 Daniele Lana
Daniele Lana Filippo Ugolini
Filippo Ugolini Maria G. Giovannini
Maria G. Giovannini- 1Department of Health Sciences, Section of Clinical Pharmacology and Oncology, University of Florence, Florence, Italy
- 2Department of Health Sciences, Section of Anatomopathology, University of Florence, Florence, Italy,
Neurons have been long regarded as the basic functional cells of the brain, whereas astrocytes and microglia have been regarded only as elements of support. However, proper intercommunication among neurons–astrocytes–microglia is of fundamental importance for the functional organization of the brain. Perturbation in the regulation of brain energy metabolism not only in neurons but also in astrocytes and microglia may be one of the pathophysiological mechanisms of neurodegeneration, especially in hypoxia/ischemia. Glial activation has long been considered detrimental for survival of neurons, but recently it appears that glial responses to an insult are not equal but vary in different brain areas. In this review, we first take into consideration the modifications of the vascular unit of the glymphatic system and glial metabolism in hypoxic conditions. Using the method of triple-labeling fluorescent immunohistochemistry coupled with confocal microscopy (TIC), we recently studied the interplay among neurons, astrocytes, and microglia in chronic brain hypoperfusion. We evaluated the quantitative and morpho-functional alterations of the neuron–astrocyte–microglia triads comparing the hippocampal CA1 area, more vulnerable to ischemia, to the CA3 area, less vulnerable. In these contiguous and interconnected areas, in the same experimental hypoxic conditions, astrocytes and microglia show differential, finely regulated, region-specific reactivities. In both areas, astrocytes and microglia form triad clusters with apoptotic, degenerating neurons. In the neuron–astrocyte–microglia triads, the cell body of a damaged neuron is infiltrated and bisected by branches of astrocyte that create a microscar around it while a microglial cell phagocytoses the damaged neuron. These coordinated actions are consistent with the scavenging and protective activities of microglia. In hypoxia, the neuron–astrocyte–microglia triads are more numerous in CA3 than in CA1, further indicating their protective effects. These data, taken from contiguous and interconnected hippocampal areas, demonstrate that glial response to the same hypoxic insult is not equal but varies significantly. Understanding the differences of glial reactivity is of great interest to explain the differential susceptibility of hippocampal areas to hypoxia/ischemia. Further studies may evidence the differential reactivity of glia in different brain areas, explaining the higher or lower sensitivity of these areas to different insults and whether glia may represent a target for future therapeutic interventions.
Introduction
Neuroglial cells were discovered over a century ago and were first stained by the silver-chromate technique, characterized and drawn by Camillo Golgi in 1903. The concept of the physiological role of glial cells for substance exchange and metabolic support to neurons remains quite valid today. Activation of glia has been long considered solely detrimental for survival of neurons. Nevertheless, more recently it appears that the intercommunication among astrocytes–microglia and neurons is different not only in health and disease but can even modulate or control neurodegenerative mechanisms and may also vary depending upon the type of insult, and the different regions of the brain. No single-cell type in the brain is likely to be solely responsible for the etiopathogenesis of different neuropathological disorders such as Alzheimer’s disease (AD), Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS), traumatic brain injury (TBI), or brain ischemia. It is thus of the utmost importance to understand in deeper details what it means for glia to engage in phenotype switches and how they can influence surrounding cells. By becoming reactive, astrocytes and microglia undergo a set of transcriptional, functional, and morphological changes that transform them into cells with different properties and functions. Disruption of the finely tuned interplay between endothelial cells, pericytes, astrocyte end-feet contacts, microglia, oligodendrocytes, and neurons, all cells that form the neurovascular unit (NVU), likely contributes to the pathophysiology of neurodegeneration in chronic hypoxia and other neurodegenerative conditions such as AD or TBI. Indeed, the modified interplay among neurons and astrocytes/microglia may shift the balance from normal physiological conditions toward neurodegeneration (De Keyser et al., 2008; Sofroniew, 2009), but the precise role of astrocyte–microglia interactions with neurons in hypoxia-dependent mechanisms of neurodegeneration has not been clearly defined yet.
Alterations of the Cerebrovascular Functionality in Chronic Hypoxia
Chronic cerebral hypoperfusion is one of the major mechanisms that cause the cognitive decline and dementia in aged patients. Chronic cerebral blood flow reduction is generally mild, with no sharp drop in the acute phase. Reduced cerebral perfusion correlates with the gravity of dementia and is a good predictor of who, among the patients with mild cognitive impairment (MCI), will later develop dementia (Gorelick et al., 2011). Clinical studies indicate that cerebrovascular pathologies are the primary causes of at least 20% of the cases of dementias and are cofactors in the pathogenesis of Alzheimer’s disease (AD) (Schneider et al., 2007; Gorelick et al., 2011; Toledo et al., 2013) and other neurodegenerative disorders such as Huntington disease (HD, Drouin-Ouellet et al., 2015) and ALS (Winkler et al., 2013). Stenosis or partial occlusion of the internal carotid arteries brings about reduction of cerebral blood flow (CBF) associated with chronic ischemia. These mild, chronic events result in impairment of memory and cognition in human patients, independently of the presence in the brain of severe lesions (Johnston et al., 2004; Marshall et al., 2012; Alosco et al., 2013; Balestrini et al., 2013; Stefansdottir et al., 2013). More intense reduction of CBF by about 40–50% causes suppression of brain activity and more profound cognitive dysfunctions, which are reversible after reestablishment of normal CBF (Tatemichi et al., 1995; Marshall et al., 1999, 2012). Even more intense, acute reductions of CBF cause ischemic stroke (Moskowitz et al., 2010), which doubles the risk for dementia. Around 30% of stroke patients develop cognitive dysfunction within 3 years (Leys et al., 2005; Pendlebury and Rothwell, 2009; Allan et al., 2011), and about 50% of patients younger than 50 years show cognitive deficits after a decade (Schaapsmeerders et al., 2013). Nowadays, the general consensus is that most cognitive impairments in the aged patients result from brain dysfunction caused by mild, chronic, cumulative damage to tissue and cells (Gorelick et al., 2011). Some published papers have revealed that the interplay between neurons and glial and vascular cells is particularly important in the prevention or development of vascular cognitive impairments (VCI) (Iadecola, 2010; Quaegebeur et al., 2011; Zlokovic, 2011). These data (Iadecola, 2010; Quaegebeur et al., 2011; Zlokovic, 2011) provide new avenues to reevaluate how modifications of cerebral blood vessels and of glia–neuron interaction contribute to neuronal dysfunctions and are responsible for cognitive impairment, calling for a reappraisal of the role of glial and cerebrovascular functionality in cognition.
The blood brain barrier (BBB) is disrupted in the course of chronic cerebral hypoperfusion (Rosenberg, 2012). Indeed, brain hypoxia is known to damage endothelial cells, pericytes, and astrocytes (Jiang et al., 2018) and cause increased leakage from the BBB (Al Ahmad et al., 2012), as demonstrated in vivo in a rat model of brain hypoperfusion produced by bilateral stenosis of the carotids (Ueno et al., 2002). Furthermore, breakdown of the BBB causes extravasation of plasma proteins, such as immunoglobulins, fibrinogen, and complement, all potent proinflammatory molecules, and increases the production of free radicals (Morgan et al., 1997; Yoshida et al., 2002; Davalos and Akassoglou, 2012; Crehan et al., 2013). Fibrinogen activates proinflammatory pathways that activate microglia and astrocytes (Davalos et al., 2012; Holland et al., 2015). Extravasation of proteins resulting from increased BBB permeability can exacerbate tissue edema, compressing blood vessels, reducing CBF, and increasing the hypoxic state of the tissue.
In addition, in rats and mice, cerebral hypoperfusion is associated with inflammation of the white matter and oxidative stress (Ihara et al., 2001; Masumura et al., 2001; Yoshizaki et al., 2008; Huang et al., 2010; Dong et al., 2011; Juma et al., 2011; Reimer et al., 2011). One of the main consequences of the oxidative and proinflammatory stress induced by hypoperfusion and consequent BBB breakdown is the damage to the myelin sheath. This causes modification of the integrity of the axons, demyelination, axonal loss, and decrease of the velocity of axon potential transmission (Franklin and Ffrench-Constant, 2008; Matute and Ransom, 2012). Axonal demyelination increases the requirements of energy of the denuded axons, aggravating the stress of the tissue. Indeed, oligodendrocytes are sensitive to increased levels of ATP and glutamate, which overactivate ionotropic glutamate receptors and P2X7 purinergic receptors (Bakiri et al., 2009; Verkhratsky et al., 2009). Overactivation of these receptors may kill oligodendrocytes by excitotoxicity (Arbeloa et al., 2012). These data may explain the studies in human patients that demonstrate a correlation between ischemic demyelination and stroke outcome (De Groot et al., 2002; Schmidt et al., 2003). All these effects amplify these pathogenic processes, exacerbating the damage to the brain tissue.
Furthermore, tissue hypoxia and oxidative stress activate transcription of many proinflammatory pathways through NF-kB, activating the expression of cytokines and adhesion molecules in vascular cells, reactive astrocytes, and activated microglia. Inflammation and oxidative stress have negative effects on the trophic interaction among the cells of the NVU. Reactive oxygen species (ROS) and inflammation suppress the prosurvival effects of endothelial cells on neurons by reducing BDNF levels (Guo et al., 2008).
The Hippocampus
The hippocampus, a region of the brain fundamental for memory encoding, shows numerous structural, morphological, and electrophysiological alterations in many neurodegenerative disorders such as AD (Thal et al., 2002; Braak et al., 2006; Mueller et al., 2010; Small et al., 2011; Bartsch et al., 2015) and in ischemia (De Jong et al., 1999; Zola et al., 2000; Liu et al., 2005). All these alterations are at the basis of memory loss typical of advanced age, of ischemia, and of AD. The hippocampus forms a unidirectional network with the tri-synaptic pathway that originates via the perforant path from the entorhinal cortex (EC) and connects the dentate gyrus (DG) to CA3 and CA1 pyramidal neurons. CA3 neurons receive inputs from the DG via the mossy fibers and send axons to CA1 pyramidal cells via the Schaffer collateral pathway, as well as to CA1 in the contralateral hippocampus via the associational commissural pathway. In addition, CA1 neurons receive inputs directly from the EC via the perforant path and send axons to the subiculum. The hippocampal areas CA3 and CA1 have both morphological and anatomical similarities and differences. For instance, the pyramidal cell layer forms a continuum from CA3 to CA1 and have parallel inputs but have distinct network architectures and diverging output pathways (Amaral and Witter, 1989; Cenquizca and Swanson, 2007; Bahar et al., 2011). CA1 and CA3 subserve different functions and contribute to the processing of specific information such as novelty detection, encoding, short-term memory, intermediate-term memory, and retrieval (Vazdarjanova and Guzowski, 2004). CA1 is fundamental for mediating the association with temporal components and is capable of maintaining short-term memories (Wiebe et al., 1997), whereas CA3 is involved in processes associated with rapid formation of spatial or contextual memory (Lee and Kesner, 2002, 2003; Kesner et al., 2004; Nakazawa et al., 2003). CA3 and CA1 hippocampal areas, although interconnected through the Schaffer collaterals, and often considered as a continuum, respond differently to ischemic/hypoxic conditions (Kirino, 2000). Nevertheless, it is still not completely clear how and why these two contiguous, interconnected hippocampal areas respond in a different way to an ischemic event. In patients with cerebral hypoxia/ischemia, CA1 pyramidal neurons are among the most vulnerable (Zola-Morgan et al., 1986; Petito et al., 1987), as also repeatedly demonstrated in experimental animal models of hypoxia/ischemia (reviewed by Schmidt-Kastner and Freund, 1991) in the gerbil (Kirino, 1982), in the rat (Pulsinelli et al., 1982), and in humans (Bartsch et al., 2010, 2015). Indeed, since hippocampal CA1 circuits are fundamental for the processes of memory formation (Bartsch et al., 2010), impairment of CA1 neurons contributes to memory deficits in patients with damages to the hippocampus (Kadar et al., 1998).
The decrease of both oxygen (O2) and glucose supply caused by cerebral hypoperfusion gives rise to signaling failure in the vulnerable neurons of CA1 hippocampus, with impairment of hippocampally mediated learning and memory mechanisms (De Jong et al., 1999; Liu et al., 2005; Farkas et al., 2006; Melani et al., 2010; Lana et al., 2013). Many different hypotheses have been brought about to explain the higher sensitivity of CA1 neurons to deprivation of O2 and glucose caused by hypoxia or ischemia. O2 and glucose deprivation in vitro (OGD) is an established and widely used experimental model that allows exploring potential differences in the responses of rat hippocampal CA1 and CA3 neurons to ischemia (Gee et al., 2006; Pugliese et al., 2006; Cimarosti and Henley, 2008; Dixon et al., 2009; Sun et al., 2010). A few minutes after the beginning of OGD, deprivation of energy reduces the levels of intracellular ATP, with consequent failure of the Na+/K+ pump, anoxic depolarization, and increased extracellular levels of glutamate that is not taken up by the neuronal or astrocytic glutamate uptake system (Jabaudon et al., 2000; Rossi et al., 2000). Indeed, minutes after an ischemic insult, increase of extracellular glutamate is detected in both CA1 and CA3 areas (Mitani et al., 1992), leading in the gerbil to activation of glutamate receptors and neurotoxicity (Urban et al., 1990; Choi and Rothman, 1990). Glutamate NMDA receptor (NR) subunits play a fundamental role in Ca2+-induced excitotoxicity and are expressed differentially in the CA1 and CA3 hippocampal areas. NR2C is less permeable to Ca2+ ions than the other subunits (Chung et al., 2016). Interestingly, GluN2C knockout mice show greater neuronal death in the CA1 hippocampus a few hours after global cerebral ischemia (Chung et al., 2016). Furthermore, the balance between kinase and phosphatase activities in CA1 is in favor of tyrosine kinases, while in CA3 it is in favor of phosphatases (Gee et al., 2006). In the gerbil, ischemia increases tyrosine phosphorylation of NR2A and NR2B subunits of NMDA receptors (Zalewska et al., 2005). Furthermore, expression of NR2 receptor subtypes and splice variants is higher in CA1 than in CA3, explaining the higher vulnerability of CA1 after ischemia (Zola-Morgan et al., 1986; Wu et al., 2008). These data, taken together, may be one of the possible explanations of the higher sensitivity of CA1 to ischemia.
Neurons consume 75–80% of total brain energy (Hyder et al., 2013) for restoration of neuronal membrane potentials after depolarization (Harris et al., 2012), for neurotransmitter synthesis, vesicle packaging, axoplasmic transport, and neurotransmitter release (Attwell and Laughlin, 2001; Rangaraju et al., 2014; Pathak et al., 2015). Thus, energy demand in the brain is not uniform but is higher where neurons have higher neuronal activity. CA1 pyramidal neurons of the hippocampus have a higher firing rate than CA3 neurons and have higher energy demands (Mizuseki et al., 2012). For these reasons, it is therefore possible that CA1 neurons are preferentially vulnerable (Wilde et al., 1997; Padurariu et al., 2012) during an ischemic stress (De Jong et al., 1999; Wilde et al., 1997).
Deprivation of energy reduces the levels of intracellular ATP, modifies the ionic gradients, and inverts the glutamate uptake in rat pyramidal CA1 and CA3 neurons in vitro (Jabaudon et al., 2000; Rossi et al., 2000).
In addition, the vascular architecture of CA1 and CA3 areas show anatomical differences. While CA1 is vascularized by a large ventral artery, in CA3 many capillaries are present in the vicinity of neurons (Duvernoy et al., 1983). Indeed, increased neuronal activity is one of the major determinants of the dynamic increase of CBF to supply more blood, nutrients, and O2 to active neurons. Increased blood supply depends on the concerted action of vascular cells, astrocytes, and neurons (Iadecola, 2013). Many ions and vasoactive compounds with opposing effects, such as nitric oxide (NO), metabolites of arachidonic acid, adenosine, neurotransmitters, and neuropeptides (Drake and Iadecola, 2007), act as signals to regulate the hemodynamic changes of blood supply to a brain area. All these molecules are generated by synaptic activity of the afferents to the hippocampus from the basal forebrain and brainstem, by interneurons, and by astrocytes (Drake and Iadecola, 2007; Cauli and Hamel, 2010; Kleinfeld et al., 2011). These highly coordinated signaling pathways, with such a high degree of spatial precision and temporal definition (Iadecola, 2004), are probably necessary to fulfill the higher requirements of energy of CA1 in comparison to CA3 neurons in physiological conditions and may help in explaining the more intense sensitivity of CA1 to hypoxic/ischemic conditions. Indeed, it has been demonstrated (Chip et al., 2013) that a selective vascular vulnerability is present in CA1, which in turn is responsible for the depletion of blood supply to the CA1 subregions. Disruption of the BBB causes impairment of the astrocyte–vascular communication, with consequent modification of the BBB cytoarchitecture, alteration of regional homeostasis, and energy deficiency that leads to energy crisis (Alvarez et al., 2013). In case of increased energy demand, BBB disruption affects the functionality of energy-craving neurons, specifically those in CA1 that become unable to fulfill the high requirements of energy necessary for their functional homeostasis (Muddapu et al., 2020).
Furthermore, studies demonstrate that during ischemia the levels of superoxide and ROS are higher in the CA1 than in CA3 hippocampal area (Wilde et al., 1997; Wang et al., 2005) and induce stress-activated mitochondrial transition pores (MTPs) preferentially in CA1 than in CA3 rat hippocampus (Mattiasson et al., 2003). Activation of MTPs causes Ca2+-induced mitochondrial swelling that leads to microvacuolization (Duchen, 1992) and disruption of the mitochondria, consequent release of cytochrome C (CytC) in the cytoplasm, and apoptosis (Suen et al., 2008). Furthermore, in the Mongolian gerbil, post-ischemic mitochondrial damage is more severe in CA1 than in CA3 (Radenovic et al., 2011), indicating that CA1 and CA3 pyramidal neurons respond differently to similar stress conditions. Thus, CA1 pyramidal neurons seem more sensitive than CA3 neurons to the damage caused by production of ROS by mitochondrial and oxidative stress, as demonstrated in the aged mice (Kanak et al., 2013).
From all the data reported above, it appears that the real reason for the differential sensitivity of CA1 to hypoxia is not completely understood. Therefore, the study of the differences between CA1 and CA3 hippocampal areas is of fundamental importance, because it can explain the higher sensitivity of CA1 pyramidal neurons to different types of insults observed in both animal models of neurodegeneration and in patients (Mueller et al., 2010; Small et al., 2011; Bartsch and Wulff, 2015; Ugolini et al., 2018). On these bases, we compared the changes of the interplay among astrocytes–microglia and neurons as well as the modifications of neuroinflammatory markers in CA1 and CA3 hippocampus of rats in an in vivo model of cerebral hypoperfusion.
The Neurovascular Unit and the Glymphatic System
The brain is dependent upon blood circulation in microvessels for delivering O2 and nutrients to neurons and for disposal of waste material. The NVU is composed by cells of different types and functions, all working synergistically in a highly regulated manner. Astrocyte endfeet surround the walls of the vessels, which, together with perivascular microglia and macrophages, survey the influx of molecules into the brain (Dudvarski Stankovic et al., 2016). In ischemic trauma, the disruption of blood vessels and BBB breakdown correlate with the accumulation of activated microglia, suggesting that microglia are associated with the dysfunction of blood vessels (Barkauskas et al., 2015). Disruption of the NVU is strongly associated with vascular dementia (Iadecola, 2013) and likely contributes to the early stages of AD, as shown in animal models of the disease (Iadecola et al., 1999; Iadecola, 2004; Kelly et al., 2017).
Furthermore, it has been demonstrated that in aged rats the decrease in the number of astrocytes is accompanied by decrease in VEGF expression, which further amplifies the vascular impairment (Bernal and Peterson, 2011). During ischemia, astrocytes are injured and show morphological changes such as swelling and vacuolization of the cell body and loss of their distal processes. All these modifications of astrocytes, named “clasmatodendrosis,” are caused by energy failure and acidosis (Friede and van Houten, 1961; Hulse et al., 2001). Clasmatodendrosis was first described by Cajal, as reported by Penfield (1928), and later rediscovered by Friede and van Houten (1961), and by Hulse and colleagues in hippocampal organ cultures (Hulse et al., 2001). Clasmatodendrosis is found in lesions of the white matter in patients with cerebrovascular disease and AD (Tomimoto et al., 1997). It is also present in the periventricular zone of patients with mixed dementia (Sahlas et al., 2002), in the corpus callosum of hypoperfused mice (Hase et al., 2017), in the hippocampus of rats with chronic epilepsy (Kim et al., 2011), and in aged rats (Cerbai et al., 2012). In AD and ischemia, clasmatodendrosis may represent an acute response of astrocytes to energy failure coupled with mitochondrial inhibition (Friede and van Houten, 1961; Kraig and Chesler, 1990; Hulse et al., 2001). Clasmatodendrotic morphological alterations of astrocytes are directly associated with changes in cell function (Jiang et al., 2018). Furthermore, an association between astrocyte injury and disruption of the BBB has been described in post-stroke surviving elderly patients (Chen et al., 2016). Disruption of the BBB can lead to inefficient removal and accumulation of toxins in the parenchyma, which may play a significant role in tissue damage. In subjects with MCI (Wang et al., 2006), increased permeability of the BBB has been observed at early stages of AD (Starr et al., 2009). All these findings support the idea that energy deficiency may be a cause of degeneration of neurons in ischemic conditions.
The channel aquaporin4 (AQP4), located on astrocyte endfeet, regulates the flux of water between blood and brain (Nagelhus and Ottersen, 2013) and is involved in regulation of BBB permeability (Tourdias et al., 2011). In mice, in the first few hours after ischemia, AQP4 is upregulated in the ischemic core, while the peak of its expression is found in the penumbra 48 h after ischemia (De Castro et al., 2006). Furthermore, mice deficient for AQP4 on astrocytes show significant reduction in water uptake and reduced brain edema following stroke, in comparison to wild-type animals (Haj-Yasein et al., 2011). After ischemia, reactive astrocytes increase the expression of connexin 43 and connexin 30 (Orellana et al., 2014), of AQP4 (Hirt et al., 2009; Hoshi et al., 2011), as well as of trophic factors such as BDNF (Neumann et al., 2015), which are molecules involved in neuron protection or in ischemic tolerance.
In the few hours after cerebral ischemia, hypoxia caused by the decrease of blood flow results in impairment of Na+/K+ ATPase, accumulation of intracellular Na+ which recalls water into the cell and induces cytotoxic edema (Simard et al., 2007). Astrocytes are the major cell type involved in cytotoxic edema (Kimelberg, 1995), and one of the key molecular players is AQP4 (Manley et al., 2000). The development of ischemic cellular damage causes breakdown of BBB, giving rise to leakage of plasma proteins to the extracellular space. Furthermore, swelling of astrocytes may compress the vessels in the ischemic regions, further decreasing the vessel caliber and exacerbating the hypoperfusion (Syková, 2001).
Since most of the high-affinity glutamate transporters are located on astrocyte membranes, astrocytes are the main cells involved in reuptake of glutamate at the NVU (Dallérac and Rouach, 2016). In the hippocampus, the two most expressed isoforms of glutamate transporters are excitatory amino acid transporter-1 (EAAT-1, GLAST in rodents) and excitatory amino acid transporter-2 (EAAT-2, GLT1) (Holmseth et al., 2012). In in vivo transient ischemia in the gerbil, astrocytic immunoreactivity for GLT1 is upregulated between 30 min and 12 hours after ischemia (Kim et al., 2006). In rat CA3 hippocampus, GLT1 immunoreactivity increases between 1 and 21 days after global ischemia (Bruhn et al., 2000). From these data, it can be postulated that increased uptake of glutamate and lactate may be the cause of astrocyte swelling the first hours/days after ischemia (Landis, 1994; Kimelberg, 2005; Verkhratsky et al., 2016). On the other hand, immunohistochemical studies show that GLT1 expression decreases in CA1 rat hippocampus between 2 and 4 days after ischemia reperfusion (Bruhn et al., 2000). Inconsistencies in these data may be due to differences of species or of brain regions.
Astrogliosis has been shown to be present in many neurodegenerative disorders, such as ischemia, AD, PD, ALS, and MS (for references, see Li et al., 2019). In addition, astrogliosis causes loss of AQP4 polarization in perivascular astrocytes, which potentially represents a mechanism common to NVU and glymphatic dysfunctions in many neurodegenerative diseases such as brain infarcts (Wang et al., 2017; Zbesko et al., 2018), AD (Kress et al., 2014; Xu et al., 2015), and TBI (Iliff et al., 2014; Guo et al., 2014; for references see also Rasmussen et al., 2018).
Nevertheless, reactive astrocytes seem to have both detrimental and beneficial roles (Sofroniew, 2009; Liddelow et al., 2017) in many neuropathological conditions such as ischemia (Pekny et al., 2019; Amantea et al., 2010; Barreto et al., 2011). The different responses of astrocytes to the ischemic insults depend, at least in part, on the severity of ischemia. In addition, other conditions such as age and spatial localization of astrocytes can determine whether astrocytic functions are protective or damaging, as will be discussed later. Nevertheless, in mice it has been demonstrated that activation of astrocytes is essential for induction of preconditioning, or ischemic tolerance, indicating the importance of reactive astrocytes for neuroprotection (Hirayama et al., 2015; for ref. see Koizumi et al., 2018).
As reported above, other data demonstrate that, due to metabolic stress, astrocytes located in CA1 have higher reaction to ROS, while the activity of glutamate transporters is reduced, leading to increased extracellular levels of glutamate that cause higher stress to CA1 neurons in rats after transient forebrain ischemia (Ouyang et al., 2007). Furthermore, astrocytes in pathological conditions express and release cytokines that disrupt the permeability of the BBB (Abbott, 2002). In addition, the clasmatodendrotic modifications of astrocytes that occur in ischemic conditions (Hulse et al., 2001) are further causes of BBB derangement. Disruption of BBB and of the glio-vascular network causes increase of endothelial cell permeability, reduction of glucose transport, and increased extracellular levels of toxic substances, which further increase neuroinflammation (Abbott, 2002; Zhu et al., 2007; Miyazaki et al., 2011; Freeman and Keller, 2012; Guan et al., 2013; Sweeney et al., 2018a, b).
In addition to the NVU, more recently the glia-lymphatic (glymphatic) system has come into focus as a highly specialized transport system that facilitates disposal of extracellular waste into the cervical and basal meningeal lymphatic networks or the dural sinuses. The glymphatic system consists of a network of perivascular or perineural channels supported by astrocytes (Reeves et al., 2020). Astrocytic endfeet express high levels of polarized AQP4 that facilitate not only the NVU but also the glymphatic flow, disposing of and clearing the interstitium from potentially toxic substances (Iliff et al., 2012). The glymphatic system is impaired during aging (Kress et al., 2014), and its dysfunction is involved in many neurodegenerative disorders, particularly those in which accumulation of extracellular waste such as Aβ and tau is an important pathogenetic mechanism, such as AD (Weller et al., 2009; Iliff et al., 2012; Jessen et al., 2015). Indeed, the glymphatic system represents a fundamental pathway in the net clearance of Aβ (Iliff et al., 2013; Xu et al., 2015). The clasmatodendrotic modification of astrocytes during aging (Cerbai et al., 2012) and ischemia (Hulse et al., 2001) may represent one of the causes not only of vascular but also of glymphatic dysfunction. Dysfunction of the glymphatic influx is secondary to acute ischemia, and multiple micro-infarction (Gaberel et al., 2014; Wang et al., 2017), but the astrocyte involvement on the efficiency of this system needs to be completely understood.
Physiological and Pathological Actions of Astrocytes
Astrocytes, among the most represented glial cells in the central nervous system, have distinctive morphologies that differ both between and within regions of the brain. Astrocytes have many housekeeping functions, which help maintain a healthy brain (Verkhratsky and Nedergaard, 2018), Astrocytes control the formation, maturation, and plasticity of synapses by secreting thrombospondins, hevin, and solid-phase attachment of red cells (SPARC), all proteins that regulate synapse formation (Christopherson et al., 2005; Kucukdereli et al., 2011). Astrocytes control neural circuit formation through TNF-α (Stellwagen and Malenka, 2006) and TGF-β signaling (Diniz et al., 2012, 2014a, b, 2017). In addition, healthy astrocytes envelope synapses with their processes and are indispensable for neurotransmitter homeostasis, release of gliotransmitters, and maintenance and maturation of synapses (Pfrieger, 2009; Heneka et al., 2010). Furthermore, neuronal activity excites the membrane of astrocytes and, increasing intracellular Ca2+, induces the release of gliotransmitters (Perea and Araque, 2007; Navarrete et al., 2012; Araque et al., 2014), which are necessary for synaptic plasticity, indicating that astrocytes are involved in memory formation (Verkhratsky et al., 2011; Navarrete et al., 2012). In addition, astrocytes control the levels of the neurotransmitters GABA and glutamate at the synapses, thus mediating the synaptic functions (Sofroniew and Vinters, 2010) of the so-called tripartite synapse.
As already mentioned, healthy astrocytes maintain intimate contact with endothelial cells and pericytes through gap junctions, which allow intercellular diffusion of ions, regulate water and ion homeostasis, maintain the pH, allow the diffusion of small molecules, and contribute to functionality of the BBB (Siqueira et al., 2018). In this way, astrocytes provide energy required by neurons in the form of glucose (Rouach et al., 2008) and lactate (Figley, 2011; Sotelo-Hitschfeld et al., 2015), as well as trophic factors essential for neuronal survival (Nones et al., 2012; Dezonne et al., 2013). As reported above, astrocytes form an integral part of the BBB and of the glymphatic system and regulate neurovascular coupling, vascular tone, and blood flow (Sofroniew and Vinters, 2010; Macvicar and Newman, 2015; Verkhratsky and Nedergaard, 2018; Govindpani et al., 2019).
Astrocytes are maintained actively in a resting state, but the precise molecular signals that trigger astrocytic activation at the initial phases of an insult are still not known. Recent studies have demonstrated that different CNS injuries can stimulate at least two types of astrocytes with different properties, A2 reactive astrocytes that have beneficial, neuroprotective properties, and A1 reactive astrocytes that are harmful to neurons. A2 astrocytes are predicted to promote neuronal survival, outgrowth, synaptogenesis, and phagocytosis. On the contrary, A1 neuroinflammatory reactive astrocytes have harmful effects, upregulating many genes that express proinflammatory proteins and other factors that are destructive for synapses (for ref. see Liddelow and Barres, 2017). The “detrimental” A1 astrocytes, stimulated by inflammatory stimuli, upregulate genes associated with destruction of synapses and loss of neurons (Zamanian et al., 2012), while the “helpful” A2 astrocytes, induced by an ischemic event (Zamanian et al., 2012), upregulate cytokines such as TNFα. Inhibiting the proinflammatory cytokine IL-12p40 (Zakharova and Ziegler, 2005), TNFα has anti-inflammatory properties. A2 astrocytes upregulate neurotrophic factors and thrombospondins, predicted to stimulate development of synapses and survival of neurons (Zamanian et al., 2012).
A recent hypothesis postulates that in physiological conditions astrocytes may exist as a continuum of heterogeneous, mixed populations (Zhang and Barres, 2010; Khakh and Sofroniew, 2015; Ben Haim and Rowitch, 2016; Khakh and Deneen, 2019; Pestana et al., 2020). Consequently, the states of astrocytes vary in physiopathological conditions, depending not only on the type of insult but also possibly on the brain structure in which astrocytes are located (Liddelow and Barres, 2017). For instance, it is not clear yet whether different astrocytes located in different brain areas show the same morphofunctional modifications after a similar insult or whether astrocytes react differently to the same insult. The first hypothesis indicates that astrocytic responses are controlled by intrinsic cues, while the second hypothesis indicates that astrocytic responses are controlled by external, environmental signals (Martín-López et al., 2013; Bribian et al., 2018). Some recent insights give way to the idea that there may exist an apparent continuum in the intensity of astrocyte reactions to insults, which possibly hides different, discrete reactive states. Nevertheless, astrocyte reactivity differs among different areas of the brain. For instance, it has been demonstrated in both mice and humans that during aging astrocytes located in distinct brain regions have different transcriptional profiles (Soreq et al., 2017; Boisvert et al., 2018; Clarke et al., 2018) including glial fibrillary acidic protein (GFAP) and serpin (Boisvert et al., 2018) that result in complex region-specific molecular and morphological changes (Rodríguez et al., 2014). In AD, astrocytes become hypertrophic or atrophic depending not only on the stage of the disease but also to the proximity of Aβ plaques (Rodríguez-Arellano et al., 2016).
Recent evidences show that, although microglia represent the main phagocytic cells in the brain, astrocytes can participate in phagocytosis (Wu et al., 2009; Lu et al., 2011; Iram et al., 2016; Morizawa et al., 2017) after ischemia. Since so far astrocytes have received only limited attention as phagocytes, the mechanisms of astrocytic phagocytosis are still not completely understood. Nevertheless, it has been demonstrated that astrocytes use the ATP-binding cassette transporter (ABCA1) pathway (Morizawa et al., 2017), as well as multiple EGF-like-domains 10 (MEGF10) and proto-oncogene tyrosine-protein kinase MER (MERTK) pathways for phagocytosis (Chung et al., 2013). Microglia use the classical complement pathway to recognize and prune unwanted synapses in the developing mouse brain. Other studies suggest that astrocytes express other phagocytic receptors, such as brain-specific angiogenesis inhibitor 1 (BAI1) and integrin αvβ3 or αvβ5 (Park et al., 2007). Furthermore, in a transient middle cerebral artery occlusion mouse model, most of the phagocytic activity of astrocytes has a late onset after ischemia (7 days) and is localized mainly in the penumbra around the ischemic core (Morizawa et al., 2017), where neurons are slightly damaged and still recoverable. Most of phagocytic activity of microglia has an early onset after ischemia (1–3 days) and is located in the ischemic core where astrocytes are not phagocytic and where neurons and other cells are dead (Morizawa et al., 2017). Indeed, since astrocytes are not as mobile as microglia (Nimmerjahn et al., 2005; Okada et al., 2006), they are not able to migrate from the penumbra to the ischemic core. Astrocytes polarize their distal processes without cell body migration and engulf apoptotic bodies derived from dendrites of dying neurons, while microglia migrate toward damaged neurons and completely engulf dendrites, cell bodies, and nuclei (Damisah et al., 2020). Astrocytes located in the penumbra can become phagocytic and can contribute to clearance of debris and repair of the tissue (Morizawa et al., 2017). Furthermore, phagocytic microglia engulf larger debris than astrocytes (Morizawa et al., 2017), indicating that the size of debris that can be phagocytosed by astrocytes is limited (Damisah et al., 2020). Indeed, as will be discussed below, microglia can phagocytose live neurons during injury (Brown and Neher, 2014), inducing phagoptosis, a mechanism that contributes to neuronal cell death (Fricker et al., 2012; Neher et al., 2013). Astrocytes and microglia play specialized and orchestrated roles, acting in a highly coordinated fashion with spatiotemporal differences in different brain areas that can have important physiopathological consequences (Morizawa et al., 2017).
In a less neuron-centric view of neurodegeneration, the alterations of astrocytes, such as decreased maintenance of brain homeostasis, impairment of buffering of extracellular glutamate, and reduced supply of nutrients to neurons, may contribute to the diffusion of damage to neurons and neurodegeneration (Miller et al., 2017). In vitro it has been shown that A1 astrocytes have a novel, deleterious function (Liddelow and Barres, 2017; Liddelow et al., 2017). A1 astrocytes secrete a neurotoxin that induces apoptosis in neurons and release toxic factors that target specifically motor neurons and mediate cell death in a mouse model of ALS (Re et al., 2014). We have first demonstrated in CA1 (Cerbai et al., 2012) and later in CA3 (Lana et al., 2016), as well as in DG (Lana et al., 2017b) of hypoperfused rats, that astrocytes send branches to embrace, infiltrate, and bisect apoptotic neurons (Figures 1G1,G2). This mechanism is finalized to the fragmentation of apoptotic, dying neurons to form cellular debris in order to spare and protect the surrounding tissue from the damage caused by the release of proinflammatory products in the parenchyma. Ours are the first demonstrations that astrocyte branches infiltrate the cytoplasm of apoptotic neurons, to bisect the dying neuron and form debris. We have demonstrated that neuronal debris are indeed more numerous in the CA1 and CA3 stratum radiatum (SR) of hypoperfused rats than in controls, and neuronal debris are all closely apposed to the branches of astrocytes (Figure 1B) and ready to be phagocytosed by microglia (Figure 1E). While adaptive, reactive astrogliosis has been shown to have beneficial effects, suppression of astrocyte reactivity may also increase neuronal vulnerability, exacerbating the pathology and altering regeneration (Burda and Sofroniew, 2014; Pekny et al., 2014). In addition, it is possible that when astrocyte reactivity becomes too intense, the release of neurotoxic factors, such as components of the complement cascade that enhance synaptic degeneration (Stevens et al., 2007; Hong et al., 2016; Sekar et al., 2016), and of neurotoxins that cause the death of motor neurons (Di Giorgio et al., 2007; Nagai et al., 2007; Lobsiger et al., 2007) could be responsible for increased neurotoxicity. Thus, astrocytes can behave as actors in causing or preventing neurodegeneration.

Figure 1. Characterization of neurodegeneration and alterations of the neuron–glia interplay in the bCCAo model of brain hypoperfusion in the rat. (A) Representative images showing CytC + apoptotic neurons in CA1 SP of a bCCAo rat (arrows). Scale bar: 15 μm. Adapted from Lana et al. (2014). (B) Representative image of a GFAP + astrocyte (green), a NeuN + damaged neuron (red, open arrow), and a NeuN + neuronal debris (in the circled area) in CA3 SR of a bCCAo rat. The neuronal debris appear closely apposed to the astrocyte branches. Scale bar: 10 μm. Adapted from Lana et al., 2017a. (C1–C3) Representative images showing NeuN + neurons (red) and calretinin + interneurons (green) in CA3 SP, SL, and SR of a bCCAo rat. Scale bar: 60 μm. (C2–C3) Magnifications of the framed area in (C1) showing the Calretinin + interneurons (open arrows, C2) and the NeuN + ectopic neurons (arrows, C3), demonstrating that ectopic neurons are not Calretinin+. Scale bar: 30 μm. Adapted from Lana et al., 2017a. (D1–D3) Representative images showing the colocalization of CytC (green) with NeuN (red) in the cytoplasm of an apoptotic–ectopic neuron (open arrow, D1) in the proximity of CA1 SP of a bCCAo rat. An IBA1 + microglial cell (blue, arrow, D1) projects its branches to surround the neuron. SP, appears indented in correspondence with the ectopic neuron (D1,D3, asterisk). Scale bar: 8 μm. Adapted from Lana et al. (2014). (E) Representative image showing a NeuN + ectopic neuron (open arrow) undergoing phagocytosis by an IBA1 + microglial cell (blue) in the proximity of CA1 SP of a bCCAo rat. The microglial cell resides on the top of the neuron and embraces it with its branches. Scale bar: 3 μm. Adapted from Lana et al. (2014). (F) Representative image showing two NeuN + ectopic neurons (red) surrounded by GFAP + astrocyte branches (green) and phagocytosed by IBA1 + microglial cells (blue) in CA1 SR of a bCCAo rat. In the framed area is shown a triad cluster. Scale bar: 10 μm. Adapted from Lana et al., 2014. (G1–G2) Representative image showing four GFAP + astrocytes (green, arrowheads) projecting their branches toward a NeuN + neuron (red, open arrow) undergoing phagocytosis by an IBA1 + microglial cells (blue) in CA1 SR of a bCCAo rat. Astrocyte branches form a glial “microscar” around the neuron. The digital subslicing of the neuron along the dotted line (G1) and the 45° rotation (G2) show that an astrocyte branch infiltrates the neuronal cell body (arrow, G2). Scale bar: 10 μm. Adapted from Lana et al. (2014). (H) Representative image showing a GFAP + TNFα + astrocyte (GFAP in red, TNFα in green) in CA3 SR of a bCCAo rat. Scale bar 15 μm. Adapted from Lana et al. (2017a).
The comprehension of the multiple, contrasting roles of astrocytes in the pathophysiological mechanisms of neurodegeneration, a theme that only recently entered into focus, will be of great interest in understanding the pathogenesis of many neurodegenerative disorders such as AD, PD, ALS, and TBI.
Physiological and Pathological Actions of Microglia
Microglia represent 5–10% of the brain cells and are the resident immune cells of the central nervous system (Frost and Schafer, 2016). In physiological conditions, the highly mobile processes of microglia dynamically reorganize by cyclically forming and withdrawing (Nimmerjahn et al., 2005), thus allowing microglia to patrol the brain parenchyma (Nimmerjahn et al., 2005). Under physiological circumstances, microglial cells in resting state have small somata and fine, ramified branches. Upon detection of proinflammatory stimuli, microglial cells activate rapidly and become major actors of the neuroinflammatory response (Crotti and Glass, 2015; Hickman et al., 2018). Microglial cells that rapidly undergo morphological and genetic changes upon activation are first responders to insults such as ischemic brain injury (Kettenmann et al., 2011). Disruption of brain homeostasis causes morphofunctional changes such as cell body hypertrophy and thickening of the branches. Contemporarily, many cell surface markers are upregulated, such as cluster of differentiation (CD) 45, major histocompatibility complex II, and CD68 (Ransohoff and Perry, 2009). Activated microglia have dual roles in the ischemic brain, depending on the stimulus. Microglia can acquire a spectrum of different but overlapping functional phenotypes, including the classical pro-inflammatory and the anti-inflammatory alternatively activated phenotypes. Microglia produce a plethora of cytokines and chemokines that promote inflammatory mechanisms, BBB dysregulation, and leukocyte infiltration (da Fonseca et al., 2014). Activated microglia may also have beneficial effects, phagocytosing cellular debris, and suppressing inflammatory responses, as reported by Neumann and coworkers (Neumann et al., 2006) in a model of ischemia in rat organotypic hippocampal slices in vitro. The dual roles of microglia may depend on their phenotypic polarization after ischemia (Franco and Fernández-Suárez, 2015; Hu et al., 2015; Jiang et al., 2016; Xiong et al., 2016). Microglial cells patrol the parenchyma to detect and eliminate debris or apoptotic neurons by phagocytosis (Hsieh et al., 2009; Lana et al., 2017a). It has been shown by 2-photon imaging in the zebrafish spinal cord (Morsch et al., 2015) and in mouse brain (Davalos et al., 2005; Nimmerjahn et al., 2005) that microglia exhibit ramified processes with high motility that allow a dynamic and continual survey of brain parenchyma and have an active role in the surveillance and maintenance of healthy brain. Indeed, activation of microglia is now viewed as a multistage, reversible process that generates multiple phenotypes of reactive cells with protective abilities (Hanisch and Kettenmann, 2007; Ransohoff and Perry, 2009; Kettenmann et al., 2013). Microglial projections are chemotactic sensors that extend toward injured cells in the “find-me” step of neuron phagocytosis (Hanisch and Kettenmann, 2007). Therefore, damage of microglial projections may weaken the neuroprotective activity of microglia. Reasonably, decreased microglial migration may hamper its phagocytic efficacy, favoring the accumulation of degenerating neurons and proinflammatory neuronal toxic debris (Tian et al., 2012), typical of brain aging (Cerbai et al., 2012). Nevertheless, it is still accepted that chronic, exaggerated activation of microglia, such as in chronic inflammatory diseases, can cause robust pathological alterations and neurobehavioral complications (for references see also Glass et al., 2010; Norden and Godbout, 2013). Recent work from Barres’s lab demonstrates that during inflammatory responses activated microglia modify their secretory profile, increase the release of factors such as C1q, TNF-α, IL-1α, and influence astrocyte activation (Liddelow et al., 2017). Nevertheless, microglial activation and production of cytokines have positive effects in early brain development (for ref. see Salter and Beggs, 2014), in synaptic pruning (for references see Schafer and Stevens, 2013), and in normal learning and memory in mice (Ziv et al., 2006; Derecki et al., 2010).
It is now known that two types of activated microglia exist, characterized by different phenotypes (Allen and Barres, 2009) and opposed effects. The proinflammatory M1 state occurs when microglial cells are activated by an acute insult and release proinflammatory mediators such as NO, ROS, quinolinic acid, and cytokines such as TNFα, IL-1, IL-6, and IL-18. The non-inflammatory, repairing M2 state of microglia, associated with secretion of anti-inflammatory cytokines such IL-4, IL-10, IL-13, and TNF-ß, also has a role in tissue repair. The role of microglia in neurodegeneration depends on the expression of apolipoprotein E (APOE) and triggering receptor expressed on myeloid cells 2 (TREM2) (Krasemann et al., 2017). In acute models of neurodegeneration, APOE regulates TREM2, which in turn modulates the activation of microglia (Krasemann et al., 2017).
Nevertheless, as for A1 and A2 astrocytes, this quite recent classification of microglia in M1 and M2 states (Liddelow and Barres, 2017) seems to be rather narrow, not corresponding to the variety of microglia phenotypes so far discovered in the brain. The activation profile of microglial cells is not an all or none response but can be imagined as a continuous, highly dynamic process that depends on the type of insult and is also influenced by the area(s) of the brain, the stimuli, and disease progression (De Biase et al., 2017; Keren-Shaul et al., 2017). It can also change during the progression of the pathological conditions. Indeed, the distinction between the protective or pathogenic roles of activated microglia could depend on the timing, and/or severity of insult, and/or on the location of the microglia cell. According to Halder and Milner (2019), in the mouse spinal cord during the early phase of vascular damage and leakage, microglia mediate a protective response to maintain vascular integrity. When the damage is continued, microglia may become inappropriately stimulated into a phagocytic phenotype. Indeed, in the early stages of a model of ischemic stroke in the mouse, microglia congregate around damaged blood vessels, but later microglia switch from a repair phenotype into a phagocytic one, removing the dying endothelial cells (Jolivel et al., 2015). Furthermore, in a mouse model of cerebral ischemia, decreased proliferation of microglia leads to increased neuronal death and larger lesion after stroke, indicating a protective role of microglia (Wolf et al., 2017).
Furthermore, regional differences may play an important role in the resting status and responsiveness to insults. Indeed, in a study it has been demonstrated in the mouse that the microglial transcriptome and their sensitivity and response to insults vary in a region-dependent way (Grabert et al., 2016). Furthermore, microglia express many different receptors such as the purinergic P2, toll-like, TNF-α, fractalkine R1 (CX3CR1), and TREM2 receptors, among many others (Mariani and Kielian, 2009; Gelderblom et al., 2015; Hug et al., 2018). All these receptors have a variety of functions, and their expression and the outcome of their activation depend on the functional state of the cell and on the pathological conditions. Depending on the nature of the ligand and on the receptor, downstream intracellular pathways translate their activation to detrimental or beneficial effects (for references see Gomes-Leal, 2012). For instance, ATP binds to different purinergic receptors with beneficial or detrimental effect after ischemia (Gelderblom et al., 2015). ATP, activating its receptors on microglia, stimulates the inflammasome, which causes increased secretion of proinflammatory cytokines that spread and intensify the inflammatory environment. ATP binds to purinergic P2 receptors and is involved in the formation or resolution of inflammation (Eltzschig et al., 2012). Among the P2 purinergic receptors, the metabotropic G-protein-coupled P2Y receptor (P2YR) and the nucleotide-gated ion channel P2X receptor (P2XR) are the most studied. Within the P2YR family, P2Y2R and P2Y6R both promote the phagocytic clearance of apoptotic cells or bacteria, contributing to the termination of inflammation (Koizumi et al., 2007). On the contrary, microglial P2Y12R seems to be involved in mediating damage in cerebral ischemia (Webster et al., 2013). The P2 × 7R likely participates in the cerebral damage associated with stroke (Domercq et al., 2010).
In preclinical studies, increased density and activation of microglia following hypoperfusion (Reimer et al., 2011; Manso et al., 2018) is associated with release of matrix metalloproteinase-2 (MMP-2) (Simpson et al., 1999). MMPs are proteases that degrade the extracellular matrix (ECM) and the tight junctions between endothelial cells. Furthermore, MMPs are involved in BBB breakdown after ischemia (Seo et al., 2013) and can degrade myelin (Chandler et al., 1995). Increased production of ROS by activated microglia may disrupt NO signaling, causing endothelial dysfunction (Freeman and Keller, 2012). Thus, microglia may further damage the BBB through a proinflammatory cascade that causes degradation of the ECM by production of MMPs and by oxidative damage.
Age is a key risk factor for vascular cognitive impairment and dementia (VCID) and is associated with modifications of phenotype and functions of microglia (von Bernhardi et al., 2015), which assume a more intense proinflammatory phenotype (Norden and Godbout, 2013). Aging microglia have less branchings, reduced motility, and lower migration rates that confer an altered surveillance phenotype and have a more sustained inflammatory response to damage (Damani et al., 2011). The phenotypic modifications seem to be caused by a shift in microglial metabolic pathways since activated microglial cells treated with an inflammatory stimulus show a reduction in mitochondrial oxidative phosphorylation (Nair et al., 2019). Minocycline is a potent inhibitor of inflammatory responses that has preclinical efficacy in several animal models of VCID and particularly in vascular conditions in which microglial cells are activated, including cerebral hypoperfusion (Ma et al., 2015; Manso et al., 2018). Chronic administration of minocycline reduces the number of microglial cells, restores the hypoperfusion-induced impairment in white matter function, and has protective effects (Ma et al., 2015; Manso et al., 2018).
Apoptosis is a mechanism of controlled cell death and may subserve a homeostatic function to maintain and regulate the number of cells in health and pathological conditions (Kerr et al., 1972; Becker and Bonni, 2004). Increased apoptosis is thought to be a physiological mechanism that helps maintain normal tissue homeostasis through resolution of low-grade inflammation, such as those that develop during normal aging (Gupta et al., 2006). Therefore, in the chronic, low-grade inflammatory conditions typical of chronic brain hypoperfusion, brain parenchyma surveillance by astrocytes and patrolling by microglia seem to be aimed at reducing the spreading of inflammation from apoptotic neurons and debris, preventing further damage to neighboring neurons. As a consequence, under these conditions, astrocytes and microglia have a protective role, cooperating in the disposal of neuronal debris by phagocytosis or of whole neurons by phagoptosis (Lana et al., 2017a), clearing dysfunctional synapses, controlling proinflammatory mediators, and diffusing damage to neighboring cells. How apoptosis causes neurons to be disposed of is still uncertain. The principal mechanism is probably by triggering the release of intercellular signals, such as the “find-me” signals ATP and fractalkine (CX3CL1) (Noda et al., 2011; Cerbai et al., 2012) and the “eat-me” signals such as phosphatidylserine (PS) (reviewed in Márquez-Ropero et al., 2020), which recall and activate phagocytic cells such as microglia to engulf and consume the neuron. The release of “don’t eat me” signals from neurons, such as CD47-SIRPα or CD200-CD200L, maintains microglia in a quiescent state and suppresses phagocytosis (reviewed in Michell-Robinson et al., 2015). Astrocytes and microglia express membrane receptors, such as CX3CR1, P2Y6, P2Y12, stabilin 1, SIRPα, TREM2, MerTK, and CD11b (Takahashi et al., 2005; Peri and Nüsslein-Volhard, 2008; Wakselman et al., 2008; Mazaheri et al., 2014), that recognize molecules released by damaged neurons (Harrison et al., 1998; Noda et al., 2011), causing the phagocytosis of degenerating neurons and neuronal debris. Nevertheless, in a model of hypoperfusion in the rat, no significant decrease of pyramidal neurons in both CA1 and CA3 (Cerbai et al., 2012; Lana et al., 2016) was found. The damaged neurons are possibly replaced by the continuous addition of newborn neurons by neurogenesis (see below).
Metabolism of Astrocytes and Microglia During Hypoperfusion
The brain is an energetically demanding organ, and most of energy supply, in form of ATP, is utilized by neurons to meet the high energy requirements necessary for neuronal activity such as the synthesis and release of neurotransmitters and neuromodulators and for the maintenance of the ionic gradients necessary for synaptic activity (Attwell and Laughlin, 2001). In astrocytes, energy stores are localized mainly as glycogen that represents a short-term buffer for transient energy requirements from neurons (Kong et al., 2002) that do not store energy. Brain metabolic requests and modifications strongly influence the origin and progression of many neurodegenerative disorders such as AD and PD (for references see Aldana, 2019). Nevertheless, an important fraction of energy in the brain is not directly requested by neurons for their activity. Normal brain activity mainly depends on metabolic plasticity of astrocytes and requires not only glucose supply from blood but also glycogen stored in astrocytes that fuels specific activity in the brain and that can last beyond the limits of glucose supply from blood (Brown and Ransom, 2007). Glycogen in astrocytes is essential for the survival of axons, and its depletion is related to brain dysfunctions and neurodegeneration (Wender et al., 2000). Noradrenaline and insulin regulate glycolysis and glycogenesis in astrocytes, while ATP production in the mitochondria and oxidation of fatty acids is regulated by the thyroid hormone (Morita et al., 2019). In response to inflammation or oxidative stress, astrocytes upregulate glycolysis producing ATP and lactate, which support energy metabolism to neurons but also accelerate neurodegeneration by fueling reactive astrocytes (Almeida et al., 2001). It appears therefore that the energy metabolism of reactive astrocytes is a determinant of physiological processes, and its impairments or modifications may cause significant decline of brain functions (Morita et al., 2019). It has also been shown that in hypoxic/ischemic conditions the neuroprotective A2 reactive astrocytes upregulate fatty acid oxidation (FAO) (Zamanian et al., 2012), which is neuroprotective. Upregulation of fatty acid metabolism attenuates inflammation by clearing fatty acid from the nearby parenchyma (Takahashi et al., 2014). The ketone bodies, produced by fatty acid oxidation in astrocytes, maintain the energy metabolism in neurons, compensating for the glucose metabolism damaged by ischemia and by production of ROS (Takahashi et al., 2014).
Microglia, for their role in brain development, for their continuous scanning of brain parenchyma to maintain a healthy environment, and for their harmful responses to injuries and activation of repair programs (Aldana, 2019; Engl and Attwell, 2015), have large energy demands. Furthermore, during insults and brain tissue damage, microglia are the first responders to pathological changes to homeostasis, migrate to the site of injury, modify their morphology retracting their processes, and phagocytose debris and cells (Davalos et al., 2005). It is not completely understood whether microglial motility and phagocytosis are powered by oxidative phosphorylation or by glycolytic pathways, but either one or both these processes represent a significant energy demand (Engl and Attwell, 2015).
Recent findings demonstrate that peripheral immune cells can adapt to environmental challenges, modifying their metabolic pathways in order to utilize nutrients other than glucose, such as fatty acids or amino acids (Van den Bossche et al., 2017). Recently, it has been demonstrated (Bernier et al., 2020) in models of hypoglycemia or aglycemia that activities of microglia, such as process motility and damage sensing functions, are maintained by alternative metabolic pathways such as glutaminolysis, which depend on mammalian target of rapamycin (mTOR) activation. This metabolic plasticity sustains mitochondrial metabolism even in brain neuroenergetic crisis, allowing microglia to maintain their fundamental surveillance and phagocytic roles (Bernier et al., 2020). Furthermore, in aged mice, microglia display changes in metabolic profile such as modifications of proteins involved not only in inflammatory signaling but also in mitochondrial function and cellular metabolism (Flowers et al., 2017). Indeed, oxidative stress is associated with non-pathological aging, and functional decline of mitochondria in microglia increases the production of ROS and inflammatory mediators that in turn may increase oxidative stress.
Therefore, it appears that perturbation in the regulation of brain energy metabolism in neurons, as well as in astrocytes and microglia, may be one of the pathophysiological mechanisms of neurodegenerative disorders among which AD, PD, ALS, and HD, and this can be even more important in hypoxia/ischemia-dependent pathologies. Indeed, metabolic disturbances such as high blood pressure, atherosclerosis, obesity, and diabetes are among the most important risk factors for dementia (Kivipelto et al., 2006), as shown by epidemiological studies. Deficits of energy metabolism such as hypometabolism of glucose and mitochondrial dysfunctions in both neurons and astrocytes are early indicators of neurodegenerative disorders such as AD, PD, ALS, and HD (Hoyer, 1993; Diehl-Schmid et al., 2007; Edison et al., 2013; Adiele and Adiele, 2019).
Neuron–Astrocyte–Microglia Interactions in the Hippocampus in a Rat Model of Chronic Hypoperfusion
Brain chronic hypoperfusion is a progressive, dynamic process induced by partial carotid occlusion during aging, heart failure, hypotension, atherosclerosis of large or small vessels, and carotid stenosis. Brain chronic hypoperfusion causes multiple progressive modifications that eventually lead to vascular dementia and neurodegeneration (Chmayssani et al., 2007; Farkas et al., 2007; Ozacmak et al., 2007), which may manifest with cognitive dysfunctions.
The rat model of brain chronic hypoperfusion, obtained with the permanent bilateral occlusion of the common carotid arteries (bCCAo) (Sarti et al., 2002a, b; Farkas et al., 2007; Lana et al., 2014, 2017a), represents a model of cerebrovascular stenosis in aging humans. This model is helpful in investigating the mechanisms and effects of long-term chronic cerebral hypoperfusion (Farkas et al., 2007). bCCAo in the rat causes chronic cerebral hypoperfusion mainly in the forebrain and leads to early disruption of the BBB, to white matter rarefaction with axonal and myelin damage, to neuroinflammation, and to hippocampal and cortical neuronal damage (Ciacciarelli et al., 2020). Indeed, the infarcts generated by bCCAo are seen not only in the striatum and the dorsolateral cortex but also in areas such as the hippocampus, thalamus, and hypothalamus (Ciacciarelli et al., 2020). Among the plausible explanations for the unexpected brain damages to the latter brain areas are (i) anomalies of the circle of Willis (Kitagawa et al., 1998), (ii) production of neurotoxic molecules that propagate to the hippocampus and cause ischemic damage (Xie et al., 2011), and (iii) overexcitation of the hippocampal neuronal network by glutamate produced after the occlusion. In addition, an alternative explanation is that small and deep arteries beyond the circle of Willis, such as the anterior choroidal artery (AchA), the lateral hypothalamic artery (LHA), and the ventral thalamic artery (VTA), have a function in the supply of blood to deep brain areas. The AchA provides the major blood supply to the anterior hippocampus and other deep areas. In addition, these blood vessels can originate directly from the internal carotid artery, proximally to the origin of middle cerebral artery (MCA). Therefore, whereas distal occlusions cause lesions to the striatum, the piriform cortex, and portions of the parietal–temporal cortex, proximal occlusions may produce damage to the anterior hippocampus, thalamus, and/or hypothalamus.
In our investigations in the rat model of bCCAo (Lana et al., 2014, 2017a), we highlighted and characterized the quantitative, morphological, and functional alterations on neuron, astrocyte, and microglial interactions that may be relevant for the neurodegenerative processes in CA1 and CA3 hippocampus. Our findings demonstrate the existence of common and differential features of the interplay among neurons and glia in the two hippocampal areas, which may help in explaining the higher sensitivity of pyramidal neurons in CA1 to hypoxia leading to neurodegeneration. We developed the innovative ex vivo method of the triple-labeling fluorescent immunohistochemistry coupled with confocal microscopy (TIC) and digital imaging, which we exploited to make comparisons between areas CA1 and CA3 of the hippocampus. Using this novel method, we also implemented the new technique of digital subslicing (Lana et al., 2014, 2017a), which allows to visualize the intimate interplay among different cells, and the colocalization of different antigens that can take place within the cell. With this novel method, we studied the morphological and functional alterations of the neuron–astrocyte–microglia triad as a possible mechanism responsible on one side for neuroprotection and on the other one for the neurodegeneration that characterizes animal models of neurodegenerative diseases such as AD or ischemia (Lana et al., 2014, 2017a; Fusco et al., 2018; Ugolini et al., 2018).
In CA1 and CA3 str. pyramidalis (SP) of hypoperfused rats, numerous apoptotic neurons are present, characterized by intense and uniform CytC immunostaining in the cytoplasm (Figure 1A), a hallmark of the late phases of apoptosis (Suen et al., 2008). In both CA1 and CA3 str. radiatum (SR) of hypoperfused rats in the proximity of SP, we found the presence of numerous neurons with pyramidal shape, the so-called ectopic neurons. More specifically, in area CA3 the ectopic neurons are localized mainly in str. lucidum (SL) (Figures 1C1–C3), which is an “a-neuronal” region of CA3, as paradigmatically defined by Amaral and Lavenex (2007). Ectopic neurons are not interneurons (see Figures 1C1–C3) but likely are degenerating pyramidal neurons that derive from SP and are being detached from it. Indeed, ectopic neurons infiltrated by astrocyte branches (see Figures 1G1,G2), are located in close proximity to SP, which appears indented in correspondence with the ectopic neuron (Figures 1D1,D3, asterisk). We hypothesize that apoptotic CA1 neurons are removed from CA1 SP, possibly by astrocyte branches through signaling molecules such as the Cx43 and/or CX3CL1 (Noda et al., 2011; Cerbai et al., 2012). The ectopic neurons detached from the pyramidal layer in CA1 and CA3 are apoptotic and undergo the process of phagocytosis by microglia (Figures 1D1–D3). Furthermore, as a consequence of apoptosis, ectopic neurons are fragmented to form neuronal debris, present in high density in both CA1 and CA3 SR of hypoperfused rats (circled area in Figure 1B). This is possibly a protective mechanism that avoids the diffusion of inflammatory damage to surrounding neurons in response to proinflammatory substances released in the parenchyma.
As demonstrated in other models of neurodegeneration, astrocyte branches, infiltrating the neuronal cell body, may trigger or accelerate the fragmentation of apoptotic neurons (Polazzi and Monti, 2010; Cerbai et al., 2012; Huizinga et al., 2012). Neuronal debris are located throughout SR of CA1 and CA3, not in a random position but mostly closely apposed to astrocyte branches (circled area in Figure 1B). This phenomenon is similar to that observed in normal brain aging and LPS-induced neuroinflammation (Cerbai et al., 2012; Lana et al., 2016, 2017b).
We postulate that the detachment of a neuron from the pyramidal layer to form ectopic neurons that are fragmented into neuronal debris is part of a common mechanism of neuronal death in the hippocampus in a milieu of hypoxia/ischemia, in acute or chronic inflammatory conditions that can lead to neurodegeneration.
Despite all the above mechanisms, in hypoperfused rats, the quantity of CA1 and CA3 pyramidal neurons does not decrease (Lana et al., 2014, 2017a). Increased neurogenesis (Farkas et al., 2007; Dirnagl, 2012; Maraula et al., 2013) during the restitution phase of brain chronic hypoperfusion can explain this apparently contradictory result. Newborn neurons migrating from the subgranular zone of the DG to CA1 and CA3 pyramidal layers possibly reintegrate the apoptotic neurons.
Numerous phagocytic events by microglia on the ectopic neurons detached from the pyramidal layer (Figure 1E) also involve astrocytes that form peculiar clusters of cells with neurons and microglia that we define “triads.” In a triad, one or more astrocyte(s) project its branches toward a neuron that is phagocytosed by a microglial cell (Figures 1F,G1). According to our findings, the branches of astrocytes involved in the triad exert a fundamental role. They form a protective microscar around the neuron that is undergoing phagocytosis, to prevent the spread in the surrounding tissue of proinflammatory mediators and of neuronal debris, which could initiate an inflammatory response potentially harmful for other healthy neurons. Astrocyte branches exert a noxious effect on the neuron that is involved in the triad: they can infiltrate the neuronal body to help or accelerate its fragmentation (see Figures 1G1,G2). The mechanism of infiltration and fragmentation of the neuronal cell body by astrocytes branches, first demonstrated in our laboratory in models of normal brain aging and acute inflammation (Cerbai et al., 2012), was later confirmed in a model of ALS (Re et al., 2014) and in other papers published by our group (Lana et al., 2014, 2016). The triad formation and the astrocyte-mediated fragmentation of the ectopic-apoptotic neurons to form debris (Cerbai et al., 2012; Lana et al., 2014, 2016, 2017a, b) could be a common mechanism of neuronal death in a neurodegenerative milieu in the hippocampus. We also hypothesize that this could be a general mechanism of neuronal death also valid in healthy tissue for senescent neurons. In the hippocampus of hypoperfused rats, the expression of TNF-α increases in astrocytes and dendrites of pyramidal neurons in both CA1 and CA3 SR (Figure 1H). Microglia and astrocytes can both recognize danger signals, including those released by cellular debris produced from apoptotic cells, and can cooperate and help clearing apoptotic neurons or neuronal debris (Medzhitov and Janeway, 2002; Milligan and Watkins, 2009). This concerted action can prevent or reduce release of proinflammatory mediators and consequent injury to neighboring neurons (Nguyen et al., 2002; Turrin and Rivest, 2006).
Some events reflect a common response of CA1 and CA3 to the hypoperfusion, but the two areas show different responses to the ischemic insult. In particular, these differences are related mainly to behavior of glial cells, confirming their primary function in influencing the pathophysiology of the brain. In CA1 SR of hypoperfused rats, astrocytes show no modifications (Lana et al., 2014), whereas in CA3 SR, astrocytes significantly increase in response to hypoperfusion but do not appear hypertrophic or hyperactivated (Lana et al., 2017a). Higher demand of O2 and nutrient supply by pyramidal neurons that are in a hypoxic and hypoglycemic state due to hypoperfusion can possibly cause the increase of astrocyte density, which can balance the reduced trophic support to CA3 neurons. This response of astrocytes, contrary to that observed in CA1, is possibly a protective effect of astrocytes toward neurons.
Astrogliosis, a late-emerging event during chronic cerebral hypoperfusion (Pappas et al., 1996; Farkas et al., 2004, 2006, 2007; Schmidt-Kastner et al., 2005), has long been considered a negative phenomenon. This idea is rapidly changing, thanks to new data that suggest a more elaborated and diversified role of astrocytes upon different insults leading to neurodegenerative disorders such as AD, ALS, and stroke (Sofroniew, 2009; Verkhratsky et al., 2013; Burda and Sofroniew, 2014). Dysfunctions in the reactivity of astrocytes can be the primary cause, or can contribute to loss of normal functions of neurons, leading to or increasing neurodegeneration (Sofroniew, 2009).
Another remarkable difference is the behavior of microglia which decreases in CA1 (Lana et al., 2014) and increases in CA3 SR of hypoperfused rats (Lana et al., 2017a). These differences may explain the higher sensitivity of CA1 pyramidal cells to an ischemic insult. Recruitment and activation of microglia to the site of the insult generally is believed a negative mechanism that causes accumulation of neurotoxic phagocytes. Recently, microglial activation following neuronal injury is considered a reversible multistep process that represents mainly a protective mechanism (Minghetti and Levi, 1998; Streit et al., 1999; Polazzi and Contestabile, 2002; Hanisch and Kettenmann, 2007; Ransohoff and Perry, 2009; Kettenmann et al., 2013). In microglia-depleted organotypic cultures, CA1 pyramidal cell death increases, suggesting a neuroprotective role of microglia (Montero et al., 2009). Apoptotic neurons can release diffusible signals that enhance microglia neuroprotective properties. In turn, microglia release molecules that can rescue neurons from apoptosis (Polazzi et al., 2001). Phagocytosis of apoptotic cells by microglia decreases the production of pro-inflammatory cytokines, such as TNF-α and IL-12, without affecting the secretion of anti-inflammatory, potentially neuroprotective molecules, such as IL-10 and TGF-ßl (Magnus et al., 2001). During ischemia, microglia are responsible for the phenomenon of phagoptosis (also called primary phagocytosis), first defined by Brown and Neher (2012) as “death caused by being devoured.” Phagoptosis represents the phagocytosis of whole neurons that show no sign of neurodegeneration (Zhang et al., 2015). It is triggered by a stressful stimulus which is too mild to cause cell death, too intense to allow restoration of the healthy neuron, but sufficient to release “find-me” signals that activate and recruit microglia and astrocytes for phagocytosis (Ravichandran, 2011; Kao et al., 2011; Brown and Neher, 2012).
Thus, formation of triads appears as a specific mechanism for clearance of neurons under degeneration, not only through the mechanism of phagocytosis but also through phagoptosis (Brown and Neher, 2014). The increase of microglia that help in the disposal of damaged neurons (Nathan and Ding, 2010) may result in anti-inflammatory and neuroprotective effects (Liesz et al., 2009). Increased microglia in area CA3 of hypoperfused rats during the restitution phase (Farkas et al., 2007; Dirnagl, 2012) may release anti-inflammatory cytokines that trigger an anti-inflammatory milieu (Spite and Serhan, 2010). Microglia in CA3 SR (Beynon and Walker, 2012) phagocytose apoptotic pyramidal neurons or neuronal debris, promoting tissue repair and the resolution of inflammation. Taken together, our results demonstrate that effects of astrocytes and microglia in pathological conditions may contribute to neuronal damage but may also be a mechanism of protection to control the proinflammatory process and the diffusion of the cellular damage to the surrounding cells.
Conclusion
The data of this review contribute to deepening the concept that the interactions that occur among the different cell populations of the CNS give rise to reciprocal networks of morphological and functional reliance and dependency. To comprehend the peculiar aspects of the onset and progression of neurodegeneration, it is necessary to consider that any tissue, and mainly the nervous tissue, is not composed by a collection of independent elements but rather by interdependent cell populations that interact and cooperate to maintain the homeostasis and functionality of the organ. Different types of insults that affect one population modifying its functionality reasonably reverberate to the others, either favoring or dysregulating their activities. In the neuron–astrocyte–microglia triads, the cell body of a damaged neuron is infiltrated and bisected by astrocyte branches that form a microscar around it and is embraced by microglia cells for the purpose of phagocytosing it. While activation of glia has long been considered as a detrimental mechanism for neuron survival, recently it is emerging that this is not always the case. Nevertheless, in contiguous, interconnected hippocampal areas, the responses of glia to the same insult are not equal but vary significantly from CA1 area to CA3.
The comparison between the response of areas CA1 and CA3, shown schematically in Figure 2, helps to understand the differential reactivities of the two areas in hypoxia and to understand why pyramidal neurons in CA3 show higher adaptation than those in CA1 to hypoxia/ischemia (Lana et al., 2014, 2017a). The increase of apoptotic neurons is similar in the two regions, but in CA3 the density of ectopic pyramidal neurons is significantly higher than in CA1. The translocation of neurons from their competence, pyramidal layer to the contiguous SL or SR allows the damaged neurons to be fragmented by astrocytes to form neuronal debris that can be phagocytosed by microglial cells in the triad clusters. All these mechanisms are protective effect toward the surrounding neurons. In addition, the migration of neurons from the SP can decrease the diffusion of toxic/proinflammatory compounds to neighboring vital neurons. Likely, in CA3 the scavenging mechanism on degenerating pyramidal neurons is more active, explaining the lower sensitivity of the hippocampal CA3 area to the ischemic insult. Indeed, in CA3 of ischemic rats microglia is significantly higher than in the CA1 area where microglia decrease. As mentioned earlier, since activation of microglia is considered a protective mechanism (Hanisch and Kettenmann, 2007; Ransohoff and Perry, 2009; Kettenmann et al., 2013), the increase of microglia in CA3 may explain the better response of this area to an ischemic insult, in comparison to CA1. Lastly, in CA3 there is a significant increase of astrocytes, a phenomenon that can depend on the increased trophic request of the tissue and may represent a sign of a better response of CA3 to the ischemic insult. Finally, the neuron–astrocyte–microglial triads are significantly more numerous in CA3 than in CA1 of ischemic rats, further indicating the protective effects of the triads. Taken together, these results demonstrate that in CA3 astrocytes and microglia interplay with apoptotic neurons may represent a protective mechanism able to control the inflammatory process and the diffusion of the cellular damage to the neighboring tissues.
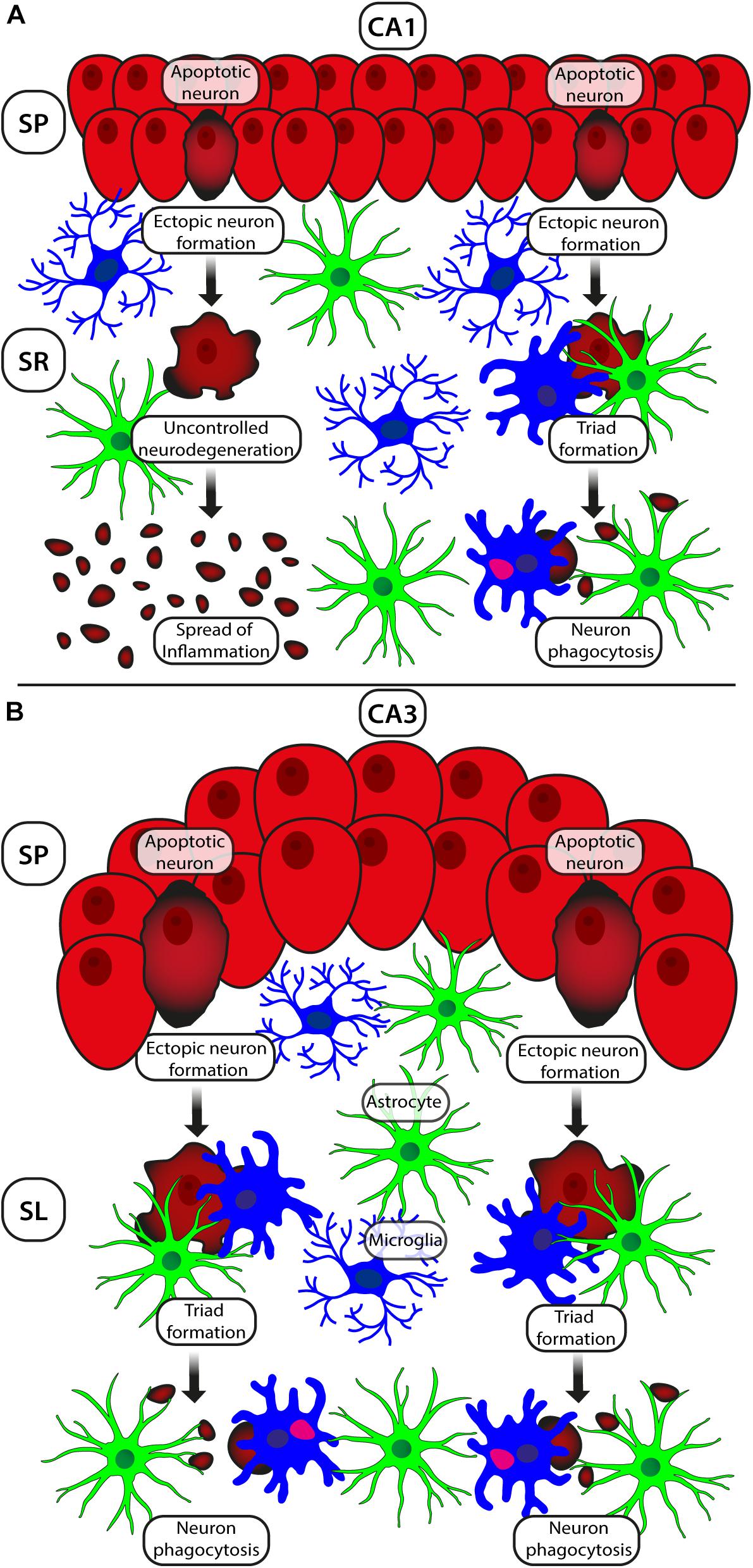
Figure 2. Schematic representation of the different responses of neurons and astrocytes–microglia to ischemia/hypoxia in areas CA1 (A) and CA3 (B). The increase of apoptotic neurons (shown in dark red) is comparable in the two regions, but the density of ectopic pyramidal neurons in CA3 (dark red) is higher than in CA1. Pyramidal apoptotic neurons translocate from their competence layer to the underlying SR (CA1) or SL (CA3) to form triad clusters, be fragmented by astrocytes and be phagocytosed by microglia. This mechanism is finalized to the protection of the surrounding neurons in CA1 SP. In CA3 of ischemic rats, microglia and triads are significantly more numerous than in CA1, a further sign of better responsiveness of CA3 to hypoxia. Astrocytes are more numerous in CA3, possibly because of the increased trophic request of the tissue. This effect may be a further mechanism of a better response of CA3 to the ischemic insult. In CA3, astrocytes and microglia may help control the inflammatory process and the ensuing diffusion of the cellular damage to the surrounding tissue.
It is of great interest to verify if the differences of glial reactivity in these two contiguous hippocampal areas are mirrored in many other areas of the brain, reflecting the higher or lower sensitivity to different insults, and whether they may represent targets for future therapeutic interventions.
Author Contributions
MG and DL wrote the manuscript. FU contributed to the preparation of the figures. MG edited and revised the manuscript. All authors read and approved the final manuscript.
Funding
This work was partly funded by University of Florence (FFO). DL is a Research Fellow funded by Intesa San Paolo (cod B/2019/0236).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
ABCA1, ATP-binding cassette transporter; AD, Alzheimer’s disease; ALS, Amyotrophic lateral sclerosis; APOE, Apolipoprotein E; BAI1, Brain-specific angiogenesis inhibitor 1; bCCAo, Bilateral occlusion of the common carotid arteries; BDNF, Brain-derived neurotrophic factor; CBF, Cerebral blood flow; CNS, Central nervous system; CX3CL1, Fractalkine; CX3CR1, Fractalkine receptor1; Cx43, Connexin43; CytC, Cytochrome C; FAO, Fatty acid oxidation; GFAP, Glial fibrillary acidic protein; GLAST, Excitatory amino acid transporter-1, EAAT-1; GLT1, Excitatory amino acid transporter-2, EAAT-2; HD, Huntington’s disease; IBA1, Ionized calcium binding adaptor molecule 1; LPS, Lipopolysaccharide; MCI, Mild cognitive impairment; MEGF10, Multiple EGF-like-domains 10; MERTK, Proto-oncogene tyrosine-protein kinase MER; MHC, Major histocompatibility complex; MMP, Matrix metalproteinases; MS, Multiple sclerosis; mTOR, Mammalian target of rapamycin; MTP, Mitochondrial transition pore; NeuN, Neuronal nuclei; NF-kB, Nuclear factor-kappa B; OGD, Oxygen and glucose deprivation; PD, Parkinson’s disease; ROS, Reactive oxygen species; SL, Stratum lucidum; SP, Stratum pyramidalis; SPARC, solid-phase attachment of red cells; SR, Stratum radiatum; TBI, Traumatic brain injury; TGF- β, Transforming growth factor-beta; TIC, Triple-labeling fluorescent immunohistochemistry coupled with confocal microscopy; TNF- α, Tumor necrosis factor- α; TNF, Tumor necrosis factor; VCI, Vascular cognitive impairments; VCID, Vascular cognitive impairment and dementia; VEGF, Vascular endothelial growth factor.
References
Abbott, N. J. (2002). Astrocyte-endothelial interactions and blood-brain barrier permeability. J. Anat. 200, 629–638. doi: 10.1046/j.1469-7580.2002.00064.x
Adiele, R. C., and Adiele, C. A. (2019). Metabolic defects in multiple sclerosis. Mitochondrion 44, 7–14. doi: 10.1016/j.mito.2017.12.005
Al Ahmad, A., Gassmann, M., and Ogunshola, O. O. (2012). Involvement of oxidative stress in hypoxia-induced blood-brain barrier breakdown. Microvasc. Res. 84, 222–225. doi: 10.1016/j.mvr.2012.05.008
Aldana, B. I. (2019). Microglia-Specific Metabolic Changes in Neurodegeneration. J. Mol. Biol. 431, 1830–1842. doi: 10.1016/j.jmb.2019.03.006
Allan, L. M., Rowan, E. N., Firbank, M. J., Thomas, A. J., Parry, W., Polvikoski, T. M., et al. (2011). Long term incidence of dementia, predictors of mortality and pathological diagnosis in older stroke survivors. Brain 134, 3716–3727.
Allen, N. J., and Barres, B. A. (2009). Neuroscience: Glia - more than just brain glue. Nature 457, 675–677. doi: 10.1038/457675a
Almeida, A., Almeida, J., Bolaños, J. P., and Moncada, S. (2001). Different responses of astrocytes and neurons to nitric oxide: The role of glycolytically generated ATP in astrocyte protection. Proc. Natl. Acad. Sci. U S A. 98, 15294–15299. doi: 10.1073/pnas.261560998
Alosco, M. L., Brickman, A. M., Spitznagel, M. B., Garcia, S. L., Narkhede, A., Griffith, E. Y., et al. (2013). Cerebral perfusion is associated with white matter hyperintensities in older adults with heart failure. Congest. Hear. Fail 19, E29–34. doi: 10.1111/chf.12025
Alvarez, J. I., Katayama, T., and Prat, A. (2013). Glial influence on the blood brain barrier. Glia 61, 1939–1958. doi: 10.1002/glia.22575
Amantea, D., Bagetta, G., Tassorelli, C., Mercuri, N. B., and Corasaniti, M. T. (2010). Identification of distinct cellular pools of interleukin-1β during the evolution of the neuroinflammatory response induced by transient middle cerebral artery occlusion in the brain of rat. Brain Res. 1313, 259–269. doi: 10.1016/j.brainres.2009.12.017
Amaral, D. G., and Lavenex, P. (2007). “Hippocampal Neuroanatomy,” in The Hippocampus Book, eds P. Andersen, R. Morris, D. Amaral, and J. O’Keefe (New York: Oxford University Press), 37–114.
Amaral, D. G., and Witter, M. P. (1989). The three-dimensional organization of the hippocampal formation: A review of anatomical data. Neuroscience 31, 571–591. doi: 10.1016/0306-4522(89)90424-7
Araque, A., Carmignoto, G., Haydon, P. G., Oliet, S. H. R., Robitaille, R., and Volterra, A. (2014). Gliotransmitters travel in time and space. Neuron 81, 728–739. doi: 10.1016/j.neuron.2014.02.007
Arbeloa, J., Pérez-Samartín, A., Gottlieb, M., and Matute, C. (2012). P2X7 receptor blockade prevents ATP excitotoxicity in neurons and reduces brain damage after ischemia. Neurobiol. Dis. 45, 954–961. doi: 10.1016/j.nbd.2011.12.014
Attwell, D., and Laughlin, S. B. (2001). An energy budget for signaling in the grey matter of the brain. J. Cereb. Blood Flow Metab. 21, 1133–1145. doi: 10.1097/00004647-200110000-00001
Bahar, A. S., Shirvalkar, P. R., and Shapiro, M. L. (2011). Memory-guided learning: CA1 and CA3 neuronal ensembles differentially encode the commonalities and differences between situations. J. Neurosci. 31, 12270–12281. doi: 10.1523/JNEUROSCI.1671-11.2011
Bakiri, Y., Burzomato, V., Frugier, G., Hamilton, N. B., Káradóttir, R., and Attwell, D. (2009). Glutamatergic signaling in the brain’s white matter. Neuroscience 158, 266–274. doi: 10.1016/j.neuroscience.2008.01.015
Balestrini, S., Perozzi, C., Altamura, C., Vernieri, F., Luzzi, S., Bartolini, M., et al. (2013). Severe carotid stenosis and impaired cerebral hemodynamics can influence cognitive deterioration. Neurology 80, 2145–2150. doi: 10.1212/WNL.0b013e318295d71a
Barkauskas, D. S., Dixon Dorand, R., Myers, J. T., Evans, T. A., Barkauskas, K. J., Askew, D., et al. (2015). Focal transient CNS vessel leak provides a tissue niche for sequential immune cell accumulation during the asymptomatic phase of EAE induction. Exp. Neurol. 266, 74–85. doi: 10.1016/j.expneurol.2015.02.018
Barreto, G., White, R. E., Ouyang, Y., Xu, L., and Giffard, R. G. (2011). Astrocytes: Targets for Neuroprotection in Stroke. Cent. Nerv. Syst. Agents Med. Chem 11, 164–173. doi: 10.2174/187152411796011303
Bartsch, T., and Wulff, P. (2015). The hippocampus in aging and disease: From plasticity to vulnerability. Neuroscience 309, 1–16. doi: 10.1016/j.neuroscience.2015.07.084
Bartsch, T., Döhring, J., Reuter, S., Finke, C., Rohr, A., Brauer, H., et al. (2015). Selective neuronal vulnerability of human hippocampal CA1 neurons: Lesion evolution, temporal course, and pattern of hippocampal damage in diffusion-weighted MR imaging. J. Cereb. Blood Flow Metab. 35, 1836–1845. doi: 10.1038/jcbfm.2015.137
Bartsch, T., Schönfeld, R., Müller, F. J., Alfke, K., Leplow, B., Aldenhoff, J., et al. (2010). Focal lesions of human hippocampal CA1 neurons in transient global amnesia impair place memory. Science 328, 1412–1415. doi: 10.1126/science.1188160
Becker, E. B. E., and Bonni, A. (2004). Cell cycle regulation of neuronal apoptosis in development and disease. Prog. Neurobiol. 72, 1–25. doi: 10.1016/j.pneurobio.2003.12.005
Ben Haim, L., and Rowitch, D. H. (2016). Functional diversity of astrocytes in neural circuit regulation. Nat. Rev. Neurosci. 18, 31–41. doi: 10.1038/nrn.2016.159
Bernal, G. M., and Peterson, D. A. (2011). Phenotypic and gene expression modification with normal brain aging in GFAP-positive astrocytes and neural stem cells. Aging Cell 10, 466–482. doi: 10.1111/j.1474-9726.2011.00694.x
Bernier, L. P., York, E. M., Kamyabi, A., Choi, H. B., Weilinger, N. L., and MacVicar, B. A. (2020). Microglial metabolic flexibility supports immune surveillance of the brain parenchyma. Nat. Commun. 11:1559. doi: 10.1038/s41467-020-15267-z
Beynon, S. B., and Walker, F. R. (2012). Microglial activation in the injured and healthy brain: What are we really talking about? Practical and theoretical issues associated with the measurement of changes in microglial morphology. Neuroscience 225, 162–171. doi: 10.1016/j.neuroscience.2012.07.029
Boisvert, M. M., Erikson, G. A., Shokhirev, M. N., and Allen, N. J. (2018). The Aging Astrocyte Transcriptome from Multiple Regions of the Mouse Brain. Cell Rep. 22, 269–285. doi: 10.1016/j.celrep.2017.12.039
Braak, H., Alafuzoff, I., Arzberger, T., Kretzschmar, H., and Tredici, K. (2006). Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 112, 389–404. doi: 10.1007/s00401-006-0127-z
Bribian, A., Pérez-Cerdá, F., Matute, C., and López-Mascaraque, L. (2018). Clonal glial response in a multiple sclerosis mouse model. Front. Cell. Neurosci. 12:375. doi: 10.3389/fncel.2018.00375
Brown, A. M., and Ransom, B. R. (2007). Astrocyte glycogen and brain energy metabolism. Glia 55, 1263–1271. doi: 10.1002/glia.20557
Brown, G. C., and Neher, J. J. (2012). Eaten alive! Cell death by primary phagocytosis: “Phagoptosis.”. Trends Biochem. Sci. 37, 325–332. doi: 10.1016/j.tibs.2012.05.002
Brown, G. C., and Neher, J. J. (2014). Microglial phagocytosis of live neurons. Nat. Rev. Neurosci. 15, 209–216. doi: 10.1038/nrn3710
Bruhn, T., Levy, L. M., Nielsen, M., Christensen, T., Johansen, F. F., and Diemer, N. H. (2000). Ischemia induced changes in expression of the astrocyte glutamate transporter GLT1 in hippocampus of the rat. Neurochem. Int. 37, 277–285. doi: 10.1016/S0197-0186(00)00029-2
Burda, J. E., and Sofroniew, M. V. (2014). Reactive gliosis and the multicellular response to CNS damage and disease. Neuron 81, 229–248. doi: 10.1016/j.neuron.2013.12.034
Cauli, B., and Hamel, E. (2010). Revisiting the role of neurons in neurovascular coupling. Front. Neuroenergetics 2:9. doi: 10.3389/fnene.2010.00009
Cenquizca, L. A., and Swanson, L. W. (2007). Spatial organization of direct hippocampal field CA1 axonal projections to the rest of the cerebral cortex. Brain Res. Rev. 56, 1–26. doi: 10.1016/j.brainresrev.2007.05.002
Cerbai, F., Lana, D., Nosi, D., Petkova-Kirova, P., Zecchi, S., Brothers, H. M., et al. (2012). The Neuron-Astrocyte-Microglia Triad in Normal Brain Ageing and in a Model of Neuroinflammation in the Rat Hippocampus. PLoS One 7:e45250. doi: 10.1371/journal.pone.0045250
Chandler, S., Coates, R., Gearing, A., Lury, J., Wells, G., and Bone, E. (1995). Matrix metalloproteinases degrade myelin basic protein. Neurosci. Lett. 201, 223–226. doi: 10.1016/0304-3940(95)12173-0
Chen, A., Akinyemi, R. O., Hase, Y., Firbank, M. J., Ndung’u, M. N., Foster, V., et al. (2016). Frontal White Matter Hyperintensities. Clasmatodendrosis and Gliovascular Abnormalities in Ageing and Post-Stroke Dementia - PubMed. Brain 139, 242–258. doi: 10.1093/brain/awv328
Chip, S., Nitsch, C., Wellmann, S., and Kapfhammer, J. P. (2013). Subfield-specific neurovascular remodeling in the entorhino-hippocampal- organotypic slice culture as a response to oxygen-glucose deprivation and excitotoxic cell death. J. Cereb. Blood Flow Metab. 33, 508–518. doi: 10.1038/jcbfm.2012.190
Chmayssani, M., Festa, J. R., and Marshall, R. S. (2007). Chronic Ischemia and Neurocognition. Neuroimaging Clin. N. Am. 17, 313–324. doi: 10.1016/j.nic.2007.03.002
Choi, D. W., and Rothman, S. M. (1990). The role of glutamate neurotoxicity in hypoxic-ischemic neuronal death. Annu. Rev. Neurosci. 13, 171–182. doi: 10.1146/annurev.ne.13.030190.001131
Christopherson, K. S., Ullian, E. M., Stokes, C. C. A., Mullowney, C. E., Hell, J. W., Agah, A., et al. (2005). Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell 120, 421–433. doi: 10.1016/j.cell.2004.12.020
Chung, C., Marson, J. D., Zhang, Q. G., Kim, J., Wu, W. H., Brann, D. W., et al. (2016). Neuroprotection Mediated through GluN2C-Containing N-methyl-D-aspartate (n.d.) Receptors Following Ischemia. Sci. Rep. 6:37033. doi: 10.1038/srep37033
Chung, W. S., Clarke, L. E., Wang, G. X., Stafford, B. K., Sher, A., Chakraborty, C., et al. (2013). Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 504, 394–400. doi: 10.1038/nature12776
Ciacciarelli, A., Sette, G., Giubilei, F., and Orzi, F. (2020). Chronic cerebral hypoperfusion: An undefined, relevant entity. J. Clin. Neurosci. 73, 8–12. doi: 10.1016/j.jocn.2020.01.026
Cimarosti, H., and Henley, J. M. (2008). Investigating the mechanisms underlying neuronal death in ischemia using in vitro oxygen-glucose deprivation: Potential involvement of protein SUMOylation. Neuroscientist 14, 626–636. doi: 10.1177/1073858408322677
Clarke, L. E., Liddelow, S. A., Chakraborty, C., Münch, A. E., Heiman, M., and Barres, B. A. (2018). Normal aging induces A1-like astrocyte reactivity. Proc. Natl. Acad. Sci. U S A. 115, E1896–E1905. doi: 10.1073/pnas.1800165115
Crehan, H., Hardy, J., and Pocock, J. (2013). Blockage of CR1 prevents activation of rodent microglia. Neurobiol. Dis. 54, 139–149. doi: 10.1016/j.nbd.2013.02.003
Crotti, A., and Glass, C. K. (2015). The choreography of neuroinflammation in Huntington’s disease. Trends Immunol. 36, 364–373. doi: 10.1016/j.it.2015.04.007
da Fonseca, A. C. C., Matias, D., Garcia, C., Amaral, R., Geraldo, L. H., Freitas, C., et al. (2014). The impact of microglial activation on blood-brain barrier in brain diseases. Front. Cell. Neurosci. 8:1–13. doi: 10.3389/fncel.2014.00362
Dallérac, G., and Rouach, N. (2016). Astrocytes as new targets to improve cognitive functions. Prog. Neurobiol. 144, 48–67. doi: 10.1016/j.pneurobio.2016.01.003
Damani, M. R., Zhao, L., Fontainhas, A. M., Amaral, J., Fariss, R. N., and Wong, W. T. (2011). Age-related alterations in the dynamic behavior of microglia. Aging Cell 10, 263–276. doi: 10.1111/j.1474-9726.2010.00660.x
Damisah, E. C., Hill, R. A., Rai, A., Chen, F., Rothlin, C. V., Ghosh, S., et al. (2020). Astrocytes and microglia play orchestrated roles and respect phagocytic territories during neuronal corpse removal in vivo. Sci. Adv 6:eaba3239. doi: 10.1126/sciadv.aba3239
Davalos, D., and Akassoglou, K. (2012). Fibrinogen as a key regulator of inflammation in disease. Semin. Immunopathol. 34, 43–62. doi: 10.1007/s00281-011-0290-8
Davalos, D., Grutzendler, J., Yang, G., Kim, J. V., Zuo, Y., Jung, S., et al. (2005). ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 8, 752–758. doi: 10.1038/nn1472
Davalos, D., Kyu Ryu, J., Merlini, M., Baeten, K. M., Le Moan, N., Petersen, M. A., et al. (2012). Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation. Nat. Commun. 3:1227. doi: 10.1038/ncomms2230
De Biase, L. M., Schuebel, K. E., Fusfeld, Z. H., Jair, K., Hawes, I. A., Cimbro, R., et al. (2017). Local Cues Establish and Maintain Region-Specific Phenotypes of Basal Ganglia Microglia. Neuron 95, 341.e–356.e. doi: 10.1016/j.neuron.2017.06.020
De Castro, S., Rasura, M., Di Angelantonio, E., Beccia, M., Passaseo, I., Di Lisi, F., et al. (2006). Distribution of potential cardiac sources of embolism in young and older stroke patients: Implications for recurrent vascular events. J. Cardiovasc. Med. 7, 191–196. doi: 10.2459/01.JCM.0000215272.32273.b9
De Groot, J. C., De Leeuw, F. E., Oudkerk, M., Van Gijn, J., Hofman, A., Jolles, J., et al. (2002). Periventricular cerebral white matter lesions predict rate of cognitive decline. Ann. Neurol. 52, 335–341. doi: 10.1002/ana.10294
De Jong, G. I., Farkas, E., Stienstra, C. M., Plass, J. R. M., Keijser, J. N., De La Torre, J. C., et al. (1999). Cerebral hypoperfusion yields capillary damage in the hippocampal CA1 area that correlates with spatial memory impairment. Neuroscience 91, 203–210. doi: 10.1016/S0306-4522(98)00659-9
De Keyser, J., Mostert, J. P., and Koch, M. W. (2008). Dysfunctional astrocytes as key players in the pathogenesis of central nervous system disorders. J. Neurol. Sci. 267, 3–16. doi: 10.1016/j.jns.2007.08.044
Derecki, N. C., Cardani, A. N., Yang, C. H., Quinnies, K. M., Crihfield, A., Lynch, K. R., et al. (2010). Regulation of learning and memory by meningeal immunity: A key role for IL-4. J. Exp. Med. 207, 1067–1080. doi: 10.1084/jem.20091419
Dezonne, R. S., Stipursky, J., Araujo, A. P. B., Nones, J., Pavão, M. S. G., Porcionatto, M., et al. (2013). Thyroid hormone treated astrocytes induce maturation of cerebral cortical neurons through modulation of proteoglycan levels. Front. Cell. Neurosci. 7:125. doi: 10.3389/fncel.2013.00125
Di Giorgio, F. P., Carrasco, M. A., Siao, M. C., Maniatis, T., and Eggan, K. (2007). Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS model. Nat. Neurosci. 10, 608–614. doi: 10.1038/nn1885
Diehl-Schmid, J., Grimmer, T., Drzezga, A., Bornschein, S., Riemenschneider, M., Förstl, H., et al. (2007). Decline of cerebral glucose metabolism in frontotemporal dementia: a longitudinal 18F-FDG-PET-study. Neurobiol. Aging 28, 42–50. doi: 10.1016/j.neurobiolaging.2005.11.002
Diniz, L. P., Almeida, J. C., Tortelli, V., Lopes, C. V., Setti-Perdigão, P., Stipursky, J., et al. (2012). Astrocyte-induced synaptogenesis is mediated by transforming growth factor β signaling through modulation of d-serine levels in cerebral cortex neurons. J. Biol. Chem. 287, 41432–41445. doi: 10.1074/jbc.M112.380824
Diniz, L. P., Matias, I. C. P., Garcia, M. N., and Gomes, F. C. A. (2014a). Astrocytic control of neural circuit formation: Highlights on TGF-beta signaling. Neurochem. Int. 78, 18–27. doi: 10.1016/j.neuint.2014.07.008
Diniz, L. P., Tortelli, V., Garcia, M. N., Araújo, A. P. B., Melo, H. M., Seixas, et al. (2014b). Astrocyte transforming growth factor beta 1 promotes inhibitory synapse formation via CaM kinase II signaling. Glia 62, 1917–1931. doi: 10.1002/glia.22713
Diniz, L. P., Tortelli, V., Matias, I., Morgado, J., Araujo, A. P. B., Melo, H. M., et al. (2017). Astrocyte transforming growth factor beta 1 protects synapses against Aβ oligomers in Alzheimer’s disease model. J. Neurosci. 37, 6797–6809. doi: 10.1523/JNEUROSCI.3351-16.2017
Dirnagl, U. (2012). Pathobiology of injury after stroke: The neurovascular unit and beyond. Ann. N. Y. Acad. Sci. 1268, 21–25. doi: 10.1111/j.1749-6632.2012.06691.x
Dixon, R. M., Mellor, J. R., and Hanley, J. G. (2009). PICK1-mediated glutamate receptor subunit 2 (GluR2) trafficking contributes to cell death in oxygen/glucose-deprivedhippocampal neurons. J. Biol. Chem. 284, 14230–14235. doi: 10.1074/jbc.M901203200
Domercq, M., Perez-Samartin, A., Aparicio, D., Alberdi, E., Pampliega, O., and Matute, C. (2010). P2X7 receptors mediate ischemic damage to oligodendrocytes. Glia 58, 730–740. doi: 10.1002/glia.20958
Dong, Y. F., Kataoka, K., Toyama, K., Sueta, D., Koibuchi, N., Yamamoto, E., et al. (2011). Attenuation of brain damage and cognitive impairment by direct renin inhibition in mice with chronic cerebral hypoperfusion. Hypertension 58, 635–642. doi: 10.1161/HYPERTENSIONAHA.111.173534
Drake, C. T., and Iadecola, C. (2007). The role of neuronal signaling in controlling cerebral blood flow. Brain Lang. 102, 141–152. doi: 10.1016/j.bandl.2006.08.002
Drouin-Ouellet, J., Sawiak, S. J., Cisbani, G., Lagacé, M., Kuan, W. L., Saint-Pierre, M., et al. (2015). Cerebrovascular and blood-brain barrier impairments in Huntington’s disease: Potential implications for its pathophysiology. Ann. Neurol. 78, 160–177. doi: 10.1002/ana.24406
Duchen, L. W. (1992). “General pathology of neurons and neuroglia,” in Greenfield’s neuropathology, eds J. H. Adams and L. W. Duchen (New York, NY: Oxford University Press), 1–68.
Dudvarski Stankovic, N., Teodorczyk, M., Ploen, R., Zipp, F., and Schmidt, M. H. H. (2016). Microglia–blood vessel interactions: a double-edged sword in brain pathologies. Acta Neuropathol. 131, 347–363. doi: 10.1007/s00401-015-1524-y
Duvernoy, H., Delon, S., and Vannson, J. L. (1983). The vascularization of the human cerebellar cortex. Brain Res. Bull. 11, 419–480. doi: 10.1016/0361-9230(83)90116-8
Edison, P., Ahmed, I., Fan, Z., Hinz, R., Gelosa, G., Ray Chaudhuri, K., et al. (2013). Microglia, amyloid, and glucose metabolism in parkinson’s disease with and without dementia. Neuropsychopharmacology 38, 938–949. doi: 10.1038/npp.2012.255
Eltzschig, H. K., Sitkovsky, M. V., and Robson, S. C. (2012). Purinergic signaling during inflammation. N. Engl. J. Med. 367, 2322–2333. doi: 10.1056/NEJMra1205750
Engl, E., and Attwell, D. (2015). Non-signalling energy use in the brain. J. Physiol. 593, 3417–3429. doi: 10.1113/jphysiol.2014.282517
Farkas, E., Donka, G., de Vos, R. A. I., Mihály, A., Bari, F., and Luiten, P. G. M. (2004). Experimental cerebral hypoperfusion induces white matter injury and microglial activation in the rat brain. Acta Neuropathol. 108, 57–64. doi: 10.1007/s00401-004-0864-9
Farkas, E., Institóris, Á, Domoki, F., Mihály, A., and Bari, F. (2006). The effect of pre- and posttreatment with diazoxide on the early phase of chronic cerebral hypoperfusion in the rat. Brain Res. 1087, 168–174. doi: 10.1016/j.brainres.2006.02.134
Farkas, E., Luiten, P. G. M., and Bari, F. (2007). Permanent, bilateral common carotid artery occlusion in the rat: A model for chronic cerebral hypoperfusion-related neurodegenerative diseases. Brain Res. Rev. 54, 162–180. doi: 10.1016/j.brainresrev.2007.01.003
Figley, C. R. (2011). Lactate transport and metabolism in the human brain: Implications for the astrocyte-neuron lactate shuttle hypothesis. J. Neurosci. 31, 4768–4770. doi: 10.1523/JNEUROSCI.6612-10.2011
Flowers, A., Bell-Temin, H., Jalloh, A., Stevens, S. M., and Bickford, P. C. (2017). Proteomic anaysis of aged microglia: Shifts in transcription, bioenergetics, and nutrient response. J. Neuroinfl. 14:96. doi: 10.1186/s12974-017-0840-7
Franco, R., and Fernández-Suárez, D. (2015). Alternatively activated microglia and macrophages in the central nervous system. Prog. Neurobiol. 131, 65–86. doi: 10.1016/j.pneurobio.2015.05.003
Franklin, R. J. M., and Ffrench-Constant, C. (2008). Remyelination in the CNS: From biology to therapy. Nat. Rev. Neurosci. 9, 839–855. doi: 10.1038/nrn2480
Freeman, L. R., and Keller, J. N. (2012). Oxidative stress and cerebral endothelial cells: Regulation of the blood-brain-barrier and antioxidant based interventions. Biochim. Biophys. Acta Mol. Basis Dis. 1822, 822–829. doi: 10.1016/j.bbadis.2011.12.009
Fricker, M., Neher, J. J., Zhao, J. W., Théry, C., Tolkovsky, A. M., and Brown, G. C. (2012). MFG-E8 mediates primary phagocytosis of viable neurons during neuroinflammation. J. Neurosci. 32, 2657–2666. doi: 10.1523/JNEUROSCI.4837-11.2012
Friede, R. L., and van Houten, W. H. (1961). Relations between post-mortem alterations and glycolytic metabolism in the brain. Exp. Neurol. 4, 197–204. doi: 10.1016/0014-4886(61)90041-3
Frost, J. L., and Schafer, D. P. (2016). Microglia: Architects of the Developing Nervous System. Trends Cell Biol. 26, 587–597. doi: 10.1016/j.tcb.2016.02.006
Fusco, I., Ugolini, F., Lana, D., Coppi, E., Dettori, I., Gaviano, L., et al. (2018). The selective antagonism of adenosine A2B receptors reduces the synaptic failure and neuronal death induced by oxygen and glucose deprivation in rat CA1 hippocampus in vitro. Front. Pharmacol. 9:399. doi: 10.3389/fphar.2018.00399
Gaberel, T., Gakuba, C., Goulay, R., De Lizarrondo, S. M., Hanouz, J. L., Emery, E., et al. (2014). Impaired glymphatic perfusion after strokes revealed by contrast-enhanced MRI: A new target for fibrinolysis? Stroke 45, 3092–3096. doi: 10.1161/STROKEAHA.114.006617
Gee, C. E., Benquet, P., Raineteau, O., Rietschin, L., Kirbach, S. W., and Gerber, U. (2006). NMDA receptors and the differential ischemic vulnerability of hippocampal neurons. Eur. J. Neurosci. 23, 2595–2603. doi: 10.1111/j.1460-9568.2006.04786.x
Gelderblom, M., Sobey, C. G., Kleinschnitz, C., and Magnus, T. (2015). Danger signals in stroke. Ageing Res. Rev. 24, 77–82. doi: 10.1016/j.arr.2015.07.004
Glass, C. K., Saijo, K., Winner, B., Marchetto, M. C., and Gage, F. H. (2010). Mechanisms Underlying Inflammation in Neurodegeneration. Cell 140, 918–934. doi: 10.1016/j.cell.2010.02.016
Gomes-Leal, W. (2012). Microglial physiopathology: How to explain the dual role of microglia after acute neural disorders? Brain Behav. 2, 345–356. doi: 10.1002/brb3.51
Gorelick, P. B., Scuteri, A., Black, S. E., Decarli, C., Greenberg, S. M., Iadecola, C., et al. (2011). Vascular contributions to cognitive impairment and dementia: A statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 42, 2672–2713. doi: 10.1161/STR.0b013e3182299496
Govindpani, K., McNamara, L. G., Smith, N. R., Vinnakota, C., Waldvogel, H. J., Faull, R. L., et al. (2019). Vascular Dysfunction in Alzheimer’s Disease: A Prelude to the Pathological Process or a Consequence of It? J. Clin. Med. 8:651. doi: 10.3390/jcm8050651
Grabert, K., Michoel, T., Karavolos, M. H., Clohisey, S., Kenneth Baillie, J., Stevens, M. P., et al. (2016). Microglial brain regionâ ’dependent diversity and selective regional sensitivities to aging. Nat. Neurosci. 19, 504–516. doi: 10.1038/nn.4222
Guan, J., Pavlovic, D., Dalkie, N., Waldvogel, H. J., O’Carroll, S. J., Green, C. R., et al. (2013). Vascular degeneration in parkinsons disease. Brain Pathol. 23, 154–164. doi: 10.1111/j.1750-3639.2012.00628.x
Guo, S., Kim, W. J., Lok, J., Lee, S. R., Besancon, E., Luo, B. H., et al. (2008). Neuroprotection via matrix-trophic coupling between cerebral endothelial cells and neurons. Proc. Natl. Acad. Sci. U S A. 105, 7582–7587. doi: 10.1073/pnas.0801105105
Guo, Z., Zhang, L., Wu, Z., Chen, Y., Wang, F., and Chen, G. (2014). In vivo direct reprogramming of reactive glial cells into functional neurons after brain injury and in an Alzheimer’s disease model. Cell Stem Cell 14, 188–202. doi: 10.1016/j.stem.2013.12.001
Gupta, S., Agrawal, A., Agrawal, S., Su, H., and Gollapudi, S. (2006). A paradox of immunodeficiency and inflammation in human aging: Lessons learned from apoptosis. Immun. Ageing 3:5. doi: 10.1186/1742-4933-3-5
Haj-Yasein, N. N., Vindedal, G. F., Eilert-Olsen, M., Gundersen, G. A., Skare, Ø, Laake, P., et al. (2011). Glial-conditional deletion of aquaporin-4 (Aqp4) reduces blood-brain water uptake and confers barrier function on perivascular astrocyte endfeet. Proc. Natl. Acad. Sci. U S A 108, 17815–17820. doi: 10.1073/pnas.1110655108
Halder, S. K., and Milner, R. (2019). A critical role for microglia in maintaining vascular integrity in the hypoxic spinal cord. Proc. Natl. Acad. Sci. U S A. 116, 26029–26037. doi: 10.1073/pnas.1912178116
Hanisch, U. K., and Kettenmann, H. (2007). Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 10, 1387–1394. doi: 10.1038/nn1997
Harris, J. J., Jolivet, R., and Attwell, D. (2012). Synaptic Energy Use and Supply. Neuron 75, 762–777. doi: 10.1016/j.neuron.2012.08.019
Harrison, J. K., Jiang, Y., Chen, S., Xia, Y., Maciejewski, D., Mcnamara, R. K., et al. (1998). Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc. Natl. Acad. Sci. U S A. 95, 10896–10901. doi: 10.1073/pnas.95.18.10896
Hase, Y., Craggs, L., Hase, M., Stevenson, W., Slade, J., Lopez, D., et al. (2017). Effects of environmental enrichment on white matter glial responses in a mouse model of chronic cerebral hypoperfusion. J. Neuroinfl. 14:81. doi: 10.1186/s12974-017-0850-5
Heneka, M. T., Rodríguez, J. J., and Verkhratsky, A. (2010). Neuroglia in neurodegeneration. Brain Res. Rev. 63, 189–211. doi: 10.1016/j.brainresrev.2009.11.004
Hickman, S., Izzy, S., Sen, P., Morsett, L., and El Khoury, J. (2018). Microglia in neurodegeneration. Nat. Neurosci. 21, 1359–1369. doi: 10.1038/s41593-018-0242-x
Hirayama, Y., Ikeda-Matsuo, Y., Notomi, S., Enaida, H., Kinouchi, H., and Koizumi, S. (2015). Astrocyte-mediated ischemic tolerance. J. Neurosci. 35, 3794–3805. doi: 10.1523/JNEUROSCI.4218-14.2015
Hirt, L., Ternon, B., Price, M., Mastour, N., Brunet, J. F., and Badaut, J. (2009). Protective role of early Aquaporin 4 induction against postischemic edema formation. J. Cereb. Blood Flow Metab. 29, 423–433. doi: 10.1038/jcbfm.2008.133
Holland, P. R., Searcy, J. L., Salvadores, N., Scullion, G., Chen, G., Lawson, G., et al. (2015). Gliovascular disruption and cognitive deficits in a mouse model with features of small vessel disease. J. Cereb. Blood Flow Metab. 35, 1005–1014. doi: 10.1038/jcbfm.2015.12
Holmseth, S., Dehnes, Y., Huang, Y. H., Follin-Arbelet, V. V., Grutle, N. J., Mylonakou, M. N., et al. (2012). The density of EAAC1 (EAAT3) glutamate transporters expressed by neurons in the mammalian CNS. J. Neurosci. 32, 6000–6013. doi: 10.1523/JNEUROSCI.5347-11.2012
Hong, S., Beja-Glasser, V. F., Nfonoyim, B. M., Frouin, A., Li, S., Ramakrishnan, S., et al. (2016). Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716. doi: 10.1126/science.aad8373
Hoshi, A., Yamamoto, T., Shimizu, K., Sugiura, Y., and Ugawa, Y. (2011). Chemical preconditioning-induced reactive astrocytosis contributes to the reduction of post-ischemic edema through aquaporin-4 downregulation. Exp. Neurol. 227, 89–95. doi: 10.1016/j.expneurol.2010.09.016
Hoyer, S. (1993). Abnormalities in Brain Glucose Utilization and Its Impact on Cellular and Molecular Mechanisms in Sporadic Dementia of Alzheimer Type. Ann. N. Y. Acad. Sci. 695, 77–80. doi: 10.1111/j.1749-6632.1993.tb23032.x
Hsieh, C. L., Koike, M., Spusta, S. C., Niemi, E. C., Yenari, M., Nakamura, M. C., et al. (2009). A role for TREM2 ligands in the phagocytosis of apoptotic neuronal cells by microglia. J. Neurochem. 109, 1144–1156. doi: 10.1111/j.1471-4159.2009.06042.x
Hu, X., Leak, R. K., Shi, Y., Suenaga, J., Gao, Y., Zheng, P., et al. (2015). Microglial and macrophage polarization - New prospects for brain repair. Nat. Rev. Neurol. 11, 56–64. doi: 10.1038/nrneurol.2014.207
Huang, Y., Zhang, W., Lin, L., Feng, J., Chen, F., Wei, W., et al. (2010). Is endothelial dysfunction of cerebral small vessel responsible for white matter lesions after chronic cerebral hypoperfusion in rats? J. Neurol. Sci. 299, 72–80. doi: 10.1016/j.jns.2010.08.035
Hug, H., Mohajeri, M. H., and La Fata, G. (2018). Toll-like receptors: Regulators of the immune response in the human gut. Nutrients 10:203. doi: 10.3390/nu10020203
Huizinga, R., van der Star, B. J., Kipp, M., Jong, R., Gerritsen, W., Clarner, T., et al. (2012). Phagocytosis of neuronal debris by microglia is associated with neuronal damage in multiple sclerosis. Glia 60, 422–431. doi: 10.1002/glia.22276
Hulse, R. E., Winterfield, J., Kunkler, P., and Kraig, R. P. (2001). Astrocytic Clasmatodendrosis in Hippocampal Organ Culture. Glia 33, 169–179. doi: 10.1002/1098-1136(200102)33:2<169::AID-GLIA1016<3.0.CO;2-B
Hyder, F., Rothman, D. L., and Bennett, M. R. (2013). Cortical energy demands of signaling and nonsignaling components in brain are conserved across mammalian species and activity levels. Proc. Natl. Acad. Sci. U S A. 110, 3549–3554. doi: 10.1073/pnas.1214912110
Iadecola, C. (2004). Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat. Rev. Neurosci. 5, 347–360. doi: 10.1038/nrn1387
Iadecola, C. (2010). The overlap between neurodegenerative and vascular factors in the pathogenesis of dementia. Acta Neuropathol. 120, 287–296. doi: 10.1007/s00401-010-0718-6
Iadecola, C. (2013). The Pathobiology of Vascular Dementia. Neuron 80, 844–866. doi: 10.1016/j.neuron.2013.10.008
Iadecola, C., Zhang, F., Niwa, K., Eckman, C., Turner, S. K., Fischer, E., et al. (1999). SOD1 rescues cerebral endothelial dysfunction in mice overexpressing amyloid precursor protein. Nat. Neurosci. 2, 157–161. doi: 10.1038/5715
Ihara, M., Tomimoto, H., Kinoshita, M., Oh, J., Noda, M., Wakita, H., et al. (2001). Chronic cerebral hypoperfusion induces MMP-2 but not MMP-9 expression in the microglia and vascular endothelium of white matter. J. Cereb. Blood Flow Metab. 21, 828–834. doi: 10.1097/00004647-200107000-00008
Iliff, J. J., Chen, M. J., Plog, B. A., Zeppenfeld, D. M., Soltero, M., Yang, L., et al. (2014). Impairment of glymphatic pathway function promotes tau pathology after traumatic brain injury. J. Neurosci. 34, 16180–16193. doi: 10.1523/JNEUROSCI.3020-14.2014
Iliff, J. J., Wang, M., Liao, Y., Plogg, B. A., Peng, W., Gundersen, G. A., et al. (2012). A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci. Transl. Med. 4:147ra111. doi: 10.1126/scitranslmed.3003748
Iliff, J. J., Wang, M., Zeppenfeld, D. M., Venkataraman, A., Plog, B. A., Liao, Y., et al. (2013). Cerebral arterial pulsation drives paravascular CSF-Interstitial fluid exchange in the murine brain. J. Neurosci. 33, 18190–18199. doi: 10.1523/JNEUROSCI.1592-13.2013
Iram, T., Ramirez-Ortiz, Z., Byrne, M. H., Coleman, U. A., Kingery, N. D., Means, T. K., et al. (2016). Megf10 Is a receptor for C1Q that mediates clearance of apoptotic cells by astrocytes. J. Neurosci. 36, 5185–5192. doi: 10.1523/JNEUROSCI.3850-15.2016
Jabaudon, D., Scanziani, M., Gähwiler, B. H., and Gerber, U. (2000). Acute decrease in net glutamate uptake during energy deprivation. Proc. Natl. Acad. Sci. U S A. 97, 5610–5615. doi: 10.1073/pnas.97.10.5610
Jessen, N. A., Munk, A. S. F., Lundgaard, I., and Nedergaard, M. (2015). The Glymphatic System: A Beginner’s Guide. Neurochem. Res. 40, 2583–2599. doi: 10.1007/s11064-015-1581-6
Jiang, X., Andjelkovic, A. V., Zhu, L., Yang, T., Bennett, M. V. L., Chen, J., et al. (2018). Blood-brain barrier dysfunction and recovery after ischemic stroke. Prog. Neurobiol. 16, 144–171. doi: 10.1016/j.pneurobio.2017.10.001
Jiang, X., Pu, H., Hu, X., Wei, Z., Hong, D., Zhang, W., et al. (2016). A Post-stroke Therapeutic Regimen with Omega-3 Polyunsaturated Fatty Acids that Promotes White Matter Integrity and Beneficial Microglial Responses after Cerebral Ischemia. Transl. Stroke Res. 7, 548–561. doi: 10.1007/s12975-016-0502-6
Johnston, S. C., O’Meara, E. S., Manolio, T. A., Lefkowitz, D., O’Leary, D. H., Goldstein, S., et al. (2004). Cognitive Impairment and Decline Are Associated with Carotid Artery Disease in Patients without Clinically Evident Cerebrovascular Disease. Ann. Intern. Med. 140, 237–247. doi: 10.7326/0003-4819-140-4-200402170-00005
Jolivel, V., Bicker, F., Binamé, F., Ploen, R., Keller, S., Gollan, R., et al. (2015). Perivascular microglia promote blood vessel disintegration in the ischemic penumbra. Acta Neuropathol. 129, 279–295. doi: 10.1007/s00401-014-1372-1
Juma, W. M., Lira, A., Marzuk, A., Marzuk, Z., Hakim, A. M., and Thompson, C. S. (2011). C-reactive protein expression in a rodent model of chronic cerebral hypoperfusion. Brain Res. 1414, 85–93. doi: 10.1016/j.brainres.2011.07.047
Kadar, T., Dachir, S., Shukitt-Hale, B., and Levy, A. (1998). Sub-regional hippocampal vulnerability in various animal models leading to cognitive dysfunction. J. Neural Transm. 105, 987–1004. doi: 10.1007/s007020050107
Kanak, D. J., Rose, G. M., Zaveri, H. P., and Patrylo, P. R. (2013). Altered Network Timing in the CA3-CA1 Circuit of Hippocampal Slices from Aged Mice. PLoS One 8:e61364. doi: 10.1371/journal.pone.0061364
Kao, A. W., Eisenhut, R. J., Martens, L. H., Nakamura, A., Huang, A., Bagley, J. A., et al. (2011). A neurodegenerative disease mutation that accelerates the clearance of apoptotic cells. Proc. Natl. Acad. Sci. U S A. 108, 4441–4446. doi: 10.1073/pnas.1100650108
Kelly, P., Denver, P., Satchell, S. C., Ackermann, M., Konerding, M. A., and Mitchell, C. A. (2017). Microvascular ultrastructural changes precede cognitive impairment in the murine APPswe/PS1dE9 model of Alzheimer’s disease. Angiogenesis 20, 567–580. doi: 10.1007/s10456-017-9568-3
Keren-Shaul, H., Spinrad, A., Weiner, A., Matcovitch-Natan, O., Dvir-Szternfeld, R., Ulland, T. K., et al. (2017). A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 169, 1276.e–1290.e. doi: 10.1016/j.cell.2017.05.018
Kerr, J. F. R., Wyllie, A. H., and Currie, A. R. (1972). Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 26, 239–257. doi: 10.1038/bjc.1972.33
Kesner, R. P., Lee, I., and Gilbert, P. (2004). A behavioral assessment of hippocampal function based on a subregional analysis. Rev. Neurosci. 15, 333–351. doi: 10.1515/REVNEURO.2004.15.5.333
Kettenmann, H., Hanisch, U. K., Noda, M., and Verkhratsky, A. (2011). Physiology of microglia. Physiol. Rev. 91, 461–553. doi: 10.1152/physrev.00011.2010
Kettenmann, H., Kirchhoff, F., and Verkhratsky, A. (2013). Microglia: New Roles for the Synaptic Stripper. Neuron 77, 10–18. doi: 10.1016/j.neuron.2012.12.023
Khakh, B. S., and Deneen, B. (2019). The Emerging Nature of Astrocyte Diversity. Annu. Rev. Neurosci. 42, 187–207. doi: 10.1146/annurev-neuro-070918-050443
Khakh, B. S., and Sofroniew, M. V. (2015). Diversity of astrocyte functions and phenotypes in neural circuits. Nat. Neurosci. 18, 942–952. doi: 10.1038/nn.4043
Kim, D. S., Kwak, S. E., Kim, J. E., Jung, J. Y., Won, M. H., Choi, S. Y., et al. (2006). Transient ischaemia affects plasma membrane glutamate transporter, not vesicular glutamate transporter, expressions in the gerbil hippocampus. J. Vet. Med. Ser. C Anat. Histol. Embryol. 35, 265–270. doi: 10.1111/j.1439-0264.2005.00677.x
Kim, J. E., Ryu, H. J., Yeo, S., Il, and Kang, T. C. (2011). P2X7 receptor differentially modulates astroglial apoptosis and clasmatodendrosis in the rat brain following status epilepticus. Hippocampus 21, 1318–1333. doi: 10.1002/hipo.20850
Kimelberg, H. K. (1995). Current concepts of brain edema: Review of laboratory investigations. J. Neurosurg. 83, 1051–1059. doi: 10.3171/jns.1995.83.6.1051
Kimelberg, H. K. (2005). Astrocytic swelling in cerebral ischemia as a possible cause of injury and target for therapy. Glia 50, 389–397. doi: 10.1002/glia.20174
Kirino, T. (1982). Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res. 239, 57–69. doi: 10.1016/0006-8993(82)90833-2
Kirino, T. (2000). Delayed neuronal death. Neuropathology 20, 95–97. doi: 10.1046/j.1440-1789.2000.00306.x
Kitagawa, K., Matsumoto, M., Yang, G., Mabuchi, T., Yagita, Y., Hori, M., et al. (1998). Cerebral ischemia after bilateral carotid artery occlusion and intraluminal suture occlusion in mice: Evaluation of the patency of the posterior communicating artery. J. Cereb. Blood Flow Metab. 18, 570–579. doi: 10.1097/00004647-199805000-00012
Kivipelto, M., Ngandu, T., Laatikainen, T., Winblad, B., Soininen, H., and Tuomilehto, J. (2006). Risk score for the prediction of dementia risk in 20 years among middle aged people: a longitudinal, population-based study. Lancet Neurol. 5, 735–741. doi: 10.1016/S1474-4422(06)70537-3
Kleinfeld, D., Blinder, P., Drew, P. J., Driscoll, J. D., Muller, A., Tsai, P. S., et al. (2011). A guide to delineate the logic of neurovascular signaling in the brain. Front. Neuroenergetics 3:1–9. doi: 10.3389/fnene.2011.00001
Koizumi, S., Hirayama, Y., and Morizawa, Y. M. (2018). New roles of reactive astrocytes in the brain; an organizer of cerebral ischemia. Neurochem. Int. 119, 107–114. doi: 10.1016/j.neuint.2018.01.007
Koizumi, S., Shigemoto-Mogami, Y., Nasu-Tada, K., Shinozaki, Y., Ohsawa, K., Tsuda, M., et al. (2007). UDP acting at P2Y6 receptors is a mediator of microglial phagocytosis. Nature 446, 1091–1095. doi: 10.1038/nature05704
Kong, J., Shepel, P. N., Holden, C. P., Mackiewicz, M., Pack, A. I., and Geiger, J. D. (2002). Brain glycogen decreases with increased periods of wakefulness: Implications for homeostatic drive to sleep. J. Neurosci. 22, 5581–5587. doi: 10.1523/jneurosci.22-13-05581.2002
Kraig, R. P., and Chesler, M. (1990). Astrocytic acidosis in hyperglycemic and complete ischemia. J. Cereb. Blood Flow Metab. 10, 104–114. doi: 10.1038/jcbfm.1990.13
Krasemann, S., Madore, C., Cialic, R., Baufeld, C., Calcagno, N., El Fatimy, R., et al. (2017). The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 47, 566.e–581.e. doi: 10.1016/j.immuni.2017.08.008
Kress, B. T., Iliff, J. J., Xia, M., Wang, M., Wei, Bs, H. S., et al. (2014). Impairment of paravascular clearance pathways in the aging brain. Ann. Neurol. 76, 845–861. doi: 10.1002/ana.24271
Kucukdereli, H., Allen, N. J., Lee, A. T., Feng, A., Ozlu, M. I., Conatser, L. M., et al. (2011). Control of excitatory CNS synaptogenesis by astrocyte-secreted proteins hevin and SPARC. Proc. Natl. Acad. Sci. U S A. 108, E440–E449. doi: 10.1073/pnas.1104977108
Lana, D., Cerbai, F., Di Russo, J., Boscaro, F., Giannetti, A., Petkova-Kirova, P., et al. (2013). Hippocampal long term memory: Effect of the cholinergic system on local protein synthesis. Neurobiol. Learn. Mem. 106, 246–257. doi: 10.1016/j.nlm.2013.09.013
Lana, D., Iovino, L., Nosi, D., Wenk, G. L., and Giovannini, M. G. (2016). The neuron-astrocyte-microglia triad involvement in neuroinflammaging mechanisms in the CA3 hippocampus of memory-impaired aged rats. Exp. Gerontol. 83, 71–88. doi: 10.1016/j.exger.2016.07.011
Lana, D., Melani, A., Pugliese, A. M., Cipriani, S., Nosi, D., Pedata, F., et al. (2014). The neuron-astrocyte-microglia triad in a rat model of chronic cerebral hypoperfusion: Protective effect of dipyridamole. Front. Aging Neurosci. 6:322. doi: 10.3389/fnagi.2014.00322
Lana, D., Ugolini, F., Melani, A., Nosi, D., Pedata, F., and Giovannini, M. G. (2017a). The neuron-astrocyte-microglia triad in CA3 after chronic cerebral hypoperfusion in the rat: Protective effect of dipyridamole. Exp. Gerontol. 96, 46–62. doi: 10.1016/j.exger.2017.06.006
Lana, D., Ugolini, F., Nosi, D., Wenk, G. L., and Giovannini, M. G. (2017b). Alterations in the interplay between neurons, astrocytes and microglia in the rat dentate gyrus in experimental models of neurodegeneration. Front. Aging Neurosci. 9:296. doi: 10.3389/fnagi.2017.00296
Landis, D. M. D. (1994). The Early Reactions of Non-Neuronal Cells to Brain Injury. Annu. Rev. Neurosci. 17, 133–151. doi: 10.1146/annurev.ne.17.030194.001025
Lee, I., and Kesner, R. P. (2002). Differential contribution of NMDA receptors in hippocampal subregions to spatial working memory. Nat. Neurosci. 5, 162–168. doi: 10.1038/nn790
Lee, I., and Kesner, R. P. (2003). Differential roles of dorsal hippocampal subregions in spatial working memory with short versus intermediate delay. Behav. Neurosci. 117, 1044–1053. doi: 10.1037/0735-7044.117.5.1044
Leys, D., Hénon, H., Mackowiak-Cordoliani, M. A., and Pasquier, F. (2005). Poststroke dementia. Lancet Neurol. 4, 752–759. doi: 10.1016/S1474-4422(05)70221-0
Li, K., Li, J., Zheng, J., and Qin, S. (2019). Reactive Astrocytes in Neurodegenerative Diseases. Aging Dis. 10:664. doi: 10.14336/ad.2018.0720
Liddelow, S. A., and Barres, B. A. (2017). Reactive Astrocytes: Production. Function, and Therapeutic Potential. Immunity 46, 957–967. doi: 10.1016/j.immuni.2017.06.006
Liddelow, S. A., Guttenplan, K. A., Clarke, L. E., Bennett, F. C., Bohlen, C. J., Schirmer, L., et al. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. doi: 10.1038/nature21029
Liesz, A., Suri-Payer, E., Veltkamp, C., Doerr, H., Sommer, C., Rivest, S., et al. (2009). Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat. Med. 15, 192–199. doi: 10.1038/nm.1927
Liu, H. X., Zhang, J. J., Zheng, P., and Zhang, Y. (2005). Altered expression of MAP-2, GAP-43, and synaptophysin in the hippocampus of rats with chronic cerebral hypoperfusion correlates with cognitive impairment. Mol. Brain Res. 139, 169–177. doi: 10.1016/j.molbrainres.2005.05.014
Lobsiger, C. S., Boillée, S., and Cleveland, D. W. (2007). Toxicity from different SOD1 mutants dysregulates the complement system and the neuronal regenerative response in ALS motor neurons. Proc. Natl. Acad. Sci. U S A. 104, 7319–7326. doi: 10.1073/pnas.0702230104
Lu, Z., Elliott, M. R., Chen, Y., Walsh, J. T., Klibanov, A. L., Ravichandran, K. S., et al. (2011). Phagocytic activity of neuronal progenitors regulates adult neurogenesis. Nat. Cell Biol. 13, 1076–1084. doi: 10.1038/ncb2299
Ma, J., Zhang, J., Hou, W. W., Wu, X. H., Liao, R. J., Chen, Y., et al. (2015). Early treatment of minocycline alleviates white matter and cognitive impairments after chronic cerebral hypoperfusion. Sci. Rep. 5:12079. doi: 10.1038/srep12079
Macvicar, B. A., and Newman, E. A. (2015). Astrocyte regulation of blood flow in the brain. Cold Spring Harb. Perspect. Biol. 7, 1–15. doi: 10.1101/cshperspect.a020388
Magnus, T., Chan, A., Grauer, O., Toyka, K. V., and Gold, R. (2001). Microglial Phagocytosis of Apoptotic Inflammatory T Cells Leads to Down-Regulation of Microglial Immune Activation. J. Immunol. 167, 5004–5010. doi: 10.4049/jimmunol.167.9.5004
Manley, G. T., Fujimura, M., Ma, T., Noshita, N., Filiz, F., Bollen, A. W., et al. (2000). Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat. Med. 6, 159–163. doi: 10.1038/72256
Manso, Y., Holland, P. R., Kitamura, A., Szymkowiak, S., Duncombe, J., Hennessy, E., et al. (2018). Minocycline reduces microgliosis and improves subcortical white matter function in a model of cerebral vascular disease. Glia 66, 34–46. doi: 10.1002/glia.23190
Maraula, G., Traini, C., Mello, T., Coppi, E., Galli, A., Pedata, F., et al. (2013). Effects of oxygen and glucose deprivation on synaptic transmission in rat dentate gyrus: Role of A2A adenosine receptors. Neuropharmacology 67, 511–520. doi: 10.1016/j.neuropharm.2012.12.002
Mariani, M. M., and Kielian, T. (2009). Microglia in infectious diseases of the central nervous system. J. Neuroimmune Pharmacol. 4, 448–461. doi: 10.1007/s11481-009-9170-6
Márquez-Ropero, M., Benito, E., Plaza-Zabala, A., and Sierra, A. (2020). Microglial Corpse Clearance: Lessons From Macrophages. Front. Immunol. 11:506. doi: 10.3389/fimmu.2020.00506
Marshall, R. S., Festa, J. R., Cheung, Y. K., Chen, R., Pavol, M. A., Derdeyn, C. P., et al. (2012). Cerebral hemodynamics and cognitive impairment: Baseline data from the RECON trial. Neurology 78, 250–255. doi: 10.1212/WNL.0b013e31824365d3
Marshall, R. S., Lazar, R. M., Mohr, J. P., Pile-Spellman, J., Hacein-Bey, L., Duong, D. H., et al. (1999). Higher cerebral function and hemispheric blood flow during awake carotid artery balloon test occlusions. J. Neurol. Neurosurg. Psychiatry 66, 734–738. doi: 10.1136/jnnp.66.6.734
Martín-López, E., García-Marques, J., Núñez-Llaves, R., and López-Mascaraque, L. (2013). Clonal Astrocytic Response to Cortical Injury. PLoS One 8:74039. doi: 10.1371/journal.pone.0074039
Masumura, M., Hata, R., Nagai, Y., and Sawada, T. (2001). Oligodendroglial cell death with DNA fragmentation in the white matter under chronic cerebral hypoperfusion: Comparison between normotensive and spontaneously hypertensive rats. Neurosci. Res. 39, 401–412. doi: 10.1016/S0168-0102(01)00195-X
Mattiasson, G., Friberg, H., Hansson, M., Elmér, E., and Wieloch, T. (2003). Flow cytometric analysis of mitochondria from CA1 and CA3 regions of rat hippocampus reveals differences in permeability transition pore activation. J. Neurochem. 87, 532–544. doi: 10.1046/j.1471-4159.2003.02026.x
Matute, C., and Ransom, B. R. (2012). Roles of white matter in central nervous system pathophysiologies. ASN Neuro 4, 89–101. doi: 10.1042/AN20110060
Mazaheri, F., Breus, O., Durdu, S., Haas, P., Wittbrodt, J., Gilmour, D., et al. (2014). Distinct roles for BAI1 and TIM-4 in the engulfment of dying neurons by microglia. Nat. Commun. 5:4046. doi: 10.1038/ncomms5046
Medzhitov, R., and Janeway, C. A. (2002). Decoding the patterns of self and nonself by the innate immune system. Science 296, 298–300. doi: 10.1126/science.1068883
Melani, A., Cipriani, S., Corti, F., and Pedata, F. (2010). Effect of intravenous administration of dipyridamole in a rat model of chronic cerebral ischemia. Ann. N Y. Acad. Sci. 1207, 89–96. doi: 10.1111/j.1749-6632.2010.05732.x
Michell-Robinson, M. A., Touil, H., Healy, L. M., Owen, D. R., Durafourt, B. A., Bar-Or, A., et al. (2015). Roles of microglia in brain development, tissue maintenance and repair. Brain 138, 1138–1159. doi: 10.1093/brain/awv066
Miller, A. P., Shah, A. S., Aperi, B. V., Kurpad, S. N., Stemper, B. D., and Glavaski-Joksimovic, A. (2017). Acute death of astrocytes in blast-exposed rat organotypic hippocampal slice cultures. PLoS One 12:73167. doi: 10.1371/journal.pone.0173167
Milligan, E. D., and Watkins, L. R. (2009). Pathological and protective roles of glia in chronic pain. Nat. Rev. Neurosci. 10, 23–36. doi: 10.1038/nrn2533
Minghetti, L., and Levi, G. (1998). Microglia as effector cells in brain damage and repair: Focus on prostanoids and nitric oxide. Prog. Neurobiol. 54, 99–125. doi: 10.1016/S0301-0082(97)00052-X
Mitani, A., Andou, Y., and Kataoka, K. (1992). Selective vulnerability of hippocampal CA1 neurons cannot be explained in terms of an increase in glutamate concentration during ischemia in the gerbil: Brain microdialysis study. Neuroscience 48, 307–313. doi: 10.1016/0306-4522(92)90492-K
Miyazaki, K., Ohta, Y., Nagai, M., Morimoto, N., Kurata, T., Takehisa, Y., et al. (2011). Disruption of neurovascular unit prior to motor neuron degeneration in amyotrophic lateral sclerosis. J. Neurosci. Res. 89, 718–728. doi: 10.1002/jnr.22594
Mizuseki, K., Royer, S., Diba, K., and Buzsáki, G. (2012). Activity dynamics and behavioral correlates of CA3 and CA1 hippocampal pyramidal neurons. Hippocampus 22, 1659–1680. doi: 10.1002/hipo.22002
Montero, M., González, B., and Zimmer, J. (2009). Immunotoxic depletion of microglia in mouse hippocampal slice cultures enhances ischemia-like neurodegeneration. Brain Res. 1291, 140–152. doi: 10.1016/j.brainres.2009.06.097
Morgan, T. E., Rozovsky, I., Goldsmith, S. K., Stone, D. J., Yoshida, T., and Finch, C. E. (1997). Increased transcription of the astrocyte gene GFAP during middle-age is attenuated by food restriction: Implications for the role of oxidative stress. Free Radic. Biol. Med. 23, 524–528. doi: 10.1016/S0891-5849(97)00120-2
Morita, M., Ikeshima-Kataoka, H., Kreft, M., Vardjan, N., Zorec, R., and Noda, M. (2019). Metabolic plasticity of astrocytes and aging of the brain. Int. J. Mol. Sci. 20:941. doi: 10.3390/ijms20040941
Morizawa, Y. M., Hirayama, Y., Ohno, N., Shibata, S., Shigetomi, E., Sui, Y., et al. (2017). Reactive astrocytes function as phagocytes after brain ischemia via ABCA1-mediated pathway. Nat. Commun. 8:28. doi: 10.1038/s41467-017-00037-1
Morsch, M., Radford, R., Lee, A., Don, E. K., Badrock, A. P., Hall, T. E., et al. (2015). In vivo characterization of microglial engulfment of dying neurons in the zebrafish spinal cord. Front. Cell. Neurosci. 9:321. doi: 10.3389/fncel.2015.00321
Moskowitz, M. A., Lo, E. H., and Iadecola, C. (2010). The science of stroke: Mechanisms in search of treatments. Neuron 67, 181–198. doi: 10.1016/j.neuron.2010.07.002
Muddapu, V. R., Dharshini, S. A. P., Chakravarthy, V. S., and Gromiha, M. M. (2020). Neurodegenerative Diseases – Is Metabolic Deficiency the Root Cause? Front. Neurosci. 14:213. doi: 10.3389/fnins.2020.00213
Mueller, S. G., Schuff, N., Yaffe, K., Madison, C., Miller, B., and Weiner, M. W. (2010). Hippocampal atrophy patterns in mild cognitive impairment and alzheimer’s disease. Hum. Brain Mapp. 31, 1339–1347. doi: 10.1002/hbm.20934
Nagai, M., Re, D. B., Nagata, T., Chalazonitis, A., Jessell, T. M., Wichterle, H., et al. (2007). Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat. Neurosci. 10, 615–622. doi: 10.1038/nn1876
Nagelhus, E. A., and Ottersen, O. P. (2013). Physiological roles of Aquaporin-4 in brain. Physiol. Rev. 93, 1543–1562. doi: 10.1152/physrev.00011.2013
Nair, S., Sobotka, K. S., Joshi, P., Gressens, P., Fleiss, B., Thornton, C., et al. (2019). Lipopolysaccharide-induced alteration of mitochondrial morphology induces a metabolic shift in microglia modulating the inflammatory response in vitro and in vivo. Glia 67, 1047–1061. doi: 10.1002/glia.23587
Nakazawa, K., Sun, L. D., Quirk, M. C., Rondi-Reig, L., Wilson, M. A., and Tonegawa, S. (2003). Hippocampal CA3 NMDA receptors are crucial for memory acquisition of one-time experience. Neuron 38, 305–315. doi: 10.1016/S0896-6273(03)00165-X
Nathan, C., and Ding, A. (2010). Snapshot: Reactive oxygen intermediates (ROI). Cell 140, 951–951.e2. doi: 10.1016/j.cell.2010.03.008
Navarrete, M., Perea, G., de Sevilla, D. F., Gómez-Gonzalo, M., Núñez, A., Martín, E. D., et al. (2012). Astrocytes mediate in vivo cholinergic-induced synaptic plasticity. PLoS Biol. 10:1259. doi: 10.1371/journal.pbio.1001259
Neher, J. J., Emmrich, J. V., Fricker, M., Mander, P. K., Théry, C., and Brown, G. C. (2013). Phagocytosis executes delayed neuronal death after focal brain ischemia. Proc. Natl. Acad. Sci. U S A 110, E4098–107. doi: 10.1073/pnas.1308679110
Neumann, J. T., Thompson, J. W., Raval, A. P., Cohan, C. H., Koronowski, K. B., and Perez-Pinzon, M. A. (2015). Increased BDNF protein expression after ischemic or PKC epsilon preconditioning promotes electrophysiologic changes that lead to neuroprotection. J. Cereb. Blood Flow Metab. 35, 121–130. doi: 10.1038/jcbfm.2014.185
Neumann, J., Gunzer, M., Gutzeit, H. O., Ullrich, O., Reymann, K. G., and Dinkel, K. (2006). Microglia provide neuroprotection after ischemia. FASEB J. 20, 714–716. doi: 10.1096/fj.05-4882fje
Nguyen, M. D., Julien, J. P., and Rivest, S. (2002). Innate immunity: The missing link in neuroprotection and neurodegeneration? Nat. Rev. Neurosci. 3, 216–227. doi: 10.1038/nrn752
Nimmerjahn, A., Kirchhoff, F., and Helmchen, F. (2005). Neuroscience: Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318. doi: 10.1126/science.1110647
Noda, M., Doi, Y., Liang, J., Kawanokuchi, J., Sonobe, Y., Takeuchi, H., et al. (2011). Fractalkine attenuates excito-neurotoxicity via microglial clearance of damaged neurons and antioxidant enzyme heme oxygenase-1 expression. J. Biol. Chem. 286, 2308–2319. doi: 10.1074/jbc.M110.169839
Nones, J., Spohr, T. C. L., de, S., and Gomes, F. C. A. (2012). Effects of the flavonoid hesperidin in cerebral cortical progenitors in vitro: Indirect action through astrocytes. Int. J. Dev. Neurosci. 30, 303–313. doi: 10.1016/j.ijdevneu.2012.01.008
Norden, D. M., and Godbout, J. P. (2013). Review: Microglia of the aged brain: Primed to be activated and resistant to regulation. Neuropathol. Appl. Neurobiol. 39, 19–34. doi: 10.1111/j.1365-2990.2012.01306.x
Okada, S., Nakamura, M., Katoh, H., Miyao, T., Shimazaki, T., Ishii, K., et al. (2006). Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nat. Med. 12, 829–834. doi: 10.1038/nm1425
Orellana, J. A., Avendano, B. C., and Montero, T. D. (2014). Role of Connexins and Pannexins in Ischemic Stroke. Curr. Med. Chem. 21, 2165–2182. doi: 10.2174/0929867321666131228191714
Ouyang, Y. B., Voloboueva, L. A., Xu, L. J., and Giffard, R. G. (2007). Selective dysfunction of hippocampal CA1 astrocytes contributes to delayed neuronal damage after transient forebrain ischemia. J. Neurosci. 27, 4253–4260. doi: 10.1523/JNEUROSCI.0211-07.2007
Ozacmak, V. H., Sayan, H., Cetin, A., and Akyıldız-Igdem, A. (2007). AT1 receptor blocker candesartan-induced attenuation of brain injury of rats subjected to chronic cerebral hypoperfusion. Neurochem. Res. 32, 1314–1321. doi: 10.1007/s11064-007-9305-1
Padurariu, M., Ciobica, A., Mavroudis, I., Fotiou, D., and Baloyannis, S. (2012). Hippocampal Neuronal Loss in the CA1 and CA3 Areas of Alzheimer’s Disease Patients - PubMed. Psychiatr. Danub 24, 152–158.
Pappas, B. A., De La Torre, J. C., Davidson, C. M., Keyes, M. T., and Fortin, T. (1996). Chronic reduction of cerebral blood flow in the adult rat: Late-emerging CA1 cell loss and memory dysfunction. Brain Res. 708, 50–58. doi: 10.1016/0006-8993(95)01267-2
Park, D., Tosello-Trampont, A. C., Elliott, M. R., Lu, M., Haney, L. B., Ma, Z., et al. (2007). BAI1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature 450, 430–434. doi: 10.1038/nature06329
Pathak, D., Shields, L. Y., Mendelsohn, B. A., Haddad, D., Lin, W., Gerencser, A. A., et al. (2015). The role of mitochondrially derived ATP in synaptic vesicle recycling. J. Biol. Chem. 290, 22325–22336. doi: 10.1074/jbc.M115.656405
Pekny, M., Wilhelmsson, U., and Pekna, M. (2014). The dual role of astrocyte activation and reactive gliosis. Neurosci. Lett. 565, 30–38. doi: 10.1016/j.neulet.2013.12.071
Pekny, M., Wilhelmsson, U., Tatlisumak, T., and Pekna, M. (2019). Astrocyte activation and reactive gliosis—A new target in stroke? Neurosci. Lett. 689, 45–55. doi: 10.1016/j.neulet.2018.07.021
Pendlebury, S. T., and Rothwell, P. M. (2009). Prevalence, incidence, and factors associated with pre-stroke and post-stroke dementia: a systematic review and meta-analysis. Lancet Neurol. 8, 1006–1018. doi: 10.1016/S1474-4422(09)70236-4
Penfield, S. W. (1928). “Neuroglia and microglia - the interstitial tissue of the central nervous system,” in Special cytology: the form and functions of the cell in health and disease, ed. E. V. Cowdry (New York, NY: Hoeber), 1033–1068.
Perea, G., and Araque, A. (2007). Astrocytes potentiate transmitter release at single hippocampal synapses. Science 317, 1083–1086. doi: 10.1126/science.1144640
Peri, F., and Nüsslein-Volhard, C. (2008). Live Imaging of Neuronal Degradation by Microglia Reveals a Role for v0-ATPase a1 in Phagosomal Fusion In Vivo. Cell 133, 916–927. doi: 10.1016/j.cell.2008.04.037
Pestana, F., Edwards-Faret, G., Belgard, T. G., Martirosyan, A., and Holt, M. G. (2020). No longer underappreciated: The emerging concept of astrocyte heterogeneity in neuroscience. Brain Sci. 10:168. doi: 10.3390/brainsci10030168
Petito, C. K., Feldmann, E., Pulsinelli, W. A., and Plum, F. (1987). Delayed hippocampal damage in humans following cardiorespiratory arrest. Neurology 37, 1281–1286. doi: 10.1212/wnl.37.8.1281
Pfrieger, F. W. (2009). Roles of glial cells in synapse development. Cell. Mol. Life Sci. 66, 2037–2047. doi: 10.1007/s00018-009-0005-7
Polazzi, E., and Contestabile, A. (2002). Reciprocal interactions between microglia and neurons: From survival to neuropathology. Rev. Neurosci. 13, 221–242. doi: 10.1515/REVNEURO.2002.13.3.221
Polazzi, E., and Monti, B. (2010). Microglia and neuroprotection: From in vitro studies to therapeutic applications. Prog. Neurobiol. 92, 293–315. doi: 10.1016/j.pneurobio.2010.06.009
Polazzi, E., Gianni, T., and Contestabile, A. (2001). Microglial cells protect cerebellar granule neurons from apoptosis: Evidence for reciprocal signaling. Glia 36, 271–280. doi: 10.1002/glia.1115
Pugliese, A. M., Coppi, E., Spalluto, G., Corradetti, R., and Pedata, F. (2006). A 3 adenosine receptor antagonists delay irreversible synaptic failure caused by oxygen and glucose deprivation in the rat CA1 hippocampus in vitro. Br. J. Pharmacol. 147, 524–532. doi: 10.1038/sj.bjp.0706646
Pulsinelli, W. A., Brierley, J. B., and Plum, F. (1982). Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann. Neurol. 11, 491–498. doi: 10.1002/ana.410110509
Quaegebeur, A., Lange, C., and Carmeliet, P. (2011). The neurovascular link in health and disease: Molecular mechanisms and therapeutic implications. Neuron 71, 406–424. doi: 10.1016/j.neuron.2011.07.013
Radenovic, L., Korenic, A., Maleeva, G., Osadchenko, I., Kovalenko, T., and Skibo, G. (2011). Comparative Ultrastructural Analysis of Mitochondria in the CA1 and CA3 Hippocampal Pyramidal Cells Following Global Ischemia in Mongolian Gerbils. Anat. Rec. 294, 1057–1065. doi: 10.1002/ar.21390
Rangaraju, V., Calloway, N., and Ryan, T. A. (2014). Activity-driven local ATP synthesis is required for synaptic function. Cell 156, 825–835. doi: 10.1016/j.cell.2013.12.042
Ransohoff, R. M., and Perry, V. H. (2009). Microglial Physiology: Unique Stimuli. Specialized Responses. Annu. Rev. Immunol. 27, 119–145. doi: 10.1146/annurev.immunol.021908.132528
Rasmussen, M. K., Mestre, H., and Nedergaard, M. (2018). The glymphatic pathway in neurological disorders. Lancet Neurol. 17, 1016–1024. doi: 10.1016/S1474-4422(18)30318-1
Ravichandran, K. S. (2011). Beginnings of a Good Apoptotic Meal: The Find-Me and Eat-Me Signaling Pathways. Immunity 35, 445–455. doi: 10.1016/j.immuni.2011.09.004
Re, D. B., Le Verche, V., Yu, C., Amoroso, M. W., Politi, K. A., Phani, S., et al. (2014). Necroptosis drives motor neuron death in models of both sporadic and familial ALS. Neuron 81, 1001–1008. doi: 10.1016/j.neuron.2014.01.011
Reeves, B. C., Karimy, J. K., Kundishora, A. J., Mestre, H., Cerci, H. M., Matouk, C., et al. (2020). Glymphatic System Impairment in Alzheimer’s Disease and Idiopathic Normal Pressure Hydrocephalus. Trends Mol. Med. 26, 285–295. doi: 10.1016/j.molmed.2019.11.008
Reimer, M. M., McQueen, J., Searcy, L., Scullion, G., Zonta, B., Desmazieres, A., et al. (2011). Rapid disruption of axon-glial integrity in response to mild cerebral hypoperfusion. J. Neurosci. 31, 18185–18194. doi: 10.1523/JNEUROSCI.4936-11.2011
Rodríguez, J. J., Yeh, C. Y., Terzieva, S., Olabarria, M., Kulijewicz-Nawrot, M., and Verkhratsky, A. (2014). Complex and region-specific changes in astroglial markers in the aging brain. Neurobiol. Aging 35, 15–23. doi: 10.1016/j.neurobiolaging.2013.07.002
Rodríguez-Arellano, J. J., Parpura, V., Zorec, R., and Verkhratsky, A. (2016). Astrocytes in physiological aging and Alzheimer’s disease. Neuroscience 323, 170–182. doi: 10.1016/j.neuroscience.2015.01.007
Rosenberg, G. A. (2012). Neurological diseases in relation to the blood-brain barrier. J. Cereb. Blood Flow Metab. 32, 1139–1151. doi: 10.1038/jcbfm.2011.197
Rossi, D. J., Oshima, T., and Attwell, D. (2000). Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature 403, 316–321. doi: 10.1038/35002090
Rouach, N., Koulakoff, A., Abudara, V., Willecke, K., and Giaume, C. (2008). Astroglial metabolic networks sustain hippocampal synaptic transmission. Science 322, 1551–1555. doi: 10.1126/science.1164022
Sahlas, D. J., Bilbao, J. M., Swartz, R. H., and Black, S. E. (2002). Clasmatodendrosis correlating with periventricular hyperintensity in mixed dementia. Ann. Neurol. 52, 378–381. doi: 10.1002/ana.10310
Salter, M. W., and Beggs, S. (2014). Sublime microglia: Expanding roles for the guardians of the CNS. Cell 158, 15–24. doi: 10.1016/j.cell.2014.06.008
Sarti, C., Pantoni, L., Bartolini, L., and Inzitari, D. (2002a). Cognitive impairment and chronic cerebral hypoperfusion: What can be learned from experimental models. J. Neurol. Sci. 2002, 263–266. doi: 10.1016/S0022-510X(02)00302-7
Sarti, C., Pantoni, L., Bartolini, L., and Inzitari, D. (2002b). Persistent impairment of gait performances and working memory after bilateral common carotid artery occlusion in the adult Wistar rat. Behav. Brain Res. 136, 13–20. doi: 10.1016/S0166-4328(02)00090-6
Schaapsmeerders, P., Maaijwee, N. A. M., Van Dijk, E. J., Rutten-Jacobs, L. C. A., Arntz, R. M., Schoonderwaldt, H. C., et al. (2013). Long-term cognitive impairment after first-ever ischemic stroke in young adults. Stroke 44, 1621–1628. doi: 10.1161/STROKEAHA.111.000792
Schafer, D. P., and Stevens, B. (2013). Phagocytic glial cells: Sculpting synaptic circuits in the developing nervous system. Curr. Opin. Neurobiol. 23, 1034–1040. doi: 10.1016/j.conb.2013.09.012
Schmidt, R., Enzinger, C., Ropele, S., Schmidt, H., and Fazekas, F. (2003). Progression of cerebral white matter lesions: 6-Year results of the Austrian Stroke Prevention Study. Lancet 361, 2046–2048. doi: 10.1016/S0140-6736(03)13616-1
Schmidt-Kastner, R., Aguirre-Chen, C., Saul, I., Yick, L., Hamasaki, D., Busto, R., et al. (2005). Astrocytes react to oligemia in the forebrain induced by chronic bilateral common carotid artery occlusion in rats. Brain Res. 1052, 28–39. doi: 10.1016/j.brainres.2005.06.018
Schmidt-Kastner, R., and Freund, T. F. (1991). Selective vulnerability of the hippocampus in brain ischemia. Neuroscience 40, 599–636. doi: 10.1016/0306-4522(91)90001-5
Schneider, J. A., Arvanitakis, Z., Bang, W., and Bennett, D. A. (2007). Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology 69, 2197–2204. doi: 10.1212/01.wnl.0000271090.28148.24
Sekar, A., Bialas, A. R., De Rivera, H., Davis, A., Hammond, T. R., Kamitaki, N., et al. (2016). Schizophrenia risk from complex variation of complement component 4. Nature 530, 177–183. doi: 10.1038/nature16549
Seo, J. H., Miyamoto, N., Hayakawa, K., Pham, L. D. D., Maki, T., Ayata, C., et al. (2013). Oligodendrocyte precursors induce early blood-brain barrier opening after white matter injury. J. Clin. Invest. 123, 782–786. doi: 10.1172/JCI65863
Simard, J. M., Kent, T. A., Chen, M., Tarasov, K. V., and Gerzanich, V. (2007). Brain oedema in focal ischaemia: molecular pathophysiology and theoretical implications. Lancet Neurol. 6, 258–268. doi: 10.1016/S1474-4422(07)70055-8
Simpson, R. A., Hemingway, D., Crowther, M., Goodall, S., and Thompson, M. M. (1999). The gelatinases, their activators and inhibitors in the progression of colorectal cancer. Color. Dis. 1, 248–255. doi: 10.1046/j.1463-1318.1999.00064.x
Siqueira, M., Francis, D., Gisbert, D., Gomes, F. C. A., and Stipursky, J. (2018). Radial Glia Cells Control Angiogenesis in the Developing Cerebral Cortex Through TGF-β1 Signaling. Mol. Neurobiol. 55, 3660–3675. doi: 10.1007/s12035-017-0557-8
Small, S. A., Schobel, S. A., Buxton, R. B., Witter, M. P., and Barnes, C. A. (2011). A pathophysiological framework of hippocampal dysfunction in ageing and disease. Nat. Rev. Neurosci. 12, 585–601. doi: 10.1038/nrn3085
Sofroniew, M. V. (2009). Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 32, 638–647. doi: 10.1016/j.tins.2009.08.002
Sofroniew, M. V., and Vinters, H. V. (2010). Astrocytes: biology and pathology. Acta Neuropathol. 119, 7–35. doi: 10.1007/s00401-009-0619-8
Soreq, L., Rose, J., Soreq, E., Hardy, J., Trabzuni, D., Cookson, M. R., et al. (2017). Major Shifts in Glial Regional Identity Are a Transcriptional Hallmark of Human Brain Aging. Cell Rep. 18, 557–570. doi: 10.1016/j.celrep.2016.12.011
Sotelo-Hitschfeld, T., Niemeyer, M. I., Mächler, P., Ruminot, I., Lerchundi, R., Wyss, M. T., et al. (2015). Channel-mediated lactate release by K+ -stimulated astrocytes. J. Neurosci. 35, 4168–4178. doi: 10.1523/JNEUROSCI.5036-14.2015
Spite, M., and Serhan, C. N. (2010). Novel lipid mediators promote resolution of acute inflammation: Impact of aspirin and statins. Circ. Res. 107, 1170–1184. doi: 10.1161/CIRCRESAHA.110.223883
Starr, J. M., Farrall, A. J., Armitage, P., McGurn, B., and Wardlaw, J. (2009). Blood-brain barrier permeability in Alzheimer’s disease: a case-control MRI study. Psychiatry Res. Neuroimaging 171, 232–241. doi: 10.1016/j.pscychresns.2008.04.003
Stefansdottir, H., Arnar, D. O., Aspelund, T., Sigurdsson, S., Jonsdottir, M. K., Hjaltason, H., et al. (2013). Atrial fibrillation is associated with reduced brain volume and cognitive function independent of cerebral infarcts. Stroke 44, 1020–1025. doi: 10.1161/STROKEAHA.12.679381
Stellwagen, D., and Malenka, R. C. (2006). Synaptic scaling mediated by glial TNF-α. Nature 440, 1054–1059. doi: 10.1038/nature04671
Stevens, B., Allen, N. J., Vazquez, L. E., Howell, G. R., Christopherson, K. S., Nouri, N., et al. (2007). The Classical Complement Cascade Mediates CNS Synapse Elimination. Cell 131, 1164–1178. doi: 10.1016/j.cell.2007.10.036
Streit, W. J., Walter, S. A., and Pennell, N. A. (1999). Reactive microgliosis. Prog. Neurobiol. 57, 563–581. doi: 10.1016/S0301-0082(98)00069-0
Suen, D. F., Norris, K. L., and Youle, R. J. (2008). Mitochondrial dynamics and apoptosis. Genes Dev. 22, 1577–1590. doi: 10.1101/gad.1658508
Sun, X., Yao, H., Douglas, R. M., Gu, X. Q., Wang, J., and Haddad, G. G. (2010). InsulinPI3K signaling protects dentate neurons from oxygen-glucose deprivation in organotypic slice cultures. J. Neurochem. 112, 377–388. doi: 10.1111/j.1471-4159.2009.06450.x
Sweeney, M. D., Kisler, K., Montagne, A., Toga, A. W., and Zlokovic, B. V. (2018a). The role of brain vasculature in neurodegenerative disorders. Nat. Neurosci. 21, 1318–1331. doi: 10.1038/s41593-018-0234-x
Sweeney, M. D., Sagare, A. P., and Zlokovic, B. V. (2018b). Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 14, 133–150. doi: 10.1038/nrneurol.2017.188
Syková, E. (2001). Glial diffusion barriers during aging and pathological states. Prog. Brain Res. 132, 339–363. doi: 10.1016/S0079-6123(01)32087-3
Takahashi, K., Rochford, C. D. P., and Neumann, H. (2005). Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J. Exp. Med. 201, 647–657. doi: 10.1084/jem.20041611
Takahashi, S., Iizumi, T., Mashima, K., Abe, T., and Suzuki, N. (2014). Roles and regulation of ketogenesis in cultured astroglia and neurons under hypoxia and hypoglycemia. ASN Neuro 6:1759091414550997. doi: 10.1177/1759091414550997
Tatemichi, T. K., Desmond, D. W., Prohovnik, I., and Eidelberg, D. (1995). Dementia associated with bilateral carotid occlusions: Neuropsychological and haemodynamic course after extracranial to intracranial bypass surgery. J. Neurol. Neurosurg. Psychiatry 58, 633–636. doi: 10.1136/jnnp.58.5.633
Thal, D. R., Rüb, U., Orantes, M., and Braak, H. (2002). Phases of Aβ-deposition in the human brain and its relevance for the development of AD. Neurology 58, 1791–1800. doi: 10.1212/WNL.58.12.1791
Tian, L., Ma, L., Kaarela, T., and Li, Z. (2012). Neuroimmune crosstalk in the central nervous system and its significance for neurological diseases. J. Neuroinfl. 9:155. doi: 10.1186/1742-2094-9-155
Toledo, J. B., Arnold, S. E., Raible, K., Brettschneider, J., Xie, S. X., Grossman, M., et al. (2013). Contribution of Cerebrovascular Disease in Autopsy Confirmed Neurodegenerative Disease Cases in the National Alzheimer’s Coordinating Centre - PubMed. Brain 136, 2697–2706. doi: 10.1093/brain/awt188
Tomimoto, H., Akiguchi, I., Wakita, H., Suenaga, T., Nakamura, S., and Kimura, J. (1997). Regressive changes of astroglia in white matter lesions in cerebrovascular disease and Alzheimer’s disease patients. Acta Neuropathol. 94, 146–152. doi: 10.1007/s004010050686
Tourdias, T., Mori, N., Dragonu, I., Cassagno, N., Boiziau, C., Aussudre, J., et al. (2011). Differential aquaporin 4 expression during edema build-up and resolution phases of brain inflammation. J. Neuroinfl. 8:143. doi: 10.1186/1742-2094-8-143
Turrin, N. P., and Rivest, S. (2006). Molecular and cellular immune mediators of neuroprotection. Mol. Neurobiol. 34, 221–242. doi: 10.1385/MN
Ueno, M., Tomimoto, H., Akiguchi, I., Wakita, H., and Sakamoto, H. (2002). Blood-brain barrier disruption in white matter lesions in a rat model of chronic cerebral hypoperfusion. J. Cereb. Blood Flow Metab. 22, 97–104. doi: 10.1097/00004647-200201000-00012
Ugolini, F., Lana, D., Nardiello, P., Nosi, D., Pantano, D., Casamenti, F., et al. (2018). Different Patterns of Neurodegeneration and Glia Activation in CA1 and CA3 Hippocampal Regions of TgCRND8 Mice. Front. Aging Neurosci. 10:372. doi: 10.3389/fnagi.2018.00372
Urban, L., Neill, K. H., Crain, B. J., Nadler, J. V., and Somjen, G. G. (1990). Effects of transient forebrain ischemia in area CA1 of the gerbil hippocampus: An in vitro study. Adv. Exp. Med. Biol. 1990, 491–500. doi: 10.1007/978-1-4684-5769-8_54
Van den Bossche, J., O’Neill, L. A., and Menon, D. (2017). Macrophage Immunometabolism: Where Are We (Going)? Trends Immunol. 38, 395–406. doi: 10.1016/j.it.2017.03.001
Vazdarjanova, A., and Guzowski, J. F. (2004). Differences in hippocampal neuronal population responses to modifications of an environmental context: Evidence for distinct, yet complementary, functions of CA3 and CA1 ensembles. J. Neurosci. 24, 6489–6496. doi: 10.1523/JNEUROSCI.0350-04.2004
Verkhratsky, A., and Nedergaard, M. (2018). Physiology of Astroglia. Physiol. Rev. 98, 239–389. doi: 10.1152/physrev.00042.2016
Verkhratsky, A., Krishtal, O. A., and Burnstock, G. (2009). Purinoceptors on neuroglia. Mol. Neurobiol. 39, 190–208. doi: 10.1007/s12035-009-8063-2
Verkhratsky, A., Parpura, V., and Rodríguez, J. J. (2011). Where the thoughts dwell: The physiology of neuronal-glial “diffuse neural net.”. Brain Res. Rev. 66, 133–151. doi: 10.1016/j.brainresrev.2010.05.002
Verkhratsky, A., Rodríguez, J. J., and Parpura, V. (2013). Astroglia in neurological diseases. Future Neurol. 8, 149–158. doi: 10.2217/fnl.12.90
Verkhratsky, A., Steardo, L., Parpura, V., and Montana, V. (2016). Translational potential of astrocytes in brain disorders. Prog. Neurobiol. 144, 188–205. doi: 10.1016/j.pneurobio.2015.09.003
von Bernhardi, R., Eugenín-von Bernhardi, L., and Eugenín, J. (2015). Microglial cell dysregulation in brain aging and neurodegeneration. Front. Aging Neurosci. 7:124. doi: 10.3389/fnagi.2015.00124
Wakselman, S., Béchade, C., Roumier, A., Bernard, D., Triller, A., and Bessis, A. (2008). Developmental neuronal death in hippocampus requires the microglial CD11b integrin and DAP12 immunoreceptor. J. Neurosci. 28, 8138–8143. doi: 10.1523/JNEUROSCI.1006-08.2008
Wang, H., Golob, E. J., and Su, M. Y. (2006). Vascular volume and blood-brain barrier permeability measured by dynamic contrast enhanced MRI in hippocampus and cerebellum of patients with MCI and normal controls. J. Magn. Reson. Imaging 24, 695–700. doi: 10.1002/jmri.20669
Wang, M., Ding, F., Deng, S. Y., Guo, X., Wang, W., Iliff, J. J., et al. (2017). Focal solute trapping and global glymphatic pathway impairment in a murine model of multiple microinfarcts. J. Neurosci. 37, 2870–2877. doi: 10.1523/JNEUROSCI.2112-16.2017
Wang, X., Pal, R., Chen, X. W., Limpeanchob, N., Kumar, K. N., and Michaelis, E. K. (2005). High intrinsic oxidative stress may underlie selective vulnerability of the hippocampal CA1 region. Mol. Brain Res. 140, 120–126. doi: 10.1016/j.molbrainres.2005.07.018
Webster, C. M., Hokari, M., McManus, A., Tang, X. N., Ma, H., Kacimi, R., et al. (2013). Microglial P2Y12 Deficiency/Inhibition Protects against Brain Ischemia. PLoS One 8:70927. doi: 10.1371/journal.pone.0070927
Weller, R. O., Djuanda, E., Yow, H. Y., and Carare, R. O. (2009). Lymphatic drainage of the brain and the pathophysiology of neurological disease. Acta Neuropathol. 117, 1–14. doi: 10.1007/s00401-008-0457-0
Wender, R., Brown, A. M., Fern, R., Swanson, R. A., Farrell, K., and Ransom, B. R. (2000). Astrocytic glycogen influences axon function and survival during glucose deprivation in central white matter. J. Neurosci. 20, 6804–6810. doi: 10.1523/jneurosci.20-18-06804.2000
Wiebe, S., Stäubli, U., and Ambros-Ingerson, J. (1997). Short-term Reverberant Memory Model of Hippocampal Field CA3. Hippocampus 7, 656–665. doi: 10.1002/(SICI)1098-106319977:6<656::AID-HIPO7<3.0.CO;2-C
Wilde, G. J. C., Pringle, A. K., Wright, P., and Iannotti, F. (1997). Differential vulnerability of the CA1 and CA3 subfields of the hippocampus to superoxide and hydroxyl radicals in vitro. J. Neurochem. 69, 883–886. doi: 10.1046/j.1471-4159.1997.69020883.x
Winkler, E. A., Sengillo, J. D., Sullivan, J. S., Henkel, J. S., Appel, S. H., and Zlokovic, B. V. (2013). Blood-spinal cord barrier breakdown and pericyte reductions in amyotrophic lateral sclerosis. Acta Neuropathol. 125, 111–120. doi: 10.1007/s00401-012-1039-8
Wolf, S. A., Boddeke, H. W. G. M., and Kettenmann, H. (2017). Microglia in Physiology and Disease. Annu. Rev. Physiol. 79, 619–643. doi: 10.1146/annurev-physiol-022516-034406
Wu, H. H., Bellmunt, E., Scheib, J. L., Venegas, V., Burkert, C., Reichardt, L. F., et al. (2009). Glial precursors clear sensory neuron corpses during development via Jedi-1, an engulfment receptor. Nat. Neurosci. 12, 1534–1541. doi: 10.1038/nn.2446
Wu, W., Brickman, A. M., Luchsinger, J., Ferrazzano, P., Pichiule, P., Yoshita, M., et al. (2008). The brain in the age of old: The hippocampal formation is targeted differentially by diseases of late life. Ann. Neurol. 64, 698–706. doi: 10.1002/ana.21557
Xie, M., Yi, C., Luo, X., Xu, S., Yu, Z., Tang, Y., et al. (2011). Glial gap junctional communication involvement in hippocampal damage after middle cerebral artery occlusion. Ann. Neurol. 70, 121–132. doi: 10.1002/ana.22386
Xiong, X. Y., Liu, L., and Yang, Q. W. (2016). Functions and mechanisms of microglia/macrophages in neuroinflammation and neurogenesis after stroke. Prog. Neurobiol. 142, 23–44. doi: 10.1016/j.pneurobio.2016.05.001
Xu, Z., Xiao, N., Chen, Y., Huang, H., Marshall, C., Gao, J., et al. (2015). Deletion of aquaporin-4 in APP/PS1 mice exacerbates brain Aβ accumulation and memory deficits. Mol. Neurodegener 10:58. doi: 10.1186/s13024-015-0056-1
Yoshida, T., Fujimori, T., and Nabeshima, Y. O. I. (2002). Mediation of unusually high concentrations of 1,25-dihydroxyvitamin D in homozygous klotho mutant mice by increased expression of renal 1α-hydroxylase gene. Endocrinology 143, 683–689. doi: 10.1210/endo.143.2.8657
Yoshizaki, K., Adachi, K., Kataoka, S., Watanabe, A., Tabira, T., Takahashi, K., et al. (2008). Chronic cerebral hypoperfusion induced by right unilateral common carotid artery occlusion causes delayed white matter lesions and cognitive impairment in adult mice. Exp. Neurol. 210, 585–591. doi: 10.1016/j.expneurol.2007.12.005
Zakharova, M., and Ziegler, H. K. (2005). Paradoxical Anti-Inflammatory Actions of TNF-α: Inhibition of IL-12 and IL-23 via TNF Receptor 1 in Macrophages and Dendritic Cells. J. Immunol. 175, 5024–5033. doi: 10.4049/jimmunol.175.8.5024
Zalewska, T., Ziemka-Nałȩcz, M., and Domańska-Janik, K. (2005). Transient forebrain ischemia effects interaction of Src, FAK, and PYK2 with the NR2B subunit of N-methyl-d-aspartate receptor in gerbil hippocampus. Brain Res. 1042, 214–223. doi: 10.1016/j.brainres.2005.02.025
Zamanian, J. L., Xu, L., Foo, L. C., Nouri, N., Zhou, L., Giffard, R. G., et al. (2012). Genomic analysis of reactive astrogliosis. J. Neurosci. 32, 6391–6410. doi: 10.1523/JNEUROSCI.6221-11.2012
Zbesko, J. C., Nguyen, T. V. V., Yang, T., Frye, J. B., Hussain, O., Hayes, M., et al. (2018). Glial scars are permeable to the neurotoxic environment of chronic stroke infarcts. Neurobiol. Dis. 112, 63–78. doi: 10.1016/j.nbd.2018.01.007
Zhang, X., Dong, H., Zhang, S., Lu, S., Sun, J., and Qian, Y. (2015). Enhancement of LPS-induced microglial inflammation response via TLR4 under high glucose conditions. Cell. Physiol. Biochem. 35, 1571–1581. doi: 10.1159/000373972
Zhang, Y., and Barres, B. A. (2010). Astrocyte heterogeneity: An underappreciated topic in neurobiology. Curr. Opin. Neurobiol. 20, 588–594. doi: 10.1016/j.conb.2010.06.005
Zhu, X., Smith, M. A., Honda, K., Aliev, G., Moreira, P. I., Nunomura, A., et al. (2007). Vascular oxidative stress in Alzheimer disease. J. Neurol. Sci. 257, 240–246. doi: 10.1016/j.jns.2007.01.039
Ziv, Y., Ron, N., Butovsky, O., Landa, G., Sudai, E., Greenberg, N., et al. (2006). Immune cells contribute to the maintenance of neurogenesis and spatial learning abilities in adulthood. Nat. Neurosci. 9, 268–275. doi: 10.1038/nn1629
Zlokovic, B. V. (2011). Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci. 12, 723–738. doi: 10.1038/nrn3114
Zola, S. M., Squire, L. R., Teng, E., Stefanacci, L., Buffalo, E. A., and Clark, R. E. (2000). Impaired recognition memory in monkeys after damage limited to the hippocampal region. J. Neurosci. 20, 451–463. doi: 10.1523/jneurosci.20-01-00451.2000
Keywords: CA1 hippocampus, CA3 hippocampus, triads, confocal microscopy, brain metabolism, neurovascular unit, glymphatic system, clasmatodendrosis
Citation: Lana D, Ugolini F and Giovannini MG (2020) An Overview on the Differential Interplay Among Neurons–Astrocytes–Microglia in CA1 and CA3 Hippocampus in Hypoxia/Ischemia. Front. Cell. Neurosci. 14:585833. doi: 10.3389/fncel.2020.585833
Received: 21 July 2020; Accepted: 09 October 2020;
Published: 11 November 2020.
Edited by:
Yannick Poitelon, Albany Medical College, United StatesReviewed by:
Lance Johnson, University of Kentucky, United StatesShane Antony Liddelow, New York University, United States
Copyright © 2020 Lana, Ugolini and Giovannini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria G. Giovannini, bWFyaWFncmF6aWEuZ2lvdmFubmluaUB1bmlmaS5pdA==