 Talia Bergaglio
Talia Bergaglio Antonio Luchicchi
Antonio Luchicchi Geert J. Schenk
Geert J. Schenk- Department of Anatomy and Neurosciences, Amsterdam Neuroscience, Amsterdam University Medical Center, Amsterdam MS Center, Amsterdam, Netherlands
Multiple Sclerosis (MS) is a complex and chronic disease of the central nervous system (CNS), characterized by both degenerative and inflammatory processes leading to axonal damage, demyelination, and neuronal loss. In the last decade, the traditional outside-in standpoint on MS pathogenesis, which identifies a primary autoimmune inflammatory etiology, has been challenged by a complementary inside-out theory. By focusing on the degenerative processes of MS, the axo-myelinic system may reveal new insights into the disease triggering mechanisms. Oxidative stress (OS) has been widely described as one of the means driving tissue injury in neurodegenerative disorders, including MS. Axonal mitochondria constitute the main energy source for electrically active axons and neurons and are largely vulnerable to oxidative injury. Consequently, axonal mitochondrial dysfunction might impair efficient axo-glial communication, which could, in turn, affect axonal integrity and the maintenance of axonal, neuronal, and synaptic signaling. In this review article, we argue that OS-derived mitochondrial impairment may underline the dysfunctional relationship between axons and their supportive glia cells, specifically oligodendrocytes and that this mechanism is implicated in the development of a primary cytodegeneration and a secondary pro-inflammatory response (inside-out), which in turn, together with a variably primed host’s immune system, may lead to the onset of MS and its different subtypes.
Introduction
Multiple Sclerosis (MS) is a complex, chronic progressive disorder of the central nervous system (CNS) and the most prominent cause of neurological disability in young adults (Noseworthy et al., 2000; Compston and Coles, 2002; Stys et al., 2012). Comprising of damage to both white and gray matter regions of the brain, evidence from histopathological findings identifies demyelination and axonal damage, as well as microglia activation, synaptic and neuronal loss as hallmarks of the disease (Geurts and Barkhof, 2008; Brinar and Braun, 2013; Klaver et al., 2013; Calabrese et al., 2015). Conflicting ideas have been proposed to explain the etiology of MS, particularly the origin of lesion formation and disease progression. The indisputable involvement of inflammatory processes has resulted in the development of the main model of MS, namely outside-in, whereby a dysregulated immune system in the periphery attacks elements within the CNS, causing demyelination and tissue damage (Stys et al., 2012; Dendrou et al., 2015). Once inflammation becomes compartmentalized within the CNS and compensatory mechanisms can no longer overcome the chronic inflammatory processes, the progressive phenotype takes over and current anti-inflammatory therapeutic strategies are no longer effective (Lassmann et al., 2012). Alternatively, MS may originate from the “inside-out,” with a primary degenerative episode, possibly evolving around the axo-myelinic synapse (AMS), disrupting the dynamic communication between axons and their myelinating oligodendrocyte, thus initiating a secondary inflammatory response due to the highly antigenic debris derived from myelin breakdown (Trapp and Nave, 2008; Stys et al., 2012). Consequently, the strength of the convolution between degenerative and inflammatory processes will derive the clinical course of the disease.
According to the inside-out model, more focus should be aimed at understanding the mechanisms underlying the neurodegenerative processes of MS. Observations of early-stage lesion formation, in both experimental models and human MS, corroborate on the presence of axonal injury in still myelinated axons, suggesting that, at least in some cases, degenerative CNS pathology precedes the peripheral inflammatory attack (Brück and Stadelmann, 2003; Barnett et al., 2009; Edgar et al., 2010; Nikić et al., 2011; Lubetzki and Stankoff, 2014). A potential mechanism driving inside-out immunopathogenesis may evolve around the myelinating unit (Stys, 2013). Here, earlier studies in mice revealed a crucial role of myelin molecules and oligodendroglial support in the maintenance of axonal integrity (Griffiths et al., 1998; Yin et al., 1998; Lappe-Siefke et al., 2003). More specifically, axonal metabolic support requires specific axo-myelinic communication, and disruptions to this relationship may induce primary axonal injury and potentially long-lasting myelin abnormalities (Micu et al., 2016).
Axonal and oligodendroglial processes are highly energy-consuming and as such, are vulnerable to metabolic challenges (Micu et al., 2018). Due to the crucial functions mitochondria play as the power plants of a cell, it is not surprising that both genetic and environmental factors altering their functions have profound downstream effects on axonal health (Campbell et al., 2014; Witte et al., 2014). One of the mechanisms that cause injury to mitochondria is Oxidative stress (OS), defined as an imbalance in Reactive Oxygen Species (ROS) production against the cell’s antioxidant defenses (Betteridge, 2000). Despite the established involvement of ROS-mediated tissue injury in MS inflammatory processes, the induction of pro-apoptotic mechanisms by OS as well as its impact on the mitochondrial respiratory chain may induce a state of energy deprivation that, if chronic, may ultimately initiate a cascade of degenerative processes, including axonal and neuronal death (Lassmann and van Horssen, 2011; Franklin et al., 2012; Stys, 2013; Ohl et al., 2016).
This mini review article aims to highlight a specific component of MS pathogenesis by lending credence to the role of OS-derived mitochondrial dysfunction as a potential key mechanism contributing to an unstable AMS, which may ultimately contribute to a primary cytodegeneration in MS pathogenesis. To do so, evidence supporting an inside-out view of MS will be presented, together with the recent insights into the function of axo-myelinic neurotransmission and the role of axonal mitochondria and OS-associated mitochondrial dysfunction. Here, we propose that OS-derived mitochondrial impairment may underline the dysfunctional relationship between axons and their supportive glia cells, which later initiates the primary cytodegeneration and secondary inflammatory processes of MS.
Multiple Sclerosis as A Primary Cytodegenerative Disease
Although an outside-in view of MS pathogenesis cannot be disregarded, the inside-out model is equally plausible and equally consistent with the majority of pathological observations. When viewing MS pathogenesis from the inside-out, axonal injury and myelin defects likely act as the initiators of the degenerative processes underlying the disease, due to their observation even in the absence of inflammation in human brain samples (Trapp et al., 1998; Traka et al., 2016). Histopathological analysis of patient material from very early stages of the disease has revealed myelin abnormalities, specifically involving the inner myelin sheath and oligodendrocyte loss with little peripheral inflammatory infiltration but with a more pronounced innate immune response (Rodriguez and Scheithauer, 1994; Aboul-Enein et al., 2003; Barnett and Prineas, 2004; Henderson et al., 2009). Moreover, the ineffectiveness of available anti-inflammatory treatments in halting disease progression further exacerbates the presence of underlying cytodegenerative processes driving MS progression (Seewann et al., 2009; Hawker, 2011; Lassmann, 2013). This evidence is corroborated by genetic studies. Immunologically relevant genes are significantly overrepresented in genome-wide association studies (Compston and Coles, 2002). Sawcer et al. (2011) found a strong correlation with immune-related genes within patients with RRMS mainly, which is to be expected due to the great inflammatory character of this MS variant and may explain symptomatic heterogeneity due to a variably primed host’s immune system (Stys et al., 2012). Interestingly, however, when a subgroup analysis of only PPMS patients was performed, no associations with genes related to the immune system were found, further indicating that these immune-related factors may only determine the intensity of autoimmune response to a degenerative brain (Stys and Tsutsui, 2019). Instead, a general state of chronic excitotoxicity seems to, at least in part, drive the degenerative processes of MS, whereby variations in genes related to glutamate signaling prevailed in PPMS patients (Baranzini et al., 2010; Strijbis et al., 2013). Due to the chronic and progressive fate of MS, which will rarely be fatal in the early stages, all human neuropathological studies will inevitably mirror a combination of degenerative processes and inflammatory reactions that have evolved over many months or years (Stys, 2013). Hence, it is crucial to critically acknowledge that a single histopathological snapshot in time may not be fully representative of the initial events of MS pathogenesis.
If we consider progressive MS to reflect the real pathogenic mechanisms of the disease, then the origin of lesion formation lays within episodes of axonal injury and disruptions to axo-myelinic communication (Lassmann et al., 2012; Friese et al., 2014; Mahad et al., 2015; Guttmann et al., 2016). Thus, axonal injury may start at an early, and yet not observable, stage of the disease and it does not initially manifest in neurological disability (Trapp et al., 1998). Indeed, the CNS comprises many reparative mechanisms that allow for the compensation of axonal loss (Bjartmar et al., 2003). Once acute demyelination progresses into a more chronic state, demyelinated axons hardly survive and degenerative mechanisms become more prevalent. Approximately up to 60–70% of axonal loss was estimated in chronic white matter lesions in severely disabled MS patients (Mews et al., 1998). It is still hypothetical which exact mechanism may generate damage to the axons and whether axonal injury may represent a primary degenerative process, or maybe caused by secondary, non-inflammatory, processes (Stys et al., 2012). However, disruption of the close dynamic relationship between axons and their insulating myelin sheaths has been identified as a potential mode of action leading to a state of energy deficiency and, consequently, axonal injury (Tsutsui and Stys, 2013; Simons et al., 2014).
Axon-Glia Interaction
The architecture of the axo-myelinic unit is very intricate. Although the myelin sheath supports the electrical properties of the axon, it simultaneously limits the access of the axon to its extracellular environment (Nave, 2010; Simons et al., 2014). Nevertheless, the studies cited provide strong evidence for the implication of the myelinating oligodendrocytes in sustaining the axons by providing the necessary metabolic support (Stys, 2011; Fünfschilling et al., 2012; Lee et al., 2012; Micu et al., 2016). The large and complex crosstalk network between axons and oligodendrocytes constitutes an essential part of the proper functioning of the CNS and exposes the system to a diffuse vulnerability to disorders affecting the myelin (Ortiz et al., 2016).
Despite the close relationship between axons and myelin, only recently, an activity-dependent communication between the two was proposed, namely the axo-myelinic synapse, whose arrangement presents very similar features to the traditional interneuronal synapses (Stys, 2011; Micu et al., 2016, 2018). One critical function of the AMS is thus to couple electrical activity along the axons to the metabolic output from the oligodendrocytes. Remarkably, axons may signal the oligodendrocyte, through AMPA and NMDA myelinic receptors, to supply certain metabolites to fuel the electrically active fiber in response to physiological stimuli; as a result, the glial cells will transfer such metabolites to the internodal axon, providing it with the necessary energy support for signal conductance (Stys, 2011; Micu et al., 2018). In this view, Micu et al. (2016, 2018) propose a potential mechanism of neurotransmission underlying the tightly orchestrated complex directed by the AMS, which is further described in Figure 1.
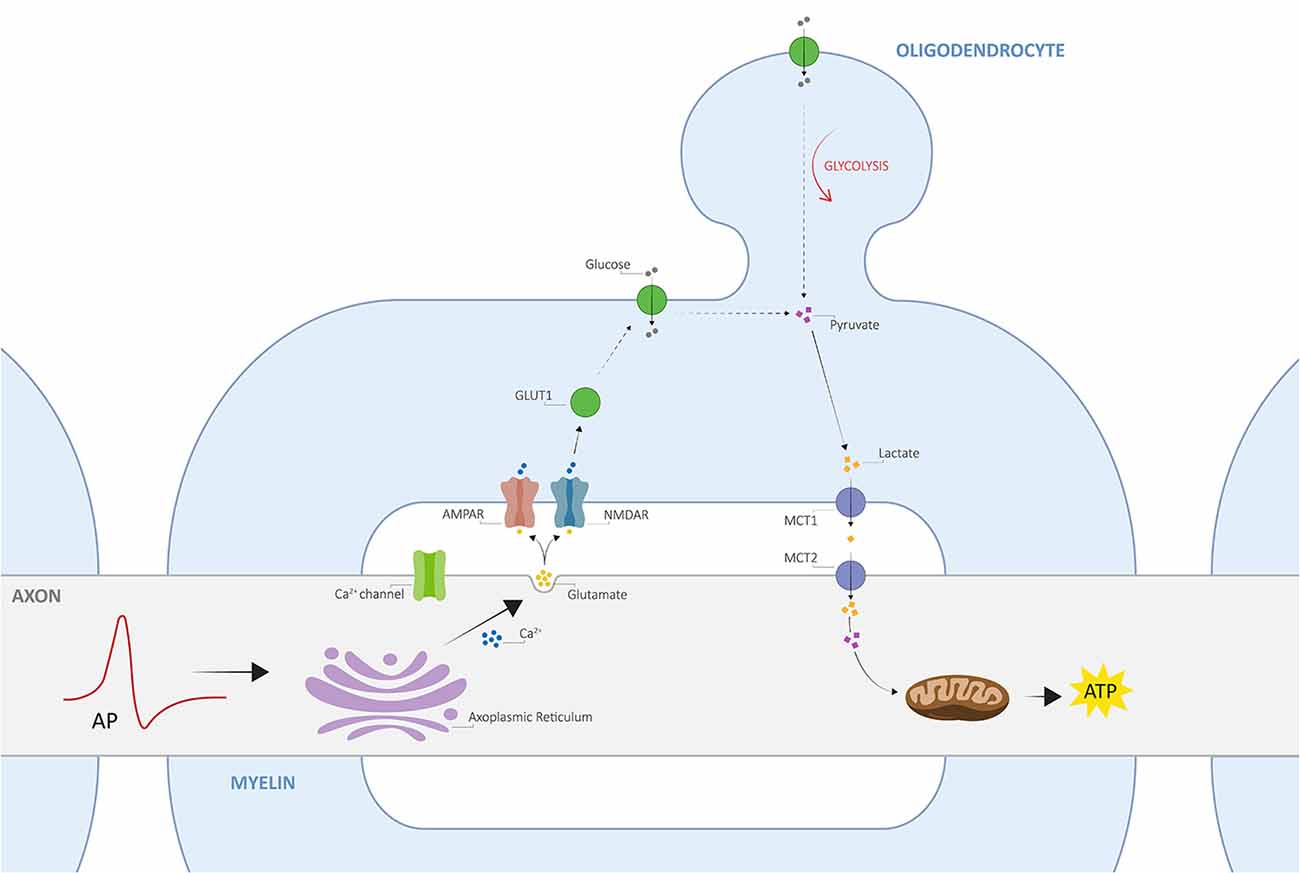
Figure 1. The architecture of axo-myelinic neurotransmission. Traversing action potentials cause depolarization of the axon. The latter is sensed by voltage-gated Ca2+ channels, which in turn activate and cause intra-axonal Ca2+ release from the axoplasmic reticulum. This stimulates the fusion of glutamatergic vesicles and the consequent release of glutamate into the periaxonal space, which in turn activates myelinic AMPA and NMDA receptors, located on the innermost myelin sheaths (AMPARs and NMDARs, respectively), finally promoting Ca2+ influx into the myelin cytoplasm. Myelin receptor activation further results in the recruitment of glucose transporter type 1 (GLUT1), increased uptake of glucose, and the stimulation of glycolysis by the oligodendrocyte, where the production of pyruvate and lactate is enhanced. Pyruvate is then used as an energy supply by myelinic mitochondria, whereas lactate is transported across the periaxonal space and into the axon by monocarboxylate transporters 1 and 2 (MCT1 and MCT2). Finally, lactate is used to fuel aerobic metabolism by axonal mitochondria for the efficient internodal production of ATP. Adapted from Micu et al. (2018); with permission from the authors and SpringerNature.
The continuous supply of energy along the entire length of the myelinated axon is crucial for the efficient conduction of action potentials as well as for the proper maintenance of neuronal functioning, including its myelinating system. Consequently, the energy supply from the myelinating oligodendrocytes is suggested to be of vital importance to fuelling the axon, following transient increases in energy demands and, more generally, to the vast dynamic range of firing frequencies of myelinated axons (Trevisiol et al., 2017). Glucose is the primary energy source in the adult brain and it fuels neuronal activity via aerobic respiration by mitochondria (Su et al., 2009). Given the crucial role of mitochondria in driving the majority of cellular processes by providing the necessary energy, it is not surprising that mitochondrial dysfunction can result in significant neuronal injury and degenerative processes (Lin and Beal, 2006; Mahad et al., 2008; Witte et al., 2010).
The balance between ROS production and antioxidant defenses under normal physiological conditions can be disrupted with an overproduction of free radicals, namely OS. Oxidants can play a dual role as both toxic and beneficial compounds, the latter due to their crucial role as essential signaling molecules (Pham-Huy et al., 2008). They are produced from both endogenous (e.g., cell metabolism) and exogenous (e.g., cigarette smoking, environmental toxins, chronic physiological stress, iron overload, inflammation, et cetera) sources and can contribute to disease via disruptions to cellular homeostasis by redox signaling (Bhattacharyya et al., 2014; Basria et al., 2019). Over time, exposure to multiple inciting factors, as well as the failure of enzymes responsible for redox homeostasis and detoxification activities, may trigger a chronic imbalance between ROS production and antioxidant defenses (Bhattacharyya et al., 2014; Basria et al., 2019). Excess free radicals can lead to mitochondrial dysfunction by inducing mitochondrial DNA mutations, damage to its respiratory chain, alteration of its membrane permeability, and by influencing Ca2+ homeostasis and mitochondrial defense systems (Guo et al., 2013). Once damaged, mitochondrial dysfunctional processes can further amplify OS and generate tissue injury via three crucial mechanisms, including energy failure, induction of apoptosis, and enhanced production of ROS (Witte et al., 2014). Energy deficiency derived from mitochondrial dysfunction poses the axon to a state of ‘virtual hypoxia’, whereby axon-glia energy metabolism would directly influence axon-myelin transmission and result in chronic and progressive damage to the axons (Lassmann et al., 2012). More specifically, when energy failure occurs, sodium ions begin to accumulate within the axon, causing, together with membrane depolarization, the reverse mode of action of the Na+-Ca2+ exchanger (Franklin et al., 2012; Campbell et al., 2014). As a result, detrimental levels of calcium ions build up to a point where they can interfere with axon survival and lead to axonal injury (Tsutsui and Stys, 2013). Through in vivo calcium imaging, Witte et al. (2019) have shown that activation of calpains, Ca2+-dependent, non-lysosomal proteases, upon increased levels of axoplasmic Ca2+, can promote the breakdown of the cytoskeleton as well as oncotic axonal swelling. Activated calpains are also responsible for the permeabilization of lysosomal membranes, which in turn causes the release of the lysosomal hydrolytic enzyme Cathepsin, an important mediator of apoptosis (Czaja, 2001). Both activation of Ca2+-dependent proteases, as well as mitochondrial impairment, have been associated with the degradation of myelin-associated glycoprotein (MAG), an adhesive protein located on the periaxonal surface of myelin sheaths and responsible for the inhibition of axon regeneration and the control of myelin formation and maintenance (Kroemer and Jäättelä, 2005; Zong and Thompson, 2006). MAG loss, together with other myelinic proteins, is thought to destabilize the AMS by dysregulating the axon cytoskeleton and by inducing myelin breakdown, further exacerbating myelin-axon communication (Schnaar and Lopez, 2009).
Mitochondrial Oxidative Stress in Axo-Myelinic Neurotransmission: A Key Player in Ms Primary Degeneration?
Here, we propose an alternative view of MS pathogenesis, whereby OS-derived mitochondrial impairment acts as the key initiator of a disrupted axo-myelinic relationship and consequently drives the onset of primary cytodegenerative and secondary inflammatory processes (Figure 2).
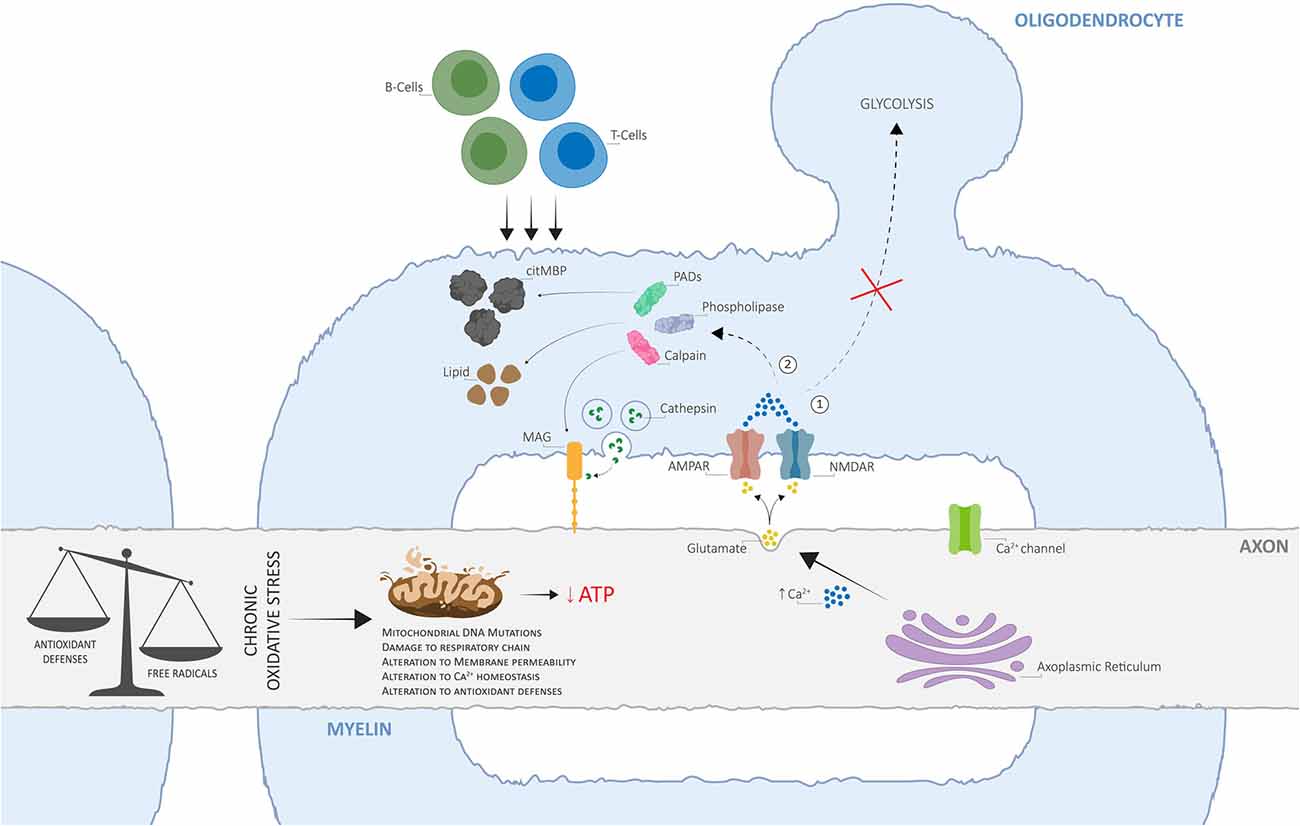
Figure 2. The alternative view of Multiple Sclerosis (MS) pathogenesis is based on dysfunctional axo-myelinic neurotransmission due to oxidative stress (OS)-derived mitochondrial impairment. Chronic OS mechanisms, originating from excess free radical production (due to cell metabolism, physiological stress, iron overload, environmental toxins, cigarette smoking, et cetera) in relation to the cell’s antioxidant defenses, can result in the inability of the axonal mitochondria to synthesize ATP, leading to a state of chronic virtual hypoxia (energy failure). As a result, the failure of ion transporters and the consequent influx of Na+ ions, activate voltage-gated Ca2+ channels and induce the excessive intra-axonal release of Ca2+. The latter stimulates the excessive vesicular release of glutamate into the periaxonal space, over-activating myelinic AMPA and NMDA receptors and resulting in excessive Ca2+ influx into the myelin cytoplasm, leading to two crucial pathological outcomes: (1) the inability of the oligodendrocyte to generate pyruvate and lactate, by glycolysis, for metabolic support to the axon; (2) activation of Ca2+-dependent calpains, phospholipases, and PADs, resulting in the degradation of myelin proteins, including myelin-associated glycoprotein (MAG), phospholipids, and in the conversion of MBP into citMBP, respectively, thus inducing focal disruptions to the myelin sheath and destabilization of the axo-myelinic synapse (AMS). The consequent release of antigenic citMBP and lipid debris can result in an adaptive immune response, driven by T- and B-cells, which can cause further inflammatory reactions. Adapted from Micu et al. (2018), with permission from the authors and SpringerNature.
Dysfunctional activation of axo-myelinic neurotransmission may be driven by complementary processes originating from excess free radical production. OS can directly affect mitochondrial functioning via different pathways, each leading to a state of energy failure within the affected axons (Guo et al., 2013). The presence of excess ROS, including hydrogen peroxide, puts pressure on cellular antioxidant defense systems, such as glutathione peroxidase (GPx), to restore metabolic balance (Carvalho et al., 2014). GPx is an intracellular antioxidant enzyme that reduces hydrogen peroxide to water to both limit its harmful effects and, indirectly, to modulate mitochondrial oxidative phosphorylation (Lubos et al., 2011). Consequently, with increasing levels of oxyradicals, the production of ATP by axonal mitochondria can be depleted via the GPx system. Additionally, the accumulation of calcium ions derived from OS processes would result in detrimental effects not only on the axon itself, via over-activation of Ca2+-dependent axonal enzymes, but also on the overlying myelin sheath, by stimulating excessive vesicular glutamate release into the periaxonal space. Elevated Ca2+ levels also inhibit axonal transport of mitochondria, thus immobilizing stationary mitochondria to the affected sites, leading to the degeneration of the entire axon and, consequently, neuronal injury (Su et al., 2009). Most importantly, over time, excessive Ca2+ entry into the myelin sheath, together with a state of energy deprivation, will cause both the myelin and the oligodendrocyte to become unable to buffer the Ca2+ loads, thus hindering the transport of lactate to the axon for metabolic support (Tsutsui and Stys, 2013; Micu et al., 2018; Poerwoatmodjo et al., 2020). A recent study presented evidence for impaired glycolysis and mitochondrial respiration during T-cell activation in RRMS patients (La Rocca et al., 2017). Here, these changes were associated with a down-regulation of GLUT1 resulting from the enhanced entry of calcium ions inside the myelin sheath. Also, the inability to buffer elevated Ca2+ levels will likely trigger a series of enzymatic pathways leading to the degradation of myelin proteins and phospholipids and thus, to axonal demyelination (Stys, 2013; Micu et al., 2018). Finally, the loss of positive charge on citrullinated myelin basic protein (citMBP), induced by the conversion of positively charged arginine residues on MBP to citrulline by peptidyl arginine deiminases (Ca2+-dependent enzymes; PADs), causes focal disruption to the myelin sheath (Caprariello et al., 2018; Micu et al., 2018). The consequent release of antigenic citMBP and lipid debris can result in an adaptive immune response in a host with a reactive immune system (Micu et al., 2018). Primary injury to axonal mitochondria and subsequent demyelination may, in turn, attract leukocytes from the bloodstream, which is in line with the observation that lesions and cell infiltration tend to occur around blood vessels (Gaitán et al., 2013; Lopes Pinheiro et al., 2016). This autoimmune attack, driven by peripheral T- and B-cells, resembles the primary immune-mediated inflammatory response widely described by supporters of the outside-in model of MS. Therefore, although the loss of metabolic support may be the initiator of progressive degeneration in MS, it is unlikely to be the only underlying mechanism; a gain of toxic function is likely to promote further, and potentially irreversible, neurological damage (Micu et al., 2018).
Given the supportive interdependence of neuronal and glial health, pathology from a single axon may spread throughout the nervous system following both a transversal pattern, as well as a process similar to Wallerian degeneration in the peripheral nervous system (PNS; Chong et al., 2012; Singh et al., 2013; Simons et al., 2014). Both spreading modalities can ultimately reflect the pattern of pathology in MS patients, whereby both a longitudinal spread across the pyramidal tracts and a more transversal spread in white and gray matter regions of the CNS can be observed (DeLuca et al., 2004). Given the comparable metabolic rate of white and gray matter, both regions are equally highly vulnerable to interruptions of energy supply (Goldberg and Ransom, 2003). Depending on the location of the primary insult, differences in clinical outcomes, as well as the extent of neurological disability, may vary.
Although the increased energy demand of an axon may not be sufficient to trigger axonal degeneration, energy deprivation likely renders neurons more vulnerable to stress (Simons et al., 2014). Moreover, age-related iron accumulation in the human brain, largely stored within the myelin sheaths, may further amplify oxidative injury to the axo-myelinic unit when it is liberated upon demyelination and may be partly responsible for the presence of activated and reactive microglia and macrophages in MS lesions (Lassmann and van Horssen, 2011; Haider, 2015). Given that processes of cytodegeneration, including early mitochondrial dysfunction associated with OS, occur during the first stages of MS, even when no apparent clinical symptom is visible, it is fair to assume that cellular antioxidant defense mechanisms may be capable of controlling the extent of oxidative damage (Fischer et al., 2012). Additionally, efficient remyelinating mechanisms may be able to sustain the effects of the oxidative injury on axonal degeneration in the early stages of the disease (Franklin and ffrench-Constant, 2008). The severity of the imbalance between OS and antioxidant defenses may contribute to the severity of MS pathology and ultimately, to neurological disability.
Discussion
To elucidate whether cytodegeneration or an autoimmune attack may represent the initial trigger of MS, we presented an alternative view on MS pathogenesis, which identifies chronic OS-derived mitochondrial dysfunction as the main initiator of a primary cytodegeneration and secondary inflammatory response. Here, we hypothesized that mitochondrial-derived energy failure caused by oxidative injury represents a potential mechanism by which the AMS might contribute to MS pathogenesis. The mitochondria can be thought of as a “car engine,” which burns fuel and recovers the energy to drive cellular processes. When dysfunctional, however, the engine not only will not be as efficient in producing energy, but it will also create toxic by-products as a result of defective combustion. This toxic debris, generated by the degradation of myelin components as well as by the release of biochemical elements from the dysfunctional mitochondria, may elicit a secondary, inflammatory response, generating a cascade of detrimental effects that will result in severe neurological and cognitive dysfunction in MS patients.
The still hypothetical model for MS pathogenesis described here is likely describing part of the inside-out process and, most importantly, it does not reject the argument for the presence of immune-mediated neuroinflammation in MS. Instead, it aims to elucidate a novel mechanism that may be implicated in driving the onset of the disease. If the proposed model were to reflect the driving mechanisms of progressive degeneration and perhaps, the causative initiator of MS, antioxidant therapies could provide novel therapeutic interventions for MS patients (Adamczyk and Adamczyk-Sowa, 2016). Over-time assessment of metabolite levels of both aerobic and glycolytic energy production in patients with RRMS and PPMS may help to resolve the role that mitochondrial dysfunction and OS play in MS. Upcoming cell-based approaches for MS should also take into consideration the crucial role that a dysfunctional and highly stressed environment may play in the mechanisms of tissue repair (Witherick et al., 2010). Hence, the direct targeting of OS processes and mitochondrial dysfunction may provide a more suitable microenvironment for successful stem cell transplantations or remyelination therapies.
Author Contributions
AL and GS conceived the study. TB performed the literature research. GS and AL provided critical revision to the manuscript. TB wrote the manuscript with input from all other authors. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aboul-Enein, F., Rauschka, H., Kornek, B., Stadelmann, C., Stefferl, A., Brück, W., et al. (2003). Preferential loss of myelin-associated glycoprotein reflects hypoxia-like white matter damage in stroke and inflammatory brain diseases. J. Neuropathol. Exp. Neurol. 62, 25–33. doi: 10.1093/jnen/62.1.25
Adamczyk, B., and Adamczyk-Sowa, M. (2016). New insights into the role of oxidative stress mechanisms in the pathophysiology and treatment of multiple sclerosis. Oxid. Med. Cell Longev. 2016:1973834. doi: 10.1155/2016/1973834
Baranzini, S. E., Mudge, J., Van Velkinburgh, J. C., Khankhanian, P., Khrebtukova, I., Miller, N. A., et al. (2010). Genome, epigenome and RNA sequences of monozygotic twins discordant for multiple sclerosis. Nature 464, 1351–1356. doi: 10.1038/nature08990
Barnett, M. H., and Prineas, J. W. (2004). Relapsing and remitting multiple sclerosis: pathology of the newly forming lesion. Ann. Neurol. 55, 458–468. doi: 10.1002/ana.20016
Barnett, M. H., Parratt, J. D. E., Pollard, J. D., and Prineas, J. W. (2009). MS: is it one disease? Int. MS J. 16, 57–65.
Basria, S. M. N., Mydin, R., and Okekpa, S. I. (2019). “Reactive oxygen species, cellular redox homeostasis and cancer”, in Homeostasis – An Integrated Vision, eds Fernanda Lasakosvitsch Castanho and Sergio Dos Anjos Garnes (London, UK: IntechOpen), 123–141. doi: 10.5772/intechopen.76096
Betteridge, D. J. (2000). What is oxidative stress. Metabolism 2, 3–8. doi: 10.1016/S0026-0495(00)80077-3
Bhattacharyya, A., Chattopadhyay, R., Mitra, S., and Crowe, S. E. (2014). Oxidative stress: an essential factor in the pathogenesis of gastrointestinal mucosal diseases. Physiol. Rev. 94, 329–354. doi: 10.1152/physrev.00040.2012
Bjartmar, C., Wujek, J. R., and Trapp, B. D. (2003). Axonal loss in the pathology of MS: consequences for understanding the progressive phase of the disease. J. Neurol. Sci. 206, 165–171. doi: 10.1016/s0022-510x(02)00069-2
Brinar, V. V., and Braun, B. (2013). Challenges in multiple sclerosis; how to define occurence of progression. Clin. Neurol. Neurosurg. 115, S30–S34. doi: 10.1016/j.clineuro.2013.09.017
Brück, W., and Stadelmann, C. (2003). Inflammation and degeneration in multiple sclerosis. Neurol. Sci. 24, S265–S267. doi: 10.1007/s10072-003-0170-7
Calabrese, M., Magliozzi, R., Ciccarelli, O., Geurts, J. J. G., Reynolds, R., and Martin, R. (2015). Exploring the origins of grey matter damage in multiple sclerosis. Nat. Rev. Neurosci. 16, 147–158. doi: 10.1038/nrn3900
Campbell, G. R., Worrall, J. T., and Mahad, D. J. (2014). The central role of mitochondria in axonal degeneration in multiple sclerosis. Mult. Scler. 20, 1806–1813. doi: 10.1177/1352458514544537
Caprariello, A. V., Rogers, J. A., Morgan, M. L., Hoghooghi, V., Plemel, J. R., Koebel, A., et al. (2018). Biochemically altered myelin triggers autoimmune demyelination. Proc. Natl. Acad. Sci. U S A 115, 5528–5533. doi: 10.1073/pnas.1721115115
Carvalho, A. N., Lim, J. L., Nijland, P. G., Witte, M. E., and van Horssen, J. (2014). Glutathione in multiple sclerosis: more than just an antioxidant? Mult. Scler. 20, 1425–1431. doi: 10.1177/1352458514533400
Chong, S. Y., Rosenberg, S. S., Fancy, S. P., Zhao, C., Shen, Y.-A. A., Hahn, A. T., et al. (2012). Neurite outgrowth inhibitor nogo-a establishes spatial segregation and extent of oligodendrocyte myelination. Proc. Natl. Acad. Sci. U S A 109, 1299–1304. doi: 10.1073/pnas.1113540109
Compston, A., and Coles, A. (2002). Multiple sclerosis. Lancet 359, 1221–1231. doi: 10.1016/S0140-6736(02)08220-X
Czaja, M. J. (2001). TNF toxicity–death from caspase or cathepsin, that is the question. Hepatology 34, 844–846. doi: 10.1053/jhep.2001.0340844
DeLuca, G. C., Ebers, G. C., and Esiri, M. M. (2004). Axonal loss in multiple sclerosis: a pathological survey of the corticospinal and sensory tracts. Brain 127, 1009–1018. doi: 10.1093/brain/awh118
Dendrou, A. A., Fugger, L., and Friese, M. A. (2015). Immunopathology of multiple sclerosis. Nat. Rev. Immunol. 15, 545–558. doi: 10.1038/nri3871
Edgar, J. M., McCulloch, M. C., Montague, P., Brown, A. M., Thilemann, S., Pratola, L., et al. (2010). Demyelination and axonal preservation in a transgenic mouse model of pelizaeus-merzbacher disease. EMBO Mol. Med. 2, 42–50. doi: 10.1002/emmm.200900057
Fünfschilling, U., Supplie, L. M., Mahad, D., Boretius, S., Saab, A. S., Edgar, J., et al. (2012). Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 485, 517–521. doi: 10.1038/nature11007
Fischer, M. T., Sharma, R., Lim, J. L., Haider, L., Frischer, J. M., Drexhage, J., et al. (2012). NADPH oxidase expression in active multiple sclerosis lesions in relation to oxidative tissue damage and mitochondrial injury. Brain 135, 886–899. doi: 10.1093/brain/aws012
Franklin, R. J. M., and ffrench-Constant, C. (2008). Remyelination in the CNS: from biology to therapy. Nat. Rev. Neurosci. 9, 839–855. doi: 10.1038/nrn2480
Franklin, R. J. M., ffrench-Constant, C., Edgar, J. M., and Smith, K. J. (2012). Neuroprotection and repair in multiple sclerosis. Nat. Rev. Neurol. 8, 624–634. doi: 10.1038/nrneurol.2012.200
Friese, M. A., Schattling, B., and Fugger, L. (2014). Mechanisms of neurodegeneration and axonal dysfunction in multiple sclerosis. Nat. Rev. Neurol. 10, 225–238. doi: 10.1038/nrneurol.2014.37
Gaitán, M. I., De Alwis, M. P., Sati, P., Nair, G., and Reich, D. S. (2013). Multiple sclerosis shrinks intralesional, and enlarges extralesional, brain parenchymal veins. Neurology 80, 145–151. doi: 10.1212/WNL.0b013e31827b916f
Geurts, J. J. G., and Barkhof, F. (2008). Grey matter pathology in multiple sclerosis. Lancet Neurol. 7, 841–851. doi: 10.1016/S1474-4422(08)70191-1
Goldberg, M. P., and Ransom, B. R. (2003). New light on white matter. Stroke 34, 330–332. doi: 10.1161/01.str.0000054048.22626.b9
Griffiths, I., Klugmann, M., Anderson, T., Yool, D., Thomson, C., Schwab, M. H., et al. (1998). Axonal swellings and degeneration in mice lacking the major proteolipid of myelin. Science 280, 1610–1613. doi: 10.1126/science.280.5369.1610
Guo, C., Sun, L., Chen, X., and Zhang, D. (2013). Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 8, 2003–2014. doi: 10.3969/j.issn.1673-5374.2013.21.009
Guttmann, C. R. G., Rousset, M., Roch, J. A., Hannoun, S., Durand-Dubief, F., Belaroussi, B., et al. (2016). Multiple sclerosis lesion formation and early evolution revisited: a weekly high-resolution magnetic resonance imaging study. Mult. Scler. 22, 761–769. doi: 10.1177/1352458515600247
Haider, L. (2015). Inflammation, iron, energy failure, and oxidative stress in the pathogenesis of multiple sclerosis. Oxid. Med. Cell Longev. 2015:725370. doi: 10.1155/2015/725370
Hawker, K. (2011). Progressive multiple sclerosis: characteristics and management. Neurol. Clin. 29, 423–434. doi: 10.1016/j.ncl.2011.01.002
Henderson, A. P., Barnett, M. H., Parratt, J. D., and Prineas, J. W. (2009). Multiple sclerosis: distribution of inflammatory cells in newly forming lesions. Ann. Neurol. 66, 739–753. doi: 10.1002/ana.21800
Klaver, R., De Vries, H. E., Schenk, G. J., and Geurts, J. J. G. (2013). Grey matter damage in multiple sclerosis: a pathology perspective. Prion 7, 66–75. doi: 10.4161/pri.23499
Kroemer, G., and Jäättelä, M. (2005). Lysosomes and autophagy in cell death control. Nat. Rev. Cancer 5, 886–897. doi: 10.1038/nrc1738
La Rocca, C., Carbone, F., De Rosa, V., Colamatteo, A., Galgani, M., Perna, F., et al. (2017). Immunometabolic profiling of t cells from patients with relapsing-remitting multiple sclerosis reveals an impairment in glycolysis and mitochondrial respiration. Metabolism 77, 39–46. doi: 10.1016/j.metabol.2017.08.011
Lappe-Siefke, C., Goebbels, S., Gravel, M., Nicksch, E., Lee, J., Braun, P. E., et al. (2003). Disruption of cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat. Genet. 33, 366–374. doi: 10.1038/ng1095
Lassmann, H. (2013). Pathology and disease mechanisms in different stages of multiple sclerosis. J. Neurol. Sci. 333, 1–4. doi: 10.1016/j.jns.2013.05.010
Lassmann, H., and van Horssen, J. (2011). The molecular basis of neurodegeneration in multiple sclerosis. FEBS Lett. 585, 3715–3723. doi: 10.1016/j.febslet.2011.08.004
Lassmann, H., van Horssen, J., and Mahad, D. (2012). Progressive multiple sclerosis: pathology and pathogenesis. Nat. Rev. Neurol. 8, 647–656. doi: 10.1038/nrneurol.2012.168
Lee, Y., Morrison, B. M., Li, Y., Lengacher, S., Farah, M. H., Hoffman, P. N., et al. (2012). Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 487, 443–448. doi: 10.1038/nature11314
Lin, M. T., and Beal, M. F. (2006). Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443, 787–795. doi: 10.1038/nature05292
Lopes Pinheiro, M. A., Kooij, G., Mizee, M. R., Kamermans, A., Enzmann, G., Lyck, R., et al. (2016). Immune cell trafficking across the barriers of the central nervous system in multiple sclerosis and stroke. Biochim. Biophys. Acta 1862, 461–471. doi: 10.1016/j.bbadis.2015.10.018
Lubetzki, C., and Stankoff, B. (2014). Demyelination in multiple sclerosis. Handb. Clin. Neurol. 122, 89–99. doi: 10.1016/B978-0-444-52001-2.00004-2
Lubos, E., Loscalzo, J., and Handy, D. E. (2011). Glutathione peroxidase-1 in health and disease: from molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 15, 1957–1997. doi: 10.1089/ars.2010.3586
Mahad, D. H., Trapp, B. D., and Lassmann, H. (2015). Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol. 14, 183–193. doi: 10.1016/S1474-4422(14)70256-X
Mahad, D., Lassmann, H., and Turnbull, D. (2008). Review: mitochondria and disease progression in multiple sclerosis. Neuropathol. Appl. Neurobiol. 34, 577–589. doi: 10.1111/j.1365-2990.2008.00987.x
Mews, I., Bergmann, M., Bunkowski, S., Gullotta, F., and Brück, W. (1998). Oligodendrocyte and axon pathology in clinically silent multiple sclerosis lesions. Mult. Scler. 4, 55–62. doi: 10.1177/135245859800400203
Micu, I., Plemel, J. R., Caprariello, A. V., Nave, K.-A., and Stys, P. K. (2018). Axo-myelinic neurotransmission: a novel mode of cell signalling in the central nervous system. Nat. Rev. Neurosci. 19, 49–58. doi: 10.1038/nrn.2017.128
Micu, I., Plemel, J. R., Lachance, C., Proft, J., Hansen, A. J., Cummins, K., et al. (2016). The molecular physiology of the axo-myelinic synapse. Exp. Neurol. 276, 41–50. doi: 10.1016/j.expneurol.2015.10.006
Nave, K. A. (2010). Myelination and support of axonal integrity by glia. Nature 468, 244–252. doi: 10.1038/nature09614
Nikić, I., Merkler, D., Sorbara, C., Brinkoetter, M., Bareyre, F. M., Brück, W., et al. (2011). A reversible form of axon damage in experimental autoimmune encephalomyelitis and multiple sclerosis. Nat. Med. 17, 495–499. doi: 10.1038/nm.2324
Noseworthy, J. H., Lucchinetti, C., Rodriguez, M., and Weinshenker, B. G. (2000). Multiple sclerosis. N. Engl J. Med. 343, 938–952. doi: 10.1056/NEJM200009283431307
Ohl, K., Tenbrock, K., and Kipp, M. (2016). Oxidative stress in multiple sclerosis: central and peripheral mode of action. Exp. Neurol. 277, 58–67. doi: 10.1016/j.expneurol.2015.11.010
Ortiz, G. G., Flores-Alvarado, L. J., Pacheco-Moisés, F. P., Mireles-Ramírez, M. A., González-Renovato, E. D., Sánchez-López, A. L., et al. (2016). Cross-talk between glial cells and neurons: relationship in multiple sclerosis. Clin. Case Rep. Rev. 2, 565–571. doi: 10.15761/CCRR.1000276
Pham-Huy, L. A., He, H., and Pham-Huy, C. (2008). Free radicals, antioxidants in disease and health. Int. J. Biomed. Sci. 4, 89–96.
Poerwoatmodjo, A., Schenk, G. J., Geurts, J. J. G., and Luchicchi, A. (2020). Cysteine proteases and mitochondrial instability: a possible vicious cycle in MS myelin? Front. Cell Neurosci. 14:612383. doi: 10.3389/fncel.2020.612383
Rodriguez, M., and Scheithauer, B. (1994). Ultrastructure of multiple sclerosis. Ultrastruct. Pathol. 18, 3–13. doi: 10.3109/01913129409016267
Sawcer, S., Hellenthal, G., Pirinen, M., Spencer, C. C. A., Patsopoulos, N. A., Moutsianas, L., et al. (2011). Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 476, 214–219. doi: 10.1038/nature10251
Schnaar, R. L., and Lopez, P. H. H. (2009). Myelin-associated glycoprotein and its axonal receptors. J. Neurosci. Res. 87, 3267–3276. doi: 10.1002/jnr.21992
Seewann, A., Vrenken, H., van der Valk, P., Blezer, E. L. A., Knol, D. L., Castelijns, J. A., et al. (2009). Diffusely abnormal white matter in chronic multiple sclerosis: imaging and histopathologic analysis. Arch. Neurol. 66, 601–609. doi: 10.1001/archneurol.2009.57
Simons, M., Misgeld, T., and Kerschensteiner, M. (2014). A unified cell biological perspective on axon-myelin injury. J. Cell Biol. 206, 335–345. doi: 10.1083/jcb.201404154
Singh, S., Metz, I., Amor, S., van der Valk, P., Stadelmann, C., and Brück, W. (2013). Microglial nodules in early multiple sclerosis white matter are associated with degenerating axons. Acta Neuropathol. 125, 595–608. doi: 10.1007/s00401-013-1082-0
Strijbis, E. M., Inkster, B., Vounou, M., Naegelin, Y., Kappos, L., Radue, E.-W., et al. (2013). Glutamate gene polymorphisms predict brain volumes in multiple sclerosis. Mult. Scler. 19, 281–288. doi: 10.1177/1352458512454345
Stys, P. K. (2011). The axo-myelinic synapse. Trends Neurosci. 34, 393–400. doi: 10.1016/j.tins.2011.06.004
Stys, P. K. (2013). Pathoetiology of multiple sclerosis: are we barking up the wrong tree? F1000Prime Rep. 5:20. doi: 10.12703/P5-20
Stys, P. K., and Tsutsui, S. (2019). Recent advances in understanding multiple sclerosis. F1000Res 8:F1000. doi: 10.12688/f1000research.20906.1
Stys, P. K., Zamponi, G. W., van Minnen, J., and Geurts, J. J. G. (2012). Will the real multiple sclerosis please stand up? Nat. Rev. Neurosci. 13, 507–514. doi: 10.1038/nrn3275
Su, K. G., Banker, G., Bourdette, D., and Forte, M. (2009). Axonal degeneration in multiple sclerosis: the mitochondrial hypothesis. Curr. Neurol. Neurosci. Rep. 9, 411–417. doi: 10.1007/s11910-009-0060-3
Traka, M., Podojil, J. R., McCarthy, D. P., Miller, S. D., and Popko, B. (2016). Oligodendrocyte death results in immune-mediated CNS demyelination. Nat. Neurosci. 19, 65–74. doi: 10.1038/nn.4193
Trapp, B. D., and Nave, K. A. (2008). Multiple sclerosis: an immune or neurodegenerative disorder? Annu. Rev. Neurosci. 31, 247–269. doi: 10.1146/annurev.neuro.30.051606.094313
Trapp, B. D., Peterson, J., Ransohoff, R. M., Rudick, R., Mörk, S., and Bö, L. (1998). Axonal transection in the lesions of multiple sclerosis. N. Engl J. Med. 338, 278–285. doi: 10.1056/NEJM199801293380502
Trevisiol, A., Saab, A. S., Winkler, U., Marx, G., Imamura, H., Möbius, W., et al. (2017). Monitoring ATP dynamics in electrically active white matter tracts. eLife 6:e24241. doi: 10.7554/eLife.24241
Tsutsui, S., and Stys, P. K. (2013). Metabolic injury to axons and myelin. Exp. Neurol. 246, 26–34. doi: 10.1016/j.expneurol.2012.04.016
Witherick, J., Wilkins, A., Scolding, N., and Kemp, K. (2010). Mechanisms of oxidative damage in multiple sclerosis and a cell therapy approach to treatment. Autoimmune Dis. 2011:164608. doi: 10.4061/2011/164608
Witte, M. E., Geurts, J. J. G., de Vries, H. E., van der Valk, P., and van Horssen, J. (2010). Mitochondrial dysfunction: a potential link between neuroinflammation and neurodegeneration? Mitochondrion 10, 411–418. doi: 10.1016/j.mito.2010.05.014
Witte, M. E., Mahad, D. J., Lassmann, H., and van Horssen, J. (2014). Mitochondrial dysfunction contributes to neurodegeneration in multiple sclerosis. Trends Mol. Med. 20, 179–187. doi: 10.1016/j.molmed.2013.11.007
Witte, M. E., Schumacher, A.-M., Mahler, C. F., Bewersdorf, J. P., Lehmitz, J., Scheiter, A., et al. (2019). Calcium influx through plasma-membrane nanoruptures drives axon degeneration in a model of multiple sclerosis. Neuron 101, 615.e5–624.e5. doi: 10.1016/j.neuron.2018.12.023
Yin, X., Crawford, T. O., Griffin, J. W., Tu, P. H., Lee, V. M. Y., Li, C., et al. (1998). Myelin-associated glycoprotein is a myelin signal that modulates the caliber of myelinated axons. J. Neurosci. 18, 1953–1962. doi: 10.1523/JNEUROSCI.18-06-01953
Keywords: oxidative stress, mitochondria, axo-myelinic synapse, multiple sclerosis, neurodegeneration
Citation: Bergaglio T, Luchicchi A and Schenk GJ (2021) Engine Failure in Axo-Myelinic Signaling: A Potential Key Player in the Pathogenesis of Multiple Sclerosis. Front. Cell. Neurosci. 15:610295. doi: 10.3389/fncel.2021.610295
Received: 25 September 2020; Accepted: 20 January 2021;
Published: 10 February 2021.
Edited by:
Xin Qi, Case Western Reserve University, United StatesReviewed by:
Julia Margaret Edgar, University of Glasgow, United KingdomShan Huang, University of California, Los Angeles, United States
Copyright © 2021 Bergaglio, Luchicchi and Schenk. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Geert J. Schenk, Zy5zY2hlbmtAYW1zdGVyZGFtdW1jLm5s