 Adonis Sfera1*
Adonis Sfera1* Carolina Osorio2
Carolina Osorio2 Carlos M. Zapata Martín del Campo3
Carlos M. Zapata Martín del Campo3 Shaniah Pereida2Steve Maurer1Jose Campo Maldonado4
Shaniah Pereida2Steve Maurer1Jose Campo Maldonado4 Zisis Kozlakidis5
Zisis Kozlakidis5- 1Patton State Hospital, San Bernardino, CA, United States
- 2Loma Linda University, Loma Linda, CA, United States
- 3Psychiatry Service, Outpatient Consultation Department, National Institute of Cardiology Ignacio Chavez, Mexico, Mexico
- 4Department of Internal Medicine, The University of Texas Rio Grande Valley, Edinburg, TX, United States
- 5International Agency for Research on Cancer (IARC), Lyon, France
Myalgic encephalomyelitis/chronic fatigue syndrome is a serious illness of unknown etiology, characterized by debilitating exhaustion, memory impairment, pain and sleep abnormalities. Viral infections are believed to initiate the pathogenesis of this syndrome although the definite proof remains elusive. With the unfolding of COVID-19 pandemic, the interest in this condition has resurfaced as excessive tiredness, a major complaint of patients infected with the SARS-CoV-2 virus, often lingers for a long time, resulting in disability, and poor life quality. In a previous article, we hypothesized that COVID-19-upregulated angiotensin II triggered premature endothelial cell senescence, disrupting the intestinal and blood brain barriers. Here, we hypothesize further that post-viral sequelae, including myalgic encephalomyelitis/chronic fatigue syndrome, are promoted by the gut microbes or toxin translocation from the gastrointestinal tract into other tissues, including the brain. This model is supported by the SARS-CoV-2 interaction with host proteins and bacterial lipopolysaccharide. Conversely, targeting microbial translocation and cellular senescence may ameliorate the symptoms of this disabling illness.
Introduction
Excessive accumulation of senescent cells in body tissues has been associated with organismal aging and fatigue as observed in older individuals and patients treated with anticancer agents (Sanoff et al., 2014; Rajeevan et al., 2018; Xu et al., 2018). On the other hand, preclinical studies have reported that the selective elimination of senescent cells could alleviate not only some chemotherapy adverse effects but also various age-related symptoms, including muscle weakness, fatigue and frailty, suggesting a potential treatment modality for myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) (Demaria et al., 2017; Short et al., 2019; Kaur et al., 2020; NCT03675724).
Long lasting myalgia and exhaustion were reported in over 40% of COVID-19 patients, indicating that the SARS-CoV-2 virus may directly invade skeletal muscles, triggering myositis (Chen G. et al., 2020; Huang et al., 2020; Kucuk et al., 2020). Indeed, as muscle cells express abundant angiotensin converting enzyme-2 (ACE-2), the virus likely exploits the myocytes, engendering disabling symptoms such as fatigue and weakness (Ferrandi et al., 2020; Jin and Tong, 2020; Mao et al., 2020). For example, preclinical studies have demonstrated that SARS-CoV-2-upregulated angiotensin II (ANG II) disrupts the muscle cell autophagy, impairing both the metabolism and contractility (Neel et al., 2013; Silva et al., 2019). However, aside from directly accessing the myocytes, this virus can induce myopathy, muscle weakness and atrophy indirectly by upregulating proinflammatory cytokines, such as interleukin 1 beta (IL-1β) and 6 (IL-6), C-reactive protein (CRP), and tumor necrosis factor (TNF) (VanderVeen et al., 2019; Guidon and Amato, 2020; Jin and Tong, 2020). When persistent, exertional and unrelieved by rest, myopathy may gradually morph into ME/CFS, a severe illness, affecting up to 2.5 million Americans (Blomberg et al., 2018; Friedman, 2019).
The National Academy of Medicine (former Institute of Medicine) 2015 ME/CFS diagnostic criteria include 6 months or longer of post-exertional malaise and unrefreshing sleep along with either cognitive impairment or orthostatic intolerance (Cortes Rivera et al., 2019). Tiredness and exhaustion are non-specific symptoms that may be experienced as central or “brain fog,” muscular and post-infectious, emphasizing the multifactorial nature of this condition (Greenberg, 2002; Yamashita, 2020). Indeed, the ME/CFS etiology includes genetic pre-disposition, inflammation, metabolic dysfunction, gastrointestinal pathology, autoimmunity, and viral infections (Jason et al., 1999; Ortega-Hernandez and Shoenfeld, 2009; Scherbakov et al., 2020).
During the COVID-19 pandemic, interest in ME/CFS has resurfaced as disabling fatigue, experienced by many patients, often lingers long after recovery, lowering the life quality (Wilson, 2020). Indeed, novel studies have reported ME/CFS in the aftermath of influenza and coronavirus infections, connecting viruses to the pathogenesis of severe exhaustion (Moldofsky and Patcai, 2011; Mohabbat et al., 2020; Poole-Wright et al., 2020). Along these lines, COVID-19 was demonstrated to alter several pathways previously associated with ME/CFS, suggesting that a better understanding of the virus/host interactome may elucidate the molecular underpinnings of this disease. Indeed, the viral crosstalk with several human proteins expressed by the intestinal epithelial cells (IECs) and endothelial cells (ECs) may alter the intestinal barrier, enabling microbial translocation from the gastro-intestinal (GI) tract into other tissues, including the brain (Maes and Leunis, 2008; Maes et al., 2014; Navaneetharaja et al., 2016; Proal and Marshall, 2018; Figure 1).
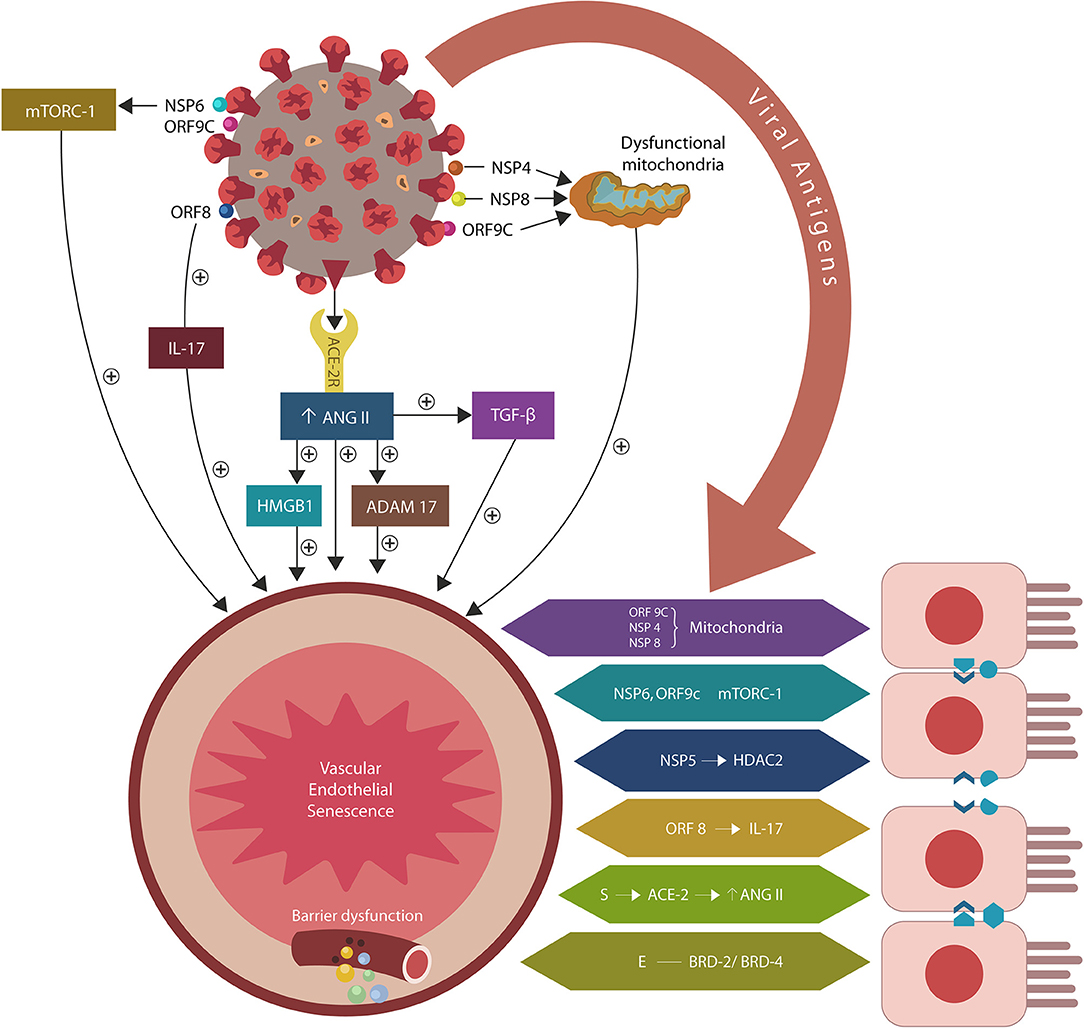
Figure 1. SARS-CoV-2/host protein-protein interactions. Viral crosstalk with several host proteins disrupts both endothelial cells (ECs) and intestinal epithelial cells (IECs), damaging the gut barrier and facilitating microbial and/or lipopolysaccharide (LPS) translocation into host tissues, including the skeletal muscle and the brain. Endothelial senescence also contributes to the disruption of blood-brain barrier (BBB), allowing microbial and/LPS access into the CNS. It is noteworthy that the cross talk between viral E antigen and host bromodomains (BRDs) 2 and 4 triggers macrophage senescence, impairing the elimination (efferocytosis) of aging cells. Viral S antigen attachment to ACE-2 receptor (ACE-2R) is followed by ANG II upregulation. This augments TGF-β, HMGB1, ADAM17, and ICAM-1 (not shown), inflicting endothelial and intestinal cells dysfunction with barrier disruption.
In an earlier paper, we proposed that premature EC senescence could increase the permeability of intestinal and blood-brain barrier (BBB), contributing to COVID-19 critical illness and its long-term sequelae (Sfera et al., 2020; Figure 1). Subsequent studies endorsed this model by demonstrating that aside from ACE-2, the SARS-CoV-2 virus can access host cells via dipeptidyl peptidase-4 (DPP4) and neuropilin-1 (NRP-1), receptors associated with ECs senescence (Kim et al., 2017; Issitt et al., 2019; Cantuti-Castelvetri et al., 2020; Chen Z. et al., 2020; Solerte et al., 2020). Further proof came from the reports linking the renin-angiotensin system (RAS) to accelerated aging via ANG II-induced telomere attrition demonstrated in both COVID-19 critical illness and Hutchinson-Gilford progeria (HGP) (Regenass et al., 1994; Herbert et al., 2008; Gerhard-Herman et al., 2012; Amraei and Rahimi, 2020; Aviv, 2020; Benetos et al., 2020; Bidault et al., 2020; Libby and Lüscher, 2020).
In the present hypothesis article, we surmise that COVID-19 sequelae, including the ME/CFS, may be caused by ANG II-inflicted fragmentation of biological barriers with subsequent microbial and/or lipopolysaccharide (LPS) translocation from the GI tract into various tissues, including the central nervous system (CNS). We focus primarily on the interaction between various SARS-CoV-2 antigens and host proteins expressed by ECs, IECs and immune cells that may increase barrier permeability, facilitating microbial translocation. If confirmed, this paradigm may open novel treatment opportunities in ME/CFS, including senotherapeutics as well as LPS and efferocytosis-targeting agents.
The Overall Presentation of Biological Model of Action
According to this paradigm, the ME/CFS pathology is initiated by cellular senescence and barrier disruption promoted by the SARS-CoV-2-upregulated ANG II or by direct viral interaction with host proteins (Figure 1). When intestinal repair is delayed (due to specific host factors), the prolonged microbial translocation results in aberrant immune responses characteristic of both ME/CFS and COVID-19 critical illness (Loebel et al., 2016; Morris et al., 2019; Wang E. Y. et al., 2020; Wang L. et al., 2020).
The model presented here is supported by the SARS-CoV-2 interactome and the cross talk between microbial LPS and viral S protein that disrupt biological barriers (Gaab et al., 2005; Maes and Leunis, 2008; Giloteaux et al., 2016; Petruk et al., 2020). Indeed, recent studies in both humans and rodents linked LPS to the unexplained fatigue, myopathy, muscle wasting and memory impairment, likely implicating the endotoxin in the pathogenesis of ME/CFS (Langhans et al., 2014; Friedrich et al., 2015; Zhang et al., 2016; Batista et al., 2019; Lasselin et al., 2020). With the same token, LPS was recently connected to neurodegenerative disorders, indicating that this pathology may be initiated by the translocation of microbes and/or their molecules into the CNS (Pretorius et al., 2018; Zhan et al., 2018).
These aspects are presented below in more detail and in relation to the SARS-COV-2 virus, starting with the interactome and cellular senescence (both for ANG II-dependent and ANG II-independent molecular changes); ANG II and defective efferocytosis; and building the case toward the connection with gut biology. Lastly, the reviewed evidence is synthesized for potential therapeutic interventions, including senotherapeutic strategies.
The Interactome and Cellular Senescence
The SARS-CoV-2 is an enveloped, positive-sense, single-stranded RNA virus that enters host cells via several receptors, including ACE-2, DPP-4 and NRP-1, expressed by various human tissues, including the gut, lung, muscle and ECs (Cantuti-Castelvetri et al., 2020; Fadini et al., 2020). The virus contains a genome of 30 kb that encodes for 29 proteins divided into structural, non-structural (NSP) and open-reading frame (ORF). These proteins were demonstrated to interact with as many as 332 human molecules, altering numerous pathways (Gordon et al., 2020).
The attachment of SARS-CoV-2 virus to ACE-2 receptors engenders ANG II dependent and independent molecular changes as reviewed below. The former include activation of ADAM17 (a disintegrin and metalloproteinase 17), ACE-2 downregulation and upregulation of transforming growth factor beta (TGF-β), high mobility group box 1 protein (HMBG1), toll-like receptor 4 (TLR4) and intercellular adhesion molecule 1 (ICAM-1). ANG II independent changes are comprised of direct interactions between viral antigens and host proteins that alter endothelia, skeletal muscle repair and immunological tolerance, especially the regulatory T cells (Tregs) and phagocytes [macrophages and natural killer cells (NKCs)] (Figure 1, Table 1).
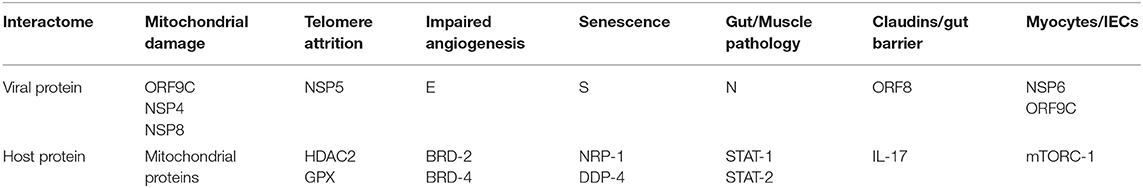
Table 1. Angiotensin II independent changes: viral antigens interact directly with host proteins, altering several senescence pathways, including the metabolism (mitochondrial damage), telomeres and angiogenesis that disrupt biological barriers and immunity, likely contributing to ME/CFS.
ANG II Dependent Molecular Changes
The SARS-CoV-2 attachment to ACE-2 receptors likely impairs ANG II hydrolysis, contributing to its unchecked accumulation. In addition, ADAM17 shedding of ACE-2 from the cell plasma membrane generates inactive, soluble ACE-2 (sACE-2) incapable of physiological functions (Patel et al., 2014). This engenders a vicious circle as excess ANG II activates ADAM17 that in turn upregulates ANG II by inhibiting its degradation. This may contribute to COVID-19 critical illness as overactive ADAM17 can lower the ACE-2 function by generating inactive sACE-2. Indeed, ACE-2 downregulation was associated with unfavorable COVID-19 prognosis, suggesting that sACE-2 along with the attached SARS-CoV-2 virus likely disseminates the infection throughout the body (Sfera et al., 2020; Verdecchia et al., 2020).
Dysfunctional renin-angiotensin system (RAS) was previously associated with accelerated aging as excess ANG II induces telomere attrition as demonstrated in both severe COVID-19 illness and children with Hutchinson-Gilford progeria, a syndrome of accelerated aging (Regenass et al., 1994; Herbert et al., 2008; Gerhard-Herman et al., 2012; Amraei and Rahimi, 2020; Benetos et al., 2020; Bidault et al., 2020; Libby and Lüscher, 2020). Moreover, ANG II-shortened telomeres and cellular senescence were documented in ME/CFS and cardiovascular disease, linking dysfunctional RAS to premature aging (Lieberman and Bell, 1993; Minamino and Komuro, 2002; Vasan et al., 2008; Fyhrquist et al., 2013; Squassina et al., 2019). Conversely, the telomere-repairing enzyme, telomerase, was reported not only to reverse ANG II-induced telomeres damage but also to ameliorate the symptoms of ME/CFS, further connecting this disorder to premature cellular senescence (Imanishi et al., 2005; Findeisen et al., 2011; Ho et al., 2012; Ait-Aissa et al., 2018). In addition, losartan, an ANG II receptor blocker (ARB), was demonstrated to restore the integrity of intestinal barrier as well as improve many symptoms of ME/CFS, further supporting the link between dysfunctional RAS and aging (Feng et al., 2011; Kumar et al., 2015; Nozu et al., 2020). Moreover, endothelial senescence and Alzheimer's disease (AD) were associated with ANG II-activated ADAM17, connecting both proteins to microvascular aging and disrupted biological barriers (Shatanawi et al., 2011; Morancho et al., 2015; Qian et al., 2016; Dou et al., 2017; Li et al., 2019; Shalaby et al., 2020). Indeed, impaired intestinal barrier with subsequent endotoxemia were demonstrated in both AD and Parkinson's diseases (PD) patients, suggesting that LPS could reach the CNS and initiate neurodegeneration (Hoban et al., 2013; Sun et al., 2016; Zhao et al., 2017; Shigemoto-Mogami et al., 2018). Along these lines, earlier studies demonstrated that LPS could access other organs, including the lung (via ANGII/ADAM17-increased vascular permeability) and trigger gram-negative pneumonias, connecting dysfunctional gut barrier to organ-specific pathology (Dreymueller et al., 2012; Morancho et al., 2015).
Aside from activating ADAM17, ANG II also upregulates intracellular TGF-β, HMBG1, TLR4, and ICAM-1, promoting inflammation, fibrosis and oxidative stress that in turn, disrupt the biological barriers and immunity (Ribadeneira et al., 1996; Kunieda et al., 2006; Crowley and Rudemiller, 2017; Cooper et al., 2018; Figure 2). ANG II also inhibits the muscle tissue repair, generating myopathy and atrophy as well as endothelial and immune damage, further implicating dysfunctional RAS in ME/CFS symptoms (Ferrario et al., 2005; Turowski et al., 2005; Cabello-Verrugio et al., 2012; Morris et al., 2014; Wyller et al., 2016; Cooper et al., 2018; Tirone et al., 2018; Monteil et al., 2020; Figure 1). Indeed, defective endothelia were directly correlated with the severity of ME/CFS symptoms, emphasizing the role of defective vascular barrier in the pathogenesis of this disease (Yamazaki et al., 2016; Obrenovich, 2018; Scherbakov et al., 2020).
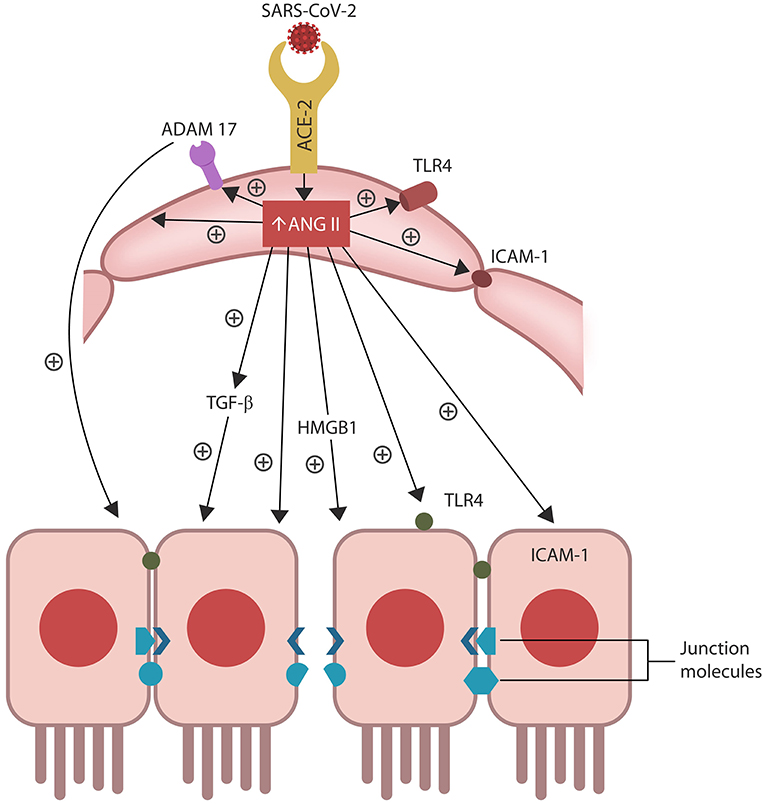
Figure 2. ANG II accumulation in ECs alters several senescence-associated pathways, including ADAM17-downregulation of ACE-2 in ECs and IECs. ANG II-upregulated pathways include TGF-β, HMGB1, TLR4, ICAM-1, and SASP (not shown). Together these molecules induce cellular senescence, increasing intestinal and BBB permeability likely contributing to ME/CFS.
Senescent cells permanently exit the cell cycle but remain metabolically active, releasing pro-inflammatory cytokines that comprise a specific secretome, the senescence-associated secretory phenotype (SASP). ANG II upregulates SASP, while ADAM17 sheds these molecules from the cell plasma membranes, facilitating their dissemination throughout the body (Effenberger et al., 2014; Rajeevan et al., 2018; Aviv, 2020; Simões et al., 2020; Song et al., 2020; Takeshita et al., 2020).
Preclinical studies have reported that aside from disrupting ECs and the vascular barrier, ANG II can trigger IECs apoptosis, increasing also the permeability of the epithelial barrier, a condition often referred to as dysbiosis or “leaky gut” (Koga et al., 2008; Shi et al., 2016; Tanaka and Itoh, 2019; Figure 2). Indeed, a bidirectional relationship was found between RAS dysfunction and intestinal dysbiosis, suggesting that ARBs may be therapeutic by optimizing the barrier integrity (Lu et al., 2018; Viana et al., 2020).
ANG II Independent Changes
The SARS-CoV-2/host interactome has revealed senescence-inducing mechanisms that can be activated by the direct cross talk between various viral antigens and host proteins. For example, mitochondrial damage can be inflicted indirectly by ANG II or by the direct cross talk of SARS-CoV-2 antigens ORF9C, NSP4, and NSP8 with mitochondrial proteins (Gordon et al., 2020; Table 1, Figure 1). In addition, aside from ANG II, telomere attrition can be triggered by the SARS-CoV-2 protein NSP5 interaction with human histone deacetylases 2 (HDAC2) or glutathione peroxidase (GPX) (Takakura et al., 2001; Månsson et al., 2019; Table 1, Figure 1).
Moreover, the dialog of SARS-CoV-2 antigen NSP6 or ORF9C with the human mammalian target of rapamycin complex 1 (mTORC-1) disrupts both IECs and myocytes, suggesting a role in the pathogenesis of ME/CFS (Yoon, 2017; Kaur and Moreau, 2019; Gordon et al., 2020). Likewise, the cross talk between viral protein ORF8 and host interleukin 17 (IL-17) was shown to alter the claudin molecule, disrupting intestinal barrier (Lee J. S. et al., 2015; Matsumoto et al., 2017; Veldhoen, 2017; Gordon et al., 2020). Indeed, SARS-CoV-2 manipulation of IL-17 may contribute directly to the pathogenesis of ME/CFS as this cytokine is crucial for muscle contractility and its depletion characterizes Duchenne muscular dystrophy (De Pasquale et al., 2012; Wang et al., 2019). Furthermore, the interaction between the N antigen of SARS-CoV-2 and host signal transducer and activator of transcription (STAT) 1 and 2, can disrupt both the function of skeletal muscle and anti-viral defenses, indicating that the virus exploits several pathways associated with weakness, fatigue and the susceptibility to infections (Moresi et al., 2019; Mu et al., 2020; Table 1).
ANG II and Defective Efferocytosis
Novel studies have shown that aside from inducing cellular senescence, COVID-19 disrupts the clearance or efferocytosis of aging or dying cells, leading to their unchecked accumulation. Excessive buildup of senescent cells has been associated with exhaustion, fatigue, sarcopenia, frailty, immune dysfunction and cognitive impairment, symptoms that also characterize ME/CFS (Jeyapalan and Sedivy, 2008; LeBrasseur et al., 2015; Lee, 2019; Nelke et al., 2019; Martínez-Cué and Rueda, 2020). For example, chemotherapy-receiving breast cancer patients with elevated levels of p16INK4a senescent marker were found to experience most fatigue (Sanoff et al., 2014; Rajeevan et al., 2018). On the other hand, enhanced clearance of senescent cells was associated with improved physical activity and reduced fatigue in animal models (Demaria et al., 2017). For this reason, various senotherapeutic strategies targeting frailty and exhaustion in individuals on cancer therapy are currently in clinical trials, suggesting that these agents may also benefit ME/CFS patients (Lewis-McDougall et al., 2019; Short et al., 2019).
Under normal circumstances, billions of cells in the human body undergo apoptosis each day and are promptly removed by professional and non-professional phagocytes, including macrophages, NKCs and ECs (facultative phagocytes) (Vann and Proctor, 1990; Kirsch et al., 2007; Qingxian et al., 2010; Seeberg et al., 2019; Zhou et al., 2019). Macrophages, the primary efferocytosis executors, also participate in endothelial repair, linking the elimination of senescent cells to the pathology of biological barriers (Kearns et al., 2012; Zhu et al., 2016). On the other hand, ANG II signaling via angiotensin II type 1 receptors (AT-1Rs) disrupts both efferocytosis and ECs function as this peptide shifts macrophages from the phagocytic (M2) to proinflammatory (M1) phenotype, a pattern consistent with autoimmune inflammation (Belline et al., 2004; Yamamoto et al., 2011). ANG II disrupts the phagocytic function of macrophages and efferocytosis in addition to inducing ECs senescence and barrier dysfunction, defects encountered in both ME/CFS and COVID-19 critical illness (Zhang et al., 2019; Schulte-Schrepping et al., 2020). Conversely, the macrophage-activating factor was shown to restore normal efferocytosis as well as ameliorate many ME/CFS symptoms, suggesting a therapeutic potential (Inui et al., 2015). Moreover, both hyperinflammation and endotoxin tolerance, were demonstrated in ME/CFS and severe COVID-19, linking aberrant immune responses to the accumulation of senescent cell (Monneret and Venet, 2012; Fenwick et al., 2019; Zheng et al., 2020). Indeed, dysfunctional macrophages can generate a sepsis-like immune pattern marked by an initial hyperinflammation followed by immunosuppression, endotoxin tolerance and exhausted lymphocytes (Pena et al., 2011; Elder and Emmerson, 2020). As aberrant immune responses were associated with both ME/CFS and COVID-19 critical illness, dysfunctional efferocytosis is a likely contributor to both disorders (Manestar-Blazić and Volf, 2009; Morris and Maes, 2013; Fukushima et al., 2018; Rajeevan et al., 2018; Silva et al., 2019; Zhou T. et al., 2020; Kruglikov and Scherer, 2021).
Efferocytosis: The Molecular Mechanisms
Efferocytosis is initiated by phagocytes responding to the “eat me” signals expressed on the plasma membrane of senescent or dying cells, marking them as “ready” for clearance (Barth et al., 2017; Karaji and Sattentau, 2017; Kale et al., 2020). Defective elimination of senescent ECs and IECs may increase the permeability of intestinal barrier, promoting dysbiosis and microbial translocation.
In general, efferocytosis occurs without immunogenicity as phagocytes engulf target cells without “spillage” of intracellular material into the extracellular milieu. When cytosolic content “escapes” cell confinement, it acts as a damage-associated molecular pattern (DAMP) that activates immunity, engendering inflammation (Abdolmaleki et al., 2018; Kawano and Nagata, 2018). For example, extracellular HMGB1 (a molecule upregulated by ANG II) is a potent DAMP associated with COVID-19 cytokine storm and several autoimmune disorders (Friggeri et al., 2010; Banerjee et al., 2011; Harris et al., 2012; Magna and Pisetsky, 2014; Tsung et al., 2014; Chen R. et al., 2020; Mangalmurti and Hunter, 2020; Figure 1). Interestingly, excessive HMBG1 was linked to unexplained fatigue, chronic pain, exhaustion, and muscle dysfunction, suggesting a role in the pathogenesis of ME/CFS (Morris and Maes, 2013; Zong et al., 2013; Wan et al., 2016; Nguyen et al., 2017; Figure 2).
Efferocytosis molecular sensors, including MerTK, recognize the externalized phosphatidylserine (PS), a major “eat me” signal, expressed by senescent or dying cell, earmarking them for clearance. MerTK is shed and inactivated by ADAM17, disrupting efferocytosis and triggering senescent cells-mediated pathology (Thorp et al., 2011; Dransfield et al., 2015; de Couto et al., 2019; Palau et al., 2020; Sfera et al., 2020; Figure 2). Indeed, overactive ADAM17 with excessive shedding of MerTK and ACE-2 could comprise a significant pathogenetic mechanism of COVID-19 and ME/CFS (Casciola-Rosen et al., 2020; Chaudhary, 2020; Megremis et al., 2020; Miesbach, 2020; Pagliaro and Penna, 2020). Along these lines, studies in athletes found a direct correlation between ACE-2 downregulation and poor muscle performance, further linking ADAM17 to the pathogenesis of ME/CFS (Motta-Santos et al., 2016). Interestingly, PS externalization was shown to promote ADAM17 activation and ACE-2 shedding, suggesting that the SARS-CoV-2 virus likely exploits this mechanism (Sommer et al., 2016).
The SARS-CoV-2 Virus and Apoptotic Mimicry
It has been established that some viruses exploit PS signaling to directly invade the host phagocytes, a process known as apoptotic mimicry (Mercer and Helenius, 2010; Morizono and Chen, 2014; Segawa and Nagata, 2015). Emerging evidence indicates that the SARS-CoV-2 virus may disrupt efferocytosis by accessing host phagocytic cells by this route. Indeed, anti-PS antibodies, PS-containing extracellular vesicles (EVs) and platelets with externalized PS were demonstrated in COVID-19 critical illness (Urciuoli and Peruzzi, 2020; Zaid et al., 2020; Zhou Y. et al., 2020; Lind, 2021). Moreover, since ECs (non-professional phagocytes) express PS receptors, the SARS-CoV-2 virus may usurp these proteins to directly invade host endothelia (Setty and Betal, 2008). In addition, neutrophils with externalized PS were reported in ME/CFS patients, suggesting that viruses capable of apoptotic mimicry may contribute to this disorder (Kennedy et al., 2004). Furthermore, dysfunctional NKCs (professional phagocytes) were demonstrated in ME/CFS patients, further connecting this illness to apoptotic mimicry-impaired efferocytosis (Maher et al., 2005; Eaton-Fitch et al., 2019).
The SARS-CoV-2/host interactome found that the crosstalk between viral protein E (envelope) and human BRD-2 and BRD-4 triggers macrophage senescence and efferocytosis disruption (Gordon et al., 2020; Wang H. et al., 2020; Table 1). Interestingly, the SARS-CoV-2 spike (S) protein interacts with microbial LPS, inducing macrophage senescence, indicating that the virus may utilize several parallel mechanisms for disabling the efferocytosis (Petruk et al., 2020). Since BDR-4 is an established driver of angiogenesis and microvascular repair, viral exploitation of this protein likely alters the biological barriers, enabling microbial translocation and the ME/CFS pathogenesis (Maes and Leunis, 2008; Maes et al., 2014; Zhou Z. et al., 2020). Furthermore, BRD-4 depletion was associated with generalized muscle weakness, suggesting that post-exertional malaise, a well-established ME/CFS marker, may be engendered through this mechanism (Segatto et al., 2017).
Taken together, this data shows that the SARS-CoV-2 virus likely utilizes apoptotic mimicry to disrupt efferocytosis, leading to the accumulation of senescent cells that in turn triggers hyperinflammation which exhausts the immune system, facilitating viral infection. Impaired efferocytosis in the intestinal barrier and failure to eliminate senescent ECs and IECs may increase the gut permeability, allowing microbial/LPS translocation.
Cellular Senescence and the Gut
Intestinal barrier separates the gut lumen from the rest of the body, preventing migration of microbes or molecules outside the GI tract, while at the same time, ensuring adequate nutrient absorption. A single layer of IECs covered by abundant mucus comprise the epithelial portion of the gut barrier, while ECs constitute the vascular component (Thomas, 2016). Aside from the barrier function, IECs mediate the interaction between gut microorganisms and resident immune cells, balancing the immunological acceptance of intestinal microbes with pathogen rejection (Mizrahi and Ilan, 2009; Edelblum et al., 2017; Poggi et al., 2019). At the molecular level, IECs are kept together by junction molecules, including the claudins, that control barrier permeability by regulating the size of intercellular spaces (Garcia-Hernandez et al., 2017; Garcia et al., 2018; Figure 2). Interestingly, preclinical studies have reported that ANG II, acting via AT-1R, increases the permeability of intestinal barrier by altering the expression of claudin-7 (Shi et al., 2016; Takashina et al., 2020). In addition, IL-17, a cytokine directly usurped by the SARS-CoV-2 viral protein ORF8, influences the gut permeability via claudins (Lee J. S. et al., 2015; Andrews et al., 2018; Table 1). Moreover, the SARS-CoV-2 antigens NSP4, NSP8, and ORF9C interact with IECs mitochondria increasing the permeability of intestinal barrier by an alternative mechanism (Lee J. H. et al., 2015; JanssenDuijghuijsen et al., 2017; Figure 1, Table 1). Moreover, ICAM-1, an ANG II-controlled protein, is essential for maintaining intestinal and endothelial integrity, suggesting that the SARS-CoV-2 virus can also alter the gut barrier by manipulating RAS (Sumagin et al., 2014; Sarelius and Glading, 2015; Figure 2).
The Permeability-Immunity Axis
In the GI tract, intestinal permeability is tightly intertwined with local immunity as IECs, ECs and gut resident immune cells regulate the barrier function, nutrient absorption and gut microbial composition. For example, intestinal ACE-2 receptors also function as neutral amino acid transporters (B0AT1 or SLC6A19), therefore a dysfunctional RAS can trigger barrier disruption, dysbiosis and amino acid malabsorption (Cheng et al., 2017; Jando et al., 2017; Viana et al., 2020). For this reason, it is not surprising that COVID-19 and ME/CFS have been associated with both aberrant immune responses and dysfunctional intestinal permeability (Gaab et al., 2005; Maes and Leunis, 2008; Maes et al., 2014; Morris et al., 2019). Conversely, normalization of gut barrier was found to ameliorate immunity in COVID-19 and the symptoms of ME/CFS patients (Maes and Leunis, 2008; Maes et al., 2014; Du Preez et al., 2018; Mandarano et al., 2018; Cardinale et al., 2020). Along these lines, the permeability-immunity connection may explain the higher prevalence of inflammatory bowel disease (IBD) in ME/CFS patients as both conditions are marked by dysfunctional gut barrier and immune responses (Newton et al., 2012; Gravina et al., 2018; Mandarano et al., 2018; Tsai et al., 2019; Scherbakov et al., 2020). With the same token, active IBD was associated with unfavorable COVID-19 outcomes, suggesting that the SARS-CoV-2 attachment to ACE-2 (abundantly expressed on IECs and resident Tregs) disrupts both the intestinal barrier and the local immunity (Bezzio et al., 2020; Lin et al., 2020). In addition, the premature senescence of intestinal endothelia and increased microbial translocation may contribute to body-wide immune changes demonstrated in ME/CFS and COVID-19 critical illness (Poujol et al., 2015; Huth et al., 2016; Kasper et al., 2016; Mandarano et al., 2018; Galván-Peña et al., 2020; Li et al., 2020; van Eeden et al., 2020; Zheng et al., 2020).
In the GI tract, commensals are immunologically protected by the resident Tregs that mediate luminal immunosuppression. However, outside the gut, microbes are vigorously attacked by the host immune system that often generates hyperinflammatory responses that may exhaust the lymphocytes, generating endotoxin tolerance (Ramos et al., 2016; Sotzny et al., 2018; Sepúlveda et al., 2019; Fine et al., 2020).
At the molecular level, Tregs-induced immunosuppression is engendered by the NRP-1/IL-10 signaling that requires the presence of vascular endothelial growth factor (VEGF) (Ohm et al., 2003; Wang et al., 2015; Chuckran et al., 2020). The SARS-CoV-2 attachment to NRP-1 lowers VEGF, leading to excessive immune tolerance that helps the virus avert detection (Yin et al., 2020; Mayi et al., 2021). In addition, as VEGF also regulates angiogenesis, endothelial senescence and efferocytosis, the disruption of NRP-1/IL-10/VEGF axis likely triggers the ME/CFS and COVID-19 pathogenesis (Watanabe et al., 1997; Hasan et al., 2011; Kearns et al., 2012). Moreover, SARS-CoV-2/NRP-1 attachment and dysfunctional VEGF in IECs, myocytes, ECs and Tregs was associated with disabling fatigue, further connecting these proteins to ME/CFS (Yadav et al., 2012; Wang et al., 2015; Yamaji et al., 2015; Issitt et al., 2019; Cantuti-Castelvetri et al., 2020; Davies et al., 2020; Moutal et al., 2021; Table 1). Interestingly, earlier studies have reported VEGF downregulation in ME/CFS patients, linking this growth factor to disabling fatigue (Landi et al., 2016; Dai et al., 2017; Petruk et al., 2020). Moreover, as the S antigen of SARS-CoV-2 virus binds microbial LPS, and LPS-activated Tregs inhibit immune responses, the ME/CFS-associated endotoxin tolerance may be triggered via this mechanism (Lewkowicz et al., 2006; Morris et al., 2019; Table 2). Furthermore, the SARS-CoV-2 virus can also generate endotoxin tolerance in other manner, including ANG II-upregulation of intracellular HMGB1 and TGF-β, NSP6/ORF9C interaction with host mTORC-1, and antigen E cross talk with BRD-2 and BRD-4 (Aneja et al., 2008; Yang et al., 2015; Sun et al., 2018; Copsel et al., 2019; Figure 1, Table 2).

Table 2. The SARS-CoV-2 immunological tolerance-inducing mechanisms.
Autoantibodies in ME/CFS
Another manifestation of the ME/CFS-associated immune dysregulation is the presence of autoantibodies directed against cholinergic and β2 adrenergic receptors (β2AdRs) documented by numerous studies (Loebel et al., 2016; Wirth and Scheibenbogen, 2020). Autoantibodies are believed to reflect the presence of altered “self” proteins that are unrecognized and therefore attacked by the immune system.
An alternative explanation could fathom autoantibodies in ME/CFS patients as being directed at microbial molecules translocated from the host GI tract. Indeed, many gut microbes express muscarinic and adrenergic receptors, suggesting that autoantibodies may target these molecules (Furukawa and Haga, 2000; Karavolos et al., 2013; Moreira et al., 2016). Along these lines, translocated microorganisms that express β2AdRs, may elicit antibodies cross-reacting with host's own adrenergic receptors. For example, antibodies against β1 and β2AdRs of translocated Escherichia coli may cross react with their human receptors (Freissmuth et al., 1991). Interestingly, recent studies have demonstrated that many ME/CFS patients present with significantly higher norepinephrine plasma levels, indicating that anti-β2AdR antibodies may aim at downregulating adrenergic transmission and restore homeostasis (Wyller et al., 2016).
Under normal circumstances, the anti-β2AdR antibodies activate β2AdR receptors, however (probably due to elevated norepinephrine) this response is attenuated in ME/CFS patients, suggesting once more the compensatory role of autoantibodies (Hartwig et al., 2020). Other pathogens, including the reovirus type 3, demonstrated molecular mimicry with human β2AdR, promoting adaptive autoantibodies that eliminate the virus-infected cells by targeting these receptors (Co et al., 1985). Moreover, a recent study demonstrated that the microbial metabolite phenylacetylglutamine (PAGIn) excessively upregulates human cardiac adrenergic signaling, promoting heart disease, a condition that autoantibodies could preempt (Nemet et al., 2020). With the same token, viruses that populate the human GI tract were shown to modify various host molecules, transforming them into immune targets (Campbell, 2014; Mukhopadhya et al., 2019). For example, the hemagglutinin antigen (HA) of H1N1 influenza virus can alter host hypocretin molecule, turning it into an antigen significant for the pathogenesis of autoimmune narcolepsy (Luo et al., 2018). On the other hand, increased tryptophan absorption due to dysfunctional intestinal barrier and overactivation of brain kynurenine pathway (KP) was demonstrated in ME/CFS patients, further linking the dysfunctional GI tract to this disease (Georgiades et al., 2003; Yamashita, 2020). Interestingly, ARBs were demonstrated to inhibit KP, probably by normalizing intestinal permeability, limiting tryptophan absorption (Blankfield, 2011; Zakrocka et al., 2017).
Potential Interventions for Barrier Dysfunction
This section focuses on three types of interventions in line with the hypothesis presented here: restoration of adequate intestinal permeability, LPS lowering or removal and efferocytosis optimization. Most agents operate by more than one mechanism of action, indicating that superior efficacy may be achieved by combining therapeutic modalities. Indeed, some ME/CFS drugs currently in use directly or indirectly restore the function of biological barriers, ameliorate efferocytosis and lower LPS. For example, rituximab, a monoclonal antibody often utilized in ME/CFS, improves the phagocytic function of macrophages and efferocytosis, in addition to its established actions on antibodies and B lymphocytes (Toubi et al., 2007; Djaldetti et al., 2019). Another example is escitalopram, a frequently prescribed drug to ME/CFS patients with depressed mood, that aside from its antidepressant action, also promotes endothelial restoration, optimizing the permeability of biological barriers (Lopez-Vilchez et al., 2016). Other ME/CFS therapies, including the combination of coenzyme Q10 and nicotinamide adenine dinucleotide (NAD), improve endothelial function, demonstrating an alternative mechanism of action (Gao et al., 2012; Castro-Marrero et al., 2016; Mateuszuk et al., 2020). Conversely, drugs that restore endothelial integrity, including beta blockers, are often beneficial to ME/CFS patients, emphasizing the role of dysfunctional biological barriers in the pathogenesis of this illness (Su, 2015; Wyller et al., 2016). Mildronate, an anti-ischemic drug, often helpful to ME/CFS patients, also restores endothelial integrity by upregulating nitric oxide (Sjakste et al., 2005; Comhaire and Deslypere, 2020). Yet other endothelium-protecting drugs, such as ARBs, may offer relief to ME/CFS patients as they improve muscle strength, exercise capacity, and cognition (Nade et al., 2015; Coelho et al., 2016). In addition, ARBs optimize intestinal permeability and macrophage-mediated efferocytosis, indicating more than one action mechanism (Villapol and Saavedra, 2015; Shi et al., 2016). Moreover, a cross-reaction was demonstrated between AT-1Rs and β2AdRs, as they are inhibited by a single antagonist, suggesting that ARBs and β-blockers may be equally effective in ME/CFS (Blumenfeld et al., 1999; Barki-Harrington et al., 2003).
Considering the hypothesis presented here, several drugs not currently utilized in ME/CFS may emerge as potential therapies. These include short chain fatty acids (SCFAs), milk fat globule membranes (MFGM), β-glucan, VEGF-agonists, fecal microbial transplantation and senolytic agents, including Navitoclax and fistein.
Microbial products, such as SCFAs are derived from the fermentation of dietary fiber in the GI tract. They may be beneficial to ME/CFS patients as they promote intestinal barrier restoration, correct dysbiosis and local immunity (Chen et al., 2017; Newberry et al., 2018; Yang et al., 2020).
Fecal microbial transplantation (FMT), has been suggested as a treatment modality for ME/CFS after being promoted by a few uncontrolled studies, that found long lasting improvement (Castro-Marrero et al., 2016). However, at present, the application protocols, optimal donors and the long-term risks of FMT are not entirely clear (Evrensel and Ceylan, 2016).
Milk fat globule membranes (MFGM) and β-glucan were found to decrease both microbial translocation and fatigue in murine models (Vetvicka and Vetvickova, 2015; Vetvicka et al., 2019; Yu et al., 2020). Indeed, aside from restoring the intestinal barrier, β-glucan also reverses endotoxin tolerance, correcting the aberrant immune responses that characterize ME/CFS (Wu et al., 2018).
Upon crossing from the GI tract into the systemic circulation, LPS may access the brain via disrupted BBB or areas of high physiological permeability, such as the circumventricular organs. Inside the brain, endotoxin binds to microglial TLR4, activating these cells, probably contributing to cognitive impairment, or “brain fog” experienced by many ME/CFS patients (Qin et al., 2007; Theoharides et al., 2015; Tsukamoto et al., 2018). β-glucan inhibits TLR4 endocytosis, preventing LPS activation of microglia and the subsequent cognitive impairment (Novakovic et al., 2016). Interestingly, LPS-binding protein (LBP), an acute phase reactant, can remove LPS by attaching to it tightly, neutralizing its actions, suggesting a therapeutic potential in ME/CFS (Mathison et al., 1992; Goldblum et al., 1994; Giloteaux et al., 2016). Moreover, β-glucan was demonstrated to augment macrophage-mediated efferocytosis in animal model, indicating that by decreasing the accumulation of senescent cells it could help the ME/CFS patients (Fatima et al., 2017). Others have suggested that metformin protects the gut barrier by lowering LPS-inflicted damage, indicating a mechanism of action in line with the hypothesis presented here (Brown et al., 2018; Wu et al., 2018). Aside from safeguarding the GI tract, metformin also augments microglia-mediated efferocytosis, protecting the CNS against senescent cell pileup (Tabuzek et al., 2010).
A recent open-label, pilot study in human subjects utilized senotherapeutics in patients with pulmonary fibrosis, demonstrating good tolerability and indicating potential therapeutic benefits in ME/CFS (Justice et al., 2019). Indeed, several senolytic drugs were recently tested, including dasatinib (FDA-approved for chronic myeloid leukemia), hyperoside, quercetin, fistein and the BCL-2 inhibitor, Navitoclax (Kirkland et al., 2017; Mohamad Anuar et al., 2020). As ECs are dependent on BCL-2, Navitoclax should be tested for ME/CFS (Zhu et al., 2016; Wissler Gerdes et al., 2020). Fistein, currently in phase 2 clinical trials for the frail elderly syndrome, has demonstrated good tolerability, indicating potential beneficial effects in ME/CFS (AFFIRM-LITE trial NCT03675724).
Conclusions
The data presented above supports the hypothesis that ME/CFS pathology likely commences with a pathogen-induced intestinal barrier disruption and subsequent microbial translocation. These in turn trigger aberrant immune responses in various host tissues, ranging from autoimmune inflammation (cytokine storm) to excessive tolerance, likely engendering the COVID-19 and ME/CFS pathologies.
Restoration of adequate intestinal permeability, LPS lowering and efferocytosis optimization are the suggested interventions in line with the hypothesis presented here.
Limitations
This is not a systematic review hence additional publications might be identified as relevant to the subject. Secondly, while this hypothesis has a clear biological foundation, it would still require to be validated at a minimum through retrospective analysis of clinical data where possible. Thirdly, as the SARS-CoV-2 interactome has demonstrated, systemic infections present a complicated landscape or pathways that may act synergistically or in parallel. As such this hypothesis can be viewed as a starting point toward an increased understanding of the relationship between viral infections, chronic inflammation (including that of the gut) and ME/CFS.
Future Perspectives
ME/CFS is a serious illness with unclear etiology and non-specific treatments. In addition, as it is often dismissed by both the public and healthcare workers, patients with this condition are frequently stigmatized and may avoid seeking help. Decreasing stigmatization by educating the public and clinicians on the biological aspects of this condition is therefore very important. In this regard, the molecular hypothesis presented here may contribute to this goal.
Future research will likely probe deeper into the interface between gut microbes and the local immune system, elucidating not only the pathogenesis of ME/CFS, but also of other illnesses associated with dysfunctional immune tolerance or activation, including autoimmune, neuropsychiatric and degenerative disorders.
Studying the virus/host interactome and associated pathology will contribute to a better understanding of the largest immune compartment in the body, the GI tract, and its role in the immunological acceptance of gut microbes. This will likely contribute to the development of antigen-specific immunosuppressant therapies, such human Tregs. Indeed, allogeneic T cells are currently in clinical trials for COVID-19 hyperinflammatory syndrome (NCT04482699). Unlike non-specific immunosuppressive agents that impact many tissues and organs, generating adverse effects, Tregs offers specificity and precision that could benefit not only the patents with ME/CFS, but also those with allergies, transplants and infectious diseases.
Furthermore, elucidation of the molecular underpinnings of microbial translocation will undoubtedly lead to more specific treatments for restoring the adequate permeability of the GI tract, that would benefit patients with inflammatory bowel disease (IBS). As gut microbes alter both the intestinal and blood-brain barrier, manipulation of microorganismal translocation will likely contribute to the development of treatments for some CNS diseases, including the neurodegenerative disorders, such as Alzheimer's and Parkinson's disease (Osorio et al., 2019).
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Disclaimer
Where authors are identified as personnel of the International Agency for Research on Cancer/WHO, the authors alone are responsible for the views expressed in this article and they do not necessarily represent the decisions, policy or views of the International Agency for Research on Cancer/WHO.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abdolmaleki, F., Farahani, N., Gheibi Hayat, S. M., Pirro, M., Bianconi, V., Barreto, G. E., et al. (2018). The role of efferocytosis in autoimmune diseases. Front. Immunol. 9:1645. doi: 10.3389/fimmu.2018.01645
Ait-Aissa, K., Kadlec, A. O., Hockenberry, J., Gutterman, D. D., and Beyer, A. M. (2018). Telomerase reverse transcriptase protects against angiotensin II-induced microvascular endothelial dysfunction. Am. J. Physiol. Heart Circ. Physiol. 314, H1053–H1060. doi: 10.1152/ajpheart.00472.2017
Amraei, R., and Rahimi, N. (2020). COVID-19, renin-angiotensin system and endothelial dysfunction. Cells 9:1652. doi: 10.3390/cells9071652
Andrews, C., McLean, M. H., and Durum, S. K. (2018). Cytokine tuning of intestinal epithelial function. Front. Immunol. 9:1270. doi: 10.3389/fimmu.2018.01270
Aneja, R. K., Tsung, A., Sjodin, H., Gefter, J. V., Delude, R. L., Billiar, T. R., et al. (2008). Preconditioning with high mobility group box 1 (HMGB1) induces lipopolysaccharide (LPS) tolerance. J. Leukoc. Biol. 84, 1326–1334. doi: 10.1189/jlb.0108030
Banerjee, S., de Freitas, A., Friggeri, A., Zmijewski, J. W., Liu, G., and Abraham, E. (2011). Intracellular HMGB1 negatively regulates efferocytosis. J. Immunol. 187, 4686–4694. doi: 10.4049/jimmunol.1101500
Barki-Harrington, L., Luttrell, L. M., and Rockman, H. A. (2003). Dual inhibition of beta-adrenergic and angiotensin II receptors by a single antagonist: a functional role for receptor-receptor interaction in vivo. Circulation 108, 1611–1618. doi: 10.1161/01.CIR.0000092166.30360.78
Barth, N. D., Marwick, J. A., Vendrell, M., Rossi, A. G., and Dransfield, I. (2017). The “phagocytic synapse” and clearance of apoptotic cells. Front. Immunol. 8:1708. doi: 10.3389/fimmu.2017.01708
Batista, C. R. A., Gomes, G. F., Candelario-Jalil, E., Fiebich, B. L., and de Oliveira, A. C. P. (2019). Lipopolysaccharide-Induced neuroinflammation as a bridge to understand neurodegeneration. Int. J. Mol. Sci. 20:2293. doi: 10.3390/ijms20092293
Belline, P., da Melo, P. S., Haun, M., Palhares, F. B., Boer, P. A., Gontijo, J. A., et al. (2004). Effect of angiotensin II and losartan on the phagocytic activity of peritoneal macrophages from Balb/C mice. Mem. Inst. Oswaldo Cruz 99, 167–172. doi: 10.1590/S0074-02762004000200009
Benetos, A., Lai, T. P., Toupance, S., Martindale, J. L., Yang, X., Indiget, F. E., et al. (2020). A Mechanism for severity of disease in older patients with COVID-19: the nexus between telomere length and lymphopenia. medRxiv. 83, 1–5. doi: 10.1101/2020.10.01.20205393
Bezzio, C., Saibeni, S., Variola, A., Allocca, M., Massari, A., Gerardi, V., et al. (2020). Outcomes of COVID-19 in 79 patients with IBD in Italy: an IG-IBD study. Gut 69, 1213–1217. doi: 10.1136/gutjnl-2020-321411
Bidault, G., Garcia, M., Capeau, J., Morichon, R., Vigouroux, C., and Béréziat, V. (2020). Progerin expression induces inflammation, oxidative stress and senescence in human coronary endothelial cells. Cells 9:1201. doi: 10.3390/cells9051201
Blankfield, A. (2011). Kynurenine pathway Hypothesis: the nature of the chronic Fatigue syndrome (cFs) Revisited. Int. J. Tryptophan Res. 4, 47–48. doi: 10.4137/IJTR.S7898
Blomberg, J., Gottfries, C. G., Elfaitouri, A., Rizwan, M., and Rosén, A. (2018). Infection elicited autoimmunity and myalgic encephalomyelitis/chronic fatigue syndrome: an explanatory model. Front. Immunol. 9:229. doi: 10.3389/fimmu.2018.00229
Blumenfeld, J. D., Sealey, J. E., Mann, S. J., Bragat, A., Marion, R., Pecker, M. S., et al. (1999). Beta-adrenergic receptor blockade as a therapeutic approach for suppressing the renin-angiotensin-aldosterone system in normotensive and hypertensive subjects. Am. J. Hypertens. 12, 451–459. doi: 10.1016/S0895-7061(99)00005-9
Brown, A. E., Dibnah, B., Fisher, E., Newton, J. L., and Walker, M. (2018). Pharmacological activation of AMPK and glucose uptake in cultured human skeletal muscle cells from patients with ME/CFS. Biosci. Rep. 38:BSR20180242. doi: 10.1042/BSR20180242
Cabello-Verrugio, C., Córdova, G., and Salas, J. D. (2012). Angiotensin II: role in skeletal muscle atrophy. Curr. Protein. Pept. Sci. 13, 560–569. doi: 10.2174/138920312803582933
Campbell, A. W. (2014). Autoimmunity and the gut. Autoimmune Dis. 2014:152428. doi: 10.1155/2014/152428
Cantuti-Castelvetri, L., Ojha, R., Pedro, L. D., Djannatian, M., Franz, J., Kuivanen, S., et al. (2020). Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science 370, 856–860. doi: 10.1126/science.abd2985
Cardinale, V., Capurso, G., Ianiro, G., Gasbarrini, A., Arcidiacono, P. G., and Alvaro, D. (2020). Intestinal permeability changes with bacterial translocation as key events modulating systemic host immune response to SARS-CoV-2: a working hypothesis. Dig Liver Dis. 52, 1383–1389. doi: 10.1016/j.dld.2020.09.009
Casciola-Rosen, L., Thiemann, D. R., Andrade, F., Zambrano, T. M. E., Hooper, J. E., Leonard, E., et al. (2020). IgM autoantibodies recognizing ACE2 are associated with severe COVID-19. medRxiv. doi: 10.1101/2020.10.13.20211664
Castro-Marrero, J., Sáez-Francàs, N., Segundo, M. J., Calvo, N., Faro, M., Aliste, L., et al. (2016). Effect of coenzyme Q10 plus nicotinamide adenine dinucleotide supplementation on maximum heart rate after exercise testing in chronic fatigue syndrome - a randomized, controlled, double-blind trial. Clin. Nutr. 35, 826–834. doi: 10.1016/j.clnu.2015.07.010
Chaudhary, M. (2020). COVID-19 susceptibility: potential of ACE2 polymorphisms. Egypt J. Med. Hum. Genet. 21:54. doi: 10.1186/s43042-020-00099-9
Chen, G., Wu, D., Guo, W., Cao, Y., Huang, D., Wang, H., et al. (2020). Clinical and immunological features of severe and moderate coronavirus disease 2019. J. Clin. Invest. 130, 2620–2629. doi: 10.1172/JCI137244
Chen, R., Huang, Y., Quan, J., Liu, J., Wang, H., Billiar, T. R., et al. (2020). HMGB1 as a potential biomarker and therapeutic target for severe COVID-19. Heliyon 6:e05672. doi: 10.1016/j.heliyon.2020.e05672
Chen, T., Kim, C. Y., Kaur, A., Lamothe, L., Shaikh, M., Keshavarzian, A., et al. (2017). Dietary fibre-based SCFA mixtures promote both protection and repair of intestinal epithelial barrier function in a Caco-2 cell model. Food Funct. 8, 1166–1173. doi: 10.1039/C6FO01532H
Chen, Z., Yu, J., Fu, M., Dong, R., Yang, Y., Luo, J., et al. (2020). Dipeptidyl peptidase-4 inhibition improves endothelial senescence by activating AMPK/SIRT1/Nrf2 signaling pathway. Biochem. Pharmacol. 177:113951. doi: 10.1016/j.bcp.2020.113951
Cheng, Q., Shah, N., Bröer, A., Fairweather, S., Jiang, Y., Schmollet, D., et al. (2017). Identification of novel inhibitors of the amino acid transporter B0 AT1 (SLC6A19), a potential target to induce protein restriction and to treat type 2 diabetes. Br. J. Pharmacol. 174, 468–482. doi: 10.1111/bph.13711
Chuckran, C. A., Liu, C., Bruno, T. C., Workman, C. J., and Vignali, D. A. (2020). Neuropilin-1: a checkpoint target with unique implications for cancer immunology and immunotherapy. J. Immunother. Cancer 8:e000967. doi: 10.1136/jitc-2020-000967
Co, M. S., Gaulton, G. N., Tominaga, A., Homcy, C. J., Fields, B. N., and Greene, M. I. (1985). Structural similarities between the mammalian beta-adrenergic and reovirus type 3 receptors. Proc. Natl. Acad. Sci. U.S.A. 82, 5315–5318. doi: 10.1073/pnas.82.16.5315
Coelho, V. A., Probst, V. S., Nogari, B. M., Teixeira, D. C., Felcar, J. M., Santos, D. C., et al. (2016). Angiotensin-II blockage, muscle strength, and exercise capacity in physically independent older adults. J. Phys. Ther. Sci. 28, 547–552. doi: 10.1589/jpts.28.547
Comhaire, F., and Deslypere, J. P. (2020). News and views in myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS): the role of co-morbidity and novel treatments. Med. Hypotheses 134:109444. doi: 10.1016/j.mehy.2019.109444
Cooper, H. A., Scalia, R., Rizzo, V., and Eguchi, S. (2018). Angiotensin II- and alzheimer-type cardiovascular aging. Circ. Res. 123, 651–653. doi: 10.1161/CIRCRESAHA.118.313477
Copsel, S. N., Lightbourn, C. O., Barreras, H., Lohse, I., Wolf, D., Bader, C. S., et al. (2019). BET bromodomain inhibitors which permit treg function enable a combinatorial strategy to suppress GVHD in pre-clinical allogeneic HSCT. Front. Immunol. 9:3104. doi: 10.3389/fimmu.2018.03104
Cortes Rivera, M., Mastronardi, C., Silva-Aldana, C. T., Arcos-Burgos, M., and Lidbury, B. A. (2019). Myalgic encephalomyelitis/chronic fatigue syndrome: a comprehensive review. Diagnostics 9:91. doi: 10.3390/diagnostics9030091
Crowley, S. D., and Rudemiller, N. P. (2017). Immunologic effects of the renin-angiotensin system. J. Am. Soc. Nephrol. 28, 1350–1361. doi: 10.1681/ASN.2016101066
Dai, X., Okon, I., Liu, Z., Wu, Y., Zhu, H., Song, P., et al. (2017). A novel role for myeloid cell-specific neuropilin 1 in mitigating sepsis. FASEB J. 31, 2881–2892. doi: 10.1096/fj.201601238R
Davies, J., Randeva, H. S., Chatha, K., Hall, M., Spandidos, D. A., Karteris, E., et al. (2020). Neuropilin-1 as a new potential SARS-CoV-2 infection mediator implicated in the neurologic features and central nervous system involvement of COVID-19. Mol. Med. Rep. 22, 4221–4226. doi: 10.3892/mmr.2020.11510
de Couto, G., Jaghatspanyan, E., DeBerge, M., Liu, W., Luther, K., Wang, Y., et al. (2019). Mechanism of enhanced mertk-dependent macrophage efferocytosis by extracellular vesicles. Arterioscler. Thromb. Vasc. Biol. 39, 2082–2096. doi: 10.1161/ATVBAHA.119.313115
De Pasquale, L., D'Amico, A., Verardo, M., Petrini, S., Bertini, E., and De Benedetti, F. (2012). Increased muscle expression of interleukin-17 in duchenne muscular dystrophy. Neurology 78, 1309–1314. doi: 10.1212/WNL.0b013e3182518302
Demaria, M., O'Leary, M. N., Chang, J., Shao, L., Liu, S., Alimirah, F., et al. (2017). Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov. 7, 165–176. doi: 10.1158/2159-8290.CD-16-0241
Djaldetti, M., Leibovitch, C., Ganelin _Cohen, E., and Bessler, H. (2019). Rituximab Modifies peripheral blood mononuclear cells immune responses. Int. J. Immunol. Immunother. 6:37. doi: 10.23937/2378-3672/1410037
Dou, H., Feher, A., Davila, A. C., Romero, M. J., Patel, V. S., Kamathet, V. M., et al. (2017). Role of adipose tissue endothelial ADAM17 in age-related coronary microvascular dysfunction. Arterioscler. Thromb. Vasc. Biol. 37, 1180–1193. doi: 10.1161/ATVBAHA.117.309430
Dransfield, I., Zagórska, A., Lew, E. D., Michail, K., and Lemke, G. (2015). Mer receptor tyrosine kinase mediates both tethering and phagocytosis of apoptotic cells. Cell Death Dis. 6:e1646. doi: 10.1038/cddis.2015.18
Dreymueller, D., Martin, C., Kogel, T., Pruessmeyer, J., Hess, F. M., Horiuchi, K., et al. (2012). Lung endothelial ADAM17 regulates the acute inflammatory response to lipopolysaccharide. EMBO Mol. Med. 4, 412–423. doi: 10.1002/emmm.201200217
Du Preez, S., Corbitt, M., Cabanas, H., Eaton, N., Staines, D., and Marshall-Gradisnik, S. (2018). A systematic review of enteric dysbiosis in chronic fatigue syndrome/myalgic encephalomyelitis. Syst. Rev. 7:241. doi: 10.1186/s13643-018-0909-0
Eaton-Fitch, N., du Preez, S., Cabanas, H., Staines, D., and Marshall-Gradisnik, S. (2019). A systematic review of natural killer cells profile and cytotoxic function in myalgic encephalomyelitis/chronic fatigue syndrome. Syst. Rev. 8:279. doi: 10.1186/s13643-019-1202-6
Edelblum, K. L., Sharon, G., Singh, G., Odenwald, M. A., Sailer, A., Cao, S., et al. (2017). The microbiome activates CD4 T-cell-mediated immunity to compensate for increased intestinal permeability. Cell. Mol. Gastroenterol. Hepatol. 4, 285–297. doi: 10.1016/j.jcmgh.2017.06.001
Effenberger, T., von der Heyde, J., Bartsch, K., Garbers, C., Schulze-Osthoff, K., Chalaris, A., et al. (2014). Senescence-associated release of transmembrane proteins involves proteolytic processing by ADAM17 and microvesicle shedding. FASEB J. 28, 4847–4856. doi: 10.1096/fj.14-254565
Elder, S. S., and Emmerson, E. (2020). Senescent cells and macrophages: key players for regeneration? Open Biol. 10:200309. doi: 10.1098/rsob.200309
Evrensel, A., and Ceylan, M. E. (2016). Fecal microbiota transplantation and its usage in neuropsychiatric disorders. Clin. Psychopharmaco. Neurosci. 14, 231–237. doi: 10.9758/cpn.2016.14.3.231
Fadini, G. P., Morieri, M. L., Longato, E., Bonora, B. M., Pinelli, S., Selmin, E., et al. (2020). Exposure to dipeptidyl-peptidase-4 inhibitors and COVID-19 among people with type 2 diabetes: a case-control study. Diabetes Obes. Metab. 22, 1946–1950. doi: 10.1111/dom.14097
Fatima, N., Upadhyay, T., Sharma, D., and Sharma, R. (2017). Particulate beta-glucan induces early and late phagosomal maturation in murine macrophages. Front. Biosci. 9, 129–140. doi: 10.2741/e791
Feng, X., Wang, L., and Li, Y. (2011). Change of telomere length in angiotensin II-induced human glomerular mesangial cell senescence and the protective role of losartan. Mol. Med. Rep. 4, 255–260. doi: 10.3892/mmr.2011.436
Fenwick, C., Joo, V., Jacquier, P., Noto, A., Banga, R., Perreau, M., et al. (2019). T-cell exhaustion in HIV infection. Immunol. Rev. 292, 149–163. doi: 10.1111/imr.12823
Ferrandi, P. J., Alway, S. E., and Mohamed, J. S. (2020). The interaction between SARS-CoV-2 and ACE2 may have consequences for skeletal muscle viral susceptibility and myopathies. J Appl. Physiol. 129, 864–867. doi: 10.1152/japplphysiol.00321.2020
Ferrario, C. M., Jessup, J., and Chappell, M. C. (2005). Effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin-converting enzyme 2. Circulation 111, 2605–2610. doi: 10.1161/CIRCULATIONAHA.104.510461
Findeisen, H. M., Gizard, F., Zhao, Y., Cohn, D., Heywood, E. B., Jones, K. L., et al. (2011). Telomerase deficiency in bone marrow-derived cells attenuates angiotensin II-induced abdominal aortic aneurysm formation. Arterioscler. Thromb. Vasc. Biol. 31, 253–260. doi: 10.1161/ATVBAHA.110.218545
Fine, R. L., Manfredo Vieira, S., Gilmore, M. S., and Kriegel, M. A. (2020). Mechanisms and consequences of gut commensal translocation in chronic diseases. Gut Microbes. 11, 217–230. doi: 10.1080/19490976.2019.1629236
Freissmuth, M., Selzer, E., Marullo, S., Schütz, W., and Strosberg, A. D. (1991). Expression of two human beta-adrenergic receptors in Escherichia coli: functional interaction with two forms of the stimulatory G protein. Proc. Natl. Acad. Sci. U.S.A. 88, 8548–8552. doi: 10.1073/pnas.88.19.8548
Friedman, K. J. (2019). Advances in ME/CFS: past, present, and future. Front Pediatr. 7:131. doi: 10.3389/fped.2019.00131
Friedrich, O., Reid, M. B., Van den Berghe, G., Vanhorebeek, I., Hermans, G., Rich, M. M., et al. (2015). The sick and the weak: neuropathies/myopathies in the critically ill. Physiol. Rev. 95, 1025–1109. doi: 10.1152/physrev.00028.2014
Friggeri, A., Yang, Y., Banerjee, S., Park, Y. J., Liu, G., and Abraham, E. (2010). HMGB1 inhibits macrophage activity in efferocytosis through binding to the alphavbeta3-integrin. Am. J. Physiol. Cell Physiol. 299, C1267–C1276. doi: 10.1152/ajpcell.00152.2010
Fukushima, Y., Minato, N., and Hattori, M. (2018). The impact of senescence-associated T cells on immunosenescence and age-related disorders. Inflamm. Regen. 38:24. doi: 10.1186/s41232-018-0082-9
Furukawa, H., and Haga, T. (2000). Expression of functional M2 muscarinic acetylcholine receptor in Escherichia coli. J. Biochem. 127, 151–161. doi: 10.1093/oxfordjournals.jbchem.a022577
Fyhrquist, F., Eriksson, A., Saijonmaa, O., Nordestgaard, B. G., Kontula, K., de Faire, U., et al. (2013). Telomere length is associated with ACE I/D polymorphism in hypertensive patients with left ventricular hypertrophy. J. Renin Angiotensin Aldosterone Syst. 14, 227–234. doi: 10.1177/1470320312460292
Gaab, J., Rohleder, N., Heitz, V., Engert, V., Schad, T., Schürmeyer, T. H., et al. (2005). Stress-induced changes in LPS-induced pro-inflammatory cytokine production in chronic fatigue syndrome. Psychoneuroendocrinology 30, 188–198. doi: 10.1016/j.psyneuen.2004.06.008
Galván-Peña, S., Leon, J., Chowdhary, K., Michelson, D. A., Vijaykumar, B., Yang, L., et al. (2020). Profound treg perturbations correlate with COVID-19 severity. bioRxiv. doi: 10.1101/2020.12.11.416180
Gao, L., Mao, Q., Cao, J., Wang, Y., Zhou, X., and Fan, L. (2012). Effects of coenzyme Q10 on vascular endothelial function in humans: a meta-analysis of randomized controlled trials. Atherosclerosis 221, 311–316. doi: 10.1016/j.atherosclerosis.2011.10.027
Garcia, M. A., Nelson, W. J., and Chavez, N. (2018). Cell-Cell junctions organize structural and signaling networks. Cold Spring Harb. Perspect. Biol. 10:a029181. doi: 10.1101/cshperspect.a029181
Garcia-Hernandez, V., Quiros, M., and Nusrat, A. (2017). Intestinal epithelial claudins: expression and regulation in homeostasis and inflammation. Ann. N. Y. Acad. Sci. 1397, 66–79. doi: 10.1111/nyas.13360
Georgiades, E., Behan, W. M., Kilduff, L. P., Hadjicharalambous, M., Mackie, E. E., Wilson, J., et al. (2003). Chronic fatigue syndrome: new evidence for a central fatigue disorder. Clin. Sci. 105, 213–218. doi: 10.1042/CS20020354
Gerhard-Herman, M., Smoot, L. B., Wake, N., Kieran, M. W., Kleinman, M. E., Miller, D. T., et al. (2012). Mechanisms of premature vascular aging in children with hutchinson-gilford progeria syndrome. Hypertension 59, 92–97. doi: 10.1161/HYPERTENSIONAHA.111.180919
Giloteaux, L., Goodrich, J. K., Walters, W. A., Levine, S. M., Ley, R. E., and Hanson, M. R. (2016). Reduced diversity and altered composition of the gut microbiome in individuals with myalgic encephalomyelitis/chronic fatigue syndrome. Microbiome 4:30. doi: 10.1186/s40168-016-0171-4
Goldblum, S. E., Brann, T. W., Ding, X., Pugin, J., and Tobias, P. S. (1994). Lipopolysaccharide (LPS)-binding protein and soluble CD14 function as accessory molecules for LPS-induced changes in endothelial barrier function, in vitro. J. Clin. Invest. 93, 692–702. doi: 10.1172/JCI117022
Gordon, D. E., Jang, G. M., Bouhaddou, M., Xu, J., Obernier, K., White, K. M., et al. (2020). A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 583, 459–468. doi: 10.1038/s41586-020-2286-9
Gravina, A. G., Dallio, M., Masarone, M., Rosato, V., Aglitti, A., Persicoet, M., et al. (2018). Vascular endothelial dysfunction in inflammatory bowel diseases: pharmacological and nonpharmacological targets. Oxid. Med. Cell Longev. 2018:2568569. doi: 10.1155/2018/2568569
Greenberg, D. B. (2002). Clinical dimensions of fatigue. Prim. Care Companion J. Clin. Psychiatry 4, 90–93. doi: 10.4088/PCC.v04n0301
Guidon, A. C., and Amato, A. A. (2020). COVID-19 and neuromuscular disorders. Neurology 94, 959–969. doi: 10.1212/WNL.0000000000009566
Harris, H. E., Andersson, U., and Pisetsky, D. S. (2012). HMGB1: a multifunctional alarmin driving autoimmune and inflammatory disease. Nat. Rev. Rheumatol. 8, 195–202. doi: 10.1038/nrrheum.2011.222
Hartwig, J., Sotzny, F., Bauer, S., Heidecke, H., Riemekasten, G., Dragun, D., et al. (2020). IgG stimulated β2 adrenergic receptor activation is attenuated in patients with ME/CFS. Brain Behav. Immun. 3:100047. doi: 10.1016/j.bbih.2020.100047
Hasan, M. R., Ho, S. H., Owen, D. A., and Tai, I. T. (2011). Inhibition of VEGF induces cellular senescence in colorectal cancer cells. Int. J. Cancer 129, 2115–2123. doi: 10.1002/ijc.26179
Herbert, K. E., Mistry, Y., Hastings, R., Poolman, T., Niklason, L., and Williams, B. (2008). Angiotensin II-mediated oxidative DNA damage accelerates cellular senescence in cultured human vascular smooth muscle cells via telomere-dependent and independent pathways. Circ. Res. 102, 201–208. doi: 10.1161/CIRCRESAHA.107.158626
Ho, R. T., Chan, J. S., Wang, C. W., So, K. F., Yuen, L. P., Sham, J. S., et al. (2012). A randomized controlled trial of qigong exercise on fatigue symptoms, functioning, and telomerase activity in persons with chronic fatigue or chronic fatigue syndrome. Ann. Behav. Med. 44, 160–170. doi: 10.1007/s12160-012-9381-6
Hoban, D. B., Connaughton, E., Connaughton, C., Hogan, G., Thornton, C., Mulcahy, P., et al. (2013). Further characterisation of the LPS model of Parkinson's disease: a comparison of intra-nigral and intra-striatal lipopolysaccharide administration on motor function, microgliosis and nigrostriatal neurodegeneration in the rat. Brain Behav. Immun. 27, 91–100. doi: 10.1016/j.bbi.2012.10.001
Huang, C., Wang, Y., Li, X., Ren, L., Zhao, J., Hu, Y., et al. (2020). Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395, 497–506. doi: 10.1016/S0140-6736(20)30183-5
Huth, T. K., Brenu, E. W., Ramos, S., Nguyen, T., Broadley, S., Staines, D., et al. (2016). Pilot study of natural killer cells in chronic fatigue syndrome/myalgic encephalomyelitis and multiple sclerosis. Scand. J. Immunol. 83, 44–51. doi: 10.1111/sji.12388
Imanishi, T., Hano, T., and Nishio, I. (2005). Angiotensin II accelerates endothelial progenitor cell senescence through induction of oxidative stress. J. Hypertens. 23, 97–104. doi: 10.1097/00004872-200501000-00018
Inui, T., Kubo, K., Kuchiike, D., Uto, Y., Nishikata, T., Sakamoto, N., et al. (2015). Oral colostrum macrophage-activating factor for serious infection and chronic fatigue syndrome: three case reports. Anticancer Res. 35, 4545–4549.
Issitt, T., Bosseboeuf, E., De Winter, N., Dufton, N., Gestri, G., Senatore, V., et al. (2019). Neuropilin-1 controls endothelial homeostasis by regulating mitochondrial function and iron-dependent oxidative stress. iScience 11, 205–223. doi: 10.1016/j.isci.2018.12.005
Jando, J., Camargo, S. M. R., Herzog, B., and Verrey, F. (2017). Expression and regulation of the neutral amino acid transporter B0AT1 in rat small intestine. PLoS ONE 12:e0184845. doi: 10.1371/journal.pone.0184845
JanssenDuijghuijsen, L. M., Grefte, S., de Boer, V. C. J., Zeper, L., van Dartel, D. A. M., van der Stelt, I., et al. (2017). Mitochondrial ATP depletion disrupts caco-2 monolayer integrity and internalizes claudin 7. Front. Physiol. 8:794. doi: 10.3389/fphys.2017.00794
Jason, L. A., Richman, J. A., Rademaker, A. W., Jordan, K. M., Plioplys, A. V., Taylor, R. R., et al. (1999). A community-based study of chronic fatigue syndrome. Arch. Intern. Med. 159, 2129–2137. doi: 10.1001/archinte.159.18.2129
Jeyapalan, J. C., and Sedivy, J. M. (2008). Cellular senescence and organismal aging. Mech. Ageing Dev. 129, 467–474. doi: 10.1016/j.mad.2008.04.001
Jin, M., and Tong, Q. (2020). Rhabdomyolysis as potential late complication associated with COVID-19. Emerge. Infect. Dis. 26, 1618–1620. doi: 10.3201/eid2607.200445
Justice, J. N., Nambiar, A. M., Tchkonia, T., LeBrasseur, N. K., Pascual, R., Hashmi, S. K., et al. (2019). Senolytics in idiopathic pulmonary fibrosis: results from a first-in-human, open-label, pilot study. EBioMedicine 40, 554–563. doi: 10.1016/j.ebiom.2018.12.052
Kale, A., Sharma, A., Stolzing, A., Desprez, P. Y., and Campisi, J. (2020). Role of immune cells in the removal of deleterious senescent cells. Immun. Ageing 17:16. doi: 10.1186/s12979-020-00187-9
Karaji, N., and Sattentau, Q. J. (2017). Efferocytosis of pathogen-infected cells. Front. Immunol. 8:1863. doi: 10.3389/fimmu.2017.01863
Karavolos, M. H., Winzer, K., Williams, P., and Khan, C. M. (2013). Pathogen espionage: multiple bacterial adrenergic sensors eavesdrop on host communication systems. Mol. Microbiol. 87, 455–465. doi: 10.1111/mmi.12110
Kasper, J. Y., Hermanns, M. I., Cavelius, C., Kraegeloh, A., Jung, T., Danzebrink, R., et al. (2016). The role of the intestinal microvasculature in inflammatory bowel disease: studies with a modified Caco-2 model including endothelial cells resembling the intestinal barrier in vitro. Int. J. Nanomedicine 11, 6353–6364. doi: 10.2147/IJN.S92608
Kaur, A., Macip, S., and Stover, C. M. (2020). An appraisal on the value of using nutraceutical based senolytics and senostatics in aging. Front. Cell Dev. Biol. 8:218. doi: 10.3389/fcell.2020.00218
Kaur, H., and Moreau, R. (2019). Role of mTORC1 in intestinal epithelial repair and tumorigenesis. Cell Mol. Life Sci. 76, 2525–2546. doi: 10.1007/s00018-019-03085-6
Kawano, M., and Nagata, S. (2018). Efferocytosis and autoimmune disease. Int. Immunol. 30, 551–558. doi: 10.1093/intimm/dxy055
Kearns, M. T., Dalal, S., Horstmann, S. A., Richens, T. R., Tanaka, T., Doe, J. M., et al. (2012). Vascular endothelial growth factor enhances macrophage clearance of apoptotic cells. Am. J. Physiol. Lung Cell Mol. Physiol. 302, L711–L718. doi: 10.1152/ajplung.00116.2011
Kennedy, G., Spence, V., Khan, F., and Belch, J. J. (2004). Plasma endothelin-1 levels in chronic fatigue syndrome. Rheumatology 43, 252–3; author reply 253–4. doi: 10.1093/rheumatology/keg462
Kim, K. M., Noh, J. H., Bodogai, M., Martindale, J. L., Yang, X., Indig, F. E., et al. (2017). Identification of senescent cell surface targetable protein DPP4. Genes Dev. 31, 1529–1534. doi: 10.1101/gad.302570.117
Kirkland, J. L., Tchkonia, T., Zhu, Y., Niedernhofer, L. J., and Robbins, P. D. (2017). The clinical potential of senolytic drugs. J. Am. Geriatr. Soc. 65, 2297–2301. doi: 10.1111/jgs.14969
Kirsch, T., Woywodt, A., Beese, M., Wyss, K., Park, J. K., Erdbruegger, U., et al. (2007). Engulfment of apoptotic cells by microvascular endothelial cells induces proinflammatory responses. Blood 109, 2854–2862. doi: 10.1182/blood-2006-06-026187
Koga, H., Yang, H., Haxhija, E. Q., and Teitelbaum, D. H. (2008). The role of angiotensin II type 1a receptor on intestinal epithelial cells following small bowel resection in a mouse model. Pediatr. Surg. Int. 24, 1279–1286. doi: 10.1007/s00383-008-2277-7
Kruglikov, I. L., and Scherer, P. E. (2021). Preexisting and inducible endotoxemia as crucial contributors to the severity of COVID-19 outcomes. PLoS Pathog 17:e1009306. doi: 10.1371/journal.ppat.1009306
Kucuk, A., Cumhur Cure, M., and Cure, E. (2020). Can COVID-19 cause myalgia with a completely different mechanism? A hypothesis. Clin. Rheumatol. 39, 2103–2104. doi: 10.1007/s10067-020-05178-1
Kumar, A., Singh, B., Mishra, J., Sah, S. P., and Pottabathini, R. (2015). Neuroprotective mechanism of losartan and its interaction with nimesulide against chronic fatigue stress. Inflammopharmacology 23, 291–305. doi: 10.1007/s10787-015-0238-z
Kunieda, T., Minamino, T., Nishi, J., Tateno, K., Oyama, T., Katsuno, T., et al. (2006). Angiotensin II induces premature senescence of vascular smooth muscle cells and accelerates the development of atherosclerosis via a p21-dependent pathway. Circulation 114, 953–960. doi: 10.1161/CIRCULATIONAHA.106.626606
Landi, A., Broadhurst, D., Vernon, S. D., Tyrrell, D. L., and Houghton, M. (2016). Reductions in circulating levels of IL-16, IL-7 and VEGF-A in myalgic encephalomyelitis/chronic fatigue syndrome. Cytokine 78, 27–36. doi: 10.1016/j.cyto.2015.11.018
Langhans, C., Weber-Carstens, S., Schmidt, F., Hamati, J., Kny, M., Zhu, X., et al. (2014). Inflammation-induced acute phase response in skeletal muscle and critical illness myopathy. PLoS ONE 9:e92048. doi: 10.1371/journal.pone.0092048
Lasselin, J., Karshikoff, B., Axelsson, J., Åkerstedt, T., Benson, S, Engler, H., et al. (2020). Fatigue and sleepiness responses to experimental inflammation and exploratory analysis of the effect of baseline inflammation in healthy humans. Brain Behav. Immun. 83, 309–314. doi: 10.1016/j.bbi.2019.10.020
LeBrasseur, N. K., Tchkonia, T., and Kirkland, J. L. (2015). Cellular senescence and the biology of aging, disease, and frailty. Nestle Nutr. Inst. Workshop Ser. 83, 11–18. doi: 10.1159/000382054
Lee, J. H., Lee, Y. K., Lim, J. J., Byun, H.-O., Park, I., Kim, G.-H., et al. (2015). Mitochondrial respiratory dysfunction induces claudin-1 expression via reactive oxygen species-mediated heat shock factor 1 activation, leading to hepatoma cell invasiveness. J. Biol. Chem. 290, 21421–21431. doi: 10.1074/jbc.M115.654913
Lee, J. S. (2019). Cellular senescence, aging, and age-related disease: special issue of bmb reports in 2019. BMB Rep. 52, 1–2. doi: 10.5483/BMBRep.2019.52.1.002
Lee, J. S., Tato, C. M., Joyce-Shaikh, B., Gulen, M. F., Cayatte, C., Chen, Y., et al. (2015). Interleukin-23-independent IL-17 production regulates intestinal epithelial permeability. Immunity 43:1022. doi: 10.1016/j.immuni.2015.10.019
Lewis-McDougall, F. C., Ruchaya, P. J., Domenjo-Vila, E., Teoh, T. S., Prata, L., Cottle, B. J., et al. (2019). Aged-senescent cells contribute to impaired heart regeneration. Aging Cell 18:e12931. doi: 10.1111/acel.12931
Lewkowicz, P., Lewkowicz, N., Sasiak, A., and Tchórzewski, H. (2006). Lipopolysaccharide-activated CD4+CD25+ T regulatory cells inhibit neutrophil function and promote their apoptosis and death. J. Immunol. 177, 7155–7163. doi: 10.4049/jimmunol.177.10.7155
Li, M., Guo, W., Dong, Y., Wang, X., Dai, D., Liu, X., et al. (2020). Elevated exhaustion levels of NK and CD8+ T cells as indicators for progression and prognosis of COVID-19 disease. Front. Immunol. 11:580237. doi: 10.3389/fimmu.2020.580237
Li, R., Mi, X., Yang, S., Yang, Y., Zhang, S., Hui, R., et al. (2019). Long-term stimulation of angiotensin II induced endothelial senescence and dysfunction. Exp. Gerontol. 119, 212–220. doi: 10.1016/j.exger.2019.02.012
Libby, P., and Lüscher, T. (2020). COVID-19 is, in the end, an endothelial disease. Eur. Heart J. 41, 3038–3044. doi: 10.1093/eurheartj/ehaa623
Lieberman, J., and Bell, D. S. (1993). Serum angiotensin-converting enzyme as a marker for the chronic fatigue-immune dysfunction syndrome: a comparison to serum angiotensin-converting enzyme in sarcoidosis. Am. J. Med. 95, 407–412. doi: 10.1016/0002-9343(93)90310-L
Lin, L., Jiang, X., Zhang, Z., Huang, S., Zhang, Z., Fang, Z., et al. (2020). Gastrointestinal symptoms of 95 cases with SARS-CoV-2 infection. Gut 69, 997–1001. doi: 10.1136/gutjnl-2020-321013
Lind, S. E. (2021). Phosphatidylserine is an overlooked mediator of COVID-19 thromboinflammation. Heliyon 7:e06033. doi: 10.1016/j.heliyon.2021.e06033
Loebel, M., Grabowski, P., Heidecke, H., Bauer, S., Hanitsch, L. G., Wittke, K., et al. (2016). Antibodies to β adrenergic and muscarinic cholinergic receptors in patients with chronic fatigue syndrome. Brain Behav. Immun. 52, 32–39. doi: 10.1016/j.bbi.2015.09.013
Lopez-Vilchez, I., Diaz-Ricart, M., Navarro, V., Torramade, S., Zamorano-Leon, J., Lopez-Farre, A., et al. (2016). Endothelial damage in major depression patients is modulated by SSRI treatment, as demonstrated by circulating biomarkers and an in vitro cell model. Transl. Psychiatry 6:e886. doi: 10.1038/tp.2016.156
Lu, C. C., Ma, K. L., Ruan, X. Z., and Liu, B. C. (2018). Intestinal dysbiosis activates renal renin-angiotensin system contributing to incipient diabetic nephropathy. Int. J. Med. Sci. 15, 816–822. doi: 10.7150/ijms.25543
Luo, G., Ambati, A., Lin, L., Bonvalet, M., Partinen, M., Ji, X., et al. (2018). Autoimmunity to hypocretin and molecular mimicry to flu in type 1 narcolepsy. Proc. Natl. Acad. Sci. U.S.A. 115, E12323–E12332. doi: 10.1073/pnas.1818150116
Månsson, K. N. T., Lindqvist, D., Yang, L. L., Svanborg, C., Isung, J., Nilsonne, G., et al. (2019). Improvement in indices of cellular protection after psychological treatment for social anxiety disorder. Transl. Psychiatry 9:340. doi: 10.1038/s41398-019-0668-2
Maes, M., and Leunis, J. C. (2008). Normalization of leaky gut in chronic fatigue syndrome (CFS) is accompanied by a clinical improvement: effects of age, duration of illness and the translocation of LPS from gram-negative bacteria. Neuro. Endocrinol. Lett. 29, 902–910.
Maes, M., Leunis, J. C., Geffard, M., and Berk, M. (2014). Evidence for the existence of myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) with and without abdominal discomfort (irritable bowel) syndrome. Neuro. Endocrinol. Lett. 35, 445–453.
Magna, M., and Pisetsky, D. S. (2014). The role of HMGB1 in the pathogenesis of inflammatory and autoimmune diseases. Mol. Med. 20, 138–146. doi: 10.2119/molmed.2013.00164
Maher, K. J., Klimas, N. G., and Fletcher, M. A. (2005). Chronic fatigue syndrome is associated with diminished intracellular perforin. Clin. Exp. Immunol. 142, 505–511. doi: 10.1111/j.1365-2249.2005.02935.x
Mandarano, A. H., Giloteaux, L., Keller, B. A., Levine, S. M., and Hanson, M. R. (2018). Eukaryotes in the gut microbiota in myalgic encephalomyelitis/chronic fatigue syndrome. PeerJ 6:e4282. doi: 10.7717/peerj.4282
Manestar-Blazić, T, and Volf, M. (2009). The dynamic of senescent cells accumulation can explain the age-specific incidence of autoimmune diseases. Med. Hypotheses 73, 667–669. doi: 10.1016/j.mehy.2009.05.010
Mangalmurti, N., and Hunter, C. A. (2020). Cytokine storms: understanding COVID-19. Immunity 53, 19–25. doi: 10.1016/j.immuni.2020.06.017
Mao, L., Jin, H., Wang, M., Hu, Y., Chen, S., He, Q., et al. (2020). Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in Wuhan, China. JAMA Neurol. 77, 683–690. doi: 10.1001/jamaneurol.2020.1127
Martínez-Cué, C., and Rueda, N. (2020). Cellular senescence in neurodegenerative diseases. Front. Cell Neurosci. 14:16. doi: 10.3389/fncel.2020.00016
Mateuszuk, Ł., Campagna, R., Kutryb-Zajac, B., Kuś, K., Słominska, E. M., Smolenski, R. T., et al. (2020). Reversal of endothelial dysfunction by nicotinamide mononucleotide via extracellular conversion to nicotinamide riboside. Biochem. Pharmacol. doi: 10.1016/j.bcp.2020.114019
Mathison, J. C., Tobias, P. S., Wolfson, E., and Ulevitch, R. J. (1992). Plasma lipopolysaccharide (LPS)-binding protein. A key component in macrophage recognition of gram-negative LPS. J. Immunol. 149, 200–206.
Matsumoto, A., Pasut, A., Matsumoto, M., Yamashita, R., Fung, J., Monteleone, E., et al. (2017). mTORC1 and muscle regeneration are regulated by the LINC00961-encoded SPAR polypeptide. Nature 541, 228–232. doi: 10.1038/nature21034
Mayi, B. S., Leibowitz, J. A., Woods, A. T., Ammon, K. A., Liu, A. E., and Raja, A. (2021). The role of Neuropilin-1 in COVID-19. PLoS Pathog 17:e1009153. doi: 10.1371/journal.ppat.1009153
Megremis, S., Walker, T. D. J., He, X., Ollier, W. E. R., Chinoy, H., Hampson, L., et al. (2020). Antibodies against immunogenic epitopes with high sequence identity to SARS-CoV-2 in patients with autoimmune dermatomyositis. Ann. Rheum. Dis. 79, 1383–1386. doi: 10.1136/annrheumdis-2020-217522
Mercer, J., and Helenius, A. (2010). Apoptotic mimicry: phosphatidylserine-mediated macropinocytosis of vaccinia virus. Ann. N. Y. Acad. Sci. 1209:49–55. doi: 10.1111/j.1749-6632.2010.05772.x
Miesbach, W. (2020). Pathological role of angiotensin II in severe COVID-19. TH Open 4, e138–e144. doi: 10.1055/s-0040-1713678
Minamino, T., and Komuro, I. (2002). Role of telomere in endothelial dysfunction in atherosclerosis. Curr. Opin. Lipidol. 13, 537–543. doi: 10.1097/00041433-200210000-00010
Mizrahi, M., and Ilan, Y. (2009). The gut mucosa as a site for induction of regulatory T-cells. Curr. Pharm. Des. 15, 1191–1202. doi: 10.2174/138161209787846784
Mohabbat, A. B., Mohabbat, N. M. L., and Wight, E. C. (2020). Fibromyalgia and chronic fatigue syndrome in the age of COVID-19. Mayo Clin. Proc. Innov. Qual. Outcomes 4, 764–766. doi: 10.1016/j.mayocpiqo.2020.08.002
Mohamad Anuar, N. N., Nor Hisam, N. S., Liew, S. L., and Ugusman, A. (2020). Clinical review: navitoclax as a pro-apoptotic and anti-fibrotic agent. Front. Pharmacol. 11:564108. doi: 10.3389/fphar.2020.564108
Moldofsky, H., and Patcai, J. (2011). Chronic widespread musculoskeletal pain, fatigue, depression and disordered sleep in chronic post-SARS syndrome; a case-controlled study. BMC Neurol. 11:37. doi: 10.1186/1471-2377-11-37
Monneret, G., and Venet, F. (2012). A rapidly progressing lymphocyte exhaustion after severe sepsis. Crit. Care. 16:140. doi: 10.1186/cc11416
Monteil, V., Kwon, H., Prado, P., Hagelkrüys, A., Wimmer, R. A., Stahl, M., et al. (2020). Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell 181:905–913.e7. doi: 10.1016/j.cell.2020.04.004
Morancho, B., Martínez-Barriocanal, Á., Villanueva, J., and Arribas, J. (2015). Role of ADAM17 in the non-cell autonomous effects of oncogene-induced senescence. Breast Cancer Res. 17:106. doi: 10.1186/s13058-015-0619-7
Moreira, C. G., Russell, R., Mishra, A. A., Narayanan, S., Ritchie, J. M., Waldor, M. K., et al. (2016). Bacterial adrenergic sensors regulate virulence of enteric pathogens in the gut. mBio 7, e00826–e00816. doi: 10.1128/mBio.00826-16
Moresi, V., Adamo, S., and Berghella, L. (2019). The JAK/STAT pathway in skeletal muscle pathophysiology. Front. Physiol. 10:500. doi: 10.3389/fphys.2019.00500
Morizono, K., and Chen, I. S. (2014). Role of phosphatidylserine receptors in enveloped virus infection. J. Virol. 88, 4275–4290. doi: 10.1128/JVI.03287-13
Morris, G., Berk, M., Galecki, P., and Maes, M. (2014). The emerging role of autoimmunity in myalgic encephalomyelitis/chronic fatigue syndrome (ME/cfs). Mol. Neurobiol. 49, 741–756. doi: 10.1007/s12035-013-8553-0
Morris, G., and Maes, M. (2013). Myalgic encephalomyelitis/chronic fatigue syndrome and encephalomyelitis disseminata/multiple sclerosis show remarkable levels of similarity in phenomenology and neuroimmune characteristics. BMC Med. 11:205. doi: 10.1186/1741-7015-11-205
Morris, G., Maes, M., Berk, M., and Puri, B. K. (2019). Myalgic encephalomyelitis or chronic fatigue syndrome: how could the illness develop? Metab. Brain Dis. 34, 385–415. doi: 10.1007/s11011-019-0388-6
Motta-Santos, D., Dos Santos, R. A., Oliveira, M., Qadri, F., Poglitsch, M., Mosienko, V., et al. (2016). Effects of ACE2 deficiency on physical performance and physiological adaptations of cardiac and skeletal muscle to exercise. Hypertens. Res. 39, 506–512. doi: 10.1038/hr.2016.28
Moutal, A., Martin, L. F., Boinon, L., Gomez, K., Ran, D., Zhou, Y., et al. (2021). SARS-CoV-2 spike protein co-opts VEGF-A/neuropilin-1 receptor signaling to induce analgesia. Pain 162, 243–252. doi: 10.1097/j.pain.0000000000002097
Mu, J., Fang, Y., Yang, Q., Shu, T., Wang, A., Huang, M., et al. (2020). SARS-CoV-2 N protein antagonizes type I interferon signaling by suppressing phosphorylation and nuclear translocation of STAT1 and STAT2. Cell Discov. 6:65. doi: 10.1038/s41421-020-00208-3
Mukhopadhya, I., Segal, J. P., Carding, S. R., Hart, A. L., and Hold, G. L. (2019). The gut virome: the 'missing link' between gut bacteria and host immunity? Therap. Adv. Gastroenterol. 12:1756284819836620. doi: 10.1177/1756284819836620
Nade, V. S., Kawale, L. A., Valte, K. D., and Shendye, N. V. (2015). Cognitive enhancing effect of angiotensin-converting enzyme inhibitors and angiotensin receptor blockers on learning and memory. Indian. J. Pharmacol. 47, 263–269. doi: 10.4103/0253-7613.157114
Navaneetharaja, N., Griffiths, V., Wileman, T., and Carding, S. R. (2016). A role for the intestinal microbiota and virome in myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS)? J. Clin. Med. 5:55. doi: 10.3390/jcm5060055
Neel, B. A., Lin, Y., and Pessin, J. E. (2013). Skeletal muscle autophagy: a new metabolic regulator. Trends Endocrinol Metab. 24, 635–643. doi: 10.1016/j.tem.2013.09.004
Nelke, C., Dziewas, R., Minnerup, J., Meuth, S. G., and Ruck, T. (2019). Skeletal muscle as potential central link between sarcopenia and immune senescence. EBioMedicine 49, 381–388. doi: 10.1016/j.ebiom.2019.10.034
Nemet, I., Saha, P. P., Gupta, N., Zhu, W., Romano, K. A., Skye, S. M., et al. (2020). A cardiovascular disease-linked gut microbial metabolite acts via adrenergic receptors. Cell 180, 862–877.e22. doi: 10.1016/j.cell.2020.02.016
Newberry, F., Hsieh, S. Y., Wileman, T., and Carding, S. R. (2018). Does the microbiome and virome contribute to myalgic encephalomyelitis/chronic fatigue syndrome? Clin. Sci. 132, 523–542. doi: 10.1042/CS20171330
Newton, D. J., Kennedy, G., Chan, K. K., Lang, C. C., Belch, J. J., and Khan, F. (2012). Large and small artery endothelial dysfunction in chronic fatigue syndrome. Int. J. Cardiol. 154, 335–336. doi: 10.1016/j.ijcard.2011.10.030
Nguyen, T., Johnston, S., Chacko, A., Gibson, D., Cepon, J., Smith, P., et al. (2017). Novel characterisation of mast cell phenotypes from peripheral blood mononuclear cells in chronic fatigue syndrome/myalgic encephalomyelitis patients. Asian Pac. J. Allergy Immunol. 35, 75–81. doi: 10.12932/ap0771
Novakovic, B., Habibi, E., Wang, S. Y., Arts, R. J. W., Davar, R., Megchelenbrink, W., et al. (2016). β-Glucan reverses the epigenetic state of LPS-induced immunological tolerance. Cell 167, 1354–1368.e14. doi: 10.1016/j.cell.2016.09.034
Nozu, T., Miyagishi, S., Nozu, R., Takakusaki, K., and Okumura, T. (2020). Losartan improves visceral sensation and gut barrier in a rat model of irritable bowel syndrome. Neurogastroenterol. Motil. 32:e13819. doi: 10.1111/nmo.13819
Obrenovich, M. E. M. (2018). Leaky gut, leaky brain? Microorganisms 6:107. doi: 10.3390/microorganisms6040107
Ohm, J. E., Gabrilovich, D. I., Sempowski, G. D., Kisseleva, E., Parman, K. S., Nadaf, S., et al. (2003). VEGF inhibits T-cell development and may contribute to tumor-induced immune suppression. Blood 101, 4878–4886. doi: 10.1182/blood-2002-07-1956
Ortega-Hernandez, O. D., and Shoenfeld, Y. (2009). Infection, vaccination, and autoantibodies in chronic fatigue syndrome, cause or coincidence? Ann. N. Y. Acad. Sci. 1173, 600–609. doi: 10.1111/j.1749-6632.2009.04799.x
Osorio, C., Kanukuntla, T., Diaz, E., Jafri, N., Cummings, M., and Sfera, A. (2019). The post-amyloid era in alzheimer's disease: trust your gut feeling. Front. Aging Neurosci. 11:143. doi: 10.3389/fnagi.2019.00143
Pagliaro, P., and Penna, C. (2020). ACE/ACE2 ratio: a key also in 2019 coronavirus disease (Covid-19)? Front. Med. 7:335. doi: 10.3389/fmed.2020.00335
Palau, V., Riera, M., and Soler, M. J. (2020). ADAM17 inhibition may exert a protective effect on COVID-19. Nephrol. Dial. Transplant. 35, 1071–1072. doi: 10.1093/ndt/gfaa093
Patel, V. B., Clarke, N., Wang, Z., Fan, D., Parajuli, N., Basu, R., et al. (2014). Angiotensin II induced proteolytic cleavage of myocardial ACE2 is mediated by TACE/ADAM-17: a positive feedback mechanism in the RAS. J. Mol. Cell Cardiol. 66, 167–176. doi: 10.1016/j.yjmcc.2013.11.017
Pena, O. M., Pistolic, J., Raj, D., Fjell, C. D., and Hancock, R. E. (2011). Endotoxin tolerance represents a distinctive state of alternative polarization (M2) in human mononuclear cells. J. Immunol. 186, 7243–7254. doi: 10.4049/jimmunol.1001952
Petruk, G., Puthia, M., Petrlova, J., Strömdahl, A.-C., and Kjellström, S. (2020). Schmidtchen A. SARS-CoV-2 Spike protein binds to bacterial lipopolysaccharide and boosts proinflammatory activity. bioRxiv. doi: 10.1101/2020.06.29.175844
Poggi, A., Benelli, R., Venè, R, Costa, D., Ferrari, N., Tosetti, F., et al. (2019). Human gut-associated natural killer cells in health and disease. Front. Immunol. 10:961. doi: 10.3389/fimmu.2019.00961
Poole-Wright, K., Gaughran, F., Evans, R., and Chalder, T. (2020). Fatigue outcomes following coronavirus or influenza virus infection: a systematic review and meta-analysis. medRxiv. doi: 10.1101/2020.12.04.20244145
Poujol, F., Monneret, G., Pachot, A., Textoris, J., and Venet, F. (2015). Altered T lymphocyte proliferation upon lipopolysaccharide challenge ex vivo. PLoS ONE 10:e0144375. doi: 10.1371/journal.pone.0144375
Pretorius, E., Bester, J., Page, M. J., and Kell, D. B. (2018). The potential of LPS-binding protein to reverse amyloid formation in plasma fibrin of individuals with alzheimer-type dementia. Front. Aging Neurosci. 10:257. doi: 10.3389/fnagi.2018.00257
Proal, A., and Marshall, T. (2018). Myalgic encephalomyelitis/chronic fatigue syndrome in the era of the human microbiome: persistent pathogens drive chronic symptoms by interfering with host metabolism, gene expression, and immunity. Front. Pediatr. 6:373. doi: 10.3389/fped.2018.00373
Qian, M., Shen, X., and Wang, H. (2016). The distinct role of ADAM17 in app proteolysis and microglial activation related to alzheimer's disease. Cell Mol. Neurobiol. 36, 471–482. doi: 10.1007/s10571-015-0232-4
Qin, L., Wu, X., Block, M. L., Liu, Y., Breese, G. R., Hong, J.-S., et al. (2007). Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 55, 453–462. doi: 10.1002/glia.20467
Qingxian, L., Qiutang, L., and Qingjun, L. (2010). Regulation of phagocytosis by TAM receptors and their ligands. Front. Biol. 5, 227–237. doi: 10.1007/s11515-010-0034-5
Rajeevan, M. S., Murray, J., Oakley, L., Lin, J. S., and Unger, E. R. (2018). Association of chronic fatigue syndrome with premature telomere attrition. J. Transl. Med. 16:44. doi: 10.1186/s12967-018-1414-x
Ramos, S., Brenu, E., Broadley, S., Kwiatek, R., Ng, J., Nguyen, T., et al. (2016). Regulatory T, natural killer T and γδ T cells in multiple sclerosis and chronic fatigue syndrome/myalgic encephalomyelitis: a comparison. Asian Pac. J. Allergy Immunol. 34, 300–305. doi: 10.12932/AP0733
Regenass, S., Resink, T. J., Kern, F., Bühler, F. R., and Hahn, A. W. (1994). Angiotensin-II-induced expression of laminin complex and laminin A-chain-related transcripts in vascular smooth muscle cells. J. Vasc. Res. 31, 163–172. doi: 10.1159/000159042
Ribadeneira, M. D., Aungst, B. J., Eyermann, C. J., and Huang, S. M. (1996). Effects of structural modifications on the intestinal permeability of angiotensin II receptor antagonists and the correlation of in vitro, in situ, and in vivo absorption. Pharm. Res. 13, 227–233. doi: 10.1023/A:1016086930019
Sanoff, H. K., Deal, A. M., Krishnamurthy, J., Torrice, C., Dillon, P., Sorrentino, J., et al. (2014). Effect of cytotoxic chemotherapy on markers of molecular age in patients with breast cancer. J. Natl. Cancer Inst. 106:dju057. doi: 10.1093/jnci/dju057
Sarelius, I. H., and Glading, A. J. (2015). Control of vascular permeability by adhesion molecules. Tissue Barriers 3:e985954. doi: 10.4161/21688370.2014.985954
Scherbakov, N., Szklarski, M., Hartwig, J., Sotzny, F., Lorenz, S., Meyer, A., et al. (2020). Peripheral endothelial dysfunction in myalgic encephalomyelitis/chronic fatigue syndrome. ESC Heart Fail. 7, 1064–1071. doi: 10.1002/ehf2.12633
Schulte-Schrepping, J., Reusch, N., Paclik, D., Baßler, K., Schlickeiser, S., Zhang, B., et al. (2020). Severe COVID-19 is marked by a dysregulated myeloid cell compartment. Cell 182, 1419–1440.e23. doi: 10.1016/j.cell.2020.08.001
Seeberg, J. C., Loibl, M., Moser, F., Schwegler, M., Büttner-Herold, M., Danielet, C., et al. (2019). Non-professional phagocytosis: a general feature of normal tissue cells. Sci. Rep. 9:11875. doi: 10.1038/s41598-020-65963-5
Segatto, M., Fittipaldi, R., Pin, F., Sartori, R., Dae Ko, K., Zare, H., et al. (2017). Epigenetic targeting of bromodomain protein BRD4 counteracts cancer cachexia and prolongs survival. Nat. Commun. 8:1707. doi: 10.1038/s41467-017-01645-7
Segawa, K., and Nagata, S. (2015). An apoptotic ‘eat me' signal: phosphatidylserine exposure. Trends Cell Biol. 25, 639–650. doi: 10.1016/j.tcb.2015.08.003
Sepúlveda, N., Carneiro, J., Lacerda, E., and Nacul, L. (2019). Myalgic encephalomyelitis/chronic fatigue syndrome as a hyper-regulated immune system driven by an interplay between regulatory t cells and chronic human herpesvirus infections. Front. Immunol. 10:2684. doi: 10.3389/fimmu.2019.02684
Setty, B. N., and Betal, S. G. (2008). Microvascular endothelial cells express a phosphatidylserine receptor: a functionally active receptor for phosphatidylserine-positive erythrocytes. Blood 111, 905–914. doi: 10.1182/blood-2007-07-099465
Sfera, A., Osorio, C., Jafri, N., Diaz, E. L., and Campo Maldonado, J. E. (2020). Intoxication with endogenous Angiotensin II: a COVID-19 hypothesis. Front. Immunol. 11:1472. doi: 10.3389/fimmu.2020.01472
Shalaby, L., Thounaojam, M., Tawfik, A., Li, J., Hussein, K., Jahng, W. J., et al. (2020). Role of endothelial ADAM17 in early vascular changes associated with diabetic retinopathy. J. Clin. Med. 9:400. doi: 10.3390/jcm9020400
Shatanawi, A., Romero, M. J., Iddings, J. A., Chandra, S., Umapathy, N. S., Verin, A. D., et al. (2011). Angiotensin II-induced vascular endothelial dysfunction through RhoA/Rho kinase/p38 mitogen-activated protein kinase/arginase pathway. Am. J. Physiol. Cell Physiol. 300, C1181–C1192. doi: 10.1152/ajpcell.00328.2010
Shi, Y., Liu, T., He, L., Dougherty, U., Chen, L., Adhikariet, S., et al. (2016). Activation of the renin-angiotensin system promotes colitis development. Sci. Rep. 6:27552.doi: 10.1038/srep27552
Shigemoto-Mogami, Y., Hoshikawa, K., and Sato, K. (2018). activated microglia disrupt the blood-brain barrier and induce chemokines and cytokines in a rat in vitro model. Front. Cell Neurosci. 12:494. doi: 10.3389/fncel.2018.00494
Short, S., Fielder, E., Miwa, S., and von Zglinicki, T. (2019). Senolytics and senostatics as adjuvant tumour therapy. EBioMedicine 41, 683–692. doi: 10.1016/j.ebiom.2019.01.056
Silva, K. A. S., Ghiarone, T., Schreiber, K., Grant, D., White, T., Frisard, M. I., et al. (2019). Angiotensin II suppresses autophagy and disrupts ultrastructural morphology and function of mitochondria in mouse skeletal muscle. J. Appl. Physiol. 126, 1550–1562. doi: 10.1152/japplphysiol.00898.2018
Simões, H. G., Santos, T. R., Sousa, S. V., da Silva Aguiar, S., Motta-Santos, D., Degens, H., et al. (2020). Does longer leukocyte telomere length and higher physical fitness protect master athletes from consequences of coronavirus (SARS-CoV-2) infection? Front. Sports Act. Living 2:87. doi: 10.3389/fspor.2020.00087
Sjakste, N., Gutcaits, A., and Kalvinsh, I. (2005). Mildronate: an antiischemic drug for neurological indications. CNS Drug Rev. 11, 151–168. doi: 10.1111/j.1527-3458.2005.tb00267.x
Solerte, S. B., Di Sabatino, A., Galli, M., and Fiorina, P. (2020). Dipeptidyl peptidase-4 (DPP4) inhibition in COVID-19. Acta Diabetol. 57, 779–783. doi: 10.1007/s00592-020-01539-z
Sommer, A., Kordowski, F., Büch, J., Maretzky, T., Evers, A., Andrä, J., et al. (2016). Phosphatidylserine exposure is required for ADAM17 sheddase function. Nat. Commun. 7:11523. doi: 10.1038/ncomms11523
Song, P., An, J., and Zou, M. H. (2020). Immune clearance of senescent cells to combat ageing and chronic diseases. Cells 9:671. doi: 10.3390/cells9030671
Sotzny, F., Blanco, J., Capelli, E., Castro-Marrero, J., Steiner, S., Murovska, M., et al. (2018). Myalgic encephalomyelitis/chronic fatigue syndrome - evidence for an autoimmune disease. Autoimmun. Rev. 17, 601–609. doi: 10.1016/j.autrev.2018.01.009
Squassina, A., Pisanu, C., and Vanni, R. (2019). Mood disorders, accelerated aging, and inflammation: is the link hidden in telomeres? Cells 8:52. doi: 10.3390/cells8010052
Su, J. B. (2015). Vascular endothelial dysfunction and pharmacological treatment. World J. Cardiol. 7, 719–741. doi: 10.4330/wjc.v7.i11.719
Sumagin, R., Robin, A. Z., Nusrat, A., and Parkos, C. A. (2014). Transmigrated neutrophils in the intestinal lumen engage ICAM-1 to regulate the epithelial barrier and neutrophil recruitment. Mucosal Immunol. 7, 905–915. doi: 10.1038/mi.2013.106
Sun, I. H., Oh, M. H., Zhao, L., Patel, C. H., Arwood, M. L., Xu, W., et al. (2018). mTOR complex 1 signaling regulates the generation and function of central and effector Foxp3+ regulatory T cells. J. Immunol. 201, 481–492. doi: 10.4049/jimmunol.1701477
Sun, M., Yan, H., Zou, W., Wang, Y., Li, H., and Wang, X. (2016). [Lipopolysaccharide induces astrocyte activation and downregulates the expression of Kir4.1 channel]. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi. 32, 196–200.
Tabuzek, K., Liber, S., Gabryel, B., Adamczyk, J., and Okopie,ń, B. (2010). Metformin increases phagocytosis and acidifies lysosomal/endosomal compartments in AMPK-dependent manner in rat primary microglia. Naunyn Schmied Arch Pharmacol. 381, 171–186 doi: 10.1007/s00210-009-0477-x
Takakura, M., Kyo, S., Sowa, Y., Wang, Z., Yatabe, N., Maida, Y., et al. (2001). Telomerase activation by histone deacetylase inhibitor in normal cells. Nucleic Acids Res. 29, 3006–3011. doi: 10.1093/nar/29.14.3006
Takashina, Y., Ishizuka, N., Ikumi, N., Hayashi, H., Manabe, A., Hirota, C., et al. (2020). Upregulation of claudin-7 expression by angiotensin ii in colonic epithelial cells of mice fed with NaCl-depleted diets. Int. J. Mol. Sci. 21:1442. doi: 10.3390/ijms21041442
Takeshita, H., Yamamoto, K., Mogi, M., Nozato, S., Horiuchi, M., and Rakugi, H. (2020). Different effects of the deletion of angiotensin converting enzyme 2 and chronic activation of the renin-angiotensin system on muscle weakness in middle-aged mice. Hypertens. Res. 43, 296–304. doi: 10.1038/s41440-019-0375-7
Tanaka, M., and Itoh, H. (2019). Hypertension as a metabolic disorder and the novel role of the gut. Curr. Hypertens. Rep. 21:63. doi: 10.1007/s11906-019-0964-5
Theoharides, T. C., Stewart, J. M., Hatziagelaki, E., and Kolaitis, G. (2015). Brain “fog,” inflammation and obesity: key aspects of neuropsychiatric disorders improved by luteolin. Front. Neurosci. 9:225. doi: 10.3389/fnins.2015.00225
Thomas, H. (2016). Intestinal tract: Gut endothelial cells–another line of defence. Nat. Rev. Gastroenterol. Hepatol. 13:4. doi: 10.1038/nrgastro.2015.205
Thorp, E., Vaisar, T., Subramanian, M., Mautner, L., Blobel, C., and Tabas, I. (2011). Shedding of the Mer tyrosine kinase receptor is mediated by ADAM17 protein through a pathway involving reactive oxygen species, protein kinase Cδ, and p38 mitogen-activated protein kinase (MAPK). J. Biol. Chem. 286, 33335–33344. doi: 10.1074/jbc.M111.263020
Tirone, M., Tran, N. L., Ceriotti, C., Gorzanelli, A., Canepari, M., Bottinelli, R., et al. (2018). High mobility group box 1 orchestrates tissue regeneration via CXCR4. J. Exp. Med. 215, 303–318. doi: 10.1084/jem.20160217
Toubi, E., Kessel, A., Slobodin, G., Boulman, N., Pavlotzky, E., Zisman, D., et al. (2007). Changes in macrophage function after rituximab treatment in patients with rheumatoid arthritis. Ann. Rheum. Dis. 66, 818–820. doi: 10.1136/ard.2006.062505
Tsai, S. Y., Chen, H. J., Lio, C. F., Kuo, C. F., Kao, A. C., Wang, W. S., et al. (2019). Increased risk of chronic fatigue syndrome in patients with inflammatory bowel disease: a population-based retrospective cohort study. J. Transl Med. 17:55. doi: 10.1186/s12967-019-1797-3
Tsukamoto, H., Takeuchi, S., Kubota, K., Kobayashi, Y., Kozakai, S., Ukai, I., et al. (2018). Lipopolysaccharide (LPS)-binding protein stimulates CD14-dependent Toll-like receptor 4 internalization and LPS-induced TBK1-IKKϵ-IRF3 axis activation. J. Biol. Chem. 293, 10186–10201. doi: 10.1074/jbc.M117.796631
Tsung, A., Tohme, S., and Billiar, T. R. (2014). High-mobility group box-1 in sterile inflammation. J. Intern. Med. 276, 425–443. doi: 10.1111/joim.12276
Turowski, P., Adamson, P., and Greenwood, J. (2005). Pharmacological targeting of ICAM-1 signaling in brain endothelial cells: potential for treating neuroinflammation. Cell Mol. Neurobiol. 25, 153–170. doi: 10.1007/s10571-004-1380-0
Urciuoli, E., and Peruzzi, B. (2020). Inhibiting extracellular vesicle trafficking as antiviral approach to corona virus disease 2019 infection. Front. Pharmacol. 11:580505. doi: 10.3389/fphar.2020.580505
van Eeden, C., Khan, L., Osman, M. S., and Cohen Tervaert, J. W. (2020). Natural killer cell dysfunction and its role in COVID-19. Int. J. Mol. Sci. 21:6351. doi: 10.3390/ijms21176351
VanderVeen, B. N., Fix, D. K., Montalvo, R. N., Brittany R Counts, B. R., Smuder, A. J., Murphy, A., et al. (2019). The regulation of skeletal muscle fatigability and mitochondrial function by chronically elevated interleukin-6. Exp. Physiol. 104, 385–397. doi: 10.1113/EP087429
Vann, J. M., and Proctor, R. A. (1990). “Phagocytosis of bacteria by endothelial cells,” in Pathogenesis of Wound and Biomaterial-Associated Infections, eds T. Wadström, I. Eliasson, I. Holder, A. Ljungh (London: Springer). doi: 10.1007/978-1-4471-3454-1_9
Vasan, R. S., Demissie, S., Kimura, M., Cupples, L. A., Rifai, N., White, C., et al. (2008). Association of leukocyte telomere length with circulating biomarkers of the renin-angiotensin-aldosterone system: the framingham heart study. Circulation 117, 1138–1144. doi: 10.1161/CIRCULATIONAHA.107.731794
Veldhoen, M. (2017). Interleukin 17 is a chief orchestrator of immunity. Nat. Immunol. 18, 612–621. doi: 10.1038/ni.3742
Verdecchia, P., Cavallini, C., Spanevello, A., and Angeli, F. (2020). The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur. J. Intern. Med. 76, 14–20. doi: 10.1016/j.ejim.2020.04.037
Vetvicka, V., Vannucci, L., Sima, P., and Richter, J. (2019). Beta glucan: supplement or drug? From laboratory to clinical trials. Molecules 24:1251. doi: 10.3390/molecules24071251
Vetvicka, V., and Vetvickova, J. (2015). Beta-Glucan attenuates chronic fatigue syndrome in murine model. J. Nat. Sci. 1:e112.
Viana, S. D., Nunes, S., and Reis, F. (2020). ACE2 imbalance as a key player for the poor outcomes in COVID-19 patients with age-related comorbidities–role of gut microbiota dysbiosis. Ageing Res Rev. 62:101123. doi: 10.1016/j.arr.2020.101123
Villapol, S., and Saavedra, J. M. (2015). Neuroprotective effects of angiotensin receptor blockers. Am. J. Hypertens. 28, 289–299. doi: 10.1093/ajh/hpu197
Wan, W., Cao, L., Khanabdali, R., Kalionis, B., Tai, X., and Xia, S. (2016). The emerging role of HMGB1 in neuropathic pain: a potential therapeutic target for neuroinflammation. J. Immunol. Res. 2016:6430423. doi: 10.1155/2016/6430423
Wang, E. Y., Mao, T., Klein, J., Dai, Y., Huck, J. D., Liu, F., et al. (2020). Diverse functional autoantibodies in patients with COVID-19. medRxiv 2020.12.10.20247205. doi: 10.1101/2020.12.10.20247205
Wang, H., Fu, H., Zhu, R., Wu, X., Ji, X., Li, X., et al. (2020). BRD4 contributes to LPS-induced macrophage senescence and promotes progression of atherosclerosis-associated lipid uptake. Aging 12, 9240–9259. doi: 10.18632/aging.103200
Wang, J., Wang, S., Li, M., Wu, D., Liu, F., Yang, R., et al. (2015). The neuropilin-1 inhibitor, ATWLPPR peptide, prevents experimental diabetes-induced retinal injury by preserving vascular integrity and decreasing oxidative stress. PLoS ONE 10:e0142571. doi: 10.1371/journal.pone.0142571
Wang, L., Muneer, A., Xie, L., Zhang, F., Wu, B., Mei, L., et al. (2020). Novel gene-specific translation mechanism of dysregulated, chronic inflammation reveals promising, multifaceted COVID-19. bioRxiv 2020.11.14.382416. doi: 10.1101/2020.11.14.382416
Wang, X., Sun, R., Hao, X., Lian, Z. X., Wei, H., and Tian, Z. (2019). IL-17 constrains natural killer cell activity by restraining IL-15-driven cell maturation via SOCS3. Proc. Natl. Acad. Sci. U.S.A. 116, 17409–17418. doi: 10.1073/pnas.1904125116
Watanabe, Y., Lee, S. W., Detmar, M., Ajioka, I., and Dvorak, H. F. (1997). Vascular permeability factor/vascular endothelial growth factor (VPF/VEGF) delays and induces escape from senescence in human dermal microvascular endothelial cells. Oncogene 14, 2025–2032. doi: 10.1038/sj.onc.1201033
Wilson, C. (2020). Concern coronavirus may trigger post-viral fatigue syndromes. New Sci. 246, 10–11. doi: 10.1016/S0262-4079(20)30746-6
Wirth, K., and Scheibenbogen, C. (2020). A unifying hypothesis of the pathophysiology of myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS): recognitions from the finding of autoantibodies against ß2-adrenergic receptors. Autoimmun. Rev. 19:102527. doi: 10.1016/j.autrev.2020.102527
Wissler Gerdes, E. O., Zhu, Y., Tchkonia, T., and Kirkland, J. L. (2020). Discovery, development, and future application of senolytics: theories and predictions. FEBS J. 287, 2418–2427. doi: 10.1111/febs.15264
Wu, W., Wang, S., Liu, Q., Shan, T., and Wang, Y. (2018). Metformin protects against LPS-induced intestinal barrier dysfunction by activating AMPK pathway. Mol. Pharm. 15, 3272–3284. doi: 10.1021/acs.molpharmaceut.8b00332
Wyller, V. B., Vitelli, V., Sulheim, D., Fagermoen, E., Winger, A., Godang, K., et al. (2016). Altered neuroendocrine control and association to clinical symptoms in adolescent chronic fatigue syndrome: a cross-sectional study. J. Transl. Med. 14:121. doi: 10.1186/s12967-016-0873-1
Xu, M., Pirtskhalava, T., Farr, J. N., Weigand, B. M., Palmer, A. K., Weivoda, M. M., et al. (2018). Senolytics improve physical function and increase lifespan in old age. Nat. Med. 24, 1246–1256. doi: 10.1038/s41591-018-0092-9
Yadav, M., Louvet, C., Davini, D., Gardner, J. M., Martinez-Llordella, M., Bailey-Bucktrout, S., et al. (2012). Neuropilin-1 distinguishes natural and inducible regulatory T cells among regulatory T cell subsets in vivo. J. Exp. Med. 209, 1713–S19. doi: 10.1084/jem.20120822
Yamaji, M., Mahmoud, M., Evans, I. M., and Zachary, I. C. (2015). Neuropilin 1 is essential for gastrointestinal smooth muscle contractility and motility in aged mice. PLoS ONE 10:e0115563. doi: 10.1371/journal.pone.0115563
Yamamoto, S., Yancey, P. G., Zuo, Y., Ma, L. J., Kaseda, R., Fogo, A. B., et al. (2011). Macrophage polarization by angiotensin II-type 1 receptor aggravates renal injury-acceleration of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 31, 2856–2864. doi: 10.1161/ATVBAHA.111.237198
Yamashita, M. (2020). Potential role of neuroactive tryptophan metabolites in central fatigue: establishment of the fatigue circuit. Int. J. Tryptophan Res. 13:1178646920936279. doi: 10.1177/1178646920936279
Yamazaki, Y., Baker, D. J., Tachibana, M., Liu, C. C., van Deursen, J. M., Brott, T. G., et al. (2016). Vascular cell senescence contributes to blood-brain barrier breakdown. Stroke 47, 1068–1077. doi: 10.1161/STROKEAHA.115.010835
Yang, W., Yu, T., Huang, X., Bilotta, A. J., Xu, L., Luet, Y., et al. (2020). Intestinal microbiota-derived short-chain fatty acids regulation of immune cell IL-22 production and gut immunity. Nat. Commun. 11:4457. doi: 10.1038/s41467-020-18262-6
Yang, Y., Sun, H., Li, X., Ding, Q., Wei, P., and Zhou, J. (2015). Transforming growth factor beta-induced is essential for endotoxin tolerance induced by a low dose of lipopolysaccharide in human peripheral blood mononuclear cells. Iran J. Allergy Asthma Immunol. 14, 321–330.
Yin, X. X., Zheng, X. R., Peng, W., Wu, M. L., and Mao, X. Y. (2020). Vascular endothelial growth factor (VEGF) as a vital target for brain inflammation during the COVID-19 outbreak. ACS Chem. Neurosci. 11, 1704–1705. doi: 10.1021/acschemneuro.0c00294
Yoon, M. S. (2017). mTOR as a key regulator in maintaining skeletal muscle mass. Front. Physiol. 8:788. doi: 10.3389/fphys.2017.00788
Yu, Z., Li, Y., Niu, Y., Tang, Q., and Wu, J. (2020). Milk fat globule membrane enhances colonic-mucus-barrier function in a rat model of short-bowel syndrome. JPEN J. Parenter. Enteral. Nutr.
Zaid, Y., Puhm, F., Allaeys, I., Naya, A., Oudghiri, M., Khalki, L., et al. (2020). Platelets can associate with SARS-Cov-2 RNA and are hyperactivated in COVID-19. Circ. Res. 127, 1404–1418. doi: 10.1161/CIRCRESAHA.120.317703
Zakrocka, I., Targowska-Duda, K. M., Wnorowski, A., Kocki, T., Józwiak, K., and Turski, W. A. (2017). Angiotensin II type 1 receptor blockers inhibit KAT II activity in the brain-its possible clinical applications. Neurotox. Res. 32, 639–648. doi: 10.1007/s12640-017-9781-2
Zhan, X., Stamova, B., and Sharp, F. R. (2018). Lipopolysaccharide associates with amyloid plaques, neurons and oligodendrocytes in alzheimer's disease brain: a review. Front. Aging Neurosci. 10:42. doi: 10.3389/fnagi.2018.00042
Zhang, Y., Wang, Y., Zhou, D., Zhang, L. S., Deng, F. X., Shu, S., et al. (2019). Angiotensin II deteriorates advanced atherosclerosis by promoting MerTK cleavage and impairing efferocytosis through the AT1R/ROS/p38 MAPK/ADAM17 pathway. Am. J. Physiol. Cell Physiol. 317, C776–C787. doi: 10.1152/ajpcell.00145.2019
Zhang, Z. T., Du, X. M., Ma, X. J., Zong, Y., Chen, J.-K., Yu, C.-L., et al. (2016). Activation of the NLRP3 inflammasome in lipopolysaccharide-induced mouse fatigue and its relevance to chronic fatigue syndrome. J. Neuroinflammation 13:71. doi: 10.1186/s12974-016-0539-1
Zhao, Y., Cong, L., and Lukiw, W. J. (2017). Lipopolysaccharide (LPS) accumulates in neocortical neurons of alzheimer's disease (AD) brain and impairs transcription in human neuronal-glial primary co-cultures. Front. Aging Neurosci. 9:407. doi: 10.3389/fnagi.2017.00407
Zheng, M., Gao, Y., Wang, G., Song, G., Liu, S., Sun, D., et al. (2020). Functional exhaustion of antiviral lymphocytes in COVID-19 patients. Cell Mol. Immunol. 17, 533–535. doi: 10.1038/s41423-020-0402-2
Zhou, T., Su, T. T., Mudianto, T., and Wang, J. (2020). Immune asynchrony in COVID-19 pathogenesis and potential immunotherapies. J. Exp. Med. 217:e20200674. doi: 10.1084/jem.20200674
Zhou, T., Zheng, Y., Sun, L., Badea, S. R., Jin, Y., Liu, Y., et al. (2019). Microvascular endothelial cells engulf myelin debris and promote macrophage recruitment and fibrosis after neural injury. Nat. Neurosci. 22, 421–435. doi: 10.1038/s41593-018-0324-9
Zhou, Y., Yao, Y., Deng, Y., and Shao, A. (2020). Regulation of efferocytosis as a novel cancer therapy. Cell Commun. Signal 18:71. doi: 10.1186/s12964-020-00542-9
Zhou, Z., Li, X., Liu, Z., Huang, L., Yao, Y., Li, L., et al. (2020). A bromodomain-containing protein 4 (BRD4) inhibitor suppresses angiogenesis by regulating AP-1 expression. Front. Pharmacol. 11:1043. doi: 10.3389/fphar.2020.01043
Zhu, Y., Tchkonia, T., Fuhrmann-Stroissnigg, H., Dai, H. M., Ling, Y. Y., Stout, M. B., et al. (2016). Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell 15, 428–435. doi: 10.1111/acel.12445
Keywords: endothelial cells, cellular senescence, gut microbial community, endotoxin tolerance, microbial translocation
Citation: Sfera A, Osorio C, Zapata Martín del Campo CM, Pereida S, Maurer S, Maldonado JC and Kozlakidis Z (2021) Endothelial Senescence and Chronic Fatigue Syndrome, a COVID-19 Based Hypothesis. Front. Cell. Neurosci. 15:673217. doi: 10.3389/fncel.2021.673217
Received: 27 February 2021; Accepted: 25 May 2021;
Published: 25 June 2021.
Edited by:
Aurel Popa-Wagner, University of Medicine and Pharmacy of Craiova, RomaniaReviewed by:
Ravi Kant Narayan, All India Institute of Medical Sciences (Patna), IndiaAshutosh Kumar, All India Institute of Medical Sciences (Patna), India
Copyright © 2021 Sfera, Osorio, Zapata Martín del Campo, Pereida, Maurer, Maldonado and Kozlakidis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Adonis Sfera, ZHIuc2ZlcmFAZ21haWwuY29t