 María José Castellanos-Montiel
María José Castellanos-Montiel Mathilde Chaineau
Mathilde Chaineau Thomas M. Durcan
Thomas M. Durcan- Early Drug Discovery Unit (EDDU), Montreal Neurological Institute-Hospital, McGill University, Montreal, QC, Canada
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease that selectively affects motor neurons (MNs) of the cortex, brainstem, and spinal cord. Several genes have been linked to both familial (fALS) and sporadic (sALS) cases of ALS. Among all the ALS-related genes, a group of genes known to directly affect cytoskeletal dynamics (ALS2, DCTN1, PFN1, KIF5A, NF-L, NF-H, PRPH, SPAST, and TUBA4A) is of high importance for MN health and survival, considering that MNs are large polarized cells with axons that can reach up to 1 m in length. In particular, cytoskeletal dynamics facilitate the transport of organelles and molecules across the long axonal distances within the cell, playing a key role in synapse maintenance. The majority of ALS-related genes affecting cytoskeletal dynamics were identified within the past two decades, making it a new area to explore for ALS. The purpose of this review is to provide insights into ALS-associated cytoskeletal genes and outline how recent studies have pointed towards novel pathways that might be impacted in ALS. Further studies making use of extensive analysis models to look for true hits, the newest technologies such as CRIPSR/Cas9, human induced pluripotent stem cells (iPSCs) and axon sequencing, as well as the development of more transgenic animal models could potentially help to: differentiate the variants that truly act as a primary cause of the disease from the ones that act as risk factors or disease modifiers, identify potential interactions between two or more ALS-related genes in disease onset and progression and increase our understanding of the molecular mechanisms leading to cytoskeletal defects. Altogether, this information will give us a hint on the real contribution of the cytoskeletal ALS-related genes during this lethal disease.
Introduction
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease characterized by the progressive loss of motor neurons (MNs) within the brain cortex, brainstem, and spinal cord. As MNs degenerate, the synaptic connections with their target muscles are lost, leading to muscle spasticity, weakness, and atrophy. Currently, there is no cure for ALS and available treatments only help to relieve symptoms. Typically, ALS patients die of respiratory failure within 2–5 years after diagnosis due to diaphragm paralysis. About 90–95% of ALS cases are sporadic (sALS) while the remaining 5–10% are familial (fALS) (Brown and Al-Chalabi, 2017; Volk et al., 2018).The clinical presentation of ALS is heterogeneous and sometimes it can be misdiagnosed with other MN diseases. For instance, the population of MNs involved and the survival can vary depending on the mutated gene and the specific mutation present. Additionally, sALS is more complex than that of fALS because the cause of the disease is attributed to an additive effect of hundreds of common variants that increase the risk of developing the disease. Even more, some sALS cases cannot be attributed to genetic or biologic factors instead they are ascribed to environmental and undefined factors (Brown and Al-Chalabi, 2017). Many ALS patients show cognitive and behavioral changes characteristic of frontotemporal dementia (FTD), a neurodegenerative disease that shares neuropathological and genetic features with ALS. Thus, it has been suggested that both FTD and ALS are a continuum of the same phenotypic spectrum (Lipton et al., 2004; Vance et al., 2006; Volk et al., 2018).
Ever since the first causative gene for fALS, Cu/Zn superoxide dismutase 1 (SOD1), was discovered in 1993 (Rosen et al., 1993), over 30 genes have been linked to fALS as well as being identified as the molecular cause in certain sALS cases. The list of ALS-related genes is continuously growing, however, SOD1, chromosome open reading frame 72 (C9orf72), TARDBP (transactive response DNA-binding protein) and FUS (fused in sarcoma) are the most well-studied, mainly because they account for the majority of both fALS and sALS cases (Brown and Al-Chalabi, 2017). On the whole, ALS-related genes can be broadly categorized into four groups depending on the cellular pathways in which they are involved: (1) protein homeostasis; (2) RNA homeostasis and trafficking; (3) cytoskeletal dynamics; and (4) mitochondrial function (Mathis et al., 2019). The purpose of this review is to provide insight into several ALS-related genes linked to the disruption of cytoskeletal dynamics. Specially, we will focus on how the disruption of such dynamics potentially triggers axonal degeneration in MNs, impairing their ability to maintain synapses.
ALS-Related Genes Affecting Cytoskeletal Dynamics
MNs are known to be the largest polarized cells in the human body and axons of spinal MNs can reach a meter in length in adults. The significant length of such axons makes them highly dependent on proper cytoskeletal architecture, whose integrity is essential for the axonal transport necessary to maintain synapse integrity. Both anterograde (from the cell body to the periphery) and retrograde (from the periphery to the cell body) microtubule (MT)-dependent transport are key mediators for MN survival, maintenance, and functionality (Chevalier-Larsen and Holzbaur, 2006). Thus, disruption of cytoskeleton integrity and/or MT-dependent transport mechanisms could translate into an inability of MNs to supply their synapses with essential components and/or to convey information back to the cell body, potentially triggering degeneration processes. Recently, a few genes known to play a role in cytoskeletal dynamics have been linked to ALS (Figure 1): alsin rho guanine nucleotide exchange factor (ALS2), dynactin subunit 1 (DCTN1), kinesin family member 5A (KIF5A), neurofilament light (NF-L), neurofilament heavy (NF-H), peripherin (PRPH), profilin 1 (PFN1), spastin (SPAST) and tubulin alpha 4a (TUBA4A). Below, we discuss each gene and outline how ALS-associated mutations (Supplementary Table 1) might affect the normal function of each protein, and the implications for the axonal cytoskeletal system.
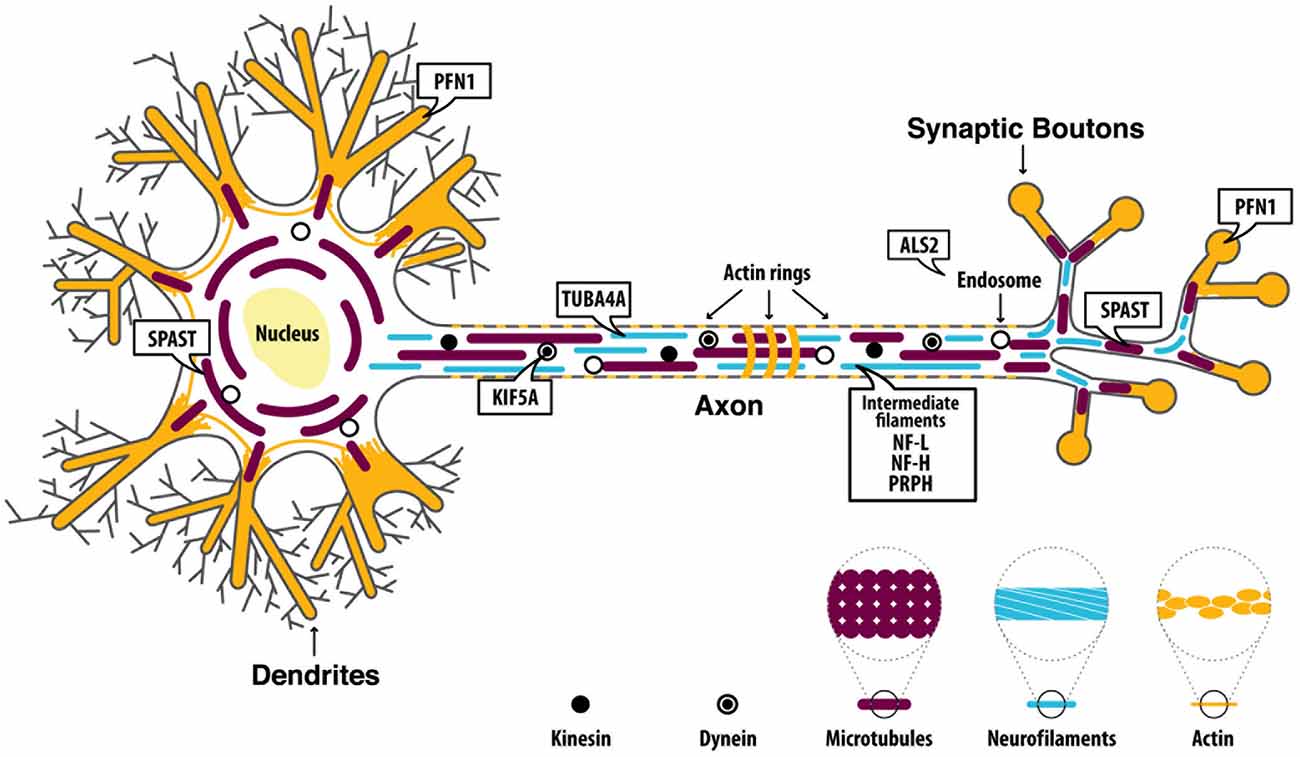
Figure 1. Proteins are encoded by amyotrophic lateral sclerosis (ALS)-related genes affecting cytoskeletal dynamics. ALS2 is mainly implicated in the regulation of endosomal dynamics and it is also found at the growth cone of neurons where it promotes neurite outgrowth. Dynactin Subunit 1 (DCTN1) is involved in microtubules (MTs) anterograde and retrograde transport.Neurofilament light (NF-L), neurofilament heavy (NF-H), andperipherin (PRPH) are the main components of motor neuron (MN) intermediate filaments (IFs). Kinesin family member 5A (KIF5A) encodes a kinesin protein which mainly mediates anterograde transport along MTs. Profilin 1 (PFN1) is involved in the polymerization of actin filaments. Spastin (SPAST) is involved in MTs disassembly and therefore plays a key role in regulating MTs dynamics. Tubulin alpha 4a (TUBA4A) encodes for an α-tubulin subunit which is assembled within the MT filaments.
Alsin Rho Guanine Nucleotide Exchange Factor (ALS2)
ALS2, which encodes the protein alsin, is one of the few ALS-related genes that exhibits a recessive pattern of inheritance, often linked to juvenile onset-ALS (Hadano et al., 2001; Yang et al., 2001). Nevertheless, mutations in ALS2 have also been linked to other diseases selectively affecting MNs, namely infantile ascending hereditary spastic paraplegia (IAHSP) and juvenile primary lateral sclerosis (PLS), which have different clinical profiles to ALS. For instance, in IAHSP and PLS the degeneration is restricted to upper MNs, while both upper and lower MNs are affected in ALS. This broad range of phenotypes arising from mutations in ALS2 adds complexity to its study (Hand et al., 2003; Simone et al., 2018). Importantly, only two studies report the development of ALS in two independent families as a result of an ALS2 mutation, making it an extremely rare cause for the development of ALS (Hadano et al., 2001; Yang et al., 2001).
The identified mutations causing an ALS phenotype are deletions producing a premature stop codon, suggesting that alsin loss of function might be triggering disease mechanisms. However, ALS2 knockout (ALS2−/−) mice lack an overt pathological phenotype, suggesting that ALS2 might not contribute to ALS pathology through either a loss of function or a dominant-negative mechanism (Cai et al., 2008). The existence of an alsin-related protein (ALS2CL) with homology to the C-terminal domain of alsin found in mice and humans, suggests the possibility of a compensatory mechanism to cope with the loss of function of alsin. However, this hypothesis remains controversial since a few studies imply that the alsin-related protein has a distinct function from alsin (Hadano et al., 2004) and/or it binds to alsin to function as a complex thereby playing a role in other pathways (Suzuki-Utsunomiya et al., 2007). While the loss of alsin appears to be insufficient to trigger MN degeneration (Cai et al., 2005, 2008), its absence can increase MN susceptibility to oxidative stress induced by environmental factors (e.g., paraquat) in vivo and in vitro (Cai et al., 2005).
Alsin contains three domains that act as GTPase regulators (Hadano et al., 2001): (1) a regulator of chromosome condensation 1 (RCC1) like domain (termed RLD); (2) a diffuse B cell lymphoma (Dbl) homology/pleckstrin homology domain (termed DH/PH); and (3) a vacuolar protein sorting 9 domain (termed VPS9). All three domains of alsin have been implicated in the regulation of endosomal dynamics (Chandran et al., 2007). It has been shown that the genetic ablation of ALS2 in SOD1H46R mice exacerbates the disruption of endolysosomal trafficking (Hadano et al., 2010), although more studies are needed to analyze the possible interactions between SOD1 and alsin in causing an ALS phenotype. ALS2 has also been linked to glutamate-mediated excitotoxicity, an ALS synaptic disease mechanism. The RCC1 domain can interact with several domains of the glutamate receptor-interacting protein 1 (GRIP1), which mediates GluR2 subunit transport to different cellular compartments. GluR2 is important for AMPA receptors as it makes them calcium impermeable. In ALS2−/− neurons, the subcellular localization of GRIP1 protein is altered, which translates into a reduction of GluR2 at the synaptic surface, making MNs more vulnerable to excitotoxicity (Lai et al., 2006). Alsin has also been located at the growth cone of neurons, where it promotes neurite outgrowth (Tudor et al., 2005). Furthermore, when ALS2 was specifically knocked down in rat embryonic spinal MNs, increased cell death, and reduced neurite outgrowth was observed (Jacquier et al., 2006).
Remarkably, alsin has been found to also have neuroprotective effects in MNs within an ALS context. One study showed that the DH/PH domain of alsin can bind mutated SOD1 and suppress its neurotoxic effects (Kanekura et al., 2004). Currently, no animal models are available to study ALS-related mutations in ALS2 in vivo. Such models would give a broad perspective into the neuroprotective and neurotoxic effects of alsin in ALS.
Dynactin Subunit 1 (DCTN1)
The dynactin complex contains more than 20 subunits, corresponding to 11 different proteins. The largest subunit of dynactin, dynactin subunit 1 or p150Glued (DCTN1), directly interacts with cytoplasmic dynein-1 motor (dynein) and MTs to promote the retrograde transport of vesicles, organelles, RNAs, and different binding proteins (Laird et al., 2008; Moughamian and Holzbaur, 2012; Urnavicius et al., 2015). An initial study identified a missense mutation (G59S) in DCTN1 as the cause of an autosomal dominant form of lower MN disease in all the affected members of one family (Puls et al., 2003). The G59S substitution is localized in the cytoskeleton-associated protein glycine-rich (CAP-Gly) domain of DCTN1 which mediates the binding of dynactin to MTs. Consistently, subsequent studies showed that G59S mutation decreases the ability of dynactin to bind MTs (Puls et al., 2003; Levy et al., 2006; Lai et al., 2007). Several other mutations in DCTN1 have now been identified in fALS (Münch et al., 2004, 2005; Liu et al., 2014), and some studies have revealed the presence of DCTN1 variants in sALS cases. Such variants were able to induce abnormal morphological changes when overexpressed in an in vitro system (Stockmann et al., 2013). However, DCTN1-mRNA has been observed to be reduced in the motor cortex and spinal cord of sALS patients (Jiang et al., 2007; Ikenaka et al., 2013; Kuźma-Kozakiewicz et al., 2013), raising the question of whether DCTN1 variants may contribute to disease onset through either a loss or gain of function in ALS.
Transgenic animal models with mutated or ablated DCTN1 have been generated to elucidate the potential role of DCTN1 in the degeneration of MNs. The intracellular trafficking of autophagosomes (Laird et al., 2008; Ikenaka et al., 2013) and the bidirectional transport of motor proteins kinesins (Hsu et al., 2011) are disrupted in vivo. Remarkably, DCTN1 has also shown to play a key role within the synaptic processes of MNs. For instance, DCTN1 depletion leads to neuromuscular junction (NMJ) instability, functional abnormalities, and locomotion defects in a zebrafish model (Bercier et al., 2019). Similarly, the ablation of DCTN1 in the postnatal neurons of aged mice resulted in the preferential degeneration of spinal MNs, accompanied by increased gliosis, NMJ disintegration, and muscle atrophy (Yu et al., 2018). When DCTN1 was mutated in Drosophila in the DCTN1 homolog Glued, the formation and maturation of MN synapses were impaired (Allen et al., 1999). Additionally, the dynactin complex was shown to have a local role within the MN presynaptic terminal by controlling synapse stabilization by decreasing the rate of presynaptic retraction (Eaton et al., 2002). Thus, mutations in DCTN1 can disrupt the local role of dynactin at the MN presynaptic terminal. Taken together, all these studies point towards an inability of MNs to form synapses when DCTN1 is mutated or ablated. However, the exact mechanisms leading to such deficits remain unknown.
Intermediate Filament Proteins
Mature MNs express different types of intermediate filament (IF) genes that code for proteins which contribute to the maintenance of the cytoskeletal architecture and signaling within the cells: neurofilament (NF) light (NF-L), medium (NF-M), and heavy (NF-H) chains, as well as α-internexin (INA) and peripherin (PRPH). These proteins assemble into complex structures and undergo several post-translational modifications that include glycosylation and phosphorylation, particularly NF-H, with the presence of numerous lysine-serine-proline (KSP) repeats in the C-terminal tail domain of the protein that is heavily phosphorylated. Abnormal accumulation of NFs in the spinal cord of patients with sALS was first reported in the 80s (Hirano et al., 1984) and since then, growing evidence has demonstrated that NF abnormalities could be an early pathological feature of ALS in patients, a phenotype that can also be recapitulated in animal models. Perikaryal and axonal inclusion bodies (also termed spheroids) containing IF proteins are a hallmark of degenerating spinal MNs in ALS patients. NF-L and NF-H subunits, as well as peripherin, are particularly enriched in such spheroids, and how they interact has been gaining attention in the last few years.
In 1994, Bergeron and colleagues discovered that NF-L-mRNA was decreased by 60% in the spinal MNs of ALS patients (Bergeron et al., 1994). To further study the contribution of NF-L in ALS pathogenesis, a transgenic mouse model expressing a low level of human NF-L with a point mutation was created [NF-L(Pro)]. These mice exhibited distinct hallmarks of ALS including selective degeneration of MNs, perikaryal and axonal swellings with the presence of IF spheroids, and NMJ denervation presenting as muscle atrophy (Lee et al., 1994). However, there are no known point mutations in NF-L associated with ALS, and NF-L subunit accumulation is thought to be a secondary effect of other primary disease mechanisms.
In contrast to NF-L, mutations in the region of the gene NF-H coding for the highly phosphorylated C-terminal domain of the NF-H protein have been reported in several patients with sALS (Figlewicz et al., 1994). Similar to patients, transgenic mice overexpressing human NF-H present with features of ALS pathology, including swellings of proximal axons in the spinal cord, progressive axonopathy, and atrophy of muscle fibers (Côté et al., 1993). In these mice, it is not only the axonal transport of NF proteins that are altered but to a lesser extent, also that of actin and tubulin. Also, in degenerating spinal MNs, mitochondria are found within the perikaryon, next to NF aggregates, supporting the idea that disorganization of the NFs architecture can affect the axonal integrity of MNs by altering the transport of other essential components (Collard et al., 1995). These hallmarks of ALS pathology have also been reported in transgenic mice overexpressing various mutations of human SOD1 and NF involvement in the pathology observed in these models has been previously discussed (Julien, 1997). For instance, transgenic SOD1G93A mice that develop an ALS clinical phenotype, present pathological hallmarks of the disease, such as the presence of some NF-rich spheroids containing NF proteins and phosphorylated NF-H and NF-M subunits, as well as α-internexin and peripherin. However, in this case, it seems that only the NF cytoskeleton is altered as no immunoreactivity against actin or tubulin was observed in these spheroids (Tu et al., 1996). To reconcile the role of NF in the SOD1-mediated pathology, a transgenic SOD1G37R mouse overexpressing human NF-H has been generated. Surprisingly, the lifespan of these animals was increased by NF-H overexpression (Couillard-Després et al., 1998). In these mice, the NF proteins were primarily localized in the perikaryal area of neurons where NF-H might exert its neuroprotective effect.
While understanding the regulation of IFs can help elucidate the underlying causes of ALS, examining the levels of NF-L and the phosphorylated form of NF-H in cerebrospinal fluid (CSF), plasma, and blood of patients can be used as a diagnostic tool for and as a predictor of disease progression. Indeed, several recent studies with a cohort of ALS patients have reported an increase in these levels in both patients with fALS and sALS. Given their potential as biomarkers for ALS, considerable efforts have recently been made in the development of precise and reliable detection techniques for both NF-L and the phosphorylated form of NF-H (for review, see Poesen and Van Damme, 2018).
Peripherin, encoded by the gene PRPH, is a component of MN spheroids in both transgenic mouse models (Beaulieu et al., 2000; Robertson et al., 2003) and ALS patients (Corbo and Hays, 1992; Migheli et al., 1993; Keller et al., 2012). Since ALS patients typically show decreased levels of NF-L-mRNA, a transgenic mouse model overexpressing peripherin but with NF-L knocked out (TPer;L−/−) was created. The TPer;L−/− mouse model exhibited a 46% loss in MNs with spheroids mainly found in the axons and, in the absence of NF-L, there was an increase in the formation of NF-H heterodimers with peripherin and α-internexin (Beaulieu et al., 1999, 2000). Such spheroids can trigger MN degeneration by disrupting the axonal transport of the other IF proteins as observed in a less complex transgenic mouse model only overexpressing peripherin (Per mouse; Beaulieu et al., 1999; Millecamps et al., 2006). Importantly, the overexpression of NF-H within the TPer;L−/− genetic background (hH:TPer;L−/−) can protect against the neurotoxicity exerted by peripherin. In these mice, the intracellular inclusion bodies were re-localized to the perikaryal of the spinal MNs, suggesting that excess NF-H sequesters peripherin and by doing so, prevents its accumulation within the axon. This supports the hypothesis that the composition of the IF protein inclusions determines their localization within the cell as well as their role as neuroprotective or neurotoxic structures (Beaulieu and Julien, 2003).
In mice, PRPH gives rise to three splice variants (Per 56, 58, 61). The SOD1G37R mouse model, in which MNs carry IF spheroids containing peripherin similar to ALS patients, was used to demonstrate that Per61 was the splice variant enriched within the spheroids. However, the existence of the Per61 splice variant in humans remains controversial since it is argued that the splicing event that happens in mice cannot occur in humans (Xiao et al., 2008). Additionally, only one study has been able to find Per61 within lumbar degenerating MNs of two ALS patients (Robertson et al., 2003). Instead, a different splice variant, Per28, is thought to be the equivalent of Per61 in humans and it is upregulated at the mRNA and protein level, in patients with sALS. The analysis of spinal cord sections of these patients showed Per28 aggregates within MNs, and its subsequent overexpression in an in vitro system showed Per28 had neurotoxic effects (Xiao et al., 2008). However, the exact role of Per28 in ALS onset remains controversial as it has also been found to have cytoprotective effects against oxidative stress (McLean et al., 2014).
Until 2011, it was thought that peripherin neurotoxicity was a secondary disease mechanism associated with other events like a SOD1 mutation. However, the identification of several ALS-associated point mutations (Gros-Louis et al., 2004; Leung et al., 2004; Corrado et al., 2011) and a frameshift deletion (Gros-Louis et al., 2004) in PRPH raised the question as to whether these mutations are drivers of the ALS phenotype. However, the absence of mouse models or human iPSCs with mutations in PRPH has prevented further testing of this hypothesis.
Kinesin Family Member 5A (KIF5A)
Recently, the kinesin family member 5A (KIF5A) was confirmed as an ALS-related gene (Nicolas et al., 2018). Kinesins are the microtubule-based motor proteins involved in the anterograde transport of cargos. Currently, it is unknown if KIF5A mutations themselves are sufficient to cause ALS but genome-wide analysis has identified KIF5A mutations as low or high-risk factors for the development of ALS (Nicolas et al., 2018). The mechanisms through which KIF5A mutations would be contributing to ALS onset have not been studied yet, but several hypotheses exist focused on the central role of kinesins in axonal transport. For instance, KIF5A knockout mice (KIF5A−/−) display abnormal transport of NF proteins (Xia et al., 2003), which has been proposed as a causative mechanism of NF accumulation, an ALS hallmark, as discussed previously (Chevalier-Larsen and Holzbaur, 2006). Additionally, primary motor neurons (PMNs) derived from KIF5A−/− mice showed transport deficits, reduced axonal outgrowth, and reduced survival. In particular, such transport deficits were observed for mitochondria (Karle et al., 2012). The impairment of mitochondrial transport was observed in both anterograde and retrograde direction, consistent with previous findings in Drosophila models lacking the KIF5A homolog khc (Martin et al., 1999). Importantly, deficits in mitochondria transport and function have also been identified as hallmarks of ALS (Chevalier-Larsen and Holzbaur, 2006; Smith et al., 2019). KIF5A also has been shown to affect neurite outgrowth through its interaction with protrudin in the mouse brain, which plays a role in the regulation of vesicular transport in neurons (Matsuzaki et al., 2011). Impaired neurite outgrowth could potentially affect the ability of MNs to form synaptic connections, which are lost in ALS.
Profilin 1 (PFN1)
Profilin 1 (PFN1) is an essential protein for the polymerization of filamentous (F)-actin through binding of monomeric (G)-actin. Early studies assessing the impact of PFN1 mutations in different neuronal cell types showed that a profilin mutant (H119E), which conserved its ability to bind all its target proteins except actin, blocked neurite formation in vitro (Suetsugu et al., 1998). Later, in vivo studies in Drosophila showed growth cone arrest and reduced axon outgrowth in embryonic MNs carrying a mutation in chickadee, the homolog to PFN1 (Wills et al., 1999). In 2012, the exome sequencing of two large ALS families displaying a dominant pattern of inheritance and the subsequent screening of a larger cohort revealed a link between ALS and several mutations in PFN1 (C71G, M114T, E117G, G118V). In the same study and consistent with early findings, PMNs overexpressing mutated PFN1 (G118V) demonstrated a reduction in levels of bound actin relative to wild-type PFN1, inhibition of axonal outgrowth, and growth cone size reduction (Wu et al., 2012). Since then, additional ALS cohorts have been analyzed and further mutations in the PFN1 gene have been identified, not only in fALS but also in sALS (Ingre et al., 2013; Tiloca et al., 2013; Yang et al., 2013; Smith et al., 2015).
Aside from dysregulation of actin dynamics that disrupt axonal growth and promote growth cone arrest, PFN1 has been linked to other ALS features that include abnormalities in autophagy (Nguyen et al., 2019) and cytoplasmatic aggregations (Wu et al., 2012; Smith et al., 2015). PFN1 aggregates co-stained for the transactive response DNA-binding protein 43 (TDP-43) in vitro. Interestingly, abnormal PFN1 pathology was not observed in a cohort of sALS patients with TDP-43 pathology, suggesting that whereas mutant PFN1 can induce aggregation of TDP-43, PFN1 aggregation does not occur in patients with TDP-43 pathology. The mechanisms inducing TDP-43 accumulation through mutant PFN1 remains unknown (Wu et al., 2012). TDP-43 is encoded by TARDBP, another ALS-related gene ubiquitously expressed in the majority of cells, which plays a key role in the regulation of RNA metabolism in different subcellular compartments (Brown and Al-Chalabi, 2017; Mathis et al., 2019). Further studies are needed to analyze the possible interaction between these two genes in disease onset in sALS.
Transgenic mice models of PFN1 have been generated to study the contribution of this gene to ALS pathology. For instance, mice harboring the PFN1 G118V variant display several clinical and pathological characteristics of ALS, including loss of lower and upper MNs, loss of NMJs, and profilin aggregation. Consistent with profilin function and results obtained in PMNs overexpressing the same mutation in PFN1, these mice also show an abnormal G/F actin ratio in the spinal cord (Fil et al., 2017). Similarly, another mouse model in which mutated PFN1 (C71G) was restricted to MNs during development showed abnormal G/F actin ratio in the spinal cord and significant motor deficits (Brettle et al., 2019).
Spastin (SPAST)
Spastin protein, coded by the gene SPAST (or SPG4) is a member of the ATPases associated with diverse cellular activities (AAA) family that can induce MT severing in vitro, thereby influencing MT dynamics (Errico et al., 2002). In support of this observation, overexpression of spastin increases MT disassembly, negatively affecting axonal transport (Kasher et al., 2009). Over 150 mutations within the SPAST gene have been identified to date, most of them being causative of hereditary spastic paraplegia (HSP) but two mutations have been associated with an ALS phenotype (Meyer et al., 2005; Münch et al., 2008). The first ALS reported case linked to the SPG4 gene was the result of a duplication mutation within exon 1 (Meyer et al., 2005). Unlike the duplication mutation that gave rise to an early-onset but slowly progressing ALS, a missense mutation in SPAST (S44L) gave rise to a rapidly progressive adult-onset ALS (Münch et al., 2008). Excitability studies were performed to identify the cortical excitability changes in HSP, ALS, and PLS patients. The three diseases were shown to have different patterns of cortical excitability, ALS is characterized by cortical hyperexcitability. Nevertheless, the molecular mechanisms behind the different excitability found in HSP and ALS remain unknown (Geevasinga et al., 2015). So far, there are no studies of spastin in ALS. However, SPAST mutations leading to an HSP phenotype display a dying-back axonopathy of the affected neurons, especially the corticospinal MNs. Moreover, such axon abnormalities have been identified in iPSC-derived neurons (Denton et al., 2014, 2016), which are known to be a powerful tool to study human neurons in vitro.
Tubulin Alpha 4a (TUBA4A)
Genetic screening of several fALS (Smith et al., 2014; Li et al., 2018) and sALS (Pensato et al., 2015) patients has revealed several mutations in the TUBA4A gene, encoding the tubulin alpha 4a protein, a ubiquitously expressed MT protein highly enriched in the nervous system but lacking a known role in MNs (Rustici et al., 2013; Smith et al., 2014). Through several in vitro experiments, Smith et al. (2014) demonstrated that most of the identified variants showed the inefficient formation of α-/β- tubulin dimers, decreased incorporation into MTs, and inhibited MT network stability. However, it remains controversial whether the proteins resulting from TUBA4A mutations can form aggregates since only one familial ALS mutation (W407X) has been shown to trigger the formation of small ubiquitinated cytoplasmic inclusions in vitro in PMNs and HEK293T cells (Smith et al., 2014).
Unlike other MT proteins linked to neurological disorders, TUBA4A expression increases dramatically (>50-fold) with age in humans, potentially explaining why a mutation in this gene may promote a late-onset disease (Tischfield et al., 2011; Smith et al., 2014; Clark et al., 2016). Interestingly, decreased levels of TUBA4A-mRNA have been found in the brain and spinal cord of sALS and fALS patients with mutations in SOD1 and C9orf72 (Helferich et al., 2018). Unfortunately, the impact of TUBA4A mutations in MT dynamics and how its expression changes over time have not been assessed in vivo. Recently, a neuron-like cell line with transient overexpression of ALS-related mutated forms (R320C and A383T) of TUBA4A showed altered neurite length and MT defects after exposure to selenium (Maraldi et al., 2019). This novel study highlighted a potential link between environmental factors and TUBA4A mutations in triggering ALS onset. The development of animal models harboring ALS mutations in the TUBA4A gene will help lead to understanding the role of TUBA4A in ALS.
Targeting Cytoskeletal Dynamics to Treatment for ALS
Currently, neither the FDA-approved drugs for ALS nor the drugs at different stages of clinical trials are known to have a direct effect on the dysfunction of cytoskeletal dynamics. Besides the IF proteins (NF-L and NF-H subunits, and peripherin) which aggregate to form perikaryal or axonal spheroids (Hirano et al., 1984; Corbo and Hays, 1992; Côté et al., 1993), no other connection between any of the aforementioned proteins has been described. For many years, the dysfunction of most of these proteins was thought to be a result of mutations in other genes (e.g., SOD1) that trigger the disruption of several cellular processes within an MN (Julien, 1997; Couillard-Després et al., 1998; Xiao et al., 2008; Hadano et al., 2010). However, this perspective is now challenged. The discovery of mutations able to trigger disease mechanisms, in the different genes affecting cytoskeletal dynamics, has increased the consensus on a greater contribution of such genes in the development of ALS. Therefore, in addition to NF-L and the phosphorylated form of NF-H that have recently been identified as disease markers (Poesen and Van Damme, 2018), these genes and their products have the potential to be studied as clinical targets.
Conclusion
The discovery of ALS-related genes that affect cytoskeletal dynamics is very recent and their study in an ALS-related context does not extend more than 20 years. The available studies are mainly descriptive reports of isolated subjects or one family with a few members. Only a couple of genetic studies count with large cohorts of patients and/or an extensive study model (e.g., GWAS), meaning that the population presence of most of the mutations remains unknown. It is still unclear which of these genes are acting as the primary cause in the onset of ALS. Some studies identified mutations in genes only in patients with sALS or fALS and not in controls. In contrast, others identified some mutations in patients as well as in non-affected individuals, while others have failed to report any significant disease-associated mutations in the same genes. For example, variants in NF genes are mainly considered a risk factor but a larger cohort of patients might have to be considered to conclude if specific variants are the primary cause of ALS or simply a risk factor. Instead of triggering disease onset or acting as risk factors, different mutations in ALS-related genes can also act as modifiers of the disease by changing the age of clinical presentation (early VS late-onset), the evolution of the disease (e.g., fast VS slow progression), the development of certain profiles (e.g., mutations on ALS2 or mutations on DCTN1 observed within the ALS/FTD family case), the molecular changes observed, etc. Additionally, the interaction between two or more of these genes to trigger and/or modify the clinical presentation of the disease has to be taken into account. It is also important to mention that not all the studies systematically looked at several ALS-related genes in the same study, meaning that the possibility exists that a mutation might be associated as the cause of the disease while not accounting for the presence of other mutations in other genes that might be playing a more significant role in the development of ALS. As to the molecular mechanisms through which the different variants of these genes might be contributing to ALS onset and progression, most of them remain poorly understood. Therefore, animal models harboring ALS-related mutations have been generated to study disease mechanisms, however, they exist for only a few of these genes. Similarly, there are almost no studies making use of novel approaches such as iPSC technology and CRISPR-Cas9 to study the impact of these mutations in ALS pathology. Due to their large size, MNs highly rely on cytoskeletal dynamics to maintain axonal transport and synapse integrity which allows them to function properly. Efforts to study how the ALS-related genes are linked to abnormalities of cytoskeletal dynamics should be increased to better understand the mechanisms underlying this lethal disease.
Author Contributions
MC-M contributed with the conception and design of the manuscript, as well as in the drafting and substantial revision. MC contributed to the drafting and substantial revision of the manuscript. TD contributed with substantial revision of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
TD was supported by funding from the Structural Genomics Consortium (SGC), McGill Healthy Brains, Healthy Lives (HBHL), CQDM, and ALS-RAP, with funding from ALS-Canada, ALS Alliance, and the Motor Neuron Disease Association. MC-M was supported through funding from a Canadian Mitacs Accelerate fellowship, in partnership with Enuvio.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge León Felipe Castellanos Morales for designing the figure of this manuscript and Dr. Lenore K. Beitel for editing the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fncel.2020.594975/full#supplementary-material.
References
Allen, M. J., Shan, X., Caruccio, P., Froggett, S. J., Moffat, K. G., and Murphey, R. K. (1999). Targeted expression of truncated glued disrupts giant fiber synapse formation in Drosophila. J. Neurosci. 19, 9374–9384. doi: 10.1523/JNEUROSCI.19-21-09374.1999
Beaulieu, J. M., Jacomy, H., and Julien, J. P. (2000). Formation of intermediate filament protein aggregates with disparate effects in two transgenic mouse models lacking the neurofilament light subunit. J. Neurosci. 20, 5321–5328. doi: 10.1523/JNEUROSCI.20-14-05321.2000
Beaulieu, J. M., and Julien, J. P. (2003). Peripherin-mediated death of motor neurons rescued by overexpression of neurofilament NF-H proteins. J. Neurochem. 85, 248–256. doi: 10.1046/j.1471-4159.2003.01653.x
Beaulieu, J. M., Nguyen, M. D., and Julien, J. P. (1999). Late onset of motor neurons in mice overexpressing wild-type peripherin. J. Cell Biol. 147, 531–544. doi: 10.1083/jcb.147.3.531
Bercier, V., Hubbard, J. M., Fidelin, K., Duroure, K., Auer, T. O., Revenu, C., et al. (2019). Dynactin1 depletion leads to neuromuscular synapse instability and functional abnormalities. Mol. Neurodegener. 14:27. doi: 10.1186/s13024-019-0327-3
Bergeron, C., Beric-Maskarel, K., Muntasser, S., Weyer, L., Somerville, M. J., and Percy, M. E. (1994). Neurofilament light and polyadenylated mRNA levels are decreased in amyotrophic lateral sclerosis motor neurons. J. Neuropathol. Exp. Neurol. 53, 221–230. doi: 10.1097/00005072-199405000-00002
Brettle, M., Stefen, H., Djordjevic, A., Fok, S. Y. Y., Chan, J. W., van Hummel, A., et al. (2019). Developmental expression of mutant PFN1 in motor neurons impacts neuronal growth and motor performance of young and adult mice. Front. Mol. Neurosci. 12:231. doi: 10.3389/fnmol.2019.00231
Brown, R. H., and Al-Chalabi, A. (2017). Amyotrophic lateral sclerosis. N. Engl. J. Med. 377, 162–172. doi: 10.1056/NEJMra1603471
Cai, H., Lin, X., Xie, C., Laird, F. M., Lai, C., Wen, H., et al. (2005). Loss of ALS2 function is insufficient to trigger motor neuron degeneration in knock-out mice but predisposes neurons to oxidative stress. J. Neurosci. 25, 7567–7574. doi: 10.1523/JNEUROSCI.1645-05.2005
Cai, H., Shim, H., Lai, C., Xie, C., Lin, X., Yang, W. J., et al. (2008). ALS2/alsin knockout mice and motor neuron diseases. Neurodegener. Dis. 5, 359–366. doi: 10.1159/000151295
Chandran, J., Ding, J., and Cai, H. (2007). Alsin and the molecular pathways of amyotrophic lateral sclerosis. Mol. Neurobiol. 36, 224–231. doi: 10.1007/s12035-007-0034-x
Chevalier-Larsen, E., and Holzbaur, E. L. (2006). Axonal transport and neurodegenerative disease. Biochim. Biophys. Acta 1762, 1094–1108. doi: 10.1016/j.bbadis.2006.04.002
Clark, J. A., Yeaman, E. J., Blizzard, C. A., Chuckowree, J. A., and Dickson, T. C. (2016). A case for microtubule vulnerability in amyotrophic lateral sclerosis: altered dynamics during disease. Front. Cell. Neurosci. 10:204. doi: 10.3389/fncel.2016.00204
Collard, J. F., Côté, F., and Julien, J. P. (1995). Defective axonal transport in a transgenic mouse model of amyotrophic lateral sclerosis. Nature 375, 61–64. doi: 10.1038/375061a0
Corbo, M., and Hays, A. P. (1992). Peripherin and neurofilament protein coexist in spinal spheroids of motor neuron disease. J. Neuropathol. Exp. Neurol. 51, 531–537. doi: 10.1097/00005072-199209000-00008
Corrado, L., Carlomagno, Y., Falasco, L., Mellone, S., Godi, M., Cova, E., et al. (2011). A novel peripherin gene (PRPH) mutation identified in one sporadic amyotrophic lateral sclerosis patient. Neurobiol. Aging 32, 552.e1–556.e6. doi: 10.1016/j.neurobiolaging.2010.02.011
Côté, F., Collard, J. F., and Julien, J. P. (1993). Progressive neuronopathy in transgenic mice expressing the human neurofilament heavy gene: a mouse model of amyotrophic lateral sclerosis. Cell 73, 35–46. doi: 10.1016/0092-8674(93)90158-m
Couillard-Després, S., Zhu, Q., Wong, P. C., Price, D. L., Cleveland, D. W., and Julien, J. P. (1998). Protective effect of neurofilament heavy gene overexpression in motor neuron disease induced by mutant superoxide dismutase. Proc. Natl. Acad. Sci. U S A 95, 9626–9630. doi: 10.1073/pnas.95.16.9626
Denton, K. R., Lei, L., Grenier, J., Rodionov, V., Blackstone, C., and Li, X. J. (2014). Loss of spastin function results in disease-specific axonal defects in human pluripotent stem cell-based models of hereditary spastic paraplegia. Stem Cells 32, 414–423. doi: 10.1002/stem.1569
Denton, K. R., Xu, C., Shah, H., and Li, X. J. (2016). Modeling axonal defects in hereditary spastic paraplegia with human pluripotent stem cells. Front. Biol. 11, 339–354. doi: 10.1007/s11515-016-1416-0
Eaton, B. A., Fetter, R. D., and Davis, G. W. (2002). Dynactin is necessary for synapse stabilization. Neuron 34, 729–741. doi: 10.1016/s0896-6273(02)00721-3
Errico, A., Ballabio, A., and Rugarli, E. I. (2002). Spastin, the protein mutated in autosomal dominant hereditary spastic paraplegia, is involved in microtubule dynamics. Hum. Mol. Genet. 11, 153–163. doi: 10.1093/hmg/11.2.153
Figlewicz, D. A., Krizus, A., Martinoli, M. G., Meininger, V., Dib, M., Rouleau, G. A., et al. (1994). Variants of the heavy neurofilament subunit are associated with the development of amyotrophic lateral sclerosis. Hum. Mol. Genet. 3, 1757–1761. doi: 10.1093/hmg/3.10.1757
Fil, D., DeLoach, A., Yadav, S., Alkam, D., MacNicol, M., Singh, A., et al. (2017). Mutant Profilin1 transgenic mice recapitulate cardinal features of motor neuron disease. Hum. Mol. Genet. 26, 686–701. doi: 10.1093/hmg/ddw429
Geevasinga, N., Menon, P., Sue, C. M., Kumar, K. R., Ng, K., Yiannikas, C., et al. (2015). Cortical excitability changes distinguish the motor neuron disease phenotypes from hereditary spastic paraplegia. Eur. J. Neurol. 22, 826.e57-8–831.e57-8. doi: 10.1111/ene.12669
Gros-Louis, F., Larivière, R., Gowing, G., Laurent, S., Camu, W., Bouchard, J. P., et al. (2004). A frameshift deletion in peripherin gene associated with amyotrophic lateral sclerosis. J. Biol. Chem. 279, 45951–45956. doi: 10.1074/jbc.M408139200
Hadano, S., Hand, C. K., Osuga, H., Yanagisawa, Y., Otomo, A., Devon, R. S., et al. (2001). A gene encoding a putative GTPase regulator is mutated in familial amyotrophic lateral sclerosis 2. Nat. Genet. 29, 166–173. doi: 10.1038/ng1001-166
Hadano, S., Otomo, A., Kunita, R., Suzuki-Utsunomiya, K., Akatsuka, A., Koike, M., et al. (2010). Loss of ALS2/Alsin exacerbates motor dysfunction in a SOD1-expressing mouse ALS model by disturbing endolysosomal trafficking. PLoS One 5:e9805. doi: 10.1371/journal.pone.0009805
Hadano, S., Otomo, A., Suzuki-Utsunomiya, K., Kunita, R., Yanagisawa, Y., Showguchi-Miyata, J., et al. (2004). ALS2CL, the novel protein highly homologous to the carboxy-terminal half of ALS2, binds to Rab5 and modulates endosome dynamics. FEBS Lett. 575, 64–70. doi: 10.1016/j.febslet.2004.07.092
Hand, C. K., Devon, R. S., Gros-Louis, F., Rochefort, D., Khoris, J., Meininger, V., et al. (2003). Mutation screening of the ALS2 gene in sporadic and familial amyotrophic lateral sclerosis. Arch. Neurol. 60, 1768–1771. doi: 10.1001/archneur.60.12.1768
Helferich, A. M., Brockmann, S. J., Reinders, J., Deshpande, D., Holzmann, K., Brenner, D., et al. (2018). Dysregulation of a novel miR-1825/TBCB/TUBA4A pathway in sporadic and familial ALS. Cell. Mol. Life Sci. 75, 4301–4319. doi: 10.1007/s00018-018-2873-1
Hirano, A., Donnenfeld, H., Sasaki, S., and Nakano, I. (1984). Fine structural observations of neurofilamentous changes in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 43, 461–470. doi: 10.1097/00005072-198409000-00001
Hsu, C. C., Moncaleano, J. D., and Wagner, O. I. (2011). Sub-cellular distribution of UNC-104(KIF1A) upon binding to adaptors as UNC-16(JIP3), DNC-1(DCTN1/Glued) and SYD-2(Liprin-α) in C. elegans neurons. Neuroscience 176, 39–52. doi: 10.1016/j.neuroscience.2010.12.044
Ikenaka, K., Kawai, K., Katsuno, M., Huang, Z., Jiang, Y. M., Iguchi, Y., et al. (2013). dnc-1/dynactin 1 knockdown disrupts transport of autophagosomes and induces motor neuron degeneration. PLoS One 8:e54511. doi: 10.1371/journal.pone.0054511
Ingre, C., Landers, J. E., Rizik, N., Volk, A. E., Akimoto, C., Birve, A., et al. (2013). A novel phosphorylation site mutation in profilin 1 revealed in a large screen of US, Nordic and German amyotrophic lateral sclerosis/frontotemporal dementia cohorts. Neurobiol. Aging 34, 1708.e1–1708.e6. doi: 10.1016/j.neurobiolaging.2012.10.009
Jacquier, A., Buhler, E., Schäfer, M. K., Bohl, D., Blanchard, S., Beclin, C., et al. (2006). Alsin/Rac1 signaling controls survival and growth of spinal motoneurons. Ann. Neurol. 60, 105–117. doi: 10.1002/ana.20886
Jiang, Y. M., Yamamoto, M., Tanaka, F., Ishigaki, S., Katsuno, M., Adachi, H., et al. (2007). Gene expressions specifically detected in motor neurons (dynactin 1, early growth response 3, acetyl-CoA transporter, death receptor 5 and cyclin C) differentially correlate to pathologic markers in sporadic amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 66, 617–627. doi: 10.1097/nen.0b013e318093ece3
Julien, J. P. (1997). Neurofilaments and motor neuron disease. Trends Cell Biol. 7, 243–249. doi: 10.1016/S0962-8924(97)01049-0
Kanekura, K., Hashimoto, Y., Niikura, T., Aiso, S., Matsuoka, M., and Nishimoto, I. (2004). Alsin, the product of ALS2 gene, suppresses SOD1 mutant neurotoxicity through RhoGEF domain by interacting with SOD1 mutants. J. Biol. Chem. 279, 19247–19256. doi: 10.1074/jbc.M313236200
Karle, K. N., Möckel, D., Reid, E., and Schöls, L. (2012). Axonal transport deficit in a KIF5A−/− mouse model. Neurogenetics 13, 169–179. doi: 10.1007/s10048-012-0324-y
Kasher, P. R., De Vos, K. J., Wharton, S. B., Manser, C., Bennett, E. J., Bingley, M., et al. (2009). Direct evidence for axonal transport defects in a novel mouse model of mutant spastin-induced hereditary spastic paraplegia (HSP) and human HSP patients. J. Neurochem. 110, 34–44. doi: 10.1111/j.1471-4159.2009.06104.x
Keller, B. A., Volkening, K., Droppelmann, C. A., Ang, L. C., Rademakers, R., and Strong, M. J. (2012). Co-aggregation of RNA binding proteins in ALS spinal motor neurons: evidence of a common pathogenic mechanism. Acta Neuropathol. 124, 733–747. doi: 10.1007/s00401-012-1035-z
Kuźma-Kozakiewicz, M., Chudy, A., Kaźmierczak, B., Dziewulska, D., Usarek, E., and Barańczyk-Kuźma, A. (2013). Dynactin deficiency in the CNS of humans with sporadic ALS and mice with genetically determined motor neuron degeneration. Neurochem. Res. 38, 2463–2473. doi: 10.1007/s11064-013-1160-7
Lai, C., Lin, X., Chandran, J., Shim, H., Yang, W. J., and Cai, H. (2007). The G59S mutation in p150(glued) causes dysfunction of dynactin in mice. J. Neurosci. 27, 13982–13990. doi: 10.1523/JNEUROSCI.4226-07.2007
Lai, C., Xie, C., McCormack, S. G., Chiang, H. C., Michalak, M. K., Lin, X., et al. (2006). Amyotrophic lateral sclerosis 2-deficiency leads to neuronal degeneration in amyotrophic lateral sclerosis through altered AMPA receptor trafficking. J. Neurosci. 26, 11798–11806. doi: 10.1523/JNEUROSCI.2084-06.2006
Laird, F. M., Farah, M. H., Ackerley, S., Hoke, A., Maragakis, N., Rothstein, J. D., et al. (2008). Motor neuron disease occurring in a mutant dynactin mouse model is characterized by defects in vesicular trafficking. J. Neurosci. 28, 1997–2005. doi: 10.1523/JNEUROSCI.4231-07.2008
Lee, M. K., Marszalek, J. R., and Cleveland, D. W. (1994). A mutant neurofilament subunit causes massive, selective motor neuron death: implications for the pathogenesis of human motor neuron disease. Neuron 13, 975–988. doi: 10.1016/0896-6273(94)90263-1
Leung, C. L., He, C. Z., Kaufmann, P., Chin, S. S., Naini, A., Liem, R. K., et al. (2004). A pathogenic peripherin gene mutation in a patient with amyotrophic lateral sclerosis. Brain Pathol. 14, 290–296. doi: 10.1111/j.1750-3639.2004.tb00066.x
Levy, J. R., Sumner, C. J., Caviston, J. P., Tokito, M. K., Ranganathan, S., Ligon, L. A., et al. (2006). A motor neuron disease-associated mutation in p150Glued perturbs dynactin function and induces protein aggregation. J. Cell Biol. 172, 733–745. doi: 10.1083/jcb.200511068
Li, J., He, J., Tang, L., Chen, L., Ma, Y., and Fan, D. (2018). Screening for TUBA4A mutations in a large Chinese cohort of patients with ALS: re-evaluating the pathogenesis of TUBA4A in ALS. J. Neurol. Neurosurg. Psychiatry 89, 1350–1352. doi: 10.1136/jnnp-2017-317560
Lipton, A. M., White, C. L., and 3rd and Bigio, E. H. (2004). Frontotemporal lobar degeneration with motor neuron disease-type inclusions predominates in 76 cases of frontotemporal degeneration. Acta Neuropathol. 108, 379–385. doi: 10.1007/s00401-004-0900-9
Liu, Z. J., Li, H. F., Tan, G. H., Tao, Q. Q., Ni, W., Cheng, X. W., et al. (2014). Identify mutation in amyotrophic lateral sclerosis cases using HaloPlex target enrichment system. Neurobiol. Aging 35, 2881.e11–2881.e11. doi: 10.1016/j.neurobiolaging.2014.07.003
Maraldi, T., Beretti, F., Anselmi, L., Franchin, C., Arrigoni, G., Braglia, L., et al. (2019). Influence of selenium on the emergence of neuro tubule defects in a neuron-like cell line and its implications for amyotrophic lateral sclerosis. Neurotoxicology 75, 209–220. doi: 10.1016/j.neuro.2019.09.015
Martin, M., Iyadurai, S. J., Gassman, A., Gindhart, J. G. Jr., Hays, T. S., and Saxton, W. M. (1999). Cytoplasmic dynein, the dynactin complex and kinesin are interdependent and essential for fast axonal transport. Mol. Biol. Cell 10, 3717–3728. doi: 10.1091/mbc.10.11.3717
Mathis, S., Goizet, C., Soulages, A., Vallat, J. M., and Masson, G. L. (2019). Genetics of amyotrophic lateral sclerosis: a review. J. Neurol. Sci. 399, 217–226. doi: 10.1016/j.jns.2019.02.030
Matsuzaki, F., Shirane, M., Matsumoto, M., and Nakayama, K. I. (2011). Protrudin serves as an adaptor molecule that connects KIF5 and its cargoes in vesicular transport during process formation. Mol. Biol. Cell 22, 4602–4620. doi: 10.1091/mbc.E11-01-0068
McLean, J. R., Smith, G. A., Rocha, E. M., Osborn, T. M., Dib, S., Hayes, M. A., et al. (2014). ALS-associated peripherin spliced transcripts form distinct protein inclusions that are neuroprotective against oxidative stress. Exp. Neurol. 261, 217–229. doi: 10.1016/j.expneurol.2014.05.024
Meyer, T., Schwan, A., Dullinger, J. S., Brocke, J., Hoffmann, K. T., Nolte, C. H., et al. (2005). Early-onset alS with long-term survival associated with spastin gene mutation. Neurology 65, 141–143. doi: 10.1212/01.wnl.0000167130.31618.0a
Migheli, A., Pezzulo, T., Attanasio, A., and Schiffer, D. (1993). Peripherin immunoreactive structures in amyotrophic lateral sclerosis. Lab. Invest. 68, 185–191.
Millecamps, S., Robertson, J., Lariviere, R., Mallet, J., and Julien, J. P. (2006). Defective axonal transport of neurofilament proteins in neurons overexpressing peripherin. J. Neurochem. 98, 926–938. doi: 10.1111/j.1471-4159.2006.03932.x
Moughamian, A. J., and Holzbaur, E. L. (2012). Dynactin is required for transport initiation from the distal axon. Neuron 74, 331–343. doi: 10.1016/j.neuron.2012.02.025
Münch, C., Rolfs, A., and Meyer, T. (2008). Heterozygous S44L missense change of the spastin gene in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 9, 251–253. doi: 10.1080/17482960801900172
Münch, C., Rosenbohm, A., Sperfeld, A. D., Uttner, I., Reske, S., Krause, B. J., et al. (2005). Heterozygous R1101K mutation of the DCTN1 gene in a family with ALS and FTD. Ann. Neurol. 58, 777–780. doi: 10.1002/ana.20631
Münch, C., Sedlmeier, R., Meyer, T., Homberg, V., Sperfeld, A. D., Kurt, A., et al. (2004). Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology 63, 724–726. doi: 10.1212/01.wnl.0000134608.83927.b1
Nguyen, D. K. H., Thombre, R., and Wang, J. (2019). Autophagy as a common pathway in amyotrophic lateral sclerosis. Neurosci. Lett. 697, 34–48. doi: 10.1016/j.neulet.2018.04.006
Nicolas, A., Kenna, K. P., Renton, A. E., Ticozzi, N., Faghri, F., Chia, R., et al. (2018). Genome-wide analyses identify KIF5A as a novel ALS gene. Neuron 97, 1268.e6–1283.e6. doi: 10.1016/j.neuron.2018.02.027
Pensato, V., Tiloca, C., Corrado, L., Bertolin, C., Sardone, V., Del Bo, R., et al. (2015). TUBA4A gene analysis in sporadic amyotrophic lateral sclerosis: identification of novel mutations. J. Neurol. 262, 1376–1378. doi: 10.1007/s00415-015-7739-y
Poesen, K., and Van Damme, P. (2018). Diagnostic and prognostic performance of neurofilaments in ALS. Front. Neurol. 9:1167. doi: 10.3389/fneur.2018.01167
Puls, I., Jonnakuty, C., LaMonte, B. H., Holzbaur, E. L., Tokito, M., Mann, E., et al. (2003). Mutant dynactin in motor neuron disease. Nat. Genet. 33, 455–456. doi: 10.1038/ng1123
Robertson, J., Doroudchi, M. M., Nguyen, M. D., Durham, H. D., Strong, M. J., Shaw, G., et al. (2003). A neurotoxic peripherin splice variant in a mouse model of ALS. J. Cell Biol. 160, 939–949. doi: 10.1083/jcb.200205027
Rosen, D. R., Siddique, T., Patterson, D., Figlewicz, D. A., Sapp, P., Hentati, A., et al. (1993). Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62. doi: 10.1038/362059a0
Rustici, G., Kolesnikov, N., Brandizi, M., Burdett, T., Dylag, M., Emam, I., et al. (2013). ArrayExpress update—trends in database growth and links to data analysis tools. Nucleic Acids Res. 41, D987–D990. doi: 10.1093/nar/gks1174
Simone, M., Trabacca, A., Panzeri, E., Losito, L., Citterio, A., and Bassi, M. T. (2018). KIF5A and ALS2 variants in a family with hereditary spastic paraplegia and amyotrophic lateral sclerosis. Front. Neurol. 9:1078. doi: 10.3389/fneur.2018.01078
Smith, E. F., Shaw, P. J., and De Vos, K. J. (2019). The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 710:132933. doi: 10.1016/j.neulet.2017.06.052
Smith, B. N., Ticozzi, N., Fallini, C., Gkazi, A. S., Topp, S., Kenna, K. P., et al. (2014). Exome-wide rare variant analysis identifies TUBA4A mutations associated with familial ALS. Neuron 84, 324–331. doi: 10.1016/j.neuron.2014.09.027
Smith, B. N., Vance, C., Scotter, E. L., Troakes, C., Wong, C. H., Topp, S., et al. (2015). Novel mutations support a role for Profilin 1 in the pathogenesis of ALS. Neurobiol. Aging 36:1602.e17.–1627.e27. doi: 10.1016/j.neurobiolaging.2014.10.032
Stockmann, M., Meyer-Ohlendorf, M., Achberger, K., Putz, S., Demestre, M., Yin, H., et al. (2013). The dynactin p150 subunit: cell biology studies of sequence changes found in ALS/MND and Parkinsonian syndromes. J. Neural Transm. 120, 785–798. doi: 10.1007/s00702-012-0910-z
Suetsugu, S., Miki, H., and Takenawa, T. (1998). The essential role of profilin in the assembly of actin for microspike formation. EMBO J. 17, 6516–6526. doi: 10.1093/emboj/17.22.6516
Suzuki-Utsunomiya, K., Hadano, S., Otomo, A., Kunita, R., Mizumura, H., Osuga, H., et al. (2007). ALS2CL, a novel ALS2-interactor, modulates ALS2-mediated endosome dynamics. Biochem. Biophys. Res. Commun. 354, 491–497. doi: 10.1016/j.bbrc.2006.12.229
Tiloca, C., Ticozzi, N., Pensato, V., Corrado, L., Del Bo, R., Bertolin, C., et al. (2013). Screening of the PFN1 gene in sporadic amyotrophic lateral sclerosis and in frontotemporal dementia. Neurobiol. Aging 34:1517.e9.–1517.e10. doi: 10.1016/j.neurobiolaging.2012.09.016
Tischfield, M. A., Cederquist, G. Y., Gupta, M. L. Jr., and Engle, E. C. (2011). Phenotypic spectrum of the tubulin-related disorders and functional implications of disease-causing mutations. Curr. Opin. Genet. Dev. 21, 286–294. doi: 10.1016/j.gde.2011.01.003
Tu, P. H., Raju, P., Robinson, K. A., Gurney, M. E., Trojanowski, J. Q., and Lee, V. M. (1996). Transgenic mice carrying a human mutant superoxide dismutase transgene develop neuronal cytoskeletal pathology resembling human amyotrophic lateral sclerosis lesions. Proc. Natl. Acad. Sci. U S A 93, 3155–3160. doi: 10.1073/pnas.93.7.3155
Tudor, E. L., Perkinton, M. S., Schmidt, A., Ackerley, S., Brownlees, J., Jacobsen, N. J., et al. (2005). ALS2/Alsin regulates Rac-PAK signaling and neurite outgrowth. J. Biol. Chem. 280, 34735–34740. doi: 10.1074/jbc.M506216200
Urnavicius, L., Zhang, K., Diamant, A. G., Motz, C., Schlager, M. A., Yu, M., et al. (2015). The structure of the dynactin complex and its interaction with dynein. Science 347, 1441–1446. doi: 10.1126/science.aaa4080
Vance, C., Al-Chalabi, A., Ruddy, D., Smith, B. N., Hu, X., Sreedharan, J., et al. (2006). Familial amyotrophic lateral sclerosis with frontotemporal dementia is linked to a locus on chromosome 9p13.2–21.3. Brain 129, 868–876. doi: 10.1093/brain/awl030
Volk, A. E., Weishaupt, J. H., Andersen, P. M., Ludolph, A. C., and Kubisch, C. (2018). Current knowledge and recent insights into the genetic basis of amyotrophic lateral sclerosis. Med. Genet. 30, 252–258. doi: 10.1007/s11825-018-0185-3
Wills, Z., Marr, L., Zinn, K., Goodman, C. S., and Van Vactor, D. (1999). Profilin and the Abl tyrosine kinase are required for motor axon outgrowth in the Drosophila embryo. Neuron 22, 291–299. doi: 10.1016/s0896-6273(00)81090-9
Wu, C. H., Fallini, C., Ticozzi, N., Keagle, P. J., Sapp, P. C., Piotrowska, K., et al. (2012). Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature 488, 499–503. doi: 10.1038/nature11280
Xia, C. H., Roberts, E. A., Her, L. S., Liu, X., Williams, D. S., Cleveland, D. W., et al. (2003). Abnormal neurofilament transport caused by targeted disruption of neuronal kinesin heavy chain KIF5A. J. Cell Biol. 161, 55–66. doi: 10.1083/jcb.200301026
Xiao, S., Tjostheim, S., Sanelli, T., McLean, J. R., Horne, P., Fan, Y., et al. (2008). An aggregate-inducing peripherin isoform generated through intron retention is upregulated in amyotrophic lateral sclerosis and associated with disease pathology. J. Neurosci. 28, 1833–1840. doi: 10.1523/JNEUROSCI.3222-07.2008
Yang, S., Fifita, J. A., Williams, K. L., Warraich, S. T., Pamphlett, R., Nicholson, G. A., et al. (2013). Mutation analysis and immunopathological studies of PFN1 in familial and sporadic amyotrophic lateral sclerosis. Neurobiol. Aging 34, 2235.e7–2235.e10. doi: 10.1016/j.neurobiolaging.2013.04.003
Yang, Y., Hentati, A., Deng, H. X., Dabbagh, O., Sasaki, T., Hirano, M., et al. (2001). The gene encoding alsin, a protein with three guanine-nucleotide exchange factor domains, is mutated in a form of recessive amyotrophic lateral sclerosis. Nat. Genet. 29, 160–165. doi: 10.1038/ng1001-160
Keywords: ALS, ALS2, DCTN1, intermediate filaments, KIF5A, PFN1, SPAST, TUBA4A
Citation: Castellanos-Montiel MJ, Chaineau M and Durcan TM (2020) The Neglected Genes of ALS: Cytoskeletal Dynamics Impact Synaptic Degeneration in ALS. Front. Cell. Neurosci. 14:594975. doi: 10.3389/fncel.2020.594975
Received: 14 August 2020; Accepted: 21 October 2020;
Published: 13 November 2020.
Edited by:
Felipe A. Court, Universidad Mayor, ChileReviewed by:
Archan Ganguly, University of Rochester, United StatesJorge Matias-Guiu, Complutense University of Madrid, Spain
Copyright © 2020 Castellanos-Montiel, Chaineau and Durcan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas M. Durcan, dGhvbWFzLmR1cmNhbkBtY2dpbGwuY2E=