 Lucy S. French
Lucy S. French Carla B. Mellough
Carla B. Mellough Fred K. Chen
Fred K. Chen Livia S. Carvalho
Livia S. Carvalho- 1Centre for Ophthalmology and Visual Sciences (incorporating Lions Eye Institute), The University of Western Australia, Nedlands, WA, Australia
- 2Department of Ophthalmology, Royal Perth Hospital, Perth, WA, Australia
- 3Department of Ophthalmology, Perth Children’s Hospital, Nedlands, WA, Australia
Usher syndrome is a genetic disorder causing neurosensory hearing loss and blindness from retinitis pigmentosa (RP). Adaptive techniques such as braille, digital and optical magnifiers, mobility training, cochlear implants, or other assistive listening devices are indispensable for reducing disability. However, there is currently no treatment to reduce or arrest sensory cell degeneration. There are several classes of treatments for Usher syndrome being investigated. The present article reviews the progress this research has made towards delivering commercial options for patients with Usher syndrome.
Introduction
Usher syndrome is a group of autosomal recessive disorders characterized by congenital neurosensory hearing loss, progressive night vision impairment, and constriction of the visual field due to retinitis pigmentosa (RP). Some forms of Usher syndrome may also have varying levels of vestibular dysfunction resulting in loss of balance. It is the most common form of inherited deaf-blindness (El-Amraoui and Petit, 2014) affecting an estimated 1 in 6,000 people worldwide (Kimberling et al., 2010). Usher subtypes (1, 2, and 3; Table 1) are graded according to the severity of symptoms and age of onset. Type 1 patients are born profoundly deaf and experience pre-pubertal onset of progressive vision loss caused by RP. The majority of type 1 patients also have developmental motor delays caused by vestibular dysfunction. Type 2 patients have mild to moderate congenital hearing loss with RP diagnosed during puberty. Hearing loss in type 3 patients is progressive and post-lingual, while RP onset may be delayed until mid-adulthood (Reiners et al., 2006). The clinical presentation of RP begins with night blindness caused by the degeneration of rod photoreceptor cells. Subsequent constriction of the visual field results in a “tunnel vision” effect caused by the centripetal progression of cone photoreceptor cell loss. In classical RP, the death of cones may be secondary to rod degeneration and this may ultimately lead to complete loss of vision in advanced age (Hartong et al., 2006). Many other inherited retinal diseases are associated with deafness (Table 2) such as cone-rod dystrophy and hearing loss-1 (CRDHL1, OMIM #617236), diabetes and deafness, maternally inherited (MIDD, OMIM #520000) and Leber congenital amaurosis (LCA) with early-onset deafness (LCAEOD, OMIM #617879). This review is limited to the combination of RP and deafness, the classical presentation of Usher syndrome.
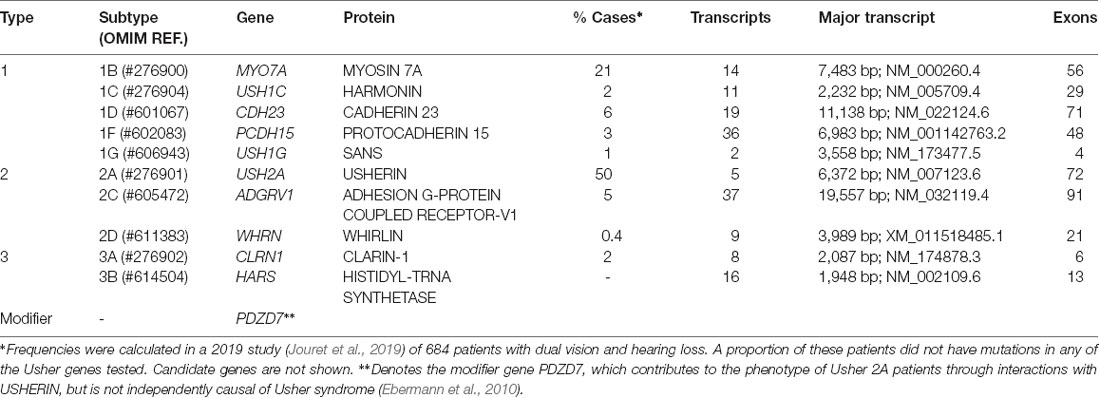
Table 1. Genes and proteins associated with various Usher syndrome subtypes.
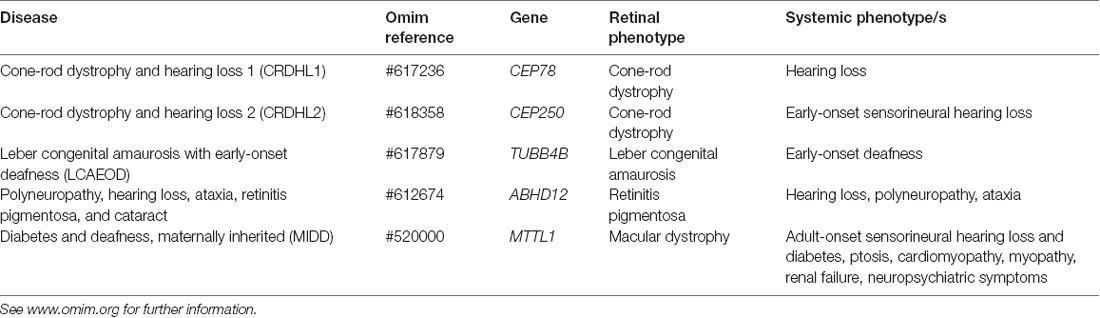
Table 2. List of non-Usher syndromes that cause hearing loss and inherited retinal disease.
The Genetics and Biology of Usher Syndrome
Usher syndrome is caused by autosomal recessive inheritance of mutations in Usher genes known to encode proteins involved in transmembrane adhesion, scaffolding and motor transport. Ten causative genes have so far been identified. Inheritance of hypomorphic alleles with missense mutations often causes non-syndromic deafness, while nonsense and cryptic splice-site mutations result in Usher syndrome (Ahmed et al., 2008; Bademci et al., 2016). Certain types of mutations in Usher genes may also cause non-syndromic RP (Seyedahmadi et al., 2004). Digenic inheritance of mutations in separate Usher genes has also been proposed to be causative of Usher syndrome (Zheng et al., 2005; Bonnet et al., 2011), but remains controversial (Jouret et al., 2019). Additionally, the disparity in phenotypes and progression rates between monozygotic twins is suggestive of environmental influence (Liu et al., 1999). A summary of identified genes and their proteins can be seen in Table 1.
Usher proteins are localized both to the inner ear and retina. In the inner ear, Usher proteins are present in the cochlea and vestibular organs, accounting for the balance and deafness phenotypes. Usher genes instruct the differentiation of mechano-sensitive hair cells and affect hair bundle organization during development. Functional Usher proteins are also essential for the transduction of electrical signals in mature stereocilia (Grati and Kachar, 2011; Cosgrove and Zallocchi, 2014; El-Amraoui and Petit, 2014; Pepermans et al., 2014; Mathur and Yang, 2015, 2019; Han et al., 2018). In the retina, Usher proteins are found in the light-sensitive photoreceptor neurons. They have been implicated in intracellular trafficking at the connecting cilium, which links the photoreceptor’s inner and outer segments, and facilitates the movement of phototransduction proteins and lipids to the outer segment (Liu et al., 1997; Mathur and Yang, 2015). Knowledge of the function of Usher proteins in the retina is limited by a lack of effective animal models (El-Amraoui and Petit, 2014), perhaps due to their association with calyceal processes, the microvilli which protrude from the apical region of the inner segment and surround the connecting cilium of human, but not murine, photoreceptors (Sahly et al., 2012; Schietroma et al., 2017).
Current Trials and Pre-clinical Studies for Usher Syndrome Treatment
Cochlear implants are often provided to patients with type 1 Usher syndrome due to the profound deafness at birth. Those with type 2 or 3 Usher syndrome may benefit from hearing aids or cochlear implant later in life if the mild congenital deafness progresses. However, there is currently no treatment available to prevent or reverse the inevitable retinal degeneration associated with RP. There is some evidence for use of dietary supplements to delay the progression of RP, including vitamin A (Berson et al., 1993, 2018), omega-3 fish oils (Berson et al., 2004, 2012), N-acetylcysteine (Campochiaro et al., 2020)1 and the antioxidant taurine (Trouillet et al., 2018). Although some of these have been disputed (Rayapudi et al., 2013; Hoffman et al., 2014), the lack of strong evidence for efficacy may be related to the heterogeneity of study cohort, small sample size, and short follow-up study design. Additionally, vector-induced expression of ciliary neurotrophic factor (CNTF) was shown to be neuroprotective in a mouse model of RP (Lipinski et al., 2015), yet long-term follow up of three RP patients treated with sustained-release CNTF delivered intravitreally via encapsulated cell technology implant (NT-501, Neurotech Pharmaceuticals Inc.), one of which had Usher-associated RP, showed no significant difference in visual acuity (Talcott et al., 2011). In this review article, we will summarise the major classes of ongoing investigations for gene and mutation-specific treatment of hearing and visual loss and tissue regeneration and the progress and challenges in delivering therapeutic outcomes to patients with Usher syndrome. The main approaches discussed can be divided into DNA intervention, RNA intervention and cell replacement and an overview of the different approaches is shown in Figure 1.
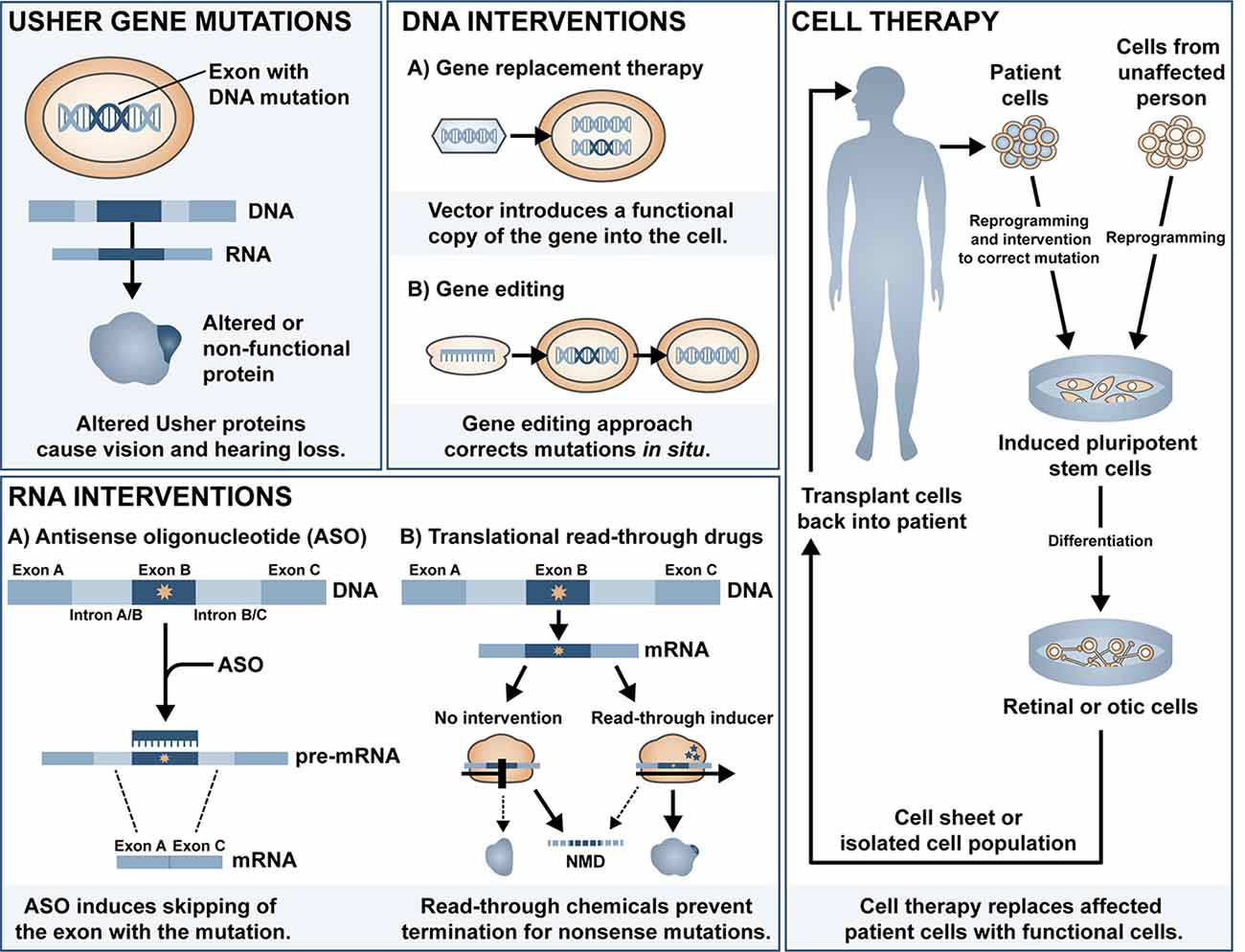
Figure 1. Overview of the different therapeutic approaches for Usher syndrome.
DNA Interventions
Non-viral Systems
DNA interventions are possible due to engineered or recombinant vectors (broadly classified as viral and non-viral) that are synthesized to carry genetic material (Sengillo et al., 2016). Non-viral vectors (nanoparticles) are non-immunogenic, readily customizable, and may package up to 20 kb of material, allowing the delivery of large therapeutic genes (Moore et al., 2018).
Lipid-based nanoparticles, which form a protective bilayer around transported genetic components, have been explored as potential treatments for hearing loss. Zou et al. (2017) investigated the functional, inflammatory, and apoptotic response to liposome nanoparticle delivery to the murine inner ear and found no adverse reactions. Additionally, Gao et al. (2018) reported that cationic lipid-mediated delivery of Cas9:guideRNA complexes to the Beethoven (Bth) mouse model of deafness selectively disrupted the dominant mutant Tmc1 allele, reducing hearing loss. Inner hair cells (IHCs) and outer hair cells (OHCs) had significantly improved survival rates and auditory brainstem response (ABR) thresholds compared to uninjected controls (Gao et al., 2018). Lipid-based nanoparticles have also been used to deliver base editing machinery in a proof-of-concept study to ameliorate hearing loss (discussed further in section Base Editing; Yeh et al., 2018).
Supraparticles are colloidal nanoparticle aggregates with a larger drug loading capacity relative to individual nanoparticles (Sperling and Gradzielski, 2017). Supraparticles have already been used to deliver the developmental neurotrophin, brain-derived neurotrophic factor (BDNF), to the inner ear of a hearing loss guinea pig model (Wang et al., 2014). BDNF is required for the maintenance of spiral ganglion neurons (SGNs; Ylikoski et al., 1993) and may serve to protect or regenerate SGNs as well as promote synaptic regeneration at the ribbon synapse (Suzuki et al., 2016). Supraparticles offer sustained longer-term release of BDNF and maintained near-wild-type numbers of SGNs in the guinea pig cochlea (Wang et al., 2014). However, delivery of supraparticles may mediate the unintentional movement of small nanoparticles from the inner ear to the cerebral spinal fuid via the cochlear aqueduct (Zhang et al., 2013).
Another significant advancement in the non-viral delivery platform was the development of the polyethylene glycol-substituted 30-mer lysine peptide (CK30-PEG) nanoparticles, which have been used successfully in a cystic fibrosis clinical trial (Konstan et al., 2004). CK30-PEG nanoparticles may also be effective for the treatment of ocular diseases, as they show a retinal targeting efficiency comparable to viral vectors up to 2 weeks post-injection (Farjo et al., 2006; Han et al., 2012a). Appreciable transgene expression mediated by CK30-PEG nanoparticle gene delivery has been reported in retinal degeneration models, including an autosomal dominant model of RP (Cai et al., 2010; Han et al., 2012b). In the autosomal RP model study, nanoparticles containing the mouse opsin promoter and the Prph2 gene were subretinally delivered to Rds mice carrying a haploinsufficiency mutation, resulting in wild-type-level recovery of cone function but a modest restoration of rod function (Cai et al., 2010). Modified CK30-PEG nanoparticles with a photoreceptor-specific promoter also led to the structural and functional rescue of Stargardt-associated pathology in the Abca4−/− mouse model of vision loss (Han et al., 2012b). Recent studies have used solid lipid nanoparticles (SLNs) as a more efficient non-viral delivery system and a study on a mouse model of x-linked juvenile retinoschisis showed transduction of retinal pigment epithelium (RPE) and photoreceptors and improved retinal phenotype (Apaolaza et al., 2016).
Though nanoparticle delivery is promising and continues to be developed, there are several roadblocks to its clinical application, including biodegradability, biocompatibility, and non-specific transfection (Yin et al., 2014; Chen et al., 2016). Topical delivery is the simplest and most patient-friendly form of administration. However, ocular barriers and the long diffusion pathway prevent therapeutic levels from reaching the retina (Bisht et al., 2018). The intraocular injection route would, therefore, be preferable, though the transient expression associated with non-viral gene delivery (Bisht et al., 2018; Huang and Chau, 2019) would necessitate repeated injections and consequently, an increase in their associated risks. Thus far, no pre-clinical studies have investigated nanoparticle delivery of Usher genes but the large packaging capacity of non-viral vectors does increase the potential of this type of platform for treatment development for Usher-causing mutations in the large genes such as CDH23, PCDH15, ADGRV1, MYO7A, and USH2A (Table 1).
Viral-Based Gene Replacement Therapy
With the advantage of increased efficiency over non-viral vectors (Nayerossadat et al., 2012), viral vectors currently represent the most promising approach to therapeutic genetic interventions. Recombinant viral vectors utilize the inherent capability of viruses for cellular transduction to deliver genetic material to donor cells in vivo. However, viral vectors come with their limitations, including limited packaging capacities. Potential immunogenicity is also an obstacle to clinical application, though advances in molecular biology have allowed for the separation of wild-type viral coding genes and cis-acting sequences. Consequently, segregation can now produce viral vectors that do not reconstitute by recombination into productive viral particles but still maintain viral infectivity capacity (Kay et al., 2001).
Gene replacement therapy involves the introduction of a non-mutant copy of the affected gene to restore expression to the host cell or tissue (Sengillo et al., 2016). One of the first described, and most frequently used vectors, adenovirus, has a large (~40 kb) packaging capacity and has been used in over 400 clinical trials (Wold and Toth, 2013). In a study by Bennett et al. (1996), an adenovirus-based vector was used to deliver the Phosphodiesterase β subunit gene to Rd1 mice, delaying photoreceptor degeneration. However, many people have circulating antibodies against adenoviruses, so their use in gene therapies is limited due to a high immunogenic response (DiCarlo et al., 2018). Furthermore, adenovirus has shown to have low tropism for photoreceptor cells in vivo (Li et al., 1994), an additional limitation that instigated the search for other types of viral vectors with improved targeting in the eye. The sections below will discuss the two most promising vectors: lentivirus and adeno-associated virus (AAV).
Lentiviral Vectors
Lentiviral vectors, derived from the human immunodeficiency virus, were also initially favored by researchers for clinical application due to their relatively large packaging capacity (reviewed in Zheng et al., 2018). However, whilst their immunogenicity is low, their tendency to integrate into the genome calls into question their clinical applicability (Zheng et al., 2018). Pioneering work by Hashimoto et al. (2007) led to the development of a lentiviral-mediated MYO7A gene delivery vector that produced wild-type protein levels in RPE cells in culture, as well as rescuing phenotypic RPE defects in vitro. In vivo, melanosome mislocalization and opsin accumulation at the photoreceptor connecting cilium was corrected in the Myo7a-deficient shaker1 mouse model of Usher 1B (Hashimoto et al., 2007). Subsequently, UshStat, a therapeutic recombinant vector expressing the human MYO7A, was developed using the equine infectious anaemia virus (Zallocchi et al., 2014), a non-pathogenic, non-primate-derived lentivirus capable of transducing human cells (Mitrophanous et al., 1999; Mazarakis et al., 2001). Subretinal delivery of UshStat to shaker1 mice was shown to protect photoreceptors from light-induced degeneration and demonstrated tolerability in non-human primates, which led to this approach progressing to a clinical trial for the prevention of RP in Usher 1B patients (NCT02065011; results pending).
Adeno-Associated Viral Vectors (AAV)
Safety is of principal concern in all viral vector technologies and as AAVs are not known to cause any human pathogenesis, they have emerged as the vector of choice for gene therapy applications worldwide. AAVs have low immunogenicity, high cellular specificity, and long-lasting gene expression. AAVs typically persist as episomes [Zheng et al., 2018; circular genomes which replicate independently of their host (Watanabe and Fukasawa, 1961)], reducing the risk of insertional mutagenesis. Vandendriessche et al. (2007) directly compared lentiviral and AAV vectors and found that AAV serotypes 8 and 9 induced higher expression of transgenic factor IX protein than lentiviruses in mouse models of hemophilia B and severe combined immunodeficiency, without interacting with proinflammatory cytokines. AAVs also hold potential for the treatment of Usher retinal degeneration as they can efficiently transduce photoreceptors and RPE (Rodrigues et al., 2018). Successful treatment of vision loss in LCA type 2 (LCA2) and choroideremia via AAV-mediated delivery of the RPE65 and CHM genes, respectively, have been demonstrated in human clinical trials (reviewed in Russell et al., 2017). The LCA2 trials have now resulted in the first-ever Food and Drug Administration (FDA)-approved and European Medicines Agency (EMA)-approved AAV-based gene therapy drug for LCA2 due to recessive RPE65 mutations (Luxturna® or Luxturna™/voretigene neparvovec-ryzl or voretigene neparvovec, Roche and Novartis).
A major disadvantage of AAV vectors is their relatively small (4.7 kb) packaging capacity (Zheng et al., 2018); however, in vivo delivery of the smaller Usher genes has been investigated using AAV vectors. Delivery of rAAV2/8 containing USH1G cDNA to the inner ear improved hearing and hair cell disorganization in the Usher 1G mice (Emptoz et al., 2017). Durable inner ear expression of Usher genes has also resulted in in vivo protein restoration of WHIRLIN (Zou et al., 2011) and CLARIN-1 in several studies (Dinculescu et al., 2016; Geng et al., 2017; Dulon et al., 2018; György et al., 2019).
Transduction efficiency is a crucial factor in the success of gene therapy and is highly cell and serotype-dependent. Several synthetically-produced AAV variants have been investigated for Usher syndrome treatments. One example is the synthetic rAAV2/Anc80L65, which may transduce close to 100% of IHCs and 90% of OHCs in mice (Landegger et al., 2017). Using rAAV2/Anc80L65, Pan et al. (2017) demonstrated gene and protein recovery of Harmonin in an Usher 1C mouse model. Deverman et al. (2016) reported a highly efficient synthetic vector, AAV9-PHP.B, which showed robust transduction efficiency in the retina of a dominant RP mouse model (Giannelli et al., 2017). Further, AAV9-PHP.B carrying a green fluorescent protein (GFP) reporter transduced the inner ear and retina of wild-type mice at a rate of 60–80% for IHCs, 30–40% for OHCs and 70–80% for photoreceptors (György et al., 2019). When used to package the Clrn-1 gene, AAV9-PHP.B mediated rescue of low-frequency hearing (4–8 kHz) in a mouse model of Usher 3A deafness (György et al., 2019). However, the tropism and potential toxicity of the AAV9-PHP.B vector in the central nervous system (CNS) of non-human primates is still under study (Hordeaux et al., 2018; reviewed in Deverman et al., 2018). The potential, therefore, for treatment of Usher syndrome using certain AAV-based vectors is high, but the large size of some Usher genes, including some of the most common forms of Usher such as USH1B (MYO7A), does present unique challenges for the field.
Oversized Adeno-Associated Viral Vectors
As mentioned above, the packaging limitation of AAVs precludes delivery of several of the larger Usher genes, including MYO7A (USH1B; Jaijo et al., 2006) and PCDH15 (USH1F; Alagramam et al., 2001). Some studies, however, have pushed the limits of AAV packaging capacity by using oversized AAV transgene constructs (fAAV). A study by Allocca et al. (2008) identified rAAV2/5 as being an efficient packager of up to 8.9 kb of genetic material, allowing, in theory, the large retinal disease genes MYO7A, ABCA4 and CEP290 to be packaged and delivered subretinally. They showed that fAAV2/5 ABCA4 delivery led to a stable improvement of morphological abnormalities and retinal dysfunction associated with a mouse model of Stargardt disease. Additionally, Colella et al. (2013) identified defects in light/dark adaptation in shaker1 mice, which was improved by fAAV2/5 delivery of MYO7A. However, other studies have shown that these vectors do not contain full-length genes, but instead tend to contain heterogeneous mixtures of gene fragments, most of which are truncated and typically less than 5 kb (Dong et al., 2010). Furthermore, Grieger and Samulski (2005) demonstrated that AAV vectors carrying genomes larger than 5 kb had less efficient transduction capacity due to a post entry preferential degradation of AAVs carrying larger genomes. Though the truncated genomes may reassemble before transcription and form full-length proteins inside the cell (Lopes et al., 2013), the lack of characterization and heterogeneity of this approach limits its clinical use.
Multi-Adeno-Associated Viral Vector Systems
As an alternative to the oversized AAV approach, different research groups have tested the use of dual and triple AAV vector systems. Multi or dual AAV systems use the inherent concatemerization of AAV genomes in vivo to form full-length cDNA which can then be transcribed into functional mRNA within the transduced cell (Trapani et al., 2014). Different methods are used to deliver dual/multivectors and are usually divided into trans-splicing, overlap, and hybrid approaches. Trans-splicing vectors, first described by Yan et al. (2000), separates the gene of interest into 5′ (left) and 3′ (right) halves, with the 3′ end of the left construct containing a splice donor site, which concatemerises with the splice acceptor site located on the 5′ end of the right construct. In the overlap approach, the 3′ end of the right half and the 5′ end of the left half contain a highly recombinogenic overlapping region which mediates homologous recombination between gene segments (Duan et al., 2001). This sequence can be from an external gene, like alkaline phosphatase, or directly from the gene of interest. Finally, hybrid vectors contain both the splice donor/acceptor sites of trans-splicing vectors and the recombinogenic properties of overlapping vectors, allowing reconstitution to occur via either method (Ghosh et al., 2008).
Several research groups have published proof-of-concept studies using multi or dual AAV systems, including for inherited retinal diseases (Colella et al., 2014; Dyka et al., 2014, 2019; McClements et al., 2019). Multi-AAV systems have been used to express MYO7A in vivo and in vitro with equal or higher efficiency than fAAV delivery (Dyka et al., 2014), and to produce full-length mRNA with 100% fidelity to the target cDNA (Dyka et al., 2014). Additionally, Maddalena et al. (2018) expanded the transfer capacity further by using a triple therapeutic vector system to incorporate large genes such as ALMS1 and the Usher 1D gene, CDH23. Truncated protein products were detected in eyes treated with CDH23 but not ALMS1 triple AAV vectors. A functional response was not recorded in CDH23-treated mice but treated Alms1−/− mice showed a non-significant increase in outer nuclear layer thickness and transient (2–6 months) improvement of electroretinogram a- and b-wave measurements (Maddalena et al., 2018).
Despite the promising potential of dual AAV approaches, most studies show very low levels of protein expression using this system. Dyka et al. (2014) quantified the relative expression of full-length cDNA mediated by dual AAV reconstitution and found hybridizing vectors to be 2–3 fold more efficient than overlap and trans-splicing counterparts in MYO7A-transfected HEK293T cells. Data published by Colella et al. (2014) found that hybrid and trans-splice dual AAV reconstitution achieved approximately one-quarter of the photoreceptor expression levels induced by single AAV vectors in the large white pig retina. In their 2018 article, Maddalena et al. (2018) reported that the co-transduction rate for triple AAV vectors was <6% of single vector expression in HEK293T cells. Interestingly, this efficiency increased when vectors were delivered subretinally to mice and pigs (photoreceptor expression = 27 ± 6% and 39 ± 17% of that induced by single AAV, respectively). In a recent study, Carvalho et al. (2017) tested the in vitro and in vivo expression of the different dual AAV approaches and found hybrid vectors to have superior rates of reconstitution. They showed that reconstitution efficiency in HEK293T cells for trans-splicing, hybrid, and overlapping vectors was 10.3%, 15.3%, and 17.4%, respectively. The efficiency of overlapping vectors was found to be gene-specific as it was dependent on the length of the recombinogenic region and showed no detectable levels in vivo. In vivo subretinal delivery to the mouse retina resulted in full-length protein expression in 9.07% and 1.78% of cells transduced with hybrid and trans-splicing vectors, respectively (Carvalho et al., 2017). Interestingly, they were the first to show a discrepancy in reconstituted mRNA and protein levels after dual AAV delivery which may be explained by transcript instability of longer mRNA sequences (Feng and Niu, 2007).
An alternative to dual AAV systems may be dual-intein splicing, which is based on protein rather than mRNA reconstitution, but is still delivered by AAV vectors. Inteins are segments of proteins that have been described as “protein introns,” as they can excise themselves from the sequence of peptides and join the flanking portions (the exteins) together. Recently, Tornabene et al. (2019) demonstrated reporter protein levels in C57BL/6J mice retinae induced by dual-intein splicing to be comparable to single AAV transduction and significantly higher than dual AAV transduction. This approach was also shown to alleviate retinal degeneration in animal models of Leber Congenital Amaurosis (Rd16 mice) and Stargardt disease (Abca4−/− mice). Physiological symptoms of retinal degeneration, including RPE lipofuscin accumulation in Abca4−/− mice, and outer nuclear layer thinning in Rd16 mice, were significantly reduced in the dual-intein treated mice. Additionally, pupillary light responses increased by ~20% in treated Rd16 mice compared to untreated Rd16 controls (Tornabene et al., 2019). However, the viability of this approach for the delivery of large genes has not yet been tested in models of Usher syndrome.
DNA Editing
While gene delivery aims to replace a defective gene with a functional copy, DNA editing attempts to directly correct the mutation in vivo, which also allows the repaired gene to be expressed under endogenous regulators. Furthermore, the size of the gene is not a limiting factor. Repair of single base transitions or transversions at the DNA level can be achieved through the induction of a DNA break to facilitate incorporation of the correct DNA base (Kim et al., 1996; Christian et al., 2010; Jinek et al., 2012; Cong et al., 2013; Mali et al., 2013) while targeted conversion of a single DNA base can reverse a transitional mutation (Komor et al., 2016; Gaudelli et al., 2017).
Gene Editing
The first attempt using gene editing for Usher syndrome was a study by Overlack et al. (2012) that used zinc finger nucleases (ZFNs) to target the p.R31X mutation in the human Ush1c gene. Their in vitro results show partial repair of the Ush1c gene and recovery of harmonin protein in a p.R31X cell line after transfection with ZFNs. In recent years, however, the discovery of the clustered, regularly interspaced, palindromic repeats (CRISPR) and CRISPR-associated protein 9 (Cas9) system have changed the field of gene editing profoundly. Their higher efficiency, simplicity, and targeting capacity have made them the preferred technology of choice for gene editing (Khan, 2019).
The CRISPR/Cas9 system uses an adapted microbial immune technique for precise editing of the genome (Jinek et al., 2012; Cong et al., 2013; Mali et al., 2013). A guide RNA is engineered to target a specific locus, which is then digested by the Cas9 endonuclease (Jinek et al., 2012; Cong et al., 2013; Mali et al., 2013). Either of two endogenous mechanisms repairs double-stranded breaks in the genome. The clinically-favorable homology-directed repair relies on an intact template strand whilst error-prone non-homologous end-joining is independent of a template sequence, and often results in insertions or deletions (Adli, 2018).
CRISPR/Cas9 has been used to study potential treatments for inherited retinal diseases. Moreno et al. (2018) used the viral delivery of CRISPR components to downregulate Nrl in mouse models of non-Usher RP. Nrl is a transcriptional regulator that indirectly determines whether photoreceptors develop into rods or cones (Cheng et al., 2004; Moreno et al., 2018). The knockdown of Nrl expression transformed rods into cone-like cells that did not experience rod-specific degeneration. This leads to a decrease in rods and therefore night blindness, but an increase in daylight vision (Moreno et al., 2018). However, mouse models of Usher 1C and 1G have cones that are sensitive to oxidative stress, indicating that increasing the number of cone-like cells may not be a viable option for Usher RP treatment (Trouillet et al., 2018).
Viral delivery of CRISPR/Cas9 to fibroblasts derived from an Usher 2A patient showed some rescue of Usherin expression, however, the efficiency of restoration was not significant enough to proceed to clinical trials (Fuster-García et al., 2017). Additionally, induced pluripotent stem cells (iPSCs) derived from a patient with a mutation in MYO7A were investigated by Tang et al. (2016). Stereocilia-like protrusions from patient iPSCs developed disorganized morphology which was rescued by CRISPR/Cas9 editing (Tang et al., 2016), indicating a potential treatment avenue for Usher 1B patients that deserves further investigation.
Recently, the CRISPR/Cas9-based tool EDIT-101 received FDA approval for a clinical trial in LCA10 patients (NCT03872479), leading to the first-ever direct human administration of CRISPR/Cas92. EDIT-101 is a gene editing therapeutic which utilizes a Staphylococcus aureus Cas9 guide RNA which has high specificity for the c.2991 + 1655A >G transversion in intron 26 (IVS26) of the CEP290 gene, limiting off-target effects (Maeder et al., 2019). Recently, the company behind EDIT-101, Editas3, has publically announced the utilization of a new therapeutic, EDIT-102, to target Usher type 2A4. Another recent update is the introduction of a new gene editing tool, “prime editing,” which utilizes a synthetic fusion of an altered Cas9 endonuclease and reverse transcriptase, directed by a gene editing guide RNA (Anzalone et al., 2019). This technology shows promising specificity and broad applicability to a large number of human pathogenic mutations, although further studies will be needed to confirm its viability for treating Usher syndrome.
Though the potential of CRISPR/Cas9 to make precise and patient-specific gene corrections with high targeting capacity is attractive, the efficacy of in vivo delivery is still in doubt due to potential off-target effects (Fu et al., 2013; Kuscu et al., 2014). Advancements in the field since its original application in gene editing, including the development of anti-CRISPR proteins to limit non-specific activity (Pawluk et al., 2016; Rauch et al., 2017; Nakamura et al., 2019), have potential to address some limitations of this technology and further studies could help validate CRISPR/Cas9-based approaches for the treatment of Usher syndrome.
Base Editing
The advent of CRISPR/Cas9 gene editing systems has also allowed for the development of base editing, which utilizes inactivated Cas nucleases and single-stranded editing enzymes to replace one nucleobase with another without creating double-stranded breaks (Rees and Liu, 2018). Two classes of base editing technology have so far been reported; cytosine base editors (CBEs; Komor et al., 2016) which convert C-G base pairs to T-A, and adenine base editors (ABEs), which convert A-T to G-C (Gaudelli et al., 2017). Both CBEs and ABEs introduce transition mutations between chemically similar base pairs, which can theoretically correct over 60% of human pathogenic point mutations (Rees and Liu, 2018). However, transversion base editing (e.g., G-C to C-G) remains an elusive target.
Base editing has already been used in proof-of-concept studies in mouse models of Duchenne muscular dystrophy (Ryu et al., 2018), phenylketonuria (Villiger et al., 2018), hereditary tyrosinemia type I (Song et al., 2020) and hypercholesterolemia through targeting the PCSK9 gene (Chadwick et al., 2017). Importantly, base editing has also been used to investigate enhanced cellular reprogramming of supporting cells to cochlea hair cells to mediate hearing loss (Yeh et al., 2018). C-T conversion of a single base in the CTNNB1 gene prevented phosphorylation and degradation of β-catenin protein, leading to a 7-fold increase of β-catenin levels in HEK293T cultures. Consequently, there was an increase in signaling to the Wnt pathway, which is crucial to the development of sensory hair cells. When delivered to mice via intracochlear injection, base editing resulted in the differentiation of cochlea supporting cells to MYO7A-expressing hair cells (Yeh et al., 2018).
RNA Intervention
RNA splicing can be altered to either restore exons lost due to aberrant splicing induced by the mutation, or induce exon skipping to remove nonsense or missense mutations in coding regions. RNA intervention resulting in altered splicing is achieved through the binding of an antisense oligonucleotide to RNA strands (reviewed by Havens et al., 2013). Base editing of RNA is also possible (Bass, 2002). Finally, translational readthrough techniques aim to suppress protein truncation by overriding mutations that cause premature termination of the translation machinery. The reader is referred to a review outlining the therapeutic progress of translational readthrough-inducing compounds in the treatment of inherited diseases (Nagel-Wolfrum et al., 2016).
RNA-Based Drug Interventions
RNA molecules designed to silence or interfere with toxic gain-of-function mutations have been investigated in the treatment of several non-Usher models of genetic hearing loss. Notably, Maeda et al. (2005) identified a short interfering RNA (siRNA) that targets the R75W allele variant in GJB2, a common cause of autosomal recessive hearing loss. The synthetic siRNA suppressed GJB2 in HEK293T cells by >80% for more than 120 h. When administered to the GJB2 mouse model, siRNA treatment suppressed GJB2 expression by >70% and prevented hearing loss (Maeda et al., 2005, 2007). Another study by Shibata et al. (2016) developed a microRNA targeting non-syndromic deafness caused by gain-of-function mutations in TMC1. The microRNA was packaged via AAV2/9 and delivered to Bth mice, resulting in significant preservation of hearing for up to 21 weeks relative to untreated controls. The animals who responded best to treatment maintained ABR thresholds 40 dB greater than untreated counterparts (Shibata et al., 2016).
Antisense Oligonucleotides
ASOs are short nucleic acid sequences that modulate gene expression via complementary binding to mRNA. ASOs are often synthesized to activate ribonuclease H (RNAse-H, which degrades mRNA) or target splicing defects (Goyal and Narayanaswami, 2018). ASOs can then be designed to target a pathogenic mutation; hence, the size of the gene is not an obstacle as with gene delivery techniques. However, the half-life of the drug means re-occurring invasive administration to the eye rather than one-off treatments, as is the case with a gene therapy approach.
ASO treatment has been widely investigated for retinal disease therapies (Huang et al., 2017). Recently, up to nine antisense-oligonucleotide variants were identified for the treatment of Stargardt disease caused by the intronic c.4539 + 2001G >A mutation in the large ABCA4 gene (Garanto et al., 2019). In a separate study, screening of ASOs led to the discovery of QR-110 (Dulla et al., 2018), a therapeutic for LCA type 10 (LCA10), caused by the c.2991 + 1655A >G mutation, in the CEP290 gene (Den Hollander et al., 2006). Like Usher syndrome, the pathogenesis of LCA10 is caused by defects in the photoreceptor connecting cilium. QR-110 was able to restore wild-type CEP290 transcript and protein expression in mutant fibroblasts and decrease ciliopathy in 3-dimensional (3D) retinal organoids (explained further in section Cell Therapy for Retinal Degeneration). The drug was also well-tolerated in mice, rabbits, and non-human primates (Dulla et al., 2018), leading to the approval of phase I/IIa clinical trial (NCT03140969) by ProQR Therapeutics5.
ASOs have also been developed for autosomal dominant RP (Naessens et al., 2019), as well as Usher 2A-associated RP (Slijkerman et al., 2016). An engineered ASO targeting a pseudo-exon-causing mutation in USH2A (c.7595–2144A >G) displayed splice-correcting properties in patient-derived fibroblasts (Slijkerman et al., 2016). The same company that funded this study, ProQR Therapeutics, has also investigated an ASO, QR-421a, for the treatment of Usher 2A-associated RP caused by a common exon 13 mutation (c.2299delG; Van Diepen et al., 2019). The efficacy of this treatment was initially demonstrated in patient-derived retinal organoids and animal models (Table 3), with exon-skipping capability maintained in cynomolgus monkeys for more than 100 days post-treatment. They also showed that Usherin protein was present in wild-type zebrafish larvae at the photoreceptor connecting cilium but absent in untreated c.2299delG zebrafish. Partial restoration of correctly localized usherin expression was observed in treated larvae as well as improved ERG recordings compared to untreated zebrafish (Van Diepen et al., 2019; conference abstract). The results of this study have allowed for advancement to a clinical trial (NCT03780257), which has already led to the first treatment to the eyes of an Usher 2A patient using QR-421a6 (preliminary findings reported on the 20th of April 2020 here: https://ir.proqr.com/news-releases/news-release-details/proqr-announces-positive-findings-interim-analysis-phase-12).
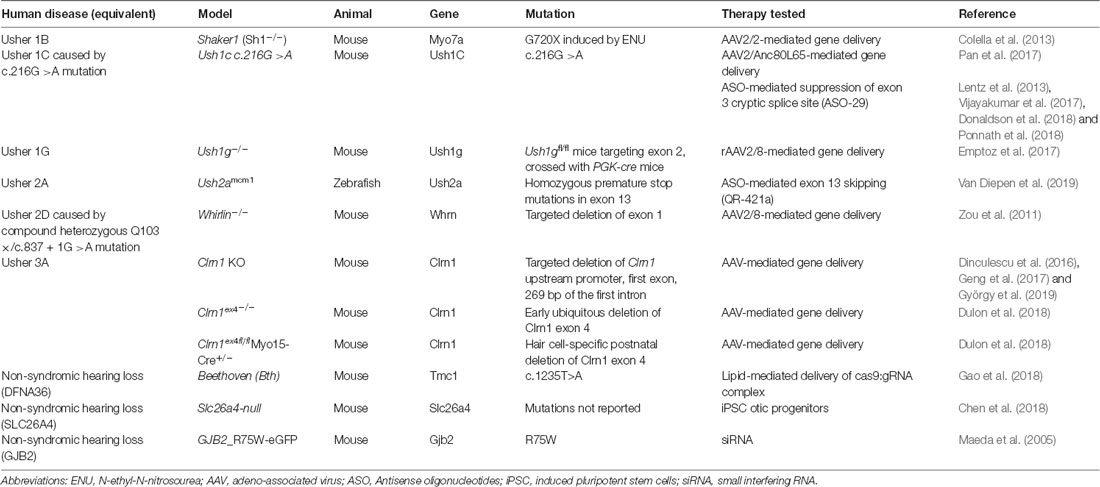
Table 3. Summary of transgenic animal disease models discussed in this review.
Several studies have investigated ASO-29 treatment of the Usher 1C, c.216G >A mouse model, which exhibits hearing loss as well as disrupted exploratory movements, indicating vestibular dysfunction. ASO-29 administration to the inner ear of c.216G >A mouse pups has been shown to improve both the auditory and vestibular response (Lentz et al., 2013; Vijayakumar et al., 2017; Donaldson et al., 2018). However, mounting evidence has demonstrated an age threshold for effective delivery, with early-treated mice responding better to treatment. Ponnath et al. (2018) demonstrated that both outer and inner hair cell function was effectively restored by ASO-29 treatment, but the threshold for effective outer hair cell treatment (post-natal day 5) was lower than that of inner hair cell treatment (post-natal day 7). Considering these promising results in the auditory and vestibular response of USH1C mice, it would be interesting to assess the efficacy of ASO-29 towards the vision loss phenotype. However, the limited visual phenotype of the USH1C mice model means alternative models will need to be used for testing ASO-29.
Translational Readthrough Inducing Drugs
Nonsense mutations cause premature termination codons (PTCs), leading to the production of a truncated polypeptide or targeting of the transcript by nonsense-mediated decay (NMD). When a translation termination codon (UAA, UGA, UAG) enters the ribosomal A site, it is recognized by eukaryotic release factor 1 (eRF1), which changes conformation when bound to eRF3, initiating a signal cascade that results in hydrolytic cleavage of the polypeptide by eRF1. Translation termination is a competition between recognition by eRF1 leading to termination and continuation of the translation process, leading to readthrough or natural suppression. Natural suppression occurs >0.1% of the time in non-mutant transcripts, but PTCs increase the rate of natural suppression to up to 1% (Keeling et al., 2012). Nonsense suppression therapies aim to increase translational readthrough, avoiding the production of a truncated protein. The two main obstacles with this type of therapy are avoiding mRNA degradation by NMD, and avoiding the promotion of non-specific readthrough (Frischmeyer and Dietz, 1999; Keeling and Bedwell, 2011; Wang and Gregory-Evans, 2015; Dabrowski et al., 2018; Campofelice et al., 2019).
Nonsense suppression has clear advantages over gene-based therapies; they are not gene- or mutation-specific, the size of the gene is not an issue, and they allow the cell to maintain expression regulation. However, they are associated with nephrotoxic (Mingeot-Leclercq and Tulkens, 1999) and ototoxic (Selimoglu, 2007) effects and often result in the incorporation of a non-cognate amino acid, which may alter protein function. A recent advancement in the field was the development of anticodon engineered transfer RNAs (ACE-tRNAs), which recognize and suppress stop codons while encoding the cognate amino acid of the non-mutant polypeptide (Lueck et al., 2019). This method induced a specific readthrough of CFTR mutations in vitro. Low levels of off-target suppression were detected, depending on the genetic environment of the mutation, but the authors suggest that endogenous cellular pathways to degrade incorrectly transcribed proteins would be sufficient to offset these effects (Lueck et al., 2019).
Several studies have investigated nonsense-mediated therapies for the treatment of Usher syndrome. Initial studies focussed on nb30, a synthetic derivative developed from the commercial aminoglycoside, paromomycin (Nudelman et al., 2006). Nb30 was shown to induce significant nonsense suppression of a common USH1C mutation (p.R31X) with reduced toxicity and increased biocompatibility compared to commercial aminoglycosides (Goldmann et al., 2010). Similarly, favorable toxicity profiles were observed in nb30-mediated suppression of PCDH15 nonsense mutations relative to traditional antibiotics (Rebibo-Sabbah et al., 2007). Subsequently, a second paromomycin derivative, nb54, was identified as a promising drug candidate for nonsense suppression which demonstrated readthrough ability in several prominent disease genes, including Usher 1F syndrome (Nudelman et al., 2009). In this study, Nudelman et al. (2009) show that nb54 is capable of inducing in vitro stop codon suppression 3–5-fold times more efficient for PCDH15 (Usher 1F) mutations p.R3X and p.R245X compared to three other aminoglycoside compounds.
PTC-124 (Ataluren) is another translational readthrough inducer which has been studied for application in cystic fibrosis and Duchenne muscular dystrophy cases (Welch et al., 2007), though success has thus far been limited in clinical trials (Wilschanski et al., 2011; Kerem et al., 2014; McDonald et al., 2017). PTC-124-induced readthrough in vitro has been demonstrated in the common p.Trp3955* mutation, which accounts for 13% of USH2A mutations (Neuhaus et al., 2017). Additionally, PTC-124 treatment induced in vitro and in vivo readthrough of USH1C mutations, leading to the recovery of protein function with superior biocompatibility to gentamicin and nb30 (Goldmann et al., 2011). Finally, Goldmann et al. (2012) compared nb30, nb54, and PTC-124 for translational readthrough of USH1C mutations and identified both PTC-124 and nb54 as ideal drug candidates. Though promising, functional rescue of Usher phenotypes in vivo will nonetheless be necessary to evaluate the efficacy of both PTC-124 and nb54.
Cell Replacement Therapy
Cellular therapies are therapeutic approaches that aim to regenerate damaged tissue by introducing replacement donor cells (Zakrzewski et al., 2019). This approach relies on the survival of connecting interneurons when the stimulus-receiving neuron has already degenerated in the cochlea or retina. Typically, progenitor cells derived from stem cells are used as an unlimited source of donor cells. Stem cells are pluripotent progenitors that can differentiate into cell lineages from each of the three germ layers. Early stem cell studies relied on the controversial use of human embryonic stem cells (hESCs; Omole and Fakoya, 2018) until the discovery in 2007 by Shinya Yamanaka and colleagues that adult human somatic cells, such as adult fibroblasts, could be reprogrammed back into a pluripotent state (Takahashi et al., 2007). They achieved this through the expression of characteristic pluripotent markers SOX2, OCT3/4, KLF4, and c-MYC, delivered to the cell via retroviral vectors (Takahashi et al., 2007). As the stem cells carry the same genetic information as the somatic cells from which they were made, this breakthrough allowed human diseases to be replicated and studied non-invasively in the laboratory for the first time (Omole and Fakoya, 2018).
Cell Therapy for Retinal Degeneration
iPSCs can aggregate to form “embryoid bodies” (aggregates of cells thought to mimic the early embryo), which can be prompted to differentiate into specific lineages (Omole and Fakoya, 2018), including 3D retinal organoids (Nakano et al., 2012; Phillips et al., 2012; Zhong et al., 2014; Mellough et al., 2015, 2019). These organoids closely resemble in vivo human eye development, forming eye field-like domains that form 3D retinal neuroepithelium and RPE. The neural retina and RPE domains transition into a pseudo optic cup-like structure and retinal progenitor cells differentiate into neurons including horizontal cells, amacrine cells, and retinal ganglion cells (Zhong et al., 2014). Organoids develop a defined outer nuclear layer containing radially-aligned photoreceptors with detectable inner and outer segments connected by a connecting cilium (Zhong et al., 2014; Mellough et al., 2015, 2019; Lowe et al., 2016; Parfitt et al., 2016). These photoreceptors express phototransduction proteins, including opsins, and can respond to a light stimulus (Zhong et al., 2014; Hallam et al., 2018; Mellough et al., 2019).
Retinal organoids are a good resource of photoreceptor progenitors for transplantation. These progenitors can be enriched before transplantation for a homogenous cell population via cell sorting for dissociated cell transplants (Lakowski et al., 2015, 2018). MacLaren et al. (2006) transplanted developing mouse photoreceptors into the retinae of degenerative mouse models with successful integration and differentiation, noting an optimal therapeutic window corresponding to the specification of rod cell fate. Subsequently, photoreceptor transplantation was reported by several research groups (Pearson et al., 2012; Barber et al., 2013; Gonzalez-Cordero et al., 2013; Singh et al., 2013; Santos-Ferreira et al., 2016), including Barber et al. (2013) who looked into six mouse models of inherited retinal degenerations (IRDs) with environmental, disease course, and genetic factors influencing the integration outcome (Barber et al., 2013). Though these initial photoreceptor transplantation experiments seemed promising, the transfer of cytoplasmic material between labeled donors and host photoreceptors has been attributed to the seemingly high rates of integration (Santos-Ferreira et al., 2016; Singh et al., 2016). Alternatively, a stem cell-derived retinal sheet may be transplanted into the recipient’s eye (Assawachananont et al., 2014). Though clinical trials have focussed on RPE transfer in patients with age-related macular degeneration (AMD) and Stargardt Disease (NCT03102138, NCT02941991, NCT01469832), none thus far have reported results on photoreceptor transplants. However, a first-in-human phase I/II dose-escalation study is currently examining the safety and tolerability of human retinal progenitor cells in patients with RP (NCT02464436, estimated completion date: July 2021). Furthermore, proof-of-principle studies in mouse and non-human primates showed survival of retinal sheets [containing hESC-derived retinal cells (Shirai et al., 2016), hiPSC-derived RPE cells (Mandai et al., 2017) or hiPSC-derived retinal progenitors (Tu et al., 2019)] post-transplantation, and improvement of light sensitivity (Shirai et al., 2016; Mandai et al., 2017; Tu et al., 2019).
Cell Therapy for Neurosensory Deafness
Stem cell therapy also has the potential to treat hearing loss by replacing cochlear hair cells, although this is a difficult environment for this approach. The hostile high potassium luminal fluid (endolymph) environment of the cochlear duct poses significant challenges for cell survival. Further, the robust tight junctions in the auditory epithelium in the organ of Corti make donor cellular integration difficult. A recent article reported the ability of iPSC-derived otic progenitors to form connections with co-cultured spiral ganglion-like cells (Chen et al., 2018). Subsequently, these cells were detached and administered to the inner ear via round window injection in hearing loss Slc26a4 mice. Cells positive for the hair cell marker, MYO7A (also causative of Usher 1B), were detected in the organ of Corti, indicating the potential of this method to deliver progenitor cells capable of migrating, differentiating and forming appropriate connections in the inner ear (Chen et al., 2018). However, the progenitors did not form an organized stereocilial arrangement, which is critical to hearing, and disrupted in Usher cases (Mathur and Yang, 2015).
Limitations of Cell Therapies
The development of cellular therapies presents a promising and broad approach to treat a host of sensory diseases regardless of genetic etiology. However, several safety concerns remain to be solved. Of principal concern is the potential for donor cells to cause neoplastic changes in the tissue, particularly those that are derived from stem cells. Mouse embryonic stem cells injected into the subretinal space have been shown to induce tumor formation (Arnhold et al., 2004), though human iPSC-derived photoreceptor progenitor delivery to rd1 mice found no evidence of abnormal cell growth (Barnea-Cramer et al., 2016). Additionally, Chen et al. (2018) injected mouse models with hiPSC-derived otic progenitors and hiPSC controls to examine the tumorigenicity of hiPSC-derived cells. They reported tumor formation in hiPSC-injected tissue, but not in tissue injected with hiPSC-derived progenitors (Chen et al., 2018). Exclusion of undifferentiated stem cells before transplantation could further decrease the risk of neoplastic changes in the host. In a recent study, Gagliardi et al. (2018) demonstrated the potential of this strategy by safely transplanting CD73+ photoreceptor precursors separated from a cell population using magnetic-activated cell sorting into the eyes of P23H-transgenic rats.
Despite the multitude of challenges in delivering stem cell therapeutics safely and effectively, regenerative medicine remains attractive to patients, industry, and commercial clinics. Several stem cell-based clinical trials are listed on clinicaltrials.gov for AMD (NCT01736059, NCT01920867) and other ocular diseases (NCT01920867), including RP (NCT02320812). Recently, the stem cell ophthalmology treatment study (SCOTS) published results from a phase I/IIa study of five Usher syndrome patients with bilaterally-treated eyes (Weiss and Levy, 2019). Autologous bone marrow-derived stem cells were separated from bone marrow aspirate and administered into patients’ eyes via a combination of retrobulbar, sub-Tenon, intravitreal, subretinal, and intra-optic injections, as chosen by the patient and depending on the extent of visual loss and relative risks. Increases in visual acuity were noted in 80% of treated eyes with a statistical significance of p < 0.01. There was no reported visual loss nor any complications up to a year post-treatment (Weiss and Levy, 2019). However, a continual follow up is needed to confirm the long-term visual acuity effects and determine whether the patients remain free of adverse events.
Conclusions
Significant advancements have been made in the last several years towards the treatment and prevention of sensory loss in Usher patients. Particularly, gene and cell therapies pose attractive, potentially one-off solutions that would reduce the burden of invasive re-administration of medications to the eye. Though the majority of studies are currently proof-of-principle treatment strategies using animal models of disease (summarized in Table 3), it is highly possible that the results from current ongoing clinical trials may translate into effective new treatments (see Table 4 for a summary of ongoing clinical trials). However, more temporary therapeutics such as ASOs and translational readthrough inhibitors may also offer a significant reduction in disability. Continuing to improve the safety and efficacy of these treatment approaches is of utmost importance to provide commercially available options for Usher syndrome patients.

Table 4. Active clinical trials for Usher patients.
Author Contributions
LF wrote the first draft of the manuscript. FC, CM, and LC contributed to manuscript revision, reading, and approval of the submitted version.
Funding
This work was supported by funding from Retina Australia (LC, FC, and CM), Usher 1 Collaborative (LC), Genetics Cures Australia (LC), Lions Eye Institute/Save Sight Foundation Brian King Fellowship (CM), Australian National Health and Medical Research Council (MRF1142962, GNT1188694, and GNT1116360; FC).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
- ^ https://tinyurl.com/r93ejqs
- ^ https://www.nature.com/articles/d41586-020-00655-8
- ^ https://www.editasmedicine.com
- ^ https://ir.editasmedicine.com/news-releases/news-release-details/editas-medicine-reports-recent-progress-jp-morgan-healthcare-0
- ^ https://www.proqr.com
- ^ https://www.globenewswire.com/news-release/2019/03/11/1751124/0/en/ProQR-Doses-First-Patient-in-Phase-1-2-STELLAR-Trial-of-QR-421a-for-Usher-Syndrome-Type-2.html
References
Adli, M. (2018). The CRISPR tool kit for genome editing and beyond. Nat. Commun. 9:1911. doi: 10.1038/s41467-018-04252-2
Ahmed, Z. M., Riazuddin, S., Aye, S., Ali, R. A., Venselaar, H., Anwar, S., et al. (2008). Gene structure and mutant alleles of PCDH15: nonsyndromic deafness DFNB23 and type 1 Usher syndrome. Hum. Genet. 124, 215–223. doi: 10.1007/s00439-008-0543-3
Alagramam, K. N., Murcia, C. L., Kwon, H. Y., Pawlowski, K. S., Wright, C. G., and Woychik, R. P. (2001). The mouse Ames waltzer hearing-loss mutant is caused by mutation of Pcdh15, a novel protocadherin gene. Nat. Genet. 27, 99–102. doi: 10.1038/83837
Allocca, M., Doria, M., Petrillo, M., Colella, P., Garcia-Hoyos, M., Gibbs, D., et al. (2008). Serotype-dependent packaging of large genes in adeno-associated viral vectors results in effective gene delivery in mice. J. Clin. Invest. 118, 1955–1964. doi: 10.1172/JCI34316
Anzalone, A. V., Randolph, P. B., Davis, J. R., Sousa, A. A., Koblan, L. W., Levy, J. M., et al. (2019). Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576, 149–157. doi: 10.1038/s41586-019-1711-4
Apaolaza, P. S., del Pozo-Rodríguez, A., Solinís, M. A., Rodríguez, J. M., Friedrich, U., Torrecilla, J., et al. (2016). Structural recovery of the retina in a retinoschisin-deficient mouse after gene replacement therapy by solid lipid nanoparticles. Biomaterials 90, 40–49. doi: 10.1016/j.biomaterials.2016.03.004
Arnhold, S., Klein, H., Semkova, I., Addicks, K., and Schraermeyer, U. (2004). Neurally selected embryonic stem cells induce tumor formation after long-term survival following engraftment into the subretinal space. Invest. Ophthalmol. Vis. Sci. 45, 4251–4255. doi: 10.1167/iovs.03-1108
Assawachananont, J., Mandai, M., Okamoto, S., Yamada, C., Eiraku, M., Yonemura, S., et al. (2014). Transplantation of embryonic and induced pluripotent stem cell-derived 3D retinal sheets into retinal degenerative mice. Stem Cell Reports 2, 662–674. doi: 10.1016/j.stemcr.2014.03.011
Bademci, G., Foster, J. II., Mahdieh, N., Bonyadi, M., Duman, D., Cengiz, F. B., et al. (2016). Comprehensive analysis via exome sequencing uncovers genetic etiology in autosomal recessive nonsyndromic deafness in a large multiethnic cohort. Genet. Med. 18, 364–371. doi: 10.1038/gim.2015.89
Barber, A. C., Hippert, C., Duran, Y., West, E. L., Bainbridge, J. W. B., Warre-Cornish, K., et al. (2013). Repair of the degenerate retina by photoreceptor transplantation. Proc. Natl. Acad. Sci. U S A 110, 354–359. doi: 10.1073/pnas.1212677110
Barnea-Cramer, A. O., Wang, W., Lu, S.-J., Singh, M. S., Luo, C., Huo, H., et al. (2016). Function of human pluripotent stem cell-derived photoreceptor progenitors in blind mice. Sci. Rep. 6:29784. doi: 10.1038/srep29784
Bass, B. L. (2002). RNA editing by adenosine deaminases that act on RNA. Annu. Rev. Biochem. 71, 817–846. doi: 10.1146/annurev.biochem.71.110601.135501
Bennett, J., Tanabe, T., Sun, D., Zeng, Y., Kjeldbye, H., Gouras, P., et al. (1996). Photoreceptor cell rescue in retinal degeneration (Rd) mice by in vivo gene therapy. Nat. Med. 2, 649–654. doi: 10.1038/nm0696-649
Berson, E. L., Rosner, B., Sandberg, M. A., Hayes, K. C., Nicholson, B. W., Weigel-DiFranco, C., et al. (1993). A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa. Arch. Ophthalmol. 111, 761–772. doi: 10.1001/archopht.1993.01090060049022
Berson, E. L., Rosner, B., Sandberg, M. A., Weigel-DiFranco, C., and Willett, W. C. (2012). Ω-3 intake and visual acuity in patients with retinitis pigmentosa receiving vitamin A. Arch. Ophthalmol. 130, 707–711. doi: 10.1001/archophthalmol.2011.2580
Berson, E. L., Rosner, B., Sandberg, M. A., Weigel-DiFranco, C., Moser, A., Brockhurst, R. J., et al. (2004). Further evaluation of docosahexaenoic acid in patients with retinitis pigmentosa receiving vitamin A treatment: subgroup analyses. Arch. Ophthalmol. 122, 1306–1314. doi: 10.1001/archopht.122.9.1306
Berson, E. L., Weigel-DiFranco, C., Rosner, B., Gaudio, A. R., and Sandberg, M. A. (2018). Association of vitamin a supplementation with disease course in children with retinitis pigmentosa. JAMA Ophthalmol. 136, 490–495. doi: 10.1001/jamaophthalmol.2018.0590
Bisht, R., Mandal, A., Jaiswal, J. K., and Rupenthal, I. D. (2018). Nanocarrier mediated retinal drug delivery: overcoming ocular barriers to treat posterior eye diseases. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 10:e1473. doi: 10.1002/wnan.1473
Bonnet, C., Grati, M. H., Marlin, S., Levilliers, J., Hardelin, J.-P., Parodi, M., et al. (2011). Complete exon sequencing of all known Usher syndrome genes greatly improves molecular diagnosis. Orphanet J. Rare Dis. 6:21. doi: 10.1186/1750-1172-6-21
Cai, X., Conley, S. M., Nash, Z., Fliesler, S. J., Cooper, M. J., and Naash, M. I. (2010). Gene delivery to mitotic and postmitotic photoreceptors via compacted DNA nanoparticles results in improved phenotype in a mouse model of retinitis pigmentosa. FASEB J. 24, 1178–1191. doi: 10.1096/fj.09-139147
Campochiaro, P. A., Iftikhar, M., Hafiz, G., Akhlaq, A., Tsai, G., Wehling, D., et al. (2020). Oral N-acetylcysteine improves cone function in retinitis pigmentosa patients in phase I trial. J. Clin. Invest. 130, 1527–1541. doi: 10.1172/JCI132990
Campofelice, A., Lentini, L., Di Leonardo, A., Melfi, R., Tutone, M., Pace, A., et al. (2019). Strategies against nonsense: oxadiazoles as translational readthrough-inducing drugs (TRIDs). Int. J. Mol. Sci. 20:3329. doi: 10.3390/ijms20133329
Carvalho, L. S., Turunen, H. T., Wassmer, S. J., Luna-Velez, M. V., Xiao, R., Bennett, J., et al. (2017). Evaluating efficiencies of dual AAV approaches for retinal targeting. Front. Neurosci. 11:503. doi: 10.3389/fnins.2017.00503
Chadwick, A. C., Wang, X., and Musunuru, K. (2017). In vivo base editing of pcsk9 (proprotein convertase subtilisin/kexin type 9) as a therapeutic alternative to genome editing. Arterioscler. Thromb. Vasc. Biol. 37, 1741–1747. doi: 10.1161/atvbaha.117.309881
Chen, J., Guo, Z., Tian, H., and Chen, X. (2016). Production and clinical development of nanoparticles for gene delivery. Mol. Ther. Methods Clin. Dev. 3:16023. doi: 10.1038/mtm.2016.23
Chen, J., Hong, F., Zhang, C., Li, L., Wang, C., Shi, H., et al. (2018). Differentiation and transplantation of human induced pluripotent stem cell-derived otic epithelial progenitors in mouse cochlea. Stem Cell Res. Ther. 9:230. doi: 10.1186/s13287-018-0967-1
Cheng, H., Khanna, H., Oh, E. C., Hicks, D., Mitton, K. P., and Swaroop, A. (2004). Photoreceptor-specific nuclear receptor NR2E3 functions as a transcriptional activator in rod photoreceptors. Hum. Mol. Genet. 13, 1563–1575. doi: 10.1093/hmg/ddh173
Christian, M., Cermak, T., Doyle, E. L., Schmidt, C., Zhang, F., Hummel, A., et al. (2010). Targeting DNA double-strand breaks with TAL effector nucleases. Genetics 186, 757–761. doi: 10.1534/genetics.110.120717
Colella, P., Sommella, A., Marrocco, E., Di Vicino, U., Polishchuk, E., Garcia Garrido, M., et al. (2013). Myosin7a deficiency results in reduced retinal activity which is improved by gene therapy. PLoS One 8:e72027. doi: 10.1371/journal.pone.0072027
Colella, P., Trapani, I., Cesi, G., Sommella, A., Manfredi, A., Puppo, A., et al. (2014). Efficient gene delivery to the cone-enriched pig retina by dual AAV vectors. Gene Ther. 21, 450–456. doi: 10.1038/gt.2014.8
Cong, L., Ran, F. A., Cox, D., Lin, S., Barretto, R., Habib, N., et al. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823. doi: 10.1126/science.1231143
Cosgrove, D., and Zallocchi, M. (2014). Usher protein functions in hair cells and photoreceptors. Int. J. Biochem. Cell Biol. 46, 80–89. doi: 10.1016/j.biocel.2013.11.001
Dabrowski, M., Bukowy-Bieryllo, Z., and Zietkiewicz, E. (2018). Advances in therapeutic use of a drug-stimulated translational readthrough of premature termination codons. Mol. Med. 24, 25–25. doi: 10.1186/s10020-018-0024-7
Den Hollander, A. I., Koenekoop, R. K., Yzer, S., Lopez, I., Arends, M. L., Voesenek, K. E. J., et al. (2006). Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. Am. J. Hum. Genet. 79, 556–561. doi: 10.1086/507318
Deverman, B. E., Pravdo, P. L., Simpson, B. P., Kumar, S. R., Chan, K. Y., Banerjee, A., et al. (2016). Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat. Biotechnol. 34, 204–209. doi: 10.1038/nbt.3440
Deverman, B. E., Ravina, B. M., Bankiewicz, K. S., Paul, S. M., and Sah, D. W. (2018). Gene therapy for neurological disorders: progress and prospects. Nat. Rev. Drug Discov. 17, 641–659. doi: 10.1038/nrd.2018.110
DiCarlo, J. E., Mahajan, V. B., and Tsang, S. H. (2018). Gene therapy and genome surgery in the retina. J. Clin. Invest. 128, 2177–2188. doi: 10.1172/jci120429
Dinculescu, A., Stupay, R. M., Deng, W. T., Dyka, F. M., Min, S. H., Boye, S. L., et al. (2016). AAV-mediated clarin-1 expression in the mouse retina: implications for USH3A gene therapy. PLoS One 11:e0148874. doi: 10.1371/journal.pone.0148874
Donaldson, T. N., Jennings, K. T., Cherep, L. A., McNeela, A. M., Depreux, F. F., Jodelka, F. M., et al. (2018). Antisense oligonucleotide therapy rescues disruptions in organization of exploratory movements associated with Usher syndrome type 1C in mice. Behav. Brain Res. 338, 76–87. doi: 10.1016/j.bbr.2017.10.012
Dong, B., Nakai, H., and Xiao, W. (2010). Characterization of genome integrity for oversized recombinant AAV vector. Mol. Ther. 18, 87–92. doi: 10.1038/mt.2009.258
Duan, D., Yue, Y., and Engelhardt, J. F. (2001). Expanding AAV packaging capacity with trans-splicing or overlapping vectors: a quantitative comparison. Mol. Ther. 4, 383–391. doi: 10.1006/mthe.2001.0456
Dulla, K., Aguila, M., Lane, A., Jovanovic, K., Parfitt, D. A., Schulkens, I., et al. (2018). Splice-modulating oligonucleotide QR-110 restores CEP290 mRNA and function in human c.2991+1655A>G LCA10 models. Mol. Ther. Nucleic Acids 12, 730–740. doi: 10.1016/j.omtn.2018.07.010
Dulon, D., Papal, S., Patni, P., Cortese, M., Vincent, P. F., Tertrais, M., et al. (2018). Clarin-1 gene transfer rescues auditory synaptopathy in model of Usher syndrome. J. Clin. Invest. 128, 3382–3401. doi: 10.1172/jci94351
Dyka, F. M., Boye, S. L., Chiodo, V. A., Hauswirth, W. W., and Boye, S. E. (2014). Dual adeno-associated virus vectors result in efficient in vitro and in vivo expression of an oversized gene, MYO7A. Hum. Gene Ther. Methods 25, 166–177. doi: 10.1089/hgtb.2013.212
Dyka, F. M., Molday, L. L., Chiodo, V. A., Molday, R. S., and Hauswirth, W. W. (2019). Dual ABCA4-AAV vector treatment reduces pathogenic retinal A2E accumulation in a mouse model of autosomal recessive stargardt disease. Hum. Gene Ther. 30, 1361–1370. doi: 10.1089/hum.2019.132
Ebermann, I., Phillips, J. B., Liebau, M. C., Koenekoop, R. K., Schermer, B., Lopez, I., et al. (2010). PDZD7 is a modifier of retinal disease and a contributor to digenic Usher syndrome. J. Clin. Invest. 120, 1812–1823. doi: 10.1172/JCI39715
El-Amraoui, A., and Petit, C. (2014). The retinal phenotype of Usher syndrome: pathophysiological insights from animal models. C. R. Biol. 337, 167–177. doi: 10.1016/j.crvi.2013.12.004
Emptoz, A., Michel, V., Lelli, A., Akil, O., Boutet de Monvel, J., Lahlou, G., et al. (2017). Local gene therapy durably restores vestibular function in a mouse model of Usher syndrome type 1G. Proc. Natl. Acad. Sci. U S A 114, 9695–9700. doi: 10.1073/pnas.1708894114
Farjo, R., Skaggs, J., Quiambao, A. B., Cooper, M. J., and Naash, M. I. (2006). Efficient non-viral ocular gene transfer with compacted DNA nanoparticles. PLoS One 1:e38. doi: 10.1371/journal.pone.0000038
Feng, L., and Niu, D.-K. (2007). Relationship between mRNA stability and length: an old question with a new twist. Biochem. Genet. 45, 131–137. doi: 10.1007/s10528-006-9059-5
Frischmeyer, P. A., and Dietz, H. C. (1999). Nonsense-mediated mRNA decay in health and disease. Hum. Mol. Genet. 8, 1893–1900. doi: 10.1093/hmg/8.10.1893
Fu, Y., Foden, J. A., Khayter, C., Maeder, M. L., Reyon, D., Joung, J. K., et al. (2013). High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 31, 822–826. doi: 10.1038/nbt.2623
Fuster-García, C., García-García, G., González-Romero, E., Jaijo, T., Sequedo, M. D., Ayuso, C., et al. (2017). USH2A gene editing using the CRISPR system. Mol. Ther. Nucleic Acids 8, 529–541. doi: 10.1016/j.omtn.2017.08.003
Gagliardi, G., Ben M’Barek, K., Chaffiol, A., Slembrouck-Brec, A., Conart, J.-B., Nanteau, C., et al. (2018). Characterization and transplantation of CD73-positive photoreceptors isolated from human iPSC-derived retinal organoids. Stem Cell Reports 11, 665–680. doi: 10.1016/j.stemcr.2018.07.005
Gao, X., Tao, Y., Lamas, V., Huang, M., Yeh, W.-H., Pan, B., et al. (2018). Treatment of autosomal dominant hearing loss by in vivo delivery of genome editing agents. Nature 553, 217–221. doi: 10.1038/nature25164
Garanto, A., Duijkers, L., Tomkiewicz, T. Z., and Collin, R. W. J. (2019). Antisense oligonucleotide screening to optimize the rescue of the splicing defect caused by the recurrent deep-intronic ABCA4 variant c.4539+2001G >A in stargardt disease. Genes 10:452. doi: 10.3390/genes10060452
Gaudelli, N. M., Komor, A. C., Rees, H. A., Packer, M. S., Badran, A. H., Bryson, D. I., et al. (2017). Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 551, 464–471. doi: 10.1038/nature24644
Geng, R., Omar, A., Gopal, S. R., Chen, D. H., Stepanyan, R., Basch, M. L., et al. (2017). Modeling and preventing progressive hearing loss in usher syndrome III. Sci. Rep. 7:13480. doi: 10.1038/s41598-017-13620-9
Ghosh, A., Yue, Y., Lai, Y., and Duan, D. (2008). A hybrid vector system expands adeno-associated viral vector packaging capacity in a transgene-independent manner. Mol. Ther. 16, 124–130. doi: 10.1038/sj.mt.6300322
Giannelli, S. G., Luoni, M., Castoldi, V., Massimino, L., Cabassi, T., Angeloni, D., et al. (2017). Cas9/sgRNA selective targeting of the P23H Rhodopsin mutant allele for treating retinitis pigmentosa by intravitreal AAV9.PHP.B-based delivery. Hum. Mol. Genet. 27, 761–779. doi: 10.1093/hmg/ddx438
Goldmann, T., Overlack, N., Möller, F., Belakhov, V., Van Wyk, M., Baasov, T., et al. (2012). A comparative evaluation of NB30, NB54 and PTC124 in translational read-through efficacy for treatment of an USH1C nonsense mutation. EMBO Mol. Med. 4, 1186–1199. doi: 10.1002/emmm.201201438
Goldmann, T., Overlack, N., Wolfrum, U., and Nagel-Wolfrum, K. (2011). PTC124-mediated translational readthrough of a nonsense mutation causing Usher syndrome type 1C. Hum. Gene Ther. 22, 537–547. doi: 10.1089/hum.2010.067
Goldmann, T., Rebibo-Sabbah, A., Overlack, N., Nudelman, I., Belakhov, V., Baasov, T., et al. (2010). Beneficial read-through of a USH1C nonsense mutation by designed aminoglycoside NB30 in the retina. Invest. Ophthalmol. Vis. Sci. 51, 6671–6680. doi: 10.1167/iovs.10-5741
Gonzalez-Cordero, A., West, E. L., Pearson, R. A., Duran, Y., Carvalho, L. S., Chu, C. J., et al. (2013). Photoreceptor precursors derived from three-dimensional embryonic stem cell cultures integrate and mature within adult degenerate retina. Nat. Biotechnol. 31, 741–747. doi: 10.1038/nbt.2643
Goyal, N., and Narayanaswami, P. (2018). Making sense of antisense oligonucleotides: a narrative review. Muscle Nerve 57, 356–370. doi: 10.1002/mus.26001
Grati, M. H., and Kachar, B. (2011). Myosin VIIa and sans localization at stereocilia upper tip-link density implicates these Usher syndrome proteins in mechanotransduction. Proc. Natl. Acad. Sci. U S A 108, 11476–11481. doi: 10.1073/pnas.1104161108
Grieger, J. C., and Samulski, R. J. (2005). Packaging capacity of adeno-associated virus serotypes: impact of larger genomes on infectivity and postentry steps. J. Virol. 15, 9933–9944. doi: 10.1128/jvi.79.15.9933-9944.2005
György, B., Meijer, E. J., Ivanchenko, M. V., Tenneson, K., Emond, F., Hanlon, K. S., et al. (2019). Gene transfer with AAV9-PHP.B rescues hearing in a mouse model of usher syndrome 3A and transduces hair cells in a non-human primate. Mol. Ther. Methods Clin. Dev. 13, 1–13. doi: 10.1016/j.omtm.2018.11.003
Hallam, D., Hilgen, G., Dorgau, B., Zhu, L., Yu, M., Bojic, S., et al. (2018). Human-induced pluripotent stem cells generate light responsive retinal organoids with variable and nutrient-dependent efficiency. Stem Cells 36, 1535–1551. doi: 10.1002/stem.2883
Han, Z., Conley, S. M., Makkia, R. S., Cooper, M. J., and Naash, M. I. (2012a). DNA nanoparticle-mediated ABCA4 delivery rescues Stargardt dystrophy in mice. J. Clin. Invest. 122, 3221–3226. doi: 10.1172/jci64833
Han, Z., Conley, S. M., Makkia, R., Guo, J., Cooper, M. J., and Naash, M. I. (2012b). Comparative analysis of DNA nanoparticles and AAVs for ocular gene delivery. PLoS One 7:e52189. doi: 10.1371/journal.pone.0052189
Han, S., Liu, X., Xie, S., Gao, M., Liu, F., Yu, S., et al. (2018). Knockout of ush2a gene in zebrafish causes hearing impairment and late onset rod-cone dystrophy. Hum. Genet. 137, 779–794. doi: 10.1007/s00439-018-1936-6
Hartong, D. T., Berson, E. L., and Dryja, T. P. (2006). Retinitis pigmentosa. Lancet 368, 1795–1809. doi: 10.1016/S0140-6736(06)69740-7
Hashimoto, T., Gibbs, D., Lillo, C., Azarian, S. M., Legacki, E., Zhang, X. M., et al. (2007). Lentiviral gene replacement therapy of retinas in a mouse model for Usher syndrome type 1B. Gene Ther. 14, 584–594. doi: 10.1038/sj.gt.3302897
Havens, M. A., Duelli, D. M., and Hastings, M. L. (2013). Targeting RNA splicing for disease therapy. Wiley Interdiscip. Rev. RNA 4, 247–266. doi: 10.1002/wrna.1158
Hoffman, D. R., Hughbanks-Wheaton, D. K., Pearson, N. S., Fish, G. E., Spencer, R., Takacs, A., et al. (2014). Four-year placebo-controlled trial of docosahexaenoic acid in X-linked retinitis pigmentosa (DHAX trial): a randomized clinical trial. JAMA Ophthalmol. 132, 866–873. doi: 10.1001/jamaophthalmol.2014.1634
Hordeaux, J., Wang, Q., Katz, N., Buza, E. L., Bell, P., and Wilson, J. M. (2018). The neurotropic properties of AAV-PHP.B are limited to C57BL/6J mice. Mol. Ther. 26, 664–668. doi: 10.1016/j.ymthe.2018.01.018
Huang, X., and Chau, Y. (2019). Intravitreal nanoparticles for retinal delivery. Drug Discov. Today 24, 1510–1523. doi: 10.1016/j.drudis.2019.05.005
Huang, D., Fletcher, S., Wilton, S. D., Palmer, N., McLenachan, S., Mackey, D. A., et al. (2017). Inherited retinal disease therapies targeting precursor messenger ribonucleic acid. Vision 1:22. doi: 10.3390/vision1030022
Jaijo, T., Aller, E., Oltra, S., Beneyto, M., Najera, C., Ayuso, C., et al. (2006). Mutation profile of the MYO7A gene in Spanish patients with Usher syndrome type I. Hum. Mutat. 27, 290–291. doi: 10.1002/humu.9404
Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A., and Charpentier, E. (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821. doi: 10.1126/science.1225829
Jouret, G., Poirsier, C., Spodenkiewicz, M., Jaquin, C., Gouy, E., Arndt, C., et al. (2019). Genetics of Usher syndrome: new insights from a meta-analysis. Ontol. Neurotol. 40, 121–129. doi: 10.1097/mao.0000000000002054
Kay, M. A., Glorioso, J. C., and Naldini, L. (2001). Viral vectors for gene therapy: the art of turning infectious agents into vehicles of therapeutics. Nat. Med. 7, 33–40. doi: 10.1038/83324
Keeling, K. M., and Bedwell, D. M. (2011). Suppression of nonsense mutations as a therapeutic approach to treat genetic diseases. Wiley Interdiscip. Rev. RNA 2, 837–852. doi: 10.1002/wrna.95
Keeling, K. M., Wang, D., Conard, S. E., and Bedwell, D. M. (2012). Suppression of premature termination codons as a therapeutic approach. Crit. Rev. Biochem. Mol. Biol. 47, 444–463. doi: 10.3109/10409238.2012.694846
Kerem, E., Konstan, M. W., De Boeck, K., Accurso, F. J., Sermet-Gaudelus, I., Wilschanski, M., et al. (2014). Ataluren for the treatment of nonsense-mutation cystic fibrosis: a randomised, double-blind, placebo-controlled phase 3 trial. Lancet Respir. Med. 2, 539–547. doi: 10.1016/S2213-2600(14)70100-6
Khan, S. H. (2019). Genome-editing technologies: concept, pros and cons of various genome-editing techniques and bioethical concerns for clinical application. Mol Ther. Nucleic Acids 16, 326–334. doi: 10.1016/j.omtn.2019.02.027
Kim, Y. G., Cha, J., and Chandrasegaran, S. (1996). Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc. Natl. Acad. Sci. U S A 93, 1156–1160. doi: 10.1073/pnas.93.3.1156
Kimberling, W. J., Hildebrand, M. S., Shearer, A. E., Jensen, M. L., Halder, J. A., Trzupek, K., et al. (2010). Frequency of Usher syndrome in two pediatric populations: implications for genetic screening of deaf and hard of hearing children. Genet Med. 12, 512–516. doi: 10.1097/gim.0b013e3181e5afb8
Komor, A. C., Kim, Y. B., Packer, M. S., Zuris, J. A., and Liu, D. R. (2016). Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424. doi: 10.1038/nature17946
Konstan, M. W., Davis, P. B., Wagener, J. S., Hilliard, K. A., Stern, R. C., Milgram, L. J. H., et al. (2004). Compacted DNA nanoparticles administered to the nasal mucosa of cystic fibrosis subjects are safe and demonstrate partial to complete cystic fibrosis transmembrane regulator reconstitution. Hum. Gene Ther. 15, 1255–1269. doi: 10.1089/hum.2004.15.1255
Kuscu, C., Arslan, S., Singh, R., Thorpe, J., and Adli, M. (2014). Genome-wide analysis reveals characteristics of off-target sites bound by the Cas9 endonuclease. Nat. Biotechnol. 32, 677–683. doi: 10.1038/nbt.2916
Lakowski, J., Gonzalez-Cordero, A., West, E. L., Han, Y.-T., Welby, E., Naeem, A., et al. (2015). Transplantation of photoreceptor precursors isolated via a cell surface biomarker panel from embryonic stem cell-derived self-forming retina. Stem Cells 33, 2469–2482. doi: 10.1002/stem.2051
Lakowski, J., Welby, E., Budinger, D., Di Marco, F., Di Foggia, V., Bainbridge, J. W. B., et al. (2018). Isolation of human photoreceptor precursors via a cell surface marker panel from stem cell-derived retinal organoids and fetal retinae. Stem Cells 36, 709–722. doi: 10.1002/stem.2775
Landegger, L. D., Pan, B., Askew, C., Wassmer, S. J., Gluck, S. D., Galvin, A., et al. (2017). A synthetic AAV vector enables safe and efficient gene transfer to the mammalian inner ear. Nat. Biotechnol. 35, 280–284. doi: 10.1038/nbt.3781
Lentz, J. J., Jodelka, F. M., Hinrich, A. J., McCaffrey, K. E., Farris, H. E., Spalitta, M. J., et al. (2013). Rescue of hearing and vestibular function by antisense oligonucleotides in a mouse model of human deafness. Nat. Med. 19, 345–350. doi: 10.1038/nm.3106
Li, T., Adamian, M., Roof, D. J., Berson, E. L., Dryja, T. P., Roessler, B. J., et al. (1994). In vivo transfer of a reporter gene to the retina mediated by an adenoviral vector. Invest. Ophthalmol. Vis. Sci. 35, 2543–2549.
Lipinski, D. M., Barnard, A. R., Singh, M. S., Martin, C., Lee, E. J., Davies, W. I. L., et al. (2015). CNTF gene therapy confers lifelong neuroprotection in a mouse model of human retinitis pigmentosa. Mol. Ther. 23, 1308–1319. doi: 10.1038/mt.2015.68
Liu, X. Z., Hope, C., Liang, C. Y., Zou, J. M., Xu, L. R., Cole, T., et al. (1999). A mutation (2314delG) in the Usher syndrome type IIA gene: high prevalence and phenotypic variation. Am. J. Hum. Genet. 64, 1221–1225. doi: 10.1086/302332
Liu, X., Vansant, G., Udovichenko, I. P., Wolfrum, U., and Williams, D. S. (1997). Myosin VIIa, the product of the Usher 1B syndrome gene, is concentrated in the connecting cilia of photoreceptor cells. Cell Motil. Cytoskeleton 37, 240–252. doi: 10.1002/(sici)1097-0169(1997)37:3<240::aid-cm6>3.0.co;2-a
Lopes, V. S., Boye, S. E., Louie, C. M., Boye, S., Dyka, F., Chiodo, V., et al. (2013). Retinal gene therapy with a large MYO7A cDNA using adeno-associated virus. Gene Ther. 20, 824–833. doi: 10.1038/gt.2013.3
Lowe, A., Harris, R., Bhansali, P., Cvekl, A., and Liu, W. (2016). Intercellular adhesion-dependent cell survival and ROCK-regulated actomyosin-driven forces mediate self-formation of a retinal organoid. Stem Cell Reports 6, 743–756. doi: 10.1016/j.stemcr.2016.03.011
Lueck, J. D., Yoon, J. S., Perales-Puchalt, A., Mackey, A. L., Infield, D. T., Behlke, M. A., et al. (2019). Engineered transfer RNAs for suppression of premature termination codons. Nat. Commun. 10:822. doi: 10.1038/s41467-019-08329-4
MacLaren, R. E., Pearson, R. A., MacNeil, A., Douglas, R. H., Salt, T. E., Akimoto, M., et al. (2006). Retinal repair by transplantation of photoreceptor precursors. Nature 444, 203–207. doi: 10.1038/nature05161
Maddalena, A., Tornabene, P., Tiberi, P., Minopoli, R., Manfredi, A., Mutarelli, M., et al. (2018). Triple vectors expand AAV transfer capacity in the retina. Mol. Ther. 26, 524–541. doi: 10.1016/j.ymthe.2017.11.019
Maeda, Y., Fukushima, K., Kawasaki, A., Nishizaki, K., and Smith, R. J. (2007). Cochlear expression of a dominant-negative GJB2 R75W construct delivered through the round window membrane in mice. Neurosci. Res. 58, 250–254. doi: 10.1016/j.neures.2007.03.006
Maeda, Y., Fukushima, K., Nishizaki, K., and Smith, R. J. H. (2005). In vitro and in vivo suppression of GJB2 expression by RNA interference. Hum. Mol. Genet. 14, 1641–1650. doi: 10.1093/hmg/ddi172
Maeder, M. L., Stefanidakis, M., Wilson, C. J., Baral, R., Barrera, L. A., Bounoutas, G. S., et al. (2019). Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat. Med. 25, 229–233. doi: 10.1038/s41591-018-0327-9
Mali, P., Yang, L., Esvelt, K. M., Aach, J., Guell, M., Dicarlo, J. E., et al. (2013). RNA-guided human genome engineering via Cas9. Science 339, 823–826. doi: 10.1126/science.1232033
Mandai, M., Fujii, M., Hashiguchi, T., Sunagawa, G. A., Ito, S.-I., Sun, J., et al. (2017). iPSC-derived retina transplants improve vision in rd1 end-stage retinal-degeneration mice. Stem Cell Reports 8, 69–83. doi: 10.1016/j.stemcr.2016.12.008
Mathur, P., and Yang, J. (2015). Usher syndrome: hearing loss, retinal degeneration and associated abnormalities. Biochim. Biophys. Acta 1852, 406–420. doi: 10.1016/j.bbadis.2014.11.020
Mathur, P. D., and Yang, J. (2019). Usher syndrome and non-syndromic deafness: functions of different whirlin isoforms in the cochlea, vestibular organs and retina. Hear. Res. 375, 14–24. doi: 10.1016/j.heares.2019.02.007
Mazarakis, N. D., Azzouz, M., Rohll, J. B., Ellard, F. M., Wilkes, F. J., Olsen, A. L., et al. (2001). Rabies virus glycoprotein pseudotyping of lentiviral vectors enables retrograde axonal transport and access to the nervous system after peripheral delivery. Hum. Mol. Genet. 10, 2109–2121. doi: 10.1093/hmg/10.19.2109
McClements, M. E., Barnard, A. R., Singh, M. S., Issa, P. C., Jiang, Z., Radu, R. A., et al. (2019). An AAV dual vector strategy ameliorates the stargardt phenotype in adult Abca4−/− mice. Hum. Gene Ther. 30, 590–600. doi: 10.1089/hum.2018.156
McDonald, C. M., Campbell, C., Torricelli, R. E., Finkel, R. S., Flanigan, K. M., Goemans, N., et al. (2017). Ataluren in patients with nonsense mutation Duchenne muscular dystrophy (ACT DMD): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 390, 1489–1498. doi: 10.1016/S0140-6736(17)31611-2
Mellough, C. B., Collin, J., Khazim, M., White, K., Sernagor, E., Steel, D. H. W., et al. (2015). IGF-1 signaling plays an important role in the formation of three-dimensional laminated neural retina and other ocular structures from human embryonic stem cells. Stem Cells 33, 2416–2430. doi: 10.1002/stem.2023
Mellough, C. B., Collin, J., Queen, R., Hilgen, G., Dorgau, B., Zerti, D., et al. (2019). Systematic comparison of retinal organoid differentiation from human pluripotent stem cells reveals stage specific, cell line and methodological differences. Stem Cells Transl. Med. 8, 694–706. doi: 10.1002/sctm.18-0267
Mingeot-Leclercq, M.-P., and Tulkens, P. M. (1999). Aminoglycosides: nephrotoxicity. Antimicrob. Agents Chemother. 43, 1003–1012. doi: 10.1128/AAC.43.5.1003
Mitrophanous, K. A., Yoon, S., Rohll, J. B., Patil, D., Wilkes, F. J., Kim, V. N., et al. (1999). Stable gene transfer to the nervous system using a non-primate lentiviral vector. Gene Ther. 6, 1808–1818. doi: 10.1038/sj.gt.3301023
Moore, N. A., Morral, N., Ciulla, T. A., and Bracha, P. (2018). Gene therapy for inherited retinal and optic nerve degenerations. Expert Opin. Biol. Ther. 18, 37–49. doi: 10.1080/14712598.2018.1389886
Moreno, A. M., Fu, X., Zhu, J., Katrekar, D., Shih, Y. V., Marlett, J., et al. (2018). in situ gene therapy via AAV-CRISPR-Cas9-mediated targeted gene regulation. Mol. Ther. 26, 1818–1827. doi: 10.1016/j.ymthe.2018.04.017
Naessens, S., Ruysschaert, L., Lefever, S., Coppieters, F., and De Baere, E. (2019). Antisense oligonucleotide-based downregulation of the g56r pathogenic variant causing NR2E3-associated autosomal dominant retinitis pigmentosa. Genes 10:363. doi: 10.3390/genes10050363
Nagel-Wolfrum, K., Moller, F., Penner, I., Baasov, T., and Wolfrum, U. (2016). Targeting nonsense mutations in diseases with translational read-through-inducing drugs (TRIDs). BioDrugs 30, 49–74. doi: 10.1007/s40259-016-0157-6
Nakamura, M., Srinivasan, P., Chavez, M., Carter, M. A., Dominguez, A. A., La Russa, M., et al. (2019). Anti-CRISPR-mediated control of gene editing and synthetic circuits in eukaryotic cells. Nat. Commun. 10:194. doi: 10.1038/s41467-018-08158-x
Nakano, T., Ando, S., Takata, N., Kawada, M., Muguruma, K., Sekiguchi, K., et al. (2012). Self-formation of optic cups and storable stratified neural retina from human ESCs. Cell Stem Cell 10, 771–785. doi: 10.1016/j.stem.2012.05.009
Nayerossadat, N., Maedeh, T., and Ali, P. A. (2012). Viral and nonviral delivery systems for gene delivery. Adv. Biomed. Res. 1:27. doi: 10.4103/2277-9175.98152
Neuhaus, C., Eisenberger, T., Decker, C., Nagl, S., Blank, C., Pfister, M., et al. (2017). Next-generation sequencing reveals the mutational landscape of clinically diagnosed Usher syndrome: copy number variations, phenocopies, a predominant target for translational read-through and PEX26 mutated in Heimler syndrome. Mol. Genet. Genomic Med. 5, 531–552. doi: 10.1002/mgg3.312
Nudelman, I., Rebibo-Sabbah, A., Cherniavsky, M., Belakhov, V., Hainrichson, M., Chen, F., et al. (2009). Development of novel aminoglycoside (NB54) with reduced toxicity and enhanced suppression of disease-causing premature stop mutations. J. Med. Chem. 52, 2836–2845. doi: 10.1021/jm801640k
Nudelman, I., Rebibo-Sabbah, A., Shallom-Shezifi, D., Hainrichson, M., Stahl, I., Ben-Yosef, T., et al. (2006). Redesign of aminoglycosides for treatment of human genetic diseases caused by premature stop mutations. Bioorg. Med. Chem. Lett. 16, 6310–6315. doi: 10.1016/j.bmcl.2006.09.013
Omole, A. E., and Fakoya, A. O. J. (2018). Ten years of progress and promise of induced pluripotent stem cells: historical origins, characteristics, mechanisms, limitations and potential applications. PeerJ 6:e4370. doi: 10.7717/peerj.4370
Overlack, N., Goldmann, T., Wolfrum, U., and Nagel-Wolfrum, K. (2012). Gene repair of an Usher syndrome causing mutation by zinc-finger nuclease mediated homologous recombination. Invest. Ophthalmol. Vis. Sci. 53, 4140–4146. doi: 10.1167/iovs.12-9812
Pan, B., Askew, C., Galvin, A., Heman-Ackah, S., Asai, Y., Indzhykulian, A. A., et al. (2017). Gene therapy restores auditory and vestibular function in a mouse model of Usher syndrome type 1c. Nat. Biotechnol. 35, 264–272. doi: 10.1038/nbt.3801
Parfitt, D. A., Lane, A., Ramsden, C., Jovanovic, K., Coffey, P. J., Hardcastle, A. J., et al. (2016). Using induced pluripotent stem cells to understand retinal ciliopathy disease mechanisms and develop therapies. Biochem. Soc. Trans. 44, 1245–1251. doi: 10.1042/bst20160156
Pawluk, A., Amrani, N., Zhang, Y., Garcia, B., Hidalgo-Reyes, Y., Lee, J., et al. (2016). Naturally occurring off-switches for CRISPR-Cas9. Cell 167, 1829.e9–1838.e9. doi: 10.1016/j.cell.2016.11.017
Pearson, R. A., Barber, A. C., Rizzi, M., Hippert, C., Xue, T., West, E. L., et al. (2012). Restoration of vision after transplantation of photoreceptors. Nature 485, 99–103. doi: 10.1038/nature10997
Pepermans, E., Michel, V., Goodyear, R., Bonnet, C., Abdi, S., Dupont, T., et al. (2014). The CD2 isoform of protocadherin-15 is an essential component of the tip-link complex in mature auditory hair cells. EMBO Mol. Med. 6, 984–992. doi: 10.15252/emmm.201403976
Phillips, M. J., Wallace, K. A., Dickerson, S. J., Miller, M. J., Verhoeven, A. D., Martin, J. M., et al. (2012). Blood-derived human iPS cells generate optic vesicle-like structures with the capacity to form retinal laminae and develop synapses. Invest. Ophthalmol. Vis. Sci. 53, 2007–2019. doi: 10.1167/iovs.11-9313
Ponnath, A., Depreux, F. F., Jodelka, F. M., Rigo, F., Farris, H. E., Hastings, M. L., et al. (2018). Rescue of outer hair cells with antisense oligonucleotides in usher mice is dependent on age of treatment. J. Assoc. Res. Otolaryngol. 19, 1–16. doi: 10.1007/s10162-017-0640-x
Rauch, B. J., Silvis, M. R., Hultquist, J. F., Waters, C. S., Mcgregor, M. J., Krogan, N. J., et al. (2017). Inhibition of CRISPR-Cas9 with bacteriophage proteins. Cell 168, 150.e10–158.e10. doi: 10.1016/j.cell.2016.12.009
Rayapudi, S., Schwartz, S. G., Wang, X., and Chavis, P. (2013). Vitamin A and fish oils for retinitis pigmentosa. Cochrane Database Syst. Rev. 12:CD008428. doi: 10.1002/14651858.cd008428
Rebibo-Sabbah, A., Nudelman, I., Ahmed, Z. M., Baasov, T., and Ben-Yosef, T. (2007). In vitro and ex vivo suppression by aminoglycosides of PCDH15 nonsense mutations underlying type 1 Usher syndrome. Hum. Genet. 122, 373–381. doi: 10.1007/s00439-007-0410-7
Rees, H. A., and Liu, D. R. (2018). Base editing: precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 19, 770–788. doi: 10.1038/s41576-018-0059-1
Reiners, J., Nagel-Wolfrum, K., Jurgens, K., Marker, T., and Wolfrum, U. (2006). Molecular basis of human Usher syndrome: deciphering the meshes of the Usher protein network provides insights into the pathomechanisms of the Usher disease. Exp. Eye Res. 83, 97–119. doi: 10.1016/j.exer.2005.11.010
Rodrigues, G. A., Shalaev, E., Karami, T. K., Cunningham, J., Slater, N. K. H., and Rivers, H. M. (2018). Pharmaceutical development of AAV-based gene therapy products for the eye. Pharm. Res. 36:29. doi: 10.1007/s11095-018-2554-7
Russell, S., Bennett, J., Wellman, J. A., Chung, D. C., Yu, Z.-F., Tillman, A., et al. (2017). Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet 390, 849–860. doi: 10.1016/S0140-6736(17)31868-8
Ryu, S.-M., Koo, T., Kim, K., Lim, K., Baek, G., Kim, S.-T., et al. (2018). Adenine base editing in mouse embryos and an adult mouse model of Duchenne muscular dystrophy. Nat. Biotechnol. 36, 536–539. doi: 10.1038/nbt.4148
Sahly, I., Dufour, E., Schietroma, C., Michel, V., Bahloul, A., Perfettini, I., et al. (2012). Localization of Usher 1 proteins to the photoreceptor calyceal processes, which are absent from mice. J. Cell Biol. 199, 381–399. doi: 10.1083/jcb.201202012
Santos-Ferreira, T., Llonch, S., Borsch, O., Postel, K., Haas, J., and Ader, M. (2016). Retinal transplantation of photoreceptors results in donor-host cytoplasmic exchange. Nat. Commun. 7:13028. doi: 10.1038/ncomms13028
Schietroma, C., Parain, K., Estivalet, A., Aghaie, A., Boutet De Monvel, J., Picaud, S., et al. (2017). Usher syndrome type 1-associated cadherins shape the photoreceptor outer segment. J. Cell Biol. 216, 1849–1864. doi: 10.1083/jcb.201612030
Selimoglu, E. (2007). Aminoglycoside-induced ototoxicity. Curr. Pharm. Des. 13, 119–126. doi: 10.2174/138161207779313731
Sengillo, J. D., Justus, S., Tsai, Y. T., Cabral, T., and Tsang, S. H. (2016). Gene and cell-based therapies for inherited retinal disorders: an update. Am. J. Med. Genet. C Semin. Med. Genet. 172, 349–366. doi: 10.1002/ajmg.c.31534
Seyedahmadi, B. J., Rivolta, C., Keene, J. A., Berson, E. L., and Dryja, T. P. (2004). Comprehensive screening of the USH2A gene in Usher syndrome type II and non-syndromic recessive retinitis pigmentosa. Exp. Eye Res. 79, 167–173. doi: 10.1016/j.exer.2004.03.005
Shibata, S. B., Ranum, P. T., Moteki, H., Pan, B., Goodwin, A. T., Goodman, S. S., et al. (2016). RNA interference prevents autosomal-dominant hearing loss. Am. J. Hum. Genet. 98, 1101–1113. doi: 10.1016/j.ajhg.2016.03.028
Shirai, H., Mandai, M., Matsushita, K., Kuwahara, A., Yonemura, S., Nakano, T., et al. (2016). Transplantation of human embryonic stem cell-derived retinal tissue in two primate models of retinal degeneration. Proc. Natl. Acad. Sci. U S A 113, E81–E90. doi: 10.1073/pnas.1512590113
Singh, M. S., Balmer, J., Barnard, A. R., Aslam, S. A., Moralli, D., Green, C. M., et al. (2016). Transplanted photoreceptor precursors transfer proteins to host photoreceptors by a mechanism of cytoplasmic fusion. Nat. Commun. 7:13537. doi: 10.1038/ncomms13537
Singh, M. S., Charbel Issa, P., Butler, R., Martin, C., Lipinski, D. M., Sekaran, S., et al. (2013). Reversal of end-stage retinal degeneration and restoration of visual function by photoreceptor transplantation. Proc. Natl. Acad. Sci. U S A 110, 1101–1106. doi: 10.1073/pnas.1119416110
Slijkerman, R. W., Vaché, C., Dona, M., García-García, G., Claustres, M., Hetterschijt, L., et al. (2016). Antisense oligonucleotide-based splice correction for USH2A-associated retinal degeneration caused by a frequent deep-intronic mutation. Mol. Ther. Nucleic Acids 5:e381. doi: 10.1038/mtna.2016.89
Song, C.-Q., Jiang, T., Richter, M., Rhym, L. H., Koblan, L. W., Zafra, M. P., et al. (2020). Adenine base editing in an adult mouse model of tyrosinaemia. Nat. Biomed. Eng. 4, 125–130. doi: 10.1038/s41551-019-0357-8
Sperling, M., and Gradzielski, M. (2017). Droplets, evaporation and a superhydrophobic surface: simple tools for guiding colloidal particles into complex materials. Gels 3:15. doi: 10.3390/gels3020015
Suzuki, J., Corfas, G., and Liberman, M. (2016). Round-window delivery of neurotrophin 3 regenerates cochlear synapses after acoustic overexposure. Sci. Rep. 6:24907. doi: 10.1038/srep24907
Takahashi, K., Tanabe, K., Ohnuki, M., Narita, M., Ichisaka, T., Tomoda, K., et al. (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872. doi: 10.1016/j.cell.2007.11.019
Talcott, K. E., Ratnam, K., Sundquist, S. M., Lucero, A. S., Lujan, B. J., Tao, W., et al. (2011). Longitudinal study of cone photoreceptors during retinal degeneration and in response to ciliary neurotrophic factor treatment. Invest. Ophthalmol. Vis. Sci. 52, 2219–2226. doi: 10.1167/iovs.10-6479
Tang, Z.-H., Chen, J.-R., Zheng, J., Shi, H.-S., Ding, J., Qian, X.-D., et al. (2016). Genetic correction of induced pluripotent stem cells from a deaf patient with MYO7A mutation results in morphologic and functional recovery of the derived hair cell-like cells. Stem Cells Transl. Med. 5, 561–571. doi: 10.5966/sctm.2015-0252
Tornabene, P., Trapani, I., Minopoli, R., Centrulo, M., Lupo, M., De Simone, S., et al. (2019). Intein-mediated protein trans-splicing expands adeno-associated virus transfer capacity in the retina. Sci. Transl. Med. 11:eaav4523. doi: 10.1126/scitranslmed.aav4523
Trapani, I., Colella, P., Sommella, A., Iodice, C., Cesi, G., De Simone, S., et al. (2014). Effective delivery of large genes to the retina by dual AAV vectors. EMBO Mol. Med. 6, 194–211. doi: 10.1002/emmm.201302948
Trouillet, A., Dubus, E., Degardin, J., Estivalet, A., Ivkovic, I., Godefroy, D., et al. (2018). Cone degeneration is triggered by the absence of USH1 proteins but prevented by antioxidant treatments. Sci. Rep. 8:1968. doi: 10.1038/s41598-018-20171-0
Tu, H.-Y., Watanabe, T., Shirai, H., Yamasaki, S., Kinoshita, M., Matsushita, K., et al. (2019). Medium- to long-term survival and functional examination of human iPSC-derived retinas in rat and primate models of retinal degeneration. EBioMedicine 39, 562–574. doi: 10.1016/j.ebiom.2018.11.028
Van Diepen, H., Dulla, K., Chan, H., Schulkens, I., Beumer, W., Vorthoren, L., et al. (2019). QR-421a, an antisense oligonucleotide, for the treatment of retinitis pigmentosa due to USH2A exon 13 mutations. Invest. Ophthalmol. Vis. Sci. 60:3250.
Vandendriessche, T., Thorrez, L., Acosta-Sanchez, A., Petrus, I., Wang, L., Ma, L., et al. (2007). Efficacy and safety of adeno-associated viral vectors based on serotype 8 and 9 vs. lentiviral vectors for hemophilia B gene therapy. J. Thromb. Haemost. 5, 16–24. doi: 10.1111/j.1538-7836.2006.02220.x
Vijayakumar, S., Depreux, F. F., Jodelka, F. M., Lentz, J. J., Rigo, F., Jones, T. A., et al. (2017). Rescue of peripheral vestibular function in Usher syndrome mice using a splice-switching antisense oligonucleotide. Hum. Mol. Genet. 26, 3482–3494. doi: 10.1093/hmg/ddx234
Villiger, L., Grisch-Chan, H. M., Lindsay, H., Ringnalda, F., Pogliano, C. B., Allegri, G., et al. (2018). Treatment of a metabolic liver disease by in vivo genome base editing in adult mice. Nat. Med. 24, 1519–1525. doi: 10.1038/s41591-018-0209-1
Wang, X., and Gregory-Evans, C. Y. (2015). Nonsense suppression therapies in ocular genetic diseases. Cell. Mol. Life Sci. 72, 1931–1938. doi: 10.1007/s00018-015-1843-0
Wang, Y., Wise, A. K., Tan, J., Maina, J. W., Shepherd, R. K., and Caruso, F. (2014). Mesoporous silica supraparticles for sustained inner-ear drug delivery. Small 10, 4244–4248. doi: 10.1002/smll.201401767
Watanabe, T., and Fukasawa, T. (1961). Episome-mediated transfer of drug resistance in Enterobacteriaceae. I. Transfer of resistance factors by conjugation. J. Bacteriol. 81, 669–678.
Weiss, J. N., and Levy, S. (2019). Stem Cell Ophthalmology Treatment Study (SCOTS): bone marrow derived stem cells in the treatment of Usher syndrome. Stem Cell Investig. 6:31. doi: 10.21037/sci.2019.08.07
Welch, E. M., Barton, E. R., Zhuo, J., Tomizawa, Y., Friesen, W. J., Trifillis, P., et al. (2007). PTC124 targets genetic disorders caused by nonsense mutations. Nature 447, 87–91. doi: 10.1038/nature05756
Wilschanski, M., Miller, L. L., Shoseyov, D., Blau, H., Rivlin, J., Aviram, M., et al. (2011). Chronic ataluren (PTC124) treatment of nonsense mutation cystic fibrosis. Eur. Respir. J. 38, 59–69. doi: 10.1183/09031936.00120910
Wold, W. S. M., and Toth, K. (2013). Adenovirus vectors for gene therapy, vaccination and cancer gene therapy. Curr. Gene Ther. 13, 421–433. doi: 10.2174/1566523213666131125095046
Yan, Z., Zhang, Y., and Duan, D. (2000). Trans-splicing vectors expand the utility of adeno-associated virus for gene therapy. Proc. Natl. Acad. Sci. U S A 97, 6716–6721. doi: 10.1073/pnas.97.12.6716
Yeh, W.-H., Chiang, H., Rees, H. A., Edge, A. S. B., and Liu, D. R. (2018). In vivo base editing of post-mitotic sensory cells. Nat. Commun. 9:2184. doi: 10.1038/s41467-018-04580-3
Yin, H., Kanasty, R. L., Eltoukhy, A. A., Vegas, A. J., Dorkin, J. R., and Anderson, D. G. (2014). Non-viral vectors for gene-based therapy. Nat. Rev. Genet. 15, 541–555. doi: 10.1038/nrg3763
Ylikoski, J., Pirvola, U., Moshnyakov, M., Palgi, J., Arumäe, U., and Saarma, M. (1993). Expression patterns of neurotrophin and their receptor mRNAs in the rat inner ear. Hear. Res. 65, 69–78. doi: 10.1016/0378-5955(93)90202-c
Zakrzewski, W., Dobrzyński, M., Szymonowicz, M., and Rybak, Z. (2019). Stem cells: past, present, and future. Stem Cell Res. Ther. 10:68. doi: 10.1186/s13287-019-1165-5
Zallocchi, M., Binley, K., Lad, Y., Ellis, S., Widdowson, P., Iqball, S., et al. (2014). EIAV-based retinal gene therapy in the shaker1 mouse model for usher syndrome type 1B: development of UshStat. PLoS One 9:e94272. doi: 10.1371/journal.pone.0094272
Zhang, X., Chen, G., Wen, L., Yang, F., Shao, A.-L., Li, X., et al. (2013). Novel multiple agents loaded PLGA nanoparticles for brain delivery via inner ear administration: in vitro and in vivo evaluation. Eur. J. Pharm. Sci. 48, 595–603. doi: 10.1016/j.ejps.2013.01.007
Zheng, C. X., Wang, S. M., Bai, Y. H., Luo, T. T., Wang, J. Q., Dai, C. Q., et al. (2018). Lentiviral vectors and adeno-associated virus vectors: useful tools for gene transfer in pain research. Anat. Rec. 301, 825–836. doi: 10.1002/ar.23723
Zheng, Q. Y., Yan, D., Ouyang, X. M., Du, L. L., Yu, H., Chang, B., et al. (2005). Digenic inheritance of deafness caused by mutations in genes encoding cadherin 23 and protocadherin 15 in mice and humans. Hum. Mol. Genet. 14, 103–111. doi: 10.1093/hmg/ddi010
Zhong, X., Gutierrez, C., Xue, T., Hampton, C., Vergara, M. N., Cao, L. H., et al. (2014). Generation of three-dimensional retinal tissue with functional photoreceptors from human iPSCs. Nat. Commun. 5:4047. doi: 10.1038/ncomms5047
Zou, J., Feng, H., Sood, R., Kinnunen, P. K. J., and Pyykko, I. (2017). Biocompatibility of liposome nanocarriers in the rat inner ear after intratympanic administration. Nanoscale Res. Lett. 12:372. doi: 10.1186/s11671-017-2142-5
Keywords: usher syndrome, gene therapy, cell therapy, ipscs, gene editing, adeno-associated virus, antisense oligonucleotides
Citation: French LS, Mellough CB, Chen FK and Carvalho LS (2020) A Review of Gene, Drug and Cell-Based Therapies for Usher Syndrome. Front. Cell. Neurosci. 14:183. doi: 10.3389/fncel.2020.00183
Received: 10 April 2020; Accepted: 28 May 2020;
Published: 09 July 2020.
Edited by:
Raymond Ching-Bong Wong, Centre for Eye Research Australia, AustraliaReviewed by:
Nicholas D. Mazarakis, Imperial College London, United KingdomWei Xiong, Tsinghua University, China
Copyright © 2020 French, Mellough, Chen and Carvalho. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Livia S. Carvalho, bGl2aWFjYXJ2YWxob0BsZWkub3JnLmF1