 Ya-Shuo Feng
Ya-Shuo Feng Zi-Xuan Tan1
Zi-Xuan Tan1 Man-Man Wang
Man-Man Wang Ying Xing
Ying Xing Feng Zhang
Feng Zhang- 1Department of Rehabilitation Medicine, The Third Hospital of Hebei Medical University, Shijiazhuang, China
- 2Department of Clinical Laboratory Medicine, The Third Hospital of Hebei Medical University, Shijiazhuang, China
- 3Hebei Provincial Orthopedic Biomechanics Key Laboratory, The Third Hospital of Hebei Medical University, Shijiazhuang, China
Stroke is one of the major devastating diseases with no effective medical therapeutics. Because of the high rate of disability and mortality among stroke patients, new treatments are urgently required to decrease brain damage following a stroke. In recent years, the inflammasome is a novel breakthrough point that plays an important role in the stroke, and the inhibition of inflammasome may be an effective method for stroke treatment. Briefly, inflammasome is a multi-protein complex that causes activation of caspase-1 and subsequent production of pro-inflammatory factors including interleukin (IL)-18 and IL-1β. Among them, the NLRP3 inflammasome is the most typical inflammasome, which can detect cell damage and mediate inflammatory response to tissue damage in ischemic stroke. The NLRP3 inflammasome has become a key mediator of post-ischemic inflammation, leading to a cascade of inflammatory reactions and cell death eventually. Thus, NLRP3 inflammasome is an ideal therapeutic target due to its important role in the inflammatory response after ischemic stroke. In this mini review article, we will summarize the structure, assembly, and regulation of NLRP3 inflammasome, the role of NLRP3 inflammasome in ischemic stroke, and several treatments targeting NLRP3 inflammasome in ischemic stroke. The further understanding of the mechanism of NLRP3 inflammasome in patients with ischemic stroke will provide novel targets for the treatment of cerebral ischemic stroke patients.
Introduction
Nowadays, stroke is a major reason for long-term disability and death worldwide, which can lead to a heavy burden on patients and the whole society, especially in low- and middle-income countries (Feigin et al., 2017; Lapchak and Zhang, 2017). It is estimated that one in four adults will experience a stroke in the course of life, and there are at least 80 million stroke survivors worldwide (Feigin et al., 2018; Lindsay et al., 2019). Stroke can cause immediate neurological dysfunction, and even in severe patients, the resultant mass effect and cerebral edema can lead to cerebral herniation and death (Shi et al., 2019). According to a report, death caused by stroke accounted for 11.8% of all deaths in 2015, which is the second leading cause of death in the world next to heart disease (Benjamin et al., 2018). There are many risk factors related to the occurrence of stroke. Recent gene studies on single-gene disorders have shown that common variants at about 35 genetic loci are associated with stroke risk (Dichgans et al., 2019). Besides, a variety of environmental risk factors for stroke have been reported, such as hypertension, smoking, high body mass index (BMI), atrial fibrillation, diabetes, history of stroke and high cholesterol (Donnan et al., 2008; Lu Y. et al., 2014; Hägg et al., 2015). Among these risk factors, hypertension is one of the most leading causes of stroke, accounting for 35% of all strokes (O’Donnell et al., 2010). Furthermore, more than 90% of the stroke burden is attributed to modifiable risk factors, and effective control of metabolic and behavioral risk factors can prevent more than three-quarters of the stroke burden worldwide (Feigin et al., 2016).
Clinically, there are two types of stroke: ischemic stroke and hemorrhagic stroke. Ischemic stroke caused by cerebral artery embolization or thromboembolism, which usually accounts for about 80% of all strokes, while hemorrhagic stroke caused by rupture of the brain’s blood vessel, which accounts for about 15% to 20% of all stroke cases (Gilgun-Sherki et al., 2002). In this mini review article, we focus on ischemic stroke because the incidence of ischemic stroke is much higher than other types of stroke. Among all types of ischemic stroke, focal ischemic stroke is the most common type, which is caused by a cerebral aortic embolization or thrombotic occlusion (transient or permanent) that results in decreased blood flow to a specific area of the brain (McAuley, 1995; Hata et al., 2000; Fann et al., 2013a). Inadequate blood supply can cause cerebral cells to lose essential glucose and oxygen, disrupting the balance of the intracellular environment and triggering pathophysiological processes such as oxidative stress, excitatory toxicity, apoptosis, inflammation, and cell death (Khoshnam et al., 2017).
Specifically, pathophysiology following ischemic stroke is a series of complicated processes, including acidosis, excitotoxicity, bioenergetic dysfunction, destruction of the blood-brain barrier (BBB), toxicity mediated by reactive oxygen species (ROS), infiltration of leukocytes, cytokine-mediated cytotoxicity, loss of cellular ion homeostasis, and production of arachidonic acid products and activation of complement (Woodruff et al., 2011). These various pathophysiological processes trigger each other to form a positive feedback loop, leading to the death of neuronal cell and brain damage eventually (Siesjo, 1992). Among diverse potential mechanisms of stroke, oxidative stress and inflammation are involved in the pathogenesis of cerebral ischemia-reperfusion (I/R) injury, and appropriate regulation of inflammation may have an important effect on the prevention and treatment of stroke (Ahmad et al., 2014).
The treatment of ischemic stroke is based on the re-establishment of blood flow in the ischemic region (Hong et al., 2019). However, the re-establishment process of blood flow can result in further injury to ischemic tissue via infiltration of neutrophils, deregulation of cell ion homeostasis, accumulation of ROS, and subsequent inflammatory response leading to cell death (Minutoli et al., 2016). Nowadays, effective treatments for acute ischemic stroke include intravenous injection of tissue-type plasminogen activator (tPA) and endovascular therapy (Schwamm et al., 2013; Yoshimura et al., 2018). However, tPA treatment has limitations because the treatment window is narrow (Schwamm et al., 2013). Besides, endovascular therapy has been reported to be effective for acute cerebral large-vascular occlusion, but its actual effect is unclear (Yoshimura et al., 2018). Based on the above mentioned issues, it is crucial to find an effective and reliable therapy method for ischemic stroke.
In recent, researchers have recognized a new inflammasome signaling pathway—NOD-like receptor pyrin domain containing 3 (NLRP3) inflammasome that may be a crucial mediator in detecting cell injury and mediating inflammation following stroke (Abulafia et al., 2009; Savage et al., 2012; Gustin et al., 2015). Therefore, treatments aiming at NLRP3 upstream and downstream signaling pathways may provide new strategies for treating stroke (Fann et al., 2013b). Here, we summarize the current understanding regarding the structure, assembly, and regulation of the NLRP3 inflammasome, its potential roles in ischemic stroke, and recent treatments targeting at suppressing NLRP3 inflammasome in stroke.
NLRP3 Inflammasome: Structure, Assembly, and Regulation
The Structure of NLRP3 Inflammasome
The body’s first line of defense against a variety of diseases is the innate immune system, which is based on various pattern recognition receptors (PRRs) that sense pathogenic microorganisms and other kinds of exogenous or endogenous pathogens, such as damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs; Schroder and Tschopp, 2010; Minutoli et al., 2016). When the innate immune system is activated, inflammatory responses can be initiated by the secretion of chemokines and cytokines, resulting in the expression of co-stimulatory and adhesion molecules that can recruit immune cells and activate adaptive immune responses (Abderrazak et al., 2015). NOD-like receptors (NLRs) are a type of PRRs that are expressed primarily in the cytoplasm and can detect signals of intracellular invaders (Martinon and Tschopp, 2005). There are different types of inflammasome-forming NLRs, including NLRP1, NLRP3, NLRP6, NLRP7, NLRP12, NLRC4, NLRC5, and AIM2 (Pedra et al., 2009). Among them, NLRP3 (also termed as cryopyrin or Nalp3) is the most characteristic and closely related to sterile inflammation, which is coded by the cold-induced auto-inflammatory syndrome-1 (CIAS-1) gene and highly expressed in the immune cells and neural cells (Cassel and Sutterwala, 2010; Liu et al., 2013). As a tripartite protein, NLRP3 includes the central NACHT (also termed NOD) domain, the N-terminal pyrin domain (PYD), and the C-terminal leucine-rich repeat (LRR) domain (Franchi et al., 2009b). The LRR domain is involved in mediating autoinhibition and putative ligands, while the NACHT domain is associated with the assembly of inflammasome and self-oligomerization (Duncan et al., 2007; Lamkanfi and Dixit, 2009).
As intracellular oligomeric multiprotein complexes, inflammasomes play an important role in inducing the body’s innate immune response to microbial and injury-related signals (Franchi et al., 2009a). The inflammasome, including the sensor molecule NLR, the pro-inflammatory caspase (pro-caspase-5, pro-caspase-1, or both), and adaptive proteins, can detect various danger signals in the cell, for instance, bacterial RNA and bacterial flagellin (Martinon et al., 2002; Hong et al., 2019). Among various inflammasomes, NLRP3 inflammasome is the most widely studied and is considered to be closely related to sterile inflammation, which is mainly distributed in the cytosol (Tschopp and Schroder, 2010; Li et al., 2018). There are three sections in the NLRP3 inflammasome: the NLRP3 protein, the inflammatory caspase-1, and the adapter protein ASC [Apoptosis-associated Speck-like protein containing a caspase activation recruitment domain (CARD); Abderrazak et al., 2015]. Full-length caspase-1 includes the central large catalytic domain (p20), the C-terminal small catalytic subunit domain (p10), and the N-terminal CARD (Swanson et al., 2019). Besides, ASC contains two protein interaction domains, including the N-terminal PYD and the C-terminal CARD (Swanson et al., 2019).
The Assembly of NLRP3 Inflammasome
As is shown in Figure 1, the activation of the NLRP3 inflammasome is considered as a two-stage process. The first stage, known as the priming stage, is induced by the recognition of PAMPs and DAMPs (Wang et al., 2020). This causes the activation of the NF-κB signaling pathway and promotes the expression of precursor proteins, including the NLRP3, pro-IL-1β, and pro-IL-18 (Shao et al., 2019). The second stage is the activation stage, which is induced by a series of stimulation that occurs during tissue injury, infections, or metabolic imbalances (Zhao and Zhao, 2020). During this stage, stimulation like K+ efflux, Na+ influx, Ca2+ mobilization, chloride efflux, lysosomal damage, ROS, and mitochondrial dysfunction can cause the assembly of the NLRP3 inflammasome (Kelley et al., 2019).
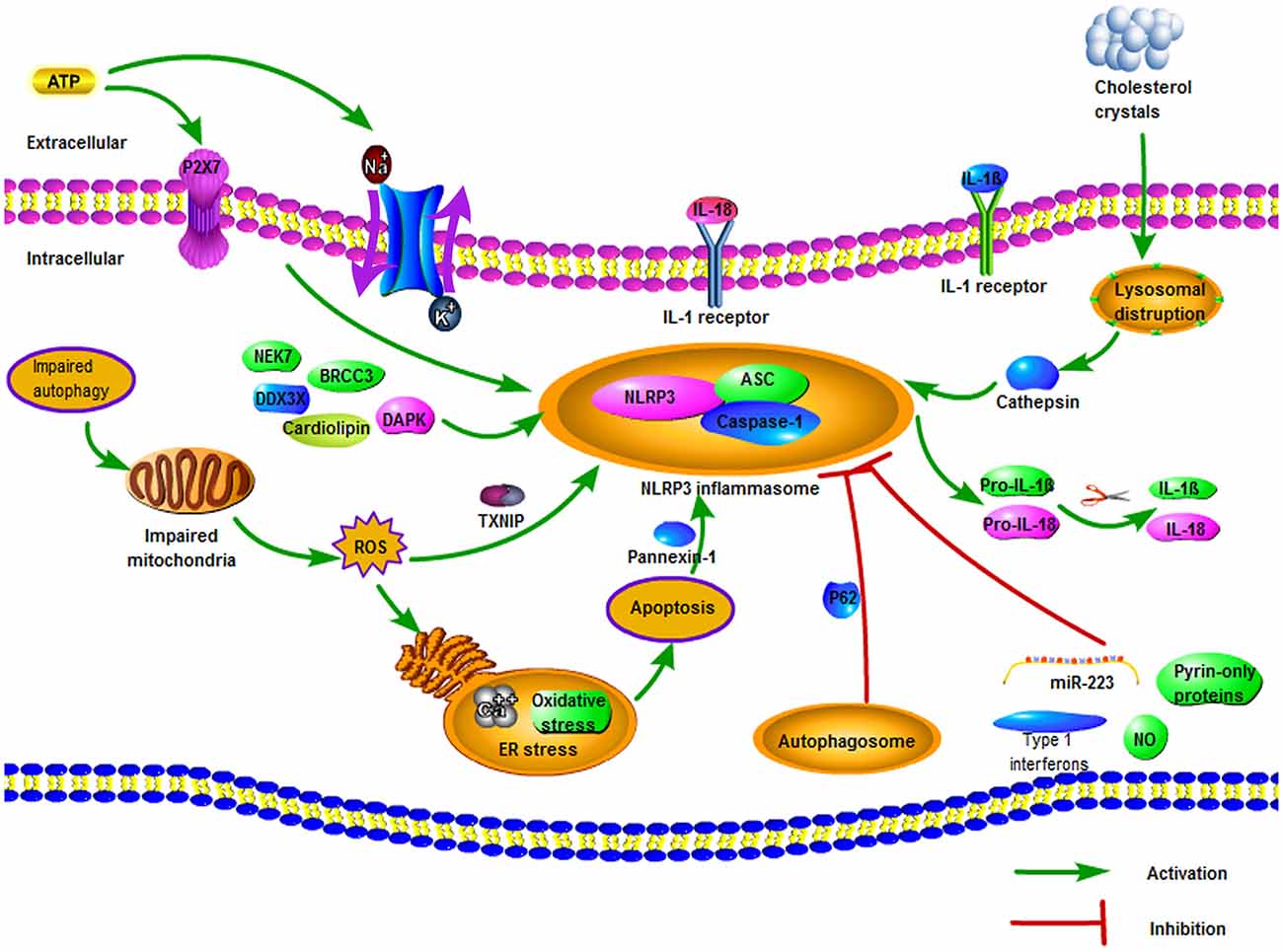
Figure 1. The regulation of NOD-like receptor pyrin domain containing 3 (NLRP3) inflammasome. There are three potential stimuli for NLRP3 inflammasome activation, including reduced intracellular K+ concentration, mitochondrial reactive oxygen species (ROS), and lysosomal membrane destruction. Extracellular ATP can promote K+ efflux via affecting the function of Na+/K+-ATPase pump and triggering autocrine and paracrine P2X7. Impaired autophagy can increase ROS levels, thereby activating NLRP3 inflammasome and resulting in secretion of Ca2+ and oxidative stress. Various particulates, such as the cholesterol crystals of atherosclerotic plaque, can disrupt the lysosomal membrane and deliver cathepsin into the cytoplasm, hence activating the NLRP3 inflammasome. Besides, several negative regulation mechanisms of NLRP3 inflammasome are also demonstrated, including autophagy, nitric oxide (NO), miR-223, type I interferons, and pyrin-only proteins.
The assembly of NLRP3 inflammasome is initiated by interaction between the pyrin domain of ASC and the pyrin domain of NLRP3 (Vajjhala et al., 2012). After detecting dangerous signals, NLRP3 monomers can be triggered and oligomerized to become definite oligomers (Lechtenberg et al., 2014). These ring structures can recruit ASC monomers to cause the ASC filaments or specks formation by interacting with homophile PYD–PYD (Lu A. et al., 2014). Then, ASC filaments/specks recruit cysteine proteases pro-caspase-1 for assembling inflammasome complexes through the interaction with CARD (Proell et al., 2013). And then, pro-caspase-1 autocatalyzes itself via cleavage and activation into active caspase-1, resulting in the subsequent processing of pro-IL-18 and pro-IL-1β into active IL-18 and IL-1β (Ozaki et al., 2015). Furthermore, activated caspase-1 can dissociate gasdermin D (GSDMD) to release its N-terminal domain (Shi et al., 2020). The N-terminal domain of GSDMD binds to phosphatidylserine and phosphatidylinositol phosphates in the cytomembrane to form a 1,020 nm pore and triggers a lytic form of cell death, called pyroptosis (Shi et al., 2015). Pyroptosis is characterized by cytosolic swelling and early rupture of the plasma membrane, which can release DAMPs to trigger inflammatory action, playing major roles in numerous types of immune diseases (Lamkanfi and Dixit, 2010).
In addition to the canonical pathway of the NLRP3 inflammasome in activation of caspase-1, there is also the non-canonical active manner of NLRP3 activated caspase-11 in mice (or the homologs caspase-5 and caspase-4 in humans; Viganò et al., 2015; Yi, 2018). In the non-canonical pathway, caspase-11 may produce abnormal protein secretion and pyroptosis in a manner independent of caspase-1 (Tan et al., 2013). Specifically, caspase-11 can directly recognize and bind to intracellular lipopolysaccharide, leading to its oligomerization and subsequent processing of activation by autoproteolytic cleavage (Kayagaki et al., 2015). And then, caspase-11 can directly cause the cleavage of GSDMD to trigger pyroptosis (Kayagaki et al., 2015; Shi et al., 2015).
The Regulation of NLRP3 Inflammasome
Positive Regulation of NLRP3 Inflammasome
So far, the exact mechanism and cellular stimulation resulting in the activation of NLRP3 inflammasome are not clear (Gao et al., 2017). Py et al. (2013) suggested that the deubiquitination mechanism plays an important role in regulating the NLRP3 inflammasome activation and BRCC3 is a key regulator of NLRP3 activity via promoting deubiquitination. Death-associated protein kinase (DAPK) is considered as a crucial molecule, which is necessary for a full generation of IL-1β and accurate assembly of NLRP3 inflammasome (Chuang et al., 2011). Recently, it has been reported that stress granule protein DDX3X can interact with NLRP3 to activate inflammasome, and assembly of stress granule could suppress the activation of NLRP3 inflammasome via the sequestration of DDX3× (Samir et al., 2019). Besides, Sharif et al. (2019) demonstrated that NIMA-related kinase 7 (NEK7) could mediate the activation of the NLRP3 inflammasome by bridging adjacent NLRP3 subunits with bipartite interactions. Upon inflammasome activation, the interaction between NEK7 and NLRP3 is enhanced to form a complex that is critical to form ASC speck and activate caspase-1 (He et al., 2016; Shi et al., 2016). Although our understanding of NLRP3 inflammasome has gradually increased in recent years, we need more studies to further clarify the detailed mechanism of NLRP3 in the process of stroke in the future.
Different activators of the NLRP3 inflammasome complex have been reported, including exogenous (such as tissue injury, infection, and metabolic imbalance) and endogenous factors (such as Aβ fibrils, hyaluronan, extracellular ATP, and uric acid crystals; Lamkanfi and Dixit, 2009; Koizumi et al., 2012). Because of the large amount of NLRP3 inflammasome activators, it seems unlikely that all of them will bind to the NLRP3 structure to form the NLRP3 inflammasome (Zhou et al., 2016). In general, researchers have reported three potential stimuli for the activation of the NLRP3 receptor: decreased intracellular K+ concentration, mitochondrial ROS, and lysosomal membrane destruction (Martinon et al., 2002; Garg, 2011).
Decreased Intracellular K+ Concentration
In most cases, decreased intracellular K+ concentration is an essential upstream event during the activation of NLRP3 inflammasome (Swanson et al., 2019). Extracellular ATP is a typical danger signal to activate NLRP3 inflammasome, which can affect the function of Na+/K+-ATPase pump, and the increased Na+ influx through aquaporin promotes the osmotic movement of water into the intracellular environment, resulting in K+ efflux (Mongin, 2007; Li et al., 2018). Besides, the secretion of ATP can also activate autocrine and paracrine P2X7, resulting in the reduction of K+ concentration in the cytoplasm, which activates NLRP3 inflammasome (Lamkanfi et al., 2009). Furthermore, the reduction of intracellular K+ level could result in the activation of NLRP3 inflammasome through pore-forming bacterial toxins or endogenous ion channels (Pétrilli et al., 2007). Additionally, the K+ channel inhibitor glibenclamide effectively inhibited the activation of inflammasome to many NLRP3 activators (Lamkanfi and Dixit, 2009).
Mitochondrial ROS
ROS has been shown to exert important effects on the activation of NLRP3 inflammasomes, which was mainly related to the function of mitochondria (Gross et al., 2011). ROS is primarily produced in the mitochondrial inner membrane, which is closely associated with the enzyme complex in the mitochondrial respiratory chain (Liu et al., 2002). Approximately 2% of the oxygen in normal mitochondria can be converted into ROS (Wang S. et al., 2019). Under normal physical conditions, the small amounts of ROS can be removed by several endogenous antioxidant systems (Sanderson et al., 2013). However, when mitochondria are impaired or oxygen supply is insufficient, a large number of ROS can be produced, further aggravating the damage of mitochondrial structure and function (Turrens, 2003). Increased level of ROS can induce oxidative stress and release of Ca2+, cause endoplasmic reticulum (ER) stress, cellular organelles injury, and lead to apoptosis eventually (Gao et al., 2017). Autophagy (i.e., mitophagy) can remove the ROS-producing mitochondria to protect cells, but this process may not occur effectively because autophagy-related proteins such as Beclin1 and microtubule-associated protein 1 light chain 3B (LC3B) are depleted in case of cellular stress and ischemic brain injury (Tian et al., 2010; Nakahira et al., 2011). Thus, impaired autophagy promotes the accumulation of impaired mitochondria in the cytoplasm, thereby increasing ROS levels that activate the NLRP3 receptor (Nakahira et al., 2011; Zhou et al., 2011). Also, the thioredoxin-interacting protein (TXNIP), as a ROS-sensitive regulator, could activate NLRP3 inflammasome (Zhou et al., 2010). In unaffected cells, TXNIP is bound to and inhibited by the oxidoreductase thioredoxin (Fann et al., 2013a). With an increased level of cytoplasmic ROS, this complex begins to dissociate and causes the TXNIP to bind to the NLRP3 receptor (mainly in LRR), resulting in activation of NLRP3 receptor (Zhou et al., 2010; Lane et al., 2013).
Lysosomal Membrane Destruction
Lysosomal disruption is also one of the well-known mechanisms of NLRP3 inflammasome activation (Li et al., 2018). NLRP3 inflammasome is activated by cathepsin delivered into cytoplasm because of lysosomal membrane damage, which is triggered by crystalline or particulate structure (Gao et al., 2017). Phagocytosis of a variety of particulates, whether self-originated particulates such as cholesterol crystals and uric acid or foreign-originated particulates such as silica, alum, and asbestos, results in lysosomal disruption and delivering the particulates into the cytoplasm (Hornung et al., 2008). For instance, the cholesterol crystals of atherosclerotic plaque in the location of occlusion can be taken by the endosomes and fuse with the lysosomes, resulting in disruption of lysosomal membranes and release of cathepsin into the cytoplasm (Duewell et al., 2010). Besides, Yamasaki et al. (2009) demonstrated that cathepsin B or cathepsin B-modified proteins are necessary for NLRP3 inflammasome activation; and the Cathepsin B inhibitor, CA-074-Me, could partially suppress NLRP3 activation (Dostert et al., 2008; Bruchard et al., 2013). Furthermore, Okada et al. (2014) suggested that the TAK1-JNK pathway was triggered via lysosome rupture and that this activation played an important role in the NLRP3 inflammasome formation via the ASC oligomerization.
Other Mechanisms
There are several other mechanisms involved in the activation of the NLRP3 inflammasome. The ER is the main intracellular organelle for protein synthesis and processing, and the main calcium reservoir for maintaining calcium homeostasis (Bauernfeind et al., 2011; McCaffrey and Braakman, 2016). ER stress can affect the activation of NLRP3 inflammasome through a variety of effects including calcium or lipid metabolism, the unfolded protein response (UPR), and the production of ROS (Chen X. et al., 2019). Recently, Piippo et al. (2018) suggested that oxidative stress strongly promoted the activation of NLRP3 inflammasome upon dysfunctional cellular clearance. Besides, NLRP3 inflammasome can also be activated by Ca2+ mobilization regulated mitochondrial injury and dysfunction (Lee et al., 2012; Murakami et al., 2012). Mitochondria related cardiolipin also plays an important role in the recruitment and activation of NLRP3 inflammasome (Iyer et al., 2013). Also, intrinsic and extrinsic apoptosis is contributed to driving the assembly of NLRP3 inflammasome via activating pannexin-1 (Chen K. W. et al., 2019).
Negative Regulation of NLRP3 Inflammasome
A series of studies demonstrated that many factors can negatively regulate the activity of NLRP3 inflammasome. Autophagy can remove injured mitochondria, prevent mtDNA, and ROS release into the cytoplasm and block assembly of NLRP3 inflammasome (Nakahira et al., 2011). What is more, autophagosome can degrade NLRP3 inflammasome via autophagy adaptor p62 (Harris et al., 2011; Shi et al., 2012). Further, Zhou et al. (2011) reported that 3-MA, an autophagy inhibitor, was able to induce the NLRP3 inflammasome activation. Besides, nitric oxide (NO) has effects on suppressing NLRP3 inflammasome activation through the stabilization of mitochondria, in both humans and mice (Mao et al., 2013). Additionally, it has been shown that NLRP3 could be directly regulated by miR-223 because NLRP3 mRNA includes a conserved miR-223 binding region in its 3’UTR (Yang et al., 2015). Other negative regulators also have been reported, such as type I interferons and pyrin-only proteins (Guarda et al., 2011; de Almeida et al., 2015).
Involvement of the NLRP3 Inflammasome in the Pathophysiological Processes Following Ischemic Stroke
As is shown in Figure 2, several mechanisms can regulate NLRP3 inflammasome after ischemic stroke. Following ischemic stroke, the generation of ROS can activate both cerebral inflammatory reactions and NLRP3 inflammasome, triggering neuronal cell injury, brain edema, and neural dysfunction (Wang et al., 2007; Minutoli et al., 2016). Mitochondrial dysfunction also exerted a crucial role in the activation of NLRP3 inflammasome after OGD/R in microglia, and mitochondrial protector was able to suppress the NLRP3 inflammasome activation in ischemic stroke rats (Gong et al., 2018). Ishrat et al. (2015) report that TXNIP could induce the activation of the NLRP3 inflammasome, resulting in the neuronal damage after ischemic stroke. Besides, serum amyloid A can contribute to the NLRP3 inflammasome activation of microglial cells in ischemic stroke (Yu et al., 2019). Bromodomain-containing protein 4 (BRD4) also plays an important role in the activation of NLRP3 inflammasome in MCAO mice (Zhou et al., 2019). TPEN, a membrane-permeant zinc chelator, can bloke the elevated levels of NLRP3 and caspase-1, suggesting that zinc is closely related to the formation of NLRP3 inflammasome (Park et al., 2020). Furthermore, TPEN inhibits the elevation of NLRP3 inflammasome in the oxygen-glucose deprivation (OGD) model, indicating that increased zinc is essential for the NLRP3 inflammasome activation in OGD models (Park et al., 2020). Besides, mitogen-activated protein kinase (MAPK) and nuclear factor-κB (NF-κB) signaling pathways played an important role in regulating the expression and activation of NLRP3 inflammasomes in brain tissue and primary cortical neurons during ischemia (Fann et al., 2018). Additionally, an in vitro research suggested that lncRNA functional intergenic repeating RNA element (FIRRE) and NF-κB could form a positive feedback loop to facilitate NLRP3 inflammasome transcription, hence cause OGD/R injury of microglia in brain (Zang et al., 2018).
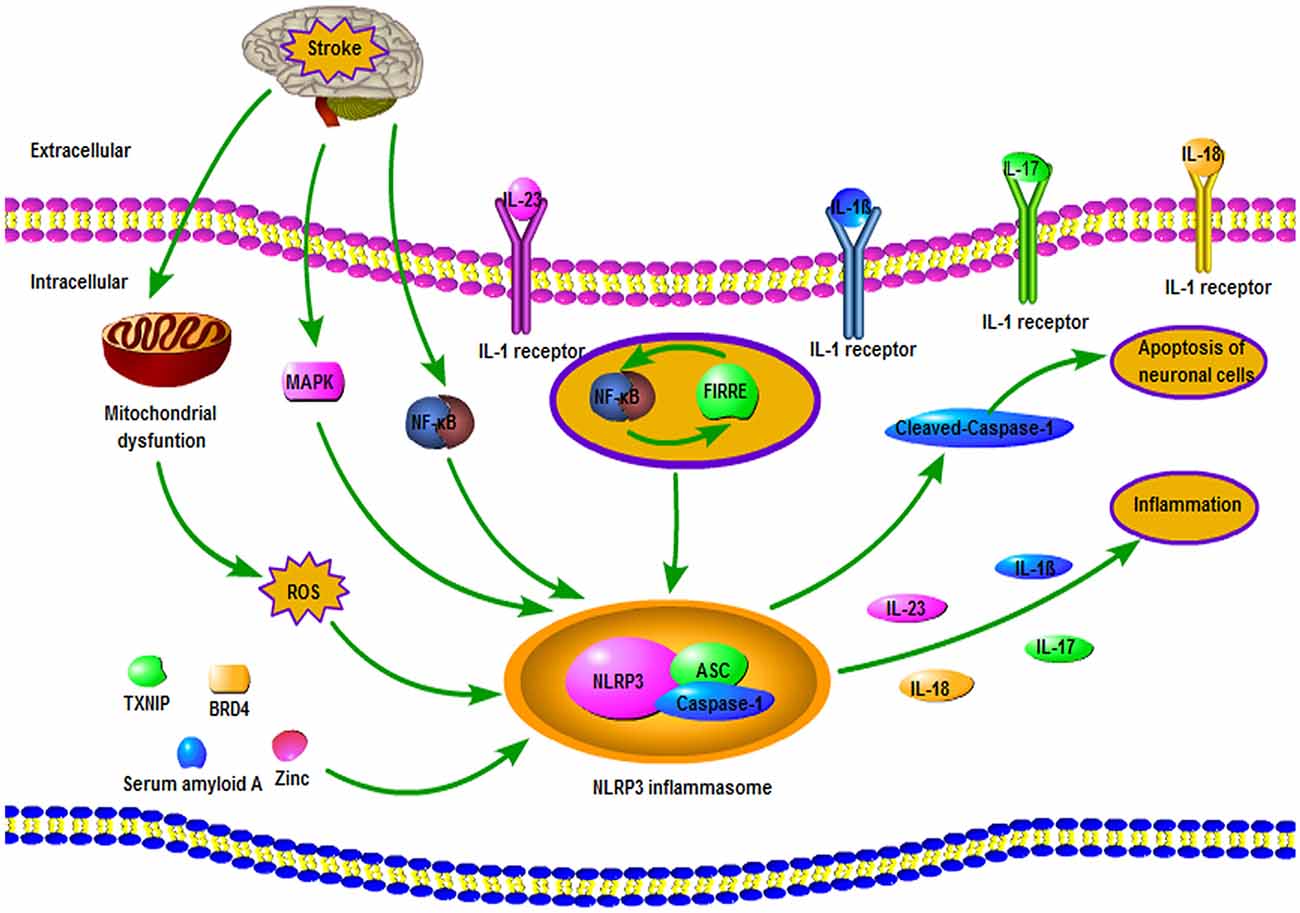
Figure 2. Involvement of the NLRP3 inflammasome in the pathophysiological processes following ischemic stroke. After a stroke, the accumulation of ROS plays an important role in the activation of the NLRP3 inflammasome. Mitogen-activated protein kinase (MAPK) and NF-κB signaling pathways are regarded as crucial mechanisms for regulating NLRP3 inflammasome. Also, a positive feedback loop formed by functional intergenic repeating RNA element (FIRRE) and NF-κB promotes the transcription of NLRP3 inflammasome. Furthermore, NLRP3 inflammasome can regulate apoptosis of neuronal cells via cleaved-caspase-1 and promote inflammation by releasing the pro-inflammatory cytokines.
Increasing evidence has suggested that NLRP3 inflammasome is a crucial mediator of neuroinflammation and plays an important role in the progression and pathogenesis of ischemic stroke (Fann et al., 2013a). Gong et al. (2018) demonstrated that the NLRP3 inflammasome was firstly activated in microglial cells after brain I/R injury onset and subsequently expressed in microvascular endothelial cells and especially in neurons (Gong et al., 2018). NLRP3 inflammasomes can regulate glial and neuronal cell death in ischemic stroke via enhancing generation and secretion of the pro-inflammatory cytokines including IL-1β and IL-18 and through pleiotropic impacts of cleaved-caspase-1 in regulating neuronal cell apoptosis (Fann et al., 2013a). Also, NLRP3 inflammasomes can cause damage to the IL-23/IL-17 axis, thus aggravating cerebral I/R injury (Wang H. et al., 2019).
The expression level changes and abnormal gene mutation of coding components of NLRP3 can impact NLRP3-regulated inflammatory response, thus disturbing the immune balance of the internal environment and the occurrence of ischemic stroke (Fann et al., 2013b; Xie et al., 2014; Yang et al., 2014). For instance, inhibition of NLRP3 significantly prevents neuronal death and reduces I/R injury in both in vitro and in vivo cerebral ischemic settings (Deroide et al., 2013; Yang et al., 2014). Besides, it was reported that polymorphism of the NLRP3 gene may affect the risk of ischemic stroke via changing plaque vulnerability in the Chinese population (Zhu et al., 2016; Cheng et al., 2018). Further, NLRP3 deficiency improved neurovascular damage in mice model following focal ischemic stroke via decreasing BBB damage and infarct volumes according to the evaluation of Evans blue permeability, magnetic resonance imaging (MRI), and electron microscopic analyses (Yang et al., 2014).
Intervention Measures and Related Mechanisms via Inhibiting NLRP3 Inflammasome in Ischemic Stroke
Medicants
Currently, there are a series of molecules that play a neuroprotective role in ischemic stroke models by inhibiting the NLRP3 inflammasome pathway. Lu et al. (2016) suggested that minocycline administrated 1 h following reperfusion improved neurological dysfunction, decreased infarct volume, and reduced cerebral edema via inhibiting activation of microglia and NLRP3 inflammasome signaling. IMM-H004, a novel coumarin derivative, could decrease the expression level of chemokine-like factor 1 (CKLF1) combining with C-C chemokine receptor 4, further inhibiting the NLRP3 inflammasome activation and inflammation, thereby exerting therapeutic effects on rats following ischemic stroke (Ai et al., 2019). Additionally, progesterone and steroids 17b-estradiol (P and E2) have positive effects on ischemic stroke. For instance, Lammerding et al. (2016) suggested that P and E2 application following cerebral ischemia decreased the expression of NLRP3, suppressed the inflammatory response, and reduced the infarct volume in transient focal ischemic rat models. Also, Zhang et al. (2017) indicated that Arctigenin pretreatment could reduce the neurological score, infarct volume, and brain water content, by suppressing the level of NLRP3, IL-1β, and IL-18 and activating SIRT1 signaling pathway. Further, these authors administrated EX527 (SIRT1 inhibitor) under oxygen-glucose deprivation (OGD) condition, and they found that EX527 could reverse the suppressive effect of Arctigenin on NLRP3 inflammasome activation, indicating that activation of SIRT1 signaling pathway plays an important role in inhibiting NLRP3 inflammasome activation induced by Arctigenin (Zhang et al., 2017). What is more, it has been reported that pretreatment with umbelliferone for 7 days could improve infarct volume, neurological outcomes, and brain edema in middle cerebral artery occlusion (MCAO) rat models by suppressing TXNIP/NLRP3 inflammasome and activating peroxisome proliferator-activated receptor-γ (PPAR-γ; Wang et al., 2015). Huang et al. (2018) demonstrated that Stachybotrys microspora triphenyl phenol-7 (SMTP-7) could reduce the expression of NLRP3, TNF-α, NF-κB, and cleaved-caspase-3 positive cells in ischemic stroke mice model. Hispidulin could reduce brain edema and infarct size, as well as provide neuroprotective effects via inhibiting NLRP3-mediated pyroptosis through regulating the AMPK/GSK3β signaling pathway both in vitro and in vivo (An et al., 2019). Also, purified anthocyanin extracts (PAEs) could decrease the cerebral infarction volume and brain damage through Toll-like receptor 4 (TLR4) /NF-κB and NLRP3 pathways (Cui et al., 2018). Besides, ruscogenin is a crucial steroid sapogenin derived from Ophiopogn japonicus, and ruscogenin could improve neurological dysfunctions of ischemic stroke mice via suppressing expression levels of NLRP3, IL-1β, caspase-1, TXNIP, MAPK, and ROS (Cao et al., 2016). Sinomenine, an effective natural anti-inflammatory, and anti-apoptotic molecule inhibited the activity of NLRP3 inflammasome in the cerebral ischemic model, and the protective effect can be reversed by AMPK inhibitors, indicating that suppressive effect of sinomenine on NLRP3 inflammasome was mediated by AMPK pathway (Qiu et al., 2016). In summary, many medicants have positive effects on improving neurological dysfunction, infarct volume, and cerebral edema in ischemic stroke model via suppressing NLRP3 pathways.
Molecular Inhibitors
MCC950
MCC950 is an NLRP3-inflammasome inhibitor that has been shown to exert positive effects on ischemic stroke models. Ismael et al. (2018) demonstrated that MCC950 could improve neurological deficits and reduce infarct volumes and edema, which was related to the suppression of cleaved-caspase-1, IL-1β, TNF-α, poly (ADP-ribose) polymerase (PARP) and cleaved-caspase-3 and paralleled less phosphorylated IκBα and NF-κB p65 expressions in ischemic stroke mouse model. Besides, Wang H. et al. (2019) suggested that MCC950 was able to inhibit the expression of IL-23 receptor and the activation of the IL-23/IL-17 axis in ischemic stroke mice model. What is more, it was also reported that MCC950 improved neurological dysfunction at 24 h after cerebral I/R and promoted 28-day survival rate in diabetic mice with ischemic stroke, involving in the mRNA transcription level changes of NLRP3, caspase-1, and IL-1β (Hong et al., 2018). Also, MCC950 exerted beneficial effects on improving the vascular integrity and cognitive dysfunction and preventing the hypoxia-regulated decrease of brain-derived neurotrophic factor (BDNF) secretion in stroke rat models with diabetes (Ward et al., 2019).
Other Molecular Inhibitors
Several other molecular inhibitors also have beneficial effects by reducing the expression of NLRP3 in ischemic stroke. NOX inhibitor apocynin and nicotinamide adenine dinucleotide phosphate (NADPH) could inhibit the level of NLRP3, ASC, caspase-1, IL-1β and IL-18 in the cortex, improve the neurological functions, and decrease the infarct volume in ischemic stroke mouse model (Qin et al., 2017). Further, the beneficial effects for the mouse model could be greatly improved by combination therapy of NADPH and NOX inhibitors (Qin et al., 2017). Nafamostat mesilate (NM), as a wide-spectrum serine protease inhibitor, has immune-modulatory impacts on ischemic stroke rats, which is related to the suppression of NLRP3 inflammasome and NF-κB signaling pathway (Li et al., 2016). NM also reduced the level of various pro-inflammatory molecules) including IL-1β, TNF-α, COX-2, and iNOS) and increased the expression of anti-inflammatory factors (including TGF-β, CD206, IL-4, and IL-10; Li et al., 2016). Besides, JQ1, a bromodomain-containing protein 4 inhibitors, has been reported to exert protective effects on ischemic stroke mice via several mechanisms, including inhibiting the expression of NLRP3, caspase-1, ASC and gasdermin D, blocking the NF-κB signaling pathway and suppressing glial activation (Zhou et al., 2019). Besides, Bruton’s tyrosine kinase (BTK), as a member of the Tec family structurally associated with spleen tyrosine kinase (Syk), which is related to ASC phosphorylation, can form ASC specks and activate AIM2 and NLRP3 inflammasomes (Hara et al., 2013). Ito et al. (2015) demonstrated that BTK inhibitor (ibrutinib) could effectively influence the activation of the NLRP3 inflammasome, attenuate infarct volume growth and improve the neurological damage, suggesting that BKT is important for the activation of NLRP3 inflammasome. Taken together, all of these molecular inhibitors can inhibit the expression of NLRP3 and have a neuroprotective effect on ischemic stroke. In Table 1, we have summarized several molecular inhibitors mentioned above that can inhibit NLRP3 inflammasome in the cerebral ischemic model.
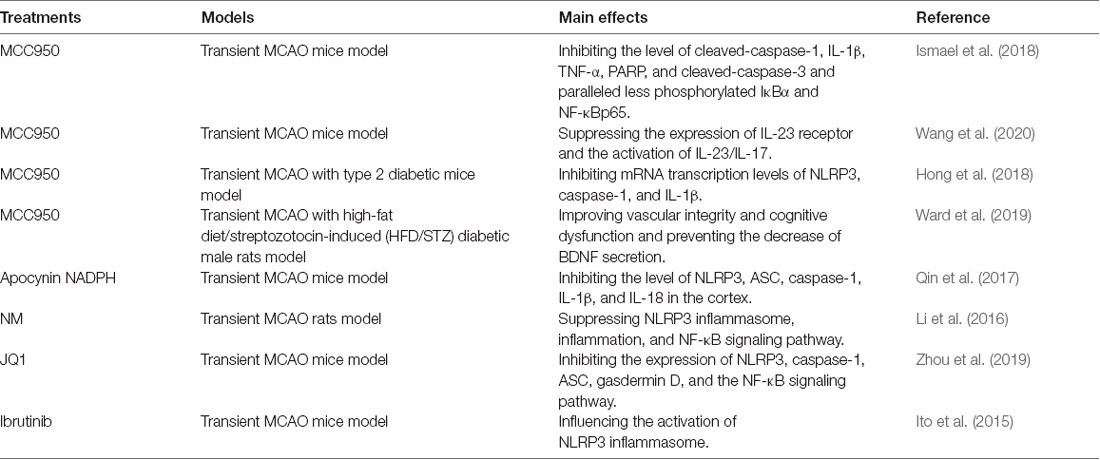
Table 1. Several molecular inhibitors via inhibiting NOD-like receptor pyrin domain containing 3 (NLRP3) inflammasome in ischemic stroke.
Intravenous Immunoglobulin
Intravenous immunoglobulin (IVIg) was used to treat a variety of inflammatory diseases, which could attenuate neuronal cell loss, apoptosis, infarct size, and improve function in ischemic stroke model (Widiapradja et al., 2012). The mechanism by which IVIg protects brain cells from ischemic injury is the inhibition of NLRP3 and NLRP1 inflammasomes, indicating that therapeutic interventions targeting inflammasome assembly and activity have obvious therapeutic benefits (Fann et al., 2013b). Furthermore, Fann et al. (2018) indicated that IVIg induced inhibition of NLRP3 and NLRP1 inflammasomes was mediated by MAPK and NF-κB signaling pathways. Besides, IVIg could also increase the expression of anti-apoptotic proteins, such as Bcl-2 and Bcl-xL (Fann et al., 2018). Therefore, IVIg is a promising therapeutic method for protecting brain cells against cerebral ischemic via inhibition of NLRP3 and NLRP1 inflammasomes, which is regulated by the MAPK and NF-κB signaling pathways.
Electroacupuncture
Electroacupuncture (EA) is a complementary and alternative medical treatment that applies electric currents to specific acupuncture points (Cai et al., 2019). It has been reported that EA treatment could decrease the inflammatory response mediated by NLRP3 inflammasome and regulate the balance between pro-inflammatory and anti-inflammatory cytokines (Jiang et al., 2019). The α-BGT, a 7nAChR antagonist, was able to reverse the EA induced suppressive effects on NLRP3 inflammasome and break the balance between pro-inflammatory and anti-inflammatory factors, suggesting that EA has neuroprotective effects on cerebral ischemic rats by regulating 7nAChR-mediated NLRP3 inflammasome (Jiang et al., 2019). Sha et al. (2019) indicated that EA remarkably reduced the neurological dysfunction and infarct volume, increased level of miR-223 and attenuated expressions of NLRP3, caspase-1, IL-18, and IL-1β. However, these beneficial effects of EA could be partially reversed by antagomiR-223, suggesting that the therapeutic effects of EA are associated with the suppression of the miR-223/NLRP3 pathway (Sha et al., 2019). In a word, EA could decrease the inflammation, infarct volume, and neurological dysfunction through suppressing the expression level of NLRP3 inflammasome mediated by 7nAChR and miR-223 in ischemic stroke.
Other Therapeutic Methods
Several other therapeutic methods have been used to treat the ischemic stroke animal models by suppressing NLRP3 inflammasome. Intermittent fasting (IF) for 4 months could suppress the inflammation and brain tissue injury in the MCAO mice model via suppressing the level of NLRP1, NLRP3, IL-1β, and IL-18, and inhibiting the up-regulation of MAPK and NF-κB signaling pathways (Fann et al., 2014). Human umbilical cord blood mononuclear cells (cbMNCs) transplantation had beneficial effects on improving the neurologic deficits, memory function and learning ability in ischemic stroke rats, which is associated with the activation of NF-κB, inhibition of NLRP3 inflammasome, increased level of vascular endothelial growth factor (VEGF) and Angiopoietin-1, and reduction of cleaved caspase-1 and mature IL-1β (Liu et al., 2018). Besides, light-emitting diode (LED) treatment could decrease neuroinflammatory reactions and brain damage after ischemic stroke via reducing cell death, decreasing IL-1β and IL-18, and inhibiting NLRP3 inflammasome, MAPK signaling, TLR-2 levels and NF-κB activation (Lee et al., 2017). Also, ketogenic diets may inhibit ER stress and protect mitochondrial integrity from ischemic brain damage via inhibiting mitochondrial transposition of dynamin-related protein 1 (Drp1), thereby suppressing activation of NLRP3 inflammasome and playing a neuroprotective role in ischemic stroke (Guo et al., 2018). Taken together, these above-mentioned treatments could improve neurological dysfunctions following ischemic stroke via inhibiting NLRP3 inflammasome.
Conclusion
In recent years, we have further deepened our understanding of the NLRP3 inflammasome. Increasing evidence has shown that inhibition of NLRP3 may significantly reduce the infarct volume and improve neurological function in cerebral ischemic animal models. In summary, the study of the effect of NLRP3 inflammasome and its potential mechanism in ischemic stroke will provide new therapeutic targets for the treatment of ischemic stroke. On the one hand, the discovery of NLRP3 inflammasome provides a new way to study the molecular mechanism of ischemic stroke. On the other hand, regulation of multiple levels of inflammation targeting NLRP3, such as its assembly, expression, and activation, may provide new ideas for saving penumbral tissue and preventing neurological deterioration after ischemia stroke.
In addition to ischemic stroke, inflammatory pathways also play an important role in the treatment of patients with cardiovascular disease (Grebe et al., 2018; Sardu et al., 2020). Numerous studies demonstrate that the activation of NLRP3 inflammasome plays a crucial role in the occurrence of cardiovascular disorders, such as atherosclerosis, hypertension, myocardial ischemia, cardiomyopathy, infectious cardiac disease, and heart failure (Pasqua et al., 2018; Wang et al., 2018). Furthermore, treatments targeting the NLRP3 inflammasome are effective for the improvement of cardiovascular disease (Wang et al., 2018). Therefore, we emphasize that there may be a correlation on the role of NLRP3 informsome between the cardiovascular and cerebrovascular networks.
However, the exact mechanisms by which NLRP3 inflammasomes perceive various activators are not fully understood and need further research in the future. Also of note, the role of NLRP3 inflammasomes in the pathogenesis of ischemic stroke is not clear and requires further investigation. Furthermore, a key focus of future research is to identify specific NLRP3 inhibitors and related mechanisms via our further study on NLRP3 inflammasome activation during a stroke. Besides, due to the complexity of the pathogenesis of ischemic stroke, it is important to determine the stages of disease at which the NLRP3 inflammasome targeted treatment is effective.
Author Contributions
Y-SF and FZ had the idea for the article. M-MW and Z-XT performed the literature search and data analysis. YX and FD drafted and critically revised the work.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abderrazak, A., Syrovets, T., Couchie, D., El Hadri, K., Friguet, B., Simmet, T., et al. (2015). NLRP3 inflammasome: from a danger signal sensor to a regulatory node of oxidative stress and inflammatory diseases. Redox Biol. 4, 296–307. doi: 10.1016/j.redox.2015.01.008
Abulafia, D. P., de Rivero Vaccari, J. P., Lozano, J. D., Lotocki, G., Keane, R. W., and Dietrich, W. D. (2009). Inhibition of the inflammasome complex reduces the inflammatory response after thromboembolic stroke in mice. J. Cereb. Blood Flow Metab. 29, 534–544. doi: 10.1038/jcbfm.2008.143
Ahmad, M., Dar, N. J., Bhat, Z. S., Hussain, A., Shah, A., Liu, H., et al. (2014). Inflammation in ischemic stroke: mechanisms, consequences and possible drug targets. CNS Neurol. Disord. Drug Targets 13, 1378–1396. doi: 10.2174/1871527313666141023094720
Ai, Q. D., Chen, C., Chu, S., Zhang, Z., Luo, Y., Guan, F., et al. (2019). IMM-H004 therapy for permanent focal ischemic cerebral injury via CKLF1/CCR4-mediated NLRP3 inflammasome activation. Transl. Res. 212, 36–53. doi: 10.1016/j.trsl.2019.05.007
An, P., Xie, J., Qiu, S., Liu, Y., Wang, J., Xiu, X., et al. (2019). Hispidulin exhibits neuroprotective activities against cerebral ischemia reperfusion injury through suppressing NLRP3-mediated pyroptosis. Life Sci. 232:116599. doi: 10.1016/j.lfs.2019.116599
Bauernfeind, F., Bartok, E., Rieger, A., Franchi, L., Núñez, G., and Hornung, V. (2011). Cutting edge: reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasome. J. Immunol. 187, 613–617. doi: 10.4049/jimmunol.1100613
Benjamin, E. J., Virani, S. S., Callaway, C. W., Chamberlain, A. M., Chang, A. R., Cheng, S., et al. (2018). Heart disease and stroke statistics-2018 update: a report from the american heart association. Circulation 137, e67–e492. doi: 10.1161/CIR.0000000000000558
Bruchard, M., Mignot, G., Derangere, V., Chalmin, F., Chevriaux, A., Vegran, F., et al. (2013). Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat. Med. 19, 57–64. doi: 10.1038/nm.2999
Cai, M., Lee, J. H., and Yang, E. J. (2019). Electroacupuncture attenuates cognition impairment via anti-neuroinflammation in an Alzheimer’s disease animal model. J. Neuroinflammation 16:264. doi: 10.1186/s12974-019-1665-3
Cao, G., Jiang, N., Hu, Y., Zhang, Y., Wang, G., Yin, M., et al. (2016). Ruscogenin attenuates cerebral ischemia-induced blood-brain barrier dysfunction by suppressing TXNIP/NLRP3 inflammasome activation and the MAPK pathway. Int. J. Mol. Sci. 17:E1418. doi: 10.3390/ijms17091418
Cassel, S. L., and Sutterwala, F. S. (2010). Sterile inflammatory responses mediated by the NLRP3 inflammasome. Eur. J. Immunol. 40, 607–611. doi: 10.1002/eji.200940207
Chen, K. W., Demarco, B., Heilig, R., Shkarina, K., Boettcher, A., Farady, C. J., et al. (2019). Extrinsic and intrinsic apoptosis activate pannexin-1 to drive NLRP3 inflammasome assembly. EMBO J. 38:e101638. doi: 10.15252/embj.2019101638
Chen, X., Guo, X., Ge, Q., Zhao, Y., Mu, H., and Zhang, J. (2019). ER stress activates the NLRP3 inflammasome: a novel mechanism of atherosclerosis. Oxid. Med. Cell. Longev. 2019:3462530. doi: 10.1155/2019/3462530
Cheng, L., Yin, R., Yang, S., Pan, X., and Ma, A. (2018). Rs4612666 polymorphism of the NLRP3 gene is associated with the occurrence of large artery atherosclerotic ischemic strokes and microembolic signals. Biomed. Res. Int. 2018:6345805. doi: 10.1155/2018/6345805
Chuang, Y. T., Lin, Y. C., Lin, K. H., Chou, T. F., Kuo, W. C., Yang, K. T., et al. (2011). Tumor suppressor death-associated protein kinase is required for full IL-1β production. Blood 117, 960–970. doi: 10.1182/blood-2010-08-303115
Cui, H. X., Chen, J. H., Li, J. W., Cheng, F. R., and Yuan, K. (2018). Protection of anthocyanin from myrica rubra against cerebral ischemia-reperfusion injury via modulation of the TLR4/NF-κB and NLRP3 pathways. Molecules 23:E1788. doi: 10.3390/molecules23071788
de Almeida, L., Khare, S., Misharin, A. V., Patel, R., Ratsimandresy, R. A., Wallin, M. C., et al. (2015). The PYRIN domain-only protein POP1 inhibits inflammasome assembly and ameliorates inflammatory disease. Immunity 43, 264–276. doi: 10.1016/j.immuni.2015.07.018
Deroide, N., Li, X., Lerouet, D., Van Vre, E., Baker, L., Harrison, J., et al. (2013). MFGE8 inhibits inflammasome-induced IL-1β production and limits postischemic cerebral injury. J. Clin. Invest. 123, 1176–1181. doi: 10.1172/jci65167
Dichgans, M., Pulit, S. L., and Rosand, J. (2019). Stroke genetics: discovery, biology, and clinical applications. Lancet Neurol. 18, 587–599. doi: 10.1016/s1474-4422(19)30043-2
Donnan, G. A., Fisher, M., Macleod, M., and Davis, S. M. (2008). Stroke. Lancet 371, 1612–1623. doi: 10.1016/S0140-6736(08)60694-7
Dostert, C., Petrilli, V., Van Bruggen, R., Steele, C., Mossman, B. T., and Tschopp, J. (2008). Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 320, 674–677. doi: 10.1126/science.1156995
Duewell, P., Kono, H., Rayner, K. J., Sirois, C. M., Vladimer, G., Bauernfeind, F. G., et al. (2010). NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464, 1357–1361. doi: 10.1038/nature08938
Duncan, J. A., Bergstralh, D. T., Wang, Y., Willingham, S. B., Ye, Z., Zimmermann, A. G., et al. (2007). Cryopyrin/NALP3 binds ATP/dATP, is an ATPase, and requires ATP binding to mediate inflammatory signaling. Proc. Natl. Acad. Sci. U S A 104, 8041–8046. doi: 10.1073/pnas.0611496104
Fann, D. Y., Lee, S. Y., Manzanero, S., Chunduri, P., Sobey, C. G., and Arumugam, T. V. (2013a). Pathogenesis of acute stroke and the role of inflammasomes. Ageing Res. Rev. 12, 941–966. doi: 10.1016/j.arr.2013.09.004
Fann, D. Y., Lee, S. Y., Manzanero, S., Tang, S. C., Gelderblom, M., Chunduri, P., et al. (2013b). Intravenous immunoglobulin suppresses NLRP1 and NLRP3 inflammasome-mediated neuronal death in ischemic stroke. Cell Death Dis. 4:e790. doi: 10.1038/cddis.2013.326
Fann, D. Y., Lim, Y. A., Cheng, Y. L., Lok, K. Z., Chunduri, P., Baik, S. H., et al. (2018). Evidence that NF-kappaB and MAPK signaling promotes NLRP inflammasome activation in neurons following ischemic stroke. Mol. Neurobiol. 55, 1082–1096. doi: 10.1007/s12035-017-0394-9
Fann, D. Y., Santro, T., Manzanero, S., Widiapradja, A., Cheng, Y. L., Lee, S. Y., et al. (2014). Intermittent fasting attenuates inflammasome activity in ischemic stroke. Exp. Neurol. 257, 114–119. doi: 10.1016/j.expneurol.2014.04.017
Feigin, V. L., Nguyen, G., Cercy, K., Johnson, C. O., Alam, T., Parmar, P. G., et al. (2018). Global, regional, and country-specific lifetime risks of stroke, 1990 and 2016. N. Engl. J. Med. 379, 2429–2437. doi: 10.1056/nejmoa1804492
Feigin, V. L., Norrving, B., and Mensah, G. A. (2017). Global burden of stroke. Circ. Res. 120, 439–448. doi: 10.1161/CIRCRESAHA.116.308413
Feigin, V. L., Roth, G. A., Naghavi, M., Parmar, P., Krishnamurthi, R., Chugh, S., et al. (2016). Global burden of stroke and risk factors in 188 countries, during 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet Neurol. 15, 913–924. doi: 10.1016/s1474-4422(16)30073-4
Franchi, L., Eigenbrod, T., Muñoz-Planillo, R., and Nuñez, G. (2009a). The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat. Immunol. 10, 241–247. doi: 10.1038/ni.1703
Franchi, L., Warner, N., Viani, K., and Nunez, G. (2009b). Function of Nod-like receptors in microbial recognition and host defense. Immunol. Rev. 227, 106–128. doi: 10.1111/j.1600-065x.2008.00734.x
Gao, L., Dong, Q., Song, Z., Shen, F., Shi, J., and Li, Y. (2017). NLRP3 inflammasome: a promising target in ischemic stroke. Inflamm. Res. 66, 17–24. doi: 10.1007/s00011-016-0981-7
Gilgun-Sherki, Y., Rosenbaum, Z., Melamed, E., and Offen, D. (2002). Antioxidant therapy in acute central nervous system injury: current state. Pharmacol. Rev. 54, 271–284. doi: 10.1124/pr.54.2.271
Gong, Z., Pan, J., Shen, Q., Li, M., and Peng, Y. (2018). Mitochondrial dysfunction induces NLRP3 inflammasome activation during cerebral ischemia/reperfusion injury. J. Neuroinflammation 15, 242–242. doi: 10.1186/s12974-018-1282-6
Grebe, A., Hoss, F., and Latz, E. (2018). NLRP3 inflammasome and the IL-1 pathway in atherosclerosis. Circ. Res. 122, 1722–1740. doi: 10.1161/CIRCRESAHA.118.311362
Gross, O., Thomas, C. J., Guarda, G., and Tschopp, J. (2011). The inflammasome: an integrated view. Immunol. Rev. 243, 136–151. doi: 10.1111/j.1600-065x.2011.01046.x
Guarda, G., Braun, M., Staehli, F., Tardivel, A., Mattmann, C., Förster, I., et al. (2011). Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity 34, 213–223. doi: 10.1016/j.immuni.2011.02.006
Guo, M., Wang, X., Zhao, Y., Yang, Q., Ding, H., Dong, Q., et al. (2018). Ketogenic diet improves brain ischemic tolerance and inhibits NLRP3 inflammasome activation by preventing Drp1-mediated mitochondrial fission and endoplasmic reticulum stress. Front. Mol. Neurosci. 11:86. doi: 10.3389/fnmol.2018.00086
Gustin, A., Kirchmeyer, M., Koncina, E., Felten, P., Losciuto, S., Heurtaux, T., et al. (2015). NLRP3 inflammasome is expressed and functional in mouse brain microglia but not in astrocytes. PLoS One 10:e0130624. doi: 10.1371/journal.pone.0130624
Hägg, S., Fall, T., Ploner, A., Mägi, R., Fischer, K., Draisma, H. H., et al. (2015). Adiposity as a cause of cardiovascular disease: a Mendelian randomization study. Int. J. Epidemiol. 44, 578–586. doi: 10.1093/ije/dyv094
Hara, H., Tsuchiya, K., Kawamura, I., Fang, R., Hernandez-Cuellar, E., Shen, Y., et al. (2013). Phosphorylation of the adaptor ASC acts as a molecular switch that controls the formation of speck-like aggregates and inflammasome activity. Nat. Immunol. 14, 1247–1255. doi: 10.1038/ni.2749
Harris, J., Hartman, M., Roche, C., Zeng, S. G., O’Shea, A., Sharp, F. A., et al. (2011). Autophagy controls IL-1β secretion by targeting pro-IL-1beta for degradation. J. Biol. Chem. 286, 9587–9597. doi: 10.1074/jbc.M110.202911
Hata, R., Maeda, K., Hermann, D., Mies, G., and Hossmann, K. A. (2000). Evolution of brain infarction after transient focal cerebral ischemia in mice. J. Cereb. Blood Flow Metab. 20, 937–946. doi: 10.1097/00004647-200006000-00006
He, Y., Zeng, M. Y., Yang, D., Motro, B., and Nunez, G. (2016). NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 530, 354–357. doi: 10.1038/nature16959
Hong, P., Gu, R.-N., Li, F.-X., Xiong, X.-X., Liang, W.-B., You, Z.-J., et al. (2019). NLRP3 inflammasome as a potential treatment in ischemic stroke concomitant with diabetes. J. Neuroinflammation 16, 121–121. doi: 10.1186/s12974-019-1498-0
Hong, P., Li, F. X., Gu, R. N., Fang, Y. Y., Lai, L. Y., Wang, Y. W., et al. (2018). Inhibition of NLRP3 inflammasome ameliorates cerebral ischemia-reperfusion injury in diabetic mice. Neural Plast. 2018:9163521. doi: 10.1155/2018/9163521
Hornung, V., Bauernfeind, F., Halle, A., Samstad, E. O., Kono, H., Rock, K. L., et al. (2008). Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 9, 847–856. doi: 10.1038/ni.1631
Huang, Y., Ohta, Y., Shang, J., Morihara, R., Nakano, Y., Fukui, Y., et al. (2018). Antineuroinflammatory effect of SMTP-7 in ischemic mice. J. Stroke Cerebrovasc. Dis. 27, 3084–3094. doi: 10.1016/j.jstrokecerebrovasdis.2018.06.039
Ishrat, T., Mohamed, I. N., Pillai, B., Soliman, S., Fouda, A. Y., Ergul, A., et al. (2015). Thioredoxin-interacting protein: a novel target for neuroprotection in experimental thromboembolic stroke in mice. Mol. Neurobiol. 51, 779–780. doi: 10.1007/s12035-014-9025-x
Ismael, S., Zhao, L., Nasoohi, S., and Ishrat, T. (2018). Inhibition of the NLRP3-inflammasome as a potential approach for neuroprotection after stroke. Sci. Rep. 8, 5971–5971. doi: 10.1038/s41598-018-24350-x
Ito, M., Shichita, T., Okada, M., Komine, R., Noguchi, Y., Yoshimura, A., et al. (2015). Bruton’s tyrosine kinase is essential for NLRP3 inflammasome activation and contributes to ischaemic brain injury. Nat. Commun. 6, 7360–7360. doi: 10.1038/ncomms8360
Iyer, S. S., He, Q., Janczy, J. R., Elliott, E. I., Zhong, Z., Olivier, A. K., et al. (2013). Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 39, 311–323. doi: 10.1016/j.immuni.2013.08.001
Jiang, T., Wu, M., Zhang, Z., Yan, C., Ma, Z., He, S., et al. (2019). Electroacupuncture attenuated cerebral ischemic injury and neuroinflammation through alpha7nAChR-mediated inhibition of NLRP3 inflammasome in stroke rats. Mol. Med. 25:22. doi: 10.1186/s10020-019-0091-4
Kayagaki, N., Stowe, I. B., Lee, B. L., O’Rourke, K., Anderson, K., Warming, S., et al. (2015). Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671. doi: 10.1038/nature15541
Kelley, N., Jeltema, D., Duan, Y., and He, Y. (2019). The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int. J. Mol. Sci. 20:E3328. doi: 10.3390/ijms20133328
Khoshnam, S. E., Winlow, W., Farzaneh, M., Farbood, Y., and Moghaddam, H. F. (2017). Pathogenic mechanisms following ischemic stroke. Neurol. Sci. 38, 1167–1186. doi: 10.1007/s10072-017-2938-1
Koizumi, Y., Toma, C., Higa, N., Nohara, T., Nakasone, N., and Suzuki, T. (2012). Inflammasome activation via intracellular NLRs triggered by bacterial infection. Cell. Microbiol. 14, 149–154. doi: 10.1111/j.1462-5822.2011.01707.x
Lamkanfi, M., and Dixit, V. M. (2009). Inflammasomes: guardians of cytosolic sanctity. Immunol. Rev. 227, 95–105. doi: 10.1111/j.1600-065x.2008.00730.x
Lamkanfi, M., and Dixit, V. M. (2010). Manipulation of host cell death pathways during microbial infections. Cell Host Microbe 8, 44–54. doi: 10.1016/j.chom.2010.06.007
Lamkanfi, M., Mueller, J. L., Vitari, A. C., Misaghi, S., Fedorova, A., Deshayes, K., et al. (2009). Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J. Cell Biol. 187, 61–70. doi: 10.1083/jcb.200903124
Lammerding, L., Slowik, A., Johann, S., Beyer, C., and Zendedel, A. (2016). Poststroke inflammasome expression and regulation in the peri-infarct area by gonadal steroids after transient focal ischemia in the rat brain. Neuroendocrinology 103, 460–475. doi: 10.1159/000439435
Lane, T., Flam, B., Lockey, R., and Kolliputi, N. (2013). TXNIP shuttling: missing link between oxidative stress and inflammasome activation. Front. Physiol. 4:50. doi: 10.3389/fphys.2013.00050
Lapchak, P. A., and Zhang, J. H. (2017). The high cost of stroke and stroke cytoprotection research. Transl. Stroke Res. 8, 307–317. doi: 10.1007/s12975-016-0518-y
Lechtenberg, B. C., Mace, P. D., and Riedl, S. J. (2014). Structural mechanisms in NLR inflammasome signaling. Curr. Opin. Struct. Biol. 29, 17–25. doi: 10.1016/j.sbi.2014.08.011
Lee, H. I., Lee, S. W., Kim, N. G., Park, K. J., Choi, B. T., Shin, Y. I., et al. (2017). Low-level light emitting diode (LED) therapy suppresses inflammasome-mediated brain damage in experimental ischemic stroke. J. Biophotonics 10, 1502–1513. doi: 10.1002/jbio.201600244
Lee, G.-S., Subramanian, N., Kim, A. I., Aksentijevich, I., Goldbach-Mansky, R., Sacks, D. B., et al. (2012). The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature 492, 123–127. doi: 10.1038/nature11588
Li, C., Wang, J., Fang, Y., Liu, Y., Chen, T., Sun, H., et al. (2016). Nafamostat mesilate improves function recovery after stroke by inhibiting neuroinflammation in rats. Brain Behav. Immun. 56, 230–245. doi: 10.1016/j.bbi.2016.03.019
Li, S.-J., Zhang, Y.-F., Ma, S.-H., Yi, Y., Yu, H.-Y., Pei, L., et al. (2018). The role of NLRP3 inflammasome in stroke and central poststroke pain. Medicine 97:e11861. doi: 10.1097/MD.0000000000011861
Lindsay, M. P., Norrving, B., Sacco, R. L., Brainin, M., Hacke, W., Martins, S., et al. (2019). World stroke organization (WSO): global stroke fact sheet 2019. Int. J. Stroke 14, 806–817. doi: 10.1177/1747493019881353
Liu, L., Cen, J., Man, Y., Li, J., Zhang, D., Wang, F., et al. (2018). Transplantation of human umbilical cord blood mononuclear cells attenuated ischemic injury in MCAO rats via inhibition of NF-kappaB and NLRP3 inflammasome. Neuroscience 369, 314–324. doi: 10.1016/j.neuroscience.2017.11.027
Liu, Y., Fiskum, G., and Schubert, D. (2002). Generation of reactive oxygen species by the mitochondrial electron transport chain. J. Neurochem. 80, 780–787. doi: 10.1046/j.0022-3042.2002.00744.x
Liu, S. B., Mi, W. L., and Wang, Y. Q. (2013). Research progress on the NLRP3 inflammasome and its role in the central nervous system. Neurosci. Bull. 29, 779–787. doi: 10.1007/s12264-013-1328-9
Lu, Y., Hajifathalian, K., Ezzati, M., Woodward, M., Rimm, E. B., and Danaei, G. (2014). Metabolic mediators of the effects of body-mass index, overweight, and obesity on coronary heart disease and stroke: a pooled analysis of 97 prospective cohorts with 1.8 million participants. Lancet 383, 970–983. doi: 10.1016/s0140-6736(13)61836-x
Lu, A., Magupalli, V. G., Ruan, J., Yin, Q., Atianand, M. K., Vos, M. R., et al. (2014). Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 156, 1193–1206. doi: 10.1016/j.cell.2014.02.008
Lu, Y., Xiao, G., and Luo, W. (2016). Minocycline suppresses NLRP3 inflammasome activation in experimental ischemic stroke. Neuroimmunomodulation 23, 230–238. doi: 10.1159/000452172
Mao, K., Chen, S., Chen, M., Ma, Y., Wang, Y., Huang, B., et al. (2013). Nitric oxide suppresses NLRP3 inflammasome activation and protects against LPS-induced septic shock. Cell Res. 23, 201–212. doi: 10.1038/cr.2013.6
Martinon, F., and Tschopp, J. (2005). NLRs join TLRs as innate sensors of pathogens. Trends Immunol. 26, 447–454. doi: 10.1016/j.it.2005.06.004
Martinon, F., Burns, K., and Tschopp, J. (2002). The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 10, 417–426. doi: 10.1016/s1097-2765(02)00599-3
McCaffrey, K., and Braakman, I. (2016). Protein quality control at the endoplasmic reticulum. Essays Biochem. 60, 227–235. doi: 10.1042/EBC20160003
Minutoli, L., Puzzolo, D., Rinaldi, M., Irrera, N., Marini, H., Arcoraci, V., et al. (2016). ROS-mediated NLRP3 inflammasome activation in brain, heart, kidney, and testis ischemia/reperfusion injury. Oxid. Med. Cell. Longev. 2016:2183026. doi: 10.1155/2016/2183026
Mongin, A. A. (2007). Disruption of ionic and cell volume homeostasis in cerebral ischemia: the perfect storm. Pathophysiology 14, 183–193. doi: 10.1016/j.pathophys.2007.09.009
Murakami, T., Ockinger, J., Yu, J., Byles, V., McColl, A., Hofer, A. M., et al. (2012). Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc. Natl. Acad. Sci. U S A 109, 11282–11287. doi: 10.1073/pnas.1117765109
Nakahira, K., Haspel, J. A., Rathinam, V. A., Lee, S. J., Dolinay, T., Lam, H. C., et al. (2011). Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 12, 222–230. doi: 10.1038/ni.1980
O’Donnell, M. J., Xavier, D., Liu, L., Zhang, H., Chin, S. L., Rao-Melacini, P., et al. (2010). Risk factors for ischaemic and intracerebral haemorrhagic stroke in 22 countries (the INTERSTROKE study): a case-control study. Lancet 376, 112–123. doi: 10.1016/S0140-6736(10)60834-3
Okada, M., Matsuzawa, A., Yoshimura, A., and Ichijo, H. (2014). The lysosome rupture-activated TAK1-JNK pathway regulates NLRP3 inflammasome activation. J. Biol. Chem. 289, 32926–32936. doi: 10.1074/jbc.M114.579961
Ozaki, E., Campbell, M., and Doyle, S. L. (2015). Targeting the NLRP3 inflammasome in chronic inflammatory diseases: current perspectives. J. Inflamm. Res. 8, 15–27. doi: 10.2147/jir.s51250
Park, H.-S., Yoo, M. H., and Koh, J.-Y. (2020). Role of zinc dyshomeostasis in inflammasome formation in cultured cortical cells following lipopolysaccharide or oxygen-glucose deprivation/reperfusion exposure. Neurobiol. Dis. 137:104771. doi: 10.1016/j.nbd.2020.104771
Pasqua, T., Pagliaro, P., Rocca, C., Angelone, T., and Penna, C. (2018). Role of NLRP-3 inflammasome in hypertension: a potential therapeutic target. Curr. Pharm. Biotechnol. 19, 708–714. doi: 10.2174/1389201019666180808162011
Pedra, J. H. F., Cassel, S. L., and Sutterwala, F. S. (2009). Sensing pathogens and danger signals by the inflammasome. Curr. Opin. Immunol. 21, 10–16. doi: 10.1016/j.coi.2009.01.006
Pétrilli, V., Papin, S., Dostert, C., Mayor, A., Martinon, F., and Tschopp, J. (2007). Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 14, 1583–1589. doi: 10.1038/sj.cdd.4402195
Piippo, N., Korhonen, E., Hytti, M., Kinnunen, K., Kaarniranta, K., and Kauppinen, A. (2018). Oxidative stress is the principal contributor to inflammasome activation in retinal pigment epithelium cells with defunct proteasomes and autophagy. Cell. Physiol. Biochem. 49, 359–367. doi: 10.1159/000492886
Proell, M., Gerlic, M., Mace, P. D., Reed, J. C., and Riedl, S. J. (2013). The CARD plays a critical role in ASC foci formation and inflammasome signalling. Biochem. J. 449, 613–621. doi: 10.1042/bj20121198
Py, B. F., Kim, M. S., Vakifahmetoglu-Norberg, H., and Yuan, J. (2013). Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol. Cell 49, 331–338. doi: 10.1016/j.molcel.2012.11.009
Qin, Y. Y., Li, M., Feng, X., Wang, J., Cao, L., Shen, X. K., et al. (2017). Combined NADPH and the NOX inhibitor apocynin provides greater anti-inflammatory and neuroprotective effects in a mouse model of stroke. Free Radic. Biol. Med. 104, 333–345. doi: 10.1016/j.freeradbiomed.2017.01.034
Qiu, J., Wang, M., Zhang, J., Cai, Q., Lu, D., Li, Y., et al. (2016). The neuroprotection of Sinomenine against ischemic stroke in mice by suppressing NLRP3 inflammasome via AMPK signaling. Int. Immunopharmacol. 40, 492–500. doi: 10.1016/j.intimp.2016.09.024
Samir, P., Kesavardhana, S., Patmore, D. M., Gingras, S., Malireddi, R. K. S., Karki, R., et al. (2019). DDX3X acts as a live-or-die checkpoint in stressed cells by regulating NLRP3 inflammasome. Nature 573, 590–594. doi: 10.1038/s41586-019-1551-2
Sanderson, T. H., Reynolds, C. A., Kumar, R., Przyklenk, K., and Hüttemann, M. (2013). Molecular mechanisms of ischemia-reperfusion injury in brain: pivotal role of the mitochondrial membrane potential in reactive oxygen species generation. Mol. Neurobiol. 2013, 9–23. doi: 10.1007/s12035-012-8344-z
Sardu, C., Paolisso, G., and Marfella, R. (2020). Molecular mechanisms and therapeutic targets of inflammatory-related Cardiovascular diseases: from molecular mechanisms to therapeutic targets. Curr. Pharm. Des. doi: 10.2174/1381612826666200213123029 [Epub ahead of print].
Savage, C. D., Lopez-Castejon, G., Denes, A., and Brough, D. (2012). NLRP3-inflammasome activating DAMPs stimulate an inflammatory response in glia in the absence of priming which contributes to brain inflammation after injury. Front. Immunol. 3:288. doi: 10.3389/fimmu.2012.00288
Schroder, K., and Tschopp, J. (2010). The inflammasomes. Cell 140, 821–832. doi: 10.1016/j.cell.2010.01.040
Schwamm, L. H., Ali, S. F., Reeves, M. J., Smith, E. E., Saver, J. L., Messe, S., et al. (2013). Temporal trends in patient characteristics and treatment with intravenous thrombolysis among acute ischemic stroke patients at Get With The Guidelines-Stroke hospitals. Circ. Cardiovasc. Qual. Outcomes 6, 543–549. doi: 10.1161/CIRCOUTCOMES.111.000303
Sha, R., Zhang, B., Han, X., Peng, J., Zheng, C., Zhang, F., et al. (2019). Electroacupuncture alleviates ischemic brain injury by Inhibiting the miR-223/NLRP3 pathway. Med. Sci. Monit. 25, 4723–4733. doi: 10.12659/msm.917213
Shao, B.-Z., Wang, S.-L., Pan, P., Yao, J., Wu, K., Li, Z.-S., et al. (2019). Targeting NLRP3 inflammasome in inflammatory bowel disease: putting out the fire of inflammation. Inflammation 42, 1147–1159. doi: 10.1007/s10753-019-01008-y
Sharif, H., Wang, L., Wang, W. L., Magupalli, V. G., Andreeva, L., Qiao, Q., et al. (2019). Structural mechanism for NEK7-licensed activation of NLRP3 inflammasome. Nature 570, 338–343. doi: 10.1038/s41586-019-1295-z
Shi, C. S., Shenderov, K., Huang, N. N., Kabat, J., Abu-Asab, M., Fitzgerald, K. A., et al. (2012). Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 13, 255–263. doi: 10.1038/ni.2215
Shi, K., Tian, D. C., Li, Z. G., Ducruet, A. F., Lawton, M. T., and Shi, F. D. (2019). Global brain inflammation in stroke. Lancet Neurol. 18, 1058–1066. doi: 10.1016/S1474-4422(19)30078-X
Shi, H., Wang, Y., Li, X., Zhan, X., Tang, M., Fina, M., et al. (2016). NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat. Immunol. 17, 250–258. doi: 10.1038/ni.3333
Shi, C., Yang, H., and Zhang, Z. (2020). Involvement of nucleotide-binding oligomerization domain-like receptor family pyrin domain containing 3 inflammasome in the pathogenesis of liver diseases. Front. Cell. Dev. Biol. 8:139. doi: 10.3389/fcell.2020.00139
Shi, J., Zhao, Y., Wang, K., Shi, X., Wang, Y., Huang, H., et al. (2015). Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665. doi: 10.1038/nature15514
Siesjo, B. K. (1992). Pathophysiology and treatment of focal cerebral ischemia. Part I: pathophysiology. J. Neurosurg. 77, 169–184. doi: 10.3171/jns.1992.77.2.0169
Swanson, K. V., Deng, M., and Ting, J. P. (2019). The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 19, 477–489. doi: 10.1038/s41577-019-0165-0
Tan, M.-S., Yu, J.-T., Jiang, T., Zhu, X.-C., and Tan, L. (2013). The NLRP3 inflammasome in Alzheimer’s disease. J. Neuroimmunol. 265, 91–95. doi: 10.1016/j.jneuroim.2013.10.002
Tian, F., Deguchi, K., Yamashita, T., Ohta, Y., Morimoto, N., Shang, J., et al. (2010). In vivo imaging of autophagy in a mouse stroke model. Autophagy 6, 1107–1114. doi: 10.4161/auto.6.8.13427
Tschopp, J., and Schroder, K. (2010). NLRP3 inflammasome activation: the convergence of multiple signalling pathways on ROS production? Nat. Rev. Immunol. 10, 210–215. doi: 10.1038/nri2725
Turrens, J. F. (2003). Mitochondrial formation of reactive oxygen species. J. Physiol. 552, 335–344. doi: 10.1038/nri2725
Vajjhala, P. R., Mirams, R. E., and Hill, J. M. (2012). Multiple binding sites on the pyrin domain of ASC protein allow self-association and interaction with NLRP3 protein. J. Biol. Chem. 287, 41732–41743. doi: 10.1074/jbc.m112.381228
Viganò, E., Diamond, C. E., Spreafico, R., Balachander, A., Sobota, R. M., and Mortellaro, A. (2015). Human caspase-4 and caspase-5 regulate the one-step non-canonical inflammasome activation in monocytes. Nat. Commun. 6:8761. doi: 10.1038/ncomms9761
Wang, Z., Hu, W., Lu, C., Ma, Z., Jiang, S., Gu, C., et al. (2018). Targeting NLRP3 (nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3) inflammasome in cardiovascular disorders. Arterioscler. Thromb. Vasc. Biol. 38, 2765–2779. doi: 10.1161/ATVBAHA.118.311916
Wang, X., Li, R., Wang, X., Fu, Q., and Ma, S. (2015). Umbelliferone ameliorates cerebral ischemia-reperfusion injury via upregulating the PPAR gamma expression and suppressing TXNIP/NLRP3 inflammasome. Neurosci. Lett. 600, 182–187. doi: 10.1016/j.neulet.2015.06.016
Wang, Q., Tang, X. N., and Yenari, M. A. (2007). The inflammatory response in stroke. J. Neuroimmunol. 184, 53–68. doi: 10.1016/j.jneuroim.2006.11.014
Wang, S., Yuan, Y.-H., Chen, N.-H., and Wang, H.-B. (2019). The mechanisms of NLRP3 inflammasome/pyroptosis activation and their role in Parkinson’s disease. Int. Immunopharmacol. 67, 458–464. doi: 10.1016/j.intimp.2018.12.019
Wang, H., Zhong, D., Chen, H., Jin, J., Liu, Q., and Li, G. (2019). NLRP3 inflammasome activates interleukin-23/interleukin-17 axis during ischaemia-reperfusion injury in cerebral ischaemia in mice. Life Sci. 227, 101–113. doi: 10.1016/j.lfs.2019.04.031
Wang, Z., Zhang, S., Xiao, Y., Zhang, W., Wu, S., Qin, T., et al. (2020). NLRP3 inflammasome and inflammatory diseases. Oxid. Med. Cell. Longev. 2020:4063562 . doi: 10.1155/2020/4063562
Ward, R., Li, W., Abdul, Y., Jackson, L., Dong, G., Jamil, S., et al. (2019). NLRP3 inflammasome inhibition with MCC950 improves diabetes-mediated cognitive impairment and vasoneuronal remodeling after ischemia. Pharmacol. Res. 142, 237–250. doi: 10.1016/j.phrs.2019.01.035
Widiapradja, A., Vegh, V., Lok, K. Z., Manzanero, S., Thundyil, J., Gelderblom, M., et al. (2012). Intravenous immunoglobulin protects neurons against amyloid beta-peptide toxicity and ischemic stroke by attenuating multiple cell death pathways. J. Neurochem. 122, 321–332. doi: 10.1111/j.1471-4159.2012.07754.x
Woodruff, T. M., Thundyil, J., Tang, S. C., Sobey, C. G., Taylor, S. M., and Arumugam, T. V. (2011). Pathophysiology, treatment, and animal and cellular models of human ischemic stroke. Mol. Neurodegener. 6:11. doi: 10.4103/1673-5374.274324
Xie, Q., Shen, W. W., Zhong, J., Huang, C., Zhang, L., and Li, J. (2014). Lipopolysaccharide/adenosine triphosphate induces IL1β and IL-18 secretion through the NLRP3 inflammasome in RAW264.7 murine macrophage cells. Int. J. Mol. Med. 34, 341–349. doi: 10.3892/ijmm.2014.1755
Yamasaki, K., Muto, J., Taylor, K. R., Cogen, A. L., Audish, D., Bertin, J., et al. (2009). NLRP3/cryopyrin is necessary for interleukin-1beta (IL-1β) release in response to hyaluronan, an endogenous trigger of inflammation in response to injury. J. Biol. Chem. 284, 12762–12771. doi: 10.1074/jbc.m806084200
Yang, F., Wang, Z., Wei, X., Han, H., Meng, X., Zhang, Y., et al. (2014). NLRP3 deficiency ameliorates neurovascular damage in experimental ischemic stroke. J. Cereb. Blood Flow Metab. 34, 660–667. doi: 10.1038/jcbfm.2013.242
Yang, Z., Zhong, L., Xian, R., and Yuan, B. (2015). MicroRNA-223 regulates inflammation and brain injury via feedback to NLRP3 inflammasome after intracerebral hemorrhage. Mol. Immunol. 65, 267–276. doi: 10.1016/j.molimm.2014.12.018
Yi, Y.-S. (2018). Regulatory roles of the caspase-11 non-canonical inflammasome in inflammatory diseases. Immune Netw. 18:e41. doi: 10.4110/in.2018.18.e41
Yoshimura, S., Sakai, N., Uchida, K., Yamagami, H., Ezura, M., Okada, Y., et al. (2018). Endovascular therapy in ischemic stroke with acute large-vessel occlusion: recovery by endovascular salvage for cerebral ultra-acute embolism japan registry 2. J. Am. Heart Assoc. 7:e008796. doi: 10.1161/JAHA.118.008796
Yu, J., Zhu, H., Taheri, S., Mondy, W., Bonilha, L., Magwood, G. S., et al. (2019). Serum amyloid A-mediated inflammasome activation of microglial cells in cerebral ischemia. J. Neurosci. 39, 9465–9476. doi: 10.1523/jneurosci.0801-19.2019
Zang, Y., Zhou, X., Wang, Q., Li, X., and Huang, H. (2018). LncRNA FIRRE/NF-kB feedback loop contributes to OGD/R injury of cerebral microglial cells. Biochem. Biophys. Res. Commun. 501, 131–138. doi: 10.1016/j.bbrc.2018.04.194
Zhang, S., Jiang, L., Che, F., Lu, Y., Xie, Z., and Wang, H. (2017). Arctigenin attenuates ischemic stroke via SIRT1-dependent inhibition of NLRP3 inflammasome. Biochem. Biophys. Res. Commun. 493, 821–826. doi: 10.1016/j.bbrc.2017.08.062
Zhao, C., and Zhao, W. (2020). NLRP3 inflammasome-A key player in antiviral responses. Front. Immunol. 11:211. doi: 10.3389/fimmu.2020.00211
Zhou, Y., Gu, Y., and Liu, J. (2019). BRD4 suppression alleviates cerebral ischemia-induced brain injury by blocking glial activation via the inhibition of inflammatory response and pyroptosis. Biochem. Biophys. Res. Commun. 519, 481–488. doi: 10.1016/j.bbrc.2019.07.097
Zhou, K., Shi, L., Wang, Y., Chen, S., and Zhang, J. (2016). Recent advances of the NLRP3 inflammasome in central nervous system disorders. J. Immunol. Res. 2016:9238290. doi: 10.1155/2016/9238290
Zhou, R., Tardivel, A., Thorens, B., Choi, I., and Tschopp, J. (2010). Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 11, 136–140. doi: 10.1038/ni.1831
Zhou, R., Yazdi, A. S., Menu, P., and Tschopp, J. (2011). A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–225. doi: 10.1038/nature09663
Keywords: NLRP3 inflammasome, ischemic stroke, inflammatory reaction, death—associated protein kinase, reactive oxygen species
Citation: Feng Y-S, Tan Z-X, Wang M-M, Xing Y, Dong F and Zhang F (2020) Inhibition of NLRP3 Inflammasome: A Prospective Target for the Treatment of Ischemic Stroke. Front. Cell. Neurosci. 14:155. doi: 10.3389/fncel.2020.00155
Received: 11 January 2020; Accepted: 11 May 2020;
Published: 03 June 2020.
Edited by:
Stefania Ceruti, University of Milan, ItalyReviewed by:
Seong-Woon Yu, Daegu Gyeongbuk Institute of Science and Technology (DGIST), South KoreaCarmine Rocca, University of Calabria, Italy
Copyright © 2020 Feng, Tan, Wang, Xing, Dong and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Feng Zhang, emprMjAwMTlAMTI2LmNvbQ==