: 10.1038/30639
Dere, E., De Souza-Silva, M. A., Frisch, C., Teubner, B., Sohl, G., Willecke, K., et al. (2003). Connexin30-deficient mice show increased emotionality and decreased rearing activity in the open-field along with neurochemical changes. Eur. J. Neurosci. 18, 629–638. doi: 10.1046/j.1460-9568.2003.02784.x
Dermietzel, R., Hwang, T. K., and Spray, D. S. (1990). The gap junction family: structure, function and chemistry. Anat. Embryol. 182, 517–528. doi: 10.1007/bf00186458
Eiberger, J., Kibschull, M., Strenzke, N., Schober, A., Bussow, H., Wessig, C., et al. (2006). Expression pattern and functional characterization of connexin29 in transgenic mice. Glia 53, 601–611. doi: 10.1002/glia.20315
Fallon, R. F., and Goodenough, D. A. (1981). Five-hour half-life of mouse liver gap-junction protein. J. Cell Biol. 90, 521–526. doi: 10.1083/jcb.90.2.521
Forge, A., Becker, D., Casalotti, S., Edwards, J., Evans, W. H., Lench, N., et al. (1999). Gap junctions and connexin expression in the inner ear. Novartis Found. Symp. 219, 134–150; discussion 151–136. doi: 10.1002/9780470515587.ch9
Forge, A., Becker, D., Casalotti, S., Edwards, J., Marziano, N., and Nevill, G. (2003). Gap junctions in the inner ear: comparison of distribution patterns in different vertebrates and assessement of connexin composition in mammals. J. Comp. Neurol. 467, 207–231. doi: 10.1002/cne.10916
Forge, A., Jagger, D. J., Kelly, J. J., and Taylor, R. R. (2013). Connexin30-mediated intercellular communication plays an essential role in epithelial repair in the cochlea. J. Cell Sci. 126, 1703–1712. doi: 10.1242/jcs.125476
Gao, X., Tao, Y., Lamas, V., Huang, M., Yeh, W. H., Pan, B., et al. (2018). Treatment of autosomal dominant hearing loss by in vivo delivery of genome editing agents. Nature 553, 217–221. doi: 10.1038/nature25164
Grifa, A., Wagner, C. A., D’Ambrosio, L., Melchionda, S., Bernardi, F., Lopez-Bigas, N., et al. (1999). Mutations in GJB6 cause nonsyndromic autosomal dominant deafness at DFNA3 locus. Nat. Genet. 23, 16–18. doi: 10.1038/12612
Güiza, J., Barria, I., Sáez, J. C., and Vega, J. L. (2018). Innexins: expression, regulation and functions. Front. Physiol. 9:1414. doi: 10.3389/fphys.2018.01414
Hesketh, G. G., Van Eyk, J. E., and Tomaselli, G. F. (2009). Mechanisms of gap junction traffic in health and disease. J. Cardiovasc. Pharmacol. 54, 263–272. doi: 10.1097/fjc.0b013e3181ba0811
Hoang Dinh, E., Ahmad, S., Chang, Q., Tang, W., Stong, B., and Lin, X. (2009). Diverse deafness mechanisms of connexin mutations revealed by studies using in vitro approaches and mouse models. Brain Res. 1277, 52–69. doi: 10.1016/j.brainres.2009.02.008
Isgrig, K., Shteamer, J. W., Belyantseva, I. A., Drummond, M. C., Fitzgerald, T. S., Vijayakumar, S., et al. (2017). Gene therapy restores balance and auditory functions in a mouse model of usher syndrome. Mol. Ther. 25, 780–791. doi: 10.1016/j.ymthe.2017.01.007
Iurato, S., Franke, K. D., Luciano, L., Wermbter, G., Pannese, F., and Reale, E. (1977). The junctional complexes among the cells of the organ of Corti as revealed by freeze-fracturing. Adv. Otorhinolaryngol. 22, 76–80. doi: 10.1159/000399490
Jagger, D. J., and Forge, A. (2015). Connexins and gap junctions in the inner ear—it’s not just about K(+) recycling. Cell Tissue Res. 360, 633–644. doi: 10.1007/s00441-014-2029-z
Jahnke, K. (1975). The fine structure of freeze-fractured intercellular junctions in the guinea pig inner ear. Acta Otolaryngol. Suppl. 336, 1–40. doi: 10.3109/00016487509125512
Jun, A. I., McGuirt, W. T., Hinojosa, R., Green, G. E., Fischel-Ghodsian, N., and Smith, R. J. (2000). Temporal bone histopathology in connexin 26-related hearing loss. Laryngoscope 110, 269–275. doi: 10.1097/00005537-200002010-00016
Kagiava, A., Theophilidis, G., Sargiannidou, I., Kyriacou, K., and Kleopa, K. A. (2015). Oxaliplatin-induced neurotoxicity is mediated through gap junction channels and hemichannels and can be prevented by octanol. Neuropharmacology 97, 289–305. doi: 10.1016/j.neuropharm.2015.05.021
Kameritsch, P., Khandoga, N., Nagel, W., Hundhausen, C., Lidington, D., and Pohl, U. (2005). Nitric oxide specifically reduces the permeability of Cx37-containing gap junctions to small molecules. J. Cell. Physiol. 203, 233–242. doi: 10.1002/jcp.20218
Kamiya, K., Yum, S. W., Kurebayashi, N., Muraki, M., Ogawa, K., Karasawa, K., et al. (2014). Assembly of the cochlear gap junction macromolecular complex requires connexin 26. J. Clin. Invest. 124, 1598–1607. doi: 10.1172/JCI67621
Kammen-Jolly, K., Ichiki, H., Scholtz, A. W., Gsenger, M., Kreczy, A., and Schrott-Fischer, A. (2001). Connexin 26 in human fetal development of the inner ear. Hear. Res. 160, 15–21. doi: 10.1016/s0378-5955(01)00310-0
Kelly, J. J., Abitbol, J. M., Hulme, S., Press, E. R., Laird, D. W., and Allman, B. L. (2019). The connexin 30 A88V mutant reduces cochlear gap junction expression and confers long-term protection against hearing loss. J. Cell Sci. 132:jcs224097. doi: 10.1242/jcs.224097
Kelly, J. J., Shao, Q., Jagger, D. J., and Laird, D. W. (2015). Cx30 exhibits unique characteristics including a long half-life when assembled into gap junctions. J. Cell Sci. 128, 3947–3960. doi: 10.1242/jcs.174698
Kikuchi, T., Kimura, R. S., Paul, D. L., and Adams, J. C. (1995). Gap junctions in the rat cochlea: immunohistochemical and ultrastructural analysis. Anat. Embryol. 191, 101–118. doi: 10.1007/bf00186783
Kilpatrick, L. A., Li, Q., Yang, J., Goddard, J. C., Fekete, D. M., and Lang, H. (2011). Adeno-associated virus-mediated gene delivery into the scala media of the normal and deafened adult mouse ear. Gene Ther. 18, 569–578. doi: 10.1038/gt.2010.175
Kim, M. A., Cho, H. J., Bae, S. H., Lee, B., Oh, S. K., Kwon, T. J., et al. (2016). Methionine sulfoxide reductase b3-targeted in utero gene therapy rescues hearing function in a mouse model of congenital sensorineural hearing loss. Antioxid. Redox Signal. 24, 590–602. doi: 10.1089/ars.2015.6442
Kronengold, J., Trexler, E. B., Bukauskas, F. F., Bargiello, T. A., and Verselis, V. K. (2003). Single-channel SCAM identifies pore-lining residues in the first extracellular loop and first transmembrane domains of Cx46 hemichannels. J. Gen. Physiol. 122, 389–405. doi: 10.1085/jgp.200308861
Kurioka, T., Mizutari, K., Niwa, K., Fukumori, T., Inoue, M., Hasegawa, M., et al. (2016). Hyaluronic acid pretreatment for Sendai virus-mediated cochlear gene transfer. Gene Ther. 23, 187–195. doi: 10.1038/gt.2015.94
Laird, D. W., and Lampe, P. D. (2018). Therapeutic strategies targeting connexins. Nat. Rev. Drug Discov. 17, 905–921. doi: 10.1038/nrd.2018.138
Lampe, P. D., and Lau, A. F. (2000). Regulation of gap junctions by phosphorylation of connexins. Arch. Biochem. Biophys. 384, 205–215. doi: 10.1006/abbi.2000.2131
Lautermann, J., ten Cate, W. J., Altenhoff, P., Grummer, R., Traub, O., Frank, H., et al. (1998). Expression of the gap-junction connexins 26 and 30 in the rat cochlea. Cell Tissue Res. 294, 415–420. doi: 10.1007/s004410051192
Li, J., Habbes, H. W., Eiberger, J., Willecke, K., Dermietzel, R., and Meier, C. (2007). Analysis of connexin expression during mouse Schwann cell development identifies connexin29 as a novel marker for the transition of neural crest to precursor cells. Glia 55, 93–103. doi: 10.1002/glia.20427
Liu, F., Arce, F. T., Ramachandran, S., and Lal, R. (2006). Nanomechanics of hemichannel conformations: connexin flexibility underlying channel opening and closing. J. Biol. Chem. 281, 23207–23217. doi: 10.1074/jbc.m605048200
Liu, W., Boström, M., Kinnefors, A., and Rask-Andersen, H. (2009). Unique expression of connexins in the human cochlea. Hear. Res. 250, 55–62. doi: 10.1016/j.heares.2009.01.010
Liu, W., Edin, F., Blom, H., Magnusson, P., Schrott-Fischer, A., Glueckert, R., et al. (2016). Super-resolution structured illumination fluorescence microscopy of the lateral wall of the cochlea: the connexin26/30 proteins are separately expressed in man. Cell Tissue Res. 365, 13–27. doi: 10.1007/s00441-016-2359-0
Liu, X. Z., Xia, X. J., Adams, J., Chen, Z. Y., Welch, K. O., Tekin, M., et al. (2001). Mutations in GJA1 (connexin 43) are associated with non-syndromic autosomal recessive deafness. Hum. Mol. Genet. 10, 2945–2951. doi: 10.1093/hmg/10.25.2945
Liu, X. Z., Yuan, Y., Yan, D., Ding, E. H., Ouyang, X. M., Fei, Y., et al. (2009). Digenic inheritance of non-syndromic deafness caused by mutations at the gap junction proteins Cx26 and Cx31. Hum. Genet. 125, 53–62. doi: 10.1007/s00439-008-0602-9
Liu, Y. P., and Zhao, H. B. (2008). Cellular characterization of connexin26 and Connnexin30 expression in the cochlear lateral wall. Cell Tissue Res. 333, 395–403. doi: 10.1007/s00441-008-0641-5
Locher, H., de Groot, J. C., van Iperen, L., Huisman, M. A., Frijns, J. H., and Chuva de Sousa Lopes, S. M. (2015). Development of the stria vascularis and potassium regulation in the human fetal cochlea: insights into hereditary sensorineural hearing loss. Dev. Neurobiol. 75, 1219–1240. doi: 10.1002/dneu.22279
López-Bigas, N., Arbonés, M. L., Estivill, X., and Simonneau, L. (2002). Expression profiles of the connexin genes, Gjb1 and Gjb3, in the developing mouse cochlea. Mech. Dev. 119, S111–S115. doi: 10.1016/s0925-4773(03)00102-3
López-Bigas, N., Olivé, M., Rabionet, R., Ben-David, O., Martinez-Matos, J. A., Bravo, O., et al. (2001). Connexin 31 (GJB3) is expressed in the peripheral and auditory nerves and causes neuropathy and hearing impairment. Hum. Mol. Genet. 10, 947–952. doi: 10.1093/hmg/10.9.947
Lukashkina, V. A., Yamashita, T., Zuo, J., Lukashkin, A. N., and Russell, I. J. (2017). Amplification mode differs along the length of the mouse cochlea as revealed by connexin 26 deletion from specific gap junctions. Sci. Rep. 7:5185. doi: 10.1038/s41598-017-04279-3
Mammano, F. (2011). Ca2+ homeostasis defects and hereditary hearing loss. Biofactors 37, 182–188. doi: 10.1002/biof.150
Mammano, F. (2018). Inner ear connexin channels: roles in development and maintenance of cochlear function. Cold Spring Harb. Perspect. Med. 9:a033233. doi: 10.1101/cshperspect.a033233
Martin, P. E., Steggles, J., Wilson, C., Ahmad, S., and Evans, W. H. (2000). Targeting motifs and functional parameters governing the assembly of connexins into gap junctions. Biochem. J. 349, 281–287. doi: 10.1042/0264-6021:3490281
Matsuyama, W., Nakagawa, M., Moritoyo, T., Takashima, H., Umehara, F., Hirata, K., et al. (2001). Phenotypes of X-linked Charcot-Marie-Tooth disease and altered trafficking of mutant connexin 32 (GJB1). J. Hum. Genet. 46, 307–313. doi: 10.1007/s100380170064
Maw, M. A., Allen-Powell, D. R., Goodey, R. J., Stewart, I. A., Nancarrow, D. J., Hayward, N. K., et al. (1995). The contribution of the DFNB1 locus to neurosensory deafness in a Caucasian population. Am. J. Hum. Genet. 57, 629–635.
Mei, L., Chen, J., Zong, L., Zhu, Y., Liang, C., Jones, R. O., et al. (2017). A deafness mechanism of digenic Cx26 (GJB2) and Cx30 (GJB6) mutations: reduction of endocochlear potential by impairment of heterogeneous gap junctional function in the cochlear lateral wall. Neurobiol. Dis. 108, 195–203. doi: 10.1016/j.nbd.2017.08.002
Meşe, G., Richard, G., and White, T. W. (2007). Gap junctions: basic structure and function. J. Invest. Dermatol. 127, 2516–2524. doi: 10.1038/sj.jid.5700770
Minami, S. B., Mutai, H., Nakano, A., Arimoto, Y., Taiji, H., Morimoto, N., et al. (2013). GJB2-associated hearing loss undetected by hearing screening of newborns. Gene 532, 41–45. doi: 10.1016/j.gene.2013.08.094
Minoda, R., Miwa, T., Ise, M., and Takeda, H. (2015). Potential treatments for genetic hearing loss in humans: current conundrums. Gene Ther. 22, 603–609. doi: 10.1038/gt.2015.27
Miwa, T., Minoda, R., Ise, M., Yamada, T., and Yumoto, E. (2013). Mouse otocyst transuterine gene transfer restores hearing in mice with connexin 30 deletion-associated hearing loss. Mol. Ther. 21, 1142–1150. doi: 10.1038/mt.2013.62
Morell, R. J., Kim, H. J., Hood, L. J., Goforth, L., Friderici, K., Fisher, R., et al. (1998). Mutations in the connexin 26 gene (GJB2) among Ashkenazi Jews with nonsyndromic recessive deafness. N. Engl. J. Med. 339, 1500–1505. doi: 10.1056/nejm199811193392103
Musa, H., Fenn, E., Crye, M., Gemel, J., Beyer, E. C., and Veenstra, R. D. (2004). Amino terminal glutamate residues confer spermine sensitivity and affect voltage gating and channel conductance of rat connexin40 gap junctions. J. Physiol. 557, 863–878. doi: 10.1113/jphysiol.2003.059386
Neijssen, J., Herberts, C., Drijfhout, J. W., Reits, E., Janssen, L., and Neefjes, J. (2005). Cross-presentation by intercellular peptide transfer through gap junctions. Nature 434, 83–88. doi: 10.1038/nature03290
Nin, F., Hibino, H., Doi, K., Suzuki, T., Hisa, Y., and Kurachi, Y. (2008). The endocochlear potential depends on two K+ diffusion potentials and an electrical barrier in the stria vascularis of the inner ear. Proc. Natl. Acad. Sci. U S A 105, 1751–1756. doi: 10.1073/pnas.0711463105
Norris, V. W., Arnos, K. S., Hanks, W. D., Xia, X., Nance, W. E., and Pandya, A. (2006). Does universal newborn hearing screening identify all children with GJB2 (connexin 26) deafness? Penetrance of GJB2 deafness. Ear Hear. 27, 732–741. doi: 10.1097/01.aud.0000240492.78561.d3
Ouyang, X. M., Yan, D., Aslan, I., Du, L. L., Tekin, M., and Liu, X. Z. (2011). Mutation screening of the GJA7 (Cx45) gene in a large international series of probands with nonsyndromic hearing impairment. Genet. Test. Mol. Biomarkers. 15, 333–336. doi: 10.1089/gtmb.2010.0085
Penuela, S., Bhalla, R., Gong, X. Q., Cowan, K. N., Celetti, S. J., Cowan, B. J., et al. (2007). Pannexin 1 and pannexin 3 are glycoproteins that exhibit many distinct characteristics from the connexin family of gap junction proteins. J. Cell Sci. 120, 3772–3783. doi: 10.1242/jcs.009514
Phelan, P., Bacon, J. P., Davies, J. A., Stebbings, L. A., Todman, M. G., Avery, L., et al. (1998). Innexins: a family of invertebrate gap-junction proteins. Trends Genet. 14, 348–349. doi: 10.1016/s0168-9525(98)01547-9
Pietak, A., and Levin, M. (2018). Bioelectrical control of positional information in development and regeneration: a review of conceptual and computational advances. Prog. Biophys. Mol. Biol. 137, 52–68. doi: 10.1016/j.pbiomolbio.2018.03.008
Prazma, J. (1975). Electroanatomy of the lateral wall of the cochlea. Arch. Otorhinolaryngol. 209, 1–13. doi: 10.1007/bf00454023
Purnick, P. E., Benjamin, D. C., Verselis, V. K., Bargiello, T. A., and Dowd, T. L. (2000). Structure of the amino terminus of a gap junction protein. Arch. Biochem. Biophys. 381, 181–190. doi: 10.1006/abbi.2000.1989
Qu, Y., Tang, W., Zhou, B., Ahmad, S., Chang, Q., Li, X., et al. (2012). Early developmental expression of connexin26 in the cochlea contributes to its dominate functional role in the cochlear gap junctions. Biochem. Biophys. Res. Commun. 417, 245–250. doi: 10.1016/j.bbrc.2011.11.093
Rabionet, R., Gasparini, P., and Estivill, X. (2000). Molecular genetics of hearing impairment due to mutations in gap junction genes encoding beta connexins. Hum. Mutat. 16, 190–202. doi: 10.1002/1098-1004(200009)16:3<190::AID-HUMU2>3.0.CO;2-I
Riazuddin, S., Castelein, C. M., Ahmed, Z. M., Lalwani, A. K., Mastroianni, M. A., Naz, S., et al. (2000). Dominant modifier DFNM1 suppresses recessive deafness DFNB26. Nat. Genet. 26, 431–434. doi: 10.1038/82558
Sacheli, R., Delacroix, L., Vandenackerveken, P., Nguyen, L., and Malgrange, B. (2013). Gene transfer in inner ear cells: a challenging race. Gene Ther. 20, 237–247. doi: 10.1038/gt.2012.51
Sadanaga, M., and Morimitsu, T. (1995). Development of endocochlear potential and its negative component in mouse cochlea. Hear. Res. 89, 155–161. doi: 10.1016/0378-5955(95)00133-x
Sahu, G., Sukumaran, S., and Bera, A. K. (2014). Pannexins form gap junctions with electrophysiological and pharmacological properties distinct from connexins. Sci. Rep. 4:4955. doi: 10.1038/srep04955
Sanchez, H. A., and Verselis, V. K. (2014). Aberrant Cx26 hemichannels and keratitis-ichthyosis-deafness syndrome: insights into syndromic hearing loss. Front. Cell. Neurosci. 8:354. doi: 10.3389/fncel.2014.00354
Scherer, S. S., Xu, Y. T., Nelles, E., Fischbeck, K., Willecke, K., and Bone, L. J. (1998). Connexin32-null mice develop demyelinating peripheral neuropathy. Glia 24, 8–20. doi: 10.1002/(sici)1098-1136(199809)24:1<8::aid-glia2>3.0.co;2-3
Shao, Q., Lindstrom, K., Shi, R., Kelly, J., Schroeder, A., Juusola, J., et al. (2016). A germline variant in the PANX1 gene has reduced channel function and is associated with multisystem dysfunction. J. Biol. Chem. 291, 12432–12443. doi: 10.1074/jbc.M116.717934
Shu, Y., Tao, Y., Li, W., Shen, J., Wang, Z., and Chen, Z. Y. (2016). Adenovirus vectors target several cell subtypes of mammalian inner ear in vivo. Neural Plast. 2016:9409846. doi: 10.1155/2016/9409846
Slivko-Koltchik, G. A., Kuznetsov, V. P., and Panchin, Y. V. (2019). Are there gap junctions without connexins or pannexins? BMC Evol. Biol. 19:46. doi: 10.1186/s12862-019-1369-4
Söhl, G., Nielsen, P. A., Eiberger, J., and Willecke, K. (2003). Expression profiles of the novel human connexin genes hCx30.2, hCx40.1 and hCx62 differ from their putative mouse orthologues. Cell Commun. Adhes. 10, 27–36. doi: 10.1080/15419060302063
Srinivas, M., Verselis, V. K., and White, T. W. (2018). Human diseases associated with connexin mutations. Biochim. Biophys. Acta Biomembr. 1860, 192–201. doi: 10.1016/j.bbamem.2017.04.024
Stong, B. C., Chang, Q., Ahmad, S., and Lin, X. (2006). A novel mechanism for connexin 26 mutation linked deafness: cell death caused by leaky gap junction hemichannels. Laryngoscope 116, 2205–2210. doi: 10.1097/01.mlg.0000241944.77192.d2
Sun, J., Ahmad, S., Chen, S., Tang, W., Zhang, Y., Chen, P., et al. (2005). Cochlear gap junctions coassembled from Cx26 and 30 show faster intercellular Ca2+ signaling than homomeric counterparts. Am. J. Physiol. Cell Physiol. 288, C613–C623. doi: 10.1152/ajpcell.00341.2004
Sun, Y., Tang, W., Chang, Q., Wang, Y., Kong, W., and Lin, X. (2009). Connexin30 null and conditional connexin26 null mice display distinct pattern and time course of cellular degeneration in the cochlea. J. Comp. Neurol. 516, 569–579. doi: 10.1002/cne.22117
Suzuki, T., Takamatsu, T., and Oyamada, M. (2003). Expression of gap junction protein connexin43 in the adult rat cochlea: comparison with connexin26. J. Histochem. Cytochem. 51, 903–912. doi: 10.1177/002215540305100705
Tan, F., Chu, C., Qi, J., Li, W., You, D., Li, K., et al. (2019). AAV-ie enables safe and efficient gene transfer to inner ear cells. Nat. Commun. 10:3733. doi: 10.1038/s41467-019-11687-8
Tang, W., Ahmad, S., Shestopalov, V. I., and Lin, X. (2008). Pannexins are new molecular candidates for assembling gap junctions in the cochlea. Neuroreport 19, 1253–1257. doi: 10.1097/WNR.0b013e32830891f5
Tang, W., Zhang, Y., Chang, Q., Ahmad, S., Dahlke, I., Yi, H., et al. (2006). Connexin29 is highly expressed in cochlear Schwann cells and it is required for the normal development and function of the auditory nerve of mice. J. Neurosci. 26, 1991–1999. doi: 10.1523/JNEUROSCI.5055-05.2006
Teubner, B., Michel, V., Pesch, J., Lautermann, J., Cohen-Salmon, M., Söhl, G., et al. (2003). Connexin30 (Gjb6)-deficiency causes severe hearing impairment and lack of endocochlear potential. Hum. Mol. Genet. 12, 13–21. doi: 10.1093/hmg/ddg001
Teunissen, B. E., Jansen, A. T., Mutsaers, N. A., Vuerhard, M. J., Vos, M. A., and Bierhuizen, M. F. (2007). Primary structure, organization and expression of the rat connexin45 gene. DNA Cell Biol. 26, 108–115. doi: 10.1089/dna.2006.0540
Verselis, V. K. (2019). Connexin hemichannels and cochlear function. Neurosci. Lett. 695, 40–45. doi: 10.1016/j.neulet.2017.09.020
Verselis, V. K., Ginter, C. S., and Bargiello, T. A. (1994). Opposite voltage gating polarities of two closely related connexins. Nature 368, 348–351. doi: 10.1038/368348a0
Wang, D., Tai, P. W. L., and Gao, G. (2019). Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 18, 358–378. doi: 10.1038/s41573-019-0012-9
Wang, S. L., Yu, L. G., Liu, R. P., Zhu, W. Z., Gao, W. M., Xue, L. P., et al. (2014). Gene-gene interaction of GJB2, SOD2 and CAT on occupational noise-induced hearing loss in Chinese Han population. Biomed. Environ. Sci. 27, 965–968. doi: 10.3967/bes2014.131
Wang, X. H., Streeter, M., Liu, Y. P., and Zhao, H. B. (2009). Identification and characterization of pannexin expression in the mammalian cochlea. J. Comp. Neurol. 512, 336–346. doi: 10.1002/cne.21898
Wang, Y., Chang, Q., Tang, W., Sun, Y., Zhou, B., Li, H., et al. (2009). Targeted connexin26 ablation arrests postnatal development of the organ of Corti. Biochem. Biophys. Res. Commun. 385, 33–37. doi: 10.1016/j.bbrc.2009.05.023
White, T. W. (2002). Unique and redundant connexin contributions to lens development. Science 295, 319–320. doi: 10.1126/science.1067582
White, T. W., and Bruzzone, R. (1996). Multiple connexin proteins in single intercellular channels: connexin compatibility and functional consequences. J. Bioenerg. Biomembr. 28, 339–350. doi: 10.1007/bf02110110
White, T. W., Deans, M. R., Kelsell, D. P., and Paul, D. L. (1998). Connexin mutations in deafness. Nature 394, 630–631. doi: 10.1038/29202
Wingard, J. C., and Zhao, H. B. (2015). Cellular and deafness mechanisms underlying connexin mutation-induced hearing loss-a common hereditary deafness. Front. Cell. Neurosci. 9:202. doi: 10.3389/fncel.2015.00202
Wu, X., Wang, Y., Sun, Y., Chen, S., Zhang, S., Shen, L., et al. (2014). Reduced expression of connexin26 and its DNA promoter hypermethylation in the inner ear of mimetic aging rats induced by d-galactose. Biochem. Biophys. Res. Commun. 452, 340–346. doi: 10.1016/j.bbrc.2014.08.063
Xia, A., Katori, Y., Oshima, T., Watanabe, K., Kikuchi, T., and Ikeda, K. (2001). Expression of connexin 30 in the developing mouse cochlea. Brain Res. 898, 364–367. doi: 10.1016/s0006-8993(01)02216-8
Xia, A. P., Ikeda, K., Katori, Y., Oshima, T., Kikuchi, T., and Takasaka, T. (2000). Expression of connexin 31 in the developing mouse cochlea. Neuroreport 11, 2449–2453. doi: 10.1097/00001756-200008030-00022
Xia, J. H., Liu, C. Y., Tang, B. S., Pan, Q., Huang, L., Dai, H. P., et al. (1998). Mutations in the gene encoding gap junction protein beta-3 associated with autosomal dominant hearing impairment. Nat. Genet. 20, 370–373. doi: 10.1038/3845
Yang, J. J., Huang, S. H., Chou, K. H., Liao, P. J., Su, C. C., and Li, S. Y. (2007). Identification of mutations in members of the connexin gene family as a cause of nonsyndromic deafness in Taiwan. Audiol. Neurootol. 12, 198–208. doi: 10.1159/000099024
Yu, Q., Wang, Y., Chang, Q., Wang, J., Gong, S., Li, H., et al. (2014). Virally expressed connexin26 restores gap junction function in the cochlea of conditional Gjb2 knockout mice. Gene Ther. 21, 71–80. doi: 10.1038/gt.2013.59
Zhang, W., Kim, S. M., Wang, W., Cai, C., Feng, Y., Kong, W., et al. (2018). Cochlear gene therapy for sensorineural hearing loss: current status and major remaining hurdles for translational success. Front. Mol. Neurosci. 11:221. doi: 10.3389/fnmol.2018.00221
Zhang, Y., Tang, W., Ahmad, S., Sipp, J. A., Chen, P., and Lin, X. (2005). Gap junction-mediated intercellular biochemical coupling in cochlear supporting cells is required for normal cochlear functions. Proc. Natl. Acad. Sci. U S A 102, 15201–15206. doi: 10.1073/pnas.0501859102
Zhao, H. B. (2016). Expression and function of pannexins in the inner ear and hearing. BMC Cell Biol. 17:16. doi: 10.1186/s12860-016-0095-7
Zhao, H. B. (2017). Hypothesis of K(+)-Recycling defect is not a primary deafness mechanism for Cx26 (GJB2) deficiency. Front. Mol. Neurosci. 10:162. doi: 10.3389/fnmol.2017.00162
Zhao, H. B., and Yu, N. (2006). Distinct and gradient distributions of connexin26 and connexin30 in the cochlear sensory epithelium of guinea pigs. J. Comp. Neurol. 499, 506–518. doi: 10.1002/cne.21113
Zhao, H. B., Kikuchi, T., Ngezahayo, A., and White, T. W. (2006). Gap junctions and cochlear homeostasis. J. Membr. Biol. 209, 177–186. doi: 10.1007/s00232-005-0832-x
Zhao, H. B., Zhu, Y., Liang, C., and Chen, J. (2015). Pannexin 1 deficiency can induce hearing loss. Biochem. Biophys. Res. Commun. 463, 143–147. doi: 10.1016/j.bbrc.2015.05.049
Zhou, X. X., Chen, S., Xie, L., Ji, Y. Z., Wu, X., Wang, W. W., et al. (2016). Reduced connexin26 in the mature cochlea increases susceptibility to noise-induced hearing lossin mice. Int. J. Mol. Sci. 17:301. doi: 10.3390/ijms17030301
Zhu, Y., Chen, J., Liang, C., Zong, L., Jones, R. O., and Zhao, H. B. (2015a). Connexin26 (GJB2) deficiency reduces active cochlear amplification leading to late-onset hearing loss. Neuroscience 284, 719–729. doi: 10.1016/j.neuroscience.2014.10.061
Zhu, Y., Zong, L., Mei, L., and Zhao, H. B. (2015b). Connexin26 gap junction mediates miRNA intercellular genetic communication in the cochlea and is required for inner ear development. Sci. Rep. 5:15647. doi: 10.1038/srep15647
Zhu, Y., Liang, C., Chen, J., Zong, L., Chen, G. D., and Zhao, H. B. (2013). Active cochlear amplification is dependent on supporting cell gap junctions. Nat. Commun. 4:1786. doi: 10.1038/ncomms2806
Zong, L., Chen, J., Zhu, Y., and Zhao, H. B. (2017). Progressive age-dependence and frequency difference in the effect of gap junctions on active cochlear amplification and hearing. Biochem. Biophys. Res. Commun. 489, 223–227. doi: 10.1016/j.bbrc.2017.05.137
Zong, L., Zhu, Y., Liang, R., and Zhao, H. B. (2016). Gap junction mediated miRNA intercellular transfer and gene regulation: a novel mechanism for intercellular genetic communication. Sci. Rep. 6:19884. doi: 10.1038/srep19884
Keywords: gap junction, connexin, cochlea, structure and function, gene therapy, deafness
Citation: Wu X, Zhang W, Li Y and Lin X (2019) Structure and Function of Cochlear Gap Junctions and Implications for the Translation of Cochlear Gene Therapies. Front. Cell. Neurosci. 13:529. doi: 10.3389/fncel.2019.00529
Received: 18 September 2019; Accepted: 13 November 2019;
Published: 27 November 2019.
Edited by:
Julie S. Haas, Lehigh University, United StatesReviewed by:
Vytas K. Verselis, Albert Einstein College of Medicine, United StatesPing Liu, Huazhong University of Science and Technology, China
Copyright © 2019 Wu, Zhang, Li and Lin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xuewen Wu, eHd3dTg0MDkwM0Bob3RtYWlsLmNvbQ==; Xi Lin, eGxpbjJAZW1vcnkuZWR1