 Yu-Ju Liu
Yu-Ju Liu Yijuang Chern
Yijuang Chern- Division of Neuroscience, Institute of Biomedical Sciences, Academia Sinica, Taipei, Taiwan
Impaired energy homeostasis and aberrant translational control have independently been implicated in the pathogenesis of neurodegenerative diseases. AMP kinase (AMPK), regulated by the ratio of cellular AMP and ATP, is a major gatekeeper for cellular energy homeostasis. Abnormal regulation of AMPK has been reported in several neurodegenerative diseases, including Alzheimer’s disease (AD) and amyotrophic lateral sclerosis (ALS). Most importantly, AMPK activation is known to suppress the translational machinery by inhibiting the mechanistic target of rapamycin complex 1 (mTORC1), activating translational regulators, and phosphorylating nuclear transporter factors. In this review, we describe recent findings on the emerging role of protein translation impairment caused by energy dysregulation in neurodegenerative diseases.
Introduction
Despite the tremendous effort that has been devoted to the investigation of neurodegenerative diseases in the past decades, effective treatments remain limited. Among the common features of most degenerative diseases, impaired energy homeostasis due to mitochondrial defects and reduced protein translation efficiency are commonly reported. Ample evidence has shown that boosting bioenergetics in neurons may provide beneficial effects in various experimental models of neurodegenerative diseases, suggesting the importance of energy deficiency in diseased brains. Because the AMP-activated protein kinase (AMPK) is a key sensor of cellular energy status and an upstream regulator of multiple cellular machineries, its functions during energy stresses have been well investigated. Conversely, the protein translation process is highly energy- dependent and tightly regulated by hundreds of proteins at multiple steps under various pathophysiological conditions. In particular, gradual loss of protein translation efficiency along the progression of neurodegenerative diseases has been reported. This mini- review summarizes the current understanding of the role of energetic stress-mediated translational dysfunction in neurodegenerative diseases. Our focus is on the molecules and/or pathways in protein translation that are disturbed by energy deficiency and aberrant AMPK activation in neurodegenerative diseases, with a specific focus on Alzheimer’s disease (AD) and amyotrophic lateral sclerosis (ALS).
Protein Translation Is Energy- Dependent and Tightly Regulated at Multiple Steps
Eukaryotic protein translation is tightly controlled by hundreds of proteins at four steps (i.e., initiation, elongation, termination, and ribosome recycling (Hershey et al., 2019; Figure 1A). For the sake of clarity, only those sensitive to energy homeostasis are elaborated in the following sections. The main protein translation apparatus in mammalian cells is the 80S ribosome, which is composed of 40S and 60S ribosomal subunits. These ribosomal subunits contain four ribosomal RNA species and 79 ribosomal proteins (Kressler et al., 2010). The assembly of ribosomes occurs mainly in the nucleus, with some exceptions in the axon (Bohnsack and Bohnsack, 2019; Shigeoka et al., 2019). Controlling the appropriate levels and cellular distribution of proteins needed for ribosome maturation is important and highly energy-dependent (Bohnsack and Bohnsack, 2019). Abnormal ribosome assembly caused by various pathophysiological conditions (such as changes in nutrient levels, stresses, genetic mutations) may result in various diseases (Wong et al., 2011; Pelava et al., 2016; Bohnsack and Bohnsack, 2019; Shen et al., 2019) and can be affected by many factors, including aging and genetic defects (Correll et al., 2019). Regulatory degradation of ribosomes by ribophagy (Oka and Yoneda, 2018) and the subsequent suppression of protein translation upon stresses (e.g., starvation) are also very important for cellular survival (von Walden, 2019; Yao et al., 2020).
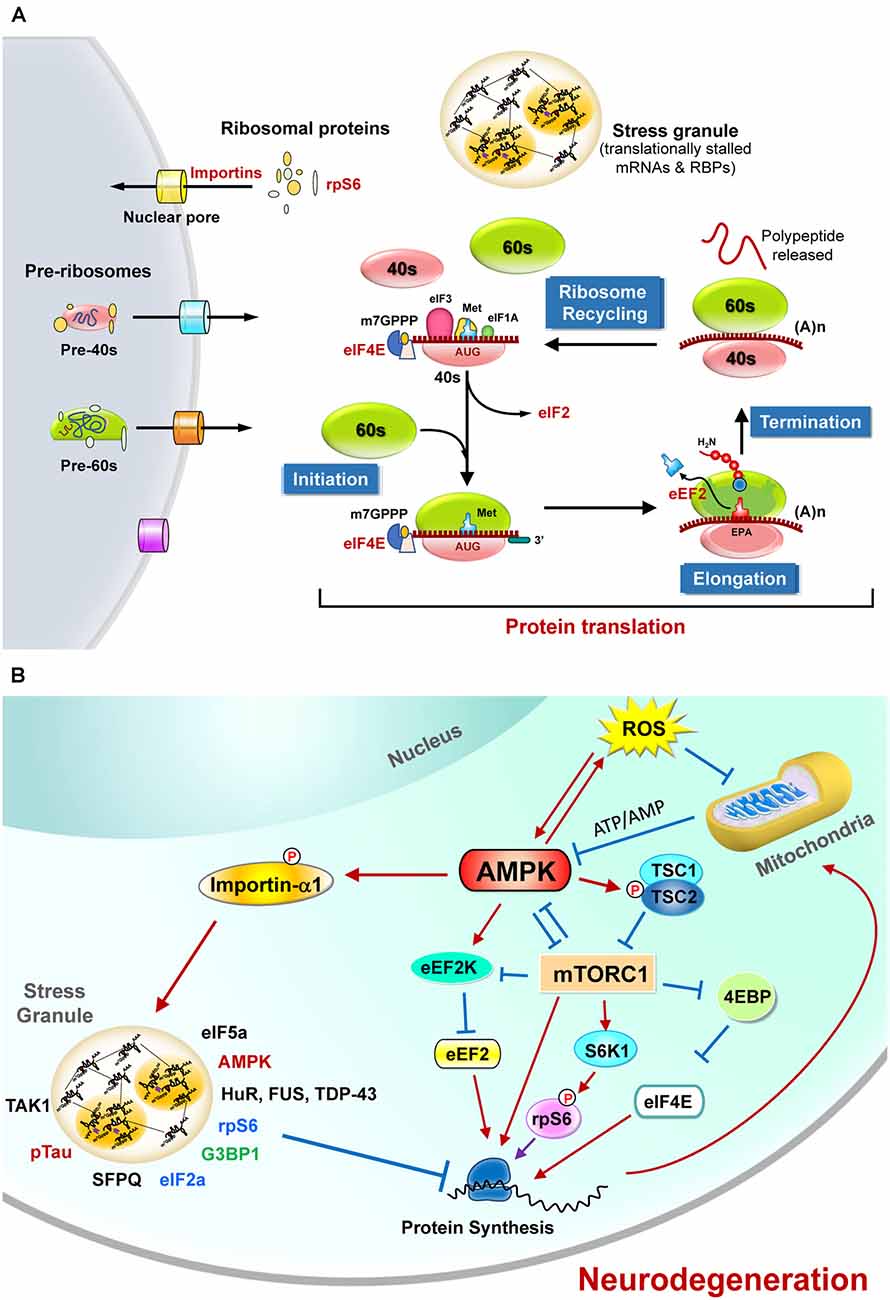
Figure 1. The regulation of protein translation by energy deficiency and AMPK activation at multiple steps. (A) Schematic representation of the four critical steps to control eukaryotic protein translation. For the sake of clarity, only those sensitive to energy homeostasis are listed here. The initiation stage involves the start codon recognition and the joining of the ribosomal subunits (40S and 60S), followed by the elongation stage with repetitive binding of aa-tRNA and translocation. When the ribosome complex encounters stop codons (UAA, UGA, or UAG), protein synthesis is terminated with the help of a protein release factor at the termination stage. The ribosome complex is then dissociated into ribosomal subunits in the ribosome recycling stage and transported into nuclei by importins. These steps are controlled by hundreds of proteins including various initiation factors (eIF) and elongation factors (eEF). In response to stress stimuli that reduce protein translation efficiency, the formation of stress granules (SGs) containing translationally stalled mRNAs and RNA-binding proteins (RBPs) can be observed. (B) The protein translation process is regulated by mitochondrial defect and AMPK activation. Briefly, elevated reactive oxygen species (ROS) and the mitochondrial defect may cause AMPK activation and subsequently inhibit protein translation by suppressing the functions of eIF4E, rpS6, s and eEF2. Activation of AMPK also causes phosphorylation of importin-α1 and interrupts its function in mediating nucleocytoplasmic transport of several RBPs (including TDP-43, HuR). During these stresses, importin-α1 can be found in SGs and is believed to play an important role in the assembly of SGs for the temporary repression of protein translation. A wide variety of proteins (including the components of translation apparatus, RBPs, ribosomal proteins, and pTau) are also recruited to SGs under stresses that cause neurodegeneration. See text for additional details.
Many initiation and elongation factors (e.g., eIF2, eIF4E, and eEF2) needed for protein translation from mRNA are known to be regulated by posttranslational modifications such as protein phosphorylation. Likewise, phosphorylation of the ribosomal protein S6 (rpS6), a component of the 40S ribosomal subunit, was reported many years ago (Gressner and Wool, 1974) and has been extensively investigated since then. It is an indicator of neuronal activity in many experimental models of neuronal plasticity and can be mediated by multiple kinases [including the p70 ribosomal protein S6 kinases (S6K1, S6K2), the p90 ribosomal protein S6 kinases (RSK), and protein kinase A (PKA); Knight et al., 2012; Meyuhas, 2015)]. Ample evidence suggests that the phosphorylation of rpS6 enhances protein translation. Additional extratranslational and extraribosomal roles of rpS6 and other ribosomal proteins have also been proposed (Biever et al., 2015).
Extensive studies have shown that hundreds of proteins are tightly coordinated to accomplish a successful protein translation process. These regulations are all energy-consuming processes (Roux and Topisirovic, 2018; Correll et al., 2019). For example, the functions of many enzymes (such as ATP-dependent RNA helicases, AAA-ATPases, GTPases, and kinases) involved in ribosome biogenesis and protein translation are highly energy-dependent (Kressler et al., 2010). Moreover, these processes can be fine-tuned during a wide variety of pathophysiological conditions (e.g., proliferation, differentiation, apoptosis, and degeneration). Poor translation efficiency consequently affects proteostasis, impairs cellular functions, and causes various diseases, including neurodegenerative diseases (Lehmkuhl and Zarnescu, 2018; Tahmasebi et al., 2018).
Regulation of Protein Translation by AMPK
In response to energy stresses, AMPK is activated to restore energy homeostasis by the promotion of catabolic processes or/and the suppression of anabolic processes. It is a serine/threonine kinase that senses the cellular AMP/ATP ratio and positively controls its activation (Kemp et al., 1999). AMPK holoenzyme is composed of a catalytic α subunit and two regulatory subunits (β and γ), and can be regulated by the adenine nucleotide/energy sensing machinery or other upstream kinases (e.g., CamKKβ, TAK1) that are involved in metabolic responses under various pathophysiological conditions, including elevated levels of reactive oxygen species (ROS) that suppress mitochondrial ATP synthesis (Hardie et al., 2012; Ross et al., 2016; Gonzalez et al., 2020). Earlier studies showed that AMPK activity is high in the brain during development (Ramamurthy and Ronnett, 2006; Ronnett et al., 2009). This is of great interest because AMPK is activated following synaptic activation to maintain metabolic plasticity and energy level by enhancing glycolysis and mitochondrial respiration, suggesting a critical role of AMPK in controlling the neuroenergetic plasticity (Marinangeli et al., 2018). Dysregulation of AMPK either positively or negatively disrupts this synaptic plasticity in response to neuronal activation and may induce synaptic loss and impair cognitive functions (Marinangeli et al., 2018; Domise et al., 2019; Wang et al., 2019). Given that aberrant AMPK activation due to energy deficiency has been reported in degenerating neurons of several neurodegenerative diseases (including Alzheimer’s disease AD; Chen et al., 2009; Mairet-Coello et al., 2013; Ma et al., 2014; Chang et al., 2021), Huntington’s disease (HD; Ju et al., 2011, 2014), and amyotrophic lateral sclerosis (ALS; Lim et al., 2012; Liu et al., 2015a,b), a better understanding of the function and regulation of AMPK during neurodegeneration may provide potential targets for the development of new treatments for neurodegeneration diseases.
The role of AMPK in the control of protein translation has also attracted much attention lately. Given that AMPK activation inhibits protein translation by multiple pathways (Figure 1B) as detailed in the following sections, inferior protein translation in neurodegenerative diseases is a critical issue worthy of further investigation.
Direct Regulation of Protein Translation by AMPK
Many studies have demonstrated that the protein translation process is controlled by energy homeostasis in a coordinated manner (Leibovitch and Topisirovic, 2018). Suppression of protein translation by AMPK at multiple steps has been reported. For example, the γ2-containing AMPK complex protected cells from ischemia-evoked injury by entering the nuclei to suppress the production of pre-rRNA and ribosome biosynthesis (Cao et al., 2017). In addition, AMPK directly phosphorylates eukaryotic elongation factor 2 kinase (eEF2K), which in turn phosphorylates eEF2 to suppress peptide translocation during protein translation and thus inhibits the rate of protein translation (Browne et al., 2004).
Regulation of the Mechanistic Target of Rapamycin Complex 1 (mTORC1) by AMPK
The mammalian target of rapamycin (mTOR) signaling pathway is a key pathway that integrates multiple signals for cell growth and differentiation and controls protein translation (Laplante and Sabatini, 2009). mTOR is a serine-threonine kinase and a member of the PI3K kinase family. It interacts with four other proteins (including raptor) and forms mTOR complex 1 (mTORC1). During activation, mTORC1 activates S6K1 by phosphorylation and disinhibits eIF-4E by suppressing its repressor (4E-binding protein 1, 4E-BP; Lekmine et al., 2003), which subsequently facilitates protein translation (Figure 1B). Many of these regulatory pathways are linked to energy-dependent pathways. Specifically, mTORC1 plays a critical role in coordinating protein translation and ATP production (Leprivier et al., 2013). Activation of mTORC1 suppresses 4E-BPs and leads to selective translation of mitochondrial proteins and enhanced mitochondrial biogenesis, thus forming a link between protein translation and mitochondrial function (Morita et al., 2013).
Ample evidence suggests that AMPK indirectly suppresses the initiation of protein translation via the inhibition of mTORC1 signaling through the phosphorylation of mTOR, raptor, and a negative regulator of mTORC1 (i.e., TSC2; Bolster et al., 2002; Inoki et al., 2003; Gwinn et al., 2008). Conversely, under pathological conditions (e.g., nutrient stress), TORC1 directly phosphorylates the catalytic subunits of AMPK and inhibits AMPK activation (Ling et al., 2020). Such reciprocal feedback regulation between two major metabolic signaling pathways provides an emerging opportunity for the development of therapeutic strategies for various human diseases (Holczer et al., 2019; Gonzalez et al., 2020; Ling et al., 2020).
Stress Granules and AMPK
In response to various stresses, eukaryotic cells form membraneless stress granules (SGs) containing a wide variety of translationally stalled mRNAs and mRNA-binding proteins (RBPs) in the cytoplasm to suppress protein translation (Buchan and Parker, 2009; Protter and Parker, 2016) and to control cellular metabolism and survival (Advani and Ivanov, 2020). These SGs appear to be heterogeneous and contain proteins of various functions, including those involved in translation initiation, mRNA degradation machinery, and other regulatory pathways for RNA metabolism. Many proteins enriched in SGs contain intrinsically disordered regions (IDRs), which not only facilitate diverse protein-protein and protein-RNA interactions but also control the assembly of SGs (Mittag and Parker, 2018). Accumulating evidence suggests that the dynamics of SGs may contribute to neurodegeneration (Advani and Ivanov, 2020).
Activation of AMPK is directly linked with SG formation. During elevated oxidative stress, AMPKα2 and the βγ subunits associate with G3BP1, a core component of SGs, and facilitate the formation of SGs. Recent studies suggest that, in response to stresses, G3BP1 coordinates SG assembly in an RNA-dependent, liquid-liquid phase separation (LLPS)-mediated manner (Yang W. et al., 2020). Suppression of AMPK using a specific inhibitor (i.e., compound C) or expression of a dominant-negative mutant of AMPKα2 demonstrated that AMPK activity is required for the formation of SGs (Mahboubi et al., 2015a,b; Kuo et al., 2020). Although AMPK activation enhances SG biogenesis, it limits the fusion of SGs and thus restrains the size of SGs (Mahboubi et al., 2016). Collectively, AMPK activation upon stress initiates the reprogramming of protein translation and RNA metabolism by facilitating SG assembly. It is interesting to note that G3BP1 also interacts with 40S ribosomal subunit proteins (e.g., rpS6) in SGs and may contribute to the global suppression of protein translation (Kedersha et al., 2016; Panas et al., 2016).
Among the many substrates of AMPK, importin-α1 plays a critical role in nucleocytoplasmic transport (Oka and Yoneda, 2018). A recent study reported that transient arsenite-mediated stress may trigger the formation of cytosolic liquid droplets of TDP-43, a pathological hallmark of ALS, and recruit importin-α1 and other components of nucleocytoplasmic transport to suppress the function of nucleocytoplasmic trafficking (Gasset-Rosa et al., 2019). Phosphorylation of importin-α1 at Ser105 by AMPK blunts its ability to transport a few nuclear RNA-binding proteins (including HuR and FUS) and to alter their cellular distributions (Wang et al., 2004; Liu et al., 2015b). HuR is an RNA-binding protein that binds with and stabilizes ARE-containing mRNA and is also a marker for SGs (Gallouzi et al., 2000). Importantly, importin-α1 has also been implicated in SG assembly during various stresses (e.g., elevated oxidative stress, sodium arsenite, and heat shock). It indirectly binds to poly(A)-containing RNA and localizes to SGs during stress for the preservation of cellular functions (such as cell survival and stress resistance; Fujimura et al., 2010; Mahboubi et al., 2013).
Impaired Translational Control Resulting from Energy Dysfunction in Neurodegenerative Diseases
The mitochondrial defect that causes energy deficiency in neurons is a major pathological feature of most neurodegenerative diseases and has been extensively reviewed recently (Monzio Compagnoni et al., 2020; Yan et al., 2020; Goyal and Chaturvedi, 2021; Johnson et al., 2021; Lee et al., 2021; Madruga et al., 2021; Nakagawa and Yamada, 2021). We have previously reviewed the evidence linking metabolic impairment and mitochondrial defect in neurodegenerative diseases (Liu and Chern, 2015; Liu et al., 2017). The current review focuses on an emerging interest in the abnormal modulation of protein translation, a key element of proteostasis, by energy dysfunction in the context of two degenerative diseases (i.e., ALS and AD) as detailed in Table 1 and in the text below.
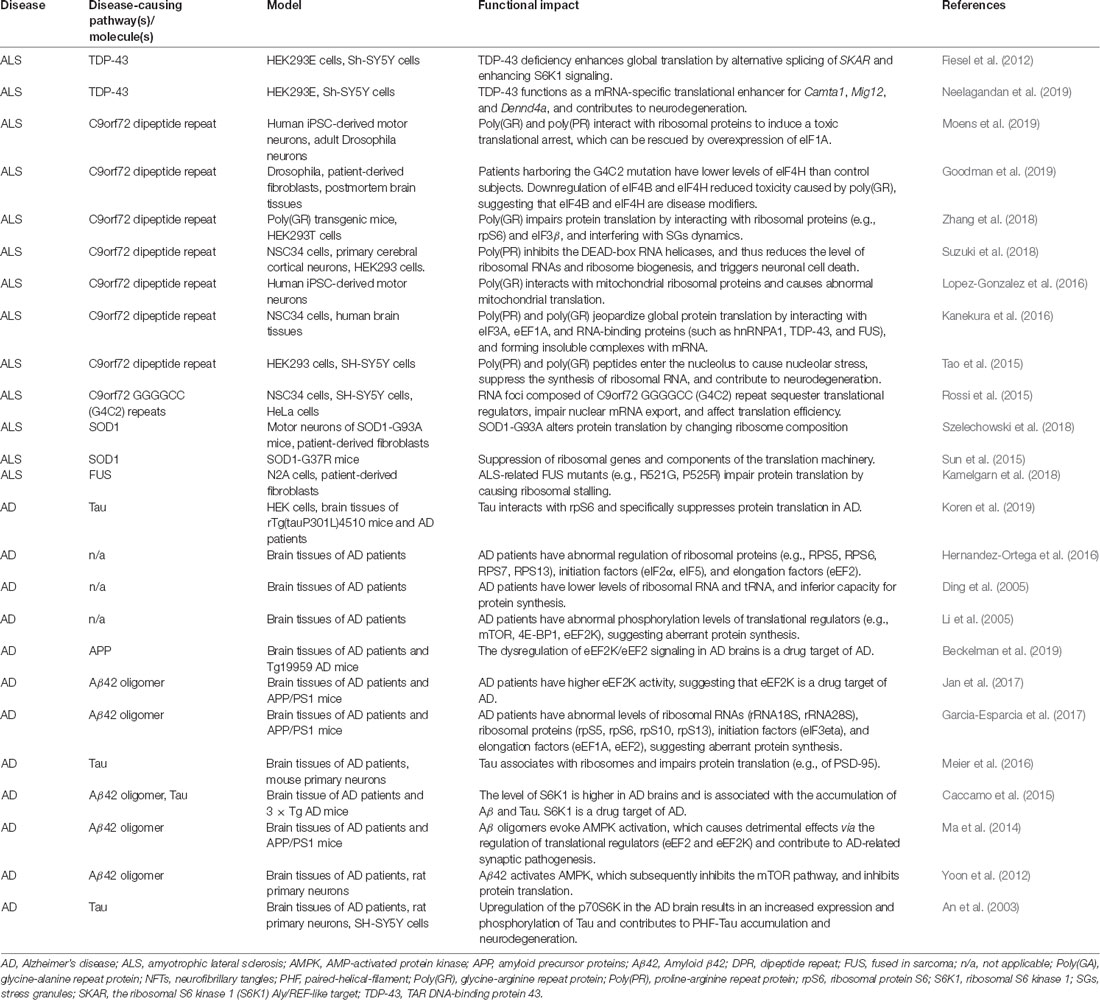
Table 1. Aberrant translational control in ALS and AD.
Amyotrophic Lateral Sclerosis (ALS)
ALS is a progressive motor neuron disease characterized by the degeneration of motor neurons. The loss of motor neurons leads to muscle weakness, disability, and shortened life span. The common hallmark of familiar and sporadic ALS is the mislocalization and accumulation of cytoplasmic inclusions containing the TAR DNA-binding protein 43 (TDP-43) in motor neurons. TDP-43 has diverse functions, including RNA splicing, mRNA transport, protein translation, and storage of specific mRNAs in SGs (Buratti et al., 2001; Prasad et al., 2019). For example, TDP-43 is known to facilitate miRNA biogenesis that is critical for neuronal outgrowth by interacting with Drosha and Dicer complexes (Kawahara and Mieda-Sato, 2012). The mislocalization of TDP-43 in motor neurons is increased that is associated with energy dysfunction and AMPK activation (Liu et al., 2015a). The cellular redistribution of TDP-43 has been implicated in the impairment of translation machinery. For example, in a Drosophila model of ALS, TDP-43 binds to futsch mRNAs and switches actively translating polysomes to nontranslating ribonuclear protein particles (Coyne et al., 2014). The mislocalized mutant TDP-43 in the cytoplasm also disrupts translational processing of hsc70-4 by recruiting mRNA from translating ribosomes (Coyne et al., 2017). Such translational inhibition also caused the impairment of synaptic vesicle endocytosis (Estes et al., 2011). In mouse hippocampal neurons, TDP-43 mediates neuronal spinogenesis via translational repression of Rac1 (Majumder et al., 2012). Interestingly, TDP-43 can regulate global translation by controlling the expression of S6 kinase 1 Aly/REF-like target (SKAR) in human embryonic kidney cells. SKAR is a downstream molecule of the mTOR/S6K1 pathway, which plays a critical role in controlling cellular translation (Fiesel et al., 2012). This is of great interest because AMPK can regulate translation by controlling the activity of mTOR (Garza-Lombo et al., 2018). Most importantly, AMPK is abnormally activated in the motor neurons of patients and two mouse models with ALS, which causes the mislocalization of TDP-43 (Lim et al., 2012; Liu et al., 2015a). It is very likely that, in addition to the AMPK-mTOR axis, AMPK might also mediate cellular translation machinery by altering TDP43-mediated translation in ALS.
In addition to TDP-43, C9orf72-encoded hexanucleotide repeat expansions have been demonstrated to be a critical molecule in ALS pathology (DeJesus-Hernandez et al., 2011; Renton et al., 2011). The toxicity mediated by dipeptide repeats (DPRs) encoded by C9orf72 via repeat-associated non-AUG (RAN) translation has attracted much attention. The RAN translation of C9-G4C2 repeats results in poly(PA), poly(PR), poly(GA), poly(GP), and poly(GR) dipeptides. Translational initiation factors (such as eIF4B and eIF4H) are important for the RAN translation of poly-GR production (Goodman et al., 2019). These C9-DPRs have been directly associated with the impairment of translation in ALS. For example, Zhang et al. (2018) have shown that C9-poly(GR) DPRs impair protein translation by interacting with ribosomal protein (rpS6) and translational initiation factor eIF3β in poly(GR) transgenic mice and ALS patients. In a Drosophila ALS model and human iPSC-derived motor neurons, C9 arginine-rich dipeptide repeats disrupted ribosomal biogenesis by interacting with ribosomal proteins such as rpS23, rpS30, rpL5, and rpL36 (Moens et al., 2019). Furthermore, poly(GR) and poly(PR) also caused a toxic translational arrest that can be restored by exogenous expression of eIF1A (Moens et al., 2019). In a motor neuron cell line, Kanekura et al. (2016) demonstrated that C9-arginine-rich DPRs impaired protein translation by sequestering translation factors (eIF3A, eEF1A; Simov et al., 2016). Moreover, in NSC34 cells, Suzuki et al. (2018) also indicated that C9-poly(PR) disrupted translational machinery by reducing ribosomal RNA expression levels. In addition to altering the levels of ribosomal RNAs, this study also shows that C9-poly(PR) impaired ribosome biogenesis and triggered neuronal toxicity by interacting with DEAD-box RNA helicases (Suzuki et al., 2018). Of note, the nucleolus is an important cellular organelle of ribosome assembly that produces ribosomal RNA (rRNA) and forms pre-ribosome subunits. Abnormal nucleolar stress may impair translation machinery by reducing rRNA biogenesis, ribonucleoprotein binding, and ribosome assembly (Yang et al., 2018). Expression of C9-DPRs leads to nucleolar stress by being localized to the nucleolus and suppressing ribosomal RNA biogenesis (Tao et al., 2015), suggesting that C9-DPRs may also impair translational machinery by inducing nucleolar stress.
It is of great interest to note that C9-mediated DPRs have been implicated in the disruption of cellular energy homeostasis by damaging mitochondrial functions (Onesto et al., 2016; Choi et al., 2019). For example, C9-mediated poly(GR) selectively binds to ATP5A1 (a mitochondrial complex V component) to reduce its expression and causes toxicity in neurons of patients and mice with C9-poly(GR) (Choi et al., 2019). In addition, C9orf72 is a mitochondrial protein. The loss of C9orf72 impairs its function to maintain the integrity of the respiratory chain (Wang et al., 2021). Recent studies also showed that motor neurons derived from iPSCs of C9orf72-ALS patients have inferior mitochondrial bioenergetic functions, poor calcium buffering ability, and shorter axons (Dafinca et al., 2020; Mehta et al., 2021). Collectively, mitochondrial defects and the resultant defective energy metabolism is believed to be a major driver that plays a critical role in ALS pathogenesis and was extensively discussed in recent reviews (Calió et al., 2020; Dafinca et al., 2021; Jhanji et al., 2021; Madruga et al., 2021; Nakagawa and Yamada, 2021). The possible role of energy deficiency and AMPK activation in the impairment of protein translation in ALS warrants further research.
The formation of SGs has attracted much attention in ALS. SGs are transient organelles that regulate cellular RNA homeostasis and translational pathways (Buchan and Parker, 2009). SGs have been indicated to cause the inactivation of protein translation by sequestering RBPs, mRNA, and ribosomal proteins under stress conditions (Buchan and Parker, 2009; Protter and Parker, 2016). Many studies have suggested a critical relationship between SGs and ALS pathology. First, several RBPs associated with ALS are localized in SGs. For example, FUS has been shown to be localized to SGs under oxidative stress or heat shock conditions (Bosco et al., 2010). Interestingly, TDP-43 can also be sequestered into SGs during several forms of stress (i.e., oxidative stress, ER stress; McDonald et al., 2011). In the spinal cords of ALS patients, TDP-43 aggregates were found to be colocalized with SG markers (such as TIA-1 and eIF3; Liu-Yesucevitz et al., 2010). Fang et al. (2019) reported that transient SG formation prevents persistent TDP-43 accumulation in iPS-MNs from ALS patients by recruiting TDP-43 into SGs. In addition, a recent study showed that cytoplasmic TDP-43 forms liquid droplets to inhibit nucleocytoplasmic transport, which is independent of SGs (Gasset-Rosa et al., 2019). Moreover, ALS-related ribonucleoproteins, such as hnRNP A1 and hnRNP A2B1, are components of SGs under stress stimulation (Guil et al., 2006; Kim et al., 2013). Notably, TDP-43 also has a physiological function in SG assembly (McDonald et al., 2011). Downregulation of TDP-43 can delay SG formation, while the loss of this function is associated with ALS pathology (Aulas et al., 2012). In addition to RBPs, another disease-causing protein (mutant SOD1) is also localized in SGs. mSOD1 interacts with G3BP1 and then interferes with the assembly of SGs (Gal et al., 2016). Moreover, C9-DPRs can interact with an SG marker (G3BP1) to impair the assembly and function of SGs (Lee et al., 2016). Importantly, the poly-PR and poly-GR of C9-DPRs also enhance SG formation and delay cellular protein translation (Wen et al., 2014; Kanekura et al., 2016). Collectively, SG formation is an important feature of ALS pathology, which is associated with the dysfunction of RBPs and the impairment of cellular translation machinery.
Alzheimer’s Disease (AD)
Protein translation is required for maintaining long-lasting synaptic plasticity and memory (Klann and Dever, 2004). Accumulating evidence suggests that poor protein translation is associated with dementia. Abnormal activation of AMPK was reported in the hippocampus of aged mice (Yang et al., 2019). Moreover, reduced expression of many ribosome-related proteins (ribosomal protein large and small subunits) was differentially regulated in the aged hippocampus of primates (Meng et al., 2020), suggesting tight control of protein translation, particularly local translation in synapses, during the aging process.
AD is the most common neurodegenerative disease, contributing to 60–70% of dementia in the world (Organization, 2020). Clinical presentations include (but are not limited to) the loss of recent memory, mood swings, and disorientation. Major pathological hallmarks of AD are amyloid plaques, hyperphosphorylation-Tau containing neurofibrillary tangles, vascular impairment, and chronic inflammation. Dysfunction of protein synthesis has been reported in brains associated with AD. For example, lower ribosomal functions and reduced ribosomal levels were shown in the cortical areas of MCI and AD patients, suggesting that impaired protein synthesis is an early AD pathology and may contribute to the development of AD (Ding et al., 2005). Altered levels of ribosomal proteins (e.g., rpS5 and rpS6) and initiation factors (e.g., eEF2) were also observed in the brains of mice and in patients with AD (Hernandez-Ortega et al., 2016; Garcia-Esparcia et al., 2017). A combination of translating ribosome affinity purification (TRAP) and RNA sequencing analyses also revealed that ribosomal processing pathways are downregulated in a mouse model of AD (McKeever et al., 2017). Collectively, protein synthesis gradually becomes abnormal during AD progression.
Abnormal regulation of AMPK and dysregulated mTOR pathways were both found in brains affected by AD. Amyloid β42 impairs translation by AMPK activation, thereby blocking the mTOR pathway (Yoon et al., 2012). Furthermore, the mTOR downstream target S6K1 increases the translation of tau mRNA by phosphorylating rpS6 in SH-SY5Y cells (Pei et al., 2006). Conversely, the reduction in S6K1 levels in an AD mouse model results in the inhibition of Tau translation. Such a reducing effect of S6K1 can improve spatial memory and synaptic plasticity (Caccamo et al., 2015). Moreover, eEF2 and its kinase eEF2K have been indicated to be the key downstream molecules of AMPK in AD pathology. eEF2K is known to phosphorylate and suppress eEF2 so that the rate of protein synthesis is reduced (Proud, 2015). Genetic inhibition of eEF2K enhances the translation machinery and improves compromised long-term potentiation (LTP) in AD mice (Beckelman et al., 2019). Importantly, treatment with an AMPK inhibitor (compound C) improves the decreased synaptic plasticity in AD mice (Ma et al., 2014). Because enhanced AMPK activity is associated with poor neuronal plasticity, suppression of AMPK activity was proposed to be a therapeutic strategy (Wang et al., 2019).
AMPK also directly phosphorylates Tau (Domise et al., 2016). While hyperphosphorylated Tau drives the formation of neurofibrillary tangles as a major hallmark of AD, Tau was found to impair protein translation by disrupting ribosome biogenesis by interacting with rpS6 in the brains of AD patients and mice with tauopathy (Koren et al., 2019). Moreover, phosphorylated Tau protein (pTau) interacts with the splicing factor proline and glutamine rich (SFPQ) and is localized in SGs in the brains affected by rapidly progressive AD (Younas et al., 2020). In addition, pathogenic Tau has been shown to associate with several ribosomal proteins (e.g., rpS5) and to selectively affect protein translation (Meier et al., 2016; Koren et al., 2019). For example, the synthesis of PSD95 has been reduced by Tau (Meier et al., 2016).
SGs play critical roles in AD pathology and are involved in RNA metabolism and translational control. The formation of SGs controls the translation process by sequestering cellular RNA and RBPs (Ivanov et al., 2019). The accumulation of Tau proteins accelerates SG formation by interacting with TIA-1 (Vanderweyde et al., 2016). The expression of human Tau in vitro is known to cause the downregulation of an RNA-binding protein (splicing factor proline and glutamine rich, SFPG). Under oxidative stress, SFPQ is recruited to SGs and colocalizes with Tau. Such dysfunction of SFPQ is associated with rapidly progressive AD patients (Younas et al., 2020). In line with the importance of SGs in AD, chronic stress and high glucocorticoid (GC) levels disrupt protein homeostasis, enhance the accumulation of RNA binding proteins residing in SGs, and facilitate Tau aggregation in a transgenic Tau mouse model of AD (Silva et al., 2019). Taken together, ample evidence demonstrates that abnormal translation control, AMPK activation, and SGs are involved in AD pathology. Further studies are required to explore the possible involvement of abnormal interplay between energy homeostasis and protein translation in AD pathogenesis.
Conclusion Remarks
The protein translation process is complex and highly energy- dependent. In the past three decades, the translation machinery has been extensively studied at the molecular level. Dysregulated protein translation in neurodegenerative diseases may occur by a wide variety of complex mechanisms that lead to inferior ribosome biogenesis and poor protein synthesis efficiency. Recently, interventions that enhance protein translation efficiency have been linked with improved neuronal plasticity and functions (e.g., cognitive activities) in many experimental models of neurodegenerative diseases. AMPK is considered a master regulator of energy homeostasis and its activation has been reported in various stages of neurodegenerative diseases. To date, many direct or indirect activators of AMPK have been developed, and some are currently being evaluated for their contribution to human diseases and disorders (mostly metabolic diseases, inflammation-associated diseases, and cancers; Steinberg and Carling, 2019; de Souza Almeida Matos et al., 2019). For AD, the effects of AMPK activators remain controversial. As reviewed recently by Assefa et al. (2020), most AMPK activators evaluated in AD are indirect activators (such as resveratrol and metformin). In addition to AMPK, these compounds also have other targets important for neuronal functions and survival. Furthermore, different AMPK subtypes (e.g., α1 vs. α2) were recently shown to play seemingly opposite roles in the control of cognitive functions (Yang P. et al., 2020; Zimmermann et al., 2020). Such complex factors may contribute to the observations that both positive and negative effects of AMPK activators on AD pathogenesis (including amyloid metabolism and tauopathy) have been reported. These exciting findings suggest that in-depth investigation into the roles of AMPK in the context of neurodegenerative diseases is required. Moreover, the upstream regulators of AMPK may also serve as excellent drug targets. For example, a new antioxidant drug (Edaravone) was recently approved for ALS (Cho and Shukla, 2020; Shakkour et al., 2021). Many natural antioxidants (such as curcumin, ginkgo biloba) have also been tested in preclinical studies and clinical trials of AD (Arslan et al., 2020). Future detailed investigations in the context of the AMPK-mediated control of protein translation efficiency will better illuminate the specific steps/pathways of protein synthesis that are sensitive to energy deficiency as well as the spectrum of strategies that can be applied to the development of novel therapies for neurodegenerative diseases. To this end, the identification of mislocalized proteins caused by the AMPK-mediated impairment of nuclear–cytoplasmic shuttling, the molecular understanding of stalled translation complexes by disease-causing proteins in SGs, and the discovery of genetic modulations/pharmacological tools to preserve sufficient protein synthesis in an energy crisis will provide critical features of the association between energy dysfunction and neurodegeneration, and may pave the way to identify new drugs for neurodegenerative disease.
Author Contributions
Y-JL and YC both wrote parts of the mini review. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the grants from the Academia Sinica, Taipei, Taiwan (AS-106-TP-B13, AS-TP-109-LM08).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Advani, V. M., and Ivanov, P. (2020). Stress granule subtypes: an emerging link to neurodegeneration. Cell Mol. Life Sci. 77, 4827–4845. doi: 10.1007/s00018-020-03565-0
An, W. L., Cowburn, R. F., Li, L., Braak, H., Alafuzoff, I., Iqbal, K., et al. (2003). Up-regulation of phosphorylated/activated p70 S6 kinase and its relationship to neurofibrillary pathology in Alzheimer’s disease. Am. J. Pathol. 163, 591–607. doi: 10.1016/S0002-9440(10)63687-5
Arslan, J., Jamshed, H., and Qureshi, H. (2020). Early detection and prevention of Alzheimer’s disease: role of oxidative markers and natural antioxidants. Front. Aging Neurosci. 12:231. doi: 10.3389/fnagi.2020.00231
Assefa, B. T., Tafere, G. G., Wondafrash, D. Z., and Gidey, M. T. (2020). The bewildering effect of AMPK activators in Alzheimer’s disease: review of the current evidence. Biomed. Res. Int. 2020:9895121. doi: 10.1155/2020/9895121
Aulas, A., Stabile, S., and Vande Velde, C. (2012). Endogenous TDP-43, but not FUS, contributes to stress granule assembly via G3BP. Mol. Neurodegener. 7:54. doi: 10.1186/1750-1326-7-54
Beckelman, B. C., Yang, W., Kasica, N. P., Zimmermann, H. R., Zhou, X., Keene, C. D., et al. (2019). Genetic reduction of eEF2 kinase alleviates pathophysiology in Alzheimer’s disease model mice. J. Clin. Invest. 129, 820–833. doi: 10.1172/JCI122954
Biever, A., Valjent, E., and Puighermanal, E. (2015). Ribosomal protein S6 phosphorylation in the nervous system: from regulation to function. Front. Mol. Neurosci. 8:75. doi: 10.3389/fnmol.2015.00075
Bohnsack, K. E., and Bohnsack, M. T. (2019). Uncovering the assembly pathway of human ribosomes and its emerging links to disease. EMBO J. 38:e100278. doi: 10.15252/embj.2018100278
Bolster, D. R., Crozier, S. J., Kimball, S. R., and Jefferson, L. S. (2002). AMP-activated protein kinase suppresses protein synthesis in rat skeletal muscle through down-regulated mammalian target of rapamycin (mTOR) signaling. J. Biol. Chem. 277, 23977–23980. doi: 10.1074/jbc.C200171200
Bosco, D. A., Lemay, N., Ko, H. K., Zhou, H., Burke, C., Kwiatkowski, T. J., Jr., et al. (2010). Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum. Mol. Genet. 19, 4160–4175. doi: 10.1093/hmg/ddq335
Browne, G. J., Finn, S. G., and Proud, C. G. (2004). Stimulation of the AMP-activated protein kinase leads to activation of eukaryotic elongation factor 2 kinase and to its phosphorylation at a novel site, serine 398. J. Biol. Chem. 279, 12220–12231. doi: 10.1074/jbc.M309773200
Buchan, J. R., and Parker, R. (2009). Eukaryotic stress granules: the ins and outs of translation. Mol. Cell 36, 932–941. doi: 10.1016/j.molcel.2009.11.020
Buratti, E., Dork, T., Zuccato, E., Pagani, F., Romano, M., and Baralle, F. E. (2001). Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. EMBO J. 20, 1774–1784. doi: 10.1093/emboj/20.7.1774
Caccamo, A., Branca, C., Talboom, J. S., Shaw, D. M., Turner, D., Ma, L., et al. (2015). Reducing ribosomal protein S6 kinase 1 expression improves spatial memory and synaptic plasticity in a mouse model of Alzheimer’s disease. J. Neurosci. 35, 14042–14056. doi: 10.1523/JNEUROSCI.2781-15.2015
Calió, M. L., Henriques, E., Siena, A., Bertoncini, C. R. A., Gil-Mohapel, J., and Rosenstock, T. R. (2020). Mitochondrial dysfunction, neurogenesis and epigenetics: putative implications for amyotrophic lateral sclerosis neurodegeneration and treatment. Front. Neurosci. 14:679. doi: 10.3389/fnins.2020.00679
Cao, Y., Bojjireddy, N., Kim, M., Li, T., Zhai, P., Nagarajan, N., et al. (2017). Activation of γ2-AMPK suppresses ribosome biogenesis and protects against myocardial ischemia/reperfusion injury. Circ. Res. 121, 1182–1191. doi: 10.1161/CIRCRESAHA.117.311159
Chang, C. P., Chang, Y. G., Chuang, P. Y., Nguyen, T. N. A., Wu, K. C., Chou, F. Y., et al. (2021). Equilibrative nucleoside transporter 1 inhibition rescues energy dysfunction and pathology in a model of tauopathy. Acta Neuropathol. Commun. 9:112. doi: 10.1186/s40478-021-01213-7
Chen, Y., Zhou, K., Wang, R., Liu, Y., Kwak, Y. D., Ma, T., et al. (2009). Antidiabetic drug metformin (GlucophageR) increases biogenesis of Alzheimer’s amyloid peptides via up-regulating BACE1 transcription. Proc. Natl. Acad. Sci. U S A 106, 3907–3912. doi: 10.1073/pnas.0807991106
Cho, H., and Shukla, S. (2020). Role of edaravone as a treatment option for patients with amyotrophic lateral sclerosis. Pharmaceuticals 14:29. doi: 10.3390/ph14010029
Choi, S. Y., Lopez-Gonzalez, R., Krishnan, G., Phillips, H. L., Li, A. N., Seeley, W. W., et al. (2019). C9ORF72-ALS/FTD-associated poly(GR) binds Atp5a1 and compromises mitochondrial function in vivo. Nat. Neurosci. 22, 851–862. doi: 10.1038/s41593-019-0397-0
Correll, C. C., Bartek, J., and Dundr, M. (2019). The nucleolus: a multiphase condensate balancing ribosome synthesis and translational capacity in health, aging and ribosomopathies. Cells 8:869. doi: 10.3390/cells8080869
Coyne, A. N., Lorenzini, I., Chou, C. C., Torvund, M., Rogers, R. S., Starr, A., et al. (2017). Post-transcriptional inhibition of Hsc70–4/HSPA8 expression leads to synaptic vesicle cycling defects in multiple models of ALS. Cell Rep. 21, 110–125. doi: 10.1016/j.celrep.2017.09.028
Coyne, A. N., Siddegowda, B. B., Estes, P. S., Johannesmeyer, J., Kovalik, T., Daniel, S. G., et al. (2014). Futsch/MAP1B mRNA is a translational target of TDP-43 and is neuroprotective in a Drosophila model of amyotrophic lateral sclerosis. J. Neurosci. 34, 15962–15974. doi: 10.1523/JNEUROSCI.2526-14.2014
Dafinca, R., Barbagallo, P., and Talbot, K. (2021). The role of mitochondrial dysfunction and ER stress in TDP-43 and C9ORF72 ALS. Front. Cell. Neurosci. 15:653688. doi: 10.3389/fncel.2021.653688
Dafinca, R., Barbagallo, P., Farrimond, L., Candalija, A., Scaber, J., Ababneh, N. A., et al. (2020). Impairment of mitochondrial calcium buffering links mutations in C9ORF72 and TARDBP in iPS-Derived motor neurons from patients with ALS/FTD. Stem Cell Rep. 14, 892–908. doi: 10.1016/j.stemcr.2020.03.023
de Souza Almeida Matos, A. L., Oakhill, J. S., Moreira, J., Loh, K., Galic, S., and Scott, J. W. (2019). Allosteric regulation of AMP-activated protein kinase by adenylate nucleotides and small-molecule drugs. Biochem. Soc. Trans. 47, 733–741. doi: 10.1042/BST20180625
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. doi: 10.1016/j.neuron.2011.09.011
Ding, Q., Markesbery, W. R., Chen, Q., Li, F., and Keller, J. N. (2005). Ribosome dysfunction is an early event in Alzheimer’s disease. J. Neurosci. 25, 9171–9175. doi: 10.1523/JNEUROSCI.3040-05.2005
Domise, M., Sauve, F., Didier, S., Caillerez, R., Begard, S., Carrier, S., et al. (2019). Neuronal AMP-activated protein kinase hyper-activation induces synaptic loss by an autophagy-mediated process. Cell Death Dis. 10:221. doi: 10.1038/s41419-019-1464-x
Domise, M., Didier, S., Marinangeli, C., Zhao, H., Chandakkar, P., Buee, L., et al. (2016). AMP-activated protein kinase modulates tau phosphorylation and tau pathology in vivo. Sci. Rep. 6:26758. doi: 10.1038/srep26758
Estes, P. S., Boehringer, A., Zwick, R., Tang, J. E., Grigsby, B., and Zarnescu, D. C. (2011). Wild-type and A315T mutant TDP-43 exert differential neurotoxicity in a Drosophila model of ALS. Hum. Mol. Genet. 20, 2308–2321. doi: 10.1093/hmg/ddr124
Fang, M. Y., Markmiller, S., Vu, A. Q., Javaherian, A., Dowdle, W. E., Jolivet, P., et al. (2019). Small-molecule modulation of TDP-43 recruitment to stress granules prevents persistent TDP-43 accumulation in ALS/FTD. Neuron 103, 802–819.e11. doi: 10.1016/j.neuron.2019.05.048
Fiesel, F. C., Weber, S. S., Supper, J., Zell, A., and Kahle, P. J. (2012). TDP-43 regulates global translational yield by splicing of exon junction complex component SKAR. Nucleic Acids Res. 40, 2668–2682. doi: 10.1093/nar/gkr1082
Fujimura, K., Suzuki, T., Yasuda, Y., Murata, M., Katahira, J., and Yoneda, Y. (2010). Identification of importin α1 as a novel constituent of RNA stress granules. Biochim. Biophys. Acta 1803, 865–871. doi: 10.1016/j.bbamcr.2010.03.020
Gal, J., Kuang, L., Barnett, K. R., Zhu, B. Z., Shissler, S. C., Korotkov, K. V., et al. (2016). ALS mutant SOD1 interacts with G3BP1 and affects stress granule dynamics. Acta Neuropathol. 132, 563–576. doi: 10.1007/s00401-016-1601-x
Gallouzi, I. E., Brennan, C. M., Stenberg, M. G., Swanson, M. S., Eversole, A., Maizels, N., et al. (2000). HuR binding to cytoplasmic mRNA is perturbed by heat shock. Proc. Natl. Acad. Sci. U S A 97, 3073–3078. doi: 10.1073/pnas.97.7.3073
Garcia-Esparcia, P., Sideris-Lampretsas, G., Hernandez-Ortega, K., Grau-Rivera, O., Sklaviadis, T., Gelpi, E., et al. (2017). Altered mechanisms of protein synthesis in frontal cortex in Alzheimer disease and a mouse model. Am. J. Neurodegener. Dis. 6, 15–25.
Garza-Lombo, C., Schroder, A., Reyes-Reyes, E. M., and Franco, R. (2018). mTOR/AMPK signaling in the brain: cell metabolism, proteostasis and survival. Curr. Opin. Toxicol. 8, 102–110. doi: 10.1016/j.cotox.2018.05.002
Gasset-Rosa, F., Lu, S., Yu, H., Chen, C., Melamed, Z., Guo, L., et al. (2019). Cytoplasmic TDP-43 De-mixing independent of stress granules drives inhibition of nuclear import, loss of nuclear TDP-43 and cell death. Neuron 102, 339–357.e7. doi: 10.1016/j.neuron.2019.02.038
Gonzalez, A., Hall, M. N., Lin, S. C., and Hardie, D. G. (2020). AMPK and TOR: the yin and yang of cellular nutrient sensing and growth control. Cell Metab. 31, 472–492. doi: 10.1016/j.cmet.2020.01.015
Goodman, L. D., Prudencio, M., Srinivasan, A. R., Rifai, O. M., Lee, V. M., Petrucelli, L., et al. (2019). eIF4B and eIF4H mediate GR production from expanded G4C2 in a Drosophila model for C9orf72-associated ALS. Acta Neuropathol. Commun. 7:62. doi: 10.1186/s40478-019-0711-9
Goyal, S., and Chaturvedi, R. K. (2021). Mitochondrial protein import dysfunction in pathogenesis of neurodegenerative diseases. Mol. Neurobiol. 58, 1418–1437. doi: 10.1007/s12035-020-02200-0
Gressner, A. M., and Wool, I. G. (1974). The phosphorylation of liver ribosomal proteins in vivo. evidence that only a single small subunit protein (S6) is phosphorylated. J. Biol. Chem. 249, 6917–6925.
Guil, S., Long, J. C., and Caceres, J. F. (2006). hnRNP A1 relocalization to the stress granules reflects a role in the stress response. Mol. Cell. Biol. 26, 5744–5758. doi: 10.1128/MCB.00224-06
Gwinn, D. M., Shackelford, D. B., Egan, D. F., Mihaylova, M. M., Mery, A., Vasquez, D. S., et al. (2008). AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 30, 214–226. doi: 10.1016/j.molcel.2008.03.003
Hardie, D. G., Ross, F. A., and Hawley, S. A. (2012). AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 13, 251–262. doi: 10.1038/nrm3311
Hernandez-Ortega, K., Garcia-Esparcia, P., Gil, L., Lucas, J. J., and Ferrer, I. (2016). Altered machinery of protein synthesis in Alzheimer’s: from the nucleolus to the ribosome. Brain Pathol. 26, 593–605. doi: 10.1111/bpa.12335
Hershey, J. W. B., Sonenberg, N., and Mathews, M. B. (2019). Principles of translational control. Cold Spring Harb Perspect Biol. 11:a032607. doi: 10.1101/cshperspect.a032607
Holczer, M., Hajdu, B., Lorincz, T., Szarka, A., Banhegyi, G., and Kapuy, O. (2019). A double negative feedback loop between mTORC1 and AMPK kinases guarantees precise autophagy induction upon cellular stress. Int. J. Mol. Sci. 20:5543. doi: 10.3390/ijms20225543
Inoki, K., Zhu, T., and Guan, K. L. (2003). TSC2 mediates cellular energy response to control cell growth and survival. Cell 115, 577–590. doi: 10.1016/s0092-8674(03)00929-2
Ivanov, P., Kedersha, N., and Anderson, P. (2019). Stress granules and processing bodies in translational control. Cold Spring Harb. Perspect. Biol. 11:a032813. doi: 10.1101/cshperspect.a032813
Jan, A., Jansonius, B., Delaidelli, A., Somasekharan, S. P., Bhanshali, F., Vandal, M., et al. (2017). eEF2K inhibition blocks Aβ42 neurotoxicity by promoting an NRF2 antioxidant response. Acta Neuropathol. 133, 101–119. doi: 10.1007/s00401-016-1634-1
Jhanji, R., Behl, T., Sehgal, A., and Bungau, S. (2021). Mitochondrial dysfunction and traffic jams in amyotrophic lateral sclerosis. Mitochondrion 58, 102–110. doi: 10.1016/j.mito.2021.02.008
Johnson, J., Mercado-Ayon, E., Mercado-Ayon, Y., Dong, Y. N., Halawani, S., Ngaba, L., et al. (2021). Mitochondrial dysfunction in the development and progression of neurodegenerative diseases. Arch. Biochem. Biophys. 702:108698. doi: 10.1016/j.abb.2020.108698
Ju, T.-C., Chen, H.-M., Chen, Y.-C., Chang, C.-P., Chang, C., and Chern, Y. (2014). AMPK-α1 functions downstream of oxidative stress to mediate neuronal atrophy in Huntington’s disease. Biochim. Biophys. Acta 1842, 1668–1680. doi: 10.1016/j.bbadis.2014.06.012
Ju, T.-C., Chen, H.-M., Lin, J.-T., Chang, C.-P., Chang, W.-C., Kang, J.-J., et al. (2011). Nuclear translocation of AMPK-α1 potentiates striatal neurodegeneration in Huntington’s disease. J. Cell Biol. 194, 209–227. doi: 10.1083/jcb.201105010
Kamelgarn, M., Chen, J., Kuang, L., Jin, H., Kasarskis, E. J., and Zhu, H. (2018). ALS mutations of FUS suppress protein translation and disrupt the regulation of nonsense-mediated decay. Proc. Natl. Acad. Sci. U S A 115, E11904–E11913. doi: 10.1073/pnas.1810413115
Kanekura, K., Yagi, T., Cammack, A. J., Mahadevan, J., Kuroda, M., Harms, M. B., et al. (2016). Poly-dipeptides encoded by the C9ORF72 repeats block global protein translation. Hum. Mol. Genet. 25, 1803–1813. doi: 10.1093/hmg/ddw052
Kawahara, Y., and Mieda-Sato, A. (2012). TDP-43 promotes microRNA biogenesis as a component of the Drosha and Dicer complexes. Proc. Natl. Acad. Sci. U S A 109, 3347–3352. doi: 10.1073/pnas.1112427109
Kedersha, N., Panas, M. D., Achorn, C. A., Lyons, S., Tisdale, S., Hickman, T., et al. (2016). G3BP-Caprin1-USP10 complexes mediate stress granule condensation and associate with 40S subunits. J. Cell Biol. 212, 845–860. doi: 10.1083/jcb.201508028
Kemp, B. E., Mitchelhill, K. I., Stapleton, D., Michell, B. J., Chen, Z. P., and Witters, L. A. (1999). Dealing with energy demand: the AMP-activated protein kinase. Trends Biochem. Sci. 24, 22–25. doi: 10.1016/s0968-0004(98)01340-1
Kim, H. J., Kim, N. C., Wang, Y.-D., Scarborough, E. A., Moore, J., Diaz, Z., et al. (2013). Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 495, 467–473. doi: 10.1038/nature11922
Klann, E., and Dever, T. E. (2004). Biochemical mechanisms for translational regulation in synaptic plasticity. Nat. Rev. Neurosci. 5, 931–942. doi: 10.1038/nrn1557
Knight, Z. A., Tan, K., Birsoy, K., Schmidt, S., Garrison, J. L., Wysocki, R. W., et al. (2012). Molecular profiling of activated neurons by phosphorylated ribosome capture. Cell 151, 1126–1137. doi: 10.1016/j.cell.2012.10.039
Koren, S. A., Hamm, M. J., Meier, S. E., Weiss, B. E., Nation, G. K., Chishti, E. A., et al. (2019). Tau drives translational selectivity by interacting with ribosomal proteins. Acta Neuropathol. 137, 571–583. doi: 10.1007/s00401-019-01970-9
Kressler, D., Hurt, E., and Bassler, J. (2010). Driving ribosome assembly. Biochim. Biophys. Acta 1803, 673–683. doi: 10.1016/j.bbamcr.2009.10.009
Kuo, C.-T., You, G.-T., Jian, Y.-J., Chen, T.-S., Siao, Y.-C., Hsu, A.-L., et al. (2020). AMPK-mediated formation of stress granules is required for dietary restriction-induced longevity in Caenorhabditis elegans. Aging Cell 19:e13157. doi: 10.1111/acel.13157
Laplante, M., and Sabatini, D. M. (2009). mTOR signaling at a glance. J. Cell Sci. 122, 3589–3594. doi: 10.1242/jcs.051011
Lee, Y. M., He, W., and Liou, Y.-C. (2021). The redox language in neurodegenerative diseases: oxidative post-translational modifications by hydrogen peroxide. Cell Death Dis. 12:58. doi: 10.1038/s41419-020-03355-3
Lee, K.-H., Zhang, P., Kim, H. J., Mitrea, D. M., Sarkar, M., Freibaum, B. D., et al. (2016). C9orf72 dipeptide repeats impair the assembly, dynamics, and function of membrane-less organelles. Cell 167, 774–788.e17. doi: 10.1016/j.cell.2016.10.002
Lehmkuhl, E. M., and Zarnescu, D. C. (2018). Lost in translation: evidence for protein synthesis deficits in ALS/FTD and related neurodegenerative diseases. Adv. Neurobiol. 20, 283–301. doi: 10.1007/978-3-319-89689-2_11
Leibovitch, M., and Topisirovic, I. (2018). Dysregulation of mRNA translation and energy metabolism in cancer. Adv. Biol. Regul. 67, 30–39. doi: 10.1016/j.jbior.2017.11.001
Lekmine, F., Uddin, S., Sassano, A., Parmar, S., Brachmann, S. M., Majchrzak, B., et al. (2003). Activation of the p70 S6 kinase and phosphorylation of the 4E-BP1 repressor of mRNA translation by type I interferons. J. Biol. Chem. 278, 27772–27780. doi: 10.1074/jbc.M301364200
Leprivier, G., Remke, M., Rotblat, B., Dubuc, A., Mateo, A.-R. F., Kool, M., et al. (2013). The eEF2 kinase confers resistance to nutrient deprivation by blocking translation elongation. Cell 153, 1064–1079. doi: 10.1016/j.cell.2013.04.055
Li, X., Alafuzoff, I., Soininen, H., Winblad, B., and Pei, J. J. (2005). Levels of mTOR and its downstream targets 4E-BP1, eEF2 and eEF2 kinase in relationships with tau in Alzheimer’s disease brain. FEBS J. 272, 4211–4220. doi: 10.1111/j.1742-4658.2005.04833.x
Lim, M. A., Selak, M. A., Xiang, Z., Krainc, D., Neve, R. L., Kraemer, B. C., et al. (2012). Reduced activity of AMP-activated protein kinase protects against genetic models of motor neuron disease. J. Neurosci. 32, 1123–1141. doi: 10.1523/JNEUROSCI.6554-10.2012
Ling, N. X. Y., Kaczmarek, A., Hoque, A., Davie, E., Ngoei, K. R. W., Morrison, K. R., et al. (2020). mTORC1 directly inhibits AMPK to promote cell proliferation under nutrient stress. Nat. Metab. 2, 41–49. doi: 10.1038/s42255-019-0157-1
Liu, Y.-J., and Chern, Y. (2015). AMPK-mediated regulation of neuronal metabolism and function in brain diseases. J. Neurogenet. 29, 50–58. doi: 10.3109/01677063.2015.1067203
Liu, Y.-J., Ju, T.-C., Chen, H.-M., Jang, Y.-S., Lee, L.-M., Lai, H.-L., et al. (2015a). Activation of AMP-activated protein kinase α1 mediates mislocalization of TDP-43 in amyotrophic lateral sclerosis. Hum. Mol. Genet. 24, 787–801. doi: 10.1093/hmg/ddu497
Liu, Y.-J., Lee, L.-M., Lai, H.-L., and Chern, Y. (2015b). Aberrant activation of AMP-activated protein kinase contributes to the abnormal distribution of HuR in amyotrophic lateral sclerosis. FEBS Lett. 589, 432–439. doi: 10.1016/j.febslet.2014.12.029
Liu, Y. J., Tsai, P. Y., and Chern, Y. (2017). Energy homeostasis and abnormal RNA metabolism in amyotrophic lateral sclerosis. Front. Cell. Neurosci. 11:126. doi: 10.3389/fncel.2017.00126
Liu-Yesucevitz, L., Bilgutay, A., Zhang, Y.-J., Vanderweyde, T., Citro, A., Mehta, T., et al. (2010). Tar DNA binding protein-43 (TDP-43) associates with stress granules: analysis of cultured cells and pathological brain tissue. PLoS One 5:e13250. doi: 10.1371/journal.pone.0013250
Lopez-Gonzalez, R., Lu, Y., Gendron, T. F., Karydas, A., Tran, H., Yang, D., et al. (2016). Poly(GR) in C9ORF72-related ALS/FTD compromises mitochondrial function and increases oxidative stress and DNA damage in iPSC-derived motor neurons. Neuron 92, 383–391. doi: 10.1016/j.neuron.2016.09.015
Ma, T., Chen, Y., Vingtdeux, V., Zhao, H., Viollet, B., Marambaud, P., et al. (2014). Inhibition of AMP-activated protein kinase signaling alleviates impairments in hippocampal synaptic plasticity induced by amyloid β. J. Neurosci. 34, 12230–12238. doi: 10.1523/JNEUROSCI.1694-14.2014
Madruga, E., Maestro, I., and Martinez, A. (2021). Mitophagy modulation, a new player in the race against ALS. Int. J. Mol. Sci. 22:740. doi: 10.3390/ijms22020740
Mahboubi, H., Barisé, R., and Stochaj, U. (2015a). 5′-AMP-activated protein kinase α regulates stress granule biogenesis. Biochim. Biophys. Acta 1853, 1725–1737. doi: 10.1016/j.bbamcr.2015.03.015
Mahboubi, H., Barisé, R., and Stochaj, U. (2015b). Data in support of 5’AMP-activated protein kinase α regulates stress granule biogenesis. Data Brief 4, 54–59. doi: 10.1016/j.dib.2015.04.010
Mahboubi, H., Koromilas, A. E., and Stochaj, U. (2016). AMP kinase activation alters oxidant-induced stress granule assembly by modulating cell signaling and microtubule organization. Mol. Pharmacol. 90, 460–468. doi: 10.1124/mol.116.105494
Mahboubi, H., Seganathy, E., Kong, D., and Stochaj, U. (2013). Identification of novel stress granule components that are involved in nuclear transport. PLoS One 8:e68356. doi: 10.1371/journal.pone.0068356
Mairet-Coello, G., Courchet, J., Pieraut, S., Courchet, V., Maximov, A., and Polleux, F. (2013). The CAMKK2-AMPK kinase pathway mediates the synaptotoxic effects of Aβ oligomers through Tau phosphorylation. Neuron 78, 94–108. doi: 10.1016/j.neuron.2013.02.003
Majumder, P., Chen, Y.-T., Bose, J. K., Wu, C.-C., Cheng, W.-C., Cheng, S.-J., et al. (2012). TDP-43 regulates the mammalian spinogenesis through translational repression of Rac1. Acta Neuropathol. 124, 231–245. doi: 10.1007/s00401-012-1006-4
Marinangeli, C., Didier, S., Ahmed, T., Caillerez, R., Domise, M., Laloux, C., et al. (2018). AMP-activated protein kinase is essential for the maintenance of energy levels during synaptic activation. iScience 9, 1–13. doi: 10.1016/j.isci.2018.10.006
McDonald, K. K., Aulas, A., Destroismaisons, L., Pickles, S., Beleac, E., Camu, W., et al. (2011). TAR DNA-binding protein 43 (TDP-43) regulates stress granule dynamics via differential regulation of G3BP and TIA-1. Hum. Mol. Genet. 20, 1400–1410. doi: 10.1093/hmg/ddr021
McKeever, P. M., Kim, T., Hesketh, A. R., MacNair, L., Miletic, D., Favrin, G., et al. (2017). Cholinergic neuron gene expression differences captured by translational profiling in a mouse model of Alzheimer’s disease. Neurobiol. Aging 57, 104–119. doi: 10.1016/j.neurobiolaging.2017.05.014
Mehta, A. R., Gregory, J. M., Dando, O., Carter, R. N., Burr, K., Nanda, J., et al. (2021). Mitochondrial bioenergetic deficits in C9orf72 amyotrophic lateral sclerosis motor neurons cause dysfunctional axonal homeostasis. Acta Neuropathol. 141, 257–279. doi: 10.1007/s00401-020-02252-5
Meier, S., Bell, M., Lyons, D. N., Rodriguez-Rivera, J., Ingram, A., Fontaine, S. N., et al. (2016). Pathological tau promotes neuronal damage by impairing ribosomal function and decreasing protein synthesis. J. Neurosci. 36, 1001–1007. doi: 10.1523/JNEUROSCI.3029-15.2016
Meng, S., Xia, W., Pan, M., Jia, Y., He, Z., and Ge, W. (2020). Proteomics profiling and pathway analysis of hippocampal aging in rhesus monkeys. BMC Neurosci. 21:2. doi: 10.1186/s12868-020-0550-4
Meyuhas, O. (2015). Ribosomal protein S6 phosphorylation: four decades of research. Int. Rev. Cell Mol. Biol. 320, 41–73. doi: 10.1016/bs.ircmb.2015.07.006
Mittag, T., and Parker, R. (2018). Multiple modes of protein-protein interactions promote RNP granule assembly. J. Mol. Biol. 430, 4636–4649. doi: 10.1016/j.jmb.2018.08.005
Moens, T. G., Niccoli, T., Wilson, K. M., Atilano, M. L., Birsa, N., Gittings, L. M., et al. (2019). C9orf72 arginine-rich dipeptide proteins interact with ribosomal proteins in vivo to induce a toxic translational arrest that is rescued by eIF1A. Acta Neuropathol. 137, 487–500. doi: 10.1007/s00401-018-1946-4
Monzio Compagnoni, G., Di Fonzo, A., Corti, S., Comi, G. P., Bresolin, N., and Masliah, E. (2020). The role of mitochondria in neurodegenerative diseases: the lesson from Alzheimer’s disease and Parkinson’s disease. Mol. Neurobiol. 57, 2959–2980. doi: 10.1007/s12035-020-01926-1
Morita, M., Gravel, S. P., Chénard, V., Sikström, K., Zheng, L., Alain, T., et al. (2013). mTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell Metab. 18, 698–711. doi: 10.1016/j.cmet.2013.10.001
Nakagawa, Y., and Yamada, S. (2021). A novel hypothesis on metal dyshomeostasis and mitochondrial dysfunction in amyotrophic lateral sclerosis: potential pathogenetic mechanism and therapeutic implications. Eur. J. Pharmacol. 892:173737. doi: 10.1016/j.ejphar.2020.173737
Neelagandan, N., Gonnella, G., Dang, S., Janiesch, P. C., Miller, K. K., Kuchler, K., et al. (2019). TDP-43 enhances translation of specific mRNAs linked to neurodegenerative disease. Nucleic Acids Res. 47, 341–361. doi: 10.1093/nar/gky972
Oka, M., and Yoneda, Y. (2018). Importin α: functions as a nuclear transport factor and beyond. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 94, 259–274. doi: 10.2183/pjab.94.018
Onesto, E., Colombrita, C., Gumina, V., Borghi, M. O., Dusi, S., Doretti, A., et al. (2016). Gene-specific mitochondria dysfunctions in human TARDBP and C9ORF72 fibroblasts. Acta Neuropathol. Commun. 4:47. doi: 10.1186/s40478-016-0316-5
Organization, W. H. (2020). Dementia Fact Sheets. Available online at: https://www.who.int/news-room/fact-sheets/detail/dementia.
Panas, M. D., Ivanov, P., and Anderson, P. (2016). Mechanistic insights into mammalian stress granule dynamics. J. Cell Biol. 215, 313–323. doi: 10.1083/jcb.201609081
Pei, J.-J., An, W.-L., Zhou, X.-W., Nishimura, T., Norberg, J., Benedikz, E., et al. (2006). P70 S6 kinase mediates tau phosphorylation and synthesis. FEBS Lett. 580, 107–114. doi: 10.1016/j.febslet.2005.11.059
Pelava, A., Schneider, C., and Watkins, N. J. (2016). The importance of ribosome production and the 5S RNP-MDM2 pathway, in health and disease. Biochem. Soc. Trans. 44, 1086–1090. doi: 10.1042/BST20160106
Prasad, A., Bharathi, V., Sivalingam, V., Girdhar, A., and Patel, B. K. (2019). Molecular mechanisms of TDP-43 misfolding and pathology in amyotrophic lateral sclerosis. Front. Mol. Neurosci. 12:25. doi: 10.3389/fnmol.2019.00025
Protter, D. S. W., and Parker, R. (2016). Principles and properties of stress granules. Trends Cell Biol. 26, 668–679. doi: 10.1016/j.tcb.2016.05.004
Proud, C. G. (2015). Regulation and roles of elongation factor 2 kinase. Biochem. Soc. Trans. 43, 328–332. doi: 10.1042/BST20140323
Ramamurthy, S., and Ronnett, G. V. (2006). Developing a head for energy sensing: AMP-activated protein kinase as a multifunctional metabolic sensor in the brain. J. Physiol. 574, 85–93. doi: 10.1113/jphysiol.2006.110122
Renton, A. E., Majounie, E., Waite, A., Simon-Sanchez, J., Rollinson, S., Gibbs, J. R., et al. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268. doi: 10.1016/j.neuron.2011.09.010
Ronnett, G. V., Ramamurthy, S., Kleman, A. M., Landree, L. E., and Aja, S. (2009). AMPK in the brain: its roles in energy balance and neuroprotection. J. Neurochem. 109, 17–23. doi: 10.1111/j.1471-4159.2009.05916.x
Ross, F. A., Jensen, T. E., and Hardie, D. G. (2016). Differential regulation by AMP and ADP of AMPK complexes containing different γ subunit isoforms. Biochem. J. 473, 189–199. doi: 10.1042/BJ20150910
Rossi, S., Serrano, A., Gerbino, V., Giorgi, A., Di Francesco, L., Nencini, M., et al. (2015). Nuclear accumulation of mRNAs underlies G4C2-repeat-induced translational repression in a cellular model of C9orf72 ALS. J. Cell Sci. 128, 1787–1799. doi: 10.1242/jcs.165332
Roux, P. P., and Topisirovic, I. (2018). Signaling pathways involved in the regulation of mRNA translation. Mol. Cell. Biol. 38:e00070-18. doi: 10.1128/MCB.00070-18
Shakkour, Z., Issa, H., Ismail, H., Ashekyan, O., Habashy, K. J., Nasrallah, L., et al. (2021). Drug repurposing: promises of edaravone target drug in traumatic brain injury. Curr. Med. Chem. 28, 2369–2391. doi: 10.2174/0929867327666200812221022
Shen, A., Chen, Y., Liu, L., Huang, Y., Chen, H., Qi, F., et al. (2019). EBF1-mediated upregulation of ribosome assembly factor PNO1 contributes to cancer progression by negatively regulating the p53 signaling pathway. Cancer Res. 79, 2257–2270. doi: 10.1158/0008-5472.CAN-18-3238
Shigeoka, T., Koppers, M., Wong, H. H., Lin, J. Q., Cagnetta, R., Dwivedy, A., et al. (2019). On-site ribosome remodeling by locally synthesized ribosomal proteins in axons. Cell Rep. 29, 3605.e10–3619.e10. doi: 10.1016/j.celrep.2019.11.025
Silva, J. M., Rodrigues, S., Sampaio-Marques, B., Gomes, P., Neves-Carvalho, A., Dioli, C., et al. (2019). Dysregulation of autophagy and stress granule-related proteins in stress-driven Tau pathology. Cell Death Differ. 26, 1411–1427. doi: 10.1038/s41418-018-0217-1
Simov, V., Deshmukh, S. V., Dinsmore, C. J., Elwood, F., Fernandez, R. B., Garcia, Y., et al. (2016). Structure-based design and development of (benz)imidazole pyridones as JAK1-selective kinase inhibitors. Bioorg. Med. Chem. Lett. 26, 1803–1808. doi: 10.1016/j.bmcl.2016.02.035
Steinberg, G. R., and Carling, D. (2019). AMP-activated protein kinase: the current landscape for drug development. Nat. Rev. Drug Discov. 18, 527–551. doi: 10.1038/s41573-019-0019-2
Sun, S., Sun, Y., Ling, S. C., Ferraiuolo, L., McAlonis-Downes, M., Zou, Y., et al. (2015). Translational profiling identifies a cascade of damage initiated in motor neurons and spreading to glia in mutant SOD1-mediated ALS. Proc. Natl. Acad. Sci. U S A 112, E6993–7002. doi: 10.1073/pnas.1520639112
Suzuki, H., Shibagaki, Y., Hattori, S., and Matsuoka, M. (2018). The proline-arginine repeat protein linked to C9-ALS/FTD causes neuronal toxicity by inhibiting the DEAD-box RNA helicase-mediated ribosome biogenesis. Cell Death Dis. 9:975. doi: 10.1038/s41419-018-1028-5
Szelechowski, M., Amoedo, N., Obre, E., Leger, C., Allard, L., Bonneu, M., et al. (2018). Metabolic reprogramming in amyotrophic lateral sclerosis. Sci. Rep. 8:3953. doi: 10.1038/s41598-018-22318-5
Tahmasebi, S., Khoutorsky, A., Mathews, M. B., and Sonenberg, N. (2018). Translation deregulation in human disease. Nat. Rev. Mol. Cell Biol. 19, 791–807. doi: 10.1038/s41580-018-0034-x
Tao, Z., Wang, H., Xia, Q., Li, K., Jiang, X., Xu, G., et al. (2015). Nucleolar stress and impaired stress granule formation contribute to C9orf72 RAN translation-induced cytotoxicity. Hum. Mol. Genet. 24, 2426–2441. doi: 10.1093/hmg/ddv005
Vanderweyde, T., Apicco, D. J., Youmans-Kidder, K., Ash, P. E. A., Cook, C., Lummertz da Rocha, E., et al. (2016). Interaction of tau with the RNA-binding protein TIA1 regulates tau pathophysiology and toxicity. Cell Rep. 15, 1455–1466. doi: 10.1016/j.celrep.2016.04.045
von Walden, F. (2019). Ribosome biogenesis in skeletal muscle: coordination of transcription and translation. J. Appl. Physiol. 127, 591–598. doi: 10.1152/japplphysiol.00963.2018
Wang, T., Liu, H., Itoh, K., Oh, S., Zhao, L., Murata, D., et al. (2021). C9orf72 regulates energy homeostasis by stabilizing mitochondrial complex I assembly. Cell Metab. 33, 531–546.e9. doi: 10.1016/j.cmet.2021.01.005
Wang, W., Yang, X., Kawai, T., López de Silanes, I., Mazan-Mamczarz, K., Chen, P., et al. (2004). AMP-activated protein kinase-regulated phosphorylation and acetylation of importin α1: involvement in the nuclear import of RNA-binding protein HuR. J. Biol. Chem. 279, 48376–48388. doi: 10.1074/jbc.M409014200
Wang, X., Zimmermann, H. R., and Ma, T. (2019). Therapeutic potential of AMP-activated protein kinase in Alzheimer’s disease. J. Alzheimers Dis. 68, 33–38. doi: 10.3233/JAD-181043
Wen, X., Tan, W., Westergard, T., Krishnamurthy, K., Markandaiah, S. S., Shi, Y., et al. (2014). Antisense proline-arginine RAN dipeptides linked to C9ORF72-ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron 84, 1213–1225. doi: 10.1016/j.neuron.2014.12.010
Wong, C. C., Traynor, D., Basse, N., Kay, R. R., and Warren, A. J. (2011). Defective ribosome assembly in Shwachman-Diamond syndrome. Blood 118, 4305–4312. doi: 10.1182/blood-2011-06-353938
Yan, X., Wang, B., Hu, Y., Wang, S., and Zhang, X. (2020). Abnormal mitochondrial quality control in neurodegenerative diseases. Front. Cell. Neurosci. 14:138. doi: 10.3389/fncel.2020.00138
Yang, P., Mathieu, C., Kolaitis, R. M., Zhang, P., Messing, J., Yurtsever, U., et al. (2020). G3BP1 is a tunable switch that triggers phase separation to assemble stress granules. Cell 181, 325–345.e28. doi: 10.1016/j.cell.2020.03.046
Yang, K., Yang, J., and Yi, J. (2018). Nucleolar stress: hallmarks, sensing mechanism and diseases. Cell Stress 2, 125–140. doi: 10.15698/cst2018.06.139
Yang, W., Zhou, X., and Ma, T. (2019). Memory decline and behavioral inflexibility in aged mice are correlated with dysregulation of protein synthesis capacity. Front. Aging Neurosci. 11:246. doi: 10.3389/fnagi.2019.00246
Yang, W., Zhou, X., Zimmermann, H. R., and Ma, T. (2020). Brain-specific suppression of AMPKα2 isoform impairs cognition and hippocampal LTP by PERK-mediated eIF2α phosphorylation. Mol. Psychiatry doi: 10.1038/s41380-020-0739-z [Epub ahead of print].
Yao, R.-Q., Ren, C., Xia, Z.-F., and Yao, Y.-M. (2020). Organelle-specific autophagy in inflammatory diseases: a potential therapeutic target underlying the quality control of multiple organelles. Autophagy 17, 385–401. doi: 10.1080/15548627.2020.1725377
Yoon, S. O., Park, D. J., Ryu, J. C., Ozer, H. G., Tep, C., Shin, Y. J., et al. (2012). JNK3 perpetuates metabolic stress induced by Aβ peptides. Neuron 75, 824–837. doi: 10.1016/j.neuron.2012.06.024
Younas, N., Zafar, S., Shafiq, M., Noor, A., Siegert, A., Arora, A. S., et al. (2020). SFPQ and Tau: critical factors contributing to rapid progression of Alzheimer’s disease. Acta Neuropathol. 140, 317–339. doi: 10.1007/s00401-020-02178-y
Zhang, Y.-J., Gendron, T. F., Ebbert, M. T. W., O’Raw, A. D., Yue, M., Jansen-West, K., et al. (2018). Poly(GR) impairs protein translation and stress granule dynamics in C9orf72-associated frontotemporal dementia and amyotrophic lateral sclerosis. Nat. Med. 24, 1136–1142. doi: 10.1038/s41591-018-0071-1
Keywords: AMP kinase, protein translation, Alzheimer’s disease, amyotrophic lateral sclerosis, stress granules
Citation: Liu Y-J and Chern Y (2021) Contribution of Energy Dysfunction to Impaired Protein Translation in Neurodegenerative Diseases. Front. Cell. Neurosci. 15:668500. doi: 10.3389/fncel.2021.668500
Received: 16 February 2021; Accepted: 09 July 2021;
Published: 28 July 2021.
Edited by:
Ho Yin Edwin Chan, The Chinese University of Hong Kong, ChinaReviewed by:
Rodrigo A. Quintanilla, Pontificia Universidad Católica de Chile, ChileChiung-Mei Chen, Linkou Chang Gung Memorial Hospital, Taiwan
Copyright © 2021 Liu and Chern. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yijuang Chern, Ym15Y2hlcm5AaWJtcy5zaW5pY2EuZWR1LnR3