 Satomi Kikuta1,2,3
Satomi Kikuta1,2,3 Yoshio Iguchi4
Yoshio Iguchi4 Toshikazu Kakizaki5
Toshikazu Kakizaki5 Kazuto Kobayashi4Yuchio Yanagawa5Masahiko Takada2
Kazuto Kobayashi4Yuchio Yanagawa5Masahiko Takada2 Makoto Osanai1,6*
Makoto Osanai1,6*- 1Department of Radiological Imaging and Informatics, Tohoku University Graduate School of Medicine, Sendai, Japan
- 2Systems Neuroscience Section, Primate Research Institute, Kyoto University, Inuyama, Japan
- 3Research Fellow of Japan Society for the Promotion of Science, Tokyo, Japan
- 4Department of Molecular Genetics, Institute of Biomedical Sciences, Fukushima Medical University School of Medicine, Fukushima, Japan
- 5Department of Genetic and Behavioral Neuroscience, Gunma University Graduate School of Medicine, Maebashi, Japan
- 6Laboratory for Physiological Functional Imaging, Department of Medical Physics and Engineering, Division of Health Sciences, Osaka University Graduate School of Medicine, Suita, Japan
The striatum plays an important role in linking cortical activity to basal ganglia output. Striatal neurons exhibit spontaneous slow Ca2+ oscillations that result from Ca2+ release from the endoplasmic reticulum (ER) induced by the mGluR5-IP3R signaling cascade. The maximum duration of a single oscillatory event is about 300 s. A major question arises as to how such a long-duration Ca2+ elevation is maintained. Store-operated calcium channels (SOCCs) are one of the calcium (Ca2+)-permeable ion channels. SOCCs are opened by activating the metabotropic glutamate receptor type 5 and inositol 1,4,5-trisphosphate receptor (mGluR5-IP3R) signal transduction cascade and are related to the pathophysiology of several neurological disorders. However, the functions of SOCCs in striatal neurons remain unclear. Here, we show that SOCCs exert a functional role in striatal GABAergic neurons. Depletion of calcium stores from the ER induced large, sustained calcium entry that was blocked by SKF96365, an inhibitor of SOCCs. Moreover, the application of SKF96365 greatly reduced the frequency of slow Ca2+ oscillations. The present results indicate that SOCCs contribute to Ca2+ signaling in striatal GABAergic neurons, including medium spiny projection neurons (MSNs) and GABAergic interneurons, through elevated Ca2+ due to spontaneous slow Ca2+ oscillations.
Introduction
The calcium ion (Ca2+) is an important messenger for signal transduction, and intracellular Ca2+ concentration ([Ca2+]i) changes in response to various physiological stimuli in both excitable and non-excitable cells (Pasti et al., 1997; Smetters et al., 1999; Berridge et al., 2000). Intracellular Ca2+ can modulate the functions of proteins such as enzymes and receptors, gene expressions, and morphological changes in cellular processes. The endoplasmic reticulum (ER) is a source of [Ca2+]i and is crucial for second messenger-induced intracellular Ca2+ signaling (Blaustein and Golovina, 2001; Berridge, 2002). Therefore, Ca2+-release from the ER contributes to the modulation of neuronal signal processing in the central nervous system (Kostyuk and Verkhratsky, 1994). In the basal ganglia, the striatum receives inputs from the cortex and is thought to play crucial roles in controlling somatic motor movements, behavioral patterns, cognition, learning, and memory (Graybiel, 1995; Chesselet and Delfs, 1996). However, the cellular Ca2+ signaling in striatal neurons still remains unclear. We previously reported long-lasting spontaneous intracellular Ca2+ oscillations in rodent striatal neurons (Osanai et al., 2006; Tamura et al., 2014), which lasted as long as 300 s. These slow Ca2+ oscillations were not induced by action potentials, but by Ca2+ release from the ER. Although both the metabotropic glutamate receptor type 5 and inositol 1,4,5-trisphosphate receptor (mGluR5-IP3R) signal transduction cascade were involved in these slow Ca2+ oscillations (Tamura et al., 2014), an important issue arises as to how such a long elevation in Ca2+ is maintained.
Store-operated calcium channels (SOCCs) are one of the Ca2+ -permeable plasmalemmal ion channels through which Ca2+ flows into the cell from the extracellular space when ER stores are depleted (Brini et al., 2014; Majewski and Kuznicki, 2015; Wegierski and Kuznicki, 2018). Although SOCCs have been most thoroughly studied in non-neuronal cells and knowledge of their function in the brain is still poor, data concerning their physiological and pathological functions in neuronal cells are accumulating (Baba et al., 2003; Bouron et al., 2004; Shideman et al., 2009; Zhang et al., 2015; Secondo et al., 2018; Wegierski and Kuznicki, 2018; Ong et al., 2019).
The very long-lasting [Ca2+]i elevations that we observed in the form of spontaneous slow Ca2+ oscillations in the striatum (Osanai et al., 2006; Tamura et al., 2014) led us to hypothesize that Ca2+ in the ER may be depleted and that SOCCs may then be opened when the slow Ca2+ oscillations occur. This opening of SOCCs may be involved in the mechanism of maintaining high [Ca2+]i for a long time in striatal neurons exhibiting these slow Ca2+ oscillations. The present study was designed to examine this hypothesis.
Materials and Methods
Mice
In the Ca2+ imaging study, to discriminate striatal GABAergic neurons, we used heterozygous GAD67-GFP knock-in mice (GAD67-GFP mice), in which enhanced GFP is selectively expressed under the control of the endogenous GAD67 gene promoter (Tamamaki et al., 2003). The colony was maintained by crossing male GAD67-GFP mice with female C57BL/6 mice (Clea Japan). All mice were housed and maintained at 22–24°C on a 12-h light/ dark cycle and permitted ad libitum access to food and water. A total of 10 mice were used in the studies.
Ca2+ Imaging
The methods for Ca2+ imaging were described previously (Osanai et al., 2006; Tamura et al., 2014; Kikuta et al., 2015). Briefly, postnatal day 12 (P12) to P17 GAD67-GFP mice of either sex were anesthetized with isoflurane (Mylan) and decapitated. The brain was rapidly isolated and placed in ice-cold artificial cerebrospinal fluid (ACSF) bubbled with 95% O2–5% CO2. The composition of normal ACSF was as follows (in mM): 137 NaCl, 2.5 KCl, 0.58 NaH2PO4, 1.2 MgCl2, 2.5 CaCl2, 21 NaHCO3, and 10 glucose. Ca2+-free ACSF was made by omitting CaCl2 and adding 7.5 mM NaCl. Corticostriatal sagittal slices (300 μm thick) were prepared using a vibratome tissue slicer (VT-1200S, Leica Microsystems) and incubated at room temperature in a submerged chamber containing gassed ACSF for at least 60 min prior to the Ca2+-sensitive Fura-2 LR/AM (Calbiochem) fluorescent dye, loading.
As previously described (Kikuta et al., 2015), [Ca2+]i elevation and manganese ion (Mn2+) quenching was measured in striatal cells loaded with the ratiometric Ca2+ sensitive dye Fura-2 LR/AM. The dye-loading methods used were as previously described (Osanai et al., 2006; Tamura et al., 2014). In brief, the corticostriatal slice was placed in a small plastic chamber containing 100 μl ACSF with 20 μM Fura-2 LR/AM, 1 μM sulforhodamine 101 (Sigma), and 0.02% Cremophor EL (Sigma). The dish was incubated at 35°C for 45 min in the small chamber, and then washed with 100 μl ACSF at 35°C for 15 min. To ensure that the [Ca2+]i change was attributed to a neuronal event, sulforhodamine 101-positive cells, corresponding to astrocytes, were excluded (Nimmerjahn et al., 2004). After dye-loading, the slice was transferred to a continuously superfused (2–2.5 ml/min) chamber, and the fluorescence was observed by an epifluorescence upright microscope (BX51WI, Olympus) equipped with a 20×, NA 1.0 water-immersion objective (Olympus). The Fura-2 LR-loaded slices were excited at wavelengths of 340 or 380 nm using a filter changer (Lambda DG-4, Sutter Instruments, Novato, CA, USA) equipped with excitation filters (26-nm bandpass filter for 340 nm wavelength and 14-nm bandpass filter for 380 nm wavelength, Semrock), and fluorescent signals at 510 nm were captured (F340 or F380) every 2 s with an EM-CCD camera (DU-885, Andor Technology, Belfast, UK). Ca2+ imaging was performed in the presence of 1 μM tetrodotoxin (TTX, Nacalai tesque, San Diego, CA, USA) to avoid Ca2+ elevations caused by the opening of voltage-gated Ca2+ channels due to action potentials. The experiments were performed at 30 ± 1°C.
We identified GFP-positive cells (i.e., GABAergic neurons) by observing green fluorescence excited at 488 nm (6-nm bandpass filter, Semrock) and quantified the average fluorescence (F340 and F380) within the region of interest (ROI) of these cells as a function of time. [Ca2+]i elevations in a striatal cell were estimated by the fluorescence ratio (R = F340/F380) from each imaged cell. The criterion for identifying neurons with the slow Ca2+ oscillations was whether they had a frequency of occurrence of spontaneous Ca2+ elevation above 0.001 Hz. The total recording duration was more than 4,200 s. All equipment was controlled by iQ software (Andor Technology, Belfast, UK). The analyses of the imaging data were performed with ImageJ software (Schneider et al., 2012) and custom-made programs (Supplementary Material) written in MATLAB (MathWorks, Natick, MA, USA).
Mn2+ Quench Experiment
Mn2+ can pass through opened Ca2+-permeable channels and quenches the Fura-2 LR fluorescence emission (Amano et al., 1997; Kikuta et al., 2015). Thus, to evaluate Ca2+ influx from the extracellular space, the rate of the quench by Mn2+ was quantified as Δ F/F at 380 nm (Uehara et al., 2002; Tu et al., 2009; Kikuta et al., 2015). To evaluate the influx rate of Mn2+, the time constant of quenching (τq) after MnCl2 perfusion was calculated by fitting with the following equation:
where t is the time from the start of MnCl2 administration, T is the time between the onset of recording and the start of MnCl2 administration, Ab, τb, and Bb are amplitude, time constant, and baseline level of the fluorescence bleaching, respectively, and Aq, τq, and Bq are amplitude, time constant and baseline level of the fluorescence quenching, respectively. The curve fitting was performed using SciPlot (M. Wesemann, 1991–95).
Immunohistochemistry
Mice were anesthetized with sodium pentobarbital (50 mg/kg, intraperitoneal) and perfused transcardially with PBS, followed by fixation with 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4). The sections (30 μm thick) were incubated with primary antibody for STIM1 (rabbit, 1:250, Alomone lab, Jerusalem, Israel), STIM2 (rabbit, 1:500, Alomone lab, Jerusalem, Israel), Orai1 (rabbit, 1:500, Alomone lab, Jerusalem, Israel), Orai2 (rabbit, 1:500, Alomone lab, Jerusalem, Israel), or Orai3 (rabbit, 1:500, ProSci, Poway, CA, USA), along with anti-GFP antibody (goat, 1:5,000, Frontier Institute, Hokkaido, Japan). These sections were incubated with species-specific secondary antibodies conjugated to Alexa488 (1:200, Invitrogen, Carlsbad, CA, USA) and Cy3 (1:200, Jackson ImmunoResearch, West Grove, PA, USA). Cellular nuclei were counterstained with DAPI (Sigma-Aldrich, St. Louis, MO, USA). Fluorescent images were visualized under a confocal laser-scanning microscope (A1R, Nikon, Tokyo, Japan) equipped with proper filter cube specifications.
For cell counts, three sections through the dorsolateral striatum along the rostrocaudal axis between −1.10 and −0.38 (mm) from the bregma were prepared from each mouse. The number of immunoreactive cells in the regions of interest (210 × 210 μm) was counted in a computer-assisted manner (Adobe Photoshop CC2018, San Jose, CA, USA), and the number of double-labeled cells was divided by that of the total GFP-positive cells in each section. The mean percentage obtained from the three sections was calculated. We used four mutant mice for quantification in each immunostaining condition.
Statistics
Statistical analyses were performed using JMP Pro 11 (SAS Institute, Cary, NC, USA). Statistically significant differences (p < 0.05) were assessed by the Mann–Whitney U test. All data are presented as mean ± SEM unless stated otherwise.
Drugs
All drugs were applied by perfusion. TTX (1 μM) was used to block action potentials. Thapsigargin (Nacalai Tesque; 2 μM) was used to deplete ER Ca2+ by blocking smooth ER Ca2+-ATPase. SKF96365 (Tocris; 10 μM) was used to inhibit SOCCs. All other chemicals were purchased from Nacalai Tesque except for immunohistochemical agents.
Results
SOCCs in Striatal GABAergic Neurons
To elucidate whether SOCCs are present in striatal GABAergic neurons, we performed fluorescence imaging with Fura-2 LR in acute slice preparations obtained from GAD67-GFP knock-in mice in which GFP was expressed in GABAergic neurons (Figure 1A). As more than 95% of neurons in the striatum are GABAergic projection neurons (Gerfen and Bolam, 2010), GFP-positive striatal cells in these mice were mostly projection neurons.
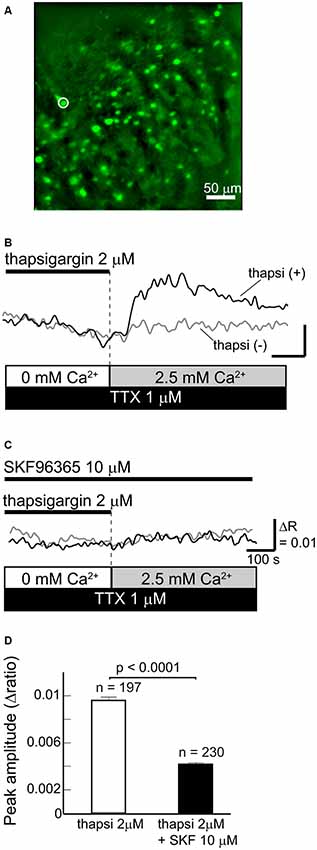
Figure 1. Functional store-operated calcium channels (SOCCs) existed in striatal GABAergic neurons. (A) Fluorescence image of a striatal slice from a GAD67-GFP mouse. Striatal GABAergic neurons were identified by GFP fluorescence (green) and ROIs were placed on the GFP-positive somata for quantifying fluorescence changes. Typical [Ca2+]i time courses presented in (B,C) were obtained from the cell marked with a white circle on the fluorescence photo. (B) [Ca2+]i elevation was induced by pretreatment with thapsigargin [black line, thapsi(+)] for more than 5 min in Ca2+-free conditions and subsequent perfusion of 2.5 mM Ca2+ (gray bar). The scale is the same as in (C). (C) The [Ca2+]i elevation induced by thapsigargin was blocked by 10 μM SKF96365. (D) The effect of SKF96365 on the peak amplitude of Ca2+ elevations induced by endoplasmic reticulum (ER) depletion.
To deplete Ca2+ from the ER, Ca2+-free ACSF with 2 μM thapsigargin, an ER Ca2+-ATPase inhibitor, was perfused for more than 5 min. Subsequently, when the extracellular Ca2+ concentration increased to 2.5 mM following normal ACSF perfusion, GABAergic neurons exhibited remarkable elevations of [Ca2+]i (Figures 1B,D). The [Ca2+]i elevation was not observed when the ER was not depleted of Ca2+ by thapsigargin administration (Figure 1B). The [Ca2+]i elevation induced by ER depletion was significantly reduced after the application of 10 μM SKF96365, a SOCC inhibitor (Baba et al., 2003; Bouron et al., 2004; Shideman et al., 2009; Figures 1C,D). The peak amplitudes of [Ca2+]i elevation without and with SKF96365 were 0.0096 ± 0.0003 (n = 197 cells, three slices, three mice) and 0.0042 ± 0.0002 (n = 230 cells, three slices, two mice; p < 0.0001), respectively. These results indicate that SOCCs exist in striatal GABAergic neurons and exert some functional role.
Involvement of SOCCs in Spontaneous Slow Ca2+ Oscillations
To examine whether SOCCs are responsible for maintaining high [Ca2+]i in striatal GABAergic neurons exhibiting spontaneous slow Ca2+ oscillations, we first tested the contribution of Ca2+-influx from the extracellular space in striatal GABAergic neurons with and without slow Ca2+ oscillations by quantifying the time constant of fluorescence quench (Figures 2A,B). More Mn2+ entry from the extracellular space caused faster fluorescence quench (Amano et al., 1997; Uehara et al., 2002; Tu et al., 2009). The Fura-2 LR fluorescence quench was observed after MnCl2 (50 μM) administration in the neurons with slow Ca2+ oscillations (Figure 2B). The time constants τq of fluorescence quench were 211 ± 4 s (n = 23) in the neurons without slow Ca2+ oscillations and 193 ± 4 s (n = 9) in those with these oscillations (Figure 2C, p < 0.01). These results suggest that more Ca2+ enters those striatal neurons with spontaneous slow Ca2+ oscillations than those without the oscillations.
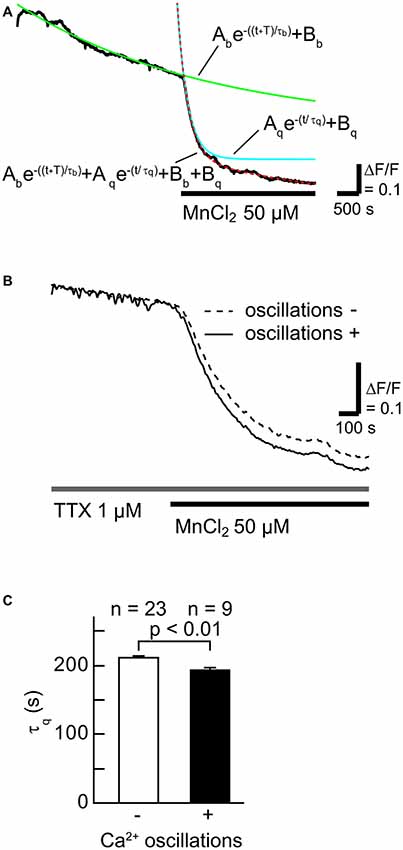
Figure 2. The rate of Ca2+ influx in neurons with slow Ca2+ oscillations was faster than in those without slow Ca2+ oscillations. (A) A typical time course of Mn2+ quench of Fura-2 LR fluorescence (black line). To evaluate the rate of Ca2+ channel opening, the fluorescence decay was fitted with the double-exponential function (dotted red line) consisting of a fluorescence bleaching component (green line) and a quenching component (blue line). (B) Typical time courses of Mn2+ quench in neurons with (solid line) and without (broken line) slow Ca2+ oscillations. (C) Summary data of the time constant of the quenching component (τq) in neurons without (open) and with (filled) slow Ca2+ oscillations.
To clarify the contribution of SOCCs to spontaneous slow Ca2+ oscillations we blocked the channels with SKF96365 (Figure 3). To avoid [Ca2+]i elevation due to action potentials, TTX was applied during the experiments. The slow Ca2+ oscillations nearly disappeared in the presence of SKF96365 (Figures 3A,B). The frequency of the oscillations in the control condition was 1.27 ± 0.16 × 10−3 Hz, whereas in the presence of SKF96365 the frequency was 0.109 ± 0.078 × 10−3 Hz (n = 10 cells, five slices, four mice, p < 0.005). Thus, SOCCs clearly contribute to the occurrence of spontaneous slow Ca2+ oscillations in striatal GABAergic neurons.
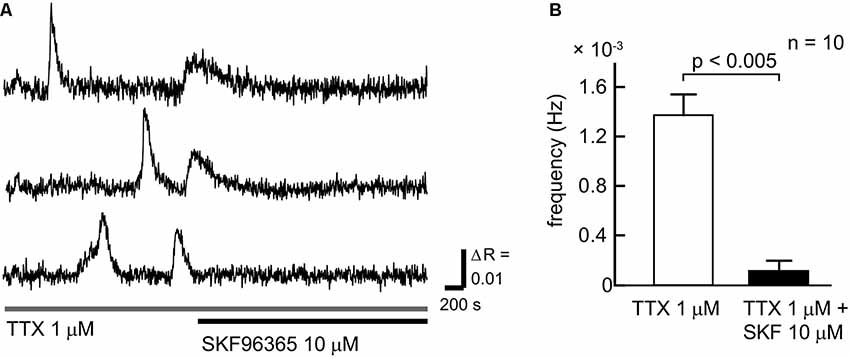
Figure 3. SOCCs were involved in the spontaneous Ca2+ oscillations. (A) Typical time courses of spontaneous slow Ca2+ oscillations during the administration of 1 μM TTX (gray bar) and 10 μM SKF96365 (black bar). Spontaneous Ca2+ elevations were not present following the application of SKF96365. (B) Frequencies of [Ca2+]i elevations in the absence (open bar) or presence (solid bar) of SKF96365. The frequency of spontaneous Ca2+ oscillations was significantly reduced by SKF96365 administration.
Molecular Components of SOCE in Striatal GABAergic Neurons
The major molecular components of the store-operated Ca2+ entry (SOCE) are STIM (STIM1 and 2) and Orai (Orai1, 2, and 3; Kraft, 2015; Moccia et al., 2015). Through immunohistochemical analyses, we examined the expression of Orai1, 2, and 3, the pore-forming subunit of the “Ca2+-release-activated Ca2+ (CRAC)” channels (Kraft, 2015; Moccia et al., 2015), in striatal GABAergic neurons. In striatal GABAergic neurons, strong expression of Orai2 and moderate expression of Orai3 were observed, while Orai1 expression was detected only in a few cells (Figure 4). The percentages of the Orai1-, Orai2-, and Orai3-positive cells relative to the total GFP-positive cells were 4.7 ± 2.3, 97.8 ± 0.9, and 62.5 ± 1.6, respectively (%, n = 4 mice). We also examined the expression of STIM1 and 2, the ER Ca2+ sensor activating CRAC channels on the plasma membrane (Kraft, 2015; Moccia et al., 2015). Both of STIM1 and 2 were expressed in striatal GABAergic neurons (Figure 4). The percentages of the STIM1- and STIM2-positive cells relative to the total GFP-positive cells were 76.1 ± 3.8 and 75.5 ± 3.3, respectively (%, n = 4 mice). These results suggest that in striatal GABAergic neurons, Orai2 is a major molecular component of SOCCs, and both STIM1 and STIM2 contribute to the activation of this Orai protein.
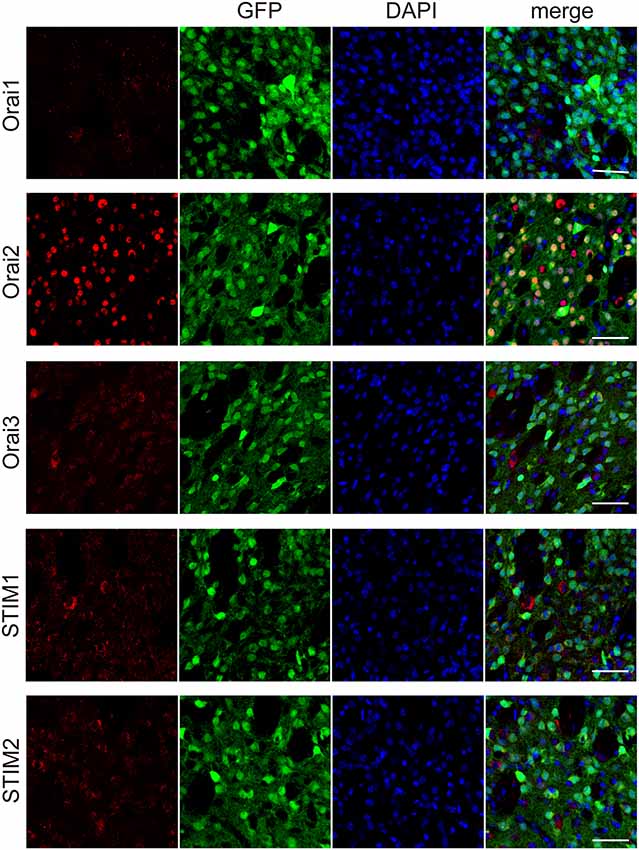
Figure 4. Each of the molecular components of SOCE expressed at different levels in the striatal GFP-positive cells. Confocal microscopy images of the sections through the dorsal striatum obtained from GAD67-GFP mice, which were stained for Orai1, Orai 2, Orai3, STIM1, or STIM2 (red), along with GFP (green). Blue staining represents DAPI. Scale bar: 50 μm.
Discussion
In the present study, we demonstrated that striatal GABAergic neurons exhibit SOCE (see Figure 1), and that blocking this Ca2+entry greatly reduces the frequency of spontaneous slow Ca2+ oscillations in these neurons (see Figure 3). Wu et al. (2016) observed elevations in [Ca2+]i after depleting Ca2+ from the ER, which is considered to be ascribed to SOCE (Majewski and Kuznicki, 2015), in corticostriatal co-culture preparations. The time course of this Ca2+ elevation was similar to that observed in our work. The Ca2+ elevation after the ER Ca2+ depletion in striatal GABAergic neurons was almost completely blocked by SKF96365, an inhibitor of SOCCs (see Figure 1). Taken together, these findings indicate that functional SOCCs exist on the plasma membrane of striatal GABAergic neurons.
The major molecular components of the SOCE pathway are STIM (STIM1 and 2) and Orai (Orai1, 2, and 3) proteins (Kraft, 2015; Moccia et al., 2015; Secondo et al., 2018; Wegierski and Kuznicki, 2018). Therefore, we investigated which molecules were responsible for the SOCE in striatal GABAergic neurons. In the immunohistochemical study, STIM1 and 2 were expressed equally in striatal GABAergic neurons (see Figure 4). This observation is consistent with previous data showing that both STIM1 and 2 are widely expressed in the rodent brain (Kraft, 2015; Moccia et al., 2015; Wegierski and Kuznicki, 2018). On the other hand, Orai2 was strongly expressed in striatal GABAergic neurons, whereas Orai1 was expressed in only a few GABAergic neurons (see Figure 4). Although Orai1 is known to be expressed extensively in the rodent brain (Moccia et al., 2015), its expression level in striatal GABAergic neurons, which is the principal component of striatal neurons, has not yet been reported. Thus, Orai2 but not Orai1 may be a major pore-forming protein of SOCE in striatal GABAergic neurons. In addition, Orai3 was also expressed in striatal GABAergic neurons, albeit its immunoreactivity was not so intense as that for Orai2 (see Figure 4). Further investigations are needed to elucidate a molecular substrate for SOCE in striatal GABAergic neurons.
We previously observed spontaneous slow Ca2+ oscillations in striatal neurons and astrocytes (Osanai et al., 2006; Tamura et al., 2014) that were blocked by mGluR5 or IP3R inhibition (Tamura et al., 2014). In addition, the mGluR5-dependent sustained Ca2+ elevation was strongly blocked by Zn2+, which is thought to be a SOCC blocker (Uehara et al., 2002), in rat cortical neurons and glia (Prothero et al., 1998). It was also shown that group I mGluRs including mGluR5 were related to SOCE in midbrain dopaminergic neurons (Tozzi et al., 2003), and that the application of the SOCC blocker 2-aminoethoxy-diphenyl borane (2-APB) totally abolished the response to DHPG (group I mGluR agonist) in about half of midbrain auditory neurons (Martinez-Galan et al., 2012). Therefore, activation of mGluR5 may lead to SOCE. In the present experiments, we took advantage of the fact that Mn2+ ions readily enter cells through Ca2+-conducting channels (Uehara et al., 2002; Tu et al., 2009; Chen et al., 2015) and quench Fura-2 LR fluorescence to verify that SOCCs contribute to the spontaneous slow Ca2+ oscillations that depend on the mGluR5-IP3R signal transduction cascade. The fluorescence was quenched faster in the neurons with the slow Ca2+ oscillations than in those without these oscillations (see Figure 2), indicating that the amount of extracellular Ca2+ influx in the neurons with the slow Ca2+ oscillations was larger than in those without the oscillations, and thereby suggesting that more SOCCs open in striatal neurons exhibiting spontaneous slow Ca2+ oscillations.
In our previous observations, the maximum duration of a single event of the slow Ca2+ oscillations was approximately 300 s (Osanai et al., 2006; Tamura et al., 2014). However, how such a long-duration Ca2+ elevation is maintained remains to be resolved. If SOCCs contribute to maintain high Ca2+ concentrations in single oscillatory events, the duration of the Ca2+ elevation must be shortened by SOCC blockade. However, the frequency of the slow Ca2+ oscillations in striatal GABAergic neurons was dramatically reduced (see Figure 3). This, together with the fact that Ca2+ flows into the cell through SOCCs when ER stores are depleted, implies that SOCCs are responsible for the initiation of Ca2+ elevation in spontaneous slow Ca2+ oscillations, and SOCCs have a potential for maintaining the long-duration Ca2+ elevation.
SOCCs have been most thoroughly studied in non-neuronal cells. Although their physiological and pathological roles have been argued, accumulated evidence suggests that dysregulation of SOCE triggers perturbation of intracellular Ca2+ signaling that participates in key physiological functions, including synaptic plasticity, axonal growth, and synaptic formation, and in the pathogenesis of neurodegenerative diseases, including Alzheimer’s disease (AD), Huntington’s disease (HD), and Parkinson’s disease (Bardo et al., 2006; Majewski and Kuznicki, 2015; Secondo et al., 2018; Wegierski and Kuznicki, 2018). Moreover, abnormal mGluR5-IP3R signal transduction is thought to be involved in SOCE related to perturbation of intracellular Ca2+ signaling in AD and HD (Amano et al., 1997; Secondo et al., 2018). Wu et al. (2016) reported that the increase in steady-state IP3R activity resulted in overactivation of SOCE in cultured medium spiny projection neurons (MSNs) of the striatum in an HD mouse model characterized by age-dependent dendritic spine loss in MSNs, and that inhibition of SOCE rescued such a spine loss. These results indicate that the striatal SOCCs may contribute to neuronal degeneration in an HD model. As the present study was conducted in normal mice, we cannot conclude that spontaneous slow Ca2+ oscillations generate abnormal Ca2+ signals induced by overactivation of SOCCs in the striatum.
Intracellular Ca2+ can modulate protein functions, gene expressions, and morphological changes in cellular processes, thus leading to modified functions of neurons and neuronal circuits (Berridge et al., 2000). The SOCCs have also been implicated in synaptic plasticity (Baba et al., 2003; Bardo et al., 2006; González-Sánchez et al., 2017; Korkotian et al., 2017). Therefore, the SOCCs contributing to the occurrence and the maintenance of spontaneous slow Ca2+ oscillations in striatal GABAergic neurons might regulate related network functions. Further investigations are needed to elucidate the role of SOCCs in striatal GABAergic neurons.
Data Availability Statement
All datasets generated for this study are included in the article/Supplementary Material.
Ethics Statement
The Tohoku University Committee for Animal Experiments approved all animal experiments, and the experiments were performed in accordance with the Guidelines for Animal Experiments and Related Activities of Tohoku University, the guiding principles of the Physiological Society of Japan and the National Institutes of Health (NIH), USA.
Author Contributions
SK, YI, TK, KK, and MO conceived and performed experiments. SK, YI, and MO analyzed data. YY provided the GAD67-GFP mice and information for maintaining them. SK, YI, YY, MT, and MO wrote the manuscript.
Funding
This work was supported by JSPS KAKENHI Grant Number JP16K08486 (to MO); MEXT KAKENHI (Adaptive Circuit Shift) Grant Number JP17H05543 (to MO) and JP17H05550 (to YY); MEXT KAKENHI (Non-linear Neuro-oscillology) JP18H04931 (to MO), JP15H05872 (to YY), and JP18H04944 (to MT); MEXT KAKENHI [JP16H06276 (AdAMS); to MO, YY, and MT]; Brain/MINDS [Mapping by Integrated Neurotechnologies for Disease Studies (JP19dm0207001)], AMED (to MO).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the Institute for Animal Experimentation, Tohoku University Graduate School of Medicine for the use of its facilities and their technical assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fncel.2019.00547/full#supplementary-material.
References
Amano, K., Sato, K., Hori, M., Ozaki, H., and Karaki, H. (1997). Palytoxin-induced increase in endothelial Ca2+ concentration in the rabbit aortic valve. Naunyn Schmiedebergs Arch. Pharmacol. 355, 751–758. doi: 10.1007/pl00005009
Baba, A., Yasui, T., Fujisawa, S., Yamada, R. X., Yamada, M. K., Nishiyama, N., et al. (2003). Activity-evoked capacitative Ca2+ entry: implications in synaptic plasticity. J. Neurosci. 23, 7737–7741. doi: 10.1523/jneurosci.23-21-07737.2003
Bardo, S., Cavazzini, M. G., and Emptage, N. (2006). The role of the endoplasmic reticulum Ca2+ store in the plasticity of central neurons. Trends Pharmacol. Sci. 27, 78–84. doi: 10.1016/j.tips.2005.12.008
Berridge, M. J. (2002). The endoplasmic reticulum: a multifunctional signaling organelle. Cell Calcium 32, 235–249. doi: 10.1016/s0143416002001823
Berridge, M. J., Lipp, P., and Bootman, M. D. (2000). The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 1, 11–21. doi: 10.1038/35036035
Blaustein, M. P., and Golovina, V. A. (2001). Structural complexity and functional diversity of endoplasmic reticulum Ca2+ stores. Trends Neurosci. 24, 602–608. doi: 10.1016/s0166-2236(00)01891-9
Bouron, A., Mbebi, C., Loeffler, J.-P., and De Waard, M. (2004). The β-amyloid precursor protein controls a store-operated Ca2+ entry in cortical neurons. Eur. J. Neurosci. 20, 2071–2078. doi: 10.1111/j.1460-9568.2004.03680.x
Brini, M., Cali, T., Ottolini, D., and Carafoli, E. (2014). Neuronal calcium signaling: function and dysfunction. Cell. Mol. Life Sci. 71, 2787–2814. doi: 10.1007/s00018-013-1550-7
Chen, P., Chakraborty, S., Mukhopadhyay, S., Lee, E., Paoliello, M. M. B., Bowman, A. B., et al. (2015). Manganese homeostasis in the nervous system. J. Neurochem. 134, 601–610. doi: 10.1111/jnc.13170
Chesselet, M. F., and Delfs, J. M. (1996). Basal ganglia and movement disorders: an update. Trends Neurosci. 19, 417–422. doi: 10.1016/0166-2236(96)10052-7
Gerfen, C. R., and Bolam, J. P. (2010). “The neuroanatomical organization of the basal Ganglia,” in Handbook of Basal Ganglia: Structure and Function, eds H. Steiner and K. Y. Tseng (London: Academic Press), 3–28.
González-Sánchez, P., Del Arco, A., Esteban, J. A., and Satrústegui, J. (2017). Store-operated calcium entry is required for mGluR-dependent long term depression in cortical neurons. Front. Cell. Neurosci. 11:363. doi: 10.3389/fncel.2017.00363
Graybiel, A. M. (1995). Building action repertoires: memory and learning functions of the basal ganglia. Curr. Opin. Neurobiol. 5, 733–741. doi: 10.1016/0959-4388(95)80100-6
Kikuta, S., Nakamura, Y., Yamamura, Y., Tamura, A., Homma, N., Yanagawa, Y., et al. (2015). Quantitative activation-induced manganese-enhanced MRI reveals severity of Parkinson’s disease in mice. Sci. Rep. 5:12800. doi: 10.1038/srep12800
Korkotian, E., Oni-Biton, E., and Segal, M. (2017). The role of the store-operated calcium entry channel Orai1 in cultured rat hippocampal synapse formation and plasticity. J. Physiol. 595, 125–140. doi: 10.1113/JP272645
Kostyuk, P., and Verkhratsky, A. (1994). Calcium stores in neurons and glia. Neuroscience 63, 381–404. doi: 10.1016/0306-4522(94)90537-1
Kraft, R. (2015). STIM and ORAI proteins in the nervous system. Channels 9, 244–252. doi: 10.1080/19336950.2015.1071747
Majewski, L., and Kuznicki, J. (2015). SOCE in neurons: signaling or just refilling? Biochim. Biophys. Acta 1853, 1940–1952. doi: 10.1016/j.bbamcr.2015.01.019
Martinez-Galan, J. R., Perez-Martinez, F. C., and Juiz, J. M. (2012). Signalling routes and developmental regulation of group I metabotropic glutamate receptors in rat auditory midbrain neurons. J. Neurosci. Res. 90, 1913–1923. doi: 10.1002/jnr.23087
Moccia, F., Zuccolo, E., Soda, T., Tanzi, F., Guerra, G., Mapelli, L., et al. (2015). Stim and Orai proteins in neuronal Ca2+ signaling and excitability. Front. Cell. Neurosci. 9:153. doi: 10.3389/fncel.2015.00153
Nimmerjahn, A., Kirchhoff, F., Kerr, J. D. N., and Helmchen, F. (2004). Sulforhodamine 101 as a specific marker of astroglia in the neocortex in vivo. Nat. Methods 1, 31–37. doi: 10.1038/nmeth706
Ong, H. L., Subedi, K. P., Son, G.-Y., Liu, X., and Ambudkar, I. S. (2019). Tuning store-operated calcium entry to modulate Ca2+-dependent physiological processes. Biochim. Biophys. Acta Mol. Cell. Res. 1866, 1037–1045. doi: 10.1016/j.bbamcr.2018.11.018
Osanai, M., Yamada, N., and Yagi, T. (2006). Long-lasting spontaneous calcium transients in the striatal cells. Neurosci. Lett. 402, 81–85. doi: 10.1016/j.neulet.2006.04.012
Pasti, L., Volterra, A., Pozzan, T., and Carmignoto, G. (1997). Intracellular calcium oscillations in astrocytes: a highly plastic, bidirectional form of communication between neurons and astrocytes in situ. J. Neurosci. 17, 7817–7830. doi: 10.1523/jneurosci.17-20-07817.1997
Prothero, L. S., Richards, C. D., and Mathie, A. (1998). Inhibition by inorganic ions of a sustained calcium signal evoked by activation of mGlu5 receptors in rat cortical neurons and glia. Br. J. Pharmacol. 125, 1551–1561. doi: 10.1038/sj.bjp.0702203
Schneider, C. A., Rasband, W. S., and Eliceiri, K. W. (2012). NIH image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. doi: 10.1038/nmeth.2089
Secondo, A., Bagetta, G., and Amantea, D. (2018). On the role of store-operated calcium entry in acute and chronic neurodegenerative diseases. Front. Mol. Neurosci. 11:87. doi: 10.3389/fnmol.2018.00087
Shideman, C. R., Reinardy, J. L., and Thayer, S. A. (2009). γ-secretase activity modulates store-operated Ca2+ entry into rat sensory neurons. Neurosci. Lett. 451, 124–128. doi: 10.1016/j.neulet.2008.12.031
Smetters, D., Majewska, A., and Yuste, R. (1999). Detecting action potentials in neuronal populations with calcium imaging. Methods 18, 215–221. doi: 10.1006/meth.1999.0774
Tamamaki, N., Yanagawa, Y., Tomioka, R., Miyazaki, J., Obata, K., and Kaneko, T. (2003). Green fluorescent protein expression and colocalization with calretinin, parvalbumin and somatostatin in the GAD67-GFP knock-in mouse. J. Comp. Neurol. 467, 60–79. doi: 10.1002/cne.10905
Tamura, A., Yamada, N., Yaguchi, Y., Machida, Y., Mori, I., and Osanai, M. (2014). Both neurons and astrocytes exhibited tetrodotoxin-resistant metabotropic glutamate receptor-dependent spontaneous slow Ca2+ oscillations in striatum. PLoS One 9:e85351. doi: 10.1371/journal.pone.0085351
Tozzi, A., Bengtson, C. P., Longone, P., Carignani, C., Fusco, F. R., Bernardi, G., et al. (2003). Involvement of transient receptor potential-like channels in responses to mGluR-I activation in midbrain dopamine neurons. Eur. J. Neurosci. 18, 2133–2145. doi: 10.1046/j.1460-9568.2003.02936.x
Tu, P., Kunert-Keil, C., Lucke, S., Brinkmeier, H., and Bouron, A. (2009). Diacylglycerol analogues activate second messenger-operated calcium channels exhibiting TRPC-like properties in cortical neurons. J. Neurochem. 108, 126–138. doi: 10.1111/j.1471-4159.2008.05752.x
Uehara, A., Yasukochi, M., Imanaga, I., Nishi, M., and Takeshima, H. (2002). Store-operated Ca2+ entry uncoupled with ryanodine receptor and junctional membrane complex in heart muscle cells. Cell Calcium 31, 89–96. doi: 10.1054/ceca.2001.0257
Wegierski, T., and Kuznicki, J. (2018). Neuronal calcium signaling via store-operated channels in health and disease. Cell Calcium 74, 102–111. doi: 10.1016/j.ceca.2018.07.001
Wu, J., Ryskamp, D. A., Liang, X., Egorova, P., Zakharova, O., Hung, G., et al. (2016). Enhanced store-operated calcium entry leads to striatal synaptic loss in a Huntington’s disease mouse model. J. Neurosci. 36, 125–141. doi: 10.1523/JNEUROSCI.1038-15.2016
Keywords: store-operated calcium entry, basal ganglia, striatum, calcium oscillations, fluorescence microscopy
Citation: Kikuta S, Iguchi Y, Kakizaki T, Kobayashi K, Yanagawa Y, Takada M and Osanai M (2019) Store-Operated Calcium Channels Are Involved in Spontaneous Slow Calcium Oscillations in Striatal Neurons. Front. Cell. Neurosci. 13:547. doi: 10.3389/fncel.2019.00547
Received: 09 August 2019; Accepted: 25 November 2019;
Published: 17 December 2019.
Edited by:
Sergey M. Korogod, National Academy of Sciences of Ukraine, UkraineReviewed by:
David Gall, Université libre de Bruxelles, BelgiumJacek Kuznicki, International Institute of Molecular and Cell Biology (IIMCB), Poland
Copyright © 2019 Kikuta, Iguchi, Kakizaki, Kobayashi, Yanagawa, Takada and Osanai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Makoto Osanai, b3NhbmFpQHNhaHMubWVkLm9zYWthLXUuYWMuanA=