 Elena Parmigiani
Elena Parmigiani Marta Scalera
Marta Scalera Elisabetta Mori
Elisabetta Mori Elena Tantillo
Elena Tantillo Eleonora Vannini
Eleonora Vannini- 1Embryology and Stem Cell Biology, Department of Biomedicine, University of Basel, Basel, Switzerland
- 2Neuroscience Institute, Consiglio Nazionale delle Ricerche (CNR), Pisa, Italy
- 3BIO@SNS Laboratory, Scuola Normale Superiore, Pisa, Italy
In recent years, the direct interaction between cancer cells and tumor microenvironment (TME) has emerged as a crucial regulator of tumor growth and a promising therapeutic target. The TME, including the surrounding peritumoral regions, is dynamically modified during tumor progression and in response to therapies. However, the mechanisms regulating the crosstalk between malignant and non-malignant cells are still poorly understood, especially in the case of glioma, an aggressive form of brain tumor. The presence of unique brain-resident cell types, namely neurons and glial cells, and an exceptionally immunosuppressive microenvironment pose additional important challenges to the development of effective treatments targeting the TME. In this review, we provide an overview on the direct and indirect interplay between glioma and neuronal and glial cells, introducing new players and mechanisms that still deserve further investigation. We will focus on the effects of neural activity and glial response in controlling glioma cell behavior and discuss the potential of exploiting these cellular interactions to develop new therapeutic approaches with the aim to preserve proper brain functionality.
Introduction
Gliomas are the most common primary brain tumors in adults and children. They account for almost 30% of all primary brain tumors and 80% of malignant brain tumors (Weller et al., 2015). Although new therapeutic strategies are continuously under investigation, gliomas remain associated with high morbidity and mortality, especially in the case of glioblastoma multiforme (GBM), the most aggressive form of glioma (Patel et al., 2019; Liang et al., 2020). Hence, treatment of gliomas represents one of the hardest challenges of our times for neuro-oncologists. The standard of care for GBM patients consists in a maximal safe surgical resection followed by cycles of radio- and chemotherapy. However, this therapeutic regimen gives only partial benefits to the patients. Complete glioma eradication is usually prevented by the invasive nature of these tumors and by recurrence due to therapy-resistance (Bahadur et al., 2019). In addition, GBM patients also suffer from devastating neurological deterioration induced by the growing tumor mass as well as the aggressive treatments. In most of the cases, neurodegeneration and other tumor-associated secondary effects contribute to an unfavorable prognosis in glioma patients (Gibson and Monje, 2012; Zetterling et al., 2020).
For a long time, cancer research has mainly focused on understanding the biology of glioma cells and investigating the aberrant pathways that guide tumor onset and progression. The concept that the interaction between glioma cells and the tumor microenvironment (TME) is crucial in driving tumor growth has emerged lately (Hanahan and Coussens, 2012). Understanding microenvironmental determinants that contribute to glioma progression is therefore a major focus of current research, with the aim of discovering new therapeutic targets and strategies (Johung and Monje, 2017).
In order to sustain their growth, glioma cells co-opt brain-resident cells by establishing different communication routes which include secreted molecules, gap junctions, tunneling nanotubes, and extracellular vesicles (Osswald et al., 2015; D’Alessio et al., 2019; Jung et al., 2019; Lane et al., 2019; Matarredona and Pastor, 2019; Gao et al., 2020). Some of these mechanisms have been recently unraveled and exploited to develop new therapeutic strategies. However, the complex cell-cell interactions within the glioma TME are still largely unknown. The TME is also dynamically shaped during disease progression and eventually adapts in response to therapies (Venkatesh et al., 2015; D’Alessio et al., 2019).
Neurons and glial cells (microglia, astrocytes, and oligodendrocytes) not only cooperate to maintain brain homeostasis, but also represent major TME components that substantially contribute to tumor growth. Although the role of microglia has been extensively investigated over the last decade, some important questions still remain open. In contrast, the contribution of oligodendrocytes and, to some extent, astrocytes have been poorly addressed. Important advances have been made in the study of neuronal regulation of glioma development, but some aspects remain controversial. In addition, secondary effects due to glioma growth, like epilepsy and edema, can profoundly affect neuron functionality and lead to cognitive deficits through mechanisms that are not completely known. Therefore, a better understanding of the cellular and molecular dynamics within the TME is urgently needed in order to efficiently counteract glioma progression and improve patients’ survival and quality of life.
Tumor-Associated Microglia and Macrophages: A Leading Player in the TME
Microglia cells are the main immune cell population in the brain (Korin et al., 2017). By continuously scanning the central nervous system (CNS) tissue, they act as “sentinels” that react to any type of pathogens or insult, and are implicated in virtually all CNS pathologies (Hanisch and Kettenmann, 2007). In the glioma field, microglial cells are commonly grouped together with infiltrating monocyte-derived macrophages (MDMs). Microglia and MDMs are collectively named as tumor-associated macrophages (TAMs). Accounting for up to 50% of the tumor mass, TAMs are a central component of the TME, and they play essential roles during tumor progression, recurrence and response to therapy (Gutmann and Kettenmann, 2019). For these reasons, TAM-targeting therapies have recently gained attention in the field as a promising way to treat gliomas (Kowal et al., 2019).
Microglia and MDMs are phenotypically similar and share the expression of markers such as Iba1, CD11b, F4/80, MerTK, and CSF-1R (Greter et al., 2015). Traditionally, their discrimination has been based on the different expression levels of CD45: while microglia are CD11b+CD45low-expressing cells, MDMs express high levels of CD45 (CD11b+CD45high). Thanks to gene expression profiling studies, more specific markers have been recently identified and are now widely used. These include P2RY12 (Butovsky et al., 2014), Sall1 (Buttgereit et al., 2016), Tmem119 (Bennett et al., 2016), and Hexb (Masuda et al., 2020) that are exclusively expressed by brain-resident microglia cells, while CD49d (Bowman et al., 2016) or Cxcr4 (Werner et al., 2020) are preferentially expressed by peripheral MDMs. However, activated microglial cells acquire some morphological and phenotypical characteristics of MDMs, making the discrimination of these two myeloid cell types problematic (Greter et al., 2015). Identifying the brain-resident vs. peripheral TAMs is important in light of functional differences that have been recently revealed and that possibly arise from a distinct ontogeny. Despite a common origin of microglia and MDMs in the yolk sac, MDMs derive from subsequent wave(s) of progenitors during development and are constantly replenished by bone marrow-derived monocytes in some adult tissues, while in others they maintain themselves locally (Ginhoux and Guilliams, 2016). In contrast, microglia cells originate from unique progenitors and colonize the brain early on during development. They remain in the brain tissue throughout life and self-maintain through a fine balance of proliferation and apoptosis with no need of contribution from the periphery, at least in physiological conditions (Ginhoux et al., 2010; Askew et al., 2017; Tay et al., 2017). In pathological conditions such as brain tumors, the blood brain barrier is compromised and a significant number of MDMs can colonize the brain and participate in disease evolution. Interestingly, when transplanted into a healthy brain, MDMs acquire some microglial-like traits under the influence of the host microenvironment, but they also maintain specific ontogeny-driven features (Bruttger et al., 2015; Bennett et al., 2018). A similar phenomenon has been observed in gliomas, where common and ontogeny-specific programs cooperate to educate microglia and MDMs to support tumor growth (Bowman et al., 2016). For instance, in the TME microglia usually show higher expression of pro-inflammatory genes (Bowman et al., 2016). Conversely, the expression of antigen processing and presentation molecules and of immunosuppressive cytokines is enriched in MDMs and, therefore, they might be more effective in hampering a proper T cell-mediated anti-tumor response (Bowman et al., 2016; Klemm et al., 2020). Differences in the expression profile of brain-resident microglia and peripheral MDMs have been recently confirmed also in humans, where they even vary depending on the brain tumor type. While the TME of less aggressive IDH mutant gliomas is dominated by microglia, in high grade gliomas the contribution of peripheral MDMs is higher. Moreover, TAMs with an immunosuppressive phenotype are particularly abundant in brain metastasis compared to primary gliomas (Friebel et al., 2020; Guldner et al., 2020; Klemm et al., 2020). Finally, a specific MDMs-related gene signature correlates with poor survival of glioma patients (Bowman et al., 2016; Friebel et al., 2020; Klemm et al., 2020). Different transcriptional responses have also been observed in microglia and MDMs after radiotherapy (Akkari et al., 2020) or anti-CD47 immunomodulatory treatment (Hutter et al., 2019).
Microglia activation in response to brain tumors has been extensively studied in mouse models of GBM (Bowman et al., 2016; Gutmann and Kettenmann, 2019; Friebel et al., 2020; Klemm et al., 2020) and in glioma patients (Müller et al., 2017; Sankowski et al., 2019; Friebel et al., 2020; Klemm et al., 2020). However, it remains unclear whether TAMs initially attempt to control tumor growth before being reprogrammed by tumor cells. Some studies have suggested that microglia and macrophages from healthy individuals may have the ability to delay the proliferation of brain tumor initiating cells in vitro (Sarkar et al., 2014), although these data still need to be confirmed. What is certainly known is that shortly after encountering tumor cells, TAMs are “educated” to promote and sustain tumor growth and invasion (Hambardzumyan et al., 2015). Multiple mechanisms inducing TAMs education have been described and these include tumor-released cytokines (Komohara et al., 2008; Zhou et al., 2015; Kloepper et al., 2016; De Boeck et al., 2020), metabolites (Colegio et al., 2014; Shen et al., 2016; Takenaka et al., 2019) and extracellular vesicles (Maas et al., 2020), or cell-cell contact between TAMs and malignant cells (Chia et al., 2018). Tumor cells can also produce chemokines like CCL2, CCL5, CX3CL1, and CSF1 to attract microglia and MDMs (Gutmann and Kettenmann, 2019).
Once activated, TAMs are reprogrammed to produce factors that stimulate glioma cell proliferation (Coniglio et al., 2012; Hammond et al., 2019), invasion (Markovic et al., 2009; Hu et al., 2014), and therapy resistance (Quail et al., 2016). For example, TAMs favor glioma invasion by remodeling the extracellular matrix (ECM). Factors released by both TAMs and tumor cells induce the reciprocal expression of MMP2, MMP9, and MT1-MMP, that degrade the ECM facilitating glioma migration (Wick et al., 2001; Markovic et al., 2009; Hu et al., 2014). Interestingly, MMP2 is released as an inactive molecule and requires the presence of MT1-MMP on the surface of TAMs to be activated (Markovic et al., 2009), indicating that TAMs and glioma cells can coordinate their functions in the TME. In addition, TAMs can indirectly promote glioma progression by favoring angiogenesis (De Palma et al., 2008) and immunosuppression. In vitro studies demonstrated that conditioned medium from glioma cells was sufficient to inhibit the anti-cancer properties of TAMs, including phagocytosis and T-cell stimulation (Wu et al., 2010). TAMs isolated from human glioma specimens showed an impaired ability to activate anti-tumor T cells (Hussain et al., 2006), possibly due to an unbalanced expression of T cell stimulatory vs. inhibitory molecules (Klemm et al., 2020). Therefore, upon activation, TAMs acquire a unique multifunctional activation state (Hambardzumyan et al., 2015) which has been recently confirmed and expanded by single cell RNA-sequencing (scRNA-seq) studies. A complex scenario has emerged describing an heterogeneous population of TAMs which comprises multiple activation states and distinct subpopulations characterized by specific expression profiles (Müller et al., 2017; Sankowski et al., 2019; Friebel et al., 2020; Guldner et al., 2020; Klemm et al., 2020; Pombo Antunes et al., 2021). This might be the consequence of an intrinsic heterogeneity and plasticity of CNS myeloid cells in response to different stimuli (Masuda et al., 2020) and/or it might reflect spatial heterogeneity within the TME. For example, TAMs expressing pro-inflammatory and immunostimulatory genes co-exist with anti-inflammatory TAMs populations (Guldner et al., 2020; Klemm et al., 2020). However, pro-inflammatory cytokines genes, like IL1, are more expressed by TAMs at the tumor periphery compared to the tumor core where, instead, anti-inflammatory genes like TGFβ are upregulated (Darmanis et al., 2017). Within the tumor core, a subpopulation of CD206+CD169+CD209+ macrophages was found specifically associated with blood vessels (Friebel et al., 2020) and TAMs expressing hypoxic and pro-angiogenic genes HIF1A and VEGF-A have been described in human gliomas and might be enriched in hypoxic regions (Darmanis et al., 2017; Sankowski et al., 2019). However, a more precise functional characterization of those TAM subpopulations is largely missing. In contrast to other neurodegenerative and inflammatory diseases, for which sequential microglia activation states have been described (Keren-Shaul et al., 2017; Mathys et al., 2017; Sousa et al., 2018; Hammond et al., 2019; Masuda et al., 2019), the temporal dynamics of TAMs education in brain tumors are still largely unknown. In addition, the functional differences among distinct TAM subpopulations and their clinical relevance is still poorly defined.
Interestingly, specific mutations in tumor cells can indirectly shape the immune TME and, in particular, TAMs recruitment and activation. NF1 or PTEN deletion in glioma cells increased microglia and macrophages infiltration in vitro and in mouse models (Wang et al., 2017; Chen et al., 2019). AKT1 overexpression also enhances physical interaction of microglia with AKT1+-tumor cells in a zebrafish model of brain tumors (Chia et al., 2019). In contrast, IDH mutation is associated with diminished production of chemoattractant cytokines and consequent lower numbers of TAMs in mouse models, and with a less immunosuppressive TAM phenotype in humans (Amankulor et al., 2017; Friebel et al., 2020; Klemm et al., 2020). This evidence highlights the ability of microglia and MDMs to respond and adapt to the TME and emphasize the complexity and heterogeneity of TAMs.
TAM Re-education as a Therapeutic Strategy to Cure Gliomas
Given their central role in tumor progression, TAMs represent a promising therapeutic target. Over the last years, multiple approaches have been tested in preclinical and clinical studies with the goal of re-educating TAMs to exploit their anti-tumorigenic potential (Kowal et al., 2019). These strategies include the use of inhibitors of myeloid-specific receptors like CSF-1R (Pyonteck et al., 2013) and MARCO (Georgoudaki et al., 2016), molecules targeting key factors involved in TAM polarization (Kaneda et al., 2016), or agents that boost TAMs phagocytic activity like anti-CD47 antibodies (Hutter et al., 2019). However, despite promising results in different cancer types (Mantovani et al., 2017), these approaches have failed to significantly improve the overall survival of GBM patients compared to traditional treatments (Butowski et al., 2016). Indeed, after an initial phase of tumor regression, recurrence occurred upon CSF-1R inhibitor treatment in preclinical mouse models of glioma (Quail et al., 2016). Interestingly, therapy-resistance is induced by tumor-derived factors that activate pro-survival pathways in TAMs (Quail et al., 2016), suggesting a continuous and dynamic TAMs-tumor cells crosstalk during tumor evolution in response to therapy. Moreover, a population of Lgals3+-myeloid cells resistant to CSF-1R inhibitors was recently identified in physiological conditions (Zhan et al., 2020) and also observed in mouse models of GBM (Ochocka et al., 2021). However, additional investigations are needed to elucidate the mechanisms driving therapy-resistance and develop approaches that increase the efficiency of TAM re-educating agents. Promising results come from combinatorial strategies where TAM immunomodulatory molecules significantly enhanced the efficacy of other treatments (Quail et al., 2016; Kowal et al., 2019; Akkari et al., 2020). Also, some non-cancer related FDA-approved drugs have demonstrated an unexpected ability to stimulate a TAM anti-tumor activity and might be repurposed as adjunctive treatments: examples are the antifungal Amphotericin B (Sarkar et al., 2014), the antibiotic thiostrepton (Hu et al., 2021), the iron supplement ferumoxytol (Zanganeh et al., 2016), and the vitamin B3 (Sarkar et al., 2020). Interestingly, TAM re-educating strategies were also effective at reducing astrocytes reactivity, oligodendrocytes loss and at ameliorating the neurological deficits associated with chemotherapy (Gibson et al., 2019).
Although the main line of evidence points toward a pro-tumorigenic role of TAMs and prompts the implementation of TAM re-educating therapies, there are some important exceptions that need to be considered. Maximov et al. (2019) described a surprising anti-tumorigenic role of TAMs, and in particular CCR2+ MDMs, in SHH-medulloblastoma, where TAMs deletion significantly accelerated tumor growth. These findings could be explained by the higher phagocytic activity of cerebellar microglia compared to other brain areas and potentially reflect regional-differences (Ayata et al., 2018). But they also suggest caution when designing TAM re-educating strategies for brain tumors.
Another Protagonist of the TME: Reactive Astrocytes
Astrocytes are the most abundant glial cells in the CNS. One of the main characteristics of these cells is their ability to respond to multiple types of injuries and to participate in different diseases (Buffo et al., 2010). It is therefore not surprising that they can react also to the presence of tumor cells. In a similar way to microglia, astrocytes are efficiently co-opted by tumor cells to sustain their proliferation, survival, migration and therapy resistance (Wasilewski et al., 2017; Brandao et al., 2019). GBM patients with high expression of reactive astrocytes genes show a worse prognosis and tumor cells co-transplanted with astrocytes develop more aggressive tumors in mice (Mega et al., 2020). Moreover, astrocytes can be a cell-of-origin of gliomas, as they give rise to high-grade brain tumors upon transformation (Bachoo et al., 2002; Zhu et al., 2005). Interestingly, different mutations can impose an astrocytic vs. oligodendrocytic fate to tumor cells, with the first usually associated with more aggressive tumors. For example, cells with aberrant EGFR and Ink4A/Arf deletion acquire expression of astrocytes markers (Bachoo et al., 2002), while PDGFB overexpression is sufficient to de-differentiate astrocyte cultures and to form oligodendroglioma or oligoastrocytoma in vivo (Dai et al., 2001). These findings are consistent with the differences described in human glioma subtypes, where an astrocytic gene signature is enriched in the classical subtypes characterized by EGFR amplification and Ink/Arf deletion (Verhaak et al., 2010). Also the contribution of different astrocyte subpopulations to the TME can vary among GBM subtypes (John Lin et al., 2017). Interestingly, a gene signature defining astrocytes activation upon co-culture with GBM cells was significantly associated with mesenchymal GBM (Mega et al., 2020), suggesting a prominent role of non-malignant reactive astrocytes in this tumor subtype.
Astrocytes are characterized by broad morphological and functional heterogeneity that may be the consequence of regional and ontogenetic differences (Khakh and Deneen, 2019). Astrocyte heterogeneity could also affect the tumorigenic potential of these cells, although the mechanisms behind are yet to be unraveled. It has been shown that GFAP+ and GLAST+ astrocytes, when transformed, give rise to GBM with different features: GFAP+ tumor cells are intrinsically more proliferative than GLAST-derived tumors that, instead, may rely more on the TME, as suggested by enhanced recruitment of non-malignant glial cells (Irvin et al., 2017). Marker combinations can be used to subdivide astrocytes in discrete subpopulations that co-exist in healthy brains and can give rise to aggressive gliomas. These astrocytes subpopulations and their malignant analogs display different molecular and functional properties (John Lin et al., 2017).
Whether such heterogeneity characterizes also non-malignant astrocytes in TME remains undefined. By comparing gene expression profiles of TME-associated astrocytes in low and high grade gliomas, Katz et al. (2012) identified a subpopulation of astrocytes specifically localized in perivascular regions of aggressive tumors, where also a population of CD44+ stem-like glioma cells reside. These astrocytes express high levels of osteopontin (or SPP1), a CD44 ligand that induce stemness in glioma cells (Pietras et al., 2014). Both osteopontin and CD44 correlate with a poor overall survival in patients, suggesting a role of perivascular-niche astrocytes in supporting glioma stem cells. Astrocytes expressing a phosphorylated form of PDGFRβ and associated with blood vessels favor brain metastases dissemination and can be targeted to impair metastatic growth (Gril et al., 2013). Another subpopulation of astrocytes that displays high levels of the immune checkpoint PD-L1 and activation of the immunomodulatory factor STAT3 has also been observed in the peritumoral area, where it might constitute a barrier against anti-tumor T lymphocytes (Priego et al., 2018; Henrik Heiland et al., 2019).
Peritumoral regions are implicated in tumor invasion and multiple studies have demonstrated a role of astrocytes in promoting tumor migration in different ways: by expressing the chemoattractant factor GDNF (Shabtay-Orbach et al., 2015), by releasing MMPs (Le et al., 2003) or by inducing MMPs upregulation in tumor cells (Chen W. et al., 2016). Similar to TAMs, astrocytes can cooperate with tumor cells in remodeling the ECM via a mechanism involving plasminogen activation. Plasminogen is produced by glioma cells and converted to plasmin by the protease uPA, which is activated by the uPA receptor expressed on astrocytes. Plasmin mediates MMP2 activation and ECM degradation favoring tumor invasion (Le et al., 2003). Nonetheless, in different contexts, such as the initial phase of brain metastasis, astrocyte-derived PA and plasmin can be cytotoxic for tumor cells that enter in a naïve brain parenchyma. To overcome this effect, tumor cells upregulate the PA inhibitors serpins allowing brain infiltration and metastasis dissemination (Valiente et al., 2014). Hence, although in the early phase of metastasis astrocytes seem to counteract brain invasion, later on they are converted by metastatic cells to promote tumor growth, similarly to what they do in primary glioma (Wasilewski et al., 2017). The events triggering the pro-tumorigenic education of astrocytes in primary and metastatic brain tumors are partially unknown. One described mechanism involves the establishment of direct cell-cell contacts to transfer signaling molecules from tumor cells to astrocytes. These molecules induce astrocytes to release cytokines which, in turn, promote tumor growth (Chen Q. et al., 2016). Astrocytomas, but not oligodendrogliomas, exploit similar gap junctions-mediated connections to form functional cellular networks of tumor cells and rapidly exchanged second messengers and propagate calcium signaling (Osswald et al., 2015). Using similar intercellular communication, astrocytes can also protect metastatic tumor cells from apoptosis by sequestering calcium accumulated in tumor cells upon chemotherapy treatment (Lin et al., 2010).
Immunomodulation is another critical aspect of astrocyte reactivity (Linnerbauer and Rothhammer, 2020). Astrocytes can regulate the immune TME by responding and releasing a broad spectrum of cytokines and by communicating with multiple immune cell types (Priego and Valiente, 2019). Astrocytes expressing high levels of immunosuppressive cytokines, such as TGFβ and IL10, have been recently identified in human GBM samples (Henrik Heiland et al., 2019). JAK/STAT and STAT3 inhibition efficiently reverts astrocytes-related immunosuppression and delay tumor growth, indicating a central role of this pathway in controlling astrocytes immunomodulatory properties (Priego et al., 2018; Brandao et al., 2019; Henrik Heiland et al., 2019). Intriguingly, the presence of phosphorylated-STAT3+ immunosuppressive astrocytes is promoted by tumor and microglia cells. Reactive astrocytes can also induce the upregulation of immunosuppressive and tumor-promoting molecules in microglia and macrophages, establishing a positive feedback loop between TAMs and astrocytes in the TME (Priego et al., 2018; Henrik Heiland et al., 2019). Hence, the bidirectional crosstalk between astrocytes and microglia is crucial to define the immunosuppressive microenvironment in brain tumors (Matias et al., 2018). Both cell types, once activated, release cytokines that can boost each other’s activation. Importantly, distinct types of cytokines differently influence the activation states and, consequently, the secretome of astrocytes and microglia (Liddelow et al., 2017). Therefore, the microglia-astrocytes interconnection could be a promising therapeutic target that certainly deserves further studies.
Astrocytes have a major role in inducing brain tumor resistance to therapy. Radiation, for instance, can stimulate the release of multiple factors by reactive astrocytes. These factors can support glioma stemness and promote tumor recurrence (Berg et al., 2021). Astrocytes collaborate with microglia to clear oncolytic viral particles, leading to severe implications for the therapeutic use of oncolytic viruses in gliomas (Kober et al., 2015). Also, it remains to be addressed whether astrocytes affect immunotherapy responses by virtue of their immunomodulatory properties and expression of immune checkpoint molecules (Priego et al., 2018; Henrik Heiland et al., 2019; Priego and Valiente, 2019).
Altogether, it is clear that astrocytes actively shape the TME of gliomas and metastatic brain tumors, playing mainly a tumor-promoting role. In the recent years some, although limited in number, innovative approaches targeting astrocytes in brain tumors have been tested in preclinical and clinical trials (Wasilewski et al., 2017). The JAK/STAT pathway is one of the best candidates, being at the crossroad of multiple astrocytes-secreted cytokines, like IL10 and IL6, as well as a critical regulator of astrocytes immune response. Legasil®, a nutraceutical product containing a natural polyphenolic flavonoid called Silibinin, inhibits activation of STAT3 in reactive astrocytes and is able to reduce brain metastasis and significantly increase overall survival rate (Priego et al., 2018). Although tested in a small cohort of patients only with metastatic brain tumors, the results are certainly promising. Interfering with intercellular gap junctions is another way to limit tumor-astrocytes communication. Carbenoloxone, a pan-connexin inhibitor, efficiently blocks intercellular calcium waves propagation among astrocytoma cells (Osswald et al., 2015). Beyond its anti-inflammatory function, meclofenamate was found to inhibit also Cx43 gap junctions (Harks et al., 2001) and is now under investigation in a clinical trial for brain metastatic patients. Moreover, astrocytes heterogeneity and astrocytes-specific factors might also be exploited as novel diagnostic strategies. For example, Katz et al. (2012) defined a tumor-associated astrocytes gene signature which efficiently predicts the survival of patients specifically with proneural GBM subtype. However, many biological and molecular aspects of TME-associated astrocytes still need to be untangled in order to fully exploit their therapeutic potential.
The Role of Neural Stem and Progenitor Cells
Neural stem cells (NSCs) and progenitors of the subventricular zone have been proposed as a cell-of-origin of gliomas (Alcantara Llaguno et al., 2009; Lee et al., 2018). Notably, differentiation of NSCs toward the neuronal lineage represents a hurdle for the initiation of brain tumors. Immature and mature neurons are resistant to transformation after loss of tumor suppressor genes, a condition that rapidly triggers tumor formation in neural progenitors and glial cells (Alcantara Llaguno et al., 2019).
In addition to being a cell-of-origin of tumors, neural progenitors can contribute to shape the TME. Non-malignant neural progenitor cells are strongly attracted by tumor cells and preferentially accumulate at the tumor border. Differently from the majority of cells composing the TME, their presence delays tumor growth, as demonstrated by enhanced mice survival when GBM cells are co-transplanted with neural precursors (Glass et al., 2005). Mechanistically, neural progenitors trigger cell death by secreting endovanilloids which stimulate the vanilloid receptor TRPV1, specifically expressed by mouse and human GBM cells (Stock et al., 2012). However, glioma cells might have developed a way to counteract the anti-tumorigenic effect of neural progenitors. In a recent study on mouse NCSs, Lawlor et al. (2020) discovered that, when in direct contact, malignant-NSCs induce quiescence in wild-type (wt)-NSCs via NOTCH signaling activation. In this way, they rapidly outcompete wt-NSCs in numbers. A crosstalk between tumor cells and neural progenitors remains to be confirmed in humans, but it might be particularly relevant in young patients, where progenitor cells are still present. However, it should be considered that in inflammatory contexts NSCs can interact with microglia/macrophages inducing immunosuppression (Peruzzotti-Jametti et al., 2018). Given their high tropism for tumors and their tumor-suppressive function, neural progenitors could be exploited as an alternative therapeutic strategy to counteract gliomas.
Oligodendrocytes and Oligodendrocyte Progenitor Cells: An Underestimated Player in the TME
Using multiple transgenic mouse models, different groups have demonstrated that oligodendrocyte progenitor cells (OPCs) can be a cell-of-origin of gliomas (Liu et al., 2011; Sugiarto et al., 2011; Galvao et al., 2014). The presence of oligodendrocytes with the typical “fried egg-shape” has been historically the main histopathological criteria to define human oligodendroglioma (Gupta et al., 2005). More recently, comprehensive molecular genetic profiling and scRNA-sequencing studies have confirmed the presence of OPC-like glioma cells not only in oligodendroglioma but also in other glioma entities, like astrocytoma and GBM, although with varying abundance (Venteicher et al., 2017; Neftel et al., 2019). Of note, the proneural GBM subtype is characterized by alteration of PDGFRA and high expression of oligodendrocytic markers (Verhaak et al., 2010; Wang et al., 2017).
The function of OPCs and oligodendrocytes within the TME has been neglected for a long time. A recent bioinformatic method that predicts tumor-TME interactions from scRNA-seq datasets revealed a significant infiltration of non-malignant oligodendrocytes in the TME of proneural GBM compared to other GBM subtypes (Caruso et al., 2020). The authors also identified ligand-receptor pairs that suggest crosstalk between malignant and non-malignant oligodendrocytes. For example, infiltrating oligodendrocytes express high levels of PDGFA that, via activation of its receptor PDGFRα on glioma cells, could fuel tumor proliferation (Cao, 2013; Caruso et al., 2020). Other evidence, mainly in vitro, suggests the existence of a bidirectional communication between oligodendrocytes and tumor cells within the TME. Conditioned media from human glioma cell lines increases the survival of OPCs. In turn, OPC-derived factors like EGF and FGF1, promote stemness and chemotherapy resistance of glioma cells (Hide et al., 2018). In contrast, a negative correlation between Wif-1+ non-malignant oligodendrocytes and the proliferation index of human astrocytoma cells has been reported, suggesting an anti-tumorigenic role of oligodendrocytes via Wnt inhibition (Asslaber et al., 2017). Although the function of non-malignant OPCs needs further investigation, the accumulation of these cells at the tumor border of human gliomas suggests that they might play a strategic role within the TME (Asslaber et al., 2017; Hide et al., 2018). In the tumor periphery TAMs are also particularly abundant suggesting a TAMs-OPCs crosstalk. Indeed, it has been shown that macrophage-derived soluble factors sustain OPCs viability (Hide et al., 2018). In addition, Huang and colleagues described a population of glioma-associated OPCs that promotes neovascularization and facilitates tumor growth (Huang et al., 2014). These PDGFRα+ OPCs are recruited at the tumor border through PDGFC released by TME cells, including TAMs. Interestingly, mice with either PDGFRA or PDGFC deletion display a normalized vascular network and a significant decrease in tumor volume. OPCs could also influence microglia via TGFβ, which is essential for proper microglia development and maintenance. Pharmacological and genetic depletion of NG2+ OPCs indirectly causes downregulation of TGFβ signaling in microglia with consequent alteration of microglia homeostasis. Although in the absence of a pathological condition no changes of activation markers were detected in microglia (Liu and Aguzzi, 2020), it would be interesting to investigate the effects of OPCs depletion on microglia in the context of a tumor. IGF-1 is, instead, crucial for OPCs survival and myelination during brain development, when it is mainly produced by CD11c+ microglia cells (Wlodarczyk et al., 2017), a subset of microglia also identified in human gliomas (Friebel et al., 2020). IGF-1, released by TAMs, promotes brain tumor progression and recurrence in response to prolonged administration of CSF-1R inhibitors (Quail et al., 2016; Yao et al., 2020) and might have also some still unexplored effects on OPCs/oligodendrocytes of the TME.
The crosstalk between glioma cells and oligodendrocytes is demonstrated also by the observation that tumor cells use white matter tracts to disseminate into the brain and escape radiotherapy, precluding a complete tumor eradication. Despite being a well-known characteristic of aggressive gliomas (Cuddapah et al., 2014), the mechanisms behind this phenomenon are unclear. Wang et al. (2019) proposed a NOTCH-SOX2-SOX9 axis driving tropism of glioma cells for white matter tracts. They found that CD133+ bona-fide glioma stem cells are preferentially associated with portions of nerve fibers, where myelin is lost and unconstrained axonal signals stimulate glioma cell survival. Partial demyelination nearby white matter infiltration was also recently reported by Brooks et al. (2021). The authors described, in addition, axonal degeneration, OPCs recruitment and microglia activation: all events usually occurring in demyelinated lesions (Franklin and Ffrench-Constant, 2008). Surprisingly, this white matter injured-like microenvironment is sufficient to stop proliferation of infiltrating tumor cells and to upregulated SOX10 which induces differentiation of tumor cells toward the oligodendrocyte lineage (Brooks et al., 2021). Therefore, in white matter tracts tumor cells may encounter opposite signals stimulating stemness or differentiation. What determines the final outcome is still unclear. More importantly, these reports revealed two intriguing aspects: (i) white matter tracts may play an active role in shaping the fate of tumor cells and not just be a route for tumor cell invasion; (ii) myelinating oligodendrocytes directly interact with tumor cells. Mechanism triggering myelin damage as well as signals regulating OPCs/oligodendrocytes-glioma communication need to be clarified with further investigation.
In addition, it is important to remember that neuronal electrical activity not only promotes tumor growth, as we will describe later (Venkatesh et al., 2015, 2017), but can also stimulate OPCs proliferation and restore myelination in demyelinated lesions (Gibson et al., 2014; Ortiz et al., 2019). Notably, OPCs express high levels of NLGN3 which, together with neuronal neurexin, forms axo-glial contacts that favor OPCs proliferation and myelination (Proctor et al., 2015). Activity-dependent release of NLGN3 by OPCs has been observed, but the molecular mechanism is still unknown and doesn’t involve ADAM10 cleavage, as in the case of neuronal-derived NLGN3 (Venkatesh et al., 2017). However, more studies are required to clarify the role of OPC-derived NLGN3 in the TME. OPCs possess also additional characteristics which might be relevant during tumor evolution, like the formation of functional synapses with neurons, the establishment of cellular networks that propagate calcium signaling and the ability to buffer the potassium released after neuronal activity (Menichella et al., 2006; Bergles et al., 2010; Parys et al., 2010; Venkatesh et al., 2019). All these features might allow non-malignant OPCs to functionally integrate into tumor cell networks and tune depolarizing currents in glioma cells, modulating their proliferation (Osswald et al., 2015; Venkatesh et al., 2019). Additional studies will help to understand a possible neuron-OPC-glioma interplay that could drive tumor progression.
The fact that oligodendrocytes possess immunomodulatory properties is a relative recent concept that is still poorly investigated in gliomas, but that might have important implications for disease progression and therapy response. Data from other CNS diseases have recently proven the ability of OPCs to directly interact with immune cells and to modulate inflammation. In a mouse model of preterm white matter injury, OPCs and particularly immature oligodendrocytes are capable of responding to neuroinflammation by upregulating cytokines and the innate immune response receptor TLR3. OPCs/oligodendrocytes stimulated in vitro with a natural ligand of TLR3 secrete factors that activate an inflammatory and phagocytic response in microglia (Boccazzi et al., 2021). OPCs can also upregulate MHC-I/II in response to IFNγ and are able to cross-present antigens and stimulate T cells (Falcão et al., 2018; Kirby et al., 2019). In multiple sclerosis plaques, this ability leads to presentation of myelin antigens and a consequent increase of inflammation, OPCs death and impaired remyelination (Kirby et al., 2019). However, in contexts where a T cell response is required to kill tumor cells, the ability of OPCs to activate cytotoxic T cells might be helpful to counteract tumor growth. The immunomodulatory activity of OPCs/oligodendrocytes can also be exerted through recruitment of innate immune cells. However, OPCs can also impair immune cells function and limit inflammation by upregulating factors, like Sema3A (Majed et al., 2006) or FasL (Dowling et al., 1996), inducing immune cells apoptosis. In addition, microglia phagocytosis and clearance can be inhibited by an excess of myelin debris (Shen et al., 2021) which can be a consequence of tumor infiltration along white matter tracts.
Finally, cell adhesion and ECM molecules, as well as MMPs and metalloproteases inhibitors, have been found enriched in OPCs in vitro and in vivo (Parolisi and Boda, 2018), indicating that these glial cells are an additional and crucial player in tissue remodeling. Moreover, oligodendrocytes release of MMP9 upon IL1β stimulation or after white matter injury, facilitates endothelial tube formation and angiogenesis (Pham et al., 2012), further suggesting a potential implication of oligodendroglia in neovascular remodeling within the TME.
Therefore, although increasing evidence propose OPCs and oligodendrocytes as potential key players in the TME, more work is required to elucidate their roles during disease progression.
The Multifaceted Role of Neuronal Cells in Glioma
Recent important findings unravel an active role of neurons within the TME. However, complex relationships between multiple types of neurons make this mechanism difficult to disentangle (Deisseroth et al., 2004; Ge et al., 2007). During normal development, electrical activity influences the formation of central and peripheral neural system (Gafarov, 2018). In particular, CNS maturation is shaped by patterned waves of electrical activity inducing calcium transients (Wong et al., 1995; Corlew et al., 2004). In the adult brain, excitatory neurons modulate proliferation and differentiation of stem cells in the subgranular zone of the dentate gyrus and in the subventricular zone (Deisseroth et al., 2004; Paez-Gonzalez et al., 2014). Moreover, adult neurons regulate Schwann cell proliferation and survival in the PNS (Maurel and Salzer, 2000) and glial precursors proliferation and myelin plasticity in the CNS, conditioning brain structure and function (Scholz et al., 2009; Takeuchi et al., 2010). Similarly, neurons can also affect glioma proliferation (Johung and Monje, 2017; Tantillo et al., 2020b).
Recent studies have proven that neural activity affects glioma cells behavior in a region-specific manner. Indeed, visual deprivation only affects glioma proliferation when the tumor is located in the visual cortex, but not in the motor cortex (Tantillo et al., 2020b). In addition, the activation of different neuronal populations may have a differential action on glioma growth. In the latest years it has been demonstrated that, while pyramidal cell activity promotes proliferation in both murine and patient-derived high-grade gliomas (Venkatesh et al., 2015, 2017; Tantillo et al., 2020b), the selective stimulation of parvalbumin-positive interneurons elicits the opposite effect in glioma-bearing mice (Tantillo et al., 2020b). Similarly, in other types of cancer such as breast cancer, the stimulation of parasympathetic (cholinergic) afferents reduces tumor growth and the stimulation of sympathetic (noradrenergic) nerves accelerates it (Kamiya et al., 2019). Several studies suggest also that the release of secreted molecules such as neurotransmitters and neurotrophins exert a relevant role in the neural regulation of glioma proliferation. For instance, direct interaction between glioma cell lines and neurons leads to an upregulation of functional GABA A receptor in glioma cells, which strongly correlates with the malignancy of brain tumors (Synowitz et al., 2001). However, since both neurons and glioma cells can induce activity-dependent secretion, unraveling the cellular source of these fundamental factors is technically challenging and require further investigation.
Despite the latest evidence demonstrating a tumor-promoting effect of neurons, an anti-proliferative effect of peritumoral neurons on glioma cells has also been described (Liu et al., 2013). Peritumoral neurons are capable of restraining glioma progression through the pivotal role of PD-L1, whose signaling in brain cells is important for GBM patient survival (Liu et al., 2013). High expression of PD-L1 by peritumoral neurons positively correlates with GBM prognosis, demonstrating the existence of a PD-L1-dependent interaction between peritumoral neurons and tumor cells (Liu et al., 2013). On the contrary, its expression on GBM cells promotes PD-1 receptor activation in TAM and T cells, inhibiting tumor cells phagocytosis and killing (Gordon et al., 2017; Litak et al., 2019).
All these studies depict a complex scenario of bidirectional interactions between neuronal and glioma cells. This entangled crosstalk needs to be completely dissolved in order to achieve a clear understanding of its underlying mechanisms, which may pose the foundation for the development of more effective therapeutic approaches.
The Complex Role of Neurotransmitters and Neurotrophins
It is well-known that glioma cells express functional neurotransmitter receptors, which could interfere with mitogenic pathways. Glutamate is one of the most abundant molecules of the CNS, but its extracellular levels must be maintained low, in order to prevent hyperexcitability and guarantee a normal brain function. Therefore, the scavenger role of astrocytes, through Na+-dependent glutamate transporters (i.e., EAAT1 or EAAT2), is of paramount relevance to maintain this equilibrium (Bergles and Jahr, 1997; Danbolt, 2001; Robert and Sontheimer, 2014). Similarly, glioma cells express high levels of EEAT1/2. When EEAT1/2 is absent or mislocalized glutamate uptake is compromised; this event causes high levels of glutamate in the extracellular space that leads to excitotoxicity (Ye et al., 1999; Buckingham and Robel, 2013; Robert and Sontheimer, 2014; Campbell et al., 2015).
Tumor cells can also extrude glutamate, predominantly through the cystine-glutamate antiporter (system xc-, SXC) that exchanges intracellular glutamate for extracellular cysteine, normally involved in the generation of antioxidant glutathione (de Groot and Sontheimer, 2011). The biochemical source of glutamate for glioma cells is not entirely known. Some studies suggest that glutamate may be generated from glutamine via a glutaminase reaction. Other works, instead, propose that glutamate could be accumulated due to other metabolic impairments, as the decreased conversion in α-ketoglutarate by GLUD2 enzyme (de Groot and Sontheimer, 2011; Franceschi et al., 2018). Glioma cells express both ionotropic and metabotropic glutamate receptors (de Groot and Sontheimer, 2011) and the increased extracellular glutamate levels in proximity of the tumor mass (Behrens et al., 2000) promote survival, growth and migration in part through the activation of AMPA receptors and, consequently, Rho-Akt pathway (Ishiuchi et al., 2007). The high amount of glutamate acts in a paracrine/autocrine manner on tumor cells, facilitating their spread into the brain parenchyma. This event results in excitotoxicity, which damages neurons located in the tumor vicinity. Moreover, gliomas are known to increase the neural excitability of peritumoral neurons (Venkatesh and Monje, 2017), even if the mechanism that links hyperactivity and cell proliferation has still not completely clarified.
In a normal brain, GABA is the most important inhibitory neurotransmitter of the CNS and controls stem cell proliferation in the subventricular zone and hippocampus, limiting the generation of new neuroblasts (Young and Bordey, 2009; Song et al., 2012; Giachino et al., 2014; Blanchart et al., 2017). It has been demonstrated that glioma cells express GABA receptors, whose function is still under debate. Studies on patient-derived glioma cultures show that the expression of functional GABA A receptors correlates more with low-grade gliomas and oligodendrogliomas than GBM (Labrakakis, 1998; Smits et al., 2012). On the contrary, in GBM GABA A receptors are downregulated to reduce GABA inhibition of tumor cell proliferation (Jung et al., 2019). At the same time, other in vitro studies on human GBM samples suggest the opposite (D’Urso et al., 2012). In a p16 Arf–/– PDGFB murine glioma model an endogenous ionotropic signaling has been identified within tumor cells, demonstrating that glioma cells not only respond to GABA, but they can also release it in the TME, limiting cell proliferation of both murine and patient-derived GBM cells (Blanchart et al., 2017). It has been proposed that GABA could have a role in tumor progression, as suggested by in vivo experiments using GABA B agonist (muscimol) and GABA A antagonist (bicuculline). Intriguingly, the effect of GABA is more pronounced in glioma cells with stem-like properties, suggesting a possible strategy to maintain a pool of quiescent tumor initiating cells (Blanchart et al., 2017).
Neurotrophins are a family of secreted molecules that influence many aspects of brain physiology. During malignancies, it is not clear whether they exert a pro or anti-tumorigenic effect on glioma cells (Garofalo et al., 2015; Venkatesh et al., 2015). Their controversial role is still under debate and may depend on their multifaceted functions. For instance, it is known that in pediatric tumors, such as neuroblastoma or medulloblastoma, tumor cells express neurotrophin receptors and the associated clinical outcome is favorable (Donovan et al., 1993; Nakagawara et al., 1993; Eberhart et al., 2001). In adult low-grade gliomas TrkA and TrkB are highly expressed, while their presence in GBM is weak, suggesting that those receptors might be involved only in the less aggressive phase of the disease (Wadhwa et al., 2003). Some studies have shown that the activation of the TrkA receptor through NGF treatment has an anti-mitotic and pro-differentiating effect on C6 glioma cells (Rabin et al., 1998; Kimura et al., 2002). Conversely, on human GBM cell lines, NGF seems to improve tumor proliferation via Notch signaling (Park et al., 2018). In addition, a phase II clinical trial with administration of NGF to patients with childhood optic gliomas led to a statistically significant improvement in vision respect to placebo-treated patients (Falsini et al., 2016).
Also the role of BDNF in the regulation of glioma growth remains controversial. BDNF is a peptide secreted in an activity-dependent manner (Hong et al., 2008). In vitro studies demonstrate that BDNF, through its receptor TrkB, stimulates high grade glioma cells proliferation (Xiong et al., 2013) and that BDNF release increases with neural activity (Venkatesh et al., 2015). At the same time, it has also been shown that BDNF overexpression in the hypothalamus has immune-augmenting properties, provoking an increased anti-tumor immune response and reducing the activity of proteins that would normally confer resistance to chemotherapy (Radin and Patel, 2017). Moreover, glioma-bearing mice reared in an enriched environment (EE) showed enhanced levels of BDNF together with significantly reduced tumor growth, suggesting a pivotal role for this neurotrophin in glioma proliferation (Garofalo et al., 2015). BDNF infusion was also found to reduce TAMs infiltration and activation, and to dampen glioma migration via inhibition of RhoA through the truncated TrkB.T1 receptor (Garofalo et al., 2015). These data also emphasize the idea that lifestyles and physical activity can have a direct impact on the brain microenvironment (Garofalo et al., 2015; Tantillo et al., 2020a), counteracting glioma progression.
Neuronal Alterations Induced by Glioma Growth
Emerging evidence suggests that glioma cells exert a strong influence on neurons and neuronal activity. For instance, deficits in neurocognitive functioning frequently occur in glioma patients, heavily affecting their quality of life (Aaronson et al., 2011; Seano et al., 2019). The tumor mass can hamper brain functionality not only by mechanical compression on brain structures but also by releasing excitotoxins which provoke neuronal death and perturb synaptic transmission (van Kessel et al., 2017).
Approximately 30–50% of patients with brain tumors manifest seizure as an initial symptom of disease progression (van Breemen et al., 2007), making tumor-associated epilepsy (TAE) among the most common hallmarks of comorbidity in glioma patients (Radin and Tsirka, 2020). Interestingly, the incidence of developing TAE is higher in low compared to high grade tumors (van Breemen et al., 2007; You G. et al., 2012), but the reasons of such difference are not known. TAE often manifests as focal seizures with secondary generalization and, despite its major clinical and social impact, the pathophysiological causes are poorly understood. In addition, due to the complex heterogeneity of perturbed mechanisms, this kind of epilepsy is often refractory to antiepileptic treatments (You G. et al., 2012; Cowie and Cunningham, 2014). An important aspect that determines the insurgence of seizures is the localization of the tumor mass in the brain. The proximity to the cortical gray matter is a fundamental factor: tumors that are localized closely to the cortex or in the limbic or perilimbic areas are highly epileptogenic, whereas tumors located in the inner parts of the brain are less prone to manifest seizures (Berntsson et al., 2009; Cowie and Cunningham, 2014). The pathogenesis of TAE is likely to be multifactorial and involves the interaction of genetic factors, changes in the peritumoral microenvironment, hypoxia, acidosis, and metabolic impairments that could affect neural morphology and function (You G. et al., 2012; Cowie and Cunningham, 2014; Armstrong et al., 2016). In addition, the balance between excitatory and inhibitory networks certainly plays a pivotal role in the generation of seizures (Nelson and Turrigiano, 1998; You Y. et al., 2012; MacKenzie et al., 2017). Indeed, the high amount of glutamate extruded by glioma cells overactivates both NMDA and AMPA receptors resulting in excitotoxicity, tumor invasion and hyperexcitability of neural tissues (Rzeski et al., 2001; Savaskan et al., 2008; Sontheimer, 2008; Vanhoutte and Hermans, 2008; Marcus et al., 2010; Robert and Sontheimer, 2014). The prolonged activation of NMDA receptors provokes a sustained influx of Ca2+ into the cell mediating excitotoxicity (Terunuma et al., 2010), whereas the reduction of neuronal cell density creates room for tumor expansion (Buckingham et al., 2011) and the overactivation of AMPA receptors further boosts neural excitability (Savaskan et al., 2008). In order to adapt to a glutamate-rich environment and avoid the toxic effects of glutamate, glioma cells downregulate AMPA or NMDA receptors in a grade- and spatial-dependent manner. Indeed, expression analyses of different tumor regions showed that their expression is highest at the infiltrating front of the tumors, suggesting a role in mediating tumor invasion (Radin and Tsirka, 2020).
Multiple factors are necessary to establish long-lasting hyperexcitation and concur to support the onset of peritumoral epilepsy (Campbell et al., 2015). For instance, pyramidal peritumoral neurons downregulate KCC2 and upregulate NKCC1, two transporters responsible for exporting and importing chloride, respectively. The consequent abnormal high amount of intracellular chloride causes GABA-mediated depolarization, reversing chloride potential and leading to an excitatory and detrimental action of GABA receptor in pyramidal neurons (Conti et al., 2011; Di Angelantonio et al., 2014; Pallud et al., 2014; Campbell et al., 2015; Huberfeld and Vecht, 2016; Jung et al., 2019). In addition, peritumoral regions show a significant loss of approximately 35% of GABAergic interneurons, together with a reduction in their firing rates and in synapses with pyramidal neurons (Campbell et al., 2015; Tewari et al., 2018; Yu et al., 2020). Both altered chloride homeostasis and decreased GABAergic inhibition contribute to render the peritumoral tissue more prone to seizures (Buckingham et al., 2011; Venkatesh et al., 2019).
Finally, several studies have shown that the perineuronal nets (PNNs), which surround fast spiking interneurons acting as ionic buffer, neuroprotecting and stabilizing their synaptic activity, resulted degraded by glioma-released proteases. This provokes an increased membrane capacitance and, in turn, reduces the firing rate of the remaining peritumoral inhibitory interneurons within 400 μm from tumor edge. In particular, the pro-seizure effect of PNN degradation seems to be due to the lacking role of electrostatic insulators affecting the physiological properties of fast-spiking interneurons (Buckingham et al., 2011; Tewari et al., 2018; Venkatesh et al., 2019).
Astrocytomas are able to directly interact with the surrounding environment, extending ultra-long membrane protrusions called tumor microtubes (TMs) that represent a route for brain invasion (Osswald et al., 2015). Recently, it has been discovered that glioma cells can create an intricate interconnection with neurons, forming an electrically coupled network. Within these networks neuronal activity evokes non-synaptic activity-dependent potassium currents amplified by tumor mediated gap junctions (Venkataramani et al., 2019, 2021). The presence of these structures correlates with the worst prognosis in human gliomas and confers resistance against all the available therapies (Osswald et al., 2015; Weil et al., 2017). Studies in Drosophila demonstrate that brain tumor cells protrude TMs to enwrap neurons, inhibiting the Wnt pathway through the accumulation of Fz1 receptor or through the secretion of pathway antagonists (such as Impl2), thus causing neurodegeneration (Portela et al., 2019; Jarabo et al., 2021). In addition, glioma cells interact with peritumoral excitatory neurons forming functional bona fide AMPA-mediated synapses (Venkataramani et al., 2019), through which pro-mitotic peptides as NLGN3 can stimulate glioma progression (Venkatesh et al., 2019). Surprisingly, even tumor cells of non-brain origin, like metastatic breast cancer cells, establish synaptic contacts with neurons (Zeng et al., 2019). This suggests that tumor-neuron synapses are an efficient way that tumor cells exploit in order to obtain glutamate and sustain their proliferation.
Neuronal Alterations Induced by Glial Cells in the TME
In addition to neuronal alterations directly induced by glioma cells, glial cells response can also profoundly affect brain functionality. Being coopted by tumor cells, glial cells can no longer support brain homeostasis. In addition, activated glial cells contribute to establish a TME milieu rich of cytokines, metabolites and signaling molecules that have potential detrimental effects on neuronal activity. The inflammatory status of gliomas is the result of complex interactions of different TME players. Astrocytes, TAMs and oligodendrocytes all possess immunomodulatory properties, including the production of a broad spectrum of cytokines. Beside their prominent role in stimulating or suppressing immune and glial cells activity, the cytokines present in the TME can also affect neuron survival and activity. Although there are some examples of chemokines with neuroprotective roles (Trettel et al., 2020), an excessive accumulation of pro- and anti-inflammatory cytokines has usually neurotoxic effects. Cytokines like IL1α/β, TNFα, and TGFβ, which are highly enriched within the TME, have been shown to induce neurons loss, impair neuronal morphogenesis and are associated with many neurodegenerative diseases (Glass et al., 2010; Nakashima et al., 2018).
Inflammation is also a consequence of aggressive chemo- and radiotherapies that can exacerbate a neurotoxic microglia-astrocytes crosstalk with long-term consequences on neurons integrity, myelin plasticity as well as neurogenesis (Monje et al., 2003; Gibson et al., 2019). It is well-known that anti-cancer treatments, beyond their unquestionable advantage of delaying tumor progression, are accompanied by multiple side effects that in most of the cases are short-term and temporary. However, what is less known are the long-term consequences of these therapies, especially on brain functions, with patients experiencing deficits in memory, attention, information process, and even mental health (Gibson and Monje, 2021). In addition, neurotoxicity associated with cerebral edema has been reported after immunomodulatory treatments, although the underlying mechanisms are still undefined (Gust et al., 2017; Spain et al., 2017).
Cerebral edema is a common complication of brain tumors and is generally associated with increased intracranial pressure, reduced blood flow and hypoxia, all events that lead to neuronal deficits (Roth et al., 2013). These phenomena can arise as consequence of the presence of a tumor mass, that physically compresses and remodels the brain parenchyma. For example, by migrating along blood vessels, tumor cells displace astrocytic endfeet from endothelial cells causing loss of endothelial tight junctions and breaches in the BBB (Watkins et al., 2014). Similar effects have also been described in some multiple sclerosis active plaques, where OPCs, due to a defective migration, accumulate along the blood vessels disrupting the BBB. Mechanistically, an excessive Wnt signaling in OPCs triggers perivascular accumulation and release of Wif-1 which inhibits Wnt ligand essential for endothelial cells tight junction, compromising the integrity of the endothelial barrier (Niu et al., 2019). OPCs expressing Wif-1 were also found in gliomas (Asslaber et al., 2017), suggesting that their influence on BBB homeostasis might not be limited to multiple sclerosis but might be extended to brain tumors. Disruption of the neurovascular coupling during disease progression impairs the hemodynamic response after seizures, causing hypoxia and exacerbating the seizure-related brain damages (Montgomery et al., 2020). Glial and immune cells in the TME can also actively contribute to cerebral edema. Indeed, BBB and vessels permeability are altered by cytokines and factors, like VEGF, released by tumor cells, astrocytes and TAMs (Calabrese et al., 2007; Priego et al., 2018; Sankowski et al., 2019; Klemm et al., 2020). Beside its pro-angiogenic functions, VEGF can induce expression of AQ4, a water-channel that is mainly present on astrocytes composing the BBB and that controls its permeability (Lan et al., 2017). Anti-angiogenic treatments that normalized tumor vasculature have demonstrated efficacy also in reducing brain edema in GBM patients (Batchelor et al., 2007). Corticosteroids are the drug of choice to treat glioma-induced cerebral edema for their ability to downregulate VEGF, modulate the expression of channels and tight junctions’ components and, therefore, limiting BBB permeability. However, among other side effects, corticosteroids, and in particular dexamethasone, show immunosuppression and might have additional and unknown effects on cells of the TME that deserve further scrutiny (Cenciarini et al., 2019).
As discussed earlier, PNNs degradation alters neuronal activity concurring to TAE. The contribution of glial cells to PNNs remodeling has been widely demonstrated in physiological conditions and in other diseases (Crapser et al., 2020; Nguyen et al., 2020), but it remains speculative in glioma. Indeed, TAMs, astrocytes and OPCs are important sources of MMPs and can assist tumor cells in ECM remodeling, possibly including PNNs degradation. Exposure to an EE, achieved by continuous physical, sensory and social stimulation, is also known to shape ECM and PNNs, to induce plasticity and neurogenesis and to modulate microglia and astrocytes activation (Baroncelli et al., 2010; Rodríguez et al., 2013; Kempermann, 2019; Yates, 2020). Interestingly, an EE is sufficient to significantly prolong the survival of glioma-bearing mice through re-education of TAMs toward a less immunosuppressive state and through IL15-mediated recruitment of anti-tumor NK cells, which also contribute to TAMs re-polarization (Garofalo et al., 2015, 2017).
Neuron hyperexcitability and acute seizures could be attenuated by microglia via P2RY12 (Eyo et al., 2014) which is, however, downregulated in glioma (Sankowski et al., 2019; Friebel et al., 2020; Guldner et al., 2020; Klemm et al., 2020). In a zebrafish brain tumor model, this mechanism was hijacked by tumor cells that transiently increased intracellular Ca2+ levels and ATP release, in order to coopt P2RY12+ microglia and establish direct microglia-to-tumor contacts that stimulate tumor proliferation (Chia et al., 2019). It remains to be investigated whether tumor cells can exploit also the synapse pruning ability of microglia and astrocytes to form and remodel glioma synapses, possibly at the expense of neurons (Paolicelli et al., 2011; Chung et al., 2013). Of note, specific astrocyte subpopulations have been shown to support synapse formation and their emergence in gliomas correlates with increased hyperexcitability and seizures (John Lin et al., 2017).
In the healthy brain, astrocytes are responsible for removing the excess of extracellular glutamate via EAAT1/2 transporters and converting glutamate into glutamine, which is released and re-used by neurons (Mahmoud et al., 2019). However, in gliomas, peritumoral reactive astrocytes show altered electrophysiological properties as well as decreased glutamate uptake and glutamine production, that lead to GABAergic disinhibition and neuronal hyperexcitability (Campbell et al., 2020). Therefore, reactive astrocytes might propagate the glutamate excitotoxicity and exacerbate, instead of preventing, neuronal death and glioma-induced epilepsy.
Conclusion and Future Perspectives
In physiological conditions a balance among glial cells, immune cells and neurons is necessary and established with the final goal of sustaining neuronal activity and brain homeostasis. In case of malignancy, tumor cells represent an additional player introduced in the game, forcing the equilibrium at its advantage. Indeed, tumor cells coopt neurons and glial cells to create a microenvironment that influences their growth (Figure 1). In this context, new interactions are established between tumor and brain resident cells. Interestingly, some of these interactions vary depending on the type of glioma, suggesting a preferred mode of communication that might be intrinsically related to the tumor cell-of-origin or be dictated by specific mutations (Gao et al., 2020).
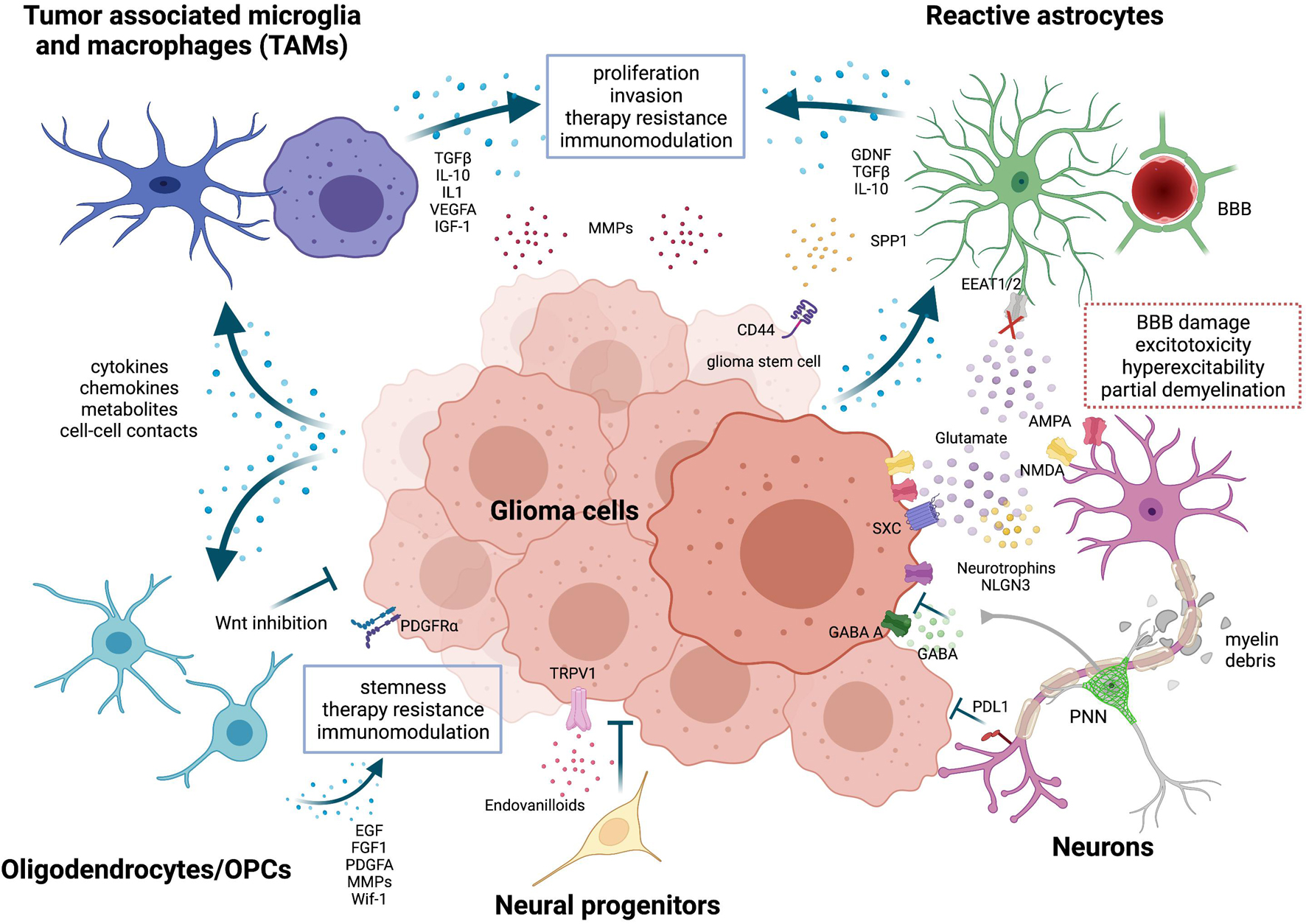
Figure 1. Neurons and glial cells in the glioma microenvironment. Tumor-associated microglia and macrophages (TAMs) are reprogrammed by tumor cells through the release of cytokines, chemokines, metabolites or through cell-cell contacts. Activated TAM produce factors that stimulate glioma proliferation, invasion, angiogenesis, and therapy resistance. Metalloproteinases (MMPs) are released by both tumor cells and TAMs to facilitate glioma cells migration. TME-associated astrocytes are efficiently coopted by tumor cells. They promote tumor migration by expressing chemoattractant factors like GDNF and support stemness via SPP1-CD44 axis. Both TAMs and astrocytes also play a role in immunomodulation, expressing high levels of immunosuppressive cytokines, such as TGFβ and IL-10. Oligodendrocytes and oligodendrocyte progenitor cells (OPCs) release factors like EGF, FGF1 and PDGFA that favor proliferation, stemness and chemotherapy resistance of glioma cells. But, at the same time, OPCs can also impair tumor growth through Wnt inhibition. Neural progenitor cells delay tumor growth and induce tumor cell death via endovanilloids stimulation of TRPV1. Neurons have a multifaceted role in glioma. Peritumoral neurons were found to counteract glioma progression through programmed death-ligand 1 (PD-L1) protein, but recent evidence has shown that the activation of peritumoral excitatory neurons was able to increase glioma proliferation. Selective activation of peritumoral parvalbumin interneurons elicited, instead, the opposite effect. In addition, tumor cells extrude glutamate outside predominantly through the overexpression of the cystine-glutamate antiporter (system xc-, SXC); this event is not harmonized by peritumoral astrocytes, which show a compromised glutamate uptake via the Na+-dependent glutamate transporters (i.e., EAAT1 or EAAT2). High amount of glutamate provokes an over-activation of both NMDA and AMPA receptors on neuronal cells, resulting in excitotoxicity and hyperexcitability of neural tissues and eventually promotes survival, growth and migration of glioma cells. Glutamatergic neurons can enhance glioma progression also by releasing NLGN3 and neurotrophins and by establishing functional bona fide AMPA-mediated synapses with tumor cells. Invading glioma cells, together with activated glial cells in the tumor microenvironment, cause also secondary alterations like perineural nets (PNNs) remodeling, partial demyelination and blood-brain-barrier (BBB) leakage. Image created with BioRender.com.
Tumor cells exploit and even strengthen pre-existing interactions within the TME. Astrocytes, TAMs and OPCs/oligodendrocytes actively communicate and modulate each other’s activation in physiological conditions, as well as during tumor progression. Yet, neuronal activity is affected by direct interaction with tumor cells and also indirectly by changes in the TME. Although some of the cellular and molecular mechanisms behind these complex networks have been recently uncovered, many aspects still need to be further investigated. For example, it still remains unclear to what extent cell-cell interactions dynamically change during disease progression and in response to treatments. In addition, cell types like OPCs, that have been so far neglected in gliomas, definitely deserve more attention.
Novel therapeutic strategies that target the TME have recently emerged, showing promising results especially when used in combination with other treatments (Kowal et al., 2019). This suggests that acting on several fronts is a successful strategy to impair cellular interplays in the TME and tumor-TME crosstalk. Nevertheless, there is still a great deal to be done. Indeed, in glioma patients, clinical trials of TME-targeting therapies, and especially immunomodulatory therapies, didn’t meet the expectations. Part of the failure is certainly due to the intrinsic therapeutic resistance of gliomas and the particularly immunosuppressed brain microenvironment (Sampson et al., 2020). However, it would be also important to understand the contribution of cells in the TME that are not directly targeted by these treatments.
Interestingly, cells in the TME can undergo senescence due to aging (Fane and Weeraratna, 2019) or upon chemo/radiotherapy (Wang et al., 2020). Cellular senescence is a complex cellular state which determines not only cell intrinsic effects, like cycle arrest and DNA damage response, but also non-cell-autonomous mechanisms that influence the surrounding microenvironment. The senescence-associated secretory phenotype (SASP) involves the secretion of pro-inflammatory factors, including cytokines and MMPs, that promote various aspects of tumor development like tumor cell proliferation (Kuilman et al., 2008) and immunosuppression (Ruhland et al., 2016). Although numerous studies have reported that non-malignant cells can acquire a senescent phenotype and promote tumor progression in multiple cancers, little is known about senescent non-malignant cells in the brain TME (Wang et al., 2020). Chemotherapy triggers in microglia and astrocytes a pro-inflammatory secretome (Gibson et al., 2019), which is similarly induced by physiological aging (Li et al., 2015; Clarke et al., 2018) or neurodegenerative diseases (Bhat et al., 2012; Liddelow et al., 2017). Progenitor cells and OPCs also show a senescence phenotype in pathological conditions (Nicaise et al., 2019; Zhang et al., 2019) and secondary detrimental effects of senescent glial cells have been reported also on neurons (Limbad et al., 2020). Senolytic therapies have been efficiently used to eliminate senescent cells in multiple diseases, including cancers (Sieben et al., 2018). JAK2/STAT3 inhibitors, beside their immunomodulatory role on tumor-associated astrocytes (Priego et al., 2018), have been used to reprogram SASP and delay prostate cancer growth (Toso et al., 2014). Moreover, CAR T cells, genetically modified T cells which usually recognize tumor cells-specific antigens, have also been recently engineered to target senescent cells, with promising results in mouse models (Amor et al., 2020). Interestingly, the senescence-specific antigen used to target senescent cells was uPAR, which is also highly upregulated by tumor-associated astrocytes (Le et al., 2003). Therefore, senolytic treatments could be considered as an alternative strategy to target the TME and might be used in combination with more classic anticancer therapies. However, further investigations are certainly necessary to unravel the mechanisms inducing senescence in brain TME cells and in order to identify the factors produced by senescent cells to sustain tumor progression.
We should also not forget secondary alterations, that can be due to direct detrimental effects of tumor cells or to indirect glial cells dysregulation in the TME. In both cases such alterations profoundly affect brain functionality and, consequently, patients’ quality of life; therefore, these events should be considered in the choice of the therapeutic intervention and in the development of alternative treatments.
Finally, some technical considerations should be made. We have here reviewed the most important recent studies that have contributed to add pieces to the complex jigsaw of the brain TME, employing different models (e.g., human, mouse, Drosophila, zebrafish) and approaches (e.g., in vitro, in vivo). Certainly, every model has its own advantages and disadvantages that must be taken into account in the interpretation of the obtained results; remarkably, using different approaches could help to tackle the issue from different points of view, producing a more complete and reliable scenario of the cellular and molecular players in the TME. Since the immune system exerts a critical role in the TME (Lei et al., 2020; Sampson et al., 2020) and, as reported in this review and in many studies, all the TME cells possess immunomodulatory functions, it would be desirable to choose models that do not exclude this important player of the TME (i.e., immunocompetent animal models). In order to address some of the current gaps of knowledge, innovative experimental approaches have been more recently developed. Patients-derived organoids or organotypic cultures are, for example, 3D in vitro models that reproduce the TME interactions and could be very useful for testing novel drugs or screening the patient-specific response and subsequently choose the proper therapeutic intervention (Pamies et al., 2020). However, due to their complexity, at the moment these types of models are of difficult adaptation to high-throughput screening. New technologies are also currently aiming at overcoming other limitations, such as the integration of microfluidics or 3D bioprinting of tissue structures to circumvent the lack of a complete vasculature system and the infiltration of peripheral immune components (Li et al., 2020; Pamies et al., 2020).
Dissecting the contribution of different cells of the TME and understanding their interactions is of paramount importance to shed light on the mechanisms that glioma cells exploit to support their growth and invasion. A profound knowledge of the TME is a puzzling but relevant issue, because it will highlight the molecules that could be targeted for the development of novel therapeutic strategies aimed at counteracting brain tumors.
Author Contributions
EP, ET, and EV: conceptualization. EP, MS, and EV: review and editing. EP, MS, EM, ET, and EV: contribution to draft preparation. MS and EM: figure making. All authors contributed to the draft preparation, have read and agreed to the published version of the manuscript.
Funding
This research was funded by AIRC, grant number IG18925, by PRIN project 20178L7WRS. EP was funded by the Swiss Cancer League (KLS-4518-08-2018) and the Forschungsfonds Nachwuchsforschende of the University of Basel.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Prof. Matteo Caleo and Dr. Claudio Giachino for the critical reading of the manuscript.
Glossary
| ADAM10 | A Disintegrin And Metalloprotease domain 10 |
| AKT | Serine/Threonine Kinase |
| AKT1 | Serine/Threonine Kinase 1 |
| AMPA | Alpha-amino-3-hydroxy-5-Methyl-4-isoxazolePropionic Acid |
| AMPAR | Alpha-amino-3-hydroxy-5-Methyl-4-isoxazolePropionic Acid Receptor |
| AQ4 | Aquaporin-4 ATP Adenosine Triphosphate BBB Blood Brain Barrier |
| BDNF | Brain-Derived Neurotrophic Factor |
| CAR T | Chimeric Antigen Receptor |
| T CCL2 | C-c motif Chemokine Ligand 2 |
| CCL5 | C-c motif Chemokine Ligand 5 |
| CCR2 | C-c motif Chemokine Receptor 2 |
| CD11b | Cluster of Differentiation molecule 11b |
| CD11c | Cluster of Differentiation molecule 11c |
| CD133 | Cluster of Differentiation molecule 133 |
| CD169 | Cluster of Differentiation molecule 169 |
| CD206 | Cluster of Differentiation molecule 206 |
| CD209 | Cluster of Differentiation molecule 209 |
| CD44 | Cluster of Differentiation molecule 44 |
| CD45 | Cluster of Differentiation molecule 45 |
| CD47 | Cluster of Differentiation molecule 47 |
| CD49d | Cluster of Differentiation molecule 49d |
| CNS | Central Nervous System |
| CSF-1R | Colony Stimulating Factor 1 Receptor |
| CSF1 | Colony Stimulating Factor 1 |
| CX3CL1 | C-X3-c motif Chemokine Ligand 1 |
| Cx43 | Connexin 43 |
| Cxcr4 | C-x-c motif chemokine receptor 4 |
| EAAT1 | Excitatory Amino Acid Transporter 1 |
| EAAT2 | Excitatory Amino Acid Transporter 2 |
| ECM | Extracellular Matrix |
| EE | Enriched Environment |
| EGF | Epidermal Growth Factor |
| EGFR | Epidermal Growth Factor Receptor |
| FasL | Fas Ligand |
| FGF1 | Fibroblast Growth Factor 1 |
| Fz1 | Frizzled class receptor 1 |
| GABA | Gamma-Aminobutyric Acid |
| GABA A | Gamma-Aminobutyric Acid type A |
| GABA B | Gamma-Aminobutyric Acid type B |
| GBM | Glioblastoma Multiforme |
| GDNF | Glial cell line-Derived Neurotrophic Factor |
| GFAP | Glial Fibrillary Acidic Protein |
| GLAST | Glutamate-ASpartate Transporter |
| GLUD2 | Glutamate Dehydrogenase 2 |
| Hexb | Hexosaminidase subunit beta |
| HIF1A | Hypoxia Inducible Factor 1 subunit Alpha |
| Iba1 | Ionized calcium-binding adapter molecule 1 |
| IDH | Isocitrate Dehydrogenase |
| IFNγ | InterFeroN Gamma |
| IGF-1 | Insuline-like Growth Factor 1 |
| IL1 | Interleukin 1 |
| IL6 | Interleukin 6 |
| IL10 | Interleukin 10 |
| IL15 | Interleukin 15 |
| IL1α | Interleukin 1 alpha |
| IL1β | Interleukin 1 beta |
| Impl2 | Imaginal morphogenesis protein-late 2 |
| Ink | Inhibitors of cyclin-dependent kinase |
| Ink4A/Arf or p16 | Inhibitors of cyclin-dependent kinase 4a/Alternate reading frame |
| JAK/STAT | Janus Kinase/Signal Transducers and Activators of Transcription |
| KCC2 | Potassium Chloride Cotransporter 2 |
| Lgals3 | Galectin 3 |
| MARCO | Macrophage Receptor with Collagenous structure |
| MDMs | Monocyte-Derived Macrophage |
| MerTK | Mer Tyrosine Kinase |
| MHC-I | Major Histocompatibility Complex class I |
| MHC-II | Major Histocompatibility Complex class II |
| MMP2 | Matrix MetalloPeptidase 2 |
| MMP9 | Matrix MetalloPeptidase 9 |
| MT1-MMP | Membrane Type 1 Matrix MetalloProteinase |
| NF1 | NeuroFibromatosis type 1 |
| NG2 | Neural/Glial antigen 2 |
| NGF | Nerve Growth Factor |
| NK | Natural Killer |
| NKCC1 | Na-K-Cl Cotransporter 1 |
| NLGN3 | Neuroligin 3 |
| NMDA | N-Methyl-D-Aspartic acid or N-Methyl-D-Aspartate |
| NMDAR | N-Methyl-D-Aspartic acid or N-Methyl-D-Aspartate Receptor |
| NSC | Neural Stem Cell |
| OPC | Oligodendrocyte Progenitor Cell |
| P2RY12 | Purinergic Receptor P2Y12 |
| PA | Plasminogen Activator |
| PD-1 | Programmed cell Death protein 1 |
| PD-L1 | Programmed Death-Ligand 1 |
| PDGFA | Platelet-Derived Growth Factor subunit A |
| PDGFB | Platelet-Derived Growth Factor subunit B |
| PDGFC | Platelet-Derived Growth Factor C |
| PDGFRA/PDGFRα | Platelet-Derived Growth Factor Receptor Alpha |
| PDGFRβ | Platelet-Derived Growth Factor Receptor beta |
| PI3K | Phosphoinositide 3-Kinase |
| PNNs | Perineuronal Nets |
| PTEN | Phosphatase and Tensin homologue |
| RhoA | Ras homolog family member A |
| Sall1 | Spalt like transcription factor 1 |
| SASP | Senescence-Associated Secretory Phenotype |
| Sema3A | Semaphorin 3A |
| SHH | Sonic HedgeHog signaling molecule |
| SOX10 | Sry-box transcription factor 10 |
| SOX2 | Sry-box transcription factor 2 |
| SOX9 | Sry-box transcription factor 9 |
| SPP1 | Secreted Phosphoprotein 1 |
| STAT3 | Signal Transducer and Activator of Transcription 3 |
| SXC | System XC- cystine/glutamate antiporter |
| TAE | Tumor-Associated Epilepsy |
| TAMs | Tumor-Associated Microglia/Macrophages |
| TGFβ | Transforming Growth Factor Beta |
| TLR3 | Toll-Like Receptor 3 |
| TME | Tumor Microenvironment |
| Tmem119 | Transmembrane protein 119 |
| TMs | Tumor Microtubes |
| TNFα | Tumor Necrosis Factor Alpha |
| TrkA | Tropomyosin receptor kinase A |
| TrkB | Tropomyosin receptor kinase B |
| TrkB.T1 | Truncated isoform of tropomyosin receptor kinase B |
| TRPV1 | Transient Receptor Potential Vanilloid 1 |
| uPA | urokinase-type Plasminogen Activator |
| uPAR | urokinase-type Plasminogen Activator Receptor |
| VEGF | Vascular Endothelial Growth Factor |
| VEGF-A | Vascular Endothelial Growth Factor A |
| Wif-1 | Wnt inhibitory factor 1 |
| Wnt | Wingless/integrated. |
References
Aaronson, N. K., Taphoorn, M. J. B., Heimans, J. J., Postma, T. J., Gundy, C. M., Beute, G. N., et al. (2011). Compromised health-related quality of life in patients with low-grade glioma. J. Clin. Oncol. 29, 4430–44350. doi: 10.1200/Jco.2011.35.5750
Akkari, L., Bowman, R. L., Tessier, J., Klemm, F., Handgraaf, S. M., de Groot, M., et al. (2020). Dynamic changes in glioma macrophage populations after radiotherapy reveal CSF-1R inhibition as a strategy to overcome resistance. Sci. Transl. Med. 12:7843. doi: 10.1126/scitranslmed.aaw7843
Alcantara Llaguno, S., Chen, J., Kwon, C. H., Jackson, E. L., Li, Y., Burns, D. K., et al. (2009). Malignant astrocytomas originate from neural stem/progenitor cells in a somatic tumor suppressor mouse model. Cancer Cell 15, 45–56. doi: 10.1016/j.ccr.2008.12.006
Alcantara Llaguno, S., Sun, D., Pedraza, A. M., Vera, E., Wang, Z., Burns, D. K., et al. (2019). Cell-of-origin susceptibility to glioblastoma formation declines with neural lineage restriction. Nat. Neurosci. 22, 545–555. doi: 10.1038/s41593-018-0333-8
Amankulor, N. M., Kim, Y., Arora, S., Kargl, J., Szulzewsky, F., Hanke, M., et al. (2017). Mutant idh1 regulates the tumor-associated immune system in gliomas. Genes Dev. 31, 774–786. doi: 10.1101/gad.294991.116
Amor, C., Feucht, J., Leibold, J., Ho, Y.-J., Zhu, C., Alonso-Curbelo, D., et al. (2020). Senolytic CAR T cells reverse senescence-associated pathologies. Nature 583, 127–132. doi: 10.1038/s41586-020-2403-9
Armstrong, T. S., Grant, R., Gilbert, M. R., Lee, J. W., and Norden, A. D. (2016). Epilepsy in glioma patients: mechanisms, management, and impact of anticonvulsant therapy. Neuro Oncol. 18, 779–789. doi: 10.1093/neuonc/nov269
Askew, K., Li, K., Olmos-Alonso, A., Garcia-Moreno, F., Liang, Y., Richardson, P., et al. (2017). Coupled proliferation and apoptosis maintain the rapid turnover of microglia in the adult brain. Cell Rep. 18, 391–405. doi: 10.1016/j.celrep.2016.12.041
Asslaber, M., Schauer, S., Gogg-Kamerer, M., Bernhart, E., Quehenberger, F., and Haybaeck, J. (2017). Native oligodendrocytes in astrocytomas might inhibit tumor proliferation by WIF1 expression. J. Neuropathol. Exp. Neurol. 76, 16–26. doi: 10.1093/jnen/nlw098
Ayata, P., Badimon, A., Strasburger, H. J., Duff, M. K., Montgomery, S. E., Loh, Y. H. E., et al. (2018). Epigenetic regulation of brain region-specific microglia clearance activity. Nat. Neurosci. 21, 1049–1060. doi: 10.1038/s41593-018-0192-3
Bachoo, R. M., Maher, E. A., Ligon, K. L., Sharpless, N. E., Chan, S. S., You, M. J., et al. (2002). Epidermal growth factor receptor and Ink4a/Arf: convergent mechanisms governing terminal differentiation and transformation along the neural stem cell to astrocyte axis. Cancer Cell 1, 269–277. doi: 10.1016/s1535-6108(02)00046-6
Bahadur, S., Kumar Sahu, A., Baghel, P., and Saha, S. (2019). Current promising treatment strategy for glioblastoma multiform: a review. Oncol. Rev. 13:417. doi: 10.4081/oncol.2019.417
Baroncelli, L., Braschi, C., Spolidoro, M., Begenisic, T., Sale, A., and Maffei, L. (2010). Nurturing brain plasticity: impact of environmental enrichment. Cell Death Differ. 17, 1092–1103. doi: 10.1038/cdd.2009.193
Batchelor, T. T., Sorensen, A. G., di Tomaso, E., Zhang, W.-T., Duda, D. G., Cohen, K. S., et al. (2007). AZD2171, a Pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell 11, 83–95. doi: 10.1016/j.ccr.2006.11.021
Behrens, P. F., Langemann, H., Strohschein, R., Draeger, J., and Hennig, J. (2000). Extracellular glutamate and other metabolites in and around RG2 rat glioma: an intracerebral microdialysis study. J. Neurooncol. 47, 11–22. doi: 10.1023/A:1006426917654
Bennett, F. C., Bennett, M. L., Yaqoob, F., Mulinyawe, S. B., Grant, G. A., Hayden Gephart, M., et al. (2018). A combination of ontogeny and CNS environment establishes microglial identity. Neuron 98, 1170.e–1183.e. doi: 10.1016/j.neuron.2018.05.014
Bennett, M. L., Bennett, F. C., Liddelow, S. A., Ajami, B., Zamanian, J. L., Fernhoff, N. B., et al. (2016). New tools for studying microglia in the mouse and human CNS. Proc. Natl. Acad. Sci. U.S.A. 113, E1738–E1746. doi: 10.1073/pnas.1525528113
Berg, T. J., Marques, C., Pantazopoulou, V., Johansson, E., von Stedingk, K., Lindgren, D., et al. (2021). The irradiated brain microenvironment supports glioma stemness and survival via astrocyte-derived Transglutaminase 2. Cancer Res. 81, 2105–2115. doi: 10.1158/0008-5472.CAN-20-1785
Bergles, D. E., Jabs, R., and Steinhäuser, C. (2010). Neuron-glia synapses in the brain. Brain Res. Rev. 63, 130–137. doi: 10.1016/j.brainresrev.2009.12.003
Bergles, D. E., and Jahr, C. E. (1997). Synaptic activation of glutamate transporters in hippocampal astrocytes. Neuron 19, 1297–1308. doi: 10.1016/S0896-6273(00)80420-1
Berntsson, S. G., Malmer, B., Bondy, M. L., Qu, M., and Smits, A. (2009). Tumor-associated epilepsy and glioma: are there common genetic pathways? Acta Oncol. (Madr.) 48, 955–963. doi: 10.1080/02841860903104145
Bhat, R., Crowe, E. P., Bitto, A., Moh, M., Katsetos, C. D., Garcia, F. U., et al. (2012). Astrocyte senescence as a component of Alzheimer’s disease. PLoS One 7:e45069. doi: 10.1371/JOURNAL.PONE.0045069
Blanchart, A., Fernando, R., Häring, M., Assaife-Lopes, N., Romanov, R. A., Andäng, M., et al. (2017). Endogenous GAB AA receptor activity suppresses glioma growth. Oncogene 36, 777–786. doi: 10.1038/onc.2016.245
Boccazzi, M., Van Steenwinckel, J., Schang, A.-L., Faivre, V., Le Charpentier, T., Bokobza, C., et al. (2021). The immune-inflammatory response of oligodendrocytes in a murine model of preterm white matter injury: the role of TLR3 activation. Cell Death Dis. 122, 1–12. doi: 10.1038/s41419-021-03446-9
Bowman, R. L., Klemm, F., Akkari, L., Pyonteck, S. M., Sevenich, L., Quail, D. F., et al. (2016). Macrophage ontogeny underlies differences in tumor-specific education in brain malignancies. Cell Rep. 17, 2445–2459. doi: 10.1016/J.CELREP.2016.10.052
Brandao, M., Simon, T., Critchley, G., and Giamas, G. (2019). Astrocytes, the rising stars of the glioblastoma microenvironment. Glia 67, 779–790. doi: 10.1002/glia.23520
Brooks, L. J., Clements, M. P., Burden, J. J., Kocher, D., Richards, L., Devesa, S. C., et al. (2021). The white matter is a pro-differentiative niche for glioblastoma. Nat. Commun. 12:2184. doi: 10.1038/s41467-021-22225-w
Bruttger, J., Karram, K., Wörtge, S., Regen, T., Marini, F., Hoppmann, N., et al. (2015). Genetic cell ablation reveals clusters of local self-renewing microglia in the mammalian central nervous system. Immunity 43, 92–106. doi: 10.1016/j.immuni.2015.06.012
Buckingham, S. C., Campbell, S. L., Haas, B. R., Montana, V., Robel, S., Ogunrinu, T., et al. (2011). Glutamate release by primary brain tumors induces epileptic activity. Nat. Med. 17, 1269–1274. doi: 10.1038/nm.2453
Buckingham, S. C., and Robel, S. (2013). Glutamate and tumor-associated epilepsy: glial cell dysfunction in the peritumoral environment. Neurochem. Int. 63, 696–701. doi: 10.1016/j.neuint.2013.01.027
Buffo, A., Rolando, C., and Ceruti, S. (2010). Astrocytes in the damaged brain: molecular and cellular insights into their reactive response and healing potential. Biochem. Pharmacol. 79, 77–89. doi: 10.1016/j.bcp.2009.09.014
Butovsky, O., Jedrychowski, M. P., Moore, C. S., Cialic, R., Lanser, A. J., Gabriely, G., et al. (2014). Identification of a unique TGF-β-dependent molecular and functional signature in microglia. Nat. Neurosci. 17, 131–143. doi: 10.1038/nn.3599
Butowski, N., Colman, H., De Groot, J. F., Omuro, A. M., Nayak, L., Wen, P. Y., et al. (2016). Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: an Ivy foundation early phase clinical trials consortium phase II study. Neuro Oncol. 18, 557–564. doi: 10.1093/neuonc/nov245
Buttgereit, A., Lelios, I., Yu, X., Vrohlings, M., Krakoski, N. R., Gautier, E. L., et al. (2016). Sall1 is a transcriptional regulator defining microglia identity and function. Nat. Immunol. 17, 1397–1406. doi: 10.1038/ni.3585
Calabrese, C., Poppleton, H., Kocak, M., Hogg, T. L., Fuller, C., Hamner, B., et al. (2007). A perivascular niche for brain tumor stem cells. Cancer Cell 11, 69–82. doi: 10.1016/j.ccr.2006.11.020
Campbell, S. C., Muñoz-Ballester, C., Chaunsali, L., Mills, W. A., Yang, J. H., Sontheimer, H., et al. (2020). Potassium and glutamate transport is impaired in scar-forming tumor-associated astrocytes. Neurochem. Int. 133:104628. doi: 10.1016/j.neuint.2019.104628
Campbell, S. L., Robel, S., Cuddapah, V. A., Robert, S., Buckingham, S. C., Kahle, K. T., et al. (2015). GABAergic disinhibition and impaired KCC2 cotransporter activity underlie tumor-associated epilepsy. Glia 63, 23–36. doi: 10.1002/glia.22730
Cao, Y. (2013). Multifarious functions of PDGFs and PDGFRs in tumor growth and metastasis. Trends Mol. Med. 19, 460–473. doi: 10.1016/j.molmed.2013.05.002
Caruso, F. P., Garofano, L., D’Angelo, F., Yu, K., Tang, F., Yuan, J., et al. (2020). A map of tumor–host interactions in glioma at single-cell resolution. Gigascience 9:giaa109. doi: 10.1093/gigascience/giaa109
Cenciarini, M., Valentino, M., Belia, S., Sforna, L., Rosa, P., Ronchetti, S., et al. (2019). Dexamethasone in glioblastoma multiforme therapy: mechanisms and controversies. Front. Mol. Neurosci. 12:65. doi: 10.3389/fnmol.2019.00065
Chen, P., Zhao, D., Li, J., Liang, X., Li, J., Chang, A., et al. (2019). Symbiotic macrophage-glioma cell interactions reveal synthetic lethality in PTEN-null glioma. Cancer Cell 35, 868.e–884.e. doi: 10.1016/j.ccell.2019.05.003
Chen, Q., Boire, A., Jin, X., Valiente, M., Er, E. E., Lopez-Soto, A., et al. (2016). Carcinoma–astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 533, 493–498. doi: 10.1038/nature18268
Chen, W., Xia, T., Wang, D., Huang, B., Zhao, P., Wang, J., et al. (2016). Human astrocytes secrete IL-6 to promote glioma migration and invasion through upregulation of cytomembrane MMP14. Oncotarget 7, 62425–62438. doi: 10.18632/oncotarget.11515
Chia, K., Keatinge, M., Mazzolini, J., and Sieger, D. (2019). Brain tumours repurpose endogenous neuron to microglia signalling mechanisms to promote their own proliferation. Elife 8:e46912. doi: 10.7554/eLife.46912
Chia, K., Mazzolini, J., Mione, M., and Sieger, D. (2018). Tumor initiating cells induce Cxcr4-mediated infiltration of pro-tumoral macrophages into the brain. Elife 7:e31918. doi: 10.7554/eLife.31918
Chung, W.-S., Clarke, L. E., Wang, G. X., Stafford, B. K., Sher, A., Chakraborty, C., et al. (2013). Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 504, 394–400. doi: 10.1038/nature12776
Clarke, L. E., Liddelow, S. A., Chakraborty, C., Münch, A. E., Heiman, M., and Barres, B. A. (2018). Normal aging induces A1-like astrocyte reactivity. Proc. Natl. Acad. Sci. U.S.A. 115, E1896–E1905. doi: 10.1073/PNAS.1800165115
Colegio, O. R., Chu, N.-Q., Szabo, A. L., Chu, T., Rhebergen, A. M., Jairam, V., et al. (2014). Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 513, 559–563. doi: 10.1038/nature13490
Coniglio, S. J., Eugenin, E., Dobrenis, K., Stanley, E. R., West, B. L., Symons, M. H., et al. (2012). Microglial stimulation of glioblastoma invasion involves epidermal growth factor receptor (EGFR) and colony stimulating factor 1 receptor (CSF-1R) signaling. Mol. Med. 18, 519–527. doi: 10.2119/molmed.2011.00217
Conti, L., Palma, E., Roseti, C., Lauro, C., Cipriani, R., De Groot, M., et al. (2011). Anomalous levels of Cl- transporters cause a decrease of GABAergic inhibition in human peritumoral epileptic cortex. Epilepsia 52, 1635–1644. doi: 10.1111/j.1528-1167.2011.03111.x
Corlew, R., Bosma, M. M., and Moody, W. J. (2004). Spontaneous, synchronous electrical activity in neonatal mouse cortical neurones. J. Physiol. 560, 377–390. doi: 10.1113/jphysiol.2004.071621
Cowie, C. J. A., and Cunningham, M. O. (2014). Peritumoral epilepsy: relating form and function for surgical success. Epilepsy Behav. 38, 53–61. doi: 10.1016/j.yebeh.2014.05.009
Crapser, J. D., Spangenberg, E. E., Barahona, R. A., Arreola, M. A., Hohsfield, L. A., and Green, K. N. (2020). Microglia facilitate loss of perineuronal nets in the Alzheimer’s disease brain. EBioMedicine 58:102919. doi: 10.1016/j.ebiom.2020.102919
Cuddapah, V. A., Robel, S., Watkins, S., and Sontheimer, H. (2014). A neurocentric perspective on glioma invasion. Nat. Rev. Neurosci. 15, 455–465. doi: 10.1038/nrn3765
Dai, C., Celestino, J. C., Okada, Y., Louis, D. N., Fuller, G. N., and Holland, E. C. (2001). PDGF autocrine stimulation dedifferentiates cultured astrocytes and induces oligodendrogliomas and oligoastrocytomas from neural progenitors and astrocytes in vivo. Genes Dev. 15, 1913–1925. doi: 10.1101/gad.903001
D’Alessio, A., Proietti, G., Sica, G., and Scicchitano, B. M. (2019). Pathological and molecular features of glioblastoma and its peritumoral tissue. Cancers (Basel) 11:469. doi: 10.3390/cancers11040469
Danbolt, N. C. (2001). Glutamate uptake. Prog. Neurobiol. 65, 1–105. doi: 10.1016/S0301-0082(00)00067-8
Darmanis, S., Sloan, S. A., Croote, D., Barres, B. A., Gephart, M. H., and Quake Correspondence, S. R. (2017). Single-cell RNA-Seq analysis of infiltrating neoplastic cells at the migrating front of human glioblastoma. Cell Rep. 21, 1399–1410. doi: 10.1016/j.celrep.2017.10.030
De Boeck, A., Ahn, B. Y., D’Mello, C., Lun, X., Menon, S. V., Alshehri, M. M., et al. (2020). Glioma-derived IL-33 orchestrates an inflammatory brain tumor microenvironment that accelerates glioma progression. Nat. Commun. 11:4997. doi: 10.1038/s41467-020-18569-4
de Groot, J., and Sontheimer, H. (2011). Glutamate and the biology of gliomas. Glia 59, 1181–1189. doi: 10.1002/glia.21113
De Palma, M., Mazzieri, R., Politi, L. S., Pucci, F., Zonari, E., Sitia, G., et al. (2008). Tumor-targeted interferon-α delivery by Tie2-expressing monocytes inhibits tumor growth and metastasis. Cancer Cell 14, 299–311. doi: 10.1016/j.ccr.2008.09.004
Deisseroth, K., Singla, S., Toda, H., Monje, M., Palmer, T. D., and Malenka, R. C. (2004). Excitation-neurogenesis coupling in adult neural stem/progenitor cells. Neuron 42, 535–552. doi: 10.1016/S0896-6273(04)00266-1
Di Angelantonio, S., Murana, E., Cocco, S., Scala, F., Bertollini, C., Molinari, M. G., et al. (2014). A role for intracellular zinc in glioma alteration of neuronal chloride equilibrium. Cell Death Dis. 5:e1501. doi: 10.1038/cddis.2014.437
Donovan, M. J., Hempstead, B. L., Horvath, C., Chao, M. V., and Schofield, D. (1993). Immunohistochemical localization of Trk receptor protein in pediatric small round blue cell tumors. Am. J. Pathol. 143, 1560–1567.
Dowling, P., Shang, G., Raval, S., Menonna, J., Cook, S., and Husar, W. (1996). Involvement of the CD95 (APO-1/Fas) receptor/ligand system in multiple sclerosis brain. J. Exp. Med. 184, 1513–1518. doi: 10.1084/jem.184.4.1513
D’Urso, P. I., D’Urso, O. F., Storelli, C., Mallardo, M., Gianfreda, C. D., Montinaro, A., et al. (2012). miR-155 is up-regulated in primary and secondary glioblastoma and promotes tumour growth by inhibiting GABA receptors. Int. J. Oncol. 41, 228–234. doi: 10.3892/ijo.2012.1420
Eberhart, C. G., Kaufman, W. E., Tihan, T., and Burger, P. C. (2001). Apoptosis, neuronal maturation, and neurotrophin expression within medulloblastoma nodules. J. Neuropathol. Exp. Neurol. 60, 462–469. doi: 10.1093/jnen/60.5.462
Eyo, U. B., Peng, J., Swiatkowski, P., Mukherjee, A., Bispo, A., and Wu, L.-J. (2014). Neuronal hyperactivity recruits microglial processes via neuronal NMDA receptors and microglial P2Y12 receptors after status epilepticus. J. Neurosci. 34, 10528–10540. doi: 10.1523/JNEUROSCI.0416-14.2014
Falcão, A. M., van Bruggen, D., Marques, S., Meijer, M., Jäkel, S., Agirre, E., et al. (2018). Disease-specific oligodendrocyte lineage cells arise in multiple sclerosis. Nat. Med. 24, 1837–1844. doi: 10.1038/s41591-018-0236-y
Falsini, B., Chiaretti, A., Rizzo, D., Piccardi, M., Ruggiero, A., Manni, L., et al. (2016). Nerve growth factor improves visual loss in childhood optic gliomas: a randomized, double-blind, phase II clinical trial. Brain 139(Pt 2), 404–414. doi: 10.1093/brain/awv366
Fane, M., and Weeraratna, A. T. (2019). How the ageing microenvironment influences tumour progression. Nat. Rev. Cancer 20, 89–106. doi: 10.1038/s41568-019-0222-9
Franceschi, S., Corsinovi, D., Lessi, F., Tantillo, E., Aretini, P., Menicagli, M., et al. (2018). Mitochondrial enzyme GLUD2 plays a critical role in glioblastoma progression. EBioMedicine 37, 56–67. doi: 10.1016/j.ebiom.2018.10.008
Franklin, R. J. M., and Ffrench-Constant, C. (2008). Remyelination in the CNS: from biology to therapy. Nat. Rev. Neurosci. 9, 839–855. doi: 10.1038/nrn2480
Friebel, E., Kapolou, K., Unger, S., Núñez, N. G., Utz, S., Rushing, E. J., et al. (2020). Single-cell mapping of human brain cancer reveals tumor-specific instruction of tissue-invading leukocytes. Cell 181, 1626.e–1642.e. doi: 10.1016/j.cell.2020.04.055
Gafarov, F. M. (2018). Neural electrical activity and neural network growth. Neural Netw. 101, 15–24. doi: 10.1016/j.neunet.2018.02.001
Galvao, R. P., Kasina, A., McNeill, R. S., Harbin, J. E., Foreman, O., Verhaak, R. G. W., et al. (2014). Transformation of quiescent adult oligodendrocyte precursor cells into malignant glioma through a multistep reactivation process. Proc. Natl. Acad. Sci. U.S.A. 111, E4214–E4223. doi: 10.1073/pnas.1414389111
Gao, X., Zhang, Z., Mashimo, T., Shen, B., Nyagilo, J., Wang, H., et al. (2020). Gliomas interact with non-glioma brain cells via extracellular vesicles. Cell Rep. 30, 2489.e–2500.e. doi: 10.1016/j.celrep.2020.01.089
Garofalo, S., D’Alessandro, G., Chece, G., Brau, F., Maggi, L., Rosa, A., et al. (2015). Enriched environment reduces glioma growth through immune and non-immune mechanisms in mice. Nat. Commun. 6:6623. doi: 10.1038/ncomms7623
Garofalo, S., Porzia, A., Mainiero, F., Di Angelantonio, S., Cortese, B., Basilico, B., et al. (2017). Environmental stimuli shape microglial plasticity in glioma. Elife 6:e33415. doi: 10.7554/eLife.33415
Ge, S., Yang, C. H., Hsu, K. S., Ming, G. L., and Song, H. (2007). A critical period for enhanced synaptic plasticity in newly generated neurons of the adult brain. Neuron 54, 559–566. doi: 10.1016/j.neuron.2007.05.002
Georgoudaki, A.-M., Prokopec, K. E., Boura, V. F., Hellqvist, E., Sohn, S., Östling, J., et al. (2016). Reprogramming tumor-associated macrophages by antibody targeting inhibits cancer progression and metastasis. Cell Rep. 15, 2000–2011. doi: 10.1016/j.celrep.2016.04.084
Giachino, C., Barz, M., Tchorz, J. S., Tome, M., Gassmann, M., Bischofberger, J., et al. (2014). GABA suppresses neurogenesis in the adult hippocampus through GABAB receptors. Development 141, 83–90. doi: 10.1242/dev.102608
Gibson, E., and Monje, M. (2012). Effect of cancer therapy on neural stem cells: implications for cognitive function. Curr. Opin. Oncol. 24, 672–678. doi: 10.1097/CCO.0b013e3283571a8e
Gibson, E. M., and Monje, M. (2021). Microglia in cancer therapy–related cognitive impairment. Trends Neurosci. 44, 441–451. doi: 10.1016/j.tins.2021.02.003
Gibson, E. M., Nagaraja, S., Ocampo, A., Tam, L. T., Wood, L. S., Pallegar, P. N., et al. (2019). Methotrexate chemotherapy induces persistent tri-glial dysregulation that underlies chemotherapy-related cognitive impairment. Cell 176, 43.e–55.e. doi: 10.1016/j.cell.2018.10.049
Gibson, E. M., Purger, D., Mount, C. W., Goldstein, A. K., Lin, G. L., Wood, L. S., et al. (2014). Neuronal activity promotes oligodendrogenesis and adaptive myelination in the mammalian brain. Science 344:1252304. doi: 10.1126/science.1252304
Ginhoux, F., Greter, M., Leboeuf, M., Nandi, S., See, P., Gokhan, S., et al. (2010). Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330, 841–845. doi: 10.1126/science.1194637
Ginhoux, F., and Guilliams, M. (2016). Review tissue-resident macrophage ontogeny and homeostasis. Immunity 44, 439–449. doi: 10.1016/j.immuni.2016.02.024
Glass, C. K., Saijo, K., Winner, B., Marchetto, M. C., and Gage, F. H. (2010). Mechanisms underlying inflammation in neurodegeneration. Cell 140, 918–934. doi: 10.1016/j.cell.2010.02.016
Glass, R., Synowitz, M., Kronenberg, G., Walzlein, J.-H., Markovic, D. S., Wang, L.-P., et al. (2005). Glioblastoma-induced attraction of endogenous neural precursor cells is associated with improved survival. J. Neurosci. 25, 2637–2646. doi: 10.1523/JNEUROSCI.5118-04.2005
Gordon, S. R., Maute, R. L., Dulken, B. W., Hutter, G., George, B. M., McCracken, M. N., et al. (2017). PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 545, 495–499. doi: 10.1038/nature22396
Greter, M., Lelios, I., and Croxford, A. L. (2015). Microglia versus myeloid cell nomenclature during brain inflammation. Front. Immunol. 6:249. doi: 10.3389/fimmu.2015.00249
Gril, B., Palmieri, D., Qian, Y., Anwar, T., Liewehr, D. J., Steinberg, S. M., et al. (2013). Pazopanib inhibits the activation of PDGFRβ-expressing astrocytes in the brain metastatic microenvironment of breast cancer cells. Am. J. Pathol. 182, 2368–2379. doi: 10.1016/j.ajpath.2013.02.043
Guldner, I. H., Wang, Q., Yang, L., Golomb, S. M., Zhao, Z., Lopez, J. A., et al. (2020). CNS-native myeloid cells drive immune suppression in the brain metastatic niche through Cxcl10. Cell 183, 1234.e–1248.e. doi: 10.1016/j.cell.2020.09.064
Gupta, M., Djalilvand, A., and Brat, D. J. (2005). Clarifying the diffuse gliomas. Am. J. Clin. Pathol. 124, 755–768. doi: 10.1309/6JNX4PA60TQ5U5VG
Gust, J., Hay, K. A., Hanafi, L.-A., Li, D., Myerson, D., Gonzalez-Cuyar, L. F., et al. (2017). Endothelial activation and blood–brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR-T cells. Cancer Discov. 7, 1404–1419. doi: 10.1158/2159-8290.CD-17-0698
Gutmann, D. H., and Kettenmann, H. (2019). Microglia/brain macrophages as central drivers of brain tumor pathobiology. Neuron 104, 442–449. doi: 10.1016/j.neuron.2019.08.028
Hambardzumyan, D., Gutmann, D. H., and Kettenmann, H. (2015). The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 19, 20–27. doi: 10.1038/nn.4185
Hammond, T. R., Dufort, C., Dissing-Olesen, L., Giera, S., Young, A., Wysoker, A., et al. (2019). Single-cell RNA sequencing of microglia throughout the mouse lifespan and in the injured brain reveals complex cell-state changes. Immunity 50, 253.e–271.e. doi: 10.1016/j.immuni.2018.11.004
Hanahan, D., and Coussens, L. M. (2012). Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell 21, 309–322. doi: 10.1016/j.ccr.2012.02.022
Hanisch, U. K., and Kettenmann, H. (2007). Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 10, 1387–1394. doi: 10.1038/nn1997
Harks, E. G. A., de Roos, A. D. G., Peters, P. H. J., Harks, L. G. A., de Roos, A. D. G., Peters, P. H. J., et al. (2001). Fenamates: a novel class of reversible gap junction blockers. J. Pharmacol. Exp. Ther. 293, 1033–1041.
Henrik Heiland, D., Ravi, V. M., Behringer, S. P., Frenking, J. H., Wurm, J., Joseph, K., et al. (2019). Tumor-associated reactive astrocytes aid the evolution of immunosuppressive environment in glioblastoma. Nat. Commun. 10:2541. doi: 10.1038/s41467-019-10493-6
Hide, T., Komohara, Y., Miyasato, Y., Nakamura, H., Makino, K., Takeya, M., et al. (2018). Oligodendrocyte progenitor cells and macrophages/microglia produce glioma stem cell niches at the tumor border. EBioMedicine 30, 94–104. doi: 10.1016/j.ebiom.2018.02.024
Hong, E. J., McCord, A. E., and Greenberg, M. E. (2008). A biological function for the neuronal activity-dependent component of Bdnf transcription in the development of cortical inhibition. Neuron 60, 610–624. doi: 10.1016/j.neuron.2008.09.024
Hu, F., Ku, M. C., Markovic, D., Dzaye, O. D., Lehnardt, S., Synowitz, M., et al. (2014). Glioma-associated microglial MMP9 expression is upregulated by TLR2 signaling and sensitive to minocycline. Int. J. Cancer 135, 2569–2578. doi: 10.1002/ijc.28908
Hu, G., Su, Y., Kang, B. H., Fan, Z., Dong, T., Brown, D. R., et al. (2021). High-throughput phenotypic screen and transcriptional analysis identify new compounds and targets for macrophage reprogramming. Nat. Commun. 12:773. doi: 10.1038/s41467-021-21066-x
Huang, Y., Hoffman, C., Rajappa, P., Kim, J.-H., Hu, W., Huse, J., et al. (2014). Oligodendrocyte progenitor cells promote neovascularization in glioma by disrupting the blood–brain barrier. Cancer Res. 74, 1011–1021. doi: 10.1158/0008-5472.Can-13-1072
Huberfeld, G., and Vecht, C. J. (2016). Seizures and gliomas–towards a single therapeutic approach. Nat. Rev. Neurol. 12, 204–216. doi: 10.1038/nrneurol.2016.26
Hussain, S. F., Yang, D., Suki, D., Aldape, K., Grimm, E., and Heimberger, A. B. (2006). The role of human glioma-infiltrating microglia/macrophages in mediating antitumor immune responses. Neuro Oncol. 8, 261–279. doi: 10.1215/15228517-2006-008
Hutter, G., Theruvath, J., Graef, C. M., Zhang, M., Schoen, M. K., Manz, E. M., et al. (2019). Microglia are effector cells of CD47-SIRPα antiphagocytic axis disruption against glioblastoma. Proc. Natl. Acad. Sci. U.S.A. 116, 997–1006. doi: 10.1073/pnas.1721434116
Irvin, D. M., McNeill, R. S., Bash, R. E., and Miller, C. R. (2017). Intrinsic astrocyte heterogeneity influences tumor growth in glioma mouse models. Brain Pathol. 27, 36–50. doi: 10.1111/bpa.12348
Ishiuchi, S., Yoshida, Y., Sugawara, K., Aihara, M., Ohtani, T., Watanabe, T., et al. (2007). Ca2+-permeable AMPA receptors regulate growth of human glioblastoma via Akt activation. J. Neurosci. 27, 7987–8001. doi: 10.1523/JNEUROSCI.2180-07.2007
Jarabo, P., de Pablo, C., Herranz, H., Martín, F. A., and Casas-Tintó, S. (2021). Insulin signaling mediates neurodegeneration in glioma. Life Sci. Alliance 4:e202000693. doi: 10.26508/lsa.202000693
John Lin, C.-C., Yu, K., Hatcher, A., Huang, T.-W., Lee, H. K., Carlson, J., et al. (2017). Identification of diverse astrocyte populations and their malignant analogs. Nat. Neurosci. 20, 396–405. doi: 10.1038/nn.4493
Johung, T., and Monje, M. (2017). Neuronal activity in the glioma microenvironment. Curr. Opin. Neurobiol. 47, 156–161. doi: 10.1016/j.conb.2017.10.009
Jung, E., Alfonso, J., Osswald, M., Monyer, H., Wick, W., and Winkler, F. (2019). Emerging intersections between neuroscience and glioma biology. Nat. Neurosci. 22, 1951–1960. doi: 10.1038/s41593-019-0540-y
Kamiya, A., Hayama, Y., Kato, S., Shimomura, A., Shimomura, T., Irie, K., et al. (2019). Genetic manipulation of autonomic nerve fiber innervation and activity and its effect on breast cancer progression. Nat. Neurosci. 22, 1289–1305. doi: 10.1038/s41593-019-0430-3
Kaneda, M. M., Messer, K. S., Ralainirina, N., Li, H., Leem, C. J., Gorjestani, S., et al. (2016). PI3Kγ is a molecular switch that controls immune suppression. Nature 539, 437–442. doi: 10.1038/nature19834
Katz, A. M., Amankulor, N. M., Pitter, K., Helmy, K., Squatrito, M., and Holland, E. C. (2012). Astrocyte-specific expression patterns associated with the PDGF-induced glioma microenvironment. PLoS One 7:e32453. doi: 10.1371/journal.pone.0032453
Kempermann, G. (2019). Environmental enrichment, new neurons and the neurobiology of individuality. Nat. Rev. Neurosci. 20, 235–245. doi: 10.1038/s41583-019-0120-x
Keren-Shaul, H., Spinrad, A., Weiner, A., Matcovitch-Natan, O., Dvir-Szternfeld, R., Ulland, T. K., et al. (2017). A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 169, 1276.e–1290.e. doi: 10.1016/j.cell.2017.05.018
Khakh, B. S., and Deneen, B. (2019). The emerging nature of astrocyte diversity. Annu. Rev. Neurosci. 42, 187–207. doi: 10.1146/annurev-neuro-070918-050443
Kimura, S., Yoshino, A., Katayama, Y., Watanabe, T., and Fukushima, T. (2002). Growth control of C6 glioma in vivo by nerve growth factor. J. Neurooncol. 59, 199–205. doi: 10.1023/A:1019919019497
Kirby, L., Jin, J., Cardona, J. G., Smith, M. D., Martin, K. A., Wang, J., et al. (2019). Oligodendrocyte precursor cells present antigen and are cytotoxic targets in inflammatory demyelination. Nat. Commun. 10:3887. doi: 10.1038/s41467-019-11638-3
Klemm, F., Maas, R. R., Bowman, R. L., Kornete, M., Soukup, K., Nassiri, S., et al. (2020). Interrogation of the microenvironmental landscape in brain tumors reveals disease-specific alterations of immune cells. Cell 181, 1643.e–1660.e. doi: 10.1016/j.cell.2020.05.007
Kloepper, J., Riedemann, L., Amoozgar, Z., Seano, G., Susek, K., Yu, V., et al. (2016). Ang-2/VEGF bispecific antibody reprograms macrophages and resident microglia to anti-tumor phenotype and prolongs glioblastoma survival. Proc. Natl. Acad. Sci. U.S.A. 113, 4476–4481. doi: 10.1073/pnas.1525360113
Kober, C., Rohn, S., Weibel, S., Geissinger, U., Chen, N. G., and Szalay, A. A. (2015). Microglia and astrocytes attenuate the replication of the oncolytic vaccinia virus LIVP 1.1.1 in murine GL261 gliomas by acting as vaccinia virus traps. J. Transl. Med. 13:216. doi: 10.1186/s12967-015-0586-x
Komohara, Y., Ohnishi, K., Kuratsu, J., and Takeya, M. (2008). Possible involvement of the M2 anti-inflammatory macrophage phenotype in growth of human gliomas. J. Pathol. 216, 15–24. doi: 10.1002/path.2370
Korin, B., Ben-Shaanan, T. L., Schiller, M., Dubovik, T., Azulay-Debby, H., Boshnak, N. T., et al. (2017). High-dimensional, single-cell characterization of the brain’s immune compartment. Nat. Neurosci. 20, 1300–1309. doi: 10.1038/nn.4610
Kowal, J., Kornete, M., and Joyce, J. A. (2019). Re-education of macrophages as a therapeutic strategy in cancer. Immunotherapy 11, 677–689. doi: 10.2217/imt-2018-0156
Kuilman, T., Michaloglou, C., Vredeveld, L. C. W., Douma, S., van Doorn, R., Desmet, C. J., et al. (2008). Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 133, 1019–1031. doi: 10.1016/J.CELL.2008.03.039
Labrakakis, C. (1998). Functional GABAA receptors on human glioma cells. Eur. J. Neurosci. 10, 231–238. doi: 10.1046/j.1460-9568.1998.00036.x
Lan, Y.-L., Wang, X., Lou, J.-C., Ma, X.-C., and Zhang, B. (2017). The potential roles of aquaporin 4 in malignant gliomas. Oncotarget 8, 32345–32355.
Lane, R., Simon, T., Vintu, M., Solkin, B., Koch, B., Stewart, N., et al. (2019). Cell-derived extracellular vesicles can be used as a biomarker reservoir for glioblastoma tumor subtyping. Commun. Biol. 2:315. doi: 10.1038/s42003-019-0560-x
Lawlor, K., Marques-Torrejon, M. A., Dharmalingham, G., El-Azhar, Y., Schneider, M. D., Pollard, S. M., et al. (2020). Glioblastoma stem cells induce quiescence in surrounding neural stem cells via Notch signaling. Genes Dev. 34, 1599–1604. doi: 10.1101/gad.336917.120
Le, D. M., Besson, A., Fogg, D. K., Choi, K.-S., Waisman, D. M., Goodyer, C. G., et al. (2003). Exploitation of Astrocytes by glioma cells to facilitate invasiveness: a mechanism involving matrix metalloproteinase-2 and the urokinase-type plasminogen activator–plasmin cascade. J. Neurosci. 23, 4034–4043. doi: 10.1523/JNEUROSCI.23-10-04034.2003
Lee, J. H., Lee, J. E., Kahng, J. Y., Kim, S. H., Park, J. S., Yoon, S. J., et al. (2018). Human glioblastoma arises from subventricular zone cells with low-level driver mutations. Nature 560, 243–247. doi: 10.1038/s41586-018-0389-3
Lei, X., Lei, Y., Li, J. K., Du, W. X., Li, R. G., Yang, J., et al. (2020). Immune cells within the tumor microenvironment: biological functions and roles in cancer immunotherapy. Cancer Lett. 470, 126–133. doi: 10.1016/J.CANLET.2019.11.009
Li, J., Parra-Cantu, C., Wang, Z., and Zhang, Y. S. (2020). Improving bioprinted volumetric tumor microenvironments in vitro. Trends in Cancer 6, 745–756. doi: 10.1016/J.TRECAN.2020.06.002
Li, M. D., Burns, T. C., Kumar, S., Morgan, A. A., Sloan, S. A., and Palmer, T. D. (2015). Aging-like changes in the transcriptome of irradiated microglia. Glia 63, 754–767. doi: 10.1002/GLIA.22782
Liang, J., Lv, X., Lu, C., Ye, X., Chen, X., Fu, J., et al. (2020). Prognostic factors of patients with gliomas- a n analysis on 335 patients with glioblastoma and other forms of gliomas. BMC Cancer 20:35. doi: 10.1186/s12885-019-6511-6
Liddelow, S. A., Guttenplan, K. A., Clarke, L. E., Bennett, F. C., Bohlen, C. J., Schirmer, L., et al. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. doi: 10.1038/nature21029
Limbad, C., Oron, T. R., Alimirah, F., Davalos, A. R., Tracy, T. E., Gan, L., et al. (2020). Astrocyte senescence promotes glutamate toxicity in cortical neurons. PLoS One 15:e0227887. doi: 10.1371/JOURNAL.PONE.0227887
Lin, Q., Balasubramanian, K., Fan, D., Kim, S. J., Guo, L., Wang, H., et al. (2010). Reactive astrocytes protect melanoma cells from chemotherapy by sequestering intracellular calcium through gap junction communication channels. Neoplasia 12, 748–754. doi: 10.1593/neo.10602
Linnerbauer, M., and Rothhammer, V. (2020). Protective functions of reactive astrocytes following central nervous system insult. Front. Immunol. 11:573256. doi: 10.3389/fimmu.2020.573256
Litak, J., Mazurek, M., Grochowski, C., Kamieniak, P., and Roliński, J. (2019). PD-L1/PD-1 axis in glioblastoma multiforme. Int. J. Mol. Sci. 20:5347. doi: 10.3390/ijms20215347
Liu, C., Sage, J. C., Miller, M. R., Verhaak, R. G. W., Hippenmeyer, S., Vogel, H., et al. (2011). Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell 146, 209–221. doi: 10.1016/j.cell.2011.06.014
Liu, Y., and Aguzzi, A. (2020). NG2 glia are required for maintaining microglia homeostatic state. Glia 68, 345–355. doi: 10.1002/GLIA.23721
Liu, Y., Carlsson, R., Ambjorn, M., Hasan, M., Badn, W., Darabi, A., et al. (2013). PD-L1 expression by neurons nearby tumors indicates better prognosis in glioblastoma patients. J. Neurosci. 33, 14231–14245. doi: 10.1523/jneurosci.5812-12.2013
Maas, S. L. N., Abels, E. R., Van De Haar, L. L., Zhang, X., Morsett, L., Sil, S., et al. (2020). Glioblastoma hijacks microglial gene expression to support tumor growth. J. Neuroinflammation 17:120. doi: 10.1186/s12974-020-01797-2
MacKenzie, G., O’Toole, K. K., Moss, J. S., and Maguire, J. (2017). Compromised GABAergic inhibition contributes to tumor-associated epilepsy. Physiol. Behav. 176, 139–148. doi: 10.1016/j.physbeh.2017.03.040
Mahmoud, S., Gharagozloo, M., Simard, C., and Gris, D. (2019). Astrocytes maintain glutamate homeostasis in the CNS by controlling the balance between glutamate uptake and release. Cells 8:184. doi: 10.3390/cells8020184
Majed, H. H., Chandran, S., Niclou, S. P., Nicholas, R. S., Wilkins, A., Wing, M. G., et al. (2006). A novel role for Sema3A in neuroprotection from injury mediated by activated microglia. J. Neurosci. 26, 1730–1738. doi: 10.1523/JNEUROSCI.0702-05.2006
Mantovani, A., Marchesi, F., Malesci, A., Laghi, L., and Allavena, P. (2017). Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 14, 399–416. doi: 10.1038/nrclinonc.2016.217
Marcus, H. J., Carpenter, K. L. H., Price, S. J., and Hutchinson, P. J. (2010). In vivo assessment of high-grade glioma biochemistry using microdialysis: a study of energy-related molecules, growth factors and cytokines. J. Neurooncol. 97, 11–23. doi: 10.1007/s11060-009-9990-5
Markovic, D. S., Vinnakota, K., Chirasani, S., Synowitz, M., Raguet, H., Stock, K., et al. (2009). Gliomas induce and exploit microglial MT1-MMP expression for tumor expansion. Proc. Natl. Acad. Sci. U.S.A. 106, 12530–12535. doi: 10.1073/pnas.0804273106
Masuda, T., Sankowski, R., Staszewski, O., Böttcher, C., Amann, L., Sagar, et al. (2019). Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature 566, 388–392. doi: 10.1038/s41586-019-0924-x
Masuda, T., Sankowski, R., Staszewski, O., and Prinz, M. (2020). Microglia heterogeneity in the single-cell era. Cell Rep. 30, 1271–1281. doi: 10.1016/j.celrep.2020.01.010
Matarredona, E. R., and Pastor, A. M. (2019). Extracellular vesicle-mediated communication between the glioblastoma and its microenvironment. Cells. 9:96. doi: 10.3390/cells9010096
Mathys, H., Adaikkan, C., Gao, F., Young, J. Z., Manet, E., Hemberg, M., et al. (2017). Temporal tracking of microglia activation in neurodegeneration at single-cell resolution. Cell Rep. 21, 366–380. doi: 10.1016/j.celrep.2017.09.039
Matias, D., Balça-Silva, J., da Graça, G. C., Wanjiru, C. M., Macharia, L. W., Nascimento, C. P., et al. (2018). Microglia/astrocytes–glioblastoma crosstalk: crucial molecular mechanisms and microenvironmental factors. Front. Cell. Neurosci. 12:235. doi: 10.3389/fncel.2018.00235
Maurel, P., and Salzer, J. L. (2000). Axonal regulation of Schwann cell proliferation and survival and the initial events of myelination requires PI 3-kinase activity. J. Neurosci. 20, 4635–4645. doi: 10.1523/jneurosci.20-12-04635.2000
Maximov, V., Chen, Z., Wei, Y., Robinson, M. H., Herting, C. J., Shanmugam, N. S., et al. (2019). Tumour-associated macrophages exhibit anti-tumoural properties in Sonic Hedgehog medulloblastoma. Nat. Commun. 10:2410. doi: 10.1038/s41467-019-10458-9
Mega, A., Hartmark Nilsen, M., Leiss, L. W., Tobin, N. P., Miletic, H., Sleire, L., et al. (2020). Astrocytes enhance glioblastoma growth. Glia 68, 316–327. doi: 10.1002/glia.23718
Menichella, D. M., Majdan, M., Awatramani, R., Goodenough, D. A., Sirkowski, E., Scherer, S. S., et al. (2006). Genetic and physiological evidence that oligodendrocyte gap junctions contribute to spatial buffering of potassium released during neuronal activity. J. Neurosci. 26, 10984–10991. doi: 10.1523/JNEUROSCI.0304-06.2006
Monje, M. L., Toda, H., and Palmer, T. D. (2003). Inflammatory blockade restores adult hippocampal neurogenesis. Science 302, 1760–1765. doi: 10.1126/science.1088417
Montgomery, M. K., Kim, S. H., Schevon, C., Canoll, P., and Hillman, E. M. C. (2020). Glioma-induced alterations in neuronal activity and neurovascular coupling during disease progression. Cell Rep. 31:107500. doi: 10.1016/j.celrep.2020.03.064
Müller, S., Kohanbash, G., Liu, S. J., Alvarado, B., Carrera, D., Bhaduri, A., et al. (2017). Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol. 18:234. doi: 10.1186/s13059-017-1362-4
Nakagawara, A., Arima-Nakagawara, M., Scavarda, N. J., Azar, C. G., Cantor, A. B., and Brodeur, G. M. (1993). Association between high levels of expression of the TRK gene and favorable outcome in human neuroblastoma. N. Engl. J. Med. 328, 847–854. doi: 10.1056/NEJM199303253281205
Nakashima, H., Tsujimura, K., Irie, K., Ishizu, M., Pan, M., Kameda, T., et al. (2018). Canonical TGF-β signaling negatively regulates neuronal morphogenesis through TGIF/smad complex-mediated CRMP2 suppression. J. Neurosci. 38, 4791–4810. doi: 10.1523/JNEUROSCI.2423-17.2018
Neftel, C., Laffy, J., Filbin, M. G., Hara, T., Shore, M. E., Rahme, G. J., et al. (2019). An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell 178, 835.e–849.e. doi: 10.1016/j.cell.2019.06.024
Nelson, S. B., and Turrigiano, G. G. (1998). Synaptic depression: a key player in the cortical balancing act. Nat. Neurosci. 1, 539–541. doi: 10.1038/2775
Nicaise, A. M., Wagstaff, L. J., Willis, C. M., Paisie, C., Chandok, H., Robson, P., et al. (2019). Cellular senescence in progenitor cells contributes to diminished remyelination potential in progressive multiple sclerosis. Proc. Natl. Acad. Sci. USA. 116, 9030–9039. doi: 10.1073/pnas.1818348116
Nguyen, P. T., Dorman, L. C., Pan, S., Vainchtein, I. D., Han, R. T., Nakao-Inoue, H., et al. (2020). Microglial remodeling of the extracellular matrix promotes synapse plasticity. Cell 182, 388.e–403.e. doi: 10.1016/j.cell.2020.05.050
Niu, J., Tsai, H.-H., Hoi, K. K., Huang, N., Yu, G., Kim, K., et al. (2019). Aberrant oligodendroglial–vascular interactions disrupt the blood–brain barrier, triggering CNS inflammation. Nat. Neurosci. 225, 709–718. doi: 10.1038/s41593-019-0369-4
Ochocka, N., Segit, P., Walentynowicz, K. A., Wojnicki, K., Cyranowski, S., Swatler, J., et al. (2021). Single-cell RNA sequencing reveals functional heterogeneity of glioma-associated brain macrophages. Nat. Commun. 12:1151. doi: 10.1038/s41467-021-21407-w
Ortiz, F. C., Habermacher, C., Graciarena, M., Houry, P. Y., Nishiyama, A., Oumesmar, B. N., et al. (2019). Neuronal activity in vivo enhances functional myelin repair. JCI Insight 4:e123434. doi: 10.1172/jci.insight.123434
Osswald, M., Jung, E., Sahm, F., Solecki, G., Venkataramani, V., Blaes, J., et al. (2015). Brain tumour cells interconnect to a functional and resistant network. Nature 528, 93–98. doi: 10.1038/nature16071
Paez-Gonzalez, P., Asrican, B., Rodriguez, E., and Kuo, C. T. (2014). Identification of distinct ChAT+ neurons and activity-dependent control of postnatal SVZ neurogenesis. Nat. Neurosci. 17, 934–942. doi: 10.1038/nn.3734
Pallud, J., Le Van Quyen, M., Bielle, F., Pellegrino, C., Varlet, P., Labussiere, M., et al. (2014). Cortical GABAergic excitation contributes to epileptic activities around human glioma. Sci. Transl. Med. 6:244ra89. doi: 10.1126/scitranslmed.3008065
Pamies, D., Zurich, M.-G., and Hartung, T. (2020). Organotypic models to study human glioblastoma: studying the beast in its ecosystem. iScience 23:101633. doi: 10.1016/J.ISCI.2020.101633
Paolicelli, R. C., Bolasco, G., Pagani, F., Maggi, L., Scianni, M., Panzanelli, P., et al. (2011). Synaptic pruning by microglia is necessary for normal brain development. Science 333, 1456–1458. doi: 10.1126/science.1202529
Park, J. C., Chang, I. B., Ahn, J. H., Kim, J. H., Song, J. H., Moon, S. M., et al. (2018). Nerve growth factor stimulates glioblastoma proliferation through Notch1 receptor signaling. J. Korean Neurosurg. Soc. 61, 441–449. doi: 10.3340/jkns.2017.0219
Parolisi, R., and Boda, E. (2018). NG2 glia: novel roles beyond Re-/Myelination. Neuroglia 1, 151–175. doi: 10.3390/NEUROGLIA1010011
Parys, B., Côté, A., Gallo, V., De Koninck, P., and Sík, A. (2010). Intercellular calcium signaling between astrocytes and oligodendrocytes via gap junctions in culture. Neuroscience 167, 1032–1043. doi: 10.1016/j.neuroscience.2010.03.004
Patel, D., Ahmad, F., Kambach, D. M., Sun, Q., Halim, A. S., Kramp, T., et al. (2019). LXRβ controls glioblastoma cell growth, lipid balance, and immune modulation independently of ABCA1. Sci. Rep. 9:15458. doi: 10.1038/s41598-019-51865-8
Peruzzotti-Jametti, L., Bernstock, J. D., Vicario, N., Costa, A. S. H., Kwok, C. K., Leonardi, T., et al. (2018). Macrophage-derived extracellular succinate licenses neural stem cells to suppress chronic neuroinflammation. Cell Stem Cell 22, 355.e–368.e. doi: 10.1016/j.stem.2018.01.020
Pham, L.-D. D., Hayakawa, K., Seo, J. H., Nguyen, M.-N., Som, A. T., Lee, B. J., et al. (2012). Crosstalk between oligodendrocytes and cerebral endothelium contributes to vascular remodeling after white matter injury. Glia 60, 875–881. doi: 10.1002/GLIA.22320
Pietras, A., Katz, A. M., Ekström, E. J., Wee, B., Halliday, J. J., Pitter, K. L., et al. (2014). Osteopontin-CD44 signaling in the glioma perivascular niche enhances cancer stem cell phenotypes and promotes aggressive tumor growth. Cell Stem Cell 14, 357–369. doi: 10.1016/j.stem.2014.01.005
Pombo Antunes, A. R., Scheyltjens, I., Lodi, F., Messiaen, J., Antoranz, A., Duerinck, J., et al. (2021). Single-cell profiling of myeloid cells in glioblastoma across species and disease stage reveals macrophage competition and specialization. Nat. Neurosci. 24, 595–610. doi: 10.1038/s41593-020-00789-y
Portela, M., Venkataramani, V., Fahey-Lozano, N., Seco, E., Losada-Perez, M., Winkler, F., et al. (2019). Glioblastoma cells vampirize WNT from neurons and trigger a JNK/MMP signaling loop that enhances glioblastoma progression and neurodegeneration. PLoS Biol 17:e3000545. doi: 10.1371/journal.pbio.3000545
Priego, N., and Valiente, M. (2019). The potential of astrocytes as immune modulators in brain tumors. Front. Immunol. 10:1314. doi: 10.3389/fimmu.2019.01314
Priego, N., Zhu, L., Monteiro, C., Mulders, M., Wasilewski, D., Bindeman, W., et al. (2018). STAT3 labels a subpopulation of reactive astrocytes required for brain metastasis article. Nat. Med. 24, 1024–1035. doi: 10.1038/s41591-018-0044-4
Proctor, D. T., Stotz, S. C., Scott, L. O. M., de la Hoz, C. L. R., Poon, K. W. C., Stys, P. K., et al. (2015). Axo-glial communication through neurexin-neuroligin signaling regulates myelination and oligodendrocyte differentiation. Glia 63, 2023–2039. doi: 10.1002/glia.22875
Pyonteck, S. M., Akkari, L., Schuhmacher, A. J., Bowman, R. L., Sevenich, L., Quail, D. F., et al. (2013). CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 19, 1264–1272. doi: 10.1038/nm.3337
Quail, D. F., Bowman, R. L., Akkari, L., Quick, M. L., Schuhmacher, A. J., Huse, J. T., et al. (2016). The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science 352:aad3018. doi: 10.1126/science.aad3018
Rabin, S. J., Tornatore, C., Baker-Cairns, B., Spiga, G., and Mocchetti, I. (1998). TrkA receptors delay C6-2B glioma cell growth in rat striatum. Mol. Brain Res. 56, 273–276. doi: 10.1016/S0169-328X(98)00020-5
Radin, D. P., and Patel, P. (2017). BDNF: an oncogene or tumor suppressor? Anticancer Res. 37, 3983–3990. doi: 10.21873/anticanres.11783
Radin, D. P., and Tsirka, S. E. (2020). Interactions between tumor cells, neurons, and microglia in the glioma microenvironment. Int. J. Mol. Sci. 21:8476. doi: 10.3390/ijms21228476
Robert, S. M., and Sontheimer, H. (2014). Glutamate transporters in the biology of malignant gliomas. Cell. Mol. Life Sci. 71, 1839–1854. doi: 10.1007/s00018-013-1521-z
Rodríguez, J. J., Terzieva, S., Olabarria, M., Lanza, R. G., and Verkhratsky, A. (2013). Enriched environment and physical activity reverse astrogliodegeneration in the hippocampus of AD transgenic mice. Cell Death Dis. 4:e678. doi: 10.1038/cddis.2013.194
Roth, P., Regli, L., Tonder, M., and Weller, M. (2013). Tumor-associated edema in brain cancer patients: pathogenesis and management. Expert Rev. Anticancer Ther. 13, 1319–1325. doi: 10.1586/14737140.2013.852473
Ruhland, M. K., Loza, A. J., Capietto, A.-H., Luo, X., Knolhoff, B. L., Flanagan, K. C., et al. (2016). Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat. Commun. 7:11762. doi: 10.1038/NCOMMS11762
Rzeski, W., Turski, L., and Ikonomidou, C. (2001). Glutamate antagonists limit tumor growth. Proc. Natl. Acad. Sci. U.S.A. 98, 6372–6377. doi: 10.1016/S0006-2952(02)01218-2
Sampson, J. H., Gunn, M. D., Fecci, P. E., and Ashley, D. M. (2020). Brain immunology and immunotherapy in brain tumours. Nat. Rev. Cancer 20, 12–25. doi: 10.1038/s41568-019-0224-7
Sankowski, R., Böttcher, C., Masuda, T., Geirsdottir, L., Sagar, Sindram, E., et al. (2019). Mapping microglia states in the human brain through the integration of high-dimensional techniques. Nat. Neurosci. 22, 2098–2110. doi: 10.1038/s41593-019-0532-y
Sarkar, S., Döring, A., Zemp, F. J., Silva, C., Lun, X., Wang, X., et al. (2014). Therapeutic activation of macrophages and microglia to suppress brain tumor-initiating cells. Nat. Neurosci. 17, 46–55. doi: 10.1038/nn.3597
Sarkar, S., Yang, R., Mirzaei, R., Rawji, K., Poon, C., Mishra, M. K., et al. (2020). Control of brain tumor growth by reactivating myeloid cells with niacin. Sci. Transl. Med. 12:eaay9924. doi: 10.1126/scitranslmed.aay9924
Savaskan, N. E., Heckel, A., Hahnen, E., Elngelhorn, T., Doerfler, A., Ganslandt, O., et al. (2008). Small interfering RNA-mediated xCT silencing in gliomas inhibits neurodegeneration and alleviates brain edema. Nat. Med. 14, 629–362. doi: 10.1038/nm1772
Scholz, J., Klein, M. C., Behrens, T. E. J., and Johansen-Berg, H. (2009). Training induces changes in white-matter architecture. Nat. Neurosci. 12, 1370–1371. doi: 10.1038/nn.2412
Seano, G., Nia, H. T., Emblem, K. E., Datta, M., Ren, J., Krishnan, S., et al. (2019). Solid stress in brain tumours causes neuronal loss and neurological dysfunction and can be reversed by lithium. Nat. Biomed. Eng. 3, 230–245. doi: 10.1038/s41551-018-0334-7
Shabtay-Orbach, A., Amit, M., Binenbaum, Y., Na’ara, S., and Gil, Z. (2015). Paracrine regulation of glioma cells invasion by astrocytes is mediated by glial-derived neurotrophic factor. Int. J. Cancer 137, 1012–1020. doi: 10.1002/ijc.29380
Shen, K., Reichelt, M., Kyauk, R. V., Ngu, H., Shen, Y. A. A., Foreman, O., et al. (2021). Multiple sclerosis risk gene Mertk is required for microglial activation and subsequent remyelination. Cell Rep. 34:108835. doi: 10.1016/j.celrep.2021.108835
Shen, X., Burguillos, M. A., Osman, A. M., Frijhoff, J., Carrillo-Jiménez, A., Kanatani, S., et al. (2016). Glioma-induced inhibition of caspase-3 in microglia promotes a tumor-supportive phenotype. Nat. Immunol. 17, 1282–1290. doi: 10.1038/ni.3545
Sieben, C. J., Sturmlechner, I., van de Sluis, B., and van Deursen, J. M. (2018). Two-step senescence-focused cancer therapies. Trends Cell Biol. 28, 723–737. doi: 10.1016/J.TCB.2018.04.006
Smits, A., Jin, Z., Elsir, T., Pedder, H., Nistér, M., Alafuzoff, I., et al. (2012). GABA-a channel subunit expression in human glioma correlates with tumor histology and clinical outcome. PLoS One 7:e37041. doi: 10.1371/journal.pone.0037041
Song, J., Zhong, C., Bonaguidi, M. A., Sun, G. J., Hsu, D., Gu, Y., et al. (2012). Neuronal circuitry mechanism regulating adult quiescent neural stem-cell fate decision. Nature 489, 150–154. doi: 10.1038/nature11306
Sontheimer, H. (2008). A role for glutamate in growth and invasion of primary brain tumors. J. Neurochem. 105, 287–295. doi: 10.1111/j.1471-4159.2008.05301.x
Sousa, C., Golebiewska, A., Poovathingal, S. K., Kaoma, T., Pires-Afonso, Y., Martina, S., et al. (2018). Single-cell transcriptomics reveals distinct inflammation-induced microglia signatures. EMBO Rep. 19:e46171. doi: 10.15252/embr.201846171
Spain, L., Walls, G., Julve, M., O’Meara, K., Schmid, T., Kalaitzaki, E., et al. (2017). Neurotoxicity from immune-checkpoint inhibition in the treatment of melanoma: a single centre experience and review of the literature. Ann. Oncol. 28, 377–385. doi: 10.1093/annonc/mdw558
Stock, K., Kumar, J., Synowitz, M., Petrosino, S., Imperatore, R., Smith, E. S. J., et al. (2012). Neural precursor cells induce cell death of high-grade astrocytomas through stimulation of TRPV1. Nat. Med. 18, 1232–1238. doi: 10.1038/nm.2827
Sugiarto, S., Persson, A. I., Munoz, E. G., Waldhuber, M., Lamagna, C., Andor, N., et al. (2011). Asymmetry-defective oligodendrocyte progenitors are glioma precursors. Cancer Cell 20, 328–340. doi: 10.1016/j.ccr.2011.08.011
Synowitz, M., Ahmann, P., Matyash, M., Kuhn, S. A., Hofmann, B., Zimmer, C., et al. (2001). GABAA-receptor expression in glioma cells is triggered by contact with neuronal cells. Eur. J. Neurosci. 14, 1294–1302. doi: 10.1046/j.0953-816X.2001.01764.x
Takenaka, M. C., Gabriely, G., Rothhammer, V., Mascanfroni, I. D., Wheeler, M. A., Chao, C., et al. (2019). Control of tumor-associated macrophages and T cells in glioblastoma via AHR and CD39. Nat. Neurosci. 22, 729–740. doi: 10.1038/s41593-019-0370-y
Takeuchi, H., Sekiguchi, A., Taki, Y., Yokoyama, S., Yomogida, Y., Komuro, N., et al. (2010). Training of working memory impacts structural connectivity. J. Neurosci. 30, 3297–3303. doi: 10.1523/JNEUROSCI.4611-09.2010
Tantillo, E., Colistra, A., Baroncelli, L., Costa, M., Caleo, M., and Vannini, E. (2020a). Voluntary physical exercise reduces motor dysfunction and hampers tumor cell proliferation in a mouse model of glioma. Int. J. Environ. Res. Public Health 17:5667. doi: 10.3390/ijerph17165667
Tantillo, E., Vannini, E., Cerri, C., Spalletti, C., Colistra, A., Mazzanti, C. M., et al. (2020b). Differential roles of pyramidal and fast-spiking, GABAergic neurons in the control of glioma cell proliferation. Neurobiol. Dis. 141:104942. doi: 10.1016/j.nbd.2020.104942
Tay, T. L., Mai, D., Dautzenberg, J., Fernández-Klett, F., Lin, G., Sagar, et al. (2017). A new fate mapping system reveals context-dependent random or clonal expansion of microglia. Nat. Neurosci. 20, 793–803. doi: 10.1038/nn.4547
Terunuma, M., Vargas, K. J., Wilkins, M. E., Ramírez, O. A., Jaureguiberry-Bravo, M., Pangalos, M. N., et al. (2010). Prolonged activation of NMDA receptors promotes dephosphorylation and alters postendocytic sorting of GABAB receptors. Proc. Natl. Acad. Sci. U.S.A. 107, 13918–13923. doi: 10.1073/pnas.1000853107
Tewari, B. P., Chaunsali, L., Campbell, S. L., Patel, D. C., Goode, A. E., and Sontheimer, H. (2018). Perineuronal nets decrease membrane capacitance of peritumoral fast spiking interneurons in a model of epilepsy. Nat. Commun. 9:4724. doi: 10.1038/s41467-018-07113-0
Toso, A., Revandkar, A., Di Mitri, D., Guccini, I., Proietti, M., Sarti, M., et al. (2014). Enhancing chemotherapy efficacy in Pten-deficient prostate tumors by activating the senescence-associated antitumor immunity. Cell Rep. 9, 75–89. doi: 10.1016/J.CELREP.2014.08.044
Trettel, F., Amalia, M., Castro, D., and Limatola, C. (2020). Chemokines: key molecules that orchestrate communication among neurons, microglia and astrocytes to preserve brain function. Neuroscience 439, 230–240. doi: 10.1016/j.neuroscience.2019.07.035
Valiente, M., Obenauf, A. C., Jin, X., Chen, Q., Zhang, X. H. F., Lee, D. J., et al. (2014). Serpins promote cancer cell survival and vascular Co-option in brain metastasis. Cell 156, 1002–1016. doi: 10.1016/j.cell.2014.01.040
van Breemen, M. S., Wilms, E. B., and Vecht, C. J. (2007). Epilepsy in patients with brain tumours: epidemiology, mechanisms, and management. Lancet Neurol. 6, 421–430. doi: 10.1016/S1474-4422(07)70103-5
van Kessel, E., Baumfalk, A. E., van Zandvoort, M. J. E., Robe, P. A., and Snijders, T. J. (2017). Tumor-related neurocognitive dysfunction in patients with diffuse glioma: a systematic review of neurocognitive functioning prior to anti-tumor treatment. J. Neurooncol. 134, 9–18. doi: 10.1007/s11060-017-2503-z
Vanhoutte, N., and Hermans, E. (2008). Glutamate-induced glioma cell proliferation is prevented by functional expression of the glutamate transporter GLT-1. FEBS Lett. 582, 1847–1852. doi: 10.1016/j.febslet.2008.04.053
Venkataramani, V., Tanev, D. I., Kuner, T., Wick, W., and Winkler, F. (2021). Synaptic input to brain tumors: clinical implications. Neuro. Oncol. 23, 23–33. doi: 10.1093/neuonc/noaa158
Venkataramani, V., Tanev, D. I., Strahle, C., Studier-Fischer, A., Fankhauser, L., Kessler, T., et al. (2019). Glutamatergic synaptic input to glioma cells drives brain tumour progression. Nature 573, 532–538. doi: 10.1038/s41586-019-1564-x
Venkatesh, H., and Monje, M. (2017). Neuronal activity in ontogeny and oncology. Trends Cancer 3, 89–112. doi: 10.1016/j.trecan.2016.12.008
Venkatesh, H. S., Johung, T. B., Caretti, V., Noll, A., Tang, Y., Nagaraja, S., et al. (2015). Neuronal activity promotes glioma growth through neuroligin-3 secretion. Cell 161, 803–816. doi: 10.1016/j.cell.2015.04.012
Venkatesh, H. S., Morishita, W., Geraghty, A. C., Silverbush, D., Gillespie, S. M., Arzt, M., et al. (2019). Electrical and synaptic integration of glioma into neural circuits. Nature 573, 539–545. doi: 10.1038/s41586-019-1563-y
Venkatesh, H. S., Tam, L. T., Woo, P. J., Lennon, J., Nagaraja, S., Gillespie, S. M., et al. (2017). Targeting neuronal activity-regulated neuroligin-3 dependency in high-grade glioma. Nature 549, 533–537. doi: 10.1038/nature24014
Venteicher, A. S., Tirosh, I., Hebert, C., Yizhak, K., Neftel, C., Filbin, M. G., et al. (2017). Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science 355:eaai8478. doi: 10.1126/science.aai8478
Verhaak, R. G. W., Hoadley, K. A., Purdom, E., Wang, V., Qi, Y., Wilkerson, M. D., et al. (2010). Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17, 98–110. doi: 10.1016/j.ccr.2009.12.020
Wadhwa, S., Nag, T. C., Jindal, A., Kushwaha, R., Mahapatra, A. K., and Sarkar, C. (2003). Expression of the neurotrophin receptors Trk A and Trk B in adult human astrocytoma and glioblastoma. J. Biosci. 28, 181–188. doi: 10.1007/BF02706217
Wang, B., Kohli, J., and Demaria, M. (2020). Senescent cells in cancer therapy: friends or foes? Trends Cancer 6, 838–857. doi: 10.1016/J.TRECAN.2020.05.004
Wang, J., Xu, S. L., Duan, J. J., Yi, L., Guo, Y. F., Shi, Y., et al. (2019). Invasion of white matter tracts by glioma stem cells is regulated by a NOTCH1–SOX2 positive-feedback loop. Nat. Neurosci. 22, 91–105. doi: 10.1038/s41593-018-0285-z
Wang, Q., Hu, B., Hu, X., Kim, H., Squatrito, M., Scarpace, L., et al. (2017). Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell 32, 42.e–56.e. doi: 10.1016/j.ccell.2017.06.003
Wasilewski, D., Priego, N., Fustero-Torre, C., and Valiente, M. (2017). Reactive astrocytes in brain metastasis. Front. Oncol. 7:298. doi: 10.3389/fonc.2017.00298
Watkins, S., Robel, S., Kimbrough, I. F., Robert, S. M., Ellis-Davies, G., and Sontheimer, H. (2014). Disruption of astrocyte–vascular coupling and the blood–brain barrier by invading glioma cells. Nat. Commun. 5:4196. doi: 10.1038/ncomms5196
Weil, S., Osswald, M., Solecki, G., Grosch, J., Jung, E., Lemke, D., et al. (2017). Tumor microtubes convey resistance to surgical lesions and chemotherapy in gliomas. Neuro Oncol. 19, 1316–1326. doi: 10.1093/neuonc/nox070
Weller, M., Wick, W., Aldape, K., Brada, M., Berger, M., Pfister, S. M., et al. (2015). Glioma. Nat. Rev. Dis. Prim. 1:15017. doi: 10.1038/nrdp.2015.17
Werner, Y., Mass, E., Ashok Kumar, P., Ulas, T., Händler, K., Horne, A., et al. (2020). Cxcr4 distinguishes HSC-derived monocytes from microglia and reveals monocyte immune responses to experimental stroke. Nat. Neurosci. 23, 351–362. doi: 10.1038/s41593-020-0585-y
Wick, W., Platten, M., and Weller, M. (2001). Glioma cell invasion: regulation of metalloproteinase activity by TGF-beta. J. Neurooncol. 53, 177–185. doi: 10.1023/a:1012209518843
Wlodarczyk, A., Holtman, I. R., Krueger, M., Yogev, N., Bruttger, J., Khorooshi, R., et al. (2017). A novel microglial subset plays a key role in myelinogenesis in developing brain. EMBO J. 36, 3292–3308. doi: 10.15252/embj.201696056
Wong, R. O. L., Chernjavsky, A., Smith, S. J., and Shatz, C. J. (1995). Early functional neural networks in the developing retina. Nature 374, 716–718. doi: 10.1038/374716a0
Wu, A., Wei, J., Kong, L. Y., Wang, Y., Priebe, W., Qiao, W., et al. (2010). Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro. Oncol. 12, 1113–1125. doi: 10.1093/neuonc/noq082
Xiong, J., Zhou, L., Lim, Y., Yang, M., Zhu, Y. H., Li, Z. W., et al. (2013). Mature BDNF promotes the growth of glioma cells in vitro. Oncol. Rep. 30, 2719–2724. doi: 10.3892/or.2013.2746
Yao, M., Ventura, P. B., Jiang, Y., Rodriguez, F. J., Wang, L., Perry, J. S. A., et al. (2020). Astrocytic trans-differentiation completes a multicellular paracrine feedback loop required for medulloblastoma tumor growth. Cell 180, 502–520e19. doi: 10.1016/j.cell.2019.12.024
Yates, D. (2020). Remodelling the matrix. Nat. Rev. Neurosci. 21:449. doi: 10.1038/s41583-020-0356-5
Ye, Z. C., Rothstein, J. D., and Sontheimer, H. (1999). Compromised glutamate transport in human glioma cells: reduction- mislocalization of sodium-dependent glutamate transporters and enhanced activity of cystine-glutamate exchange. J. Neurosci. 19, 10767–10777. doi: 10.1523/jneurosci.19-24-10767.1999
You, G., Sha, Z., and Jiang, T. (2012). The pathogenesis of tumor-related epilepsy and its implications for clinical treatment. Seizure 21, 153–159. doi: 10.1016/j.seizure.2011.12.016
You, Y., Klistorner, A., Thie, J., Gupta, V., and Graham, S. (2012). Axonal loss in a rat model of optic neuritis is closely correlated with visual evoked potential amplitudes using electroencephalogram-based scaling. Investig. Ophthalmol. Vis. Sci. 53:3662. doi: 10.1167/iovs.12-9843
Young, S. Z., and Bordey, A. (2009). GABA’s control of stem and cancer cell proliferation in adult neural and peripheral niches. Physiology 24, 171–185. doi: 10.1152/physiol.00002.2009
Yu, K., Lin, C. C. J., Hatcher, A., Lozzi, B., Kong, K., Huang-Hobbs, E., et al. (2020). PIK3CA variants selectively initiate brain hyperactivity during gliomagenesis. Nature 578, 166–171. doi: 10.1038/s41586-020-1952-2
Zanganeh, S., Hutter, G., Spitler, R., Lenkov, O., Mahmoudi, M., Shaw, A., et al. (2016). Iron oxide nanoparticles inhibit tumour growth by inducing pro-inflammatory macrophage polarization in tumour tissues. Nat. Nanotechnol. 11, 986–994. doi: 10.1038/nnano.2016.168
Zeng, Q., Michael, I. P., Zhang, P., Saghafinia, S., Knott, G., Jiao, W., et al. (2019). Synaptic proximity enables NMDAR signalling to promote brain metastasis. Nature 573, 526–531. doi: 10.1038/s41586-019-1576-6
Zetterling, M., Elf, K., Semnic, R., Latini, F., and Engström, E. R. (2020). Time course of neurological deficits after surgery for primary brain tumours. Acta Neurochir. (Wien.) 162, 3005–3018. doi: 10.1007/s00701-020-04425-3
Zhan, L., Fan, L., Kodama, L., Sohn, P. D., Wong, M. Y., Mousa, G. A., et al. (2020). A MAC2-positive progenitor-like microglial population is resistant to CSF1R inhibition in adult mouse brain. Elife 9:e51796. doi: 10.7554/eLife.51796
Zhang, P., Kishimoto, Y., Grammatikakis, I., Gottimukkala, K., Cutler, R. G., Zhang, S., et al. (2019). Senolytic therapy alleviates Aβ-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat. Neurosci. 22, 719–728. doi: 10.1038/s41593-019-0372-9
Zhou, W., Ke, S. Q., Huang, Z., Flavahan, W., Fang, X., Paul, J., et al. (2015). Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat. Cell Biol. 17, 170–182. doi: 10.1038/ncb3090
Keywords: glioblastoma, glioma, peritumoral tissue, neural activity, glial cells, tumor-associated microglia/macrophages (TAM), reactive astrocyte, OPCs
Citation: Parmigiani E, Scalera M, Mori E, Tantillo E and Vannini E (2021) Old Stars and New Players in the Brain Tumor Microenvironment. Front. Cell. Neurosci. 15:709917. doi: 10.3389/fncel.2021.709917
Received: 14 May 2021; Accepted: 04 August 2021;
Published: 06 October 2021.
Edited by:
Stefania Ceruti, University of Milan, ItalyReviewed by:
Enrica Boda, University of Turin, ItalyAngela Giangrande, Conseil National Pour La Recherche Scientifique, France
Copyright © 2021 Parmigiani, Scalera, Mori, Tantillo and Vannini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eleonora Vannini, ZS52YW5uaW5pQGluLmNuci5pdA==