 Yajin Liao
Yajin Liao Yuan Dong
Yuan Dong Jinbo Cheng
Jinbo Cheng- 1Center on Translational Neuroscience, College of Life & Environmental Science, Minzu University of China, Beijing, China
- 2Department of Biochemistry, Medical College, Qingdao University, Qingdao, China
Membrane tethering is an important communication method for membrane-packaged organelles. Mitochondria are organelles with a bilayer membrane, and the membrane contact between mitochondria and other organelles is indispensable for maintaining cellular homeostasis. Increased levels of molecular determinants that mediate the membrane contact between mitochondria and other organelles, and their functions, have been revealed in recent years. In this review article, we aim to summarize the findings on the tethering between mitochondria and other organelles in physiological or pathological conditions, and discuss their roles in cellular homeostasis, neural activity, and neurodegenerative diseases.
Introduction
Most of the organelles within cells are membrane-bound. Membrane tethering is one of the communication methods for signal/material exchanges between organelles. Membrane contact was first observed 60 years ago (Bernhard and Rouiller, 1956; Dalen et al., 1983; Grönblad and Akerman, 1984), however, the roles and molecular determinants of organelle juxtaposition were discovered only recently. The contact between two organelles is achieved by means of proteins that tether them directly. Mitochondria are vital ATP-generating organelles, and play a critical role in synapse activity, neurite outgrowth, neurogenesis, and neuronal cell death (Sun et al., 2013; Liao et al., 2015; Khacho et al., 2016; Norkett et al., 2016; Vaccaro et al., 2017). The dysfunction of mitochondria, including Ca2+ overload, excessive fission, disrupted distribution, and clearance of damaged mitochondria has been observed in multiple disorders of the nervous system (Calkins et al., 2011; Guardia-Laguarta et al., 2014; Lee et al., 2018; Berenguer-Escuder et al., 2020; Tsai et al., 2020). In previous studies, many advances have been made in the identification of the contacts among various organelles and on their structural determinants. These studies have proved that membrane contacts regulate several aspects of the biology and the behavior of mitochondria (Filadi et al., 2018; McLelland et al., 2018; Grossmann et al., 2019). Considering the example of mitofusin-1/2 (MFN1/2), their ability to control the endoplasmic reticulum (ER)-mitochondria tethering is associated with the activity of regulating the balance between mitochondrial fusion and fission (de Brito and Scorrano, 2008; Li et al., 2015; Qi et al., 2016). Either a decrease or an increase in the interaction levels between mitochondria and other organelles have been proved to induce mitochondrial dysfunction, in turn effecting energy metabolism, respiration, apoptosis, oxidative stress, and inflammation (De Vos et al., 2012; Lee et al., 2016). In addition, a growing amount of evidence indicates that the dysfunction of membrane contacts between mitochondria and other organelles is involved in the development of neural stem cell-related and neurodegenerative diseases, including Alzheimer’s disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS; Area-Gomez et al., 2012; Lee et al., 2016, 2018; Stoica et al., 2016). In this review, we mainly aim to summarize the determinants of mitochondria contacts with other organelles under different physiological or pathological conditions.
The Determinants of ER-Mitochondria Membrane Contact
General Characteristics and Functions of ER-Mitochondria Membrane Contact
One of the most well-studied membrane contacts is ER-mitochondrial tethering. The interplay between these two organelles is necessary for the proper functioning of the cell, by maintaining Ca2+ homeostasis, lipid metabolism, and autophagy (Voss et al., 2012; Wideman et al., 2013; Gomez-Suaga et al., 2017b; Hirabayashi et al., 2017; Eisenberg-Bord et al., 2019).
The ER-mitochondria encounter structure (ERMES) is the first identified complex that was found to mediate the structural interaction between the ER and mitochondria. In yeast, the ERMES is composed of mitochondrial distribution and morphology protein 10/12/34 (Mdm10/12/34), maintenance of mitochondrial morphology protein 1 (Mmm1), and mitochondrial Rho GTPase 1 (Gem1; Figure 1; Kornmann et al., 2011; Jeong et al., 2016). Among these proteins, Mdm12, Mmm1, and Mdm34 contain a conserved synaptotagmin-like mitochondrial lipid-binding protein (SMP) domain that is important for the formation of the ERMES complex and to facilitate the contacts between the ER and mitochondria (AhYoung et al., 2015). Another conserved complex, the ER membrane protein complex (EMC), also plays an important role in tethering the ER to mitochondria (Lahiri et al., 2014). By interacting with the translocase of outer membrane 5 kDa subunit (TOM5) on the outer membrane of mitochondria, EMC1-6 of the EMC complex mediates the ER-mitochondria contact necessary for phosphatidylserine transfer in yeast (Figure 1; Lahiri et al., 2014). Importantly, both ERMES- and EMC-mediated tethering is essential for cell growth.
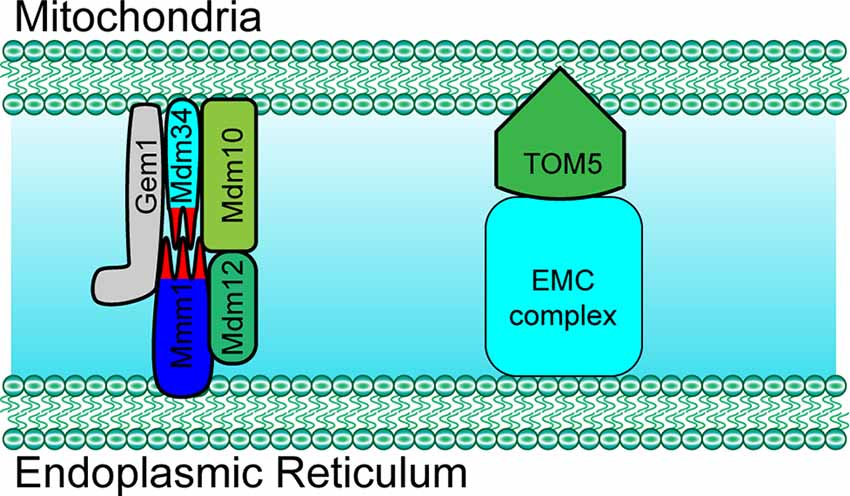
Figure 1. Paradigm of molecular determinants in endoplasmic reticulum (ER)-mitochondria tethering in yeast. Mdm10, Mdm34 and Gem1 located at the outer mitochondrial membrane (OMM) mediate ER-mitochondria tethering by forming a complex with ER membrane located proteins Mmm1 and cytosol Mdm12 in yeast. ER membrane protein complex (EMC) mediates ER-mitochondria contact by interacting with TOM5 at the mitochondrial OMM in yeast. Mdm10/34, mitochondrial distribution and morphology protein 10/34; Gem1, mitochondrial Rho GTPase 1; TOM5, translocase of outer membrane 5 kDa subunit.
In mammal cells, some proteins also localize into a subcompartment on the membrane of the ER, which makes contact with mitochondria and is referred to as the mitochondria-associated ER membrane (MAM; Vance, 1990; Rusinol et al., 1994; Csordás et al., 2006; Area-Gomez et al., 2012). MAM molecular determinants are dynamic and shape ER-mitochondria function (Poston et al., 2013; Gelmetti et al., 2017; Ma et al., 2017; Pera et al., 2017). The classic molecules that are involved in ER-mitochondria tethering include MFN1/2, phosphofurin acidic cluster sorting protein 2 (PACS2), sigma-1 receptor (Sig-1R), F-box protein FBXL2 (FBXL2), 75 kDa glucose-regulated protein (GRP75), voltage-dependent anion-selective channel protein 1/2 (VDAC1/2), and inositol 1,4,5-trisphosphate receptor type 3 (IP3R3; Figure 2 and Table 1; Simmen et al., 2005; Szabadkai et al., 2006; Hayashi and Su, 2007; de Brito and Scorrano, 2008; Mori et al., 2013; Naon et al., 2016; Kuchay et al., 2017; Veeresh et al., 2019). Both the formation of the MFN2 dimer and IP3R3-VDAC-GRP75 complex in the MAM could mediate the physical linkage between the ER and mitochondria (Csordás et al., 2006; Szabadkai et al., 2006; de Brito and Scorrano, 2008; McLelland et al., 2018). In addition, some molecules have been proven to enhance ER-mitochondria contacts in a dose-dependent manner, which promote the transfer of Ca2+ and other chemicals between the ER and mitochondria (Chen et al., 2012; Mori et al., 2013; McLelland et al., 2018; Yu et al., 2019). Furthermore, accumulative evidence suggests that the post-translational modification of these proteins is also essential for the tethering activity and dynamic balance of ER-mitochondria contacts involving the aforementioned determinants (McLelland et al., 2018). Considering the example of MFN2, a mitochondrial outer membrane protein that mediates fusion, mitophagy regulates ER-mitochondria contacts (de Brito and Scorrano, 2008; Chen et al., 2012; Naon et al., 2016). MFN2 ablation or silencing enhances ER-mitochondria close contacts and leads cells more sensitive for mitochondrial calcium overload-dependent cell death (Filadi et al., 2015). Moreover, MFN2-mediated ER-mitochondria tethering is regulated by ubiquitination. Mitochondrial ubiquitin ligase (MITOL)-mediated ubiquitination, PTEN-induced putative kinase protein 1 (PINK1) and Parkin-mediated mono-ubiquitination of MFN2 promote its tethering activity, while PINK1 and Parkin-mediated poly-ubiquitination of MFN2 result in MFN2 retrotranslocation and disrupt ER-mitochondria contacts to drive mitophagy (Figure 2; Sugiura et al., 2013; Basso et al., 2018; McLelland et al., 2018). These post-translational modifications likely explain the different findings on the role of MFN2 in keeping mitochondria and ER together (Naon et al., 2016; Filadi et al., 2017). Besides PINK1 and Parkin, other mitophagy-associated proteins, such as Beclin-1, have been found to relocalize in the MAMs during mitophagy (Gelmetti et al., 2017; McLelland et al., 2018). The relocalization of these proteins in the MAMs promotes ER-mitochondrial tethering and autophagosome formation (Gelmetti et al., 2017). Apart from post-translational modification, the interaction between the MAM components and other proteins also affects the ER-mitochondria contacts. Considering the example of IP3R3-GRP75-VDAC complex, the IP3R3-PTEN interaction and IP3R3-Sig-1R interaction promote the stability of IP3R3 and maintain the complex, while the IP3R3-BiP interaction and IP3R3-FBXL2 interaction induce the degradation of IP3R3 and disruption of the complex (Hayashi and Su, 2007; Kuchay et al., 2017). The Transglutaminase type 2 (TG2) was found to interact with GRP75 and to subsequently decrease the interaction level between GRP75 and IP3R3, thereby maintaining mitochondrial Ca2+ homeostasis. The loss of TG2 was found to increase GRP75-IP3R3 interaction levels and to decrease ER-mitochondria contact levels as well as mitochondrial Ca2+ levels (D’Eletto et al., 2018). Pyruvate dehydrogenase kinase 4 (PDK4) is another GRP75-interacting protein, and the upregulated expression of PDK4 could promote the formation of the VDAC1-GRP75-IP3R3 complex, which results in increased ER-mitochondria contact levels (Thoudam et al., 2019).
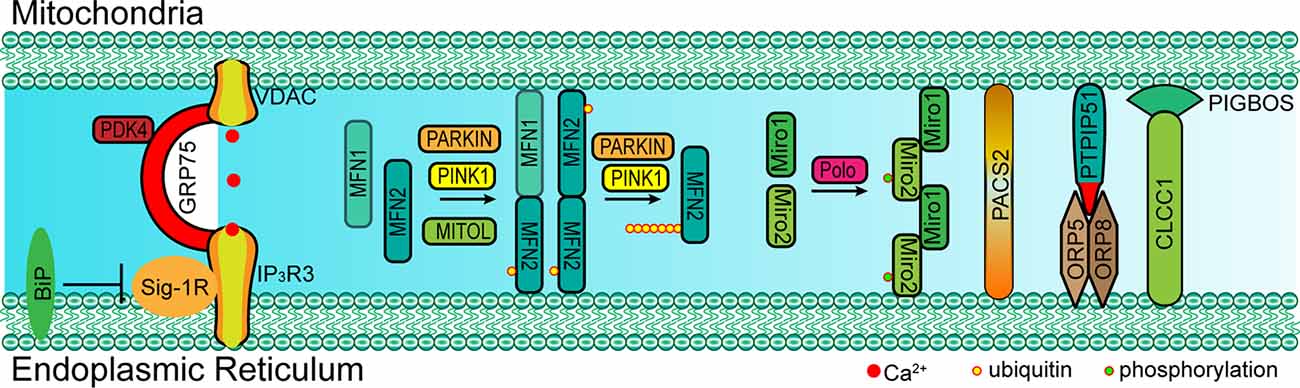
Figure 2. Paradigm of molecular determinants in ER-mitochondria tethering in mammal. Left: GRP75 interacts with both VDAC (located at OMM) and IP3R3 (located at MAM) to form a complex that mediates the ER-mitochondria contact. The interaction between Sig-1R and IP3R3 promotes the stability of IP3R3, which is essential for the IP3R3-GRP75-VDAC complex, while BiP inhibits this interaction by competitive binding to Sig-1R. Middle: MFN1/2 form a dimer at MAM and mediate ER-mitochondria tethering. PARKIN, PINK1 and MITOL mediate the mono-ubiquitination of MFN2 and promote MFN2 translocation to MAM. However, the poly-ubiquitination of MFN2 mediated via PARKIN and PINK1 leads to the tethering disruption. Right: Miro1/2 form a cluster at MAM and mediate ER-mitochondria tethering, and the phosphorylation of Miro2 by polo kinase is essential for the formation of the Miro cluster. Cytosolic PACS2 recruits to the MAMs and OMM to mediate ER-mitochondria membrane contact. The OMM-resident PTPIP51 interacts with ORP5/8 at MAM to maintain mitochondrial morphology and respiratory functions. The OMM-resident PIGBOS interacts with the ER membrane protein CLCC1 to maintain ER–mitochondrial contact. GRP75, 75 kDa glucose-regulated protein; VDAC1/2, voltage-dependent anion-selective channel protein; IP3R3, inositol 1,4,5-trisphosphate receptor type 3; MFN1/2, mitofusin-1/2; Sig-1R, sigma-1 receptor; ORP5/8, oxysterol-binding protein (OSBP)-related proteins 5/8; PTPIP51, protein tyrosine phosphatase interacting protein-51; CLCC1, chloride channel CLIC-like protein 1; PIGBOS, PIGB opposite strand 1; PACS2, phosphofurin acidic cluster sorting protein 2; Miro1/2, mitochondrial Rho 1/2; MITOL: mitochondrial ubiquitin ligase; PINK1, PTEN-induced putative kinase protein 1.

Table 1. The molecular determinant for endoplasmic reticulum (ER)-mitochondria tethering in mammal cell.
Besides these canonical molecular determinants that mediate ER-mitochondria tethering, in the last few years, several other proteins that translocate to the MAM and that mediate ER-mitochondria tethering have been identified [i.e., Mitochondrial Rho (Miro) and PIGB opposite strand 1 (PIGBOS); Chu et al., 2019; Modi et al., 2019]. In 2016, Miro1 and Miro2, two mitochondrial Rho GTPases, were discovered to mediate ER-mitochondria tethering (Lee et al., 2016). Recent studies have revealed that Miro1 and Miro2 form nanometer-sized clusters along the outer membrane of the mitochondria and are components of the mitochondrial contact site and cristae organizing system (MICOS) and ER-mitochondria contact sites (ERMCS; Figure 2; Lee et al., 2016; Modi et al., 2019). The loss of Miro1/2 results in decreased ER-mitochondria contact sites and mitochondrial Ca2+ uptake (Figure 1; Modi et al., 2019). Another study has indicated that Miro can be phosphorylated by polo-like kinase 1, leading to the activation of Miro (Lee et al., 2016). In neural stem cells, the inactivation of Miro leads to the depletion of mitochondrial Ca2+ levels resulting in a metabolic impairment, and the overexpression of Miro1/2 results in Ca2+ overload and cell death (Lee et al., 2016). PIGBOS, a novel microprotein that localizes on the outer membrane of the mitochondria (Chu et al., 2019), is found to mediate ER-mitochondria contact. The loss of PIGBOS elevates unfolded protein response (UPR) and increases cell death under stress conditions. The results of further investigation indicate that PIGBOS is able to interact with chloride channel CLIC-like protein 1 (CLCC1) on the membrane of the ER, and this interaction is essential for the function of PIGBOS (Figure 2; Chu et al., 2019). However, the interaction between PIGBOS and CLCC1 does not act as a tether between the ER and mitochondria (Chu et al., 2019). The oxysterol-binding protein (OSBP)-related proteins 5/8 (ORP5/8) mediate ER-mitochondria tethering by interacting with protein tyrosine phosphatase interacting protein-51 (PTPIP51; Figure 2). The depletion of ORP5/8 results in changes in the mitochondrial morphology and respiratory functions (Galmes et al., 2016). As the most extensively studied type of membrane contact, an increasing number of determinants that are involved in ER-mitochondria tethering is being discovered (Table 1), which promotes the understanding of the functions and regulatory mechanisms of ER-mitochondria contacts.
The Functions of ER-Mitochondria Membrane Contact in the Nervous System
In neuronal cells, some molecules that mediate the local ER-mitochondria contacts regulate dendritic Ca2+ homeostasis in order to facilitate the adaptation to synapse stimulation, thereby playing a critical role in synaptic integration properties and plasticity. For example, vesicle-associated membrane protein-associated protein B (VAPB) and PTPIP51 form a complex at the synapses and mediate ER-mitochondria tethering (Figure 3; Gómez-Suaga et al., 2019). The interaction between VAPB (localization in the ER) and PTPIP51 (localization in the mitochondria) is critical for mitochondrial Ca2+ homeostasis (De Vos et al., 2012). The loss of VAPB or PTPIP51 results in higher cytosolic Ca2+ levels in the case of dendrite postsynaptic stimulation. In addition, the number of dendritic spines is reduced in VAPB- or PTPIP51-silenced neurons (Gómez-Suaga et al., 2019). Recently, on performing cell-type-specific profiling of the brain mitochondria, a regulator of microtubule dynamics protein 3 (Rmdn3) was found to predominantly mediate ER-mitochondria tethering in the Purkinje cells (Figure 3), rather than in the astrocyte and granule cells (Fecher et al., 2019). Even if a previous study suggests that Rmdn3 mediates the ER-mitochondria tethering by interacting with VAPB, the mechanism of Rmdn3-mediated ER-mitochondria tethering in neurons is unknown (Gomez-Suaga et al., 2017a). Recently, PDZ domain-containing protein 8 (PDZD8), a SMP domain-containing protein localized in the ER membrane, has been proven to mediate ER-mitochondria tethering in the neurons (Figure 3), which is critical for dendrites activity (Hirabayashi et al., 2017). Usually, Ca2+ released from the ER can induce the subsequent mitochondrial Ca2+ uptake (Hirabayashi et al., 2017). However, the mitochondrial Ca2+ import is significantly reduced in PDZD8-deficient neurons, resulting in higher Ca2+ levels in the dendrites (Hirabayashi et al., 2017). These results suggest that particular determinants mediate the ER-mitochondria membrane contacts in the neurons, especially in the neural dendrites, and that the absence of these proteins could result in decreased synaptic plasticity and activity.
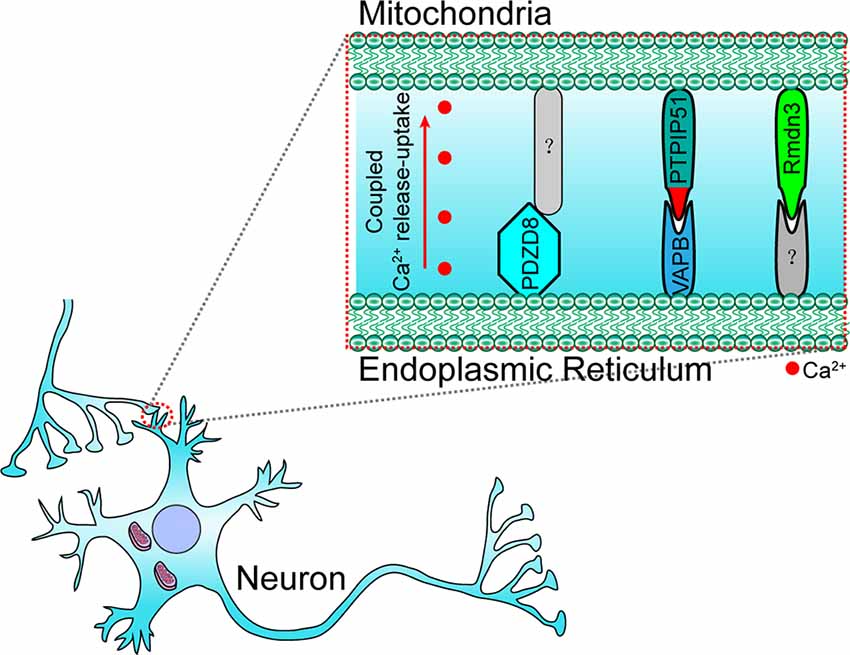
Figure 3. Paradigm of molecular determinants in ER-mitochondria tethering in the dendrite of neurons. In the dendrite, VAPB (located at ER) interacts with PTPIP51 (located at OMM) to maintain ER–mitochondria tethering and synaptic activity-coupled Ca2+ release-uptake. PDZD8 (located at ER) and Rmdn3 (located at OMM) are also involved in synaptic activity-coupled Ca2+ release-uptake and ER-mitochondria membrane contact. However, their interacting partners have still not been identified. VAPB, vesicle-associated membrane protein-associated protein B; PDZD8, PDZ domain-containing protein 8; Rmdn3, regulator of microtubule dynamics protein 3.
Besides the molecules that are present within the EMRES or MAM under normal physiological conditions, some molecules (i.e., APP-C99 and Tau) are found to localize within the ERMES or MAM under pathological conditions, resulting in excessive ER-mitochondria contacts, and mitochondrial Ca2+ overload, and cell death (Guardia-Laguarta et al., 2014; Pera et al., 2017; Cieri et al., 2018). Furthermore, excessive ER-mitochondria contacts have been recently found to be involved in neurodegenerative diseases. Based on the findings through biopsies of AD patients, the number of ER-mitochondria contact sites is positively correlated with elevated ventricular cerebrospinal fluid β-amyloid (Aβ) levels (Leal et al., 2018). A nanomolar concentration of Aβ is sufficient to increase the expression of VDAC1 and IP3R3, resulting in elevated ER-mitochondria contact levels and mitochondrial Ca2+ overload (Figure 4; Hedskog et al., 2013; Schreiner et al., 2015). The amyloid-beta precursor protein (APP)-C99, the C-terminal fragment of APP, is also found to localize in the MAM (Pera et al., 2017). The accumulation of C99 in the MAM region induces sphingolipid turnover (thereby leading to altered lipid composition in both the MAM and mitochondrial membrane), increased ER–mitochondria contacts and mitochondrial dysfunction (Figure 4; Pera et al., 2017), indicating that C99 may act as a risk factor for AD. Tau, another risk factor for AD and other types of dementia, is found to be present in the outer membrane of mitochondria and in the mitochondrial intermembrane space. Tau localization at mitochondria affects their distribution and enhances Ca2+ transfer from the ER to mitochondria (Cieri et al., 2018). Moreover, dysregulated ER-mitochondria contacts have also been observed in PD models. Synuclein-α (SNCA), a critical protein for the development of PD, has been proven to enhance the ER-mitochondria contacts, resulting in mitochondrial Ca2+ overload (Guardia-Laguarta et al., 2014; Calì et al., 2019). In neurons that were differentiated from PD-patient derived induced pluripotent stem cells (iPSC) with mutations in Parkin and PINK1, the ER-mitochondria contacts were found to be increased, resulting in elevated ER to mitochondrial lipid trafficking and the disrupted production of neuropeptide-containing vesicles (Valadas et al., 2018). Increased ER-mitochondria contacts and mitochondrial Ca2+ levels induced by PINK1 mutations lead to mitochondrial enlargement and neuronal cell death. These phenotypes could be rescued by the inhibition of Miro or components of the ERMES (Valadas et al., 2018).
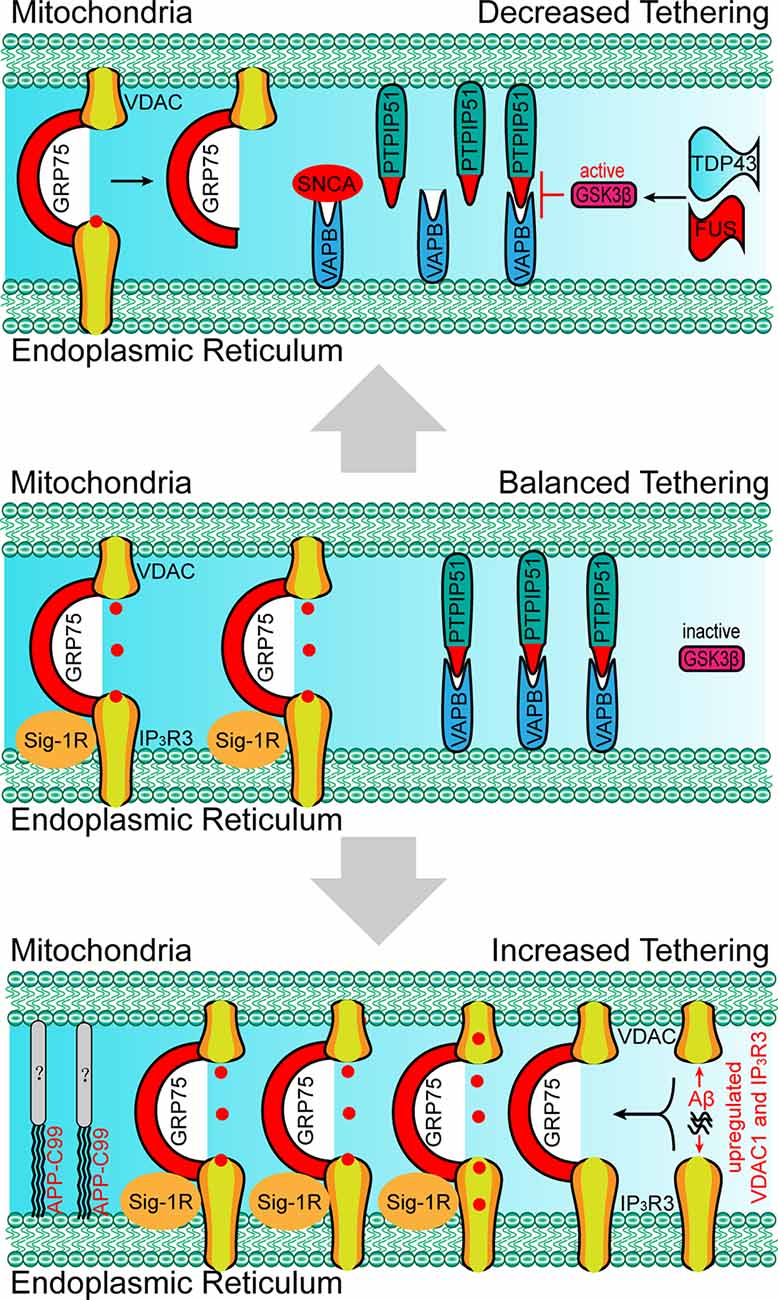
Figure 4. Paradigm of changes in molecular determinants and ER-mitochondria tethering in neurodegenerative disorders. Upper: the absence of Sig-1R at MAM leads to the de-stabilization of IP3R3, resulting in the decreased IP3R3-GRP75-VDAC complex and ER-mitochondria tethering, which is associated with the development of amyotrophic lateral sclerosis (ALS). SNCA, FUS and TDP43 disrupt the VAPB-PIPTP51 interaction and ER–mitochondria contact, which is associated with the development of Parkinson’s disease (PD) and ALS. Bottom: the accumulation of Aβ at MAM promotes the expression of VDAC and IP3R3, which leads to the increased formation of the IP3R3-GRP75-VDAC complex, ER-mitochondria tethering, and mitochondrial Ca2+ overload. APP-C99 is also observed at MAMs, which enhances the ER-mitochondrial contact. The presence of Aβ and APP-C99 at MAMs are associated with the development of Alzheimer’s disease (AD). SNCA, synuclein-α; TDP43, TAR DNA-binding protein 43; FUS, RNA-binding protein FUS; Aβ, β amyloid.
In conclusion, these results suggest that some molecules could enhance ER-mitochondria tethering by acting as membrane contact determinants or enhancers under pathological conditions, which leads to mitochondrial Ca2+ overload, oxidative stress, neuroinflammation, or apoptosis.
Apart from excessive ER-mitochondria contacts being associated with the development of diseases, decreased ER-mitochondria contacts are also found to induce the onset of abnormalities in cellular metabolism, especially in the neurons. For example, MITOL-mediated ubiquitination of MFN2 is critical for the formation of the MFN2 dimers and in facilitating the tethering of ER-mitochondria (Sugiura et al., 2013). The loss of MITOL leads to a reduction in ER-mitochondria contact sites and an increase in oxidative stress in the neurons (Nagashima et al., 2019), which also render cells more vulnerable to ER-stress induced apoptosis (Takeda et al., 2019). The mutation of Miro1 in PD patients leads to decreased ER-mitochondria contacts and low basal mitochondrial Ca2+ levels, thereby impairing energy metabolism and mitophagy (Grossmann et al., 2019). In VAPB-, PTPIP51-, or PDZD8-deficient neurons the lack of ER-mitochondria tethering at the synapses lead to decreased synaptic activities (Hirabayashi et al., 2017; Gómez-Suaga et al., 2019). Moreover, the loss of VAPB/PTPIP51-mediated ER-mitochondria contact causes a reduced number of dendritic spines in the neurons (Gómez-Suaga et al., 2019). In familial PD involving mutations in SNCA, mutated SNCA interacts with VAPB and decreases the VAPB-PTPIP51 interaction, leading to decreased dendritic ER-mitochondria contact and low synaptic activity levels (Figure 4; Paillusson et al., 2017). In addition, shrunken mitochondria were observed in neurons with mutated SNCA neurons, indicating morphological changes in the mitochondria (Little et al., 2018). Apart from SNCA mutants, the RNA-binding protein FUS can also disrupt the VAPB-PIPTP51 interaction (Figure 4). A reduction in mitochondrial adenosine triphosphate (ATP) production and Ca2+ concentration is observed in FUS-upregulated neurons, which may be associated with the development of ALS and frontotemporal dementia (FTD; Stoica et al., 2016). TAR DNA-binding protein 43 (TDP43), another ALS-associated protein, can also inhibit the VAPB-PIPTP51 interaction (Figure 4). Cells with high expression levels of TDP43 were found to exhibit the same characteristics as those of FUS-upregulated cells, including increased cytosolic Ca2+ levels and decreased mitochondrial Ca2+ levels (Figure 3; Stoica et al., 2014). In addition, the inhibition of the VAPB-PTPIP51 interaction mediated by FUS and TDP43 is associated with the activation of glycogen synthase kinase-3 beta (Stoica et al., 2014, 2016). Decreased ER-mitochondria tethering is also found in Sig-1R- and SOD1-linked ALS (Watanabe et al., 2016). Sig-1R locates in the MAM and promotes ER-mitochondria tethering by stabilizing IP3R3 (Hayashi and Su, 2007). Aberrant subcellular distribution of Sig-1R mutations is associated with ALS and distal hereditary motor neuropathy (Al-Saif et al., 2011; Almendra et al., 2018). Further studies reveal that aberrant subcellular distribution of Sig-1R results in the decrease of ER-mitochondria tethering and mitochondrial Ca2+ levels (Figure 4; Al-Saif et al., 2011; Gregianin et al., 2016). In addition, increasing ER-mitochondria tethering by activation of Sig-1R is proven to be beneficial for SOD1-linked ALS. Considered together, the above studies indicate that decreased ER-mitochondria contacts or protein interaction changes are also harmful and result in alterations in cell fate, neural activity, and synaptic plasticity in the neurons.
In summary, apart from the classic membrane contact determinants that mediate the ER-mitochondria communication and metabolism, some disease-associated abnormal proteins or peptides could also play critical roles as determinants or regulators that disrupt the contact balance between the ER and mitochondria. Enhanced ER-mitochondria contact levels in the soma of neurons induce mitochondria Ca2+ overload and neuronal cell death, while decreased ER-mitochondria contact levels in the dendrites of neurons lead to reduced synaptic activity and spine density, suggesting that dysregulated ER-mitochondria interaction are involved in the development of some neurodegenerative disorders, such as AD, PD and ALS.
The Determinants of Mitochondria-Lysosome Membrane Contact and Mitochondria-Peroxisome Membrane Contact
Mitochondria-Lysosomes Membrane Contacts
Besides mitochondria-ER contacts, determinants of mitochondria-lysosomes and of mitochondria-peroxisomes contacts have been recently discovered, further enhancing the understanding of the communication between these organelles in the cells. Here, we have mainly summarized the determinants between these organs in the neuronal system.
A lysosome is a highly dynamic organelle that mediates the degradation of unfolded proteins, damaged organelles, and pathogen-derived components. Under stress conditions or during the process of aging, the lysosome-mediated clearance of damaged mitochondria is crucial for maintaining mitochondrial homeostasis (Wang and Klionsky, 2011). The accumulation of damaged mitochondria is observed in multiple cells with dysfunction in lysosome performance, which is also involved in development neurodegenerative diseases. However, because the lysosome-mediated clearance of damaged mitochondria is believed to be autophagy-dependent, the mitochondrial-lysosome interaction has been considered indirect membrane contact.
Recently, direct mitochondria-lysosome contacts have been discovered through multiple super-resolution imaging modalities. Ras-related protein Rab-7 (RAB7) was the first identified mitochondrial-lysosome membrane contact determinant (Wong et al., 2018). The presence of RAB7 is sufficient to mediate mitochondrial-lysosome tethering. The recruitment of TBC1 domain family member 15 (TBC1D15) by mitochondrial fission 1 protein (FIS1) at the membrane of mitochondria, enhances RAB7 activity (GTP hydrolysis), which results in mitochondrial-lysosome untethering (Wong et al., 2018). The GTP- guanosine diphosphate (GDP) cycling dynamic regulates the mitochondrial-lysosome contact and fission of the mitochondria. Moreover, a deficiency in RAB7 levels in the neurons causes axonal degeneration and the development of Charcot-Marie-Tooth neuropathy type 2B (Ponomareva et al., 2016). With developments in imaging technology, apart from RAB7, we believe that other molecules mediating and controlling mitochondrial-lysosome tethering will be identified and characterized in the future.
Mitochondria-Peroxisomes Membrane Contacts
Peroxisomes are membrane-bound oxidative organelles that play an indispensable role in the metabolism of fatty acids, glyoxylate, amino acids, and reactive oxygen species (ROS; Schrader et al., 2015). The evidence generated by studies on yeast demonstrate that peroxisomes are localized to mitochondria-ER junctions and sites of acetyl-CoA synthesis (Cohen et al., 2014). This finding suggests that the mitochondria-peroxisome contacts may be important for the β-oxidation of fatty acids in the peroxisomes or mitochondria. In mammal cells, mitochondria-derived vesicles (MDVs) are considered as one of the origins of peroxisomes, as some of the components of mitochondria are selectively integrated into peroxisomes (Sugiura et al., 2017). Peroxisomal membrane protein 11 (Pex11), a peroxin that is localized within the peroxisomes, is found to interact with Mdm34 (localized in the membrane of the ER and one of the components of the ERMES) in yeast (Mattiazzi Ušaj et al., 2015). In addition, the initiation of membrane contact between peroxisomes and mitochondria mediated by Pex11 and Mdm34 is ER-dependent (Mattiazzi Ušaj et al., 2015). On systematically mapping the contact sites, Pex34 and Fzo1 were discovered to localize at the membrane of peroxisomes, which can facilitate their tethering to mitochondria. The loss of Pex34 results in reduced β-oxidation of fatty acids (Shai et al., 2018). Acyl-coenzyme A-binding domain 2 (ACBD2)/ECI2 isoform A is another newly identified protein, with a C-terminal peroxisome targeting signal-1, which aids in the localization of ACBD2/ECI2 in peroxisomes. Further, by interacting with TOM20, ACBD2/ECI2 can mediate membrane contact between peroxisomes and mitochondria (Fan et al., 2016). A functional study revealed that ACB2/ECI2-TOM20 mediated peroxisome-mitochondria tethering is essential for the biosynthesis of steroids in Leydig Cells (Fan et al., 2016). Considered together, these studies suggest that proteins localized at the membrane of mitochondria and peroxisomes regulate the mitochondria-peroxisome membrane contacts and affect the lipid metabolism in peroxisomes.
In the nervous system, although the mitochondria-lysosome membrane contact and mitochondria-peroxisome membrane contact have not been studied as much as the mitochondria-ER contact, we believe that more and more contact determinants will be discovered and characterized in the near future.
Concluding Remarks
Mitochondria exchange signals and materials with other organelles through membrane contacts. During this process, molecular determinants dynamically regulate the tethering and untethering between mitochondria and other organelles. However, in pathological conditions, some disease-associated molecules have been retrieved at MAM, where they act as membrane contact determinants (such as Aβ), disrupt physiological interactions via post-translational modification (such as PINK1), and participate in competitive interactions (such as FUS; Figure 1). These abnormal proteins present in the MAM cause dysfunctions in the tethering between mitochondria and other organelles, and thus leads to subsequent mitochondrial Ca2+ overload, lower synaptic activity levels, metabolic disorders, and finally neural cell death. An increasing amount of evidence shows that the dysregulation of mitochondria membrane contacts with other organelles cause mitochondrial dysfunctions underlying neurodegenerative diseases. However, extensive investigations are still needed to fully characterize the regulatory mechanism involved. Accordingly, in clinical practice, pharmacological modulation of contacts still has a long way to go.
Author Contributions
YL conceived the review topic, reviewed the literature, wrote the manuscript, and prepared the figure. YD and JC reviewed the manuscript. YL and JC performed a comprehensive review of the literature.
Funding
This work was supported by grants from the National Nature Science Foundation of China (81870839 and 81701187).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
MFN1/2, mitofusin-1/2; ER, endoplasmic reticulum; AD, Alzheimer’s disease; PD, Parkinson’s disease; ALS, amyotrophic lateral sclerosis; ERMES, ER-mitochondria encounter structure; Mdm10/12/34, mitochondrial distribution and morphology protein 10/12/34; Mmm1, maintenance of mitochondrial morphology protein 1; Gem1, mitochondrial Rho GTPase 1; SMP, mitochondrial lipid-binding protein; EMC, ER membrane protein complex; TOM5, translocase of outer membrane 5 kDa subunit; MAM, mitochondria-associated ER membrane; PACS2, phosphofurin acidic cluster sorting protein 2; GRP75, 75 kDa glucose-regulated protein; VDAC1/2, voltage-dependent anion-selective channel protein 1/2; IP3R3, inositol 1,4,5-trisphosphate receptor type 3; FBXL2, F-box protein FBXL2; MITOL, mitochondrial ubiquitin ligase; PINK1, PTEN-induced putative kinase protein 1; TG2, transglutaminase type 2; PDK4, pyruvate dehydrogenase kinase 4; Miro, mitochondrial Rho; PIGBOS, PIGB opposite strand; MICOS, mitochondrial contact site and cristae organizing system; ERMCS, ER-mitochondria contact sites; CLCC1, chloride channel CLIC-like protein 1; ORP5/8, oxysterol-binding protein (OSBP)-related proteins 5/8; PTPIP51, protein tyrosine phosphatase interacting protein-51; VAPB, vesicle-associated membrane protein-associated protein B; Rmdn3, regulator of microtubule dynamics protein 3; PDZD8, PDZ domain-containing protein 8; APP, amyloid-beta precursor protein; SNCA, synuclein-α; Sig-1R, sigma-1 receptor; FTD, frontotemporal dementia; TDP43, TAR DNA-binding protein 43; RAB7, Ras-related protein Rab 7; TBC1D15, TBC1 domain family member 15; FIS1, fission 1 protein; MDVs, mitochondria-derived vesicles; Pex11, Peroxisomal membrane protein 11; ACBD2/ECI2, Acyl-coenzyme A-binding domain 2/ECI2 isoform A.
References
AhYoung, A. P., Jiang, J., Zhang, J., Khoi Dang, X., Loo, J. A., Zhou, Z. H., et al. (2015). Conserved SMP domains of the ERMES complex bind phospholipids and mediate tether assembly. Proc. Natl. Acad. Sci. U S A 112, E3179–E3188. doi: 10.1073/pnas.1422363112
Almendra, L., Laranjeira, F., Fernandez-Marmiesse, A., and Negrao, L. (2018). SIGMAR1 gene mutation causing distal hereditary motor neuropathy in a portuguese family. Acta Myol. 37, 2–4.
Al-Saif, A., Al-Mohanna, F., and Bohlega, S. (2011). A mutation in sigma-1 receptor causes juvenile amyotrophic lateral sclerosis. Ann. Neurol. 70, 913–919. doi: 10.1002/ana.22534
Area-Gomez, E., Del Carmen Lara Castillo, M., Tambini, M. D., Guardia-Laguarta, C., de Groof, A. J., Madra, M., et al. (2012). Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. EMBO J. 31, 4106–4123. doi: 10.1038/emboj.2012.202
Basso, V., Marchesan, E., Peggion, C., Chakraborty, J., von Stockum, S., Giacomello, M., et al. (2018). Regulation of ER-mitochondria contacts by Parkin via Mfn2. Pharmacol. Res. 138, 43–56. doi: 10.1016/j.phrs.2018.09.006
Berenguer-Escuder, C., Grossmann, D., Antony, P., Arena, G., Wasner, K., Massart, F., et al. (2020). Impaired mitochondrial-endoplasmic reticulum interaction and mitophagy in miro1-mutant neurons in Parkinson’s disease. Hum. Mol.genetics 29, 1353–1364. doi: 10.1093/hmg/ddaa066
Bernhard, W., and Rouiller, C. (1956). Close topographical relationship between mitochondria and ergastoplasm of liver cells in a definite phase of cellular activity. J. Biophys. Biochem. Cytol. 2, 73–78. doi: 10.1083/jcb.2.4.73
Calì, T., Ottolini, D., Vicario, M., Catoni, C., Vallese, F., Cieri, D., et al. (2019). splitGFP technology reveals dose-dependent ER-mitochondria interface modulation by alpha-synuclein A53T and A30P mutants. Cells 8:1072. doi: 10.3390/cells8091072
Calkins, M. J., Manczak, M., Mao, P., Shirendeb, U., and Reddy, P. H. (2011). Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 20, 4515–4529. doi: 10.1093/hmg/ddr381
Chen, Y., Csordás, G., Jowdy, C., Schneider, T. G., Csordás, N., Wang, W., et al. (2012). Mitofusin 2-containing mitochondrial-reticular microdomains direct rapid cardiomyocyte bioenergetic responses via interorganelle Ca2+ crosstalk. Circ. Res. 111, 863–875. doi: 10.1161/circresaha.112.266585
Chen, H., Detmer, S. A., Ewald, A. J., Griffin, E. E., Fraser, S. E., and Chan, D. C. (2003). Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 160, 189–200. doi: 10.1083/jcb.200211046
Chu, Q., Martinez, T. F., Novak, S. W., Donaldson, C. J., Tan, D., Vaughan, J. M., et al. (2019). Regulation of the ER stress response by a mitochondrial microprotein. Nat. Commun. 10:4883. doi: 10.1038/s41467-019-12816-z
Cieri, D., Vicario, M., Vallese, F., D’Orsi, B., Berto, P., Grinzato, A., et al. (2018). Tau localises within mitochondrial sub-compartments and its caspase cleavage affects ER-mitochondria interactions and cellular Ca2+ handling. Biochim. Biophys. Acta Mol. Basis Dis. 1864, 3247–3256. doi: 10.1016/j.bbadis.2018.07.011
Cohen, Y., Klug, Y. A., Dimitrov, L., Erez, Z., Chuartzman, S. G., Elinger, D., et al. (2014). Peroxisomes are juxtaposed to strategic sites on mitochondria. Mol. Biosyst. 10, 1742–1748. doi: 10.1039/c4mb00001c
Csordás, G., Renken, C., Várnai, P., Walter, L., Weaver, D., Buttle, K. F., et al. (2006). Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 174, 915–921. doi: 10.1083/jcb.200604016
Dalen, H., Scheie, P., Myklebust, R., and Saetersdal, T. (1983). An ultrastructural study of cryofractured myocardial cells with special attention to the relationship between mitochondria and sarcoplasmic reticulum. J. Microsc. 131, 35–46. doi: 10.1111/j.1365-2818.1983.tb04228.x
de Brito, O. M., and Scorrano, L. (2008). Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456, 605–610. doi: 10.1038/nature07534
D’Eletto, M., Rossin, F., Occhigrossi, L., Farrace, M. G., Faccenda, D., Desai, R., et al. (2018). Transglutaminase type 2 regulates ER-mitochondria contact sites by interacting with GRP75. Cell Rep. 25, 3573.e4–3581.e4. doi: 10.1016/j.celrep.2018.11.094
De Vos, K. J., Mórotz, G. M., Stoica, R., Tudor, E. L., Lau, K. F., Ackerley, S., et al. (2012). VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum. Mol. Genet. 21, 1299–1311. doi: 10.1093/hmg/ddr559
Eisenberg-Bord, M., Tsui, H. S., Antunes, D., Fernandez-Del-Rio, L., Bradley, M. C., Dunn, C. D., et al. (2019). The endoplasmic reticulum-mitochondria encounter structure complex coordinates coenzyme Q biosynthesis. Contact 2:2515256418825409. doi: 10.1177/2515256418825409
Fan, J., Li, X., Issop, L., Culty, M., and Papadopoulos, V. (2016). ACBD2/ECI2-mediated peroxisome-mitochondria interactions in leydig cell steroid biosynthesis. Mol. Endocrinol. 30, 763–782. doi: 10.1210/me.2016-1008
Fecher, C., Trovò, L., Müller, S. A., Snaidero, N., Wettmarshausen, J., Heink, S., et al. (2019). Cell-type-specific profiling of brain mitochondria reveals functional and molecular diversity. Nat. Neurosci. 22, 1731–1742. doi: 10.1038/s41593-019-0479-z
Filadi, R., Greotti, E., Turacchio, G., Luini, A., Pozzan, T., and Pizzo, P. (2015). Mitofusin 2 ablation increases endoplasmic reticulum-mitochondria coupling. Proc. Natl. Acad. Sci. U S A 112, E2174–2181. doi: 10.1073/pnas.1504880112
Filadi, R., Greotti, E., Turacchio, G., Luini, A., Pozzan, T., and Pizzo, P. (2017). On the role of mitofusin 2 in endoplasmic reticulum-mitochondria tethering. Proc. Natl. Acad. Sci. U S A 114, E2266–E2267. doi: 10.1073/pnas.1616040114
Filadi, R., Leal, N. S., Schreiner, B., Rossi, A., Dentoni, G., Pinho, C. M., et al. (2018). TOM70 sustains cell bioenergetics by promoting IP3R3-mediated ER to mitochondria Ca2+ transfer. Curr. Biol. 28, 369.e6–382.e6. doi: 10.1016/j.cub.2017.12.047
Galmes, R., Houcine, A., van Vliet, A. R., Agostinis, P., Jackson, C. L., and Giordano, F. (2016). ORP5/ORP8 localize to endoplasmic reticulum-mitochondria contacts and are involved in mitochondrial function. EMBO Rep. 17, 800–810. doi: 10.15252/embr.201541108
Gelmetti, V., De Rosa, P., Torosantucci, L., Marini, E. S., Romagnoli, A., Di Rienzo, M., et al. (2017). PINK1 and BECN1 relocalize at mitochondria-associated membranes during mitophagy and promote ER-mitochondria tethering and autophagosome formation. Autophagy 13, 654–669. doi: 10.1080/15548627.2016.1277309
Gomez-Suaga, P., Paillusson, S., and Miller, C. C. J. (2017a). ER-mitochondria signaling regulates autophagy. Autophagy 13, 1250–1251. doi: 10.1080/15548627.2017.1317913
Gomez-Suaga, P., Paillusson, S., Stoica, R., Noble, W., Hanger, D. P., and Miller, C. C. J. (2017b). The ER-mitochondria tethering complex VAPB-PTPIP51 regulates autophagy. Curr. Biol. 27, 371–385. doi: 10.1016/j.cub.2016.12.038
Gómez-Suaga, P., Pérez-Nievas, B. G., Glennon, E. B., Lau, D. H. W., Paillusson, S., Mórotz, G. M., et al. (2019). The VAPB-PTPIP51 endoplasmic reticulum-mitochondria tethering proteins are present in neuronal synapses and regulate synaptic activity. Acta Neuropathol. Commun. 7:35. doi: 10.1186/s40478-019-0688-4
Gregianin, E., Pallafacchina, G., Zanin, S., Crippa, V., Rusmini, P., Poletti, A., et al. (2016). Loss-of-function mutations in the SIGMAR1 gene cause distal hereditary motor neuropathy by impairing ER-mitochondria tethering and Ca2+ signalling. Hum. Mol. Genet. 25, 3741–3753. doi: 10.1093/hmg/ddw220
Grönblad, M., and Akerman, K. E. (1984). Electron-dense endoplasmic reticulum-like profiles closely associated with mitochondria in glomus cells of the carotid body after fixation with oxalate. Exp. Cell Res. 152, 161–168. doi: 10.1016/0014-4827(84)90240-4
Grossmann, D., Berenguer-Escuder, C., Bellet, M. E., Scheibner, D., Bohler, J., Massart, F., et al. (2019). Mutations in RHOT1 disrupt endoplasmic reticulum-mitochondria contact sites interfering with calcium homeostasis and mitochondrial dynamics in Parkinson’s disease. Antioxid. Redox Signal. 31, 1213–1234. doi: 10.1089/ars.2018.7718
Guardia-Laguarta, C., Area-Gomez, E., Rüb, C., Liu, Y., Magrané, J., Becker, D., et al. (2014). α-synuclein is localized to mitochondria-associated ER membranes. J. Neurosci. 34, 249–259. doi: 10.1523/JNEUROSCI.2507-13.2014
Hayashi, T., and Su, T. P. (2007). Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca2+ signaling and cell survival. Cell 131, 596–610. doi: 10.1016/j.cell.2007.08.036
Hedskog, L., Pinho, C. M., Filadi, R., Ronnback, A., Hertwig, L., Wiehager, B., et al. (2013). Modulation of the endoplasmic reticulum-mitochondria interface in Alzheimer’s disease and related models. Proc. Natl. Acad. Sci. U S A 110, 7916–7921. doi: 10.1073/pnas.1300677110
Hirabayashi, Y., Kwon, S. K., Paek, H., Pernice, W. M., Paul, M. A., Lee, J., et al. (2017). ER-mitochondria tethering by PDZD8 regulates Ca2+ dynamics in mammalian neurons. Science 358, 623–630. doi: 10.1126/science.aan6009
Jeong, H., Park, J., and Lee, C. (2016). Crystal structure of Mdm12 reveals the architecture and dynamic organization of the ERMES complex. EMBO Rep. 17, 1857–1871. doi: 10.15252/embr.201642706
Khacho, M., Clark, A., Svoboda, D. S., Azzi, J., MacLaurin, J. G., Meghaizel, C., et al. (2016). Mitochondrial dynamics impacts stem cell identity and fate decisions by regulating a nuclear transcriptional program. Cell Stem Cell 19, 232–247. doi: 10.1016/j.stem.2016.04.015
Kornmann, B., Osman, C., and Walter, P. (2011). The conserved GTPase Gem1 regulates endoplasmic reticulum-mitochondria connections. Proc. Natl. Acad. Sci. U S A 108, 14151–14156. doi: 10.1073/pnas.1111314108
Kuchay, S., Giorgi, C., Simoneschi, D., Pagan, J., Missiroli, S., Saraf, A., et al. (2017). PTEN counteracts FBXL2 to promote IP3R3- and Ca2+-mediated apoptosis limiting tumour growth. Nature 546, 554–558. doi: 10.1038/nature22965
Lahiri, S., Chao, J. T., Tavassoli, S., Wong, A. K., Choudhary, V., Young, B. P., et al. (2014). A conserved endoplasmic reticulum membrane protein complex (EMC) facilitates phospholipid transfer from the ER to mitochondria. PLoS Biol. 12:e1001969. doi: 10.1371/journal.pbio.1001969
Leal, N. S., Dentoni, G., Schreiner, B., Kämäräinen, O. P., Partanen, N., Herukka, S. K., et al. (2018). Alterations in mitochondria-endoplasmic reticulum connectivity in human brain biopsies from idiopathic normal pressure hydrocephalus patients. Acta Neuropathol. Commun. 6:102. doi: 10.1186/s40478-018-0605-2
Lee, K. S., Huh, S., Lee, S., Wu, Z., Kim, A. K., Kang, H. Y., et al. (2018). Altered ER-mitochondria contact impacts mitochondria calcium homeostasis and contributes to neurodegeneration in vivo in disease models. Proc. Natl. Acad. Sci. U S A 115, E8844–E8853. doi: 10.1073/pnas.1721136115
Lee, S., Lee, K. S., Huh, S., Liu, S., Lee, D. Y., Hong, S. H., et al. (2016). Polo kinase phosphorylates miro to control ER-mitochondria contact sites and mitochondrial Ca2+ homeostasis in neural stem cell development. Dev. Cell 37, 174–189. doi: 10.1016/j.devcel.2016.03.023
Li, D., Li, X., Guan, Y., and Guo, X. (2015). Mitofusin-2-mediated tethering of mitochondria and endoplasmic reticulum promotes cell cycle arrest of vascular smooth muscle cells in G0/G1 phase. Acta Biochim. Biophys. Sin. 47, 441–450. doi: 10.1093/abbs/gmv035
Liao, Y., Hao, Y., Chen, H., He, Q., Yuan, Z., and Cheng, J. (2015). Mitochondrial calcium uniporter protein MCU is involved in oxidative stress-induced cell death. Protein Cell 6, 434–442. doi: 10.1007/s13238-015-0144-6
Little, D., Luft, C., Mosaku, O., Lorvellec, M., Yao, Z., Paillusson, S., et al. (2018). A single cell high content assay detects mitochondrial dysfunction in iPSC-derived neurons with mutations in SNCA. Sci. Rep. 8:9033. doi: 10.1038/s41598-018-27058-0
Ma, J. H., Shen, S., Wang, J. J., He, Z., Poon, A., Li, J., et al. (2017). Comparative proteomic analysis of the mitochondria-associated ER membrane (MAM) in a long-term type 2 diabetic rodent model. Sci. Rep. 7:2062. doi: 10.1038/s41598-017-02213-1
Mattiazzi Ušaj, M., Brložnik, M., Kaferle, P., Žitnik, M., Wolinski, H., Leitner, F., et al. (2015). Genome-wide localization study of yeast Pex11 identifies peroxisome-mitochondria interactions through the ERMES complex. J. Mol. Biol. 427, 2072–2087. doi: 10.1016/j.jmb.2015.03.004
McLelland, G. L., Goiran, T., Yi, W., Dorval, G., Chen, C. X., Lauinger, N. D., et al. (2018). Mfn2 ubiquitination by PINK1/parkin gates the p97-dependent release of ER from mitochondria to drive mitophagy. eLife 7:e32866. doi: 10.7554/eLife.32866
Modi, S., López-Doménech, G., Halff, E. F., Covill-Cooke, C., Ivankovic, D., Melandri, D., et al. (2019). Miro clusters regulate ER-mitochondria contact sites and link cristae organization to the mitochondrial transport machinery. Nat. Commun. 10:4399. doi: 10.1038/s41467-019-12382-4
Mori, T., Hayashi, T., Hayashi, E., and Su, T. P. (2013). Sigma-1 receptor chaperone at the ER-mitochondrion interface mediates the mitochondrion-ER-nucleus signaling for cellular survival. PLoS One 8:e76941. doi: 10.1371/journal.pone.0076941
Nagashima, S., Takeda, K., Ohno, N., Ishido, S., Aoki, M., Saitoh, Y., et al. (2019). MITOL deletion in the brain impairs mitochondrial structure and ER tethering leading to oxidative stress. Life Sci. Alliance 2:e201900308. doi: 10.26508/lsa.201900308
Naon, D., Zaninello, M., Giacomello, M., Varanita, T., Grespi, F., Lakshminaranayan, S., et al. (2016). Critical reappraisal confirms that mitofusin 2 is an endoplasmic reticulum-mitochondria tether. Proc. Natl. Acad. Sci. U S A 113, 11249–11254. doi: 10.1073/pnas.1606786113
Norkett, R., Modi, S., Birsa, N., Atkin, T. A., Ivankovic, D., Pathania, M., et al. (2016). DISC1-dependent regulation of mitochondrial dynamics controls the morphogenesis of complex neuronal dendrites. J. Biol. Chem. 291, 613–629. doi: 10.1074/jbc.m115.699447
Paillusson, S., Gomez-Suaga, P., Stoica, R., Little, D., Gissen, P., Devine, M. J., et al. (2017). α-synuclein binds to the ER-mitochondria tethering protein VAPB to disrupt Ca2+ homeostasis and mitochondrial ATP production. Acta Neuropathol. 134, 129–149. doi: 10.1007/s00401-017-1704-z
Pera, M., Larrea, D., Guardia-Laguarta, C., Montesinos, J., Velasco, K. R., Agrawal, R. R., et al. (2017). Increased localization of APP-C99 in mitochondria-associated ER membranes causes mitochondrial dysfunction in Alzheimer disease. EMBO J. 36, 3356–3371. doi: 10.15252/embj.201796797
Ponomareva, O. Y., Eliceiri, K. W., and Halloran, M. C. (2016). Charcot-Marie-Tooth 2b associated Rab7 mutations cause axon growth and guidance defects during vertebrate sensory neuron development. Neural Dev. 11:2. doi: 10.1186/s13064-016-0058-x
Poston, C. N., Krishnan, S. C., and Bazemore-Walker, C. R. (2013). In-depth proteomic analysis of mammalian mitochondria-associated membranes (MAM). J. Proteomics 79, 219–230. doi: 10.1016/j.jprot.2012.12.018
Qi, Y., Yan, L., Yu, C., Guo, X., Zhou, X., Hu, X., et al. (2016). Structures of human mitofusin 1 provide insight into mitochondrial tethering. J. Cell Biol. 215, 621–629. doi: 10.1083/jcb.201609019
Rusinol, A. E., Cui, Z., Chen, M. H., and Vance, J. E. (1994). A unique mitochondria-associated membrane fraction from rat liver has a high capacity for lipid synthesis and contains pre-Golgi secretory proteins including nascent lipoproteins. J. Biol. Chem. 269, 27494–27502.
Schrader, M., Costello, J., Godinho, L. F., and Islinger, M. (2015). Peroxisome-mitochondria interplay and disease. J. Inherit. Metab. Dis. 38, 681–702. doi: 10.1007/s10545-015-9819-7
Schreiner, B., Hedskog, L., Wiehager, B., and Ankarcrona, M. (2015). Amyloid-beta peptides are generated in mitochondria-associated endoplasmic reticulum membranes. J. Alzheimers Dis. 43, 369–374. doi: 10.3233/jad-132543
Shai, N., Yifrach, E., van Roermund, C. W. T., Cohen, N., Bibi, C., Lodewijk, I., et al. (2018). Systematic mapping of contact sites reveals tethers and a function for the peroxisome-mitochondria contact. Nat. Commun. 9:1761. doi: 10.1038/s41467-018-03957-8
Shoshan-Barmatz, V., Keinan, N., and Zaid, H. (2008). Uncovering the role of VDAC in the regulation of cell life and death. J. Bioenerg. Biomembr. 40, 183–191. doi: 10.1007/s10863-008-9147-9
Simmen, T., Aslan, J. E., Blagoveshchenskaya, A. D., Thomas, L., Wan, L., Xiang, Y., et al. (2005). PACS-2 controls endoplasmic reticulum-mitochondria communication and Bid-mediated apoptosis. EMBO J. 24, 717–729. doi: 10.1038/sj.emboj.7600559
Stoica, R., De Vos, K. J., Paillusson, S., Mueller, S., Sancho, R. M., Lau, K. F., et al. (2014). ER-mitochondria associations are regulated by the VAPB-PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat. Commun. 5:3996. doi: 10.1038/ncomms4996
Stoica, R., Paillusson, S., Gomez-Suaga, P., Mitchell, J. C., Lau, D. H., Gray, E. H., et al. (2016). ALS/FTD-associated FUS activates GSK-3beta to disrupt the VAPB-PTPIP51 interaction and ER-mitochondria associations. EMBO Rep. 17, 1326–1342. doi: 10.15252/embr.201541726
Sugiura, A., Mattie, S., Prudent, J., and McBride, H. M. (2017). Newly born peroxisomes are a hybrid of mitochondrial and ER-derived pre-peroxisomes. Nature 542, 251–254. doi: 10.1038/nature21375
Sugiura, A., Nagashima, S., Tokuyama, T., Amo, T., Matsuki, Y., Ishido, S., et al. (2013). MITOL regulates endoplasmic reticulum-mitochondria contacts via Mitofusin2. Mol. Cell 51, 20–34. doi: 10.1016/j.molcel.2013.04.023
Sun, T., Qiao, H., Pan, P. Y., Chen, Y., and Sheng, Z. H. (2013). Motile axonal mitochondria contribute to the variability of presynaptic strength. Cell Rep. 4, 413–419. doi: 10.1016/j.celrep.2013.06.040
Szabadkai, G., Bianchi, K., Varnai, P., De Stefani, D., Wieckowski, M. R., Cavagna, D., et al. (2006). Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell. Biol. 175, 901–911. doi: 10.1083/jcb.200608073
Takeda, K., Nagashima, S., Shiiba, I., Uda, A., Tokuyama, T., Ito, N., et al. (2019). MITOL prevents ER stress-induced apoptosis by IRE1alpha ubiquitylation at ER-mitochondria contact sites. EMBO J. 38:e100999. doi: 10.15252/embj.2018100999
Thoudam, T., Ha, C. M., Leem, J., Chanda, D., Park, J. S., Kim, H. J., et al. (2019). PDK4 augments ER-mitochondria contact to dampen skeletal muscle insulin signaling during obesity. Diabetes 68, 571–586. doi: 10.2337/db18-0363
Tsai, Y. L., Coady, T. H., Lu, L., Zheng, D., Alland, I., Tian, B., et al. (2020). ALS/FTD-associated protein FUS induces mitochondrial dysfunction by preferentially sequestering respiratory chain complex mRNAs. Genes Dev. 34, 785–805. doi: 10.1101/gad.335836.119
Vaccaro, V., Devine, M. J., Higgs, N. F., and Kittler, J. T. (2017). Miro1-dependent mitochondrial positioning drives the rescaling of presynaptic Ca2+ signals during homeostatic plasticity. EMBO Re. 18, 231–240. doi: 10.15252/embr.201642710
Valadas, J. S., Esposito, G., Vandekerkhove, D., Miskiewicz, K., Deaulmerie, L., Raitano, S., et al. (2018). ER lipid defects in neuropeptidergic neurons impair sleep patterns in Parkinson’s disease. Neuron 98, 1155.e6–1169.e6. doi: 10.1016/j.neuron.2018.05.022
Vance, J. E. (1990). Phospholipid synthesis in a membrane fraction associated with mitochondria. J. Biol. Chem. 265, 7248–7256.
Veeresh, P., Kaur, H., Sarmah, D., Mounica, L., Verma, G., Kotian, V., et al. (2019). Endoplasmic reticulum-mitochondria crosstalk: from junction to function across neurological disorders. Ann. N Y Acad. Sci. 1457, 41–60. doi: 10.1111/nyas.14212
Voss, C., Lahiri, S., Young, B. P., Loewen, C. J., and Prinz, W. A. (2012). ER-shaping proteins facilitate lipid exchange between the ER and mitochondria in S. cerevisiae. J. Cell Sci. 125, 4791–4799. doi: 10.1242/jcs.105635
Wang, K., and Klionsky, D. J. (2011). Mitochondria removal by autophagy. Autophagy 7, 297–300. doi: 10.4161/auto.7.3.14502
Watanabe, S., Ilieva, H., Tamada, H., Nomura, H., Komine, O., Endo, F., et al. (2016). Mitochondria-associated membrane collapse is a common pathomechanism in SIGMAR1- and SOD1-linked ALS. EMBO Mol. Med. 8, 1421–1437. doi: 10.15252/emmm.201606403
Wideman, J. G., Gawryluk, R. M., Gray, M. W., and Dacks, J. B. (2013). The ancient and widespread nature of the ER-mitochondria encounter structure. Mol. Biol. Evol. 30, 2044–2049. doi: 10.1093/molbev/mst120
Wong, Y. C., Ysselstein, D., and Krainc, D. (2018). Mitochondria-lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature 554, 382–386. doi: 10.1038/nature25486
Keywords: mitochondria, membrane contact, determinant, physiological condition, pathological condition, neurological diseases
Citation: Liao Y, Dong Y and Cheng J (2020) The Molecular Determinants of Mitochondrial Membrane Contact With ER, Lysosomes and Peroxisomes in Neuronal Physiology and Pathology. Front. Cell. Neurosci. 14:194. doi: 10.3389/fncel.2020.00194
Received: 15 February 2020; Accepted: 05 June 2020;
Published: 07 August 2020.
Edited by:
Marta Giacomello, University of Padua, ItalyCopyright © 2020 Liao, Dong and Cheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jinbo Cheng, Y2hlbmdfamluYm9AMTI2LmNvbQ==
† These authors share first authorship