 Christos Galanis
Christos Galanis Andreas Vlachos
Andreas Vlachos- 1Department of Neuroanatomy, Institute of Anatomy and Cell Biology, Faculty of Medicine, University of Freiburg, Freiburg, Germany
- 2Faculty of Biology, University of Freiburg, Freiburg, Germany
- 3Center for Basics in Neuromodulation (NeuroModulBasics), Faculty of Medicine, University of Freiburg, Freiburg, Germany
During the past 50 years, the cellular and molecular mechanisms of synaptic plasticity have been studied in great detail. A plethora of signaling pathways have been identified that account for synaptic changes based on positive and negative feedback mechanisms. Yet, the biological significance of Hebbian synaptic plasticity (= positive feedback) and homeostatic synaptic plasticity (= negative feedback) remains a matter of debate. Specifically, it is unclear how these opposing forms of plasticity, which share common downstream mechanisms, operate in the same networks, neurons, and synapses. Based on the observation that rapid and input-specific homeostatic mechanisms exist, we here discuss a model that is based on signaling pathways that may adjust a balance between Hebbian and homeostatic synaptic plasticity. Hence, “alterations” in Hebbian plasticity may, in fact, resemble “enhanced” homeostasis, which rapidly returns synaptic strength to baseline. In turn, long-lasting experience-dependent synaptic changes may require attenuation of homeostatic mechanisms or the adjustment of homeostatic setpoints at the single-synapse level. In this context, we propose a role for the proteolytic processing of the amyloid precursor protein (APP) in setting a balance between the ability of neurons to express Hebbian and homeostatic synaptic plasticity.
Introduction
The ability of neural tissue to adapt to specific stimuli through structural, functional and molecular changes plays a fundamental role in complex brain functions such as perception, decision-making, learning and memory (Citri and Malenka, 2008; Bailey et al., 2015). During the past 50 years, considerable effort has been spent to decipher and better understand the cellular and molecular mechanisms of Hebbian synaptic plasticity, which accounts for activity-dependent changes of synaptic weights based on positive feedback mechanisms (Hebb, 1949; Bliss and Lomo, 1973). It is now well-established that Hebbian plasticity resembles fast and lasting input-specific synaptic changes necessary for experience-dependent memory and learning (Bear, 1996; Chen and Tonegawa, 1997; Klintsova and Greenough, 1999). Experimentally, Hebbian mechanisms have been described in detail for excitatory pre- and postsynaptic sites (e.g., Petzoldt et al., 2016; Monday et al., 2018; Scheefhals and MacGillavry, 2018; Buonarati et al., 2019), where, for example, tetanic electrical stimulation at different frequencies results in the strengthening (long-term potentiation, LTP) or weakening (long-term depression, LTD) of neurotransmission (Bliss and Lomo, 1973; Dudek and Bear, 1992). Meanwhile, evidence has started to emerge for corresponding activity-dependent synaptic changes at GABAergic synapses (Bartos et al., 2011; Rozov et al., 2017; Chiu et al., 2019). Specifically, the plasticity of inhibitory neurotransmission seems to control the ability of neurons to express Hebbian plasticity of excitatory neurotransmission (Letzkus et al., 2015; Zhao et al., 2017).
While feedforward and feedback microcircuits dynamically match afferent excitation to recruited inhibition (Sprekeler, 2017), it has been recognized that, in the absence of physiological constraints, complex systems based solely on positive feedback mechanisms will experience instability—e.g., strong synapses will continue growing, while weakening of synapses will result in synapse elimination (Miller and Mackay, 1994). Indeed, during the past two decades, a plethora of cellular and molecular mechanisms have been identified that maintain neurons in a dynamic functional range by adjusting excitatory and inhibitory synaptic strength in a compensatory manner—i.e., based on negative feedback (Davis and Bezprozvanny, 2001; Marder and Prinz, 2003; Turrigiano and Nelson, 2004; Pozo and Goda, 2010; Keck et al., 2017). Yet, a major unresolved issue in the field concerns the interplay between Hebbian and compensatory—i.e., homeostatic—synaptic plasticity, which share common downstream mechanisms that change and/or adjust excitatory and inhibitory neurotransmission (Turrigiano et al., 1998; Feldman, 2002; Turrigiano and Nelson, 2004; Swanwick et al., 2006; Rannals and Kapur, 2011). Moreover, the biological significance of alterations in Hebbian and/or homeostatic plasticity for pathological brain states remains unclear.
In recent years, these questions have been discussed extensively by leading experts in the field (e.g., Vitureira and Goda, 2013; Fox and Stryker, 2017; Keck et al., 2017; Yee et al., 2017). It has been proposed, for example, that homeostatic plasticity operates on a longer time scale (Turrigiano, 2012; Tononi and Cirelli, 2014; Hengen et al., 2016)—thus not interfering with synaptic changes induced by Hebbian plasticity—and that all synapses of a neuron are adjusted by the same factor in the context of homeostatic “synaptic scaling” to preserve the relative differences between synapses (Turrigiano et al., 1998; Turrigiano, 2008; Vitureira and Goda, 2013). Meanwhile, theoretical modeling work has emphasized the importance of fast homeostatic mechanisms for network stability (Zenke et al., 2013), and robust experimental evidence has been provided for rapid homeostatic plasticity (Keck et al., 2011; Frank, 2014; Li et al., 2014). Furthermore, solid evidence suggests that homeostatic synaptic adaptation can occur locally, in subsets of synapses (e.g., Desai et al., 2002; Kim and Tsien, 2008; Vlachos et al., 2013). These findings indicate that Hebbian and homeostatic synaptic mechanisms may operate in parallel and could thus interfere with each other in the same subset of synapses.
In light of these considerations, it is interesting to note that the effects of classic Hebbian plasticity paradigms—e.g., local tetanic electrical stimulation (Bliss and Lomo, 1973)—have not yet been systematically evaluated for their effects on homeostatic synaptic plasticity induction. Therefore, in this article, we sought to present a “homeostatic view on classic LTP/LTD experiments” by highlighting mechanisms which may rapidly affect—and hence set a balance between—Hebbian and homeostatic synaptic plasticity (Figure 1). These considerations are put into clinical perspective by discussing the potential role of α- and β-secretase-mediated processing of the amyloid precursor protein (APP) in Hebbian and homeostatic synaptic plasticity (Figure 2).
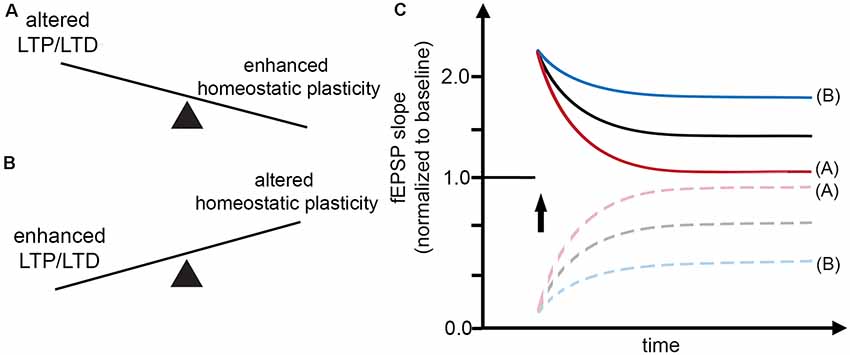
Figure 1. Interaction between Hebbian and homeostatic synaptic plasticity. (A,B) Factors may exist which rapidly set a balance between Hebbian and homeostatic synaptic plasticity, thereby affecting the induction and persistence of experience-dependent synaptic changes. (C) Alterations in Hebbian plasticity—i.e., long-term potentiation (LTP) or depression (LTD) of evoked field excitatory postsynaptic potentials (fEPSPs; red curve)—may reflect enhanced homeostatic synaptic plasticity. In turn, alterations in homeostatic synaptic plasticity may account for enhanced LTP/LTD (blue curve).
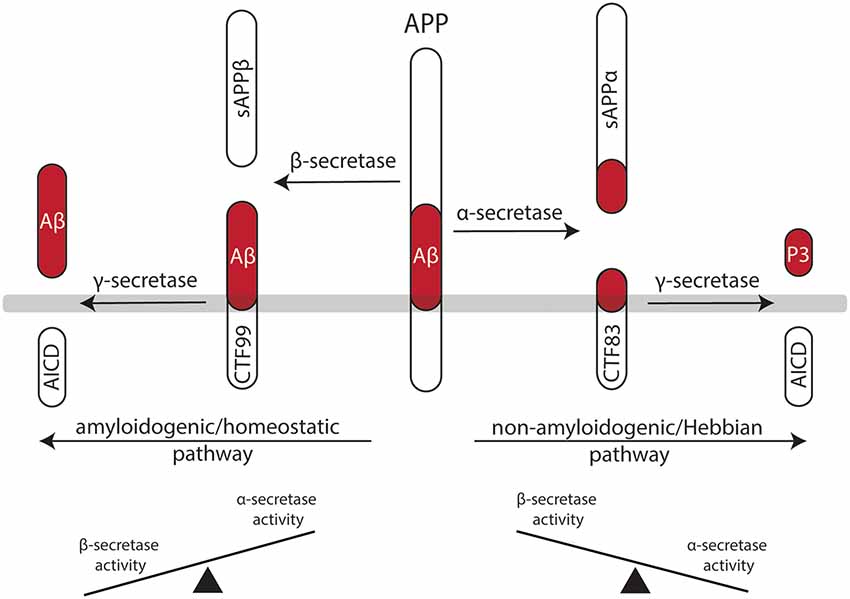
Figure 2. Processing of the amyloid precursor protein (APP) may set a balance between Hebbian and homeostatic synaptic plasticity. Work in recent years has established a firm link between the non-amyloidogenic processing pathway—i.e., APP secreted ectodomain alpha (APPsα)—and the ability of neurons to express LTP of excitatory postsynapses. Likewise, evidence has started to emerge for the role of the amyloidogenic processing pathway—i.e., amyloid-β (Aβ)—in homeostatic synaptic plasticity. Hence, differential processing of APP via α- or β-secretases may set a balance between Hebbian and homeostatic synaptic plasticity in neural networks.
Opposing Roles of Ca2+ Signaling in Hebbian and Homeostatic Synaptic Plasticity
Central mechanisms that regulate the activity-dependent strengthening (or dampening) of excitatory neurotransmission are modification, trafficking and synthesis of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPA-Rs) at excitatory postsynapses (Malinow and Malenka, 2002; Diering and Huganir, 2018). Interestingly, both Hebbian and homeostatic synaptic plasticity recruit Ca2+-dependent signaling pathways which lead to characteristic changes in synaptic AMPA-R content and function (Malinow and Malenka, 2002; Song and Huganir, 2002; Derkach et al., 2007; Turrigiano, 2008). However, Ca2+ influx via N-methyl-D-aspartate receptors (NMDA-Rs) or voltage-gated Ca2+ channels (VGCCs) can have opposing effects on postsynaptic AMPA-R content in the context of Hebbian and homeostatic synaptic plasticity (Lee et al., 2000; Diering et al., 2014; Diering and Huganir, 2018).
In the case of LTP induction, for example, tetanic electrical stimulation, which triggers Ca2+ influx, can lead to an increase in postsynaptic AMPA-R content and hence potentiation of excitatory neurotransmission (= positive feedback mechanism). Conversely, increased intracellular Ca2+ levels are expected to trigger homeostatic synaptic down-scaling, which returns AMPA-R content to baseline (= negative feedback mechanism). Considering such rapid interactions between Hebbian and homeostatic plasticity mechanisms (Figure 1), a widely used interpretation of “alterations” in Hebbian plasticity—i.e., failure to persistently change the amplitude or the slope of evoked field excitatory postsynaptic potentials (fEPSPs)—may, in fact, resemble “enhanced” homeostasis, which effectively returns fEPSPs to baseline after the LTP- or LTD-inducing “network perturbation” (see Figures 1A,C). Conversely, signaling pathways that block homeostasis or change homeostatic setpoints will result in persisting changes of excitatory neurotransmission (Figures 1B,C). We have to concede, however, that molecular signaling pathways that attenuate or adjust local homeostatic plasticity at the level of individual synapses are not well-understood. It is also interesting to speculate in this context that changes in the ability of neurons to express homeostatic plasticity per se may suffice to generate Hebbian-like associative plasticity. Indeed, a recent study employed computational modeling to demonstrate associative properties of firing-rate homeostasis in recurrent neuronal networks (Gallinaro and Rotter, 2018).
Role of Dopamine in Homeostatic Synaptic Plasticity
Based on the above considerations, we recently tested for the role of dopamine in homeostatic synaptic plasticity (Strehl et al., 2018). We reasoned that neuromodulators which promote Hebbian plasticity (Otani et al., 2003; Mu et al., 2011; Sheynikhovich et al., 2013; Broussard et al., 2016) may also act by blocking the ability of neurons to express homeostatic synaptic plasticity. Indeed, we were able to demonstrate that dopamine blocks homeostatic plasticity of excitatory neurotransmission in entorhino-hippocampal tissue cultures (Strehl et al., 2018). Pharmacological activation of D1/5 receptors, but not D2/3 receptors, mimicked the effects of dopamine on homeostatic plasticity. These findings raise the intriguing possibility that dopamine may act as a permissive factor that promotes Hebbian plasticity, at least in part, by blocking homeostasis. Interestingly, the “anti-homeostatic” effects of dopamine were only observed in immature neurons during early postnatal development (Strehl et al., 2018). Hence, specific factors may exist which adjust homeostatic plasticity in specific cells depending on the state of the neural network. It remains to be shown, however, whether dopamine indeed promotes Hebbian plasticity by attenuating homeostatic plasticity at the level of individual synapses and whether dopamine acts on neurons or glia cells (or both) to assert its differential effects on plasticity. Regardless of these considerations, these results call for a re-evaluation of the available LTP/LTD literature and a systematic assessment of well-known “LTP-/LTD-promoting or -blocking factors” in homeostatic synaptic plasticity. As an example that is of considerable clinical relevance, we here discuss the potential role of APP processing in setting a balance between Hebbian and homeostatic synaptic plasticity.
The Role of The Amyloid Precursor Protein in Synaptic Plasticity
Work in recent years has established a firm link between APP and structural and functional plasticity (comprehensively reviewed in Müller et al., 2017). These studies are based on experiments using APP-deficient mice, or mice in which the APP gene has been genetically modified (Dawson et al., 1999; Magara et al., 1999; Seabrook et al., 1999; Turner et al., 2003; Herms et al., 2004). Historically, the majority of studies in the field have focused on addressing the role of APP and its cleavage products in Hebbian plasticity. More recently, some evidence has supported its involvement in homeostatic synaptic plasticity (Jang and Chung, 2016; Styr and Slutsky, 2018).
APP is a type I transmembrane protein ubiquitously expressed in all mammalian tissues (Müller-Hill and Beyreuther, 1989; Müller et al., 2017). It is differentially processed by secretases via two pathways (Figure 2): the amyloidogenic processing pathway generates amyloid-β (Aβ) peptides, which are implicated in the pathogenesis of Alzheimer’s disease (AD), while the non-amyloidogenic processing pathway produces the neuroprotective soluble ectodomain APPsα (Turner et al., 2003). In the amyloidogenic pathway, APP is cleaved by β-site APP cleaving enzyme (BACE1), which releases APP soluble fragment beta (APPsβ), followed by γ-secretase processing, which generates Aβ fragments and the APP intracellular domain (AICD; Vassar et al., 1999; Van Der Kant and Goldstein, 2015). In contrast, the non-amyloidogenic processing pathway recruits α-secretases releasing APPsα, again followed by γ-secretases that produce the P3 peptide and AICD (O’Brien and Wong, 2011; Van Der Kant and Goldstein, 2015).
Role of The Non-Amyloidogenic Pathway in Synaptic Plasticity
APP-deficient mice show alterations in dendritic morphologies and dendritic spine counts (Perez et al., 1997; Lee et al., 2010; Tyan et al., 2012; Weyer et al., 2014). These structural defects have been linked to alterations in LTP and deficits in learning and memory (Dawson et al., 1999; Hick et al., 2015). Interestingly, APPsα rescues several of the deficits of APP−/− animals, while APPsβ does not have such a positive effect on Hebbian plasticity (Ring et al., 2007; Hick et al., 2015). Consistent with this suggestion, enhanced LTP is observed in APPsα-treated acute brain slices prepared from rats (Ishida et al., 1997), and behavioral learning is augmented when mice are injected with APPsα (Meziane et al., 1998). Moreover, pharmacologic inhibition of α-secretase activity impairs LTP in rats, which can be rescued by APPsα (Taylor et al., 2008). This line of evidence suggests that APPsα secretion seems to be activity-dependent—that is, LTP-inducing protocols lead to an increase in APPsα (Nitsch et al., 1992; Fazeli et al., 1994). Therefore, it has been proposed that the non-amyloidogenic processing pathway plays an important role in mediating Hebbian synaptic plasticity (Figure 2). However, it should be clearly stated that APPsα has not yet been tested in the context of homeostatic synaptic plasticity. It thus remains to be shown whether some of the “positive” effects of APPsα on activity-dependent structural and functional plasticity are also mediated by its ability to modulate—i.e., to attenuate—homeostatic plasticity mechanisms.
Role of The Amyloidogenic Pathway in Synaptic Plasticity
The role of APP processing via the amyloidogenic pathway has been studied in detail for its pathogenic role in neurodegeneration (Goldsworthy and Vallence, 2013; Nieweg et al., 2015; Gupta and Goyal, 2016; Chen et al., 2017; Youn et al., 2019). What remains less understood is the physiological role of the amyloidogenic processing pathway and Aβ.
It seems well-established that elevated concentrations of Aβ are “synaptotoxic” by hindering the ability of neurons to express LTP, thereby having detrimental effects on learning and memory (Chiba et al., 2009; Jo et al., 2011; Samidurai et al., 2018). In this context, it has been shown that Aβ interferes with neural Ca2+ signaling—i.e., it blocks NMDA-Rs and Ca2+/calmodulin-dependent protein kinase II (CamKII; Zhao et al., 2004; Townsend et al., 2007; Gu et al., 2009; but see the work in Opazo et al., 2018, which suggests that Aβ activates CamKII). Similar to APPsα, an increase in synaptic activity and NMDA-R stimulation can also lead to an increase in Aβ production (Kamenetz et al., 2003; Lesné et al., 2005). Thus, it has been proposed that an increase in Aβ may act as a negative feedback mechanism by blocking Hebbian synaptic plasticity. In light of the herein proposed model (Figure 1), Aβ may also act by promoting homeostatic synaptic plasticity (see Figure 1).
Indeed, evidence has started to emerge for a physiological role of Aβ in homeostatic synaptic plasticity. For example, the AMPA-R scaffolding protein PICK1 mediates homeostatic synaptic plasticity (Anggono et al., 2011) and has been linked to Aβ-mediated “alterations” in synaptic plasticity (Alfonso et al., 2014). Similar evidence exists for interaction between Aβ and PSD-95 (Roselli et al., 2005; Sun and Turrigiano, 2011), GKAP (Roselli et al., 2011; Shin et al., 2012), calcineurin (D’Amelio et al., 2011; Kim and Ziff, 2014) and STEP61 (Kurup et al., 2010). Finally, BDNF and TNFα, which have been firmly linked to homeostatic synaptic plasticity (Rutherford et al., 1998; Stellwagen and Malenka, 2006; Becker et al., 2015), seem to be dysregulated in the AD brain (Fillit et al., 1991; Phillips et al., 1991). Along this line of evidence, a role for microglia in Aβ-mediated alterations in complex brain function has been suggested (Kitazawa et al., 2004; Hansen et al., 2018; Kinney et al., 2018; Hemonnot et al., 2019). However, it is important to note that the majority of these findings are based on experiments employing transgenic mouse models of AD or high concentrations of Aβ. Hence, direct experimental evidence for a physiological role of APP/Aβ in homeostatic synaptic plasticity is currently missing (Figure 2).
Clinical Implications and Perspective
Considering the detrimental effects of Aβ in Hebbian synaptic plasticity together with promising results in experiments employing a mouse model that expressed familial mutant APP in the absence of BACE1 (Cai et al., 2001; Luo et al., 2001; Roberds et al., 2001), pharmacologic inhibition of BACE1 has been tested as a potential treatment for the cognitive decline in AD (Yan and Vassar, 2014; Coimbra et al., 2018). Indeed, BACE1 inhibitors successfully lowered Aβ levels detected in the cerebrospinal fluid of AD patients (Kennedy et al., 2016; Egan et al., 2018). However, major clinical trials were discontinued due to a series of adverse effects or no improvement and even accelerated cognitive decline in patients (Coimbra et al., 2018; Egan et al., 2019). On the same note, mice lacking BACE1 showed increased neural excitability and spontaneous seizure activity (Hitt et al., 2010; Hu et al., 2010; Zhu et al., 2018; Vnencak et al., 2019), which have been linked to impaired homeostatic mechanisms (Wondolowski and Dickman, 2013; González et al., 2015). Although it is clear that BACE1 targets several other substrates in the nervous system (Barão et al., 2016), these observations support the notion that some of the adverse effects of clinically used BACE1 inhibitors could be explained by an impairment of Aβ-mediated homeostatic synaptic plasticity.
Hence, it will be important to evaluate the significance of APP processing via the amyloidogenic and non-amyloidogenic processing pathways in homeostatic synaptic plasticity. We are confident that a systematic assessment of “pro-homeostatic” effects of Aβ and possible “anti-homeostatic” effects of APPsα will provide new and important insights into the intricate interplay between Hebbian and homeostatic synaptic plasticity. These findings may also be of relevance for the development of new therapeutic strategies in neurological and psychiatric diseases associated with alterations in APP processing or increased Aβ levels.
Author Contributions
CG and AV wrote this manuscript and prepared the figures.
Funding
This work was supported by Deutsche Forschungsgemeinschaft (CRC1080 and FOR1332 to AV).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Alfonso, S., Kessels, H. W., Banos, C. C., Chan, T. R., Lin, E. T., Kumaravel, G., et al. (2014). Synapto-depressive effects of amyloid beta require PICK 1. Eur. J. Neurosci. 39, 1225–1233. doi: 10.1111/ejn.12499.
Anggono, V., Clem, R. L., and Huganir, R. L. (2011). PICK1 loss of function occludes homeostatic synaptic scaling. J. Neurosci. 31, 2188–2196. doi: 10.1523/jneurosci.5633-10.2011
Bailey, C. H., Kandel, E. R., and Harris, K. M. (2015). Structural components of synaptic plasticity and memory consolidation. Cold Spring Harb. Perspect. Biol. 7:a021758. doi: 10.1101/cshperspect.a021758
Barão, S., Moechars, D., Lichtenthaler, S. F., and De Strooper, B. (2016). BACE1 physiological functions may limit its use as therapeutic target for Alzheimer’s disease. Trends Neurosci. 39, 158–169. doi: 10.1016/j.tins.2016.01.003
Bartos, M., Alle, H., and Vida, I. (2011). Role of microcircuit structure and input integration in hippocampal interneuron recruitment and plasticity. Neuropharmacology 60, 730–739. doi: 10.1016/j.neuropharm.2010.12.017
Bear, M. F. (1996). A synaptic basis for memory storage in the cerebral cortex. Proc. Natl. Acad. Sci. U S A 93, 13453–13459. doi: 10.1073/pnas.93.24.13453
Becker, D., Deller, T., and Vlachos, A. (2015). Tumor necrosis factor (TNF)-receptor 1 and 2 mediate homeostatic synaptic plasticity of denervated mouse dentate granule cells. Sci. Rep. 5:12726. doi: 10.1038/srep12726
Bliss, T. V., and Lomo, T. (1973). Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J. Physiology. 232, 331–356. doi: 10.1113/jphysiol.1973.sp010273
Broussard, J. I., Yang, K., Levine, A. T., Tsetsenis, T., Jenson, D., Cao, F., et al. (2016). Dopamine regulates aversive contextual learning and associated in vivo synaptic plasticity in the hippocampus. Cell Rep. 14, 1930–1939. doi: 10.1016/j.celrep.2016.01.070
Buonarati, O. R., Hammes, E. A., Watson, J. F., Greger, I. H., and Hell, J. W. (2019). Mechanisms of postsynaptic localization of AMPA-type glutamate receptors and their regulation during long-term potentiation. Sci. Signal. 12:eaar6889. doi: 10.1126/scisignal.aar6889
Cai, H., Wang, Y., Mccarthy, D., Wen, H., Borchelt, D. R., Price, D. L., et al. (2001). BACE1 is the major beta-secretase for generation of Abeta peptides by neurons. Nat. Neurosci. 4, 233–234. doi: 10.1038/85064
Chen, C., and Tonegawa, S. (1997). Molecular genetic analysis of synaptic plasticity, activity-dependent neural development, learning and memory in the mammalian brain. Annu. Rev. Neurosci. 20, 157–184. doi: 10.1146/annurev.neuro.20.1.157
Chen, G. F., Xu, T. H., Yan, Y., Zhou, Y. R., Jiang, Y., Melcher, K., et al. (2017). Amyloid beta: structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 38, 1205–1235. doi: 10.1038/aps.2017.28
Chiba, T., Yamada, M., Sasabe, J., Terashita, K., Shimoda, M., Matsuoka, M., et al. (2009). Amyloid-beta causes memory impairment by disturbing the JAK2/STAT3 axis in hippocampal neurons. Mol. Psychiatry 14, 206–222. doi: 10.1038/mp.2008.105
Chiu, C. Q., Barberis, A., and Higley, M. J. (2019). Preserving the balance: diverse forms of long-term GABAergic synaptic plasticity. Nat. Rev. Neurosci. 20, 272–281. doi: 10.1038/s41583-019-0141-5
Citri, A., and Malenka, R. C. (2008). Synaptic plasticity: multiple forms, functions and mechanisms. Neuropsychopharmacology 33, 18–41. doi: 10.1038/sj.npp.1301559
Coimbra, J. R. M., Marques, D. F. F., Baptista, S. J., Pereira, C. M. F., Moreira, P. I., Dinis, T. C. P., et al. (2018). Highlights in BACE1 inhibitors for Alzheimer’s disease treatment. Front. Chem. 6:178. doi: 10.3389/fchem.2018.00178
D’Amelio, M., Cavallucci, V., Middei, S., Marchetti, C., Pacioni, S., Ferri, A., et al. (2011). Caspase-3 triggers early synaptic dysfunction in a mouse model of Alzheimer’s disease. Nat. Neurosci. 14:69. doi: 10.1038/nn.2709
Davis, G. W., and Bezprozvanny, I. (2001). Maintaining the stability of neural function: a homeostatic hypothesis. Annu. Rev Physiol 63, 847–869. doi: 10.1146/annurev.physiol.63.1.847
Dawson, G. R., Seabrook, G. R., Zheng, H., Smith, D. W., Graham, S., O’dowd, G., et al. (1999). Age-related cognitive deficits, impaired long-term potentiation and reduction in synaptic marker density in mice lacking the beta-amyloid precursor protein. Neuroscience 90, 1–13. doi: 10.1016/s0306-4522(98)00410-2
Derkach, V. A., Oh, M. C., Guire, E. S., and Soderling, T. R. (2007). Regulatory mechanisms of AMPA receptors in synaptic plasticity. Nat. Rev. Neurosci. 8, 101–113. doi: 10.1038/nrn2055
Desai, N. S., Cudmore, R. H., Nelson, S. B., and Turrigiano, G. G. (2002). Critical periods for experience-dependent synaptic scaling in visual cortex. Nat. Neurosci. 5, 783–789. doi: 10.1038/nn878
Diering, G. H., Gustina, A. S., and Huganir, R. L. (2014). PKA-GluA1 coupling via AKAP5 controls AMPA receptor phosphorylation and cell-surface targeting during bidirectional homeostatic plasticity. Neuron 84, 790–805. doi: 10.1016/j.neuron.2014.09.024
Diering, G. H., and Huganir, R. L. (2018). The AMPA receptor code of synaptic plasticity. Neuron 100, 314–329. doi: 10.1016/j.neuron.2018.10.018
Dudek, S. M., and Bear, M. F. (1992). Homosynaptic long-term depression in area CA1 of hippocampus and effects of N-methyl-D-aspartate receptor blockade. Proc. Natl. Acad. Sci. U S A 89, 4363–4367. doi: 10.1073/pnas.89.10.4363
Egan, M. F., Kost, J., Tariot, P. N., Aisen, P. S., Cummings, J. L., Vellas, B., et al. (2018). Randomized trial of verubecestat for mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 378, 1691–1703. doi: 10.1056/NEJMoa1706441
Egan, M. F., Kost, J., Voss, T., Mukai, Y., Aisen, P. S., Cummings, J. L., et al. (2019). Randomized trial of verubecestat for prodromal Alzheimer’s disease. N. Engl. J. Med. 380, 1408–1420. doi: 10.1056/NEJMoa1812840
Fazeli, M. S., Breen, K., Errington, M. L., and Bliss, T. V. (1994). Increase in extracellular NCAM and amyloid precursor protein following induction of long-term potentiation in the dentate gyrus of anaesthetized rats. Neurosci. Lett. 169, 77–80. doi: 10.1016/0304-3940(94)90360-3
Feldman, D. E. (2002). Synapses, scaling and homeostasis in vivo. Nat. Neurosci. 5, 712–714. doi: 10.1038/nn0802-712
Fillit, H., Ding, W., Buee, L., Kalman, J., Altstiel, L., Lawlor, B., et al. (1991). Elevated circulating tumor necrosis factor levels in Alzheimer’s disease. Neurosci. Lett. 129, 318–320. doi: 10.1016/0304-3940(91)90490-k
Fox, K., and Stryker, M. (2017). Integrating hebbian and homeostatic plasticity: introduction. Philos. Trans. R. Soc. Lond. B Biol. Sci. 372:20160413. doi: 10.1098/rstb.2016.0413
Frank, C. A. (2014). Homeostatic plasticity at the Drosophila neuromuscular junction. Neuropharmacology 78, 63–74. doi: 10.1016/j.neuropharm.2013.06.015
Gallinaro, J. V., and Rotter, S. (2018). Associative properties of structural plasticity based on firing rate homeostasis in recurrent neuronal networks. Sci. Rep. 8:3754. doi: 10.1038/s41598-018-22077-3
Goldsworthy, M. R., and Vallence, A. M. (2013). The role of beta-amyloid in Alzheimer’s disease-related neurodegeneration. J. Neurosci. 33, 12910–12911. doi: 10.1523/JNEUROSCI.2252-13.2013
González, O. C., Krishnan, G. P., Chauvette, S., Timofeev, I., Sejnowski, T., and Bazhenov, M. (2015). Modeling of age-dependent epileptogenesis by differential homeostatic synaptic scaling. J. Neurosci. 35, 13448–13462. doi: 10.1523/jneurosci.5038-14.2015
Gu, Z., Liu, W., and Yan, Z. (2009). β-Amyloid impairs AMPA receptor trafficking and function by reducing Ca2+/calmodulin-dependent protein kinase II synaptic distribution. J. Biol. Chem. 284, 10639–10649. doi: 10.1074/jbc.M806508200
Gupta, A., and Goyal, R. (2016). Amyloid beta plaque: a culprit for neurodegeneration. Acta Neurol. Belg. 116, 445–450. doi: 10.1007/s13760-016-0639-9
Hansen, D. V., Hanson, J. E., and Sheng, M. (2018). Microglia in Alzheimer’s disease. J. Cell Biol. 217, 459–472. doi: 10.1083/jcb.201709069
Hebb, D. O. (1949). The Organization of Behavior; A Neuropsychological Theory. Oxford, England: Wiley.
Hemonnot, A.-L., Hua, J., Ulmann, L., and Hirbec, H. (2019). Microglia in Alzheimer disease: well-known targets and new opportunities. Front. Aging Neurosci. 11:233. doi: 10.3389/fnagi.2019.00233
Hengen, K. B., Torrado Pacheco, A., Mcgregor, J. N., Van Hooser, S. D., and Turrigiano, G. G. (2016). Neuronal firing rate homeostasis is inhibited by sleep and promoted by wake. Cell 165, 180–191. doi: 10.1016/j.cell.2016.01.046
Herms, J., Anliker, B., Heber, S., Ring, S., Fuhrmann, M., Kretzschmar, H., et al. (2004). Cortical dysplasia resembling human type 2 lissencephaly in mice lacking all three APP family members. EMBO J. 23, 4106–4115. doi: 10.1038/sj.emboj.7600390
Hick, M., Herrmann, U., Weyer, S. W., Mallm, J. P., Tschape, J. A., Borgers, M., et al. (2015). Acute function of secreted amyloid precursor protein fragment APPsalpha in synaptic plasticity. Acta Neuropathol. 129, 21–37. doi: 10.1007/s00401-014-1368-x
Hitt, B. D., Jaramillo, T. C., Chetkovich, D. M., and Vassar, R. (2010). BACE1−/− mice exhibit seizure activity that does not correlate with sodium channel level or axonal localization. Adv. Exp. Med. Biol. 5:31. doi: 10.1186/1750-1326-5-31
Hu, X., Zhou, X., He, W., Yang, J., Xiong, W., Wong, P., et al. (2010). BACE1 deficiency causes altered neuronal activity and neurodegeneration. J. Neurosci. 30, 8819–8829. doi: 10.1523/jneurosci.1334-10.2010
Ishida, A., Furukawa, K., Keller, J. N., and Mattson, M. P. (1997). Secreted form of beta-amyloid precursor protein shifts the frequency dependency for induction of LTD and enhances LTP in hippocampal slices. Neuroreport 8, 2133–2137. doi: 10.1097/00001756-199707070-00009
Jang, S. S., and Chung, H. J. (2016). Emerging link between Alzheimer’s disease and homeostatic synaptic plasticity. Neural Plast. 2016:7969272. doi: 10.1155/2016/7969272
Jo, J., Whitcomb, D. J., Olsen, K. M., Kerrigan, T. L., Lo, S. C., Bru-Mercier, G., et al. (2011). Aβ(1–42) inhibition of LTP is mediated by a signaling pathway involving caspase-3, Akt1 and GSK-3beta. Nat. Neurosci. 14, 545–547. doi: 10.1038/nn.2785
Kamenetz, F., Tomita, T., Hsieh, H., Seabrook, G., Borchelt, D., Iwatsubo, T., et al. (2003). APP processing and synaptic function. Neuron 37, 925–937. doi: 10.1016/s0896-6273(03)00124-7
Keck, T., Scheuss, V., Jacobsen, R. I., Wierenga, C. J., Eysel, U. T., Bonhoeffer, T., et al. (2011). Loss of sensory input causes rapid structural changes of inhibitory neurons in adult mouse visual cortex. Neuron 71, 869–882. doi: 10.1016/j.neuron.2011.06.034
Keck, T., Toyoizumi, T., Chen, L., Doiron, B., Feldman, D. E., Fox, K., et al. (2017). Integrating hebbian and homeostatic plasticity: the current state of the field and future research directions. Philos. Trans. R. Soc. Lond. B Biol. Sci. 372:20160158. doi: 10.1098/rstb.2016.0158
Kennedy, M. E., Stamford, A. W., Chen, X., Cox, K., Cumming, J. N., Dockendorf, M. F., et al. (2016). The BACE1 inhibitor verubecestat (MK-8931) reduces CNS β-amyloid in animal models and in Alzheimer’s disease patients. Sci. Transl. Med. 8, 363ra150–363ra150. doi: 10.1126/scitranslmed.aad9704
Kim, J., and Tsien, R. W. (2008). Synapse-specific adaptations to inactivity in hippocampal circuits achieve homeostatic gain control while dampening network reverberation. Neuron 58, 925–937. doi: 10.1016/j.neuron.2008.05.009
Kim, S., and Ziff, E. B. (2014). Calcineurin mediates synaptic scaling via synaptic trafficking of Ca2+-permeable AMPA receptors. PLoS Biol. 12:e1001900. doi: 10.1371/journal.pbio.1001900
Kinney, J. W., Bemiller, S. M., Murtishaw, A. S., Leisgang, A. M., Salazar, A. M., and Lamb, B. T. (2018). Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement. 4, 575–590. doi: 10.1016/j.trci.2018.06.014
Kitazawa, M., Yamasaki, T. R., and Laferla, F. M. (2004). Microglia as a potential bridge between the amyloid beta-peptide and tau. Ann. N. Y. Acad. Sci. 1035, 85–103. doi: 10.1196/annals.1332.006
Klintsova, A. Y., and Greenough, W. T. (1999). Synaptic plasticity in cortical systems. Curr. Opin. Neurobiol. 9, 203–208. doi: 10.1016/s0959-4388(99)80028-2
Kurup, P., Zhang, Y., Xu, J., Venkitaramani, D. V., Haroutunian, V., Greengard, P., et al. (2010). Aβ-mediated NMDA receptor endocytosis in Alzheimer’s disease involves ubiquitination of the tyrosine phosphatase STEP61. J. Neurosci. 30, 5948–5957. doi: 10.1523/JNEUROSCI.0157-10.2010
Lee, H. K., Barbarosie, M., Kameyama, K., Bear, M. F., and Huganir, R. L. (2000). Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature 405, 955–959. doi: 10.1038/35016089
Lee, K. J., Moussa, C. E.-H., Lee, Y., Sung, Y., Howell, B. W., Turner, R. S., et al. (2010). Beta amyloid-independent role of amyloid precursor protein in generation and maintenance of dendritic spines. Neuroscience 169, 344–356. doi: 10.1016/j.neuroscience.2010.04.078
Lesné, S., Ali, C., Gabriel, C., Croci, N., Mackenzie, E. T., Glabe, C. G., et al. (2005). NMDA receptor activation inhibits alpha-secretase and promotes neuronal amyloid-beta production. J. Neurosci. 25, 9367–9377. doi: 10.1523/jneurosci.0849-05.2005
Letzkus, J. J., Wolff, S. B., and Lüthi, A. (2015). Disinhibition, a circuit mechanism for associative learning and memory. Neuron 88, 264–276. doi: 10.1016/j.neuron.2015.09.024
Li, L., Gainey, M. A., Goldbeck, J. E., and Feldman, D. E. (2014). Rapid homeostasis by disinhibition during whisker map plasticity. Proc. Natl. Acad. Sci. U S A 111, 1616–1621. doi: 10.1073/pnas.1312455111
Luo, Y., Bolon, B., Kahn, S., Bennett, B. D., Babu-Khan, S., Denis, P., et al. (2001). Mice deficient in BACE1, the Alzheimer’s beta-secretase, have normal phenotype and abolished beta-amyloid generation. Nat. Neurosci. 4, 231–232. doi: 10.1038/85059
Magara, F., Müller, U., Li, Z.-W., Lipp, H.-P., Weissmann, C., Stagljar, M., et al. (1999). Genetic background changes the pattern of forebrain commissure defects in transgenic mice underexpressing the β-amyloid-precursor protein. Proc. Natl. Acad. Sci. U S A 96, 4656–4661. doi: 10.1073/pnas.96.8.4656
Malinow, R., and Malenka, R. C. (2002). AMPA receptor trafficking and synaptic plasticity. Annu. Rev. Neurosci. 25, 103–126. doi: 10.1146/annurev.neuro.25.112701.142758
Marder, E., and Prinz, A. A. (2003). Current compensation in neuronal homeostasis. Neuron 37, 2–4. doi: 10.1016/s0896-6273(02)01173-x
Meziane, H., Dodart, J. C., Mathis, C., Little, S., Clemens, J., Paul, S. M., et al. (1998). Memory-enhancing effects of secreted forms of the beta-amyloid precursor protein in normal and amnestic mice. Proc. Natl. Acad. Sci. U S A 95, 12683–12688. doi: 10.1073/pnas.95.21.12683
Miller, K. D., and Mackay, D. J. (1994). The role of constraints in Hebbian learning. Neural Compu. 6, 100–126. doi: 10.1162/neco.1994.6.1.100
Monday, H. R., Younts, T. J., and Castillo, P. E. (2018). Long-term plasticity of neurotransmitter release: emerging mechanisms and contributions to brain function and disease. Ann. Rev. Neurosci. 41, 299–322. doi: 10.1146/annurev-neuro-080317-062155
Mu, Y., Zhao, C., and Gage, F. H. (2011). Dopaminergic modulation of cortical inputs during maturation of adult-born dentate granule cells. J. Neurosci. 31, 4113–4123. doi: 10.1523/jneurosci.4913-10.2011
Müller, U. C., Deller, T., and Korte, M. (2017). Not just amyloid: physiological functions of the amyloid precursor protein family. Nat. Rev. Neurosci. 18, 281–298. doi: 10.1038/nrn.2017.29
Müller-Hill, B., and Beyreuther, K. (1989). Molecular biology of Alzheimer’s disease. Annu. Rev. Biochem. 58, 287–307. doi: 10.1146/annurev.bi.58.070189.001443
Nieweg, K., Andreyeva, A., Van Stegen, B., Tanriöver, G., and Gottmann, K. (2015). Alzheimer’s disease-related amyloid-β induces synaptotoxicity in human iPS cell-derived neurons. Cell Death Dis. 6, e1709–e1709. doi: 10.1038/cddis.2015.72
Nitsch, R. M., Slack, B. E., Wurtman, R. J., and Growdon, J. H. (1992). Release of Alzheimer amyloid precursor derivatives stimulated by activation of muscarinic acetylcholine receptors. Science 258, 304–307. doi: 10.1126/science.1411529
O’Brien, R. J., and Wong, P. C. (2011). Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 34, 185–204. doi: 10.1146/annurev-neuro-061010-113613
Opazo, P., Viana Da Silva, S., Carta, M., Breillat, C., Coultrap, S. J., Grillo-Bosch, D., et al. (2018). CaMKII metaplasticity drives Aβ oligomer-mediated synaptotoxicity. Cell Rep. 23, 3137–3145. doi: 10.1016/j.celrep.2018.05.036
Otani, S., Daniel, H., Roisin, M. P., and Crepel, F. (2003). Dopaminergic modulation of long-term synaptic plasticity in rat prefrontal neurons. Cereb. Cortex 13, 1251–1256. doi: 10.1093/cercor/bhg092
Perez, R. G., Zheng, H., Van Der Ploeg, L. H., and Koo, E. H. (1997). The β-amyloid precursor protein of Alzheimer’s disease enhances neuron viability and modulates neuronal polarity. J. Neurosci. 17, 9407–9414. doi: 10.1523/jneurosci.17-24-09407.1997
Petzoldt, A. G., Lützkendorf, J., and Sigrist, S. J. (2016). Mechanisms controlling assembly and plasticity of presynaptic active zone scaffolds. Curr. Opin. Neurobiol. 39, 69–76. doi: 10.1016/j.conb.2016.04.009
Phillips, H. S., Hains, J. M., Armanini, M., Laramee, G. R., Johnson, S. A., and Winslow, J. W. (1991). BDNF mRNA is decreased in the hippocampus of individuals with Alzheimer’s disease. Neuron 7, 695–702. doi: 10.1016/0896-6273(91)90273-3
Pozo, K., and Goda, Y. (2010). Unraveling mechanisms of homeostatic synaptic plasticity. Neuron 66, 337–351. doi: 10.1016/j.neuron.2010.04.028
Rannals, M. D., and Kapur, J. (2011). Homeostatic strengthening of inhibitory synapses is mediated by the accumulation of GABA(A) receptors. J. Neurosci. 31, 17701–17712. doi: 10.1523/jneurosci.4476-11.2011
Ring, S., Weyer, S. W., Kilian, S. B., Waldron, E., Pietrzik, C. U., Filippov, M. A., et al. (2007). The secreted beta-amyloid precursor protein ectodomain APPs alpha is sufficient to rescue the anatomical, behavioral and electrophysiological abnormalities of APP-deficient mice. J. Neurosci. 27, 7817–7826. doi: 10.1523/JNEUROSCI.1026-07.2007
Roberds, S. L., Anderson, J., Basi, G., Bienkowski, M. J., Branstetter, D. G., Chen, K. S., et al. (2001). BACE knockout mice are healthy despite lacking the primary beta-secretase activity in brain: implications for Alzheimer’s disease therapeutics. Hum. Mol. Genet. 10, 1317–1324. doi: 10.1093/hmg/10.12.1317
Roselli, F., Livrea, P., and Almeida, O. F. (2011). CDK5 is essential for soluble amyloid β-induced degradation of GKAP and remodeling of the synaptic actin cytoskeleton. PLoS One 6:e23097. doi: 10.1371/journal.pone.0023097
Roselli, F., Tirard, M., Lu, J., Hutzler, P., Lamberti, P., Livrea, P., et al. (2005). Soluble β-amyloid1–40 induces NMDA-dependent degradation of postsynaptic density-95 at glutamatergic synapses. J. Neurosci. 25, 11061–11070. doi: 10.1523/JNEUROSCI.3034-05.2005
Rozov, A. V., Valiullina, F. F., and Bolshakov, A. P. (2017). Mechanisms of long-term plasticity of hippocampal GABAergic synapses. Biochemistry 82, 257–263. doi: 10.1134/S0006297917030038
Rutherford, L. C., Nelson, S. B., and Turrigiano, G. G. (1998). BDNF has opposite effects on the quantal amplitude of pyramidal neuron and interneuron excitatory synapses. Neuron 21, 521–530. doi: 10.1016/s0896-6273(00)80563-2
Samidurai, M., Ramasamy, V. S., and Jo, J. (2018). β-amyloid inhibits hippocampal LTP through TNFR/IKK/NF-kappaB pathway. Neurol. Res. 40, 268–276. doi: 10.1080/01616412.2018.1436872
Scheefhals, N., and MacGillavry, H. D. (2018). Functional organization of postsynaptic glutamate receptors. Mol. Cell. Neurosci. 91, 82–94. doi: 10.1016/j.mcn.2018.05.002
Seabrook, G. R., Smith, D. W., Bowery, B. J., Easter, A., Reynolds, T., Fitzjohn, S. M., et al. (1999). Mechanisms contributing to the deficits in hippocampal synaptic plasticity in mice lacking amyloid precursor protein. Neuropharmacology 38, 349–359. doi: 10.1016/s0028-3908(98)00204-4
Sheynikhovich, D., Otani, S., and Arleo, A. (2013). Dopaminergic control of long-term depression/long-term potentiation threshold in prefrontal cortex. J. Neurosci. 33, 13914–13926. doi: 10.1523/jneurosci.0466-13.2013
Shin, S. M., Zhang, N., Hansen, J., Gerges, N. Z., Pak, D. T., Sheng, M., et al. (2012). GKAP orchestrates activity-dependent postsynaptic protein remodeling and homeostatic scaling. Nat. Neurosci. 15:1655. doi: 10.1038/nn.3259
Song, I., and Huganir, R. L. (2002). Regulation of AMPA receptors during synaptic plasticity. Trends Neurosci. 25, 578–588. doi: 10.1016/s0166-2236(02)02270-1
Sprekeler, H. (2017). Functional consequences of inhibitory plasticity: homeostasis, the excitation-inhibition balance and beyond. Curr. Opin. Neurobiol. 43, 198–203. doi: 10.1016/j.conb.2017.03.014
Stellwagen, D., and Malenka, R. C. (2006). Synaptic scaling mediated by glial TNF-α. Nature 440, 1054–1059. doi: 10.1038/nature04671
Strehl, A., Galanis, C., Radic, T., Schwarzacher, S. W., Deller, T., and Vlachos, A. (2018). Dopamine modulates homeostatic excitatory synaptic plasticity of immature dentate granule cells in entorhino-hippocampal slice cultures. Front. Mol. Neurosci. 11:303. doi: 10.3389/fnmol.2018.00303
Styr, B., and Slutsky, I. (2018). Imbalance between firing homeostasis and synaptic plasticity drives early-phase Alzheimer’s disease. Nat. Neurosci. 21, 463–473. doi: 10.1038/s41593-018-0080-x
Sun, Q., and Turrigiano, G. G. (2011). PSD-95 and PSD-93 play critical but distinct roles in synaptic scaling up and down. J. Neurosci. 31, 6800–6808. doi: 10.1523/jneurosci.5616-10.2011
Swanwick, C. C., Murthy, N. R., and Kapur, J. (2006). Activity-dependent scaling of GABAergic synapse strength is regulated by brain-derived neurotrophic factor. Mol. Cell. Neurosci. 31, 481–492. doi: 10.1016/j.mcn.2005.11.002
Taylor, C. J., Ireland, D. R., Ballagh, I., Bourne, K., Marechal, N. M., Turner, P. R., et al. (2008). Endogenous secreted amyloid precursor protein-alpha regulates hippocampal NMDA receptor function, long-term potentiation and spatial memory. Neurobiol. Dis. 31, 250–260. doi: 10.1016/j.nbd.2008.04.011
Tononi, G., and Cirelli, C. (2014). Sleep and the price of plasticity: from synaptic and cellular homeostasis to memory consolidation and integration. Neuron 81, 12–34. doi: 10.1016/j.neuron.2013.12.025
Townsend, M., Mehta, T., and Selkoe, D. J. (2007). Soluble Abeta inhibits specific signal transduction cascades common to the insulin receptor pathway. J. Biol. Chem. 282, 33305–33312. doi: 10.1074/jbc.m610390200
Turner, P. R., O’connor, K., Tate, W. P., and Abraham, W. C. (2003). Roles of amyloid precursor protein and its fragments in regulating neural activity, plasticity and memory. Prog. Neurobiol. 70, 1–32. doi: 10.1016/s0301-0082(03)00089-3
Turrigiano, G. (2012). Homeostatic synaptic plasticity: local and global mechanisms for stabilizing neuronal function. Cold Spring Harb. Perspect. Biol. 4:a005736. doi: 10.1101/cshperspect.a005736
Turrigiano, G. G. (2008). The self-tuning neuron: synaptic scaling of excitatory synapses. Cell 135, 422–435. doi: 10.1016/j.cell.2008.10.008
Turrigiano, G. G., and Nelson, S. B. (2004). Homeostatic plasticity in the developing nervous system. Nat. Rev. Neurosci. 5, 97–107. doi: 10.1038/nrn1327
Turrigiano, G. G., Leslie, K. R., Desai, N. S., Rutherford, L. C., and Nelson, S. B. (1998). Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature 391, 892–896. doi: 10.1038/36103
Tyan, S. H., Shih, A. Y., Walsh, J. J., Maruyama, H., Sarsoza, F., Ku, L., et al. (2012). Amyloid precursor protein (APP) regulates synaptic structure and function. Mol. Cell Neurosci. 51, 43–52. doi: 10.1016/j.mcn.2012.07.009
Van Der Kant, R., and Goldstein, L. S. (2015). Cellular functions of the amyloid precursor protein from development to dementia. Dev. Cell 32, 502–515. doi: 10.1016/j.devcel.2015.01.022
Vassar, R., Bennett, B. D., Babu-Khan, S., Kahn, S., Mendiaz, E. A., Denis, P., et al. (1999). β-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286, 735–741. doi: 10.1126/science.286.5440.735
Vitureira, N., and Goda, Y. (2013). Cell biology in neuroscience: the interplay between Hebbian and homeostatic synaptic plasticity. J. Cell Biol. 203, 175–186. doi: 10.1083/jcb.201306030
Vlachos, A., Ikenberg, B., Lenz, M., Becker, D., Reifenberg, K., Bas-Orth, C., et al. (2013). Synaptopodin regulates denervation-induced homeostatic synaptic plasticity. Proc. Natl. Acad. Sci. U S A 110, 8242–8247. doi: 10.1073/pnas.1213677110
Vnencak, M., Scholvinck, M. L., Schwarzacher, S. W., Deller, T., Willem, M., and Jedlicka, P. (2019). Lack of beta-amyloid cleaving enzyme-1 (BACE1) impairs long-term synaptic plasticity but enhances granule cell excitability and oscillatory activity in the dentate gyrus in vivo. Brain Struct. Funct. 224, 1279–1290. doi: 10.1007/s00429-019-01836-6
Weyer, S. W., Zagrebelsky, M., Herrmann, U., Hick, M., Ganss, L., Gobbert, J., et al. (2014). Comparative analysis of single and combined APP/APLP knockouts reveals reduced spine density in APP-KO mice that is prevented by APPsalpha expression. Acta Neuropathol. Commun. 2:36. doi: 10.1186/2051-5960-2-36
Wondolowski, J., and Dickman, D. (2013). Emerging links between homeostatic synaptic plasticity and neurological disease. Front. Cell. Neurosci. 7:223. doi: 10.3389/fncel.2013.00223
Yan, R., and Vassar, R. (2014). Targeting the β secretase BACE1 for Alzheimer’s disease therapy. Lancet Neurol. 13, 319–329. doi: 10.1016/S1474-4422(13)70276-X
Yee, A. X., Hsu, Y. T., and Chen, L. (2017). A metaplasticity view of the interaction between homeostatic and Hebbian plasticity. Philos. Trans. R. Soc. Lond. B Biol. Sci. 372:20160155. doi: 10.1098/rstb.2016.0155
Youn, Y. C., Kang, S., Suh, J., Park, Y. H., Kang, M. J., Pyun, J. M., et al. (2019). Blood amyloid-beta oligomerization associated with neurodegeneration of Alzheimer’s disease. Alzheimers. Res. Ther. 11:40. doi: 10.1186/s13195-019-0499-7
Zenke, F., Hennequin, G., and Gerstner, W. (2013). Synaptic plasticity in neural networks needs homeostasis with a fast rate detector. PLoS Comput. Biol. 9:e1003330. doi: 10.1371/journal.pcbi.1003330
Zhao, D., Watson, J. B., and Xie, C.-W. (2004). Amyloid β prevents activation of calcium/calmodulin-dependent protein kinase II and AMPA receptor phosphorylation during hippocampal long-term potentiation. J. Neurophysiol. 92, 2853–2858. doi: 10.1152/jn.00485.2004
Zhao, X., Huang, L., Guo, R., Liu, Y., Zhao, S., Guan, S., et al. (2017). Coordinated plasticity among glutamatergic and GABAergic neurons and synapses in the barrel cortex is correlated to learning efficiency. Front. Cell Neurosci. 11:221. doi: 10.3389/fncel.2017.00221
Keywords: hebbian plasticity, homeostatic plasticity, synaptic scaling, amyloid precursor protein, BACE1, APPsα, amyloid-β
Citation: Galanis C and Vlachos A (2020) Hebbian and Homeostatic Synaptic Plasticity—Do Alterations of One Reflect Enhancement of the Other? Front. Cell. Neurosci. 14:50. doi: 10.3389/fncel.2020.00050
Received: 04 November 2019; Accepted: 21 February 2020;
Published: 18 March 2020.
Edited by:
Lorenzo A. Cingolani, Italian Institute of Technology, ItalyReviewed by:
C. Andrew Frank, The University of Iowa, United StatesHee Jung Chung, University of Illinois at Urbana-Champaign, United States
David Stellwagen, McGill University, Canada
Copyright © 2020 Galanis and Vlachos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andreas Vlachos, YW5kcmVhcy52bGFjaG9zQGFuYXQudW5pLWZyZWlidXJnLmRl