 Stefan Passlick
Stefan Passlick Christine R. Rose2
Christine R. Rose2 Gabor C. Petzold
Gabor C. Petzold Christian Henneberger
Christian Henneberger- 1Institute of Cellular Neurosciences, Medical Faculty, University of Bonn, Bonn, Germany
- 2Institute of Neurobiology, Faculty of Mathematics and Natural Sciences, Heinrich Heine University Duesseldorf, Duesseldorf, Germany
- 3German Center for Neurodegenerative Diseases (DZNE), Bonn, Germany
- 4Division of Vascular Neurology, University Hospital Bonn, Bonn, Germany
- 5Institute of Neurology, University College London, London, United Kingdom
High-affinity, Na+-dependent glutamate transporters are the primary means by which synaptically released glutamate is removed from the extracellular space. They restrict the spread of glutamate from the synaptic cleft into the perisynaptic space and reduce its spillover to neighboring synapses. Thereby, glutamate uptake increases the spatial precision of synaptic communication. Its dysfunction and the entailing rise of the extracellular glutamate concentration accompanied by an increased spread of glutamate result in a loss of precision and in enhanced excitation, which can eventually lead to neuronal death via excitotoxicity. Efficient glutamate uptake depends on a negative resting membrane potential as well as on the transmembrane gradients of the co-transported ions (Na+, K+, and H+) and thus on the proper functioning of the Na+/K+-ATPase. Consequently, numerous studies have documented the impact of an energy shortage, as occurring for instance during an ischemic stroke, on glutamate clearance and homeostasis. The observations range from rapid changes in the transport activity to altered expression of glutamate transporters. Notably, while astrocytes account for the majority of glutamate uptake under physiological conditions, they may also become a source of extracellular glutamate elevation during metabolic stress. However, the mechanisms of the latter phenomenon are still under debate. Here, we review the recent literature addressing changes of glutamate uptake and homeostasis triggered by acute metabolic stress, i.e., on a timescale of seconds to minutes.
Introduction
Excitatory synaptic transmission in the brain is mediated by the precisely timed and highly localized release of the neurotransmitter glutamate from presynaptic release sites into the synaptic cleft and the subsequent activation of postsynaptic receptors. After the initial dilution of glutamate in the synaptic cleft (Zheng et al., 2008; Scimemi and Beato, 2009), the fast clearance of glutamate from the extracellular space (ECS) restricts its spread. Thereby, it limits the activation of extrasynaptic receptors and the amount of glutamate “spilling over” onto nearby neurons. High-affinity glutamate transporters located on the fine perisynaptic processes of astrocytes (PAPs) are believed to mediate most of the glutamate clearance in the brain under physiological conditions (Danbolt, 2001; Rose et al., 2017). Multiple studies have demonstrated the role of glutamate uptake for synaptic transmission and plasticity (for review see Tzingounis and Wadiche, 2007; Valtcheva and Venance, 2019). However, its relevance becomes particularly apparent when it is impaired (Rose et al., 2017; Pajarillo et al., 2019). The uptake of glutamate against its steep concentration gradient is mainly powered by the transmembrane driving force for Na+ at a negative membrane potential, both of which are maintained by the Na+/K+-ATPase. During conditions of metabolic stress such as ischemic stroke, the lack of ATP leads to a breakdown of ionic gradients and cellular depolarization, reducing the driving force for glutamate uptake. The resulting increased lifetime of glutamate in the ECS leads to increased activation of synaptic and extrasynaptic glutamate receptors and receptors on nearby neurons, which provokes further excitation and intracellular Ca2+ elevations, driving a vicious circle eventually culminating in irreversible excitotoxic cell death (Dirnagl et al., 1999; Grewer et al., 2008).
In an ischemic stroke, two characteristic zones can be distinguished (Rossi et al., 2007). The ischemic core is in the direct vicinity of the clogged blood vessel, where energy supply is almost completely lost and irreversible cell death occurs within minutes. The tissue surrounding the core, called the penumbra, suffers from reduced perfusion and thus lower energy levels but recovery is still possible. However, peri-infarct depolarizations (PIDs), which are waves of spreading depression that originate in the core and propagate through the penumbra into the healthy tissue, impose an additional metabolic burden on the tissue (Dirnagl et al., 1999; Rossi et al., 2007; Dreier, 2011).
Disentangling the sequence of events and identifying the relevant cellular mechanisms during this acute form of metabolic stress is vital for developing novel treatments. Indeed, multiple studies have shown that glutamate homeostasis is perturbed and, in many ways, responsible for ischemic cell death. However, some findings are still controversial (Rossi et al., 2007; Rose et al., 2017; Zhang et al., 2019; Belov Kirdajova et al., 2020) and there are multiple reasons for that. These include the complexity of the pathological condition itself, the large number of potentially relevant cellular mechanisms, and the variety of experimental conditions across different studies. In this mini-review, we discuss the impact of metabolic stress on glutamate homeostasis in the cortex, focusing on mechanisms that are activated during transient (seconds to minutes) episodes of metabolic stress, as is for instance the case in the penumbra after a stroke or during transient ischemic attacks.
Transporter Availability and Location
Expression of Glutamate Transporters
Mammalian cells express five different subtypes of excitatory amino acid transporters (EAAT1–5; Danbolt, 2001). In the hippocampus and neocortex, EAAT1 (= glutamate/aspartate transporter, GLAST) and EAAT2 (= glutamate transporter-1, GLT-1) are the predominantly expressed subtypes. However, the relative expression and functional significance for the uptake of synaptically released glutamate can vary significantly even within subregions of the neocortex (Romanos et al., 2019). EAAT1 and EAAT2 are considered to be mainly astrocytic while EAAT3 (= excitatory amino acid carrier 1, EAAC1) and EAAT4 are predominantly found in neurons (Danbolt, 2001; Massie et al., 2008; Holmseth et al., 2012; Rose et al., 2017). In the adult hippocampus, for instance, it was shown that ~10% of EAAT2 is expressed by neurons where it localizes mainly to axon terminals (Danbolt, 2001; Furness et al., 2008) while neuronal EAAT3 on the other hand is located on the postsynaptic membrane (Holmseth et al., 2012). While the density of glutamate transporters is significantly higher on astrocytes compared to neurons (for the hippocampus see for instance Holmseth et al., 2012), all subtypes are strategically located close to the synaptic glutamate release site.
Changes in glutamate transporter expression can occur on multiple levels, including transcriptional control, epigenetic DNA modifications, and regulation of translation. Furthermore, post-translational modifications such as phosphorylation, nitrosylation, and ubiquitination determine the availability of functional glutamate transporters (Pajarillo et al., 2019). For instance, EAAT2 is subject to nitrosylation, which was shown to influence uptake efficiency (Raju et al., 2015). In the case of ischemia, the extent of expression changes depends on the stage and/or severity of metabolic stress although results in the literature are heterogeneous (Zhang et al., 2019). While some studies indicate that expression of EAAT1 and/or EAAT2 is increased in the early stages of an ischemic event and decreased in later stages (Arranz et al., 2010; Liu et al., 2012; Girbovan and Plamondon, 2015), others demonstrated the opposite (Torp et al., 1995; Rao et al., 2001a; Fang et al., 2014). Intriguingly, it was also shown more recently that EAAT1 and EAAT2 mRNAs are found in astrocyte processes along with the machinery for local translation and that the distribution of mRNAs was altered after fear conditioning (Sakers et al., 2017; Mazaré et al., 2020). It is therefore tempting to speculate that changes in the translation of glutamate transporters might also occur locally in response to acute metabolic stress. However, such changes of transporter expression are likely to play a role over longer time scales than the seconds to minutes of interest here.
Localization of Glutamate Transporters
The availability of functional glutamate transporters depends, apart from transcription and translation, on the regulation of glutamate transporter insertion and internalization as well as lateral diffusion in the cell membrane. Insertion and internalization were shown to change in response to glutamate application (Duan et al., 1999) and are likely to be regulated by Ca2+ dynamics (Rose et al., 2017). Substances promoting retention of glutamate transporters in the membrane were demonstrated to have neuroprotective effects, similar to increasing expression (Martínez-Villarreal et al., 2012; Li et al., 2015). Single-molecule tracking experiments revealed that, once inside the membrane, glutamate transporters are quite mobile, their diffusion is modulated by neuronal activity and they become more stable near synapses (Murphy-Royal et al., 2015). Likewise, glutamate application was shown to affect the formation and stability of EAAT2 clusters inside the astrocyte membrane (Al Awabdh et al., 2016) while blocking synaptic activity reduced the density and perisynaptic localization of EAAT2 clusters in developing astrocytes (Benediktsson et al., 2012). Since inhibiting transporter mobility directly affected synaptic transmission (Murphy-Royal et al., 2015), there is a dynamic and plastic interplay between synaptic glutamate release and local transporter availability. Furthermore, elevated glutamate levels increased ubiquitination of EAAT2 leading to its internalization in HEK293 cells (Ibáñez et al., 2016). For these reasons, it is plausible that PIDs, associated with large extracellular glutamate and intracellular Ca2+ transients (Rakers and Petzold, 2017), may affect transporter membrane diffusion, insertion, and internalization. However, little is known on how metabolic stress precisely affects glutamate transporter trafficking. It, therefore, remains to be established if such processes play a role in, for instance, the initial stages of an ischemic stroke or during PIDs.
In addition to the density of glutamate transporters in the membrane, the physical distance between the glutamate release site and the transporters directly influences the lifetime and spread of glutamate in the ECS. Neuronal membranes facing the synaptic cleft itself contain few transporters (Furness et al., 2008). However, after escaping the synaptic cleft, the radius of action of glutamate is determined by diffusion and the hindrance thereof either by physical barriers or by binding and uptake by transporters (Zheng et al., 2008; Scimemi and Beato, 2009). Hence, glutamate transporters located close to the synaptic cleft limit the diffusion of glutamate to extrasynaptic sites, where high-affinity N-methyl-D-aspartate receptors (NMDARs) and metabotropic glutamate receptors (mGluRs) are localized, and prevent the spillover to neighboring neurons (Scimemi et al., 2004; Zheng et al., 2008; Henneberger et al., 2020; Herde et al., 2020).
Under physiological conditions, glutamate uptake is predominantly mediated by astrocytic glutamate transporters (Danbolt, 2001; Rose et al., 2017). For this reason, the perisynaptic geometry and abundance of PAPs and their role in glutamate uptake has been intensely studied. A reduction of astroglial coverage of synapses during lactation in the supraoptic nucleus, for example, was shown to increase glutamate spread leading to changes in presynaptic transmitter release (Oliet et al., 2001). In contrast, invasion of the synaptic cleft by astrocytic processes in Connexin 30 knockout mice increased the control of synaptic transmission by glutamate uptake (Pannasch et al., 2014). Along these lines, we recently demonstrated that spine size negatively correlates with the local efficacy of glutamate uptake in the hippocampus (Herde et al., 2020). Another study suggested that rearrangements of the astrocytic cytoskeleton induced changes in glutamate uptake which were linked to altered activity of the Na+/K+-ATPase (Sheean et al., 2013). Together, these results highlight that even small changes in the submicron structure of astrocytic processes can have significant consequences for the time course of extracellular glutamate transients and thus on the activation of extrasynaptic glutamate receptors and neighboring neurons. Since changes in cellular morphology as a consequence of ischemia and metabolic stress have been described for different cell types including astrocytes (Risher et al., 2009; Anderova et al., 2011), it is safe to assume that these will directly affect extracellular glutamate dynamics after the onset of metabolic stress.
Notably, we and others have also shown that astrocytic processes are mobile (Haber et al., 2006; Bernardinelli et al., 2014) and withdraw from synapses after induction of long-term potentiation (LTP) of synaptic transmission, which increases glutamate spread and spillover enhancing NMDAR activation (Henneberger et al., 2020). Interestingly, brief episodes of metabolic stress were shown to induce LTP too, a phenomenon known as ischemic LTP (Lenz et al., 2015) that also depends on NMDARs (Maggio et al., 2015). This raises the question if metabolic stress induces a similar withdrawal of astrocytic processes or whether swelling of astrocytic processes (Risher et al., 2009) is more prominent, and how either displacement of glutamate transporters changes extracellular glutamate dynamics in the perisynaptic space.
Impairment of Transporter Function
Reduced Uptake and Reversal-Ion Homeostasis and the Direction of Glutamate Transport
When compared to the sub-micromolar extracellular glutamate concentration (Herman and Jahr, 2007), the intracellular glutamate concentration in neurons and astrocytes is much higher with estimates ranging from high micromolar to low millimolar (Nedergaard et al., 2002; Rossi et al., 2007). Glutamate uptake against this steep concentration gradient is achieved by the co-transport of three Na+ and one H+ and the export of one K+ and is accompanied by an uncoupled anion flux (Danbolt, 2001; Fahlke and Nilius, 2016). Therefore, glutamate transport is very sensitive to changes of the Na+ gradient.
The Na+ gradient across the membrane is largely maintained by the Na+/K+-ATPase and therefore highly dependent on energy in the form of ATP. During metabolic stress, reduced levels of ATP lead to malfunction of the Na+/K+-ATPase and cellular depolarization, a degradation of ionic gradients, and also extracellular glutamate accumulation (Rossi et al., 2007). Cellular depolarization and the degradation of ion gradients both directly reduce the driving force for Na+ and, thereby, glutamate transport. Also, due to the high K+ membrane conductance of astrocytes, extracellular rises in K+ during metabolic stress strongly depolarize these cells, which further reduces the driving force for glutamate uptake. Furthermore, extracellular accumulation of glutamate likely accelerates the decay of ionic gradients in cells with a large contribution to glutamate uptake such as astrocytes, because of the ion transport associated with it. During extreme energy failure, e.g., in the core region of an ischemic stroke, this may lead to a reversal of glutamate uptake, i.e., to the export of glutamate from the cytosol to the ECS (Rossi et al., 2000; Grewer et al., 2008). Whether or not such a reversal occurs depends on how strongly the ionic gradients and membrane potential are degraded. Modeling studies of glutamate transport predict that determining this tipping point requires quantitative information about the intra- and extracellular concentrations of glutamate, Na+, K+, and H+ in the course of metabolic stress (Bergles et al., 2002; Rossi et al., 2007; Grewer et al., 2008), which is currently not available.
Importantly, an impairment of glutamate uptake is likely sufficient to facilitate the failure of extracellular glutamate homeostasis. Indeed, even moderate depolarization of the astrocytic membrane potential (Stephan et al., 2012) and/or increases of the Na+ concentration (Kelly et al., 2009) were shown to reduce the uptake capacity significantly (Felix et al., 2020). We recently demonstrated that even transient episodes of metabolic stress during chemical ischemia in acute brain slices as well as during PIDs in vivo induce significant increases in the neuronal as well as astrocytic Na+ concentration (Gerkau et al., 2018). Thus, acute and/or transient metabolic stress can impair glutamate uptake substantially. This will promote local glutamate accumulation and glutamate spread into the tissue (Figure 1) because inhibition of glutamate uptake increases the lifetime of glutamate in the ECS and its spread (Henneberger et al., 2020; Herde et al., 2020). However, glutamate uptake is not the only mechanism that determines extracellular glutamate concentrations and their diffusion. It is for instance well-known that ischemia and metabolic stress can induce cell swelling and a decrease of the ECS (Syková and Nicholson, 2008; Risher et al., 2009; Anderova et al., 2011). The same amount of glutamate released into a smaller ECS would intuitively lead to higher extracellular glutamate concentrations. At the same time, the tortuosity of the ECS can be increased by ischemia (Syková and Nicholson, 2008), which would on its own reduce the diffusion of glutamate in the ECS. Therefore, it will be interesting to test what the net effect of metabolic stress on glutamate spread in the ECS is.
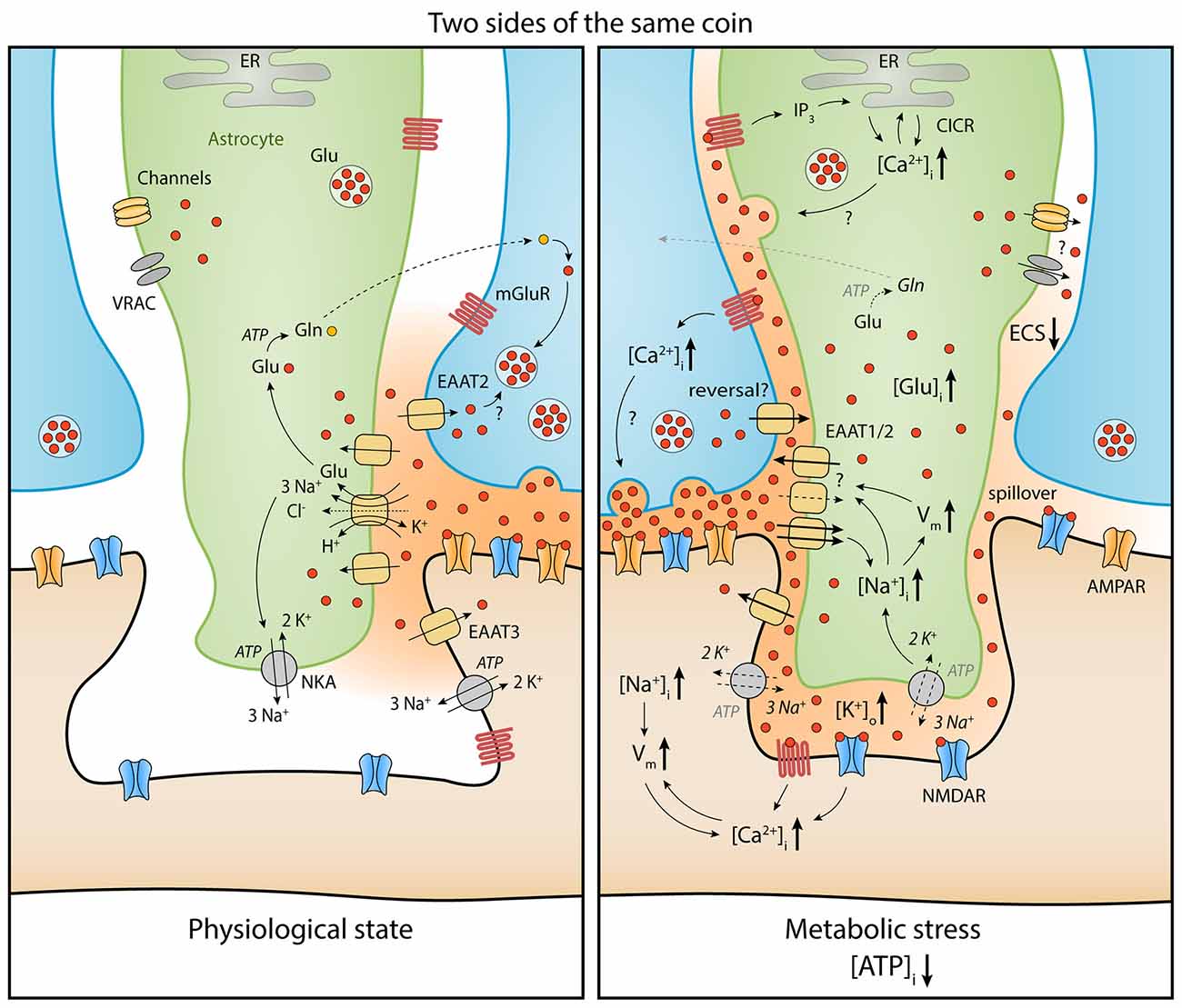
Figure 1. Comparison of extracellular glutamate (red) homeostasis under physiological conditions (left) and during metabolic stress (right) at a synapse (presynaptic bouton in blue, postsynaptic dendritic spine in brown, perisynaptic astrocyte process in green). A moderate reduction of ATP in astrocytes and neurons leads to depolarization of the membrane potential (Vm) and intracellular increases of Na+ and Ca2+ as well as extracellular increases of K+. As a consequence, the driving force for glutamate uptake is diminished and cell swelling reduces the extracellular space (ECS) fraction, which increases extracellular glutamate levels (see text). This leads to stronger activation of glutamate receptors on neurons (e.g., N-methyl-D-aspartate receptors, NMDARs) and astrocytes (e.g., metabotropic glutamate receptors, mGluRs) and a further increase in intracellular Ca2+ and Na+ concentrations and depolarization. In astrocytes, glutamate release through volume-regulated anion channels (VRACs), as well as Ca2+-dependent mechanisms, can amplify extracellular glutamate increases during acute metabolic stress. Open questions (?) regarding glutamate homeostasis are related to the quantitative role of glutamate transporter dysfunction, the relationship between acute metabolic stress and changes of presynaptic release, the role of active astrocytic signaling, and the geometrical rearrangement of astrocytes and neurons during metabolic stress and its impact on extracellular glutamate spread. Abbreviations: AMPAR, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor; CICR, Ca2+-induced Ca2+ release; EAAT, excitatory amino acid transporter; ECS, extracellular space; ER, endoplasmatic reticulum; Gln, Glutamine; Glu, Glutamate; IP3, inositol triphosphate; mGluR, metabotropic glutamate receptor; NKA, Na+/K+-ATPase; NMDAR, N-methyl-D-aspartate receptor; VRAC, volume-regulated anion channel. Note: schematic distribution of glutamate molecules not directly representative of their concentration.
Interestingly, regional variations in glutamate uptake mechanisms could lead to differential susceptibility to metabolic stress across brain regions. For instance, the differentially expressed glutamate transporters EAAT1 and EAAT2 were shown to differ in their uptake kinetics with EAAT2 being more effective (Arriza et al., 1994). Furthermore, it was shown that glutamate uptake in the cortex was slowed by increased presynaptic activity (Armbruster et al., 2016) while no such adaption was found in the hippocampus (Diamond and Jahr, 2000; Diamond, 2005). Similarly, employing the glutamate sensor iGluSnFR, other studies have found that glutamate clearance is faster in the hippocampus compared to the cortex (Pinky et al., 2018) and that its activity-dependence differs between somatosensory and frontal cortex (Romanos et al., 2019). Although the mechanisms underlying this activity-dependence are not fully established, it seems likely that they are engaged by the widespread glutamate increases accompanying PIDs (Fabricius et al., 1993; Rakers and Petzold, 2017) and, in fact, also contribute to the latter.
Once taken up by astrocytes, the enzyme glutamine synthetase (GS) converts glutamate to glutamine. The non-toxic glutamine is then shuttled back to neurons where it is converted to glutamate or GABA (Schousboe et al., 2014). The ATP-dependent conversion by GS is also inhibited during metabolic stress likely inducing glutamate accumulation in astrocytes further reducing the driving force for glutamate uptake. Consequently, it was shown that inhibiting GS increased NMDAR currents in cortical neurons indicating that accumulation of glutamate in astrocytes reduced the uptake capacity promoting glutamate spread (Trabelsi et al., 2017). Similarly, a modeling study found a direct relationship between intracellular glutamate concentration and glutamate uptake capacity (Flanagan et al., 2018).
In summary, although many links are still missing, present data indicate that acute metabolic stress affects a wide range of cellular processes that directly interfere with glutamate clearance via transporters, which in turn can affect the accumulation and spread of glutamate in the ECS (Figure 1).
Neuronal vs. Glial Glutamate Transporters
The precise differential contribution of astrocytic and neuronal glutamate transporters to glutamate clearance during physiological but also pathophysiological conditions remains a matter of debate. The commonly accepted notion is that astrocytes take up >90% of the synaptically released glutamate (Danbolt, 2001; Diamond, 2005; Rose et al., 2017). This is supported by genetic ablation studies which found that an astrocyte-specific knockout of EAAT2 showed by far the most severe phenotype of all transporter knockouts including strong seizure activity leading to neonatal death (Petr et al., 2015). However, constitutive as well as neuron-specific knockout experiments indicated that neuronal EAAT2 is particularly important for glutamate uptake into synaptosomes (Furness et al., 2008; Petr et al., 2015). It should also be noted that the knockout approach depicts the most drastic model to study protein function and compensatory effects need to be considered. Also, the relative impact of neuronal glutamate uptake might increase during acute metabolic stress when astrocytic glutamate uptake is compromised though not entirely lost.
Still, most studies identified glial glutamate transporters as the main source of glutamate via reverse transport during severe ischemia whereas increased glutamate release from neurons is mainly via vesicular release (Jabaudon et al., 2000; Andrade and Rossi, 2010). Other reports, however, suggested that the reversal of neuronal glutamate transporters is responsible for glutamate release during early phases of ischemia (Rossi et al., 2000; Hamann et al., 2002). This was supported by the observation that myelin damage in the corpus callosum during ischemic conditions is almost exclusively caused by axonal glutamate release while reverse transport by astrocytes did not contribute significantly (Doyle et al., 2018). On the other hand, selective knockdown of the mainly astrocytic glutamate transporter EAAT2 increased infarct volume in an in vivo model of stroke while knockdown of the primarily neuronal EAAT3 had no such effect (Rao et al., 2001b). Furthermore, it was recently demonstrated that brain slices from mice with astrocyte-specific EAAT2 deletion were more susceptible to spreading depression compared to EAAT1 or EAAT3 knockout mice indicating a stronger influence of glial compared to neuronal glutamate transporters (Aizawa et al., 2020).
Although the exact contributions of neuronal vs. glial glutamate transporters during metabolic stress are still under debate, it is quite likely that both are dysregulated at some stage and thus both do contribute to the failure of extracellular glutamate homeostasis. The degree to which either cell type plays a role will not only depend on its physiological contribution to glutamate uptake but also on how metabolic stress impairs concentration gradients and the membrane potential in a given cell type (Rossi et al., 2007).
Astrocytic Glutamate Release Beyond Reverse Transport
While the vesicular and non-vesicular routes of glutamate release from neurons are established (see above for glutamate transporters), mechanisms of astrocytic glutamate release are still debated. In addition to reverse transport, several studies indicate that astrocytes are capable of regulated, and in many cases Ca2+-dependent, vesicular as well as channel-mediated glutamate release (Bohmbach et al., 2018; Mahmoud et al., 2019). We recently showed that intracellular Na+ increases during acute metabolic stress induce Ca2+ elevations via reversal of the Na+/Ca2+-exchanger in neurons and astrocytes (Gerkau et al., 2018). Furthermore, we demonstrated that preventing inositol triphosphate receptor type 2-mediated release of Ca2+ from intracellular stores reduced the PID-induced Ca2+ increases in astrocytes and, at the same time, the duration of extracellular glutamate transients in vivo, leading to a secondary neuroprotective decrease of Ca2+ elevations in neurons (Rakers and Petzold, 2017). This suggests that astrocytic Ca2+ signals during PIDs could trigger astrocytic glutamate release, although the exact chain of events remains to be uncovered. Another relevant mechanism is astrocytic release via volume-regulated anion channels (VRACs), which are Ca2+-independent (Mahmoud et al., 2019; Yang et al., 2019). Importantly, VRACs were implicated in glutamate release in the penumbra while reversal of transport was suggested to be more important in the core (Feustel et al., 2004). Also, the VRAC Swell1 was recently shown to mediate ischemia-induced glutamate release from astrocytes with Swell1 knockout mice exhibiting significantly smaller infarct volumes (Yang et al., 2019). Together, these studies indicate that astrocytes can drive glutamate elevations during metabolic stress, ischemia, and stroke by mechanisms independent from high-affinity glutamate transporters.
Conclusion
Accumulating data indicate that metabolic stress acutely impairs glutamate uptake promoting glutamate accumulation and spread further aggravating excitotoxicity. Glutamate transporter dysfunction scales with the severity of metabolic stress ranging from subtle changes of uptake efficiency to a reversal of uptake. A variety of factors can potentially contribute, including lateral diffusion, localization, and abundance of glutamate transporters, their biophysical properties, and their driving forces. Given this complexity, it is no surprise that disentangling the sequence of events triggered by acute metabolic stress has remained challenging. Novel tools such as optical glutamate sensors (Marvin et al., 2013; Helassa et al., 2018) have more recently allowed researchers to gain new insights into extracellular glutamate dynamics and the role of glutamate transporters (Armbruster et al., 2016, 2020; Romanos et al., 2019; Henneberger et al., 2020; Herde et al., 2020). They also promise to provide new insights into unresolved questions regarding altered glutamate transport in metabolic stress (Rakers and Petzold, 2017). Regarding glutamate excitotoxicity, it will also be important to reveal if metabolic stress also perturbs NMDAR co-agonist supply and transport (see Henneberger et al., 2013), which could amplify the damage caused by impaired glutamate homeostasis.
Author Contributions
All authors contributed to the conception and writing of this review and approved the submitted and final published version.
Funding
Research in CH’s laboratory was supported by DFG grants SFB1089 B03, SPP1757 HE6949/1, FOR2795, HE6949/3 and HE6949/5. SP was supported by a DFG start-up funding grant within the FOR2795. CRR received support by DFG grants SPP 1757 RO2327/8-2, FOR 2795 RO2327/13-1 and 14-1. GCP was supported by DFG grant FOR 2795 PE1193/6-1 and the DZNE.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aizawa, H., Sun, W., Sugiyama, K., Itou, Y., Aida, T., Cui, W., et al. (2020). Glial glutamate transporter GLT-1 determines susceptibility to spreading depression in the mouse cerebral cortex. Glia 68, 2631–2642. doi: 10.1002/glia.23874
Al Awabdh, S., Gupta-Agarwal, S., Sheehan, D. F., Muir, J., Norkett, R., Twelvetrees, A. E., et al. (2016). Neuronal activity mediated regulation of glutamate transporter GLT-1 surface diffusion in rat astrocytes in dissociated and slice cultures. Glia 64, 1252–1264. doi: 10.1002/glia.22997
Anderova, M., Vorisek, I., Pivonkova, H., Benesova, J., Vargova, L., Cicanic, M., et al. (2011). Cell death/proliferation and alterations in glial morphology contribute to changes in diffusivity in the rat hippocampus after hypoxia-ischemia. J. Cereb. Blood Flow Metab. 31, 894–907. doi: 10.1038/jcbfm.2010.168
Andrade, A. L., and Rossi, D. J. (2010). Simulated ischaemia induces Ca2+-independent glutamatergic vesicle release through actin filament depolymerization in area CA1 of the hippocampus. J. Physiol. 588, 1499–1514. doi: 10.1113/jphysiol.2010.187609
Armbruster, M., Dulla, C. G., and Diamond, J. S. (2020). Effects of fluorescent glutamate indicators on neurotransmitter diffusion and uptake. eLife 9:e54441. doi: 10.7554/eLife.54441
Armbruster, M., Hanson, E., and Dulla, C. G. (2016). Glutamate clearance is locally modulated by presynaptic neuronal activity in the cerebral cortex. J. Neurosci. 36, 10404–10415. doi: 10.1523/JNEUROSCI.2066-16.2016
Arranz, A. M., Gottlieb, M., Pérez-Cerdá, F., and Matute, C. (2010). Increased expression of glutamate transporters in subcortical white matter after transient focal cerebral ischemia. Neurobiol. Dis. 37, 156–165. doi: 10.1016/j.nbd.2009.09.019
Arriza, J. L., Fairman, W. A., Wadiche, J. I., Murdoch, G. H., Kavanaugh, M. P., and Amara, S. G. (1994). Functional comparisons of three glutamate transporter subtypes cloned from human motor cortex. J. Neurosci. 14, 5559–5569. doi: 10.1523/JNEUROSCI.14-09-05559.1994
Belov Kirdajova, D., Kriska, J., Tureckova, J., and Anderova, M. (2020). Ischemia-triggered glutamate excitotoxicity from the perspective of glial cells. Front. Cell. Neurosci. 14:51. doi: 10.3389/fncel.2020.00051
Benediktsson, A. M., Marrs, G. S., Tu, J. C., Worley, P. F., Rothstein, J. D., Bergles, D. E., et al. (2012). Neuronal activity regulates glutamate transporter dynamics in developing astrocytes. Glia 60, 175–188. doi: 10.1002/glia.21249
Bergles, D. E., Tzingounis, A. V., and Jahr, C. E. (2002). Comparison of coupled and uncoupled currents during glutamate uptake by GLT-1 transporters. J. Neurosci. 22, 10153–10162. doi: 10.1523/JNEUROSCI.22-23-10153.2002
Bernardinelli, Y., Randall, J., Janett, E., Nikonenko, I., König, S., Jones, E. V., et al. (2014). Activity-dependent structural plasticity of perisynaptic astrocytic domains promotes excitatory synapse stability. Curr. Biol. 24, 1679–1688. doi: 10.1016/j.cub.2014.06.025
Bohmbach, K., Schwarz, M. K., Schoch, S., and Henneberger, C. (2018). The structural and functional evidence for vesicular release from astrocytes in situ. Brain Res. Bull. 136, 65–75. doi: 10.1016/j.brainresbull.2017.01.015
Danbolt, N. C. (2001). Glutamate uptake. Prog. Neurobiol. 65, 1–105. doi: 10.1016/s0301-0082(00)00067-8
Diamond, J. S. (2005). Deriving the glutamate clearance time course from transporter currents in CA1 hippocampal astrocytes: transmitter uptake gets faster during development. J. Neurosci. 25, 2906–2916. doi: 10.1523/JNEUROSCI.5125-04.2005
Diamond, J. S., and Jahr, C. E. (2000). Synaptically released glutamate does not overwhelm transporters on hippocampal astrocytes during high-frequency stimulation. J. Neurophysiol. 83, 2835–2843. doi: 10.1152/jn.2000.83.5.2835
Dirnagl, U., Iadecola, C., and Moskowitz, M. A. (1999). Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 22, 391–397. doi: 10.1016/s0166-2236(99)01401-0
Doyle, S., Hansen, D. B., Vella, J., Bond, P., Harper, G., Zammit, C., et al. (2018). Vesicular glutamate release from central axons contributes to myelin damage. Nat. Commun. 9:1032. doi: 10.1038/s41467-018-03427-1
Dreier, J. P. (2011). The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat. Med. 17, 439–447. doi: 10.1038/nm.2333
Duan, S., Anderson, C. M., Stein, B. A., and Swanson, R. A. (1999). Glutamate induces rapid upregulation of astrocyte glutamate transport and cell-surface expression of GLAST. J. Neurosci. 19, 10193–10200. doi: 10.1523/JNEUROSCI.19-23-10193.1999
Fabricius, M., Jensen, L. H., and Lauritzen, M. (1993). Microdialysis of interstitial amino acids during spreading depression and anoxic depolarization in rat neocortex. Brain Res. 612, 61–69. doi: 10.1016/0006-8993(93)91644-8
Fahlke, C., and Nilius, B. (2016). Molecular physiology of anion channels: dual function proteins and new structural motifs–a special issue. Pflugers Arch. 468, 369–370. doi: 10.1007/s00424-016-1791-z
Fang, Q., Hu, W.-W., Wang, X.-F., Yang, Y., Lou, G.-D., Jin, M.-M., et al. (2014). Histamine up-regulates astrocytic glutamate transporter 1 and protects neurons against ischemic injury. Neuropharmacology 77, 156–166. doi: 10.1016/j.neuropharm.2013.06.012
Felix, L., Delekate, A., Petzold, G. C., and Rose, C. R. (2020). Sodium fluctuations in astroglia and their potential impact on astrocyte function. Front. Physiol. 11:871. doi: 10.3389/fphys.2020.00871
Feustel, P. J., Jin, Y., and Kimelberg, H. K. (2004). Volume-regulated anion channels are the predominant contributors to release of excitatory amino acids in the ischemic cortical penumbra. Stroke 35, 1164–1168. doi: 10.1161/01.STR.0000124127.57946.a1
Flanagan, B., McDaid, L., Wade, J., Wong-Lin, K., and Harkin, J. (2018). A computational study of astrocytic glutamate influence on post-synaptic neuronal excitability. PLoS Comput. Biol. 14:e1006040. doi: 10.1371/journal.pcbi.1006040
Furness, D. N., Dehnes, Y., Akhtar, A. Q., Rossi, D. J., Hamann, M., Grutle, N. J., et al. (2008). A quantitative assessment of glutamate uptake into hippocampal synaptic terminals and astrocytes: new insights into a neuronal role for excitatory amino acid transporter 2 (EAAT2). Neuroscience 157, 80–94. doi: 10.1016/j.neuroscience.2008.08.043
Gerkau, N. J., Rakers, C., Durry, S., Petzold, G. C., and Rose, C. R. (2018). Reverse NCX attenuates cellular sodium loading in metabolically compromised cortex. Cereb. Cortex 28, 4264–4280. doi: 10.1093/cercor/bhx280
Girbovan, C., and Plamondon, H. (2015). Resveratrol downregulates type-1 glutamate transporter expression and microglia activation in the hippocampus following cerebral ischemia reperfusion in rats. Brain Res. 1608, 203–214. doi: 10.1016/j.brainres.2015.02.038
Grewer, C., Gameiro, A., Zhang, Z., Tao, Z., Braams, S., and Rauen, T. (2008). Glutamate forward and reverse transport: from molecular mechanism to transporter-mediated release after ischemia. IUBMB Life 60, 609–619. doi: 10.1002/iub.98
Haber, M., Zhou, L., and Murai, K. K. (2006). Cooperative astrocyte and dendritic spine dynamics at hippocampal excitatory synapses. J. Neurosci. 26, 8881–8891. doi: 10.1523/JNEUROSCI.1302-06.2006
Hamann, M., Rossi, D. J., Marie, H., and Attwell, D. (2002). Knocking out the glial glutamate transporter GLT-1 reduces glutamate uptake but does not affect hippocampal glutamate dynamics in early simulated ischaemia. Eur. J. Neurosci. 15, 308–314. doi: 10.1046/j.0953-816x.2001.01861.x
Helassa, N., Dürst, C. D., Coates, C., Kerruth, S., Arif, U., Schulze, C., et al. (2018). Ultrafast glutamate sensors resolve high-frequency release at Schaffer collateral synapses. Proc. Natl. Acad. Sci. U S A 115, 5594–5599. doi: 10.1073/pnas.1720648115
Henneberger, C., Bard, L., King, C., Jennings, A., and Rusakov, D. A. (2013). NMDA receptor activation: two targets for two co-agonists. Neurochem. Res. 38, 1156–1162. doi: 10.1007/s11064-013-0987-2
Henneberger, C., Bard, L., Panatier, A., Reynolds, J. P., Kopach, O., Medvedev, N. I., et al. (2020). LTP induction boosts glutamate spillover by driving withdrawal of perisynaptic astroglia. Neuron 108, 919.e11–936.e11. doi: 10.1016/j.neuron.2020.08.030
Herde, M. K., Bohmbach, K., Domingos, C., Vana, N., Komorowska-Müller, J. A., Passlick, S., et al. (2020). Local efficacy of glutamate uptake decreases with synapse size. Cell Rep. 32:108182. doi: 10.1016/j.celrep.2020.108182
Herman, M. A., and Jahr, C. E. (2007). Extracellular glutamate concentration in hippocampal slice. J. Neurosci. 27, 9736–9741. doi: 10.1523/JNEUROSCI.3009-07.2007
Holmseth, S., Dehnes, Y., Huang, Y. H., Follin-Arbelet, V. V., Grutle, N. J., Mylonakou, M. N., et al. (2012). The density of EAAC1 (EAAT3) glutamate transporters expressed by neurons in the mammalian CNS. J. Neurosci. 32, 6000–6013. doi: 10.1523/JNEUROSCI.5347-11.2012
Ibáñez, I., Díez-Guerra, F. J., Giménez, C., and Zafra, F. (2016). Activity dependent internalization of the glutamate transporter GLT-1 mediated by β-arrestin 1 and ubiquitination. Neuropharmacology 107, 376–386. doi: 10.1016/j.neuropharm.2016.03.042
Jabaudon, D., Scanziani, M., Gähwiler, B. H., and Gerber, U. (2000). Acute decrease in net glutamate uptake during energy deprivation. Proc. Natl. Acad. Sci. U S A 97, 5610–5615. doi: 10.1073/pnas.97.10.5610
Kelly, T., Kafitz, K. W., Roderigo, C., and Rose, C. R. (2009). Ammonium-evoked alterations in intracellular sodium and pH reduce glial glutamate transport activity. Glia 57, 921–934. doi: 10.1002/glia.20817
Lenz, M., Vlachos, A., and Maggio, N. (2015). Ischemic long-term-potentiation (iLTP): perspectives to set the threshold of neural plasticity toward therapy. Neural Regen Res 10, 1537–1539. doi: 10.4103/1673-5374.165215
Li, D., Hérault, K., Zylbersztejn, K., Lauterbach, M. A., Guillon, M., Oheim, M., et al. (2015). Astrocyte VAMP3 vesicles undergo Ca2+ -independent cycling and modulate glutamate transporter trafficking. J. Physiol. 593, 2807–2832. doi: 10.1113/JP270362
Liu, Y.-X., Zhang, M., Liu, L.-Z., Cui, X., Hu, Y.-Y., and Li, W.-B. (2012). The role of glutamate transporter-1a in the induction of brain ischemic tolerance in rats. Glia 60, 112–124. doi: 10.1002/glia.21252
Maggio, N., Shavit Stein, E., and Segal, M. (2015). Ischemic LTP: NMDA-dependency and dorso/ventral distribution within the hippocampus. Hippocampus 25, 1465–1471. doi: 10.1002/hipo.22467
Mahmoud, S., Gharagozloo, M., Simard, C., and Gris, D. (2019). Astrocytes maintain glutamate homeostasis in the CNS by controlling the balance between glutamate uptake and release. Cells 8:184. doi: 10.3390/cells8020184
Martínez-Villarreal, J., García Tardón, N., Ibáñez, I., Giménez, C., and Zafra, F. (2012). Cell surface turnover of the glutamate transporter GLT-1 is mediated by ubiquitination/deubiquitination. Glia 60, 1356–1365. doi: 10.1002/glia.22354
Marvin, J. S., Borghuis, B. G., Tian, L., Cichon, J., Harnett, M. T., Akerboom, J., et al. (2013). An optimized fluorescent probe for visualizing glutamate neurotransmission. Nat. Methods 10, 162–170. doi: 10.1038/nmeth.2333
Massie, A., Cnops, L., Smolders, I., McCullumsmith, R., Kooijman, R., Kwak, S., et al. (2008). High-affinity Na+/K+-dependent glutamate transporter EAAT4 is expressed throughout the rat fore- and midbrain. J. Comp. Neurol. 511, 155–172. doi: 10.1002/cne.21823
Mazaré, N., Oudart, M., Moulard, J., Cheung, G., Tortuyaux, R., Mailly, P., et al. (2020). Local translation in perisynaptic astrocytic processes is specific and changes after fear conditioning. Cell Rep. 32:108076. doi: 10.1016/j.celrep.2020.108076
Murphy-Royal, C., Dupuis, J. P., Varela, J. A., Panatier, A., Pinson, B., Baufreton, J., et al. (2015). Surface diffusion of astrocytic glutamate transporters shapes synaptic transmission. Nat. Neurosci. 18, 219–226. doi: 10.1038/nn.3901
Nedergaard, M., Takano, T., and Hansen, A. J. (2002). Beyond the role of glutamate as a neurotransmitter. Nat. Rev. Neurosci. 3, 748–755. doi: 10.1038/nrn916
Oliet, S. H., Piet, R., and Poulain, D. A. (2001). Control of glutamate clearance and synaptic efficacy by glial coverage of neurons. Science 292, 923–926. doi: 10.1126/science.1059162
Pajarillo, E., Rizor, A., Lee, J., Aschner, M., and Lee, E. (2019). The role of astrocytic glutamate transporters GLT-1 and GLAST in neurological disorders: potential targets for neurotherapeutics. Neuropharmacology 161:107559. doi: 10.1016/j.neuropharm.2019.03.002
Pannasch, U., Freche, D., Dallérac, G., Ghézali, G., Escartin, C., Ezan, P., et al. (2014). Connexin 30 sets synaptic strength by controlling astroglial synapse invasion. Nat. Neurosci. 17, 549–558. doi: 10.1038/nn.3662
Petr, G. T., Sun, Y., Frederick, N. M., Zhou, Y., Dhamne, S. C., Hameed, M. Q., et al. (2015). Conditional deletion of the glutamate transporter GLT-1 reveals that astrocytic GLT-1 protects against fatal epilepsy while neuronal GLT-1 contributes significantly to glutamate uptake into synaptosomes. J. Neurosci. 35, 5187–5201. doi: 10.1523/JNEUROSCI.4255-14.2015
Pinky, N. F., Wilkie, C. M., Barnes, J. R., and Parsons, M. P. (2018). Region- and activity-dependent regulation of extracellular glutamate. J. Neurosci. 38, 5351–5366. doi: 10.1523/JNEUROSCI.3213-17.2018
Raju, K., Doulias, P.-T., Evans, P., Krizman, E. N., Jackson, J. G., Horyn, O., et al. (2015). Regulation of brain glutamate metabolism by nitric oxide and S-nitrosylation. Sci. Signal. 8:ra68. doi: 10.1126/scisignal.aaa4312
Rakers, C., and Petzold, G. C. (2017). Astrocytic calcium release mediates peri-infarct depolarizations in a rodent stroke model. J. Clin. Invest. 127, 511–516. doi: 10.1172/JCI89354
Rao, V. L., Bowen, K. K., and Dempsey, R. J. (2001a). Transient focal cerebral ischemia down-regulates glutamate transporters GLT-1 and EAAC1 expression in rat brain. Neurochem. Res. 26, 497–502. doi: 10.1023/a:1010956711295
Rao, V. L., Dogan, A., Todd, K. G., Bowen, K. K., Kim, B. T., Rothstein, J. D., et al. (2001b). Antisense knockdown of the glial glutamate transporter GLT-1, but not the neuronal glutamate transporter EAAC1, exacerbates transient focal cerebral ischemia-induced neuronal damage in rat brain. J. Neurosci. 21, 1876–1883. doi: 10.1523/JNEUROSCI.21-06-01876.2001
Risher, W. C., Andrew, R. D., and Kirov, S. A. (2009). Real-time passive volume responses of astrocytes to acute osmotic and ischemic stress in cortical slices and in vivo revealed by two-photon microscopy. Glia 57, 207–221. doi: 10.1002/glia.20747
Romanos, J., Benke, D., Saab, A. S., Zeilhofer, H. U., and Santello, M. (2019). Differences in glutamate uptake between cortical regions impact neuronal NMDA receptor activation. Commun. Biol. 2:127. doi: 10.1038/s42003-019-0367-9
Rose, C. R., Felix, L., Zeug, A., Dietrich, D., Reiner, A., and Henneberger, C. (2017). Astroglial glutamate signaling and uptake in the hippocampus. Front. Mol. Neurosci. 10:451. doi: 10.3389/fnmol.2017.00451
Rossi, D. J., Brady, J. D., and Mohr, C. (2007). Astrocyte metabolism and signaling during brain ischemia. Nat. Neurosci. 10, 1377–1386. doi: 10.1038/nn2004
Rossi, D. J., Oshima, T., and Attwell, D. (2000). Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature 403, 316–321. doi: 10.1038/35002090
Sakers, K., Lake, A. M., Khazanchi, R., Ouwenga, R., Vasek, M. J., Dani, A., et al. (2017). Astrocytes locally translate transcripts in their peripheral processes. Proc. Natl. Acad. Sci. U S A 114, E3830–E3838. doi: 10.1073/pnas.1617782114
Schousboe, A., Scafidi, S., Bak, L. K., Waagepetersen, H. S., and McKenna, M. C. (2014). Glutamate metabolism in the brain focusing on astrocytes. Adv. Neurobiol. 11, 13–30. doi: 10.1007/978-3-319-08894-5_2
Scimemi, A., and Beato, M. (2009). Determining the neurotransmitter concentration profile at active synapses. Mol. Neurobiol. 40, 289–306. doi: 10.1007/s12035-009-8087-7
Scimemi, A., Fine, A., Kullmann, D. M., and Rusakov, D. A. (2004). NR2B-containing receptors mediate cross talk among hippocampal synapses. J. Neurosci. 24, 4767–4777. doi: 10.1523/JNEUROSCI.0364-04.2004
Sheean, R. K., Lau, C. L., Shin, Y. S., O’Shea, R. D., and Beart, P. M. (2013). Links between L-glutamate transporters, Na+/K+-ATPase and cytoskeleton in astrocytes: evidence following inhibition with rottlerin. Neuroscience 254, 335–346. doi: 10.1016/j.neuroscience.2013.09.043
Stephan, J., Haack, N., Kafitz, K. W., Durry, S., Koch, D., Hochstrate, P., et al. (2012). Kir4.1 channels mediate a depolarization of hippocampal astrocytes under hyperammonemic conditions in situ. Glia 60, 965–978. doi: 10.1002/glia.22328
Syková, E., and Nicholson, C. (2008). Diffusion in brain extracellular space. Physiol. Rev. 88, 1277–1340. doi: 10.1152/physrev.00027.2007
Torp, R., Lekieffre, D., Levy, L. M., Haug, F. M., Danbolt, N. C., Meldrum, B. S., et al. (1995). Reduced postischemic expression of a glial glutamate transporter, GLT1, in the rat hippocampus. Exp. Brain Res. 103, 51–58. doi: 10.1007/BF00241964
Trabelsi, Y., Amri, M., Becq, H., Molinari, F., and Aniksztejn, L. (2017). The conversion of glutamate by glutamine synthase in neocortical astrocytes from juvenile rat is important to limit glutamate spillover and peri/extrasynaptic activation of NMDA receptors. Glia 65, 401–415. doi: 10.1002/glia.23099
Tzingounis, A. V., and Wadiche, J. I. (2007). Glutamate transporters: confining runaway excitation by shaping synaptic transmission. Nat. Rev. Neurosci. 8, 935–947. doi: 10.1038/nrn2274
Valtcheva, S., and Venance, L. (2019). Control of long-term plasticity by glutamate transporters. Front. Synaptic Neurosci. 11:10. doi: 10.3389/fnsyn.2019.00010
Yang, J., Del Carmen, M., Chen, J., Osei-Owusu, J., Chu, J., and Qiu, Z. (2019). Glutamate-releasing SWELL1 channel in astrocytes modulates synaptic transmission and promotes brain damage in stroke. Neuron 102, 813.e6–827.e6. doi: 10.1016/j.neuron.2019.03.029
Zhang, L.-N., Hao, L., Guo, Y.-S., Wang, H.-Y., Li, L.-L., Liu, L.-Z., et al. (2019). Are glutamate transporters neuroprotective or neurodegenerative during cerebral ischemia? J. Mol. Med. 97, 281–289. doi: 10.1038/aps.2014.1
Keywords: glutamate transport, EAAT, astrocyte, metabolic stress, ischemia, stroke, excitotoxicity
Citation: Passlick S, Rose CR, Petzold GC and Henneberger C (2021) Disruption of Glutamate Transport and Homeostasis by Acute Metabolic Stress. Front. Cell. Neurosci. 15:637784. doi: 10.3389/fncel.2021.637784
Received: 04 December 2020; Accepted: 11 January 2021;
Published: 02 February 2021.
Edited by:
Annalisa Scimemi, University at Albany, United StatesReviewed by:
Christof Grewer, Binghamton University, United StatesBrian Billups, Australian National University, Australia
Copyright © 2021 Passlick, Rose, Petzold and Henneberger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stefan Passlick, c3RlZmFuLnBhc3NsaWNrQHVuaS1ib25uLmRl; Christian Henneberger, Y2hyaXN0aWFuLmhlbm5lYmVyZ2VyQHVuaS1ib25uLmRl