 Sarah K. Lotz
Sarah K. Lotz Britanie M. Blackhurst
Britanie M. Blackhurst Katie L. Reagin
Katie L. Reagin Kristen E. Funk
Kristen E. Funk- Department of Biological Sciences, University of North Carolina at Charlotte, Charlotte, NC, United States
Neurodegenerative diseases, such as Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis, comprise a family of disorders characterized by progressive loss of nervous system function. Neuroinflammation is increasingly recognized to be associated with many neurodegenerative diseases but whether it is a cause or consequence of the disease process is unclear. Of growing interest is the role of microbial infections in inciting degenerative neuroinflammatory responses and genetic factors that may regulate those responses. Microbial infections cause inflammation within the central nervous system through activation of brain-resident immune cells and infiltration of peripheral immune cells. These responses are necessary to protect the brain from lethal infections but may also induce neuropathological changes that lead to neurodegeneration. This review discusses the molecular and cellular mechanisms through which microbial infections may increase susceptibility to neurodegenerative diseases. Elucidating these mechanisms is critical for developing targeted therapeutic approaches that prevent the onset and slow the progression of neurodegenerative diseases.
Introduction
Neurodegenerative diseases, such as Alzheimer’s disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS), are clinically characterized by the progressive decline of cognitive, motor, and behavioral functions. Pathologically, these diseases exhibit significant neuronal death, brain atrophy, protein aggregation, and neuroinflammation. Despite improved understanding of disease progression, the cause or causes that initiate disease processes are not well understood. Recent genome-wide association studies have highlighted the contribution of immune molecules in many neurodegenerative diseases. Several genes with polymorphisms that increase the risk of neurodegenerative diseases, such as CD33 and TREM2 in AD, PRKN, SCNA, LRRK2, and HLA in PD, and C9ORF72 in ALS have been linked to various immune functions including phagocytosis, microglial activation, complement activation, MHC class II expression, and hematopoiesis (McGeer et al., 1988; Griciuc et al., 2013; Guerreiro et al., 2013; Jonsson et al., 2013; Dzamko et al., 2015; Burberry et al., 2016; Jimenez-Ferrer and Swanberg, 2018). Because these genetic risk factors do not cause disease in all carriers, it is hypothesized that environmental factors that induce inflammation may contribute to the etiopathogenesis of neurodegenerative diseases. Microbial infections have become of increasing interest in inciting neurodegenerative pathology, as they can invade the central nervous system (CNS) and cause significant neuroinflammation through activation of resident immune cells, such as microglia and astrocytes, as well as promote infiltration of peripheral macrophages and T cells (Vasek et al., 2016; Garber et al., 2018, 2019; Klein et al., 2019). Though these immune responses exist to protect the brain, they can cause critical damage in an attempt to clear the invading pathogen (Figure 1).
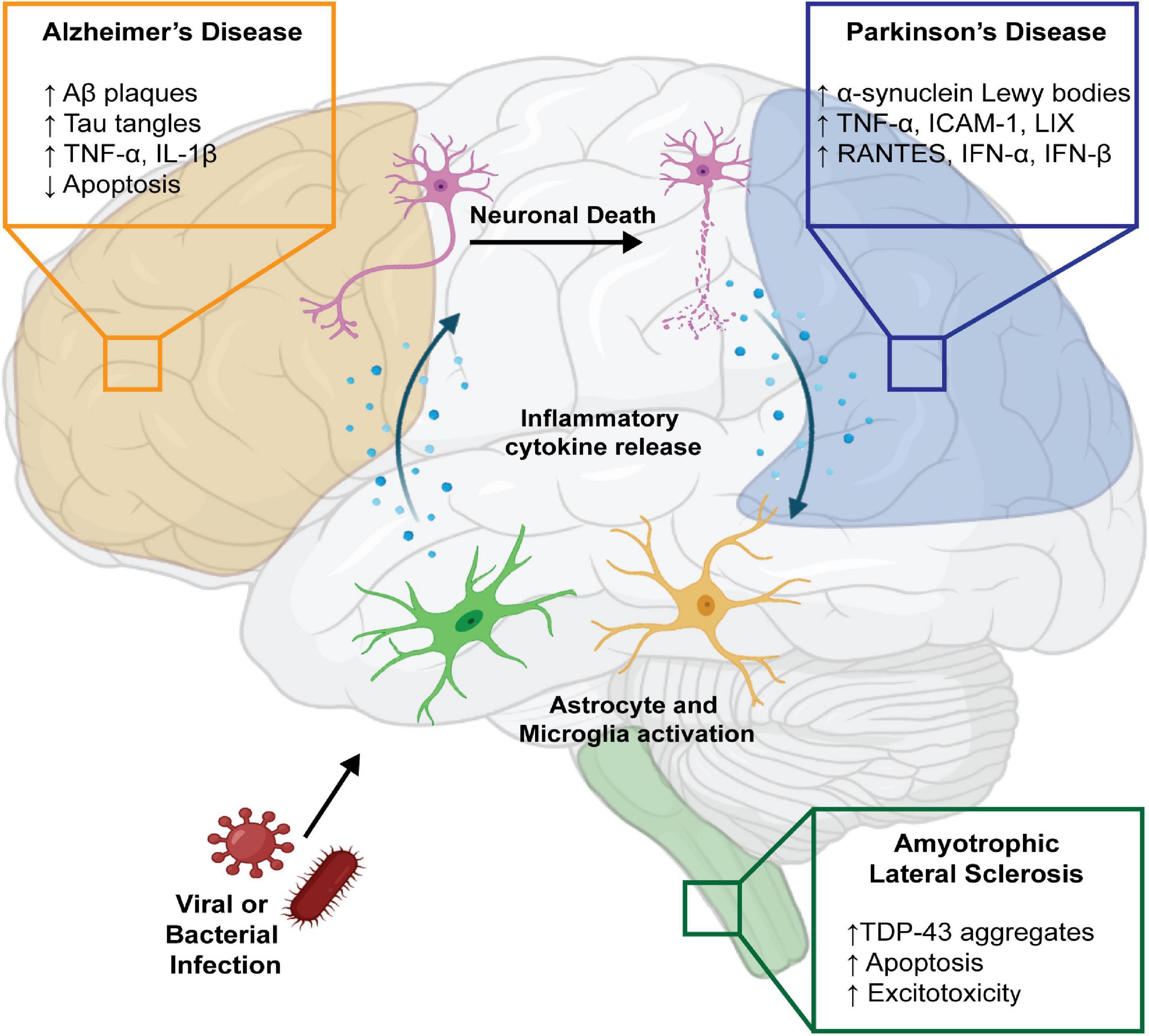
Figure 1. Infectious agents may contribute to neurodegenerative diseases directly or via immune activation. Infection by viral and bacterial pathogens can cause pro-inflammatory activation of CNS resident immune cells, including astrocytes and microglia, resulting in neuronal death. Additionally, cellular death directly caused by infectious agents and the release of damage-associated molecular patterns can exacerbate the inflammatory state through further activation of CNS immune cells, perpetuating a cycle of inflammation. In AD, this is often associated with high levels of pro-inflammatory cytokines TNF-α and IL-1β, reduced clearance of infected cells, and accumulation of neurotoxic aggregates composed of Aβ and Tau. This pro-inflammatory state has also been documented in the context of PD, where increased accumulation of neurotoxic α-synuclein is accompanied by high levels of TNF-α, ICAM-1, LIX, RANTES, IFN-α, and IFN-β, produced by infected and activated astrocytes and microglia. Additionally, some pathogens can directly infect neurons resulting in alterations in metabolism, enhanced neuronal excitotoxicity, and enhanced apoptosis, as seen in ALS. Created with BioRender.com.
Infectious agents may contribute to neurodegenerative disease pathology by eliciting an inflammatory response. Following infection, inflammation prevents damaging pathology and promotes tissue repair and regeneration; however, if uncontrolled, inflammation can become lethal to healthy cells (Chen et al., 2018). These inflammatory responses originate locally at the site of infection, but can rapidly become widespread, and in some cases, involve the CNS. Increased production of inflammatory cytokines and chemokines, including IL-1β, promote breakdown of the blood brain barrier (BBB), which typically protects the CNS resident cells from harmful agents and inflammatory mediators (Cunningham, 2013; Bendorius et al., 2018; Yong et al., 2019). However, if there is a BBB breach, these soluble mediators can stimulate CNS resident astrocytes and microglia which, upon activation, amplify inflammatory conditions in the CNS that can cause significant damage to both infected and uninfected neurons as well as resident glial cells (Holmes, 2013; Paouri and Georgopoulos, 2019; Walker et al., 2019). Importantly, neurotropic infections can lead to harmful neuroinflammation that has been identified as a potential risk factor for neurodegenerative diseases (Monastero et al., 2014; Itzhaki, 2017; Sochocka et al., 2017; Fulop et al., 2018). This review discusses recent studies linking microbial infections to neurodegenerative diseases and the cellular and molecular mechanisms through which they may increase susceptibility to disease (summarized in Table 1).
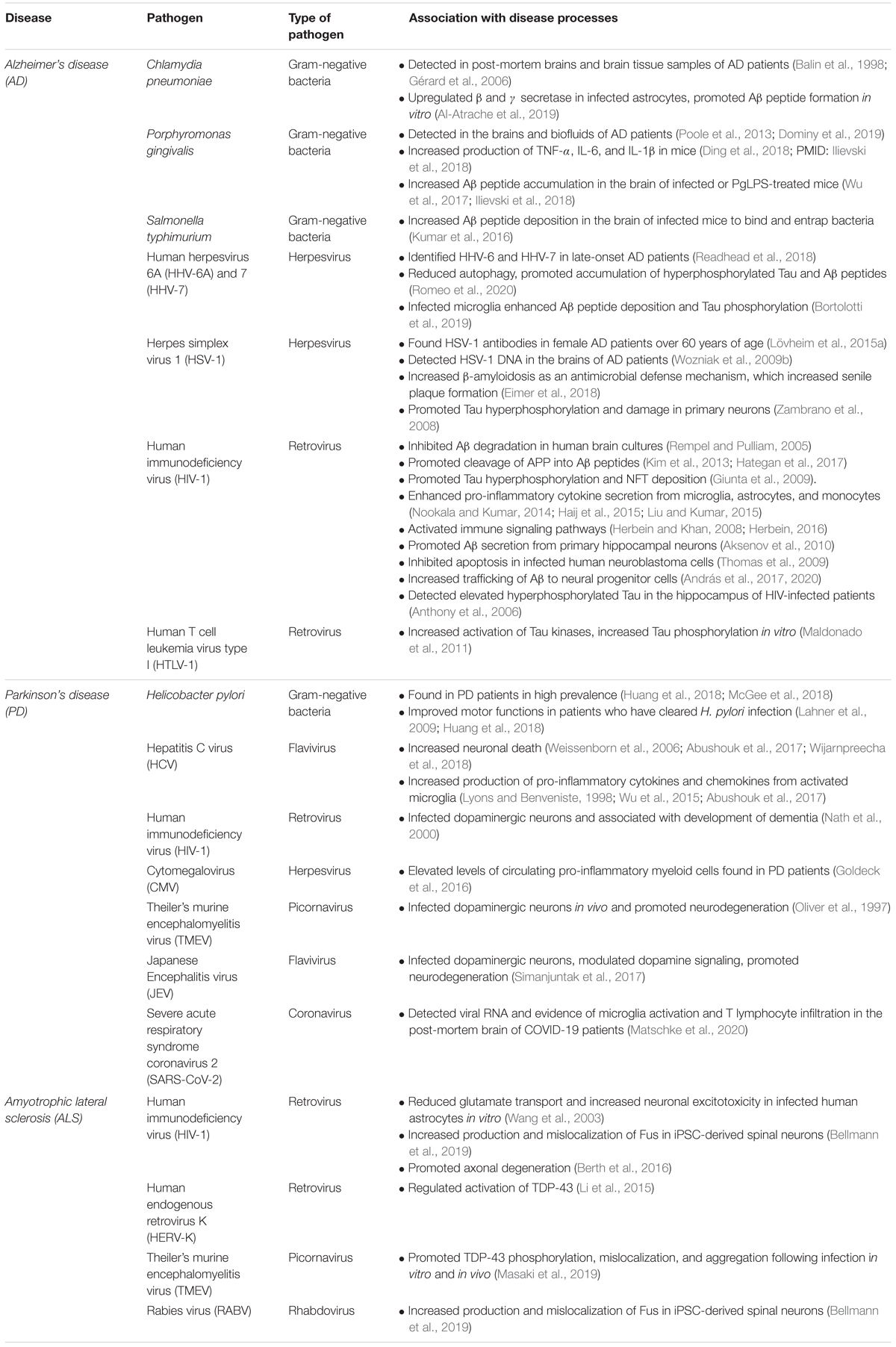
Table 1. Overview of infections associated with neurodegenerative diseases.
Alzheimer’s Disease
AD is characterized pathologically by the deposition of two proteinaceous lesions in the brain—extracellular senile plaques and intracellular neurofibrillary tangles (NFTs) (Serrano-Pozo et al., 2011). Senile plaques are extracellular aggregates composed of insoluble amyloid beta (Aβ) peptides, the proteolytic product of amyloid beta precursor protein (APP). Under homeostatic conditions, APP is cleaved by α-secretase and γ-secretase, which is facilitated by presenilin 1 (PSEN1) (Chow et al., 2010; Murphy and LeVine, 2010; Zhang et al., 2011). In AD, APP is instead cleaved by β-secretase and γ-secretase, forming the insoluble Aβ peptides, which self-aggregate into senile plaques and are believed to be toxic to neurons (O’Brien and Wong, 2011; Kelleher and Shen, 2017; Fan et al., 2019). Neurofibrillary tangles are intracellular aggregates composed of hyperphosphorylated microtubule-associated protein Tau. Tau can be phosphorylated by a number of kinases, including cyclin-dependent kinase 5 (CDK5) and glycogen synthase kinase-3β (GSK-3β) (Gong et al., 2005). Under homeostatic conditions, phosphorylation modulates the affinity of Tau for microtubules, allowing their dynamic growth and retraction (Lindwall and Cole, 1984). In AD, Tau becomes hyperphosphorylated, which decreases its affinity for microtubules and increases its propensity to self-aggregate into pathogenic NFTs (Gong et al., 2005; Gong and Iqbal, 2008; Wang et al., 2013; Miao et al., 2019).
While the mechanisms that incite Aβ and Tau aggregation are not fully understood, recent studies have suggested a role for inflammatory cytokines, including tumor necrosis factor (TNF)-α, interferon (IFN)-γ, interleukin (IL)-1β, IL-6, IL-10, and IL-18 (Griffin et al., 1995a,b, 2006; Mrak and Griffin, 2001; Li et al., 2003). For example, IL-1β is an essential mediator of the inflammatory response and has been found to be elevated near Aβ plaques (Griffin et al., 1989; Lopez-Castejon and Brough, 2011). Expression of IFN-γ, a pro-inflammatory cytokine, was elevated in transgenic mice with AD-related pathology (Roy et al., 2020), though it does not appear to be significantly elevated in human patients (Bongioanni et al., 1997; Singh and Guthikonda, 1997). The impact of IFN-γ on AD pathology is apparently diametric, as some reports indicate that IFN-γ treatment promoted Aβ clearance by microglia and macrophages, thus reducing pathological load (Chakrabarty et al., 2010; He et al., 2020). Also, overexpression of IFN-γ in transgenic mice that develop amyloid and Tau pathologies resulted in a significant decrease in Tau pathology and improved neurogenesis, suggesting elevated levels of IFN-γ can be beneficial for alleviating AD pathology within the brain (Mastrangelo et al., 2009). However, co-stimulation of primary human astrocytes in culture with IFN-γ and TNF-α induced Aβ production, and deletion of the IFN-γ receptor reduced gliosis and amyloid plaque deposition in APP transgenic mice, which would suggest elevated levels of inflammation within the CNS exacerbates AD pathology (Blasko et al., 2000; Yamamoto et al., 2007). These seemingly conflicting observations could be, in part, due to differences in the magnitude of cytokine elevation and timeframe of expression as well as other environmental and genetic factors. Altogether, they suggest that acute episodes of neuroinflammation, such as those caused by infections, may initiate pathological Aβ and Tau deposition.
Infectious microbes have long been suspected to play a role in the onset of AD, though direct evidence is still limited (Miklossy et al., 2006; Miklossy, 2011; Bu et al., 2014). Several cohort studies have examined infectious burden in patients with AD, indicating a correlation between infections and AD pathology (Balin et al., 1998; Itzhaki and Wozniak, 2004; Gérard et al., 2006; Lövheim et al., 2015b). Using multiscale networks of AD-associated virome data, integrating genomic, transcriptomic, proteomic, and histopathological information, Readhead et al. (2018) identified evidence of increased herpesvirus 6A (HHV-6A) and human herpesvirus 7 (HHV-7) in patients with late-onset AD compared to healthy controls. Additionally, a strong association was detected between the presence of herpes simplex virus 1 (HSV-1) antibodies and patients with AD, specifically in women, subjects older than 60 years of age, and when plasma samples were taken at least 6.6 years prior to dementia diagnosis (Lövheim et al., 2015a). The authors proposed that this 6.6 year lag between HSV-1 antibody detection and AD diagnosis indicates that HSV-1 plays a role primarily in early AD development (Lövheim et al., 2015a). Furthermore, HSV-1 DNA sequences and the functional HSV-1 genome, in its entirety, were detected in the brains of patients with AD (Wozniak et al., 2009b). Similarly, the presence of Chlamydia pneumoniae was detected in post mortem brain-tissue samples of patients with AD (Balin et al., 1998; Gérard et al., 2006). Additionally, serum antibody levels to common periodontal microbiota were observed to increase risk of developing AD (Sparks Stein et al., 2012; Noble et al., 2014). More recent studies have identified Porphyromonas gingivalis in the brains and biofluids of patients with AD (Poole et al., 2013; Dominy et al., 2019). Very recently, researchers reported that in a Swedish cohort of people over the age of 50, untreated herpesvirus infection [either HSV-1 or varicella zoster virus (VZV)] increased the risk of dementia by 1.5-fold. Patients diagnosed with herpesvirus infection who took antiviral medication showed reduced risk of dementia by 25% compared to those with untreated herpesvirus infection (Lopatko Lindman et al., 2021). Epidemiological data cannot prove causation between infections and AD, but collectively these studies support the hypothesis that pathogens increase the risk of developing AD.
Some of the earliest data regarding the microbial etiology hypothesis of AD implicated HSV-1, a neurotropic enveloped virus that establishes life-long latent infection in the CNS with periodic reactivation cycles. Following resolution of primary infection, HSV-1 can remain dormant, predominantly in the trigeminal ganglion, and upon reactivation induce severe acute encephalitis in the temporal and frontal cortices of the brain, known as herpes simplex encephalitis (HSE) (Mancuso et al., 2019). Ball first proposed a link between HSV-1 and AD in 1982, recognizing that similar brain regions are affected by both HSE and AD, and that people who survived HSE exhibited clinical symptoms similar to AD, including memory loss and cognitive impairment (Ball et al., 1982). Since then substantial progress has been made to understand the molecular mechanisms by which HSV-1 may contribute to the onset of AD. Zambrano et al. (2008) showed that infection of primary neurons with HSV-1 caused significant neuronal damage and death via hyperphosphorylation of Tau, increased acetylation and tyrosination of tubulin, disrupted microtubules, and damaged and shortened neurites. Similarly, HSV-1 induced GSK3-β and protein kinase A-mediated Tau hyperphosphorylation (Wozniak et al., 2009a). All of these findings are synonymous with the pathology seen in AD, suggesting that HSV-1 infection may promote AD onset.
Another virus often associated with AD pathology is human immunodeficiency virus 1 (HIV-1), a retrovirus that can become neuroinvasive and induce severe encephalitic and cognitive changes. Patients with HIV-associated neurocognitive disorder (HAND) demonstrate increased production of Aβ and development of amyloid plaques (Antinori et al., 2007; Anthony et al., 2010; Fulop et al., 2019). HIV-1 infection induced the expression of RAGE (the receptor for advanced glycation end products) in brain endothelial cells and the accumulation of Aβ in a RAGE-dependent manner. Aβ aggregates were then transferred from brain endothelial cells to neural progenitor cells, stimulating further aggregation and progenitor cell dysfunction (Deane et al., 2003; András et al., 2010). However, while much research has focused on mechanisms of Aβ production and aggregation, the total level of Aβ in the brain also depends on the mechanisms of clearance. One clearance mechanism involves the zinc-metalloprotease neprilysin, which has been shown to cleave and degrade Aβ monomers in vitro and in vivo (Shirotani et al., 2001; Marr et al., 2004; Maruyama et al., 2005; El-Amouri et al., 2008; Grimm et al., 2013; Marr and Hafez, 2014). In an in vitro assay, the HIV-1 transcription transactivator (Tat) protein inhibited activity of neprilysin by 80% and increased the soluble Aβ by 125% when applied to human brain cultures (Rempel and Pulliam, 2005). HIV-1 Tat also recruited APP in lipid rafts and stimulated its cleavage by β-secretase and γ-secretase, yielding higher levels of the Aβ peptides and causing neurotoxicity (Kim et al., 2013; Hategan et al., 2017). Furthermore, HIV-1 surface protein, gp120, promoted Aβ secretion in primary embryonic rat hippocampal neurons (Aksenov et al., 2010), inhibited apoptosis of infected human neuroblastoma cells via inhibition of the Fas-pathway (Thomas et al., 2009), and induced neurotoxicity in human neuroblastoma cells through the CXCR4 and CCR5 chemokine receptors (Catani et al., 2000; Bardi et al., 2006). Additionally, HIV-1 Tat and Nef proteins exacerbated the secretion of pro-inflammatory cytokines from surrounding microglia, astrocytes and monocytes, causing neurotoxic effects (Nookala and Kumar, 2014; Haij et al., 2015; Liu and Kumar, 2015). Furthermore, Nef can mimic TNF-α signaling by activating inflammatory pathways, such as NF-κB, AP1, JNK and AKT (Herbein and Khan, 2008; Herbein, 2016). HIV-1 infection can also promote Tau aggregation, as Anthony et al. (2006) found elevated levels of hyperphosphorylated Tau in the hippocampus of HIV-1-infected individuals compared with age-matched controls. Another study found higher expression levels of the Tau kinase CDK5 in patients with HIV encephalitis versus HIV-positive patients without neuroinvasive disease, which correlated with increased Tau hyperphosphorylation (Patrick et al., 2011). Furthermore, transgenic mice that express HIV-1 glycoprotein gp120 exhibited increased brain levels of CDK5, Tau hyperphosphorylation, and neurodegeneration, which could be rescued by either genetic knockdown or pharmacological inhibition of CDK5 (Patrick et al., 2011). Additionally, HIV-1 Tat protein was similarly found to induce hyperphosphorylation of Tau in neurons through the CDK5, resulting in accelerated NFT deposition in transgenic mice (Giunta et al., 2009). Collectively, these data indicate HIV-1 infection may induce AD pathology through several potential mechanisms.
Periodontitis has been associated with increased risk of developing AD as well as other dementias (Lee et al., 2017; Kim et al., 2020). Specifically, the bacteria P. gingivalis and its toxic proteases, called gingipains, were identified in the brains of AD patients, and their levels correlated with AD pathology (Dominy et al., 2019). Studies investigating the mechanism underlying this relationship have identified inflammatory processes, including cytokine expression and complement activation, as well as amyloid production as mediators of P. gingivalis pathogenesis (Costa et al., 2021). Murine models of P. gingivalis infection resulted in cognitive impairment in middle-aged (12 month), but not young (4 week) mice. Researchers attributed this to elevated production of proinflammatory cytokines including TNF-α, IL-6, and IL-1β in the brains of aged mice following infection (Ding et al., 2018). This was supported by additional studies in mice and primary cell cultures of microglia and hippocampal neurons, which indicated that the lysosomal protease Cathepsin B may be critical in initiating the neuroinflammatory response to repeated P. gingivalis lipopolysaccharide exposure (Wu et al., 2017). Following repeated oral application of P. gingivalis, the bacteria was detected in the hippocampus of infected mice, serving as more direct evidence of the role of P. gingivalis in AD pathology (Ilievski et al., 2018). This study also showed significantly elevated expression of inflammatory cytokines IL-6, TNF-α, and IL-1β, as well as APP and β-secretase, increased Tau phosphorylation, and neurodegeneration (Ilievski et al., 2018). Together, these data propose a mechanistic link between periodontal disease and AD pathology.
Other evidence suggests that common infectious agents may contribute to AD pathology by promoting the deposition of Tau and Aβ. C. pneumoniae is an obligate intracellular bacterium that takes residence in the nasal and pulmonary mucosa (Porritt and Crother, 2016). It has been proposed that C. pneumoniae invades the brain through the lateral entorhinal cortex, then disseminates to the frontal and temporal cortices, the same regions where Aβ plaques and NFTs are found (Hammond et al., 2010; Itzhaki et al., 2016). Subsequent in vitro studied demonstrated that infection of astrocytes with C. pneumoniae decreased activity of α-secretase and increased expression of both β-secretase and γ-secretase, yielding the aggregation-prone Aβ peptide (Al-Atrache et al., 2019). Similarly, HHV-6A, a neurovirulent pathogen, was shown to promote Aβ secretion along with Tau hyperphosphorylation in primary neurons and astrocytoma cells by reducing protein degradation via autophagy and activating the unfolded protein response (Romeo et al., 2020). Furthermore, HHV-6A infection of microglial cells in vitro induces a pro-inflammatory activation status, stimulates the production of Aβ peptides, and promotes Tau phosphorylation (Bortolotti et al., 2019). Human T-cell leukemia virus type 1 (HTLV-1) has also been shown to increase Tau phosphorylation via CDK5 and GSK-3β activation, which resulted in neurite retraction in a cell culture model (Maldonado et al., 2011). These studies suggest that many infectious agents can contribute to AD pathology, and it is likely that the composite infectious burden is more important than a single microbe.
Antimicrobial peptides are host-defense mechanisms that defend against infectious pathogens and have recently been hypothesized to initiate pathological processes that lead to neurodegeneration. Using C. elegans PVD neurons as a model, researchers showed that an epidermally-expressed antimicrobial peptide NLP-29 (neuropeptide-like protein 29) causes age-dependent dendrite degeneration and that fungal infections can induce degeneration through similar mechanisms (E et al., 2018). This NLP-29-induced degeneration could be similarly stimulated in primary cultured rat neurons, suggesting that this is an evolutionarily-conserved mechanism (E et al., 2018). A recent hypothesis posits Aβ may act as an antimicrobial peptide, providing innate immune defense against infection. Soscia et al. (2010) showed that synthetic Aβ exerts antimicrobial activity in vitro against eight common, clinically-relevant pathogens, including seven bacterial and one yeast species. Aβ also shows neutralizing activity against seasonal (H3N2) and pandemic (H1N1) strains of influenza A virus in vitro, inducing viral agglutination and preventing its infectivity in epithelial cells (White et al., 2014). Bourgade et al. (2015) showed that Aβ prevented entry of HSV-1 into fibroblast, epithelial, and neuronal cell cultures. They hypothesized that based on the sequence homology between Aβ and a proximal transmembrane region of HSV-1 glycoprotein B, Aβ may directly interfere with HSV-1 replication via insertion into the viral envelope (Bourgade et al., 2015). Kumar et al. (2016) extended these findings in vivo to mouse and nematode models of disease, demonstrating that Aβ oligomers bind the microbial cell wall of Salmonella typhimurium and Candida albicans to prevent adhesion to host cells and reduce S. typhimurium load in the brains of infected mice. They went on to show a similar effect with both HSV-1 and HHV-6A infection in a mouse model of AD, demonstrating that overexpression of Aβ in mice correlated with longer survival from HSE; however, all mice still succumbed to infection within 6 days, and authors provided no evidence of reduced viral burden in the brains Aβ overexpressing mice (Eimer et al., 2018). Altogether, these data suggest that Aβ may function in innate immunity against microbial infection. However, its role in agglutination may then seed additional amyloid deposition, initiating pathogenic plaque formation, causing persistent neuroinflammation, and ultimately, lead to neurodegeneration.
Parkinson’s Disease
PD is the second most common neurodegenerative disease, following AD, afflicting motor functions (Mhyre et al., 2012; Magrinelli et al., 2016). It is characterized by prominent dopaminergic neurodegeneration within the substantia nigra pars compacta region of the brain, which is caused by dopamine deficiency, and leads to motor neuron dysfunction (Alexander, 2004; Mhyre et al., 2012; Kalia and Lang, 2015). Patients with PD present with bradykinesia, resting tremors, gait impairment, diminished postural quality, and muscular rigidity (Antony et al., 2013; Kalia and Lang, 2015). Treatments for PD exist to manage symptoms or slow disease progression, but there is no cure. As the disease progresses, cognitive function declines and results in dementia (Aarsland et al., 2011; Kalia and Lang, 2015; Goldman et al., 2018). Though the mechanisms by which degeneration of dopaminergic neurons occurs are not fully understood, it is well established that the aggregation of misfolded α-synuclein protein in the form of Lewy bodies is a hallmark of the disease (Rocha et al., 2018). However, whether the α-synuclein aggregates themselves are neurotoxic or may be a protective mechanism to sequester the more neurotoxic protofibrils is still debated (Caughey and Lansbury, 2003). Yet another hypothesis posits that neurodegeneration is due to the loss of function of α-synuclein when it forms aggregates (Kanaan and Manfredsson, 2012). The physiological function of α-synuclein is not clear, but it appears to play an important role in dopamine biosynthesis and dopaminergic neurotransmission (Abeliovich et al., 2000; Perez et al., 2002; Liu et al., 2008). Genetic variants and post-translational modifications, including oxidation, nitration, and phosphorylation, influence the propensity of α-synuclein to aggregate; however, the physiological factors that incite these aggregation pathways are not well understood (Venda et al., 2010).
Genetic factors that cause or increase risk of developing PD include mutations in SNCA (encoding α-synuclein), PRKN, and DJ-1, among others (Hall et al., 2020). Interestingly, several of these genetic factors have been shown to contribute to immune defense against infectious agents. Polymorphisms in PRKN, a ubiquitin ligase, have been associated with increased susceptibility to intracellular pathogens, Mycobacterium leprae and Salmonella typhi (Mira et al., 2004; Ali et al., 2006). Recently PRKN was shown to limit replication of bacterial pathogens Mycobacterium tuberculosis and Listeria monocytogenes in both mice and flies by targeting them for ubiquitin-mediated autophagy (Manzanillo et al., 2013). Also, mice in which SNCA is deleted are more susceptible to West Nile virus and Venezuelan equine encephalitis, possibly by modulating ER stress signaling and thereby limiting viral replication (Beatman et al., 2016). In contrast, DJ-1 appears to negatively regulate the immune system. When DJ-1 was deleted in a mouse model of polymicrobial sepsis, mice showed improved survival and bacterial clearance. Authors showed this to be due to enhanced phagocytosis and bactericidal activity in DJ-1-deficient macrophages, adoptive transfer of which could rescue septic wildtype mice (Amatullah et al., 2016). Although genetic mutations account for only 5–15% of all PD cases (Hall et al., 2020), better understanding these genetic causes of disease have informed the pathophysiology of the more common sporadic disease cases.
Multiple environmental factors, including chemical exposure, lifestyle, and socioeconomic conditions impact the development of PD, and pathogenic infection is increasingly recognized as a possible risk factor for PD (Bu et al., 2015; Chen and Ritz, 2018). The infectious etiology hypothesis of PD was originally proposed following the presentation of PD-like symptoms in individuals infected with influenza (Maurizi, 1987). A 1963 cohort analysis identified a striking increase in PD incidence in Guam, which seemed to recede in patients born after 1920. Authors hypothesized that this transient increase in PD incidence may have been due to the influenza pandemic of 1918 (Maurizi, 1987). Another study identified three seemingly random clusters of early-onset PD patients in Canada, in which patients lacked typical genetic markers of early-onset disease (Kumar et al., 2004). This suggested that environmental factors may have increased risk of PD in these patients, and the authors hypothesized that viral infection or other toxic exposure may be an underlying cause for these clusters of disease (Kumar et al., 2004). A cohort study examining the antibody titers to common infectious pathogens found higher seropositivity to cytomegalovirus (CMV), Epstein Barr virus (EBV), HSV-1, Borrelia burdorferi, C. pneumoniae, and Helicobacter pylori in PD patients compared with healthy controls (Bu et al., 2015). A recent meta-analysis of cohort and case-controlled studies revealed that patients with H. pylori, C. pneumoniae, Hepatitis C virus (HCV), or Malassezia yeast may be at an increased risk of PD (Wang et al., 2020). While cohort studies cannot demonstrate that infections caused PD pathogenesis, together, they suggest that infection may be an important environmental risk factor for PD.
Certain viruses directly cause degeneration of dopaminergic neurons, which results in decreased dopamine availability in the CNS. Typically considered a hepatotropic virus, HCV has recently been observed to invade the CNS and disrupt dopaminergic neurotransmission, leading to neuronal death (Weissenborn et al., 2006; Abushouk et al., 2017; Wijarnpreecha et al., 2018). HCV patients are affected by neurological complications, including cognitive impairment and peripheral neuropathy (Mathew et al., 2016). HCV may gain entry to the CNS by interacting with receptors expressed by brain microvascular endothelial cells at the BBB, including CD68, CD81, and claudin-1 (Abushouk et al., 2017; Wijarnpreecha et al., 2018). Recent studies showed that once in the CNS, HCV activated resident microglia and astrocytes. This activation promoted a pro-inflammatory state through up-regulation of cytokines and chemokines, such as TNF-α and intracellular adhesion molecule-1 (ICAM-1), which caused significant damage to dopaminergic neurons (Lyons and Benveniste, 1998; Wu et al., 2015; Abushouk et al., 2017). Additionally, HCV infection was found to indirectly trigger neurotoxic effects seen in PD through IFN-α therapy. IFN-α treatment of HCV-infected murine models inhibited transmission through the nigrostriatal dopaminergic pathway, thereby reducing the levels of dihydroxyphenylacetic acid and dopamine present in the substantia nigra (Shuto et al., 1997). Furthermore, IFN-γ, which is transcriptionally upregulated in HCV-infected human brain microvascular endothelial cells (Liu et al., 2016), caused significant death of dopaminergic neurons in both in vitro murine microglia/neuron co-cultures and in vivo murine models (Mount et al., 2007). PD is generally characterized by chronic low-level systemic inflammation; however, individuals with higher infectious burden have higher levels of circulating inflammatory cytokines, including IL-1β and IL-6 (Bu et al., 2015). PD patients infected with another Herpesviridae virus, CMV, have higher frequencies of circulating pro-inflammatory myeloid dendritic cells compared with CMV-positive subjects without PD (Goldeck et al., 2016). Furthermore, when HIV-1 becomes neuroinvasive, it shows specific affinity for dopaminergic regions, including the basal ganglia, resulting in their degeneration, decreased availability of dopamine, and the development of dementia associated with acquired immunodeficiency syndrome (AIDS) (Nath et al., 2000; Kumar et al., 2009, 2011). In a mouse model of disease, Theiler’s murine encephalomyelitis virus (TMEV) was stereotaxically inoculated into the substantia nigra. TMEV specifically infected dopaminergic neurons with minimal infection or destruction to surrounding brain regions (Oliver et al., 1997). Japanese encephalitis virus (JEV) is recognized to not only target dopaminergic neuron-rich brain regions, but can also selectively manipulate dopamine signaling to increase the cell surface expression of the molecules it uses to infect the cell (Simanjuntak et al., 2017). These studies indicate that certain viruses can specifically impact populations of neurons that can lead to neurodegeneration of dopaminergic neurons directly.
Although the CNS is a primary focus of PD research, pathophysiology affects all levels of the brain-gut axis, including the autonomic and enteric nervous systems. Mulak and Bonaz (2015) recently hypothesized that α-synuclein aggregates initiate in the gut and proceed to spread along the brain-gut axis to the CNS, resulting in the motor and neuronal deficits characteristic of PD. One pathogen hypothesized to incite α-synuclein aggregation in the gut is EBV. The C-terminal region of α-synuclein is molecularly similar to a repeat region of the latent membrane protein 1, encoded by EBV (Woulfe et al., 2014). This led to their hypothesis that in genetically susceptible individuals, antibodies to the critical repeat region of the EBV latent membrane protein may cross-react with the homologous epitope on α-synuclein and induce α-synuclein oligomerization (Woulfe et al., 2014). Following this initial aggregation event, α-synuclein oligomers may spread trans-neuronally to the CNS, causing PD neuropathology, as initially proposed by Braak and Del Tredici (2016). In support of the brain-gut trans-neuronal hypothesis, researchers showed that when pre-formed α-synuclein fibrils were injected into the duodenal and pyloric muscularis layers of a mouse model, phosphorylated α-synuclein spread to regions of the CNS affected by PD, such as the locus coeruleus and substantia nigra pars compacta (Kim et al., 2019).
Another gastric microbe that is associated with increased risk of PD is the bacteria H. pylori (Bu et al., 2015; Huang et al., 2018). H. pylori is found in the intestinal endothelium and afflicts individuals with peptic ulcers, gastritis, gastric adenocarcinoma formation, and mucosal inflammation (Camilo et al., 2017). Previous studies have linked H. pylori to extra-gastrointestinal diseases, such as ischemic heart disease and neurodegenerative diseases, including AD and PD (Dobbs et al., 2000, 2012; Tan et al., 2015; Huang et al., 2018; McGee et al., 2018). A Danish population-based study found that prescriptions for H. pylori eradication treatments and proton pump inhibitors were associated with an increased risk of PD diagnosis 5 or more years later, suggesting either that chronic H. pylori infection may contribute to PD etiopathogenesis or gastritis symptoms may precede pathognomonic PD symptoms (Nielsen et al., 2012). The mechanism underlying the role of H. pylori in PD onset is not well understood; however, the benefit of treating infections in PD patients is well-documented. A prospective cohort study found that H. pylori-IgG positivity in PD patients was associated with higher daily dose of levodopa and more severe symptoms compared with H. pylori-negative patients, and were improved after H. pylori eradication treatment (Mridula et al., 2017). Several studies have shown that eradicating H. pylori infection improved motor function of PD patients by increasing oral drug absorption (Pierantozzi et al., 2006; Lahner et al., 2009). A recent cohort study showed that PD patients with successful H. pylori eradication therapy exhibited improved clinical PD symptoms, including tremors, mood, and gastrointestinal distress, compared with patients with failed H. pylori eradication therapy (Lolekha et al., 2021). Patients with active H. pylori infection had longer mean levodopa onset time, suggesting that H. pylori may interfere with the bioavailability of levodopa, possibly because of increased gastric inflammation, delayed gastric emptying, and/or impaired active transport of levodopa to the site of absorption (Lolekha et al., 2021). Though much is still unclear of the involvement of H. pylori in the etiopathogenesis of PD, these data indicate that it is prevalent in PD patients and may exacerbate the symptoms of PD by interfering with levodopa bioavailability.
The global pandemic caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and its resulting disease (COVID-19) emerged as an unprecedented worldwide healthcare crisis. In the flood of viral pneumonia and the overwhelming challenges to the healthcare systems, researchers are just beginning to understand the extent to which patients develop acute or chronic neurologic manifestations. It was reported early in the pandemic that 36% of COVID-19 patients develop neurologic symptoms, but whether these were due to CNS infection, systemic inflammatory response, or intensive care unit delirium was unknown (Mao et al., 2020). More recently, a neuropathological study found evidence of viral RNA and/or protein in the brains of 53% of autopsied COVID-19 patients; however, it is important to highlight that this study analyzed only patients who died, and thus, results are probably not generalizable to less severe cases of infection (Matschke et al., 2020). In fact, a systematic search of the literature revealed that in COVID-19 patients, SARS-CoV-2 RNA was detected in only 6.4% of those who underwent cerebrospinal fluid (CSF) PCR testing, which is likely still not representative of patients with mild infection (Farooq et al., 2021; Li et al., 2021). Nonetheless, autopsies of COVID-19 patients revealed uniform presentation of neuroimmune pathology, including microglial activation and cytotoxic T lymphocyte infiltration in the brainstem and cerebellum. This pathology was independent of the detection of virus in the brain, suggesting that CNS damage and neurological symptoms may be due to cytokine storm and neuroimmune response rather than direct viral infection (Matschke et al., 2020). Considering the importance of the cerebellum and brainstem in coordinating voluntary movement, gait, posture, and motor functions, the localization of immune cell infiltration and activation may be of particular significance to the Parkinsonian symptoms seen in some post-infectious COVID-19 patients (Cohen et al., 2020; Faber et al., 2020; Méndez-Guerrero et al., 2020). Post-encephalitic parkinsonism has been reported previously for other viruses, but whether these symptoms constitute bona fide PD is unclear (Casals et al., 1998). The three case reports describing parkinsonism following COVID-19 exhibited impaired dopaminergic nigrostriatal function, but this is not necessarily diagnostic of PD (Merello et al., 2021). Rather, these may represent a transient syndrome that eventually resolves spontaneously instead of the progressive neurodegeneration of PD (Dickson, 2012). Alternatively, it is possible that SARS-CoV-2 unmasked previously preclinical PD (Merello et al., 2021). However, given the high rate of SARS-CoV-2 infection, especially in the vulnerable aging population, the potential for developing post-infectious PD is of particular concern.
Amyotrophic Lateral Sclerosis
ALS is a neurodegenerative disease that is characterized by the loss of upper and lower motor neurons. The decrease in motor function starts as muscle weakness in the limbs and progresses to eventual paralysis of all muscular motor movements in the body (Zarei et al., 2015). Eventually, the motor neuron degeneration prevents proper functioning of the diaphragm, disrupting the proper respiratory function needed to survive (Czapliñski et al., 2003). As there are currently no pathognomonic tests for ALS, diagnosis relies on the identification of concomitant progressive upper and lower motor neuron dysfunction and the exclusion of mimicking conditions (van den Bos et al., 2019). Further complicating ALS diagnosis, is the existence of “ALS-like syndrome,” which refers to a heterogenous group of conditions in which their clinical presentation is similar to ALS (i.e., motor neuron dysfunction), but in many cases, the underlying cause of these symptoms is treatable (Ghasemi, 2016). For example, patients with HIV-1 infection presenting with ALS-like syndrome that were treated with antiretroviral therapy showed partial recovery of their motor deficit (Verma and Berger, 2006). In published reports, ALS-like syndromes cannot always be distinguished from bona fide ALS, so for the purpose of this review, we do not attempt to separate the two conditions.
There is emerging data that suggests infectious agents, including viruses and fungi, may be associated with ALS. Enteroviruses have long been suspected to play a role in ALS due to their ability to infect motor neurons in the CNS and cause meningitis and encephalitis (Ravits, 2005). However, clinical data connecting enterovirus infection and ALS have been inconclusive. Several studies have identified enterovirus RNA in spinal cord tissue of 70–80% of ALS patients (Woodall et al., 1994; Berger et al., 2000); however, others have found no detectable viral RNA in ALS patients (Swanson et al., 1995; Nix et al., 2004). Therefore, further investigation into the role of enteroviruses in ALS is necessary in order to clarify this relationship. Other recent studies have identified DNA from several Candida spp. of fungi, as well as fungal antigens in the CSF and brain tissue of ALS patients (Alonso et al., 2015). This, coupled with the detection of fungal hyphae within the motor cortex and spinal cord of ALS patients (Alonso et al., 2017), supports the idea that infection may contribute to or exacerbate ALS pathology. Numerous cellular dysfunctions associated with ALS are impacted by infectious agents, including protein aggregation and mislocalization, and glutamate excitotoxicity (Van Damme et al., 2017). Better understanding of how infectious agents may contribute to these cellular mechanisms that lead to motor neuron deficit will improve our understanding of the progressive neurodegeneration associated with ALS.
The presence of ubiquitinated protein aggregates in affected motor neurons is a central hallmark of disease; however, the composition of those aggregates varies among ALS patients (Blokhuis et al., 2013). The molecular characteristics and distribution of these protein aggregates, in many cases, are linked to the genetic mutations that cause the disease. However, proteinaceous aggregates of similar composition are also detected in non-mutation carriers, indicating a convergence of underlying cellular and pathological processes in both familial and sporadic cases of ALS (Blokhuis et al., 2013). The identification of ALS-associated mutations in two DNA/RNA binding proteins, TAR DNA-binding protein 43 (TDP-43) and protein fused in sarcoma (FUS), also implicate alterations in RNA processing as a key event in ALS pathogenesis (Lagier-Tourenne and Cleveland, 2009). Furthermore, mutations in TDP-43 lead to misfolded and truncated proteins, such as TDP-25 and TDP-35, as well as mislocalization from the nucleus to the cytoplasm (Prasad et al., 2019). Because the translocation of TDP-43 from the nucleus to the cytoplasm is tightly linked to the formation of pathological aggregates in the cytoplasm, it is difficult to decouple the consequences of its loss of function as a DNA-binding protein in the nucleus from the potentially toxic gain of function effects of the aggregates in the cytoplasm (Suk and Rousseaux, 2020). However, a similar cytoplasmic translocation occurs during HIV and fungal infection (Douville and Nath, 2017; French et al., 2019a). It is hypothesized that the release of neurotoxins, such as ochratoxin A, during fungal infection causes TDP-43 to mislocalize to the cytoplasm, leading to ALS pathogenesis (French et al., 2019b). The overall structure of TDP-43, along with its propensity to aggregate and mislocalize, is further influenced by post-translational modifications (François-Moutal et al., 2019). It has been demonstrated that infection with TMEV, both in vitro and in vivo, caused TDP-43 phosphorylation and cleavage, resulting in its cytoplasmic mislocalization and aggregation (Masaki et al., 2019). These data indicate that viral and fungal infections promote the neuropathology associated with ALS.
Interestingly, the relationship between viral infection and TDP-43 aggregation may be reciprocal in nature, as TDP-43 aggregation may enhance expression of endogenous retroviruses in the CNS. In autopsied samples of cortical and spinal neurons from ALS patients, the transcriptional expression of human endogenous retrovirus-K (HERV-K) polymerase was enhanced (Douville et al., 2011). Furthermore, in patients with sporadic ALS, HERV-K reverse transcriptase expression was correlated with TDP-43 and HERV-K long terminal repeats have four binding sites for TDP-43, which have been shown to regulate its activation (Li et al., 2015). In a Drosophila model of disease, focal glial expression of human TDP-43 triggered gypsy-ERV replication, as well as DNA damage, and neuronal apoptosis (Chang and Dubnau, 2019). Additionally, TDP-43 harbors binding sites for interferon regulatory factors (IRF) and NF-κB, which are important inflammatory mediators, causing TDP-43 to become transcriptionally upregulated in response to antiviral interferon expression (Douville et al., 2011). Together, these data suggest that HERV-K expression may be driven, in part, by TDP-43 as well as in response to local neuroinflammation (Douville et al., 2011; Li et al., 2015; Douville and Nath, 2017). In fact, TDP-43 was originally found to inhibit HIV transcription in cell culture (Ou et al., 1995), though this function is still debated and may reflect differences in cell types and model systems (Nehls et al., 2014; Manghera et al., 2016; Krug et al., 2017; Prudencio et al., 2017). Together, these studies indicate that TDP-43 aggregation and infectious agents may develop a reciprocal relationship in causing pathogenic changes that lead to ALS.
A second DNA/RNA binding protein that has been associated with familial ALS is FUS, which, when mutated, interferes with RNA metabolism, suppresses protein translation, and decreases the nonsense-mediated decay pathway (Kamelgarn et al., 2018). ALS-associated genetic mutations result in the formation of stress granules, which are composed, in part, of RNA-binding proteins, including TDP-43 and FUS (Zhang et al., 2020). The formation of FUS-containing stress granules can be stimulated by respiratory syncytial virus (RSV), as well as by poly(I:C), which is used by laboratory researchers to mimic viral double-stranded RNA (Shelkovnikova et al., 2019). It was also found that infection of induced pluripotent stem cell (iPSC)-derived spinal neurons with either rabies virus (RABV) or HIV-1 increased the production of FUS and promoted its cytoplasmic mislocalization (Bellmann et al., 2019). Furthermore, many other viruses have been shown to promote the formation of stress granules (White and Lloyd, 2012; McCormick and Khaperskyy, 2017). These studies demonstrate a link between viral infection and a key neuropathogenic hallmark of ALS.
Glutamate is a major excitatory neurotransmitter in the brain; however, excessive stimulation due to increased glutamate receptor expression or ligand availability can cause excitotoxicity and lead to neuronal death (Wang et al., 2003; Foran and Trotti, 2009). Perisynaptic astrocytes express glutamate transporters, including excitatory amino acid transporter 2 (EAAT2) and glutamate transporter-1 (GLT-1), which clear glutamate from neuronal synapses (Sheldon and Robinson, 2007). Defects in glutamate transport have been found in synaptosomes prepared from neural tissue from ALS patients (Rothstein et al., 1992). This is likely due to a combined effect of upregulation of genes that transcribe glutamate receptors in the motor cortex of ALS patients and selective loss of glutamate transporters in the motor cortex of ALS patients (Rothstein et al., 1995; Wang et al., 2006). In transgenic mice expressing mutant SOD1, GLT-1 was found to decrease in accordance with disease progression and survival could be extended by increasing expression of EAAT2 (Bruijn et al., 1997; Rosenblum et al., 2017). During viral infection, exposure of fetal human astrocytes in vitro to the HIV-1 envelope glycoprotein, gp120, caused a 40–70% decline in steady-state levels of EAAT2 RNA (Wang et al., 2003). This resulted in reduced glutamate transport and may contribute to glutamate excitotoxicity following HIV-1 infection (Wang et al., 2003). Furthermore, exposure of neurons to fungal neurotoxins caused the spontaneous release of endogenous glutamate (Bradford et al., 1990), and elevated glutamate levels have been shown to increase the toxicity associated with SOD1, as well as to promote TDP-43 translocation (Roy et al., 1998; Scofield et al., 2012). Moreover, EAAT2 expression is downregulated by TNF-α, an important cytokine involved in the antiviral response to many viruses including HIV-1, VZV, EBV, and CMV, among others (Sitcheran et al., 2005; Kim and Solomon, 2010). Thus, this excitotoxic impact from glutamate is likely common among many viral infections. Together, these data indicate that infectious diseases cause changes in glutamate signaling that can lead to excitotoxicity that is symptomatic of ALS.
Conclusion and Future Directions
Here we have reviewed recent literature linking microbial infections to neurodegenerative diseases, including AD, PD, and ALS. Although epidemiological data indicate an association between infectious agents and neurodegenerative diseases, in many cases the molecular and cellular mechanisms underlying those associations are unclear. Alternatively, patients with neurodegenerative diseases may be at increased risk of being infected with a neurotropic agent, potentially due to compromised immune systems and/or leaky BBB in affected individuals. Further research using in vitro and in vivo models will help elucidate whether infectious agents increase the risk of developing neurodegenerative diseases on their own, via anti-microbial inflammatory pathways, or other unknown mechanisms. The study of model systems, including both rodent and non-rodent models, will also improve our understanding of post-infectious neurologic and cognitive dysfunction that occurs following many systemic and neurotropic infections beyond the common neurodegenerative diseases reviewed here. Identification of molecular mechanisms common among these neurologic disorders may lead to new diagnostic biomarkers to identify individuals that may develop progressive neurocognitive or neurodegenerative diseases, as well as new therapeutic options for them.
Author Contributions
SL, BB, and KF wrote the manuscript. KR and SL generated the figure and table. All authors edited the manuscript.
Funding
This work was supported by the University of North Carolina at Charlotte, Department of Biological Sciences Start-up funds and by the National Institutes of Health grant K99/R00 AG053412 to KF.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aarsland, D., Brønnick, K., and Fladby, T. (2011). Mild cognitive impairment in Parkinson’s disease. Curr. Neurol. Neurosci. Rep. 11, 371–378. doi: 10.1007/s11910-011-0203-1
Abeliovich, A., Schmitz, Y., Fariñas, I., Choi-Lundberg, D., Ho, W. H., Castillo, P. E., et al. (2000). Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 25, 239–252. doi: 10.1016/s0896-6273(00)80886-7
Abushouk, A. I., El-Husseny, M. W. A., Magdy, M., Ismail, A., Attia, A., Ahmed, H., et al. (2017). Evidence for association between hepatitis C virus and Parkinson’s disease. Neurol. Sci. 38, 1913–1920. doi: 10.1007/s10072-017-3077-4
Aksenov, M. Y., Aksenova, M. V., Mactutus, C. F., and Booze, R. M. (2010). HIV-1 protein-mediated amyloidogenesis in rat hippocampal cell cultures. Neurosci. Lett. 475, 174–178. doi: 10.1016/j.neulet.2010.03.073
Al-Atrache, Z., Lopez, D. B., Hingley, S. T., and Appelt, D. M. (2019). Astrocytes infected with Chlamydia pneumoniae demonstrate altered expression and activity of secretases involved in the generation of β-amyloid found in Alzheimer disease. BMC Neurosci. 20:6. doi: 10.1186/s12868-019-0489-5
Alexander, G. E. (2004). Biology of Parkinson’s disease: pathogenesis and pathophysiology of a multisystem neurodegenerative disorder. Dialog. Clin. Neurosci. 6, 259–280.
Ali, S., Vollaard, A. M., Widjaja, S., Surjadi, C., van de Vosse, E., and van Dissel, J. T. (2006). PARK2/PACRG polymorphisms and susceptibility to typhoid and paratyphoid fever. Clin. Exp. Immunol. 144, 425–431. doi: 10.1111/j.1365-2249.2006.03087.x
Alonso, R., Pisa, D., Fernández-Fernández, A. M., Rábano, A., and Carrasco, L. (2017). Fungal infection in neural tissue of patients with amyotrophic lateral sclerosis. Neurobiol. Dis. 108, 249–260. doi: 10.1016/j.nbd.2017.09.001
Alonso, R., Pisa, D., Marina, A. I., Morato, E., Rábano, A., Rodal, I., et al. (2015). Evidence for fungal infection in cerebrospinal fluid and brain tissue from patients with amyotrophic lateral sclerosis. Int. J. Biol. Sci. 11, 546–558. doi: 10.7150/ijbs.11084
Amatullah, H., Shan, Y., Beauchamp, B. L., Gali, P. L., Gupta, S., Maron-Gutierrez, T., et al. (2016). DJ-1/PARK7 impairs bacterial clearance in sepsis. Am. J. Respir. Crit. Care Med. 195, 889–905. doi: 10.1164/rccm.201604-0730OC
András, I. E., Eum, S. Y., Huang, W., Zhong, Y., Hennig, B., and Toborek, M. (2010). HIV-1-induced amyloid beta accumulation in brain endothelial cells is attenuated by simvastatin. Mol. Cell. Neurosci. 43, 232–243. doi: 10.1016/j.mcn.2009.11.004
András, I. E., Garcia-Contreras, M., Yanick, C., Perez, P., Sewell, B., Durand, L., et al. (2020). Extracellular vesicle-mediated amyloid transfer to neural progenitor cells: implications for RAGE and HIV infection. Mol. Brain 13:21. doi: 10.1186/s13041-020-0562-0
András, I. E., Leda, A., Contreras, M. G., Bertrand, L., Park, M., Skowronska, M., et al. (2017). Extracellular vesicles of the blood-brain barrier: role in the HIV-1 associated amyloid beta pathology. Mol. Cell. Neurosci. 79, 12–22. doi: 10.1016/j.mcn.2016.12.006
Anthony, I. C., Norrby, K. E., Dingwall, T., Carnie, F. W., Millar, T., Arango, J. C., et al. (2010). Predisposition to accelerated Alzheimer-related changes in the brains of human immunodeficiency virus negative opiate abusers. Brain J. Neurol. 133, 3685–3698. doi: 10.1093/brain/awq263
Anthony, I. C., Ramage, S. N., Carnie, F. W., Simmonds, P., and Bell, J. E. (2006). Accelerated Tau deposition in the brains of individuals infected with human immunodeficiency virus-1 before and after the advent of highly active anti-retroviral therapy. Acta Neuropathol. (Berl.) 111, 529–538. doi: 10.1007/s00401-006-0037-0
Antinori, A., Arendt, G., Becker, J. T., Brew, B. J., Byrd, D. A., Cherner, M., et al. (2007). Updated research nosology for HIV-associated neurocognitive disorders. Neurology 69, 1789–1799. doi: 10.1212/01.WNL.0000287431.88658.8b
Antony, P. M. A., Diederich, N. J., Krüger, R., and Balling, R. (2013). The hallmarks of Parkinson’s disease. FEBS J. 280, 5981–5993. doi: 10.1111/febs.12335
Balin, B. J., Gérard, H. C., Arking, E. J., Appelt, D. M., Branigan, P. J., Abrams, J. T., et al. (1998). Identification and localization of Chlamydia pneumoniae in the Alzheimer’s brain. Med. Microbiol. Immunol. (Berl.) 187, 23–42. doi: 10.1007/s004300050071
Ball, M. J., Nuttall, K., and Warren, K. G. (1982). Neuronal and lymphocytic populations in human trigeminal ganglia: implications for ageing and for latent virus. Neuropathol. Appl. Neurobiol. 8, 177–187. doi: 10.1111/j.1365-2990.1982.tb00273.x
Bardi, G., Sengupta, R., Khan, M. Z., Patel, J. P., and Meucci, O. (2006). Human immunodeficiency virus gp120-induced apoptosis of human neuroblastoma cells in the absence of CXCR4 internalization. J. Neurovirol. 12, 211–218. doi: 10.1080/13550280600848373
Beatman, E. L., Massey, A., Shives, K. D., Burrack, K. S., Chamanian, M., Morrison, T. E., et al. (2016). Alpha-Synuclein expression restricts RNA Viral infections in the brain. J. Virol. 90, 2767–2782. doi: 10.1128/JVI.02949-15
Bellmann, J., Monette, A., Tripathy, V., Sójka, A., Abo-Rady, M., Janosh, A., et al. (2019). Viral infections exacerbate FUS-ALS phenotypes in iPSC-derived spinal neurons in a virus species-specific manner. Front. Cell. Neurosci. 13:480. doi: 10.3389/fncel.2019.00480
Bendorius, M., Po, C., Muller, S., and Jeltsch-David, H. (2018). From systemic inflammation to neuroinflammation: the case of neurolupus. Int. J. Mol. Sci. 19:3588. doi: 10.3390/ijms19113588
Berger, M. M., Kopp, N., Vital, C., Redl, B., Aymard, M., and Lina, B. (2000). Detection and cellular localization of enterovirus RNA sequences in spinal cord of patients with ALS. Neurology 54, 20–25. doi: 10.1212/wnl.54.1.20
Berth, S. H., Mesnard-Hoaglin, N., Wang, B., Kim, H., Song, Y., Sapar, M., et al. (2016). HIV glycoprotein Gp120 impairs fast axonal transport by activating Tak1 signaling pathways. ASN Neuro. 8, 1–18. doi: 10.1177/1759091416679073
Blasko, I., Veerhuis, R., Stampfer-Kountchev, M., Saurwein-Teissl, M., Eikelenboom, P., and Grubeck-Loebenstein, B. (2000). Costimulatory effects of interferon-gamma and interleukin-1beta or tumor necrosis factor alpha on the synthesis of Abeta1-40 and Abeta1-42 by human astrocytes. Neurobiol. Dis. 7, 682–689. doi: 10.1006/nbdi.2000.0321
Blokhuis, A. M., Groen, E. J. N., Koppers, M., van den Berg, L. H., and Pasterkamp, R. J. (2013). Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathol. (Berl.) 125, 777–794. doi: 10.1007/s00401-013-1125-6
Bongioanni, P., Boccardi, B., Borgna, M., Castagna, M., and Mondino, C. (1997). T-cell interferon gamma binding in patients with dementia of the Alzheimer type. Arch. Neurol. 54, 457–462. doi: 10.1001/archneur.1997.00550160085021
Bortolotti, D., Gentili, V., Rotola, A., Caselli, E., and Rizzo, R. (2019). HHV-6A infection induces amyloid-beta expression and activation of microglial cells. Alzheimers Res. Ther. 11:104. doi: 10.1186/s13195-019-0552-6
Bourgade, K., Garneau, H., Giroux, G., Le Page, A. Y., Bocti, C., Dupuis, G., et al. (2015). β-Amyloid peptides display protective activity against the human Alzheimer’s disease-associated herpes simplex virus-1. Biogerontology 16, 85–98. doi: 10.1007/s10522-014-9538-8
Braak, H., and Del Tredici, K. (2016). Potential pathways of abnormal tau and α-synuclein dissemination in sporadic Alzheimer’s and Parkinson’s diseases. Cold Spring Harb. Perspect. Biol. 8:a023630. doi: 10.1101/cshperspect.a023630
Bradford, H. F., Norris, P. J., and Smith, C. C. (1990). Changes in transmitter release patterns in vitro induced by tremorgenic mycotoxins. J. Environ. Pathol. Toxicol. Oncol. 10, 17–30.
Bruijn, L. I., Becher, M. W., Lee, M. K., Anderson, K. L., Jenkins, N. A., Copeland, N. G., et al. (1997). ALS-Linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron 18, 327–338. doi: 10.1016/S0896-6273(00)80272-X
Bu, X.-L., Wang, X., Xiang, Y., Shen, L.-L., Wang, Q.-H., Liu, Y.-H., et al. (2015). The association between infectious burden and Parkinson’s disease: a case-control study. Parkinsonism Relat. Disord. 21, 877–881. doi: 10.1016/j.parkreldis.2015.05.015
Bu, X.-L., Yao, X.-Q., Jiao, S.-S., Zeng, F., Liu, Y.-H., Xiang, Y., et al. (2014). A study on the association between infectious burden and Alzheimer’s disease. Eur. J. Neurol. 22, 1519–1525. doi: 10.1111/ene.12477
Burberry, A., Suzuki, N., Wang, J.-Y., Moccia, R., Mordes, D. A., Stewart, M. H., et al. (2016). Loss-of-function mutations in the C9ORF72 mouse ortholog cause fatal autoimmune disease. Sci. Transl. Med. 8:347ra93. doi: 10.1126/scitranslmed.aaf6038
Camilo, V., Sugiyama, T., and Touati, E. (2017). Pathogenesis of Helicobacter pylori infection. Helicobacter 22(Suppl. 1):e12405. doi: 10.1111/hel.12405
Casals, J., Elizan, T. S., and Yahr, M. D. (1998). Postencephalitic parkinsonism–a review. J. Neural Transm. (Vienna) 105, 645–676. doi: 10.1007/s007020050086
Catani, M. V., Corasaniti, M. T., Navarra, M., Nisticò, G., Finazzi-Agrò, A., and Melino, G. (2000). gp120 induces cell death in human neuroblastoma cells through the CXCR4 and CCR5 chemokine receptors. J. Neurochem. 74, 2373–2379. doi: 10.1046/j.1471-4159.2000.0742373.x
Caughey, B., and Lansbury, P. T. (2003). Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu. Rev. Neurosci. 26, 267–298. doi: 10.1146/annurev.neuro.26.010302.081142
Chakrabarty, P., Ceballos-Diaz, C., Beccard, A., Janus, C., Dickson, D., Golde, T. E., et al. (2010). IFN-gamma promotes complement expression and attenuates amyloid plaque deposition in amyloid beta precursor protein transgenic mice. J. Immunol. 184, 5333–5343. doi: 10.4049/jimmunol.0903382
Chang, Y.-H., and Dubnau, J. (2019). The gypsy endogenous retrovirus drives non-cell-autonomous propagation in a Drosophila TDP-43 Model of Neurodegeneration. Curr. Biol. 29, 3135–3152.e4. doi: 10.1016/j.cub.2019.07.071
Chen, H., and Ritz, B. (2018). The search for environmental causes of Parkinson’s disease: moving forward. J. Park. Dis. 8, S9–S17. doi: 10.3233/JPD-181493
Chen, L., Deng, H., Cui, H., Fang, J., Zuo, Z., Deng, J., et al. (2018). Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 9, 7204–7218. doi: 10.18632/oncotarget.23208
Chow, V. W., Mattson, M. P., Wong, P. C., and Gleichmann, M. (2010). An overview of app processing enzymes and products. NeuroMolecular Med. 12, 1–12. doi: 10.1007/s12017-009-8104-z
Cohen, M. E., Eichel, R., Steiner-Birmanns, B., Janah, A., Ioshpa, M., Bar-Shalom, R., et al. (2020). A case of probable Parkinson’s disease after SARS-CoV-2 infection. Lancet Neurol. 19, 804–805. doi: 10.1016/S1474-4422(20)30305-7
Costa, M. J. F., de Araújo, I. D. T., da Rocha Alves, L., da Silva, R. L., Dos Santos Calderon, P., Borges, B. C. D., et al. (2021). Relationship of Porphyromonas gingivalis and Alzheimer’s disease: a systematic review of pre-clinical studies. Clin. Oral Investig. 25, 797–806. doi: 10.1007/s00784-020-03764-w
Cunningham, C. (2013). Microglia and neurodegeneration: the role of systemic inflammation. Glia 61, 71–90. doi: 10.1002/glia.22350
Czapliñski, A., Strobel, W., Gobbi, C., Steck, A. J., Fuhr, P., and Leppert, D. (2003). Respiratory failure due to bilateral diaphragm palsy as an early manifestation of ALS. Med. Sci. Monit. 9, CS34–CS36.
Deane, R., Du Yan, S., Submamaryan, R. K., LaRue, B., Jovanovic, S., Hogg, E., et al. (2003). RAGE mediates amyloid-β peptide transport across the blood-brain barrier and accumulation in brain. Nat. Med. 9, 907–913. doi: 10.1038/nm890
Dickson, D. W. (2012). Parkinson’s disease and Parkinsonism: neuropathology. Cold Spring Harb. Perspect. Med. 2:a009258. doi: 10.1101/cshperspect.a009258
Ding, Y., Ren, J., Yu, H., Yu, W., and Zhou, Y. (2018). Porphyromonas gingivalis, a periodontitis causing bacterium, induces memory impairment and age-dependent neuroinflammation in mice. Immun. Ageing 15:6. doi: 10.1186/s12979-017-0110-7
Dobbs, R., Charlett, A., Dobbs, S. M., Weller, C., A Ibrahim, M. A., Iguodala, O., et al. (2012). Leukocyte-subset counts in idiopathic parkinsonism provide clues to a pathogenic pathway involving small intestinal bacterial overgrowth. A surveillance study. Gut Pathog. 4:12. doi: 10.1186/1757-4749-4-12
Dobbs, R. J., Charlett, A., Dobbs, S. M., Weller, C., and Peterson, D. W. (2000). Parkinsonism: differential age-trend in Helicobacter pylori antibody: helicobacter pylori antibody. Aliment. Pharmacol. Ther. 14, 1199–1205. doi: 10.1046/j.1365-2036.2000.00815.x
Dominy, S. S., Lynch, C., Ermini, F., Benedyk, M., Marczyk, A., Konradi, A., et al. (2019). Porphyromonas gingivalis in Alzheimer’s disease brains: evidence for disease causation and treatment with small-molecule inhibitors. Sci. Adv. 5:eaau3333. doi: 10.1126/sciadv.aau3333
Douville, R., Liu, J., Rothstein, J., and Nath, A. (2011). Identification of active loci of a human endogenous retrovirus in neurons of patients with amyotrophic lateral sclerosis. Ann. Neurol. 69, 141–151. doi: 10.1002/ana.22149
Douville, R. N., and Nath, A. (2017). Human endogenous retrovirus-K and TDP-43 expression bridges ALS and HIV neuropathology. Front. Microbiol. 8:1986. doi: 10.3389/fmicb.2017.01986
Dzamko, N., Geczy, C. L., and Halliday, G. M. (2015). Inflammation is genetically implicated in Parkinson’s disease. Neuroscience 302, 89–102. doi: 10.1016/j.neuroscience.2014.10.028
E, L., Zhou, T., Koh, S., Chuang, M., Sharma, R., Pujol, N., et al. (2018). An antimicrobial peptide and its neuronal receptor regulate dendrite degeneration in aging and infection. Neuron 97, 125–138.e5. doi: 10.1016/j.neuron.2017.12.001
Eimer, W. A., Vijaya Kumar, D. K., Navalpur Shanmugam, N. K., Rodriguez, A. S., Mitchell, T., Washicosky, K. J., et al. (2018). Alzheimer’s disease-associated β-amyloid is rapidly seeded by herpesviridae to protect against brain infection. Neuron 99, 56–63.e3. doi: 10.1016/j.neuron.2018.06.030
El-Amouri, S. S., Zhu, H., Yu, J., Marr, R., Verma, I. M., and Kindy, M. S. (2008). Neprilysin: an enzyme candidate to slow the progression of Alzheimer’s disease. Am. J. Pathol. 172, 1342–1354. doi: 10.2353/ajpath.2008.070620
Faber, I., Brandão, P. R. P., Menegatti, F., de Carvalho Bispo, D. D., Maluf, F. B., and Cardoso, F. (2020). Coronavirus disease 2019 and parkinsonism: a non-post-encephalitic case. Mov. Disord. 35, 1721–1722. doi: 10.1002/mds.28277
Fan, L., Mao, C., Hu, X., Zhang, S., Yang, Z., Hu, Z., et al. (2019). New insights into the pathogenesis of Alzheimer’s disease. Front. Neurol. 10:1312. doi: 10.3389/fneur.2019.01312
Farooq, H. Z., Davies, E., Brown, B., Whitfield, T., Tilston, P., McEwan, A., et al. (2021). Real-world SARS CoV-2 testing in Northern England during the first wave of the COVID-19 pandemic. J. Infect. doi: 10.1016/j.jinf.2021.04.013 [Epub ahead of print].
Foran, E., and Trotti, D. (2009). Glutamate transporters and the excitotoxic path to motor neuron degeneration in amyotrophic lateral sclerosis. Antioxid. Redox Signal. 11, 1587–1602. doi: 10.1089/ars.2009.2444
François-Moutal, L., Perez-Miller, S., Scott, D. D., Miranda, V. G., Mollasalehi, N., and Khanna, M. (2019). Structural Insights Into TDP-43 and effects of post-translational modifications. Front. Mol. Neurosci. 12:301. doi: 10.3389/fnmol.2019.00301
French, P. W., Ludowyke, R., and Guillemin, G. J. (2019a). Fungal neurotoxins and sporadic amyotrophic lateral sclerosis. Neurotox. Res. 35, 969–980. doi: 10.1007/s12640-018-9980-5
French, P. W., Ludowyke, R. I., and Guillemin, G. J. (2019b). Fungal-contaminated grass and well water and sporadic amyotrophic lateral sclerosis. Neural Regen. Res. 14, 1490–1493. doi: 10.4103/1673-5374.255959
Fulop, T., Witkowski, J. M., Bourgade, K., Khalil, A., Zerif, E., Larbi, A., et al. (2018). Can an infection hypothesis explain the beta amyloid hypothesis of Alzheimer’s disease? Front. Aging Neurosci. 10:224. doi: 10.3389/fnagi.2018.00224
Fulop, T., Witkowski, J. M., Larbi, A., Khalil, A., Herbein, G., and Frost, E. H. (2019). Does HIV infection contribute to increased beta-amyloid synthesis and plaque formation leading to neurodegeneration and Alzheimer’s disease? J. Neurovirol. 25, 634–647. doi: 10.1007/s13365-019-00732-3
Garber, C., Soung, A., Vollmer, L. L., Kanmogne, M., Last, A., Brown, J., et al. (2019). T cells promote microglia-mediated synaptic elimination and cognitive dysfunction during recovery from neuropathogenic flaviviruses. Nat. Neurosci. 22, 1276–1288. doi: 10.1038/s41593-019-0427-y
Garber, C., Vasek, M. J., Vollmer, L. L., Sun, T., Jiang, X., and Klein, R. S. (2018). Astrocytes decrease adult neurogenesis during virus-induced memory dysfunction via IL-1. Nat. Immunol. 9, 151–161. doi: 10.1038/s41590-017-0021-y
Gérard, H. C., Dreses-Werringloer, U., Wildt, K. S., Deka, S., Oszust, C., Balin, B. J., et al. (2006). Chlamydophila (Chlamydia) pneumoniae in the Alzheimer’s brain. FEMS Immunol. Med. Microbiol. 48, 355–366. doi: 10.1111/j.1574-695X.2006.00154.x
Giunta, B., Hou, H., Zhu, Y., Rrapo, E., Tian, J., Takashi, M., et al. (2009). HIV-1 Tat contributes to Alzheimer’s disease-like pathology in PSAPP mice. Int. J. Clin. Exp. Pathol. 2, 433–443.
Goldeck, D., Maetzler, W., Berg, D., Oettinger, L., and Pawelec, G. (2016). Altered dendritic cell subset distribution in patients with Parkinson’s disease: impact of CMV serostatus. J. Neuroimmunol. 290, 60–65. doi: 10.1016/j.jneuroim.2015.11.008
Goldman, J. G., Vernaleo, B. A., Camicioli, R., Dahodwala, N., Dobkin, R. D., Ellis, T., et al. (2018). Cognitive impairment in Parkinson’s disease: a report from a multidisciplinary symposium on unmet needs and future directions to maintain cognitive health. NPJ Park. Dis. 4:19. doi: 10.1038/s41531-018-0055-3
Gong, C.-X., and Iqbal, K. (2008). Hyperphosphorylation of microtubule-associated protein tau: a promising therapeutic target for Alzheimer disease. Curr. Med. Chem. 15, 2321–2328. doi: 10.2174/092986708785909111
Gong, C.-X., Liu, F., Grundke-Iqbal, I., and Iqbal, K. (2005). Post-translational modifications of tau protein in Alzheimer’s disease. J. Neural Transm. (Vienna) 112, 813–838. doi: 10.1007/s00702-004-0221-0
Griciuc, A., Serrano-Pozo, A., Parrado, A. R., Lesinski, A. N., Asselin, C. N., Mullin, K., et al. (2013). Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 78, 631–643. doi: 10.1016/j.neuron.2013.04.014
Griffin, W. S., Sheng, J. G., Roberts, G. W., and Mrak, R. E. (1995a). Interleukin-1 expression in different plaque types in Alzheimer’s disease: significance in plaque evolution. J. Neuropathol. Exp. Neurol. 54, 276–281.
Griffin, W. S., Stanley, L. C., Ling, C., White, L., MacLeod, V., Perrot, L. J., et al. (1989). Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 86, 7611–7615. doi: 10.1073/pnas.86.19.7611
Griffin, W. S., Yeralan, O., Sheng, J. G., Boop, F. A., Mrak, R. E., Rovnaghi, C. R., et al. (1995b). Overexpression of the neurotrophic cytokine S100 beta in human temporal lobe epilepsy. J. Neurochem. 65, 228–233.
Griffin, W. S. T., Liu, L., Li, Y., Mrak, R. E., and Barger, S. W. (2006). Interleukin-1 mediates Alzheimer and Lewy body pathologies. J. Neuroinflamm. 3:5. doi: 10.1186/1742-2094-3-5
Grimm, M. O. W., Mett, J., Stahlmann, C. P., Haupenthal, V. J., Zimmer, V. C., and Hartmann, T. (2013). Neprilysin and Aβ clearance: impact of the APP intracellular domain in NEP regulation and implications in Alzheimer’s disease. Front. Aging Neurosci. 5:98. doi: 10.3389/fnagi.2013.00098
Guerreiro, R., Wojtas, A., Bras, J., Carrasquillo, M., Rogaeva, E., Majounie, E., et al. (2013). TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 368, 117–127. doi: 10.1056/NEJMoa1211851
Haij, N. B., Planès, R., Leghmari, K., Serrero, M., Delobel, P., Izopet, J., et al. (2015). HIV-1 tat protein induces production of proinflammatory cytokines by human dendritic cells and monocytes/macrophages through engagement of TLR4-MD2-CD14 Complex and Activation of NF-κB pathway. PLoS One 10:e0129425. doi: 10.1371/journal.pone.0129425
Hall, A., Bandres-Ciga, S., Diez-Fairen, M., Quinn, J. P., and Billingsley, K. J. (2020). Genetic Risk Profiling in Parkinson’s disease and utilizing genetics to gain insight into disease-related biological pathways. Int. J. Mol. Sci. 21:7332. doi: 10.3390/ijms21197332
Hammond, C. J., Hallock, L. R., Howanski, R. J., Appelt, D. M., Little, C. S., and Balin, B. J. (2010). Immunohistological detection of Chlamydia pneumoniae in the Alzheimer’s disease brain. BMC Neurosci. 11:121. doi: 10.1186/1471-2202-11-121
Hategan, A., Bianchet, M. A., Steiner, J., Karnaukhova, E., Masliah, E., Fields, A., et al. (2017). HIV Tat protein and amyloid-β peptide form multifibrillar structures that cause neurotoxicity. Nat. Struct. Mol. Biol. 24, 379–386. doi: 10.1038/nsmb.3379
He, Z., Yang, Y., Xing, Z., Zuo, Z., Wang, R., Gu, H., et al. (2020). Intraperitoneal injection of IFN-γ restores microglial autophagy, promotes amyloid-β clearance and improves cognition in APP/PS1 mice. Cell Death Dis. 11:440. doi: 10.1038/s41419-020-2644-4
Herbein, G. (2016). TNF and HIV-1 Nef: an intimate interplay. EBioMedicine 13, 25–26. doi: 10.1016/j.ebiom.2016.11.009
Herbein, G., and Khan, K. A. (2008). Is HIV infection a TNF receptor signalling-driven disease? Trends Immunol. 29, 61–67. doi: 10.1016/j.it.2007.10.008
Holmes, C. (2013). Review: systemic inflammation and Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 39, 51–68. doi: 10.1111/j.1365-2990.2012.01307.x
Huang, H.-K., Wang, J.-H., Lei, W.-Y., Chen, C.-L., Chang, C.-Y., and Liou, L.-S. (2018). Helicobacter pylori infection is associated with an increased risk of Parkinson’s disease: a population-based retrospective cohort study. Parkinsonism Relat. Disord. 47, 26–31. doi: 10.1016/j.parkreldis.2017.11.331
Ilievski, V., Zuchowska, P. K., Green, S. J., Toth, P. T., Ragozzino, M. E., Le, K., et al. (2018). Chronic oral application of a periodontal pathogen results in brain inflammation, neurodegeneration and amyloid beta production in wild type mice. PLoS One 13:e0204941. doi: 10.1371/journal.pone.0204941
Itzhaki, R. F. (2017). Herpes simplex virus type 1 and Alzheimer’s disease: possible mechanisms and signposts. FASEB J. 31, 3216–3226. doi: 10.1096/fj.201700360
Itzhaki, R. F., Lathe, R., Balin, B. J., Ball, M. J., Bearer, E. L., Braak, H., et al. (2016). Microbes and Alzheimer’s disease. J. Alzheimers Dis. 51, 979–984. doi: 10.3233/JAD-160152
Itzhaki, R. F., and Wozniak, M. A. (2004). Alzheimer’s disease, the neuroimmune axis, and viral infection. J. Neuroimmunol. 156, 1–2. doi: 10.1016/j.jneuroim.2004.07.006
Jimenez-Ferrer, I., and Swanberg, M. (2018). “Immunogenetics of Parkinson’s disease,” in Parkinson’s Disease: Pathogenesis and Clinical Aspects, eds T. B. Stoker and J. C. Greenland (Brisbane QLD: Codon Publications).
Jonsson, T., Stefansson, H., Steinberg, S., Jonsdottir, I., Jonsson, P. V., Snaedal, J., et al. (2013). Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 368, 107–116. doi: 10.1056/NEJMoa1211103
Kalia, L. V., and Lang, A. E. (2015). Parkinson’s disease. Lancet 386, 896–912. doi: 10.1016/S0140-6736(14)61393-3
Kamelgarn, M., Chen, J., Kuang, L., Jin, H., Kasarskis, E. J., and Zhu, H. (2018). ALS mutations of FUS suppress protein translation and disrupt the regulation of nonsense-mediated decay. Proc. Natl. Acad. Sci. U.S.A. 115, E11904–E11913. doi: 10.1073/pnas.1810413115
Kanaan, N. M., and Manfredsson, F. P. (2012). Loss of functional alpha-synuclein: a toxic event in Parkinson’s disease? J. Park. Dis. 2, 249–267. doi: 10.3233/JPD-012138
Kelleher, R. J., and Shen, J. (2017). Presenilin-1 mutations and Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 114, 629–631. doi: 10.1073/pnas.1619574114
Kim, D. H., Jeong, S. N., and Lee, J. H. (2020). Severe periodontitis with tooth loss as a modifiable risk factor for the development of Alzheimer, vascular, and mixed dementia: National Health Insurance Service-National Health Screening retrospective Cohort 2002-2015. J. Periodontal Implant Sci. 50, 303–312. doi: 10.5051/jpis.2000600030
Kim, J., Yoon, J.-H., and Kim, Y.-S. (2013). HIV-1 Tat interacts with and regulates the localization and processing of amyloid precursor protein. PLoS One 8:e77972. doi: 10.1371/journal.pone.0077972
Kim, S., Kwon, S.-H., Kam, T.-I., Panicker, N., Karuppagounder, S. S., Lee, S., et al. (2019). Transneuronal propagation of pathologic α-synuclein from the Gut to the brain models Parkinson’s disease. Neuron 103, 627–641.e7. doi: 10.1016/j.neuron.2019.05.035
Kim, S. Y., and Solomon, D. H. (2010). Tumor necrosis factor blockade and the risk of viral infection. Nat. Rev. Rheumatol. 6, 165–174. doi: 10.1038/nrrheum.2009.279
Klein, R. S., Garber, C., Funk, K. E., Salimi, H., Soung, A., Kanmogne, M., et al. (2019). Neuroinflammation During RNA viral infections. Annu. Rev. Immunol. 37, 73–95. doi: 10.1146/annurev-immunol-042718-041417
Krug, L., Chatterjee, N., Borges-Monroy, R., Hearn, S., Liao, W.-W., Morrill, K., et al. (2017). Retrotransposon activation contributes to neurodegeneration in a Drosophila TDP-43 model of ALS. PLoS Genet. 13:e1006635. doi: 10.1371/journal.pgen.1006635
Kumar, A., Calne, S. M., Schulzer, M., Mak, E., Wszolek, Z., Van Netten, C., et al. (2004). Clustering of Parkinson disease: shared cause or coincidence? Arch. Neurol. 61, 1057–1060. doi: 10.1001/archneur.61.7.1057
Kumar, A. M., Fernandez, J. B., Singer, E. J., Commins, D., Waldrop-Valverde, D., Ownby, R. L., et al. (2009). Human immunodeficiency virus type 1 in the central nervous system leads to decreased dopamine in different regions of postmortem human brains. J. Neurovirol. 15, 257–274. doi: 10.1080/13550280902973952
Kumar, A. M., Ownby, R. L., Waldrop-Valverde, D., Fernandez, B., and Kumar, M. (2011). Human immunodeficiency virus infection in the CNS and decreased dopamine availability: relationship with neuropsychological performance. J. Neurovirol. 17, 26–40. doi: 10.1007/s13365-010-0003-4
Kumar, D. K. V., Choi, S. H., Washicosky, K. J., Eimer, W. A., Tucker, S., Ghofrani, J., et al. (2016). Amyloid-β peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci. Transl. Med 8:340ra72. doi: 10.1126/scitranslmed.aaf1059
Lagier-Tourenne, C., and Cleveland, D. W. (2009). Rethinking ALS: the FUS about TDP-43. Cell 136, 1001–1004. doi: 10.1016/j.cell.2009.03.006
Lahner, E., Annibale, B., and Delle Fave, G. (2009). Systematic review: Helicobacter pylori infection and impaired drug absorption. Aliment. Pharmacol. Ther. 29, 379–386. doi: 10.1111/j.1365-2036.2008.03906.x
Lee, Y.-T., Lee, H.-C., Hu, C.-J., Huang, L.-K., Chao, S.-P., Lin, C.-P., et al. (2017). Periodontitis as a modifiable risk factor for dementia: a Nationwide population-based cohort study. J. Am. Geriatr. Soc. 65, 301–305. doi: 10.1111/jgs.14449
Li, W., Lee, M.-H., Henderson, L., Tyagi, R., Bachani, M., Steiner, J., et al. (2015). Human endogenous retrovirus-K contributes to motor neuron disease. Sci. Transl. Med 7:307ra153. doi: 10.1126/scitranslmed.aac8201
Li, Y., Liu, L., Barger, S. W., and Griffin, W. S. T. (2003). Interleukin-1 mediates pathological effects of microglia on tau phosphorylation and on synaptophysin synthesis in cortical neurons through a p38-MAPK pathway. J. Neurosci. 23, 1605–1611.
Li, Y.-C., Zhang, Y., and Tan, B.-H. (2021). What can cerebrospinal fluid testing and brain autopsies tell us about viral neuroinvasion of SARS-CoV-2. J. Med. Virol. 93, 4247–4257. doi: 10.1002/jmv.26943
Lindwall, G., and Cole, R. D. (1984). Phosphorylation affects the ability of tau protein to promote microtubule assembly. J. Biol. Chem. 259, 5301–5305.
Liu, D., Jin, L., Wang, H., Zhao, H., Zhao, C., Duan, C., et al. (2008). Silencing α-synuclein gene expression enhances tyrosine hydroxylase activity in MN9D cells. Neurochem. Res. 33, 1401–1409. doi: 10.1007/s11064-008-9599-7
Liu, X., and Kumar, A. (2015). Differential signaling mechanism for HIV-1 Nef-mediated production of IL-6 and IL-8 in human astrocytes. Sci. Rep. 5:9867. doi: 10.1038/srep09867
Liu, Y., Chen, L., Zou, Z., Zhu, B., Hu, Z., Zeng, P., et al. (2016). Hepatitis C virus infection induces elevation of CXCL10 in human brain microvascular endothelial cells. J. Med. Virol. 88, 1596–1603. doi: 10.1002/jmv.24504
Lolekha, P., Sriphanom, T., and Vilaichone, R.-K. (2021). Helicobacter pylori eradication improves motor fluctuations in advanced Parkinson’s disease patients: a prospective cohort study (HP-PD trial). PLoS One 16:e0251042. doi: 10.1371/journal.pone.0251042
Lopatko Lindman, K., Hemmingsson, E.-S., Weidung, B., Brännström, J., Josefsson, M., Olsson, J., et al. (2021). Herpesvirus infections, antiviral treatment, and the risk of dementia-a registry-based cohort study in Sweden. Alzheimers Dement. (N. Y.) 7:e12119. doi: 10.1002/trc2.12119
Lopez-Castejon, G., and Brough, D. (2011). Understanding the mechanism of IL-1β secretion. Cytokine Growth Factor Rev. 22, 189–195. doi: 10.1016/j.cytogfr.2011.10.001
Lövheim, H., Gilthorpe, J., Adolfsson, R., Nilsson, L.-G., and Elgh, F. (2015a). Reactivated herpes simplex infection increases the risk of Alzheimer’s disease. Alzheimers Dement. 11, 593–599. doi: 10.1016/j.jalz.2014.04.522
Lövheim, H., Gilthorpe, J., Johansson, A., Eriksson, S., Hallmans, G., and Elgh, F. (2015b). Herpes simplex infection and the risk of Alzheimer’s disease: a nested case-control study. Alzheimers Dement. 11, 587–592. doi: 10.1016/j.jalz.2014.07.157
Lyons, P. D., and Benveniste, E. N. (1998). Cleavage of membrane-associated ICAM-1 from astrocytes: involvement of a metalloprotease. Glia 22, 103–112. doi: 10.1002/(sici)1098-1136(199802)22:2<103::aid-glia1<3.0.co;2-8
Magrinelli, F., Picelli, A., Tocco, P., Federico, A., Roncari, L., Smania, N., et al. (2016). Pathophysiology of motor dysfunction in Parkinson’s disease as the rationale for drug treatment and rehabilitation. Park. Dis. 2016:9832839. doi: 10.1155/2016/9832839
Maldonado, H., Ramírez, E., Utreras, E., Pando, M. E., Kettlun, A. M., Chiong, M., et al. (2011). Inhibition of cyclin-dependent kinase 5 but not of glycogen synthase kinase 3-β prevents neurite retraction and tau hyperphosphorylation caused by secretable products of human T-cell leukemia virus type I-infected lymphocytes. J. Neurosci. Res. 89, 1489–1498. doi: 10.1002/jnr.22678
Mancuso, R., Sicurella, M., Agostini, S., Marconi, P., and Clerici, M. (2019). Herpes simplex virus type 1 and Alzheimer’s disease: link and potential impact on treatment. Expert Rev. Anti Infect. Ther. 17, 715–731. doi: 10.1080/14787210.2019.1656064
Manghera, M., Ferguson-Parry, J., and Douville, R. N. (2016). TDP-43 regulates endogenous retrovirus-K viral protein accumulation. Neurobiol. Dis. 94, 226–236. doi: 10.1016/j.nbd.2016.06.017
Manzanillo, P. S., Ayres, J. S., Watson, R. O., Collins, A. C., Souza, G., Rae, C. S., et al. (2013). The ubiquitin ligase parkin mediates resistance to intracellular pathogens. Nature 501, 512–516. doi: 10.1038/nature12566
Mao, L., Jin, H., Wang, M., Hu, Y., Chen, S., He, Q., et al. (2020). Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in Wuhan, China. JAMA Neurol. 77, 683–690. doi: 10.1001/jamaneurol.2020.1127
Marr, R. A., Guan, H., Rockenstein, E., Kindy, M., Gage, F. H., Verma, I., et al. (2004). Neprilysin regulates amyloid Beta peptide levels. J. Mol. Neurosci. 22, 5–11. doi: 10.1385/JMN:22:1-2:5
Marr, R. A., and Hafez, D. M. (2014). Amyloid-beta and Alzheimer’s disease: the role of neprilysin-2 in amyloid-beta clearance. Front. Aging Neurosci. 6:187. doi: 10.3389/fnagi.2014.00187
Maruyama, M., Higuchi, M., Takaki, Y., Matsuba, Y., Tanji, H., Nemoto, M., et al. (2005). Cerebrospinal fluid neprilysin is reduced in prodromal Alzheimer’s disease. Ann. Neurol. 57, 832–842. doi: 10.1002/ana.20494
Masaki, K., Sonobe, Y., Ghadge, G., Pytel, P., and Roos, R. P. (2019). TDP-43 proteinopathy in Theiler’s murine encephalomyelitis virus infection. PLoS Pathog. 15:e1007574. doi: 10.1371/journal.ppat.1007574
Mastrangelo, M. A., Sudol, K. L., Narrow, W. C., and Bowers, W. J. (2009). Interferon-γ differentially affects Alzheimer’s disease pathologies and induces neurogenesis in triple transgenic-AD mice. Am. J. Pathol. 175, 2076–2088. doi: 10.2353/ajpath.2009.090059
Mathew, S., Faheem, M., Ibrahim, S. M., Iqbal, W., Rauff, B., Fatima, K., et al. (2016). Hepatitis C virus and neurological damage. World J. Hepatol. 8, 545–556. doi: 10.4254/wjh.v8.i12.545
Matschke, J., Lütgehetmann, M., Hagel, C., Sperhake, J. P., Schröder, A. S., Edler, C., et al. (2020). Neuropathology of patients with COVID-19 in Germany: a post-mortem case series. Lancet Neurol. 19, 919–929. doi: 10.1016/S1474-4422(20)30308-2
Maurizi, C. P. (1987). Was a neurovirulent influenza virus the cause of amyotrophic lateral sclerosis and parkinsonism-dementia on Guam? Med. Hypotheses 23, 325–326. doi: 10.1016/0306-9877(87)90024-7
McCormick, C., and Khaperskyy, D. A. (2017). Translation inhibition and stress granules in the antiviral immune response. Nat. Rev. Immunol. 17, 647–660. doi: 10.1038/nri.2017.63
McGee, D. J., Lu, X.-H., and Disbrow, E. A. (2018). Stomaching the possibility of a pathogenic role for Helicobacter pylori in Parkinson’s disease. J. Park. Dis. 8, 367–374. doi: 10.3233/JPD-181327
McGeer, P. L., Itagaki, S., and McGeer, E. G. (1988). Expression of the histocompatibility glycoprotein HLA-DR in neurological disease. Acta Neuropathol. (Berl.) 76, 550–557. doi: 10.1007/BF00689592
Méndez-Guerrero, A., Laespada-García, M. I., Gómez-Grande, A., Ruiz-Ortiz, M., Blanco-Palmero, V. A., Azcarate-Diaz, F. J., et al. (2020). Acute hypokinetic-rigid syndrome following SARS-CoV-2 infection. Neurology 95, e2109–e2118. doi: 10.1212/WNL.0000000000010282
Merello, M., Bhatia, K. P., and Obeso, J. A. (2021). SARS-CoV-2 and the risk of Parkinson’s disease: facts and fantasy. Lancet Neurol. 20, 94–95. doi: 10.1016/S1474-4422(20)30442-7
Mhyre, T. R., Boyd, J. T., Hamill, R. W., and Maguire-Zeiss, K. A. (2012). Parkinson’s disease. Subcell. Biochem. 65, 389–455. doi: 10.1007/978-94-007-5416-4_16
Miao, J., Shi, R., Li, L., Chen, F., Zhou, Y., Tung, Y. C., et al. (2019). Pathological Tau from Alzheimer’s brain induces site-specific hyperphosphorylation and SDS- and reducing agent-resistant aggregation of Tau in vivo. Front. Aging Neurosci. 11:34. doi: 10.3389/fnagi.2019.00034
Miklossy, J. (2011). Emerging roles of pathogens in Alzheimer disease. Expert Rev. Mol. Med. 13:e30. doi: 10.1017/S1462399411002006
Miklossy, J., Kis, A., Radenovic, A., Miller, L., Forro, L., Martins, R., et al. (2006). Beta-amyloid deposition and Alzheimer’s type changes induced by Borrelia spirochetes. Neurobiol. Aging 27, 228–236. doi: 10.1016/j.neurobiolaging.2005.01.018
Mira, M. T., Alcaïs, A., Van Thuc, N., Moraes, M. O., Di Flumeri, C., Hong Thai, V., et al. (2004). Susceptibility to leprosy is associated with PARK2 and PACRG. Nature 427, 636–640. doi: 10.1038/nature02326
Monastero, R., Caruso, C., and Vasto, S. (2014). Alzheimer’s disease and infections, where we stand and where we go. Immun. Ageing 11:26. doi: 10.1186/s12979-014-0026-4
Mount, M. P., Lira, A., Grimes, D., Smith, P. D., Faucher, S., Slack, R., et al. (2007). Involvement of interferon-gamma in microglial-mediated loss of dopaminergic neurons. J. Neurosci. 27, 3328–3337. doi: 10.1523/JNEUROSCI.5321-06.2007
Mrak, R. E., and Griffin, W. S. (2001). Interleukin-1, neuroinflammation, and Alzheimer’s disease. Neurobiol. Aging 22, 903–908.
Mridula, K. R., Borgohain, R., Chandrasekhar Reddy, V., Bandaru, V. C. S., and Suryaprabha, T. (2017). Association of Helicobacter pylori with Parkinson’s disease. J. Clin. Neurol. 13, 181–186. doi: 10.3988/jcn.2017.13.2.181
Mulak, A., and Bonaz, B. (2015). Brain-gut-microbiota axis in Parkinson’s disease. World J. Gastroenterol. 21, 10609–10620. doi: 10.3748/wjg.v21.i37.10609
Murphy, M. P., and LeVine, H. (2010). Alzheimer’s disease and the amyloid-beta peptide. J. Alzheimers Dis. 19, 311–323. doi: 10.3233/JAD-2010-1221
Nath, A., Anderson, C., Jones, M., Maragos, W., Booze, R., Mactutus, C., et al. (2000). Neurotoxicity and dysfunction of dopaminergic systems associated with AIDS dementia. J. Psychopharmacol. (Oxf. Engl.) 14, 222–227. doi: 10.1177/026988110001400305
Nehls, J., Koppensteiner, H., Brack-Werner, R., Floss, T., and Schindler, M. (2014). HIV-1 replication in human immune cells is independent of TAR DNA binding protein 43 (TDP-43) expression. PLoS One 9:e105478. doi: 10.1371/journal.pone.0105478
Nielsen, H. H., Qiu, J., Friis, S., Wermuth, L., and Ritz, B. (2012). Treatment for Helicobacter pylori infection and risk of Parkinson’s disease in Denmark. Eur. J. Neurol. 19, 864–869. doi: 10.1111/j.1468-1331.2011.03643.x
Nix, W. A., Berger, M. M., Oberste, M. S., Brooks, B. R., McKenna-Yasek, D. M., Brown, R. H., et al. (2004). Failure to detect enterovirus in the spinal cord of ALS patients using a sensitive RT-PCR method. Neurology 62, 1372–1377. doi: 10.1212/01.wnl.0000123258.86752.51
Noble, J. M., Scarmeas, N., Celenti, R. S., Elkind, M. S. V., Wright, C. B., Schupf, N., et al. (2014). Serum IgG antibody levels to periodontal microbiota are associated with incident Alzheimer disease. PLoS One 9:e114959. doi: 10.1371/journal.pone.0114959
Nookala, A. R., and Kumar, A. (2014). Molecular mechanisms involved in HIV-1 Tat-mediated induction of IL-6 and IL-8 in astrocytes. J. Neuroinflammation 11, 214. doi: 10.1186/s12974-014-0214-3
O’Brien, R. J., and Wong, P. C. (2011). Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 34, 185–204. doi: 10.1146/annurev-neuro-061010-113613
Oliver, K. R., Brennan, P., and Fazakerley, J. K. (1997). Specific infection and destruction of dopaminergic neurons in the substantia nigra by Theiler’s virus. J. Virol. 71, 6179–6182. doi: 10.1128/JVI.71.8.6179-6182.1997
Ou, S. H., Wu, F., Harrich, D., García-Martínez, L. F., and Gaynor, R. B. (1995). Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J. Virol. 69, 3584–3596. doi: 10.1128/JVI.69.6.3584-3596.1995
Paouri, E., and Georgopoulos, S. (2019). Systemic and CNS inflammation crosstalk: implications for Alzheimer’s disease. Curr. Alzheimer Res. 16, 559–574. doi: 10.2174/1567205016666190321154618
Patrick, C., Crews, L., Desplats, P., Dumaop, W., Rockenstein, E., Achim, C. L., et al. (2011). Increased CDK5 expression in HIV encephalitis contributes to neurodegeneration via tau phosphorylation and is reversed with Roscovitine. Am. J. Pathol. 178, 1646–1661. doi: 10.1016/j.ajpath.2010.12.033
Perez, R. G., Waymire, J. C., Lin, E., Liu, J. J., Guo, F., and Zigmond, M. J. (2002). A Role for α-Synuclein in the regulation of dopamine biosynthesis. J. Neurosci. 22, 3090–3099. doi: 10.1523/JNEUROSCI.22-08-03090.2002
Pierantozzi, M., Pietroiusti, A., Brusa, L., Galati, S., Stefani, A., Lunardi, G., et al. (2006). Helicobacter pylori eradication and l-dopa absorption in patients with PD and motor fluctuations. Neurology 66, 1824–1829. doi: 10.1212/01.wnl.0000221672.01272.ba
Poole, S., Singhrao, S. K., Kesavalu, L., Curtis, M. A., and Crean, S. (2013). Determining the presence of periodontopathic virulence factors in short-term postmortem Alzheimer’s disease brain tissue. J. Alzheimers Dis. 36, 665–677. doi: 10.3233/JAD-121918
Porritt, R. A., and Crother, T. R. (2016). Chlamydia pneumoniae infection and inflammatory diseases. Forum Immunopathol. Dis. Ther. 7, 237–254. doi: 10.1615/ForumImmunDisTher.2017020161
Prasad, A., Bharathi, V., Sivalingam, V., Girdhar, A., and Patel, B. K. (2019). Molecular mechanisms of TDP-43 misfolding and pathology in amyotrophic lateral sclerosis. Front. Mol. Neurosci. 12:25. doi: 10.3389/fnmol.2019.00025
Prudencio, M., Gonzales, P. K., Cook, C. N., Gendron, T. F., Daughrity, L. M., Song, Y., et al. (2017). Repetitive element transcripts are elevated in the brain of C9orf72 ALS/FTLD patients. Hum. Mol. Genet. 26, 3421–3431. doi: 10.1093/hmg/ddx233
Ravits, J. (2005). Sporadic amyotrophic lateral sclerosis: a hypothesis of persistent (non-lytic) enteroviral infection. Amyotroph. Lateral Scler. Mot. Neuron Disord. 6, 77–87. doi: 10.1080/14660820510027026
Readhead, B., Haure-Mirande, J.-V., Funk, C. C., Richards, M. A., Shannon, P., Haroutunian, V., et al. (2018). Multiscale analysis of independent Alzheimer’s cohorts finds disruption of molecular, genetic, and clinical networks by human herpesvirus. Neuron 99, 64–82.e7. doi: 10.1016/j.neuron.2018.05.023
Rempel, H. C., and Pulliam, L. (2005). HIV-1 Tat inhibits neprilysin and elevates amyloid beta. AIDS Lond. Engl. 19, 127–135. doi: 10.1097/00002030-200501280-00004
Rocha, E. M., De Miranda, B., and Sanders, L. H. (2018). Alpha-synuclein: pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiol. Dis. 109, 249–257. doi: 10.1016/j.nbd.2017.04.004
Romeo, M. A., Gilardini Montani, M. S., Gaeta, A., D’Orazi, G., Faggioni, A., and Cirone, M. (2020). HHV-6A infection dysregulates autophagy/UPR interplay increasing beta amyloid production and tau phosphorylation in astrocytoma cells as well as in primary neurons, possible molecular mechanisms linking viral infection to Alzheimer’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 1866:165647. doi: 10.1016/j.bbadis.2019.165647
Rosenblum, L. T., Shamamandri-Markandaiah, S., Ghosh, B., Foran, E., Lepore, A. C., Pasinelli, P., et al. (2017). Mutation of the caspase-3 cleavage site in the astroglial glutamate transporter EAAT2 delays disease progression and extends lifespan in the SOD1-G93A mouse model of ALS. Exp. Neurol. 292, 145–153. doi: 10.1016/j.expneurol.2017.03.014
Rothstein, J. D., Martin, L. J., and Kuncl, R. W. (1992). Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis. N. Engl. J. Med. 326, 1464–1468. doi: 10.1056/NEJM199205283262204
Rothstein, J. D., Van Kammen, M., Levey, A. I., Martin, L. J., and Kuncl, R. W. (1995). Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann. Neurol. 38, 73–84. doi: 10.1002/ana.410380114
Roy, E. R., Wang, B., Wan, Y., Chiu, G., Cole, A., Yin, Z., et al. (2020). Type I interferon response drives neuroinflammation and synapse loss in Alzheimer disease. J. Clin. Invest. 130, 1912–1930. doi: 10.1172/JCI133737
Roy, J., Minotti, S., Dong, L., Figlewicz, D. A., and Durham, H. D. (1998). Glutamate potentiates the toxicity of mutant Cu/Zn-superoxide dismutase in motor neurons by postsynaptic calcium-dependent mechanisms. J. Neurosci. 18, 9673–9684.
Scofield, M. D., Korutla, L., Jackson, T. G., Kalivas, P. W., and Mackler, S. A. (2012). Nucleus Accumbens 1, a Pox virus and Zinc finger/Bric-a-brac Tramtrack Broad protein binds to TAR DNA-binding protein 43 and has a potential role in amyotrophic lateral sclerosis. Neuroscience 227, 44–54. doi: 10.1016/j.neuroscience.2012.09.043
Serrano-Pozo, A., Frosch, M. P., Masliah, E., and Hyman, B. T. (2011). Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 1:a006189. doi: 10.1101/cshperspect.a006189
Sheldon, A. L., and Robinson, M. B. (2007). The role of glutamate transporters in neurodegenerative diseases and potential opportunities for intervention. Neurochem. Int. 51, 333–355. doi: 10.1016/j.neuint.2007.03.012
Shelkovnikova, T. A., An, H., Skelt, L., Tregoning, J. S., Humphreys, I. R., and Buchman, V. L. (2019). Antiviral immune response as a trigger of FUS proteinopathy in amyotrophic lateral sclerosis. Cell Rep. 29, 4496–4508.e4. doi: 10.1016/j.celrep.2019.11.094
Shirotani, K., Tsubuki, S., Iwata, N., Takaki, Y., Harigaya, W., Maruyama, K., et al. (2001). Neprilysin degrades both amyloid beta peptides 1-40 and 1-42 most rapidly and efficiently among thiorphan- and phosphoramidon-sensitive endopeptidases. J. Biol. Chem. 276, 21895–21901. doi: 10.1074/jbc.M008511200
Shuto, H., Kataoka, Y., Horikawa, T., Fujihara, N., and Oishi, R. (1997). Repeated interferon-alpha administration inhibits dopaminergic neural activity in the mouse brain. Brain Res. 747, 348–351. doi: 10.1016/s0006-8993(96)01371-6
Simanjuntak, Y., Liang, J.-J., Lee, Y.-L., and Lin, Y.-L. (2017). Japanese encephalitis virus exploits dopamine D2 Receptor-phospholipase C to target dopaminergic human neuronal cells. Front. Microbiol. 8:651. doi: 10.3389/fmicb.2017.00651
Singh, V. K., and Guthikonda, P. (1997). Circulating cytokines in Alzheimer’s disease. J. Psychiatr. Res. 31, 657–660. doi: 10.1016/s0022-3956(97)00023-x
Sitcheran, R., Gupta, P., Fisher, P. B., and Baldwin, A. S. (2005). Positive and negative regulation of EAAT2 by NF-κB: a role for N-myc in TNFα-controlled repression. EMBO J. 24, 510–520. doi: 10.1038/sj.emboj.7600555
Sochocka, M., Zwoliñska, K., and Leszek, J. (2017). The infectious etiology of Alzheimer’s disease. Curr. Neuropharmacol. 15, 996–1009. doi: 10.2174/1570159X15666170313122937
Soscia, S. J., Kirby, J. E., Washicosky, K. J., Tucker, S. M., Ingelsson, M., Hyman, B., et al. (2010). The Alzheimer’s disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS One 5:e9505. doi: 10.1371/journal.pone.0009505
Sparks Stein, P., Steffen, M. J., Smith, C., Jicha, G., Ebersole, J. L., Abner, E., et al. (2012). Serum antibodies to periodontal pathogens are a risk factor for Alzheimer’s disease. Alzheimers Dement. 8, 196–203. doi: 10.1016/j.jalz.2011.04.006
Suk, T. R., and Rousseaux, M. W. C. (2020). The role of TDP-43 mislocalization in amyotrophic lateral sclerosis. Mol. Neurodegener. 15:45. doi: 10.1186/s13024-020-00397-1
Swanson, N. R., Fox, S. A., and Mastaglia, F. L. (1995). Search for persistent infection with poliovirus or other enteroviruses in amyotrophic lateral sclerosis-motor neurone disease. Neuromuscul. Disord. 5, 457–465. doi: 10.1016/0960-8966(95)00002-5
Tan, A. H., Mahadeva, S., Marras, C., Thalha, A. M., Kiew, C. K., Yeat, C. M., et al. (2015). Helicobacter pylori infection is associated with worse severity of Parkinson’s disease. Parkinsonism Relat. Disord. 21, 221–225. doi: 10.1016/j.parkreldis.2014.12.009
Thomas, S., Mayer, L., and Sperber, K. (2009). Mitochondria influence Fas expression in gp120-induced apoptosis of neuronal cells. Int. J. Neurosci. 119, 157–165. doi: 10.1080/00207450802335537
Van Damme, P., Robberecht, W., and Van Den Bosch, L. (2017). Modelling amyotrophic lateral sclerosis: progress and possibilities. Dis. Model. Mech. 10, 537–549. doi: 10.1242/dmm.029058
van den Bos, M. A. J., Geevasinga, N., Higashihara, M., Menon, P., and Vucic, S. (2019). Pathophysiology and diagnosis of ALS: insights from advances in neurophysiological techniques. Int. J. Mol. Sci. 20:2818. doi: 10.3390/ijms20112818
Vasek, M. J., Garber, C., Dorsey, D., Durrant, D. M., Bollman, B., Soung, A., et al. (2016). A complement-microglial axis drives synapse loss during virus-induced memory impairment. Nature 534, 538–543. doi: 10.1038/nature18283
Venda, L. L., Cragg, S. J., Buchman, V. L., and Wade-Martins, R. (2010). α-Synuclein and dopamine at the crossroads of Parkinson’s disease. Trends Neurosci. 33, 559–568. doi: 10.1016/j.tins.2010.09.004
Verma, A., and Berger, J. R. (2006). ALS syndrome in patients with HIV-1 infection. J. Neurol. Sci. 240, 59–64. doi: 10.1016/j.jns.2005.09.005
Walker, K. A., Ficek, B. N., and Westbrook, R. (2019). Understanding the role of systemic inflammation in Alzheimer’s disease. ACS Chem. Neurosci. 10, 3340–3342. doi: 10.1021/acschemneuro.9b00333
Wang, H., Liu, X., Tan, C., Zhou, W., Jiang, J., Peng, W., et al. (2020). Bacterial, viral, and fungal infection-related risk of Parkinson’s disease: meta-analysis of cohort and case-control studies. Brain Behav. 10:e01549. doi: 10.1002/brb3.1549
Wang, J.-Z., Xia, Y.-Y., Grundke-Iqbal, I., and Iqbal, K. (2013). Abnormal hyperphosphorylation of tau: sites, regulation, and molecular mechanism of neurofibrillary degeneration. J. Alzheimers Dis. 33(Suppl. 1), S123–S139. doi: 10.3233/JAD-2012-129031
Wang, X.-S., Simmons, Z., Liu, W., Boyer, P. J., and Connor, J. R. (2006). Differential expression of genes in amyotrophic lateral sclerosis revealed by profiling the post mortem cortex. Amyotroph. Lateral Scler. 7, 201–210. doi: 10.1080/17482960600947689
Wang, Z., Pekarskaya, O., Bencheikh, M., Chao, W., Gelbard, H. A., Ghorpade, A., et al. (2003). Reduced expression of glutamate transporter EAAT2 and impaired glutamate transport in human primary astrocytes exposed to HIV-1 or gp120. Virology 312, 60–73. doi: 10.1016/s0042-6822(03)00181-8
Weissenborn, K., Ennen, J. C., Bokemeyer, M., Ahl, B., Wurster, U., Tillmann, H., et al. (2006). Monoaminergic neurotransmission is altered in hepatitis C virus infected patients with chronic fatigue and cognitive impairment. Gut 55, 1624–1630. doi: 10.1136/gut.2005.080267
White, J. P., and Lloyd, R. E. (2012). Regulation of stress granules in virus systems. Trends Microbiol. 20, 175–183. doi: 10.1016/j.tim.2012.02.001
White, M. R., Kandel, R., Tripathi, S., Condon, D., Qi, L., Taubenberger, J., et al. (2014). Alzheimer’s associated β-amyloid protein inhibits influenza A virus and modulates viral interactions with phagocytes. PLoS One 9:e101364. doi: 10.1371/journal.pone.0101364
Wijarnpreecha, K., Chesdachai, S., Jaruvongvanich, V., and Ungprasert, P. (2018). Hepatitis C virus infection and risk of Parkinson’s disease: a systematic review and meta-analysis. Eur. J. Gastroenterol. Hepatol. 30, 9–13. doi: 10.1097/MEG.0000000000000991
Woodall, C. J., Riding, M. H., Graham, D. I., and Clements, G. B. (1994). Sequences specific for enterovirus detected in spinal cord from patients with motor neurone disease. BMJ 308, 1541–1543. doi: 10.1136/bmj.308.6943.1541
Woulfe, J. M., Gray, M. T., Gray, D. A., Munoz, D. G., and Middeldorp, J. M. (2014). Hypothesis: a role for EBV-induced molecular mimicry in Parkinson’s disease. Parkinsonism Relat. Disord. 20, 685–694. doi: 10.1016/j.parkreldis.2014.02.031
Wozniak, M. A., Frost, A. L., and Itzhaki, R. F. (2009a). Alzheimer’s disease-specific tau phosphorylation is induced by herpes simplex virus type 1. J. Alzheimers Dis. 16, 341–350. doi: 10.3233/JAD-2009-0963
Wozniak, M. A., Mee, A. P., and Itzhaki, R. F. (2009b). Herpes simplex virus type 1 DNA is located within Alzheimer’s disease amyloid plaques. J. Pathol. 217, 131–138. doi: 10.1002/path.2449
Wu, W. Y.-Y., Kang, K.-H., Chen, S. L.-S., Chiu, S. Y.-H., Yen, A. M.-F., Fann, J. C.-Y., et al. (2015). Hepatitis C virus infection: a risk factor for Parkinson’s disease. J. Viral Hepat. 22, 784–791. doi: 10.1111/jvh.12392
Wu, Z., Ni, J., Liu, Y., Teeling, J. L., Takayama, F., Collcutt, A., et al. (2017). Cathepsin B plays a critical role in inducing Alzheimer’s disease-like phenotypes following chronic systemic exposure to lipopolysaccharide from Porphyromonas gingivalis in mice. Brain Behav. Immun. 65, 350–361. doi: 10.1016/j.bbi.2017.06.002
Yamamoto, M., Kiyota, T., Horiba, M., Buescher, J. L., Walsh, S. M., Gendelman, H. E., et al. (2007). Interferon-gamma and tumor necrosis factor-alpha regulate amyloid-beta plaque deposition and beta-secretase expression in Swedish mutant APP transgenic mice. Am. J. Pathol. 170, 680–692. doi: 10.2353/ajpath.2007.060378
Yong, H. Y. F., Rawji, K. S., Ghorbani, S., Xue, M., and Yong, V. W. (2019). The benefits of neuroinflammation for the repair of the injured central nervous system. Cell. Mol. Immunol. 16, 540–546. doi: 10.1038/s41423-019-0223-3
Zambrano, A., Solis, L., Salvadores, N., Cortés, M., Lerchundi, R., and Otth, C. (2008). Neuronal cytoskeletal dynamic modification and neurodegeneration induced by infection with herpes simplex virus type 1. J. Alzheimers Dis. 14, 259–269. doi: 10.3233/jad-2008-14301
Zarei, S., Carr, K., Reiley, L., Diaz, K., Guerra, O., Altamirano, P. F., et al. (2015). A comprehensive review of amyotrophic lateral sclerosis. Surg. Neurol. Int. 6:171. doi: 10.4103/2152-7806.169561
Zhang, X., Wang, F., Hu, Y., Chen, R., Meng, D., Guo, L., et al. (2020). In vivo stress granule misprocessing evidenced in a FUS knock-in ALS mouse model. Brain 143, 1350–1367. doi: 10.1093/brain/awaa076
Keywords: Alzheimer’s disease, amyotrophic lateral sclerosis, infection, microbes, neurodegenerative diseases, Parkinson’s disease, viruses
Citation: Lotz SK, Blackhurst BM, Reagin KL and Funk KE (2021) Microbial Infections Are a Risk Factor for Neurodegenerative Diseases. Front. Cell. Neurosci. 15:691136. doi: 10.3389/fncel.2021.691136
Received: 05 April 2021; Accepted: 08 June 2021;
Published: 07 July 2021.
Edited by:
Fabio Blandini, Neurological Institute Foundation Casimiro Mondino (IRCCS), ItalyReviewed by:
Adelaide Fernandes, University of Lisbon, PortugalE. Lezi Medical College of Wisconsin, United States
Copyright © 2021 Lotz, Blackhurst, Reagin and Funk. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kristen E. Funk, a2Z1bmtAdW5jYy5lZHU=