 Joanne L. Sharpe
Joanne L. Sharpe Nikki S. Harper1
Nikki S. Harper1 Ryan J. H. West
Ryan J. H. West- 1Division of Neuroscience and Experimental Psychology, Faculty of Biology, Medicine and Health, The University of Manchester, Manchester, United Kingdom
- 2Sheffield Institute for Translational Neuroscience, The University of Sheffield, Sheffield, United Kingdom
- 3Neuroscience Institute, The University of Sheffield, Sheffield, United Kingdom
An intronic hexanucleotide (GGGGCC) expansion in the C9orf72 gene is the most common genetic cause of frontotemporal dementia (FTD) and amyotrophic lateral sclerosis (ALS). In the decade following its discovery, much progress has been made in enhancing our understanding of how it precipitates disease. Both loss of function caused by reduced C9orf72 transcript levels, and gain of function mechanisms, triggered by the production of repetitive sense and antisense RNA and dipeptide repeat proteins, are thought to contribute to the toxicity. Drosophila models, with their unrivaled genetic tractability and short lifespan, have played a key role in developing our understanding of C9orf72-related FTD/ALS. There is no C9orf72 homolog in fly, and although this precludes investigations into loss of function toxicity, it is useful for elucidating mechanisms underpinning gain of function toxicity. To date there are a range of Drosophila C9orf72 models, encompassing different aspects of gain of function toxicity. In addition to pure repeat transgenes, which produce both repeat RNA and dipeptide repeat proteins (DPRs), RNA only models and DPR models have been generated to unpick the individual contributions of RNA and each dipeptide repeat protein to C9orf72 toxicity. In this review, we discuss how Drosophila models have shaped our understanding of C9orf72 gain of function toxicity, and address opportunities to utilize these models for further research.
Introduction
The Frontotemporal Dementia and Amyotrophic Lateral Sclerosis Spectrum
Frontotemporal dementia (FTD) and amyotrophic lateral sclerosis (ALS) are two progressive diseases of the nervous system which display significant neuropathological, genetic and clinical overlap. ALS is a motor disorder and FTD is primarily characterized by alterations to personality and behavior. However, it is estimated that 50% of ALS patients develop aspects of FTD, with 15% meeting the criteria for FTD diagnosis. Conversely 15% of FTD patients develop ALS symptoms (Ringholz et al., 2005). The two are also closely genetically linked, with a number of mutations known to cause both diseases (Al-Chalabi et al., 2012; Abramzon et al., 2020). As such FTD and ALS are commonly regarded as a spectrum of a single disease, with pure FTD and pure ALS representing distinct ends of a continuum.
Frontotemporal dementia is an umbrella term encompassing a group of clinical syndromes associated with frontotemporal lobar degeneration (FTLD), bilateral atrophy of frontal and temporal lobes (Neary et al., 1998; Snowden et al., 2002). These include semantic dementia, primary progressive aphasia and behavioral variant FTD (bvFTD). The term FTD is also commonly used to refer solely to bvFTD, the second most common early-onset dementia, with an age of onset under 65. bvFTD accounts for up to 20% of presenile dementia cases with a prevalence between 2.7 and 15.1 per 100,000 adults (Snowden et al., 2002). It most commonly occurs between the ages of 45 and 65, but can present before the age of 30 and in the elderly (Snowden et al., 2002; Harvey et al., 2003). The defining clinical characteristic of bvFTD is the alteration to behavior and character with relative preservation of memory and other instrumental functions. However, the topological pattern of atrophy is variable and this is reflected in the heterogeneity of symptoms (Snowden et al., 2002; Seelaar et al., 2011). In contrast, ALS is a motor disorder whereby denervation of upper and lower motor neurons in the brain and spinal cord leads to progressive motor deficits, muscle atrophy and eventually death due to respiratory failure (Brooks et al., 2000; Foster and Salajegheh, 2019). ALS has a prevalence of approximately 2 in 100,000 (Chio et al., 2013) and symptoms usually present between the ages of 55–75 years (Chio et al., 2009). The median survival time from onset to death ranges from 3 to 5 years and a depletion of over 50% of spinal motor neurons is usually observed is usually observed post-mortem (Hardiman et al., 2017).
Proteinopathy, a common feature of neurodegenerative disease, refers to the formation, aggregation and accumulation of misfolded proteins. FTD and ALS are heterogeneous proteinopathies that can be categorized by the predominant protein component of these inclusions. The most common protein aggregate in both ALS and FTD is the RNA-binding protein TDP-43 [trans-activation response (TAR) DNA-binding protein 43], accounting for 95–97% and 50–60% cases, respectively (Neumann et al., 2006, 2009). It was the discovery that such inclusions were present in both FTD and ALS that provided the first pathological link between the two diseases (Neumann et al., 2006). The remaining FTD cases are categorized as FTD-Tau, and less commonly FTD-FUS (fused in sarcoma) and FTD-UPS (ubiquitin-proteasome system) (Holm et al., 2009; Neumann et al., 2009; Mackenzie et al., 2010; Urwin et al., 2010). The minority of ALS cases that are TDP-43 negative are associated with mutations in, and pathological inclusions of, SOD1 (superoxide dismutase 1) or FUS (Al-Chalabi et al., 2012).
Genetically, the FTD/ALS spectrum is highly heterogeneous, with over 100 genes implicated. There is a hereditary component to both diseases, although this is more significant in FTD, where 40–50% of cases are familial (Rohrer et al., 2009; Rademakers et al., 2012), than in ALS where 5–10% of cases have a family history (Al-Chalabi et al., 2012). Crucially, the same range of pathological features are observed in both sporadic and familial forms of both FTD and ALS. As such, elucidating mechanisms associated with familial forms can highlight common mechanisms underpinning FTD/ALS spectrum disorders more generally (Al-Chalabi et al., 2012).
C9orf72
The most common genetic cause of both FTD and ALS is a hexanucleotide repeat expansion mutation in the chromosome 9 open reading frame 72 (C9orf72) gene, first discovered in 2011 (DeJesus-Hernandez et al., 2011; Renton et al., 2011). C9orf72 expansion mutations have also been reported in cases of Alzheimer’s disease (Majounie et al., 2012), Huntington’s disease phenocopy syndromes (Moss et al., 2014), parkinsonism (Lesage et al., 2013; Wilke et al., 2016) and epilepsy (Capasso et al., 2017; van den Ameele et al., 2018; Estevez-Fraga et al., 2021). The incredible variation of clinical presentation within expansion carriers suggests a substantial contribution by genetic modifiers to C9orf72 mutation-related toxicity. The mutation itself is an expansion of a GGGGCC (G4C2) sequence within the first intron of C9orf72, which in mutation carriers is typically repeated thousands of times. The length of the C9orf72 expansion in patients and its role as a potential genetic modifier has proved a contentious issue, as there is no precise threshold at which the individual will definitely present symptoms. The general consensus of what constitutes a pathogenic expansion is one between several hundred and several thousand repeat units (DeJesus-Hernandez et al., 2011; Beck et al., 2013; Garcia-Redondo et al., 2013; van Blitterswijk et al., 2013; Waite et al., 2014). This is in contrast to most healthy control cohorts in which repeat lengths of up to 24 units are observed (DeJesus-Hernandez et al., 2011; Renton et al., 2011; Gijselinck et al., 2012; Garcia-Redondo et al., 2013). Larger repeat sizes have been reported in unaffected individuals, including some over 400 repeats in length (Beck et al., 2013), likely reflecting variability in age of onset and reduced disease penetrance due to other factors. Smaller repeat lengths have also been observed in affected individuals, including one report of alleles of 20–22 repeats in one FTD family (Gomez-Tortosa et al., 2013). Furthermore, somatic instability of the mutation confounds attempts to measure repeat length in individuals; a large expansion within the central nervous system (CNS) can be found concurrently with an intermediate length repeat in blood (Nordin et al., 2015). Dissecting the role of repeat length from the tangled web of other genetic risk factors, clinical heterogeneity and variable age of onset remains an important area for investigation.
Mechanisms of Toxicity of the C9orf72 Mutation
There are three main hypotheses of how the C9orf72 repeat mutation results in neurodegeneration: (1) haploinsufficiency caused by reduced expression; (2) RNA-mediated toxicity, whereby the repeat is transcribed and the resulting repeat RNA forms toxic foci that sequester important proteins; and (3) dipeptide-repeat protein (DPR)-mediated toxicity whereby non-canonical translation of the repeat RNA produces five different DPRs that disrupt cellular processes. The mechanisms of toxicity associated with the hexanucleotide repeat are summarized in Figure 1. Although each of these mechanisms are likely to contribute toward disease it is generally accepted that DPRs are the predominant driver of toxicity (Mizielinska et al., 2014; Moens et al., 2017). However, it is unclear to what degree each different DPR is responsible, how they interact with each other, nor how they may act synergistically with other mechanisms of toxicity.
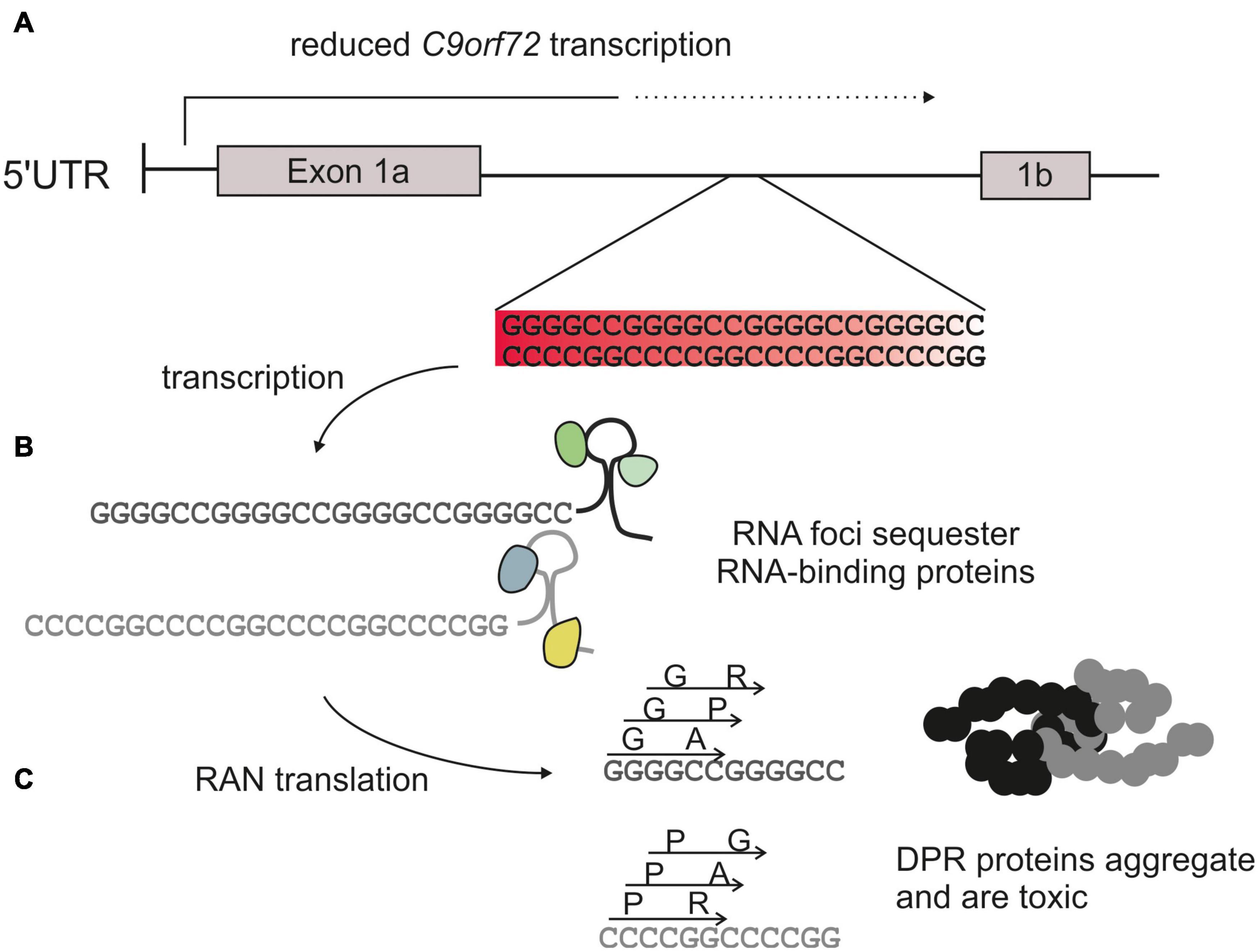
Figure 1. C9orf72 hexanucleotide expansion toxicity. The C9orf72 repeat mutation is an expansion of a GGGGCC sequence usually <30 repeats in length in healthy controls, to >1,000 repeats in affected patients. (A) The repeat is located within an intron of the C9orf72 gene. This reduces transcription and therefore production of the endogenous protein (haploinsufficiency). (B) The repeat is transcribed into sense and antisense RNA which can form foci and sequester RNA binding proteins. (C) Repeat RNA is translated via a form of non-canonical translation (repeat-associated non-AUG translation), to produce 5 different dipeptide repeat proteins: glycine-alanine (GA) and glycine-arginine (GR), alanine-proline (AP), and proline-arginine (PR) and glycine-proline (GP).
Haploinsufficiency/Loss of Function Mechanisms
The G4C2 repeat sequence is situated in the first intron of C9orf72. Here it is predicted cause early transcription abortion, hypermethylation of the repeat and adjacent CpG islands and increased histone methylation. This leads to reduced transcription of the native gene, and results in protein haploinsufficiency (Belzil et al., 2013; Gijselinck et al., 2016). The C9orf72 protein is homologous to DENN (differentially expressed in normal and neoplastic cells) proteins, suggesting that it acts as a GEF (guanine exchange factor) for Rab GTPases, key regulators of membrane trafficking events such as autophagy and endocytosis (Levine et al., 2013). It is most abundant in the brain and spinal cord, is highly soluble and is detectable in both the cytoplasm and the nucleus (DeJesus-Hernandez et al., 2011; Renton et al., 2011). In neurons it is localized to the presynaptic region where it forms a stable complex with WDR14 (WD-repeat containing protein 14) and SMCR8 (Smith-Magenis chromosome region 8), which recruits Rab proteins and thus controls autophagy from the initial recruitment of ubiquitinated substrates through to autophagosome-lysosome fusion. Loss-of-function (LOF) C9orf72 mice develop inflammatory and autoimmune phenotypes, suggesting that C9orf72 may play a role in immune homeostasis in microglia (Burberry et al., 2016). However, the lack of neurodegenerative phenotypes and absence of TDP-43 accumulation in C9orf72 knock-out mice strongly suggests that LOF is not the primary cause of neurotoxicity in FTD and ALS (Koppers et al., 2015). It is possible that haploinsufficiency may potentiate toxic RNA and DPR gain of function (GOF) mechanisms in a non-cell-autonomous manner, but it is unlikely to precipitate the disease in its own right (Jiang et al., 2016).
RNA-Mediated Toxicity
Bidirectional transcription of the G4C2 repeat produces repeat RNA prone to forming atypical secondary structures. Sense RNA is more abundant and tends to form hairpins and G-quadruplexes (Fratta et al., 2012), whereas antisense RNA forms i-motifs and protonated hairpins (Kovanda et al., 2015). Accumulations of sense and antisense RNA structures are known as RNA foci. They interact with RNA-binding proteins and disrupt gene regulation, translation and splicing (Sareen et al., 2013; Xu et al., 2013; Kharel et al., 2020). RNA foci have been demonstrated to be involved in pathogenesis in other repeat diseases; for example, in myotonic dystrophy type 1 they cause alterations in gene expression and splicing by binding and disrupting the function of RNA-binding proteins (Osborne et al., 2009). They are also a hallmark of FTD/ALS pathology, observed in multiple regions of the CNS in C9orf72 patients (DeJesus-Hernandez et al., 2011; Cooper-Knock et al., 2015). However, evidence from RNA-only models suggests that they are not a major driver of toxicity (Mizielinska et al., 2014; Tran et al., 2015).
Dipeptide-Repeat Protein (DPR)-Mediated Toxicity
The phenomenon of repeat-associated non-AUG (RAN) translation, whereby repeat RNA can be translated without a start codon, was first observed in the microsatellite expansion disease spinocerebellar ataxia type 8 (Zu et al., 2011). Subsequently, interrogation of the potential for RAN translation of C9orf72 repeat expansions confirmed the production of DPRs via this non-canonical mechanism. RAN translation of sense and anti-sense RNA across all reading frames produces five different dipeptide repeats: from the sense strand, glycine-alanine (GA) and glycine-arginine (GR); from the antisense strand, alanine-proline (AP) and proline-arginine (PR); and from both strands, glycine-proline (GP) (Mori et al., 2013a; Zu et al., 2013). RAN translation of the C9orf72 repeat is impervious to inhibition by the integrated stress response; in fact, it is selectively enhanced (Green et al., 2017), thus creating a potential positive feedback loop that contributes to neurodegeneration.
The unique pathological hallmark of C9orf72-mediated disease is TDP-43-negative, p62- and ubiquitin-positive star-shaped cytoplasmic inclusions within neurons and glia (Ash et al., 2013; Mann et al., 2013; Mori et al., 2013a). These inclusions are now known to contain the five DPR species. DPR inclusions have been observed within the cerebellum, hippocampus, basal ganglia, frontal and motor cortices and skeletal muscle (Al-Sarraj et al., 2011; Cooper-Knock et al., 2012; Hsiung et al., 2012; Mahoney et al., 2012; Ash et al., 2013; Mann et al., 2013; Cykowski et al., 2019). Additionally, although less prominent, intranuclear and para-nucleolar DPR aggregates have been observed (Wen et al., 2014; Schludi et al., 2015). Clinico-pathological studies investigating the distribution and quantities of GA in a C9orf72 cohort found GA pathology to be consistent across the cohort, regardless of clinical phenotype (Mackenzie et al., 2013, 2015). Despite some evidence to suggest a lack of correlation between DPR load and the extent of neurodegeneration (Davidson et al., 2014), these studies are based on post-mortem tissue, where only surviving cells can be analyzed. The absence of DPR pathology in end-stage disease does not, therefore, necessarily preclude their toxicity in cells already lost. Additionally, the observations were made using immunohistochemistry techniques, which do not consider any pathological burden of soluble DPR oligomers, an emerging theme in other neurodegenerative diseases such as Alzheimer’s and Parkinson’s disease (Choi and Gandhi, 2018).
Not all DPRs are equal, in terms of abundance, physical properties and likely toxicity (summarized in Table 1). GA inclusions appear most visible in C9orf72 patient brains, followed by GP, GR, PR, and AP (Mackenzie et al., 2015). However, the relative abundance of soluble and insoluble DPRs varies throughout the brain and there is a large degree of variability in DPR protein levels between individuals (Quaegebeur et al., 2020). GA, PR, and in particular GR have all been widely reported to have toxic effects in various model systems (Kwon et al., 2014; Mizielinska et al., 2014; Callister et al., 2016), but there remains little consensus on which DPRs are the main drivers of toxicity and what mechanisms they might act through. This is further complicated by the observation that different combinations of the five DPRs can be present in individual cells in patients, and the possibility that they could interact with each other, as well as act synergistically with C9orf72 haploinsufficiency and other gain-of-function mechanisms. The secondary structures formed by each DPR have been investigated in vitro, but the links between their distinct structural properties and cellular toxicity have yet to be fully elucidated. GA has been the most extensively researched in this regard due to its amyloid beta-like structure; it forms flat sheets of densely packed, ribbon-type fibrils that have been shown to have the potential to transmit between cells (Edbauer and Haass, 2016), disrupt nucleocytoplasmic transport (Zhang et al., 2016) and recruit and inhibit the proteasome (May et al., 2014; Zhang et al., 2014, 2016; Chang et al., 2016; Edbauer and Haass, 2016; Guo et al., 2018). It is this ability to form beta sheets that sets GA apart from other DPRs and could explain why, in certain models, it is more toxic than AP and GP, the other two uncharged DPRs. The unique biochemical configuration of proline precludes the formation of beta sheets in GP, AP and PR due to the central ring restricting possible confirmations of the backbone (Doig, 2017). GP and AP form flexible coils which are unable to self-aggregate into sheets in the same way as GA (Lee et al., 2016; Freibaum and Taylor, 2017). Indeed, as predicted based on their structural properties, GP and AP interact with fewer intracellular proteins (Lee et al., 2016), which is consistent with their lack of toxicity in many model systems (Mizielinska et al., 2014; Wen et al., 2014; Freibaum et al., 2015; Lee et al., 2016). PR also contains proline but unlike AP, GA, and GP, it is charged and highly polar due to the presence of arginine. This is likely why it behaves more similarly to GR in terms of toxicity. Indeed, collectively these two DPRs are often referred to as “arginine-rich” DPRs, due to the predicted importance of this residue in their toxicity. It confers a high hydrophilicity and is likely responsible for their highly interactive nature. We know that both GR and PR accumulate in the nucleus of transfected cells (Kwon et al., 2014; Callister et al., 2016) and disrupt ribosomal RNA biogenesis when overexpressed (Kwon et al., 2014). Nuclear localization signal domains tend to be rich in arginine, and it is possible that these DPRs are able to mimic this and gain access to the nucleus through transportation (Kwon et al., 2014). Additionally, multiple studies have focused on perturbed liquid-liquid phase separation (LLPS) dynamics, important in the formation and dissolution of membraneless organelles such as the nucleolus (Wen et al., 2014; Lee et al., 2016). This theory is based on the concept that arginine-containing proteins are capable of interacting with low-complexity sequence domains (LCDs) of RNA-binding proteins (RBPs) and thus alter LLPS dynamics (Lee et al., 2016; Lin et al., 2016). Perturbation of physiological LLPS by GR and PR in both the cytoplasm and nucleus (Lee et al., 2016; Boeynaems et al., 2017; Zhang et al., 2018; White et al., 2019) provides another mechanism by which arginine-rich DPRs could act to cause neurodegeneration. However, there has been limited in vivo work to confirm this. PR and GR are also capable of interacting with different cytoplasmic targets, such as translation initiation factor eIF3η and ribosomal subunits, causing translational inhibition and disrupting ribosome biogenesis and rRNA processing, respectively (Tao et al., 2015; Kanekura et al., 2016; Lee et al., 2016; Zhang et al., 2018). GR has also been shown to induce oxidative stress by interacting with mitochondrial ribosomes (Lee et al., 2016; Lopez-Gonzalez et al., 2016).
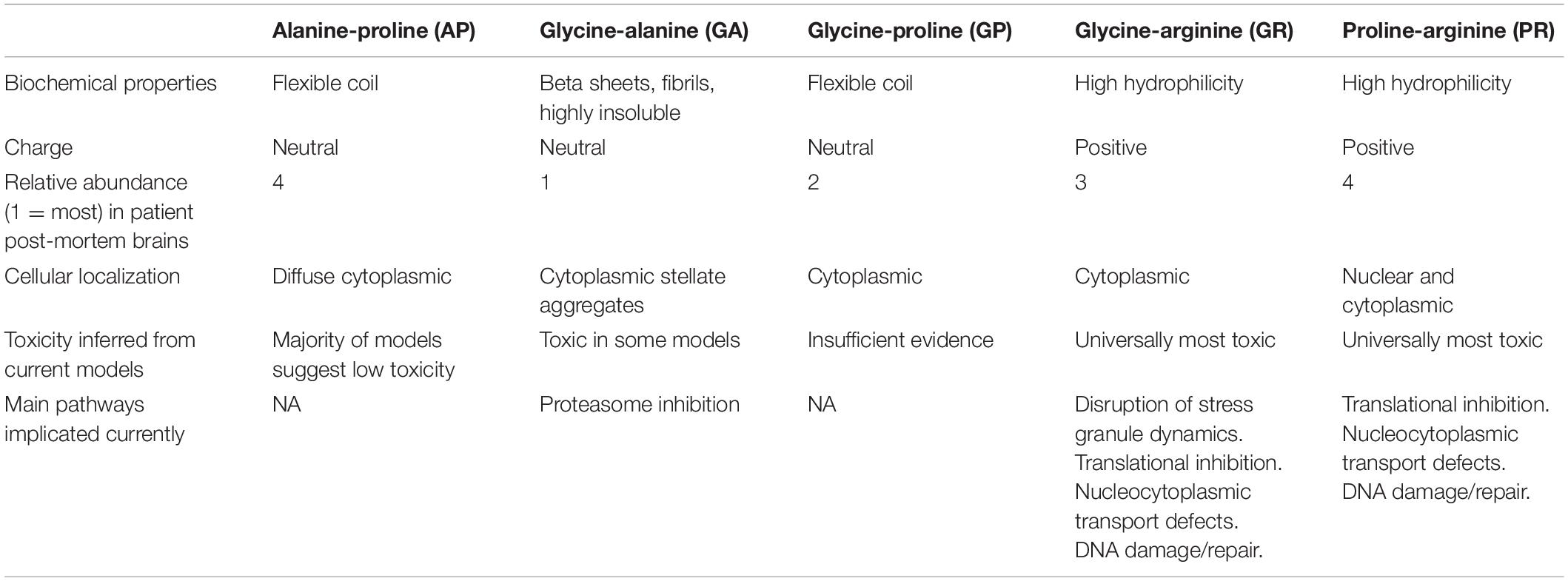
Table 1. Summary of the biochemical properties, abundance, cellular localization, toxicity, and main neuropathological pathways associated with each dipeptide repeat protein (DPR) species.
Since the initial identification of C9orf72 hexanucleotide expansion mutations as the most common genetic cause of both FTD and ALS, a number of in vitro and in vivo models have been established in order to elucidate the molecular mechanism underpinning disease. A number of these models, including mice, have been reviewed extensively elsewhere (Batra and Lee, 2017). In this review we focus on the contributions Drosophila melanogaster models have made to our understanding of C9orf72-related FTD and ALS.
Drosophila Models of C9orf72-Associated Frontotemporal Dementia/Amyotrophic Lateral Sclerosis
Drosophila have been extensively used to study a range of neurodegenerative and neurodevelopmental disorders including autism (Vilidaite et al., 2018), Alzheimer’s (Cao et al., 2008; Chakraborty et al., 2011), Parkinson’s (West et al., 2015), Huntington’s, FTD (West et al., 2020a,b), and ALS (West et al., 2018). In addition to their short lifespan, rapid generation times, low cost and ability to display complex behaviors including learning and memory, the power of Drosophila as a model organism lies in their genetic tractability. The Drosophila genome has been sequenced since 2000 and ∼75% of human genes known to cause disease have an ortholog in fly. There also exists a number of well-established tools that allow simple and elegant genetic manipulation of Drosophila. In addition to allowing detailed characterization of gene function, both endogenous and transgenic models of disease have allowed us to perform large scale genetic screens, elucidating novel mechanisms underpinning disease pathways (St. Johnston, 2002; Deal and Yamamoto, 2019). A full list of Drosophila C9orf72 models can be found in Table 2.

Table 2. Summary of Drosophila models of C9orf72-related frontotemporal dementia (FTD) and amyotrophic lateral sclerosis (ALS).
Pure Repeat Models
Pure repeat models contain the G4C2 sequence and therefore produce both repetitive RNA and all five DPRs, as in patients. Drosophila do not have a C9orf72 ortholog and whilst this precludes investigating the contribution of loss of function (Figure 2), it provides the ideal model for looking exclusively at gain-of-function toxicity. Indeed, pure repeat fly models provided the first evidence that gain of function is sufficient to cause toxicity (Xu et al., 2013; Mizielinska and Isaacs, 2014; Solomon et al., 2018; Goodman et al., 2019).
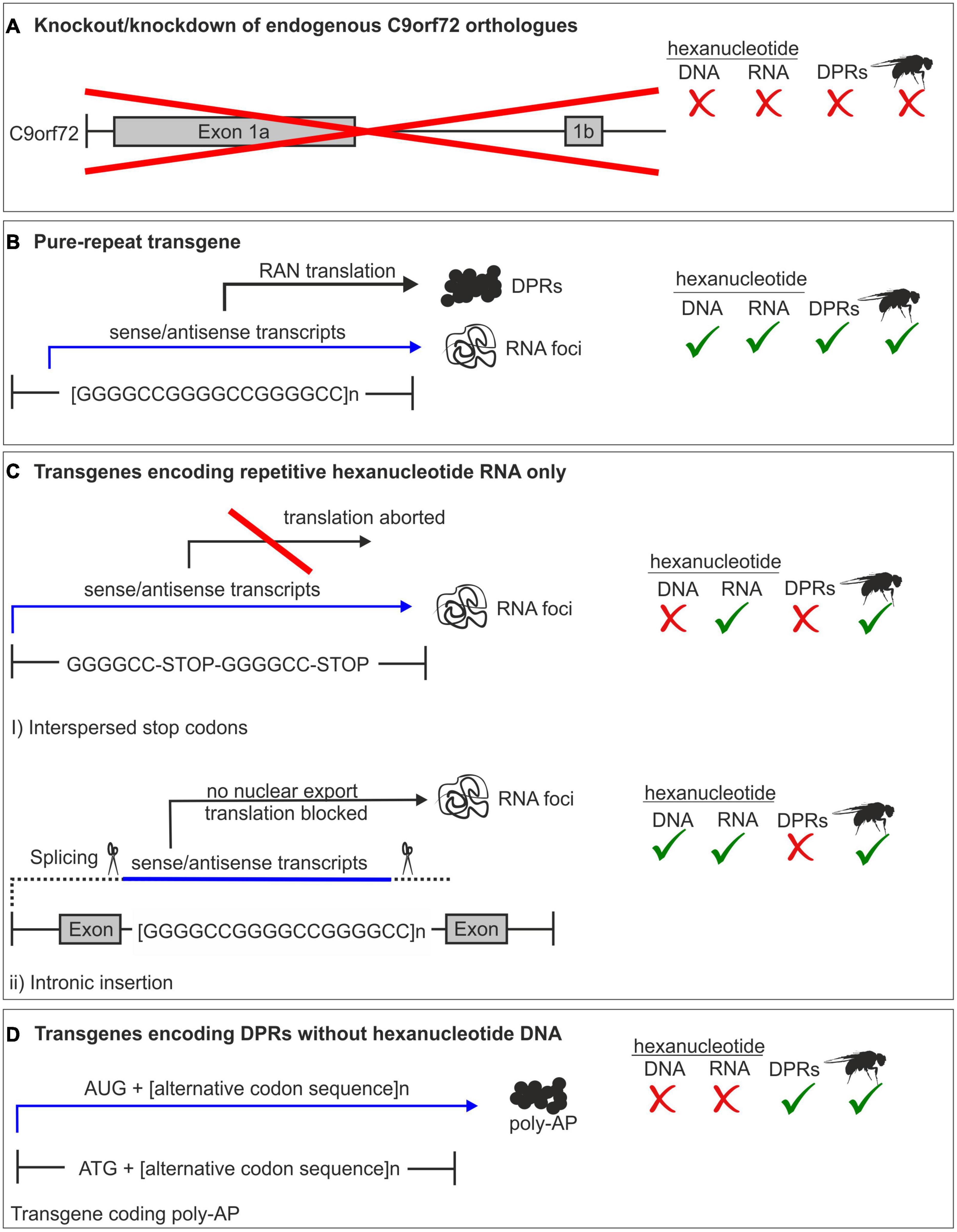
Figure 2. Summary of C9orf72 models. (A) Complete or partial knockout of the endogenous C9orf72 gene. Drosophila does not have a C9orf72 homolog, therefore a knockout model is impossible in fly. (B) Pure repeat models contain the hexanucleotide sequence, and produce all dipeptide repeat proteins (DPRs) along with repeat RNA. (C) RNA-only models are either intronic or have interspersed stop codons to prevent translation. Therefore, only repeat RNA and not DPRs are produced. (D) DPR models use an alternative codon sequence to produce one DPR protein and lack the hexanucleotide sequence.
Transgene expression within the Drosophila eye, an established system for performing genome wide modifier screens, is becoming increasingly popular as a system to compare dose- and length-dependent toxicity in C9orf72-related gain-of-function models. Expression in the fly eye, which is dispensable for survival, allows researchers to look at levels of toxicity that would be lethal if expressed either pan-neuronally or globally. Pure repeat expression in the eye, using the GMR-Gal4 driver, was shown to be toxic at a minimum repeat length of 36 repeats. Increasing the length to 103 repeats produced a more severe phenotype, implicating length as a critical factor in determining toxicity (Mizielinska et al., 2014). This is consistent with findings from multiple studies expressing pure repeats in zebrafish embryos (Lee et al., 2013), mice (Herranz-Martin et al., 2017), and yeast (Kramer et al., 2016). Furthermore, in addition to eye-specific expression, data from fly models expressing pure repeats in neurons supports a length-dependent toxicity. Larval size, synaptic bouton number and crawling ability (motor-neuronal expression) (Freibaum et al., 2015), climbing ability as adults (pan-neuronal expression) (Freibaum et al., 2015), and lifespan (adult-only neuronal expression) (Mizielinska et al., 2014), are more severely affected by expression of longer repeats. Similarly, an increased dose, conferred by homozygous expression of pure repeat transgenes, increases toxicity in the eye (GMR-Gal4), as well as perturbing larval crawling and bouton number, when expressed pan-neuronally (elav-gal4) or in motor neurons (OK371-gal4) (Freibaum et al., 2015).
A key strength of Drosophila models is the capacity for high-throughput unbiased genetic screens. Freibaum et al. (2015) utilized this to identify key genes and pathways implicated in pure repeat toxicity. A genetic modifier eye screen found that loss-of-function mutations in groups of connected genes, including many within the nucleocytoplasmic transport pathway, potentiated the eye phenotype (Freibaum et al., 2015). Consistent with results from initial eye screens, expression of 58 G4C2-repeats in Drosophila salivary glands caused nuclear envelope abnormalities and accumulation of nuclear RNA (Freibaum et al., 2015). Although physiologically less relevant than neurons for the study of FTD/ALS, their large size and accessibility makes Drosophila larval salivary glands a powerful model for imaging the localization of cellular components, in particular for assessing nuclear-cytoplasmic localization.
It is important to consider that whilst pure repeats replicate the DNA sequence in patients, their expression produces both repeat RNA and DPRs; therefore, it is not possible to distinguish between the relative contribution of each to toxicity, or through what mechanisms they each act. This is exemplified in mice. Transgenic overexpression of G4C2 repeats in mice, either on their own via somatic brain transgenesis mediated by adeno-associated virus (Chew et al., 2015) or as part of a patient-derived bacterial artificial chromosome (BAC) (O’Rourke et al., 2015; Peters et al., 2015; Liu et al., 2016), produces RNA foci and at least some DPR expression. However, the presence of TDP-43 pathology, motor and cognitive impairment and survival deficits are inconsistent. This is likely due to differences in the genetic background of the mice, which is known to have a significant impact on neurodegenerative phenotypes, rather than the nature of the repeat itself (Moens et al., 2017). Additionally, expression levels between models differ; Chew et al. (2015) and Liu et al. (2016) showed that high levels of overexpression of shorter repeats was toxic, suggesting that a threshold level of DPR and/or RNA may not have been reached in non-toxic models. However, it is unclear whether this is due to insufficient levels of DPRs or RNA, or whether the shorter lengths used precluded toxicity at lower expression levels.
To overcome the limitations of pure repeat models, RNA-only and DPR fly models have been developed, which allow the mechanisms of RNA-mediated and DPR-mediated toxicity to be investigated in isolation. Combining different approaches allows for a robust analysis of C9orf72 toxicity and the respective roles DPRs and RNA have in this. A recent example of an integrated approach used both pure repeat and DPR fly models to investigate the role of insulin signaling in GR toxicity. Data from a DPR-only model was validated using a 36 G4C2-repeat Drosophila model expressing the pure repeat in adult neurons (Atilano et al., 2021). Furthermore, a genetic screen using lifespan as a readout for toxicity identified the translation initiation factor eIF1A as capable of rescuing toxicity by enhancing translation when overexpressed in the pure repeat background. This complemented research using PR(100), PR(50), GR(100), and GR(50) models (Goodman et al., 2019). A combinatorial approach where hypotheses are tested in multiple different models would compensate for the limitations of each model and present a good option for future research.
RNA Models
In order to elucidate the contribution of repeat RNA to disease progression, fly models expressing repeat RNA only (RO) were developed. In order to produce repeat RNA without the DPRs, G4C2 of different repeat lengths was interspersed with stop codons to prevent translation (Figure 2) (Mizielinska et al., 2014). These repeats were shown to form G-quadruplexes in vitro and RNA foci in SH-SY5Y cells (Mizielinska et al., 2014). These constructs were cloned into a pUAST vector to allow generation of RO fly lines. When expressed in Drosophila using GMR-Gal4, the RO constructs at 36, 108, and 288 repeats produced RNA foci, but no antisense RNA (Mizielinska et al., 2014). Expression of RO constructs failed to produce the same eye phenotypes as seen in pure repeat models and did not have the same detrimental effect on egg-to-adult viability (Mizielinska et al., 2014). Adult longevity was unaffected by neuronal expression of RO repeats, in contrast to the shortened lifespan of pure repeats of equivalent length (Mizielinska et al., 2014). Additionally, survival deficits caused by expression of the G4C2 repeat were partially rescued by cycloheximide, a protein synthesis inhibitor (Mizielinska et al., 2014). Taken together, this strongly implicates DPR expression, not RNA, as the toxic species in C9orf72 expansions.
The first antisense repeat RNA Drosophila models were generated in 2018, along with the first expressing pathologically relevant repeat length RNA (Moens et al., 2018). In addition to models carrying interspersed stop codons within the G4C2 repeat and producing RO repeats as part of a polyadenylated transcript (Mizielinska et al., 2014), new models were generated containing 106 RO hexanucleotide repeats within a constitutively spliced artificial intron (Figure 2). In order to assess the potential for antisense repeat RNA toxicity, the original RO repeat constructs were reversed, deriving two lines expressing ∼100 repeats, one from within an intron and the other a polyadenylated transcript. In contrast to polyadenylated sense RO repeats, where a primarily cytoplasmic localization was observed, sense and antisense intronic RO repeats formed predominantly nuclear foci. Thus, the differential effects of cytoplasmic and nuclear RNA foci could be measured. Neither intronic nor polyadenylated RO repeats caused a reduction in longevity, and whilst polyadenylated sense RO repeat expression in adult neurons caused a slight climbing defect, no reduction in climbing ability was seen with intronic repeats or antisense repeats (Moens et al., 2018). When extended to ∼800 and ∼1,000 repeats, sense RO repeats formed abundant RNA foci. Expression of these longer repeats produced phenotypes comparable to those from shorter repeats. Lack of typical neurodegenerative phenotypes in both short and physiologically relevant size repeat RNA strongly suggests that RNA is not responsible for the toxicity observed with expression of the pure repeat. It was confirmed that the repeat RNA expressed in these models was sequestering RNA-binding proteins, which is thought to be a critical mediator of RNA toxicity. The fact that RO repeat RNA foci recapitulate this key property of RNA foci observed in patients, and yet do not cause overt toxicity in vivo, suggests that repeat RNA alone is insufficient to cause neurodegeneration associated with C9orf72-FTD/ALS.
In C9orf72 FTD patient brains, RNA foci are abundant in the frontal cortex, where there is greatest neuronal loss. Furthermore, a number of studies point to specific mechanisms by which RNA can be toxic (Mizielinska et al., 2013; Burguete et al., 2015). However, RO Drosophila models provide limited evidence supporting repeat RNA as a primary driver of toxicity in C9orf72-related FTD and ALS. This does not rule out a contribution for RNA in disease progression, but implicates DPRs rather than RNA as the primary driver of toxicity.
Dipeptide-Repeat Models
In order to elucidate the role of each DPR in C9orf72-related toxicity a number of models expressing each DPR in isolation have been developed. Expression of each DPR individually, without repeat RNA, is typically achieved by expression of transgenes generated using alternative codon sequences for each different DPR (Figure 2). Drosophila models expressing DPRs have proven a powerful tool to dissect the contribution of each DPR to C9orf72-releated disease. More recently they have also been used to explore the effect of concomitant expression of DPRs in vivo.
Glycine-Proline
Glycine-proline is the least studied of all the DPRs due to difficulties in cloning its sequence past 50 repeats (Callister et al., 2016). There is to date only one GP Drosophila model, which was made by Freibaum et al. (2015). Here, they found that expression of GP(47) in the eye using GMR-Gal4 did not produce a degenerative eye phenotype, and it was investigated no further (Freibaum et al., 2015). When expressed in embryonic chick spinal cord, GP(47) also proved non-toxic (Lee et al., 2017). However, due to the lack of availability of longer GP constructs, it is impossible to conclude with certainty that GP plays no role in C9orf72- related FTD/ALS. Given that AP only shows electrophysiological defects at over 1,000 repeats, it is possible that we are also missing key phenotypes associated with GP expression by relying only on these short constructs.
Glycine-Alanine
Glycine-alanines ability to form large aggregating beta-sheet fibrils sets it apart from the other DPRs, and it is easily distinguishable as it forms characteristic p62- and ubiquitin-positive stellate or fern-like cytoplasmic inclusions in patient tissue (Mann et al., 2013). However, the formation of these distinctive structures is dependent on repeat length; expression of GA of 36 and 1,020 repeats in HeLa cells showed that large fern-like inclusions only form at the longer repeat length, in contrast to the discrete spherical inclusions in GA(36) (Callister et al., 2016). Furthermore, Drosophila expressing 1,000 repeats of GA in the nervous system showed the same characteristic stellate inclusions throughout (West et al., 2020a). Such structures have not been clearly demonstrated in Drosophila models expressing shorter GA repeats, raising the question of whether short repeats have the capacity to form the distinct stellate structures observed in patients. One must also consider that differences in morphology between short and long repeat aggregates may lead to contrasting effects on downstream pathological pathways as well as on DPR spreading and localization.
There are many different Drosophila models expressing GA up to 100 repeats (Mizielinska et al., 2014; Freibaum et al., 2015; Yang et al., 2015; Boeynaems et al., 2016; Solomon et al., 2018) but only recently was a longer, potentially more physiologically relevant model developed expressing over 1,000 repeats of GA (West et al., 2020a). The discrepancies in GA toxicity across these different models highlight the need for a consistent and robust model system. Commonly, expression of GA in the eye causes no toxic effects, and this is consistent up to 1,000 repeats (Mizielinska et al., 2014; Freibaum et al., 2015; West et al., 2020a). This lack of a clear phenotype, when expressed in the eye, limits the ability to employ this model for genetic modifier screens, in particular those looking to identify suppressors. As with all of these models one must also consider the possibility that the lack of phenotype is because proteins sequestered by DPRs and pathways downstream may not all be conserved between fly and human. Furthermore, although the Drosophila eye is a robust screening tool, it is not the optimal expression system to understand how the presence of DPRs in the nervous system contribute to toxicity in a physiological, whole-organism context. Perturbations resulting from expression in the eye are largely associated with developmental effects, whereas pan-neuronal expression provides a model much more representative of DPR expression in disease. However, pan-neuronal expression of GA has led to more inconsistent observations, depending upon the model used. For example, when expressed pan-neuronally a number of the short repeat models appear to be lethal. This is in stark contrast to longer, 1,000-repeat, models. Furthermore, expression of GA(100) in adulthood using an inducible elavGeneSwitch driver, to circumvent early lethality, resulted in a late-onset decrease in survival, contrasting the finding that GA(1000), expressed pan-neuronally throughout the entire lifetime of the fly, causes a slight but significant increase in lifespan (West et al., 2020a). Comparison of expression levels between 100-repeat and 1,000-repeat fly models suggests these discrepancies do not result from significant differences in expression levels, possibly associated with different genomic locations of the transgenes; rather they may result directly from the different repeat lengths (West et al., 2020a). In this context it is important to note that while the hexanucleotide expansion is typically in the region of hundreds to thousands of repeats in length (DeJesus-Hernandez et al., 2011; Beck et al., 2013; Garcia-Redondo et al., 2013; van Blitterswijk et al., 2013; Waite et al., 2014) it remains a conflicting topic for debate as to whether repeat length correlates with age of onset, severity and disease progression. Indeed, as mentioned previously, unaffected individuals have been identified with long repeats whilst, conversely, individuals carrying repeat lengths not typically considered pathogenic have presented with disease (Beck et al., 2013; Gomez-Tortosa et al., 2013). In addition, due to the technical difficulties associated with accurately quantifying both DPR burden and length in patient tissues, the exact length of DPRs translated from the repeat expansion remains unclear. It is therefore possible that differences in age of onset, progression and severity of disease is underpinned, at least in part, by translation of different length DPRs.
Motor problems are a defining characteristic of ALS. Despite this, there is limited research testing motor function in GA-expressing flies, as much of the research focuses on GR and PR. This is largely influenced by early studies showing that GR and PR are the most toxic DPR species. However, this is arguably a short-sighted approach in terms of getting a full picture of how each DPR behaves in vivo. As GA is universally non-toxic in eye screens, it is often ignored in favor of pursuing PR and GR in further experiments. Given the relative ease and low costs associated with genetic experiments in Drosophila, there is an argument for a more consistent approach, performing experiments with all DPRs, rather than focusing on only arginine-rich DPRs. This is especially important when there is no consensus on the mechanisms and degree of toxicity of each DPR. Studies that have looked at motor function in adult flies expressing GA suggest that GA is not a major driver of acute toxicity but may contribute at a lower level when expressed at a longer repeat length. Three-day old flies expressing GA(80) in motor neurons, using OK371-Gal4, had no reduction in climbing distance over a 10s period compared to control flies (Yang et al., 2015). Similarly, a different fly model expressing GA(1000) shows a slight but significant reduction in climbing speed at 3 days post-eclosion, but when tested again at 28 days post-eclosion, they were no longer climbing more slowly than controls. This suggests a potential basal level of motor dysfunction which was not exacerbated with age (West et al., 2020a). The idea that GA alone is not overtly toxic but may contribute to neuronal dysfunction is supported by findings using primary neuronal cell culture, which show proteasome impairment, as well as ER stress and sequestration of Unc119 (May et al., 2014; Zhang et al., 2016). However, this has not been studied in fly models. Indeed, there is little work using Drosophila to elucidate the mechanisms by which GA may contribute to neurodegeneration in ALS and FTD. One study looking at the relationship between DPR expression, nucleocytoplasmic transport defects and TDP-43 pathology found that GA(64) expression in Drosophila salivary glands caused cytoplasmic accumulation of the TDP-43 homolog TBPH (Solomon et al., 2018). In this model, GA(64) was also shown to co-aggregate with TBPH (Solomon et al., 2018). This is supported by findings from West et al. (2020a) which showed that GA(1000) similarly colocalizes with TBPH in the cytoplasm (West et al., 2020a). However, in contrast to arginine rich DPRs [GR(1000) and PR(1000)] GA(1000) did not cause a significant mislocalisation of TBPH (West et al., 2020a).
A defining characteristic of GA, setting it apart from other DPRs, is its apparent propensity to form beta-sheet structures and spread between cells (Chang et al., 2016; Edbauer and Haass, 2016). This finding has been replicated in a Drosophila model expressing GA(100) and GA(200) in a distinct neuronal subset (Moron-Oset et al., 2019). Here, they showed that GA was the only DPR that could spread in vivo, spreading in a repeat length- and age-dependent manner in the fly brain (Moron-Oset et al., 2019). To what extent this changes with repeat length in much longer (e.g., 1,000 repeat) models, or when it interacts with other DPRs is yet to be investigated.
Alanine-Proline
Alanine-proline is generally considered to play little or no role in the neurodegeneration elicited by DPRs in FTD and ALS. This is consistent with findings from Drosophila models, with one notable exception (Mizielinska et al., 2014; Wen et al., 2014; Boeynaems et al., 2016; Solomon et al., 2018). Amongst the shorter repeat models, flies expressing AP in various different cell types show no eye degeneration (Mizielinska et al., 2014; Wen et al., 2014; Lee et al., 2016) and no change in egg-to-adult viability (Mizielinska et al., 2014; Wen et al., 2014; Lee et al., 2016) or longevity (Mizielinska et al., 2014). The exception is the 1,000 repeat AP model generated by West et al. (2020a), which causes a dose-dependent toxicity in the eye, significant motor impairment throughout lifespan, electrophysiological defects and neurodegenerative vacuoles in the fly brain (West et al., 2020a). This suggests that length is particularly important in AP’s toxicity. Indeed, AP has also been shown to elicit electrophysiological defects in vitro in a length-dependent manner (Callister et al., 2016), only having an effect on cellular excitability at over 1,000 repeats. Taken together, this points to subtle length-dependent phenotypes contributing to neuronal dysfunction in ALS and FTD, and raises the issue that this may be missed when working with shorter repeat models. It also further emphasizes the importance of studying all DPRs rather than focusing exclusively on the arginine-rich species.
Arginine-Rich Dipeptide Repeat Proteins: Proline-Arginine and Glycine-Arginine
There is a broad consensus that the arginine-rich DPRs, GR, and PR, exhibit the most potent neurotoxicity, as shown throughout multiple model systems (Kwon et al., 2014; Wen et al., 2014; Jovicic et al., 2015). Due to increased interest in the arginine-rich DPRs, there is a larger volume of research focusing purely on PR, GR or both, compared to the other DPRs. As a result there are also several different mechanisms that have been associated with PR and GR expression, including nucleocytoplasmic transport (Jovicic et al., 2015; Boeynaems et al., 2016; Hayes et al., 2020), DNA damage (Lopez-Gonzalez et al., 2016; Andrade et al., 2020), translational disruption (Moens et al., 2019), and stress granule dysfunction (Lee et al., 2016; Boeynaems et al., 2017). Indeed, Drosophila models of PR and GR have been instrumental in elucidating many of these toxic mechanisms, which have subsequently been validated in mouse models, iPSCs and patient tissue. An important realization, supported in a number of these models, is that in flies GR was found to be spread diffusely throughout the cytosol, and PR to be both nuclear and cytoplasmic (Yang et al., 2015; Solomon et al., 2018; West et al., 2020a), rather than localized to the nucleolus as has previously been reported using short repeats in cell culture models (Kwon et al., 2014; Wen et al., 2014). This corresponds to pathology seen in C9orf72 patients, in which GR and PR inclusions are absent from the nucleolus (Ash et al., 2013; Mori et al., 2013b) and highlights the importance of modeling DPRs in vivo.
The toxicity of GR and PR was first demonstrated in Drosophila by Mizielinska et al. (2014), where eye-specific expression of GR and PR at both 36 and 100 repeats was found to cause extensive eye degeneration and lethality (Mizielinska et al., 2014). In contrast, AP and GA had no effect. Adult-only neuronal expression of PR(100) and GR(100), using the elav-GeneSwitch inducible driver, also caused a significant reduction in lifespan, relative to other DPRs and controls (Mizielinska et al., 2014). Additionally, later research using the same model found that inclusions of GR(100) were associated with significantly enlarged nucleoli, indicative that expression of GR contributes to nucleolar stress (Mizielinska et al., 2017). This is supported by another GR(80) model where enlarged nucleoli were also observed. Crucially, this was without GR localizing to the nucleolus (Yang et al., 2015), further suggesting nucleolar localization in vitro to be an artifact and unrelated to nucleolar dysfunction. The extreme toxicity conferred by GR and PR proteins of short repeat lengths was also observed by Lee et al. (2016). In this model, expression of GFP-tagged GR(50) and PR(50) in the eye using GMR-Gal4 caused lethality to exceed 95%, and the eyes of surviving flies were severely degenerated (Freibaum et al., 2015; Lee et al., 2016). A significant limitation of this model is that expression of the GR construct is restricted to within the eye, which has less physiological and pathological relevance than pan-neuronal or global expression. Indeed, one may argue that whilst expression of transgenes in the Drosophila eye represents a useful tool for genetic screens it should not be used as a general model for all aspects of disease. For example, survival assays when DPRs are expressed in the eye offer limited insight into the true effect of DPRs upon mechanisms underpinning viability. Expressing the same GR(50) and PR(50) constructs in motor neurons using OK371-Gal4 also caused pupal lethality (Lee et al., 2016). In both systems, not altogether unexpectedly, increasing the temperature and therefore expression increased the severity of the phenotype (Lee et al., 2016). In 2020, a novel fly model expressing PR and GR at a length of 1,000 repeats was developed and the phenotypes differed significantly from previous short arginine-rich DPR models (West et al., 2020a). Although in these flies, pan-neuronal expression of GR(1000) significantly impaired longevity, when compared to wild-type and GFP controls, targeted expression of GR to the eye was not lethal, and only caused severe degenerative phenotypes when expressed homozygously or when temperatures were increased to 29°C, to increase expression and therefore DPR dose. The less acute toxicity in 1,000-repeat models, displaying more progressive, age-related neurodegeneration, perhaps offers a model more representative of disease. Furthermore, having shown that expression levels, at least in the short-DPR models tested, show no significant variance from 1,000-repeat constructs, it is likely that the nature of the repeat itself, possibly through changing protein-protein interactions or de novo synthesis rates, is responsible for the difference in toxicity observed between long- and short-repeat models.
It is well established that age is an important factor is neurodegenerative diseases, and that there are physiological changes that occur throughout healthy aging in both flies and humans, including neurodegeneration. Indeed, the average age of onset in C9orf72 carriers is ∼57 years old (Chio et al., 2012; Murphy et al., 2017). Therefore, using a model system which can be aged is important to understand the interplay between DPR dependent neurodegeneration and normal physiological aging. There are, however, limited studies looking at the effect of DPRs throughout lifetime of a model. In West et al. (2020a) the authors examined the effect of pan-neuronal expression of 1,000 repeat DPRs throughout the flies’ lifetime (West et al., 2020a). In this model, nSyb-Gal4 was used to express DPRs pan-neuronally and flies were aged for up to 42 days. Using this system, phenotypes such as motor impairments in GR(1000) flies became noticeable at around 28 days of age. In general, a fly’s lifespan is approximately 60 days, depending on temperature and other conditions, so a 28-day old fly represents middle age. Comparison of wild type and DPR-expressing flies at different ages allows the temporal effects of long-term DPR expression to be analyzed. Histological analysis and caspase-3 staining of Drosophila brains at 28 days post-eclosion revealed that GR(1000) expression causes cell death and an increase in the number of vacuoles compared to age-matched controls, highlighting its ability to drive neurodegeneration and apoptosis (West et al., 2020a). Additionally, the significant age-related decline in motor function of GR(1000) flies compared to wild type recapitulates motor dysfunction in ALS (West et al., 2020a). In contrast, PR(1000), which so often behaves similarly in shorter repeat models, shows neither significant climbing deficits nor extensive neurodegeneration at these time points.
TDP-43 is a major pathological protein in FTD/ALS, and its mislocalisation and accumulation could be a common mechanism across different genetic forms, not just C9orf72-associated disease. In the two models produced by Solomon et al. (2018) and West et al. (2020a), targeted GR(1000) and GR(64) expression, respectively, within the salivary glands was used to establish the effect of GR on the Drosophila TDP-43 homolog TBPH. Although the large size of Drosophila larval salivary glands provides a robust model to quantify the nuclear-cytoplasmic localization of TBPH/TDP-43, it is important to consider that these cells are not neurons and caution must be taken in extrapolating these findings further. Nevertheless, both models implicated GR in TDP-43 dysfunction and demonstrated that GR expression caused significant cytoplasmic mislocalisation of TBPH, although GR did not directly co-localize with it. This suggestion of a causal link between GR and TDP-43 pathology supports a recent report regarding C9orf72-ALS cases which demonstrated that the presence of GR inclusions correlated with both TDP-43 accumulation and neurodegeneration (Saberi et al., 2018). This data is particularly important in understanding how TDP-43 pathology is consistently observed in C9orf72-FTD/ALS patients, in the absence of RNA foci or DPR inclusions.
A number of approaches have been used to elucidate the mechanisms by which the arginine-rich DPRs induce toxicity in C9orf72-FTD/ALS. Using GR(50) Drosophila, previously described by Freibaum et al. (2015), RNAi eye screens revealed a range of genetic modifiers which encode components of membrane-less organelles (Lee et al., 2016). In particular, G3BP1, G3BP2, and Caprin1, promoters of stress granule assembly, enhanced GR-mediated toxicity while USP10, an inhibitor of stress granule assembly, suppressed GR toxicity (Lee et al., 2016). This link between GR-mediated toxicity and stress granule biology was strengthened by Bakthavachalu et al. (2018). Expressing the same GR(50) construct in Drosophila S2 cells revealed that GR co-localized in ribonucleoprotein granules with the RNA-binding protein ataxin-2 (ATXN2) (Bakthavachalu et al., 2018). Deletion of an intrinsically disordered region within the Drosophila ataxin-2 homolog (ATX2), was found to prevent granule formation, suppress GR toxicity in the eye and decreased the rate of pupal lethality (Bakthavachalu et al., 2018). This is consistent with earlier work which shows that ataxin-2 knockdown can suppress toxicity in yeast, Drosophila and mouse models of C9orf72-FTD/ALS (Elden et al., 2010; Becker et al., 2017). Despite this, the extreme toxicity and lethality induced by expression of these short GR constructs has thus far prevented study into how stress granule dynamics are affected throughout a lifetime of GR expression. A deeper understanding of this process would give us insight as to whether stress granules are neuroprotective or enhance neurotoxicity in the context of C9orf72-FTD/ALS.
Nucleocytoplasmic transport is another cellular process which arginine-rich DPRs are suggested to interfere with. A Drosophila RNAi screen looking for genetic modifiers of PR(25) toxicity identified several nucleocytoplasmic transport genes as enhancers and suppressors of the rough eye phenotype (Boeynaems et al., 2016). Knockdown of importins Ranbp11, Kap-alpha3, Fs(2)Ket, and Trn dramatically enhanced the phenotype, as did Rcc1 and RanGap, regulators of the Ran-GTP cycle (Boeynaems et al., 2016). In contrast, nuclear pore complex components were identified as suppressors of PR25 toxicity (Boeynaems et al., 2016). These findings support the hypothesis that nucleocytoplasmic transport defects may be responsible for the depletion of RNA-binding proteins, such as TDP-43 and FUS, from the nucleus and their accumulation in the cytoplasm. In support of this, a Drosophila model expressing GR(64) showed nuclear depletion of importins and cytoplasmic accumulation of karyopherins (Solomon et al., 2018). However, despite high levels of GR toxicity, no changes to RanGAP were identified (Solomon et al., 2018). The authors propose a feedback loop between DPRs, TDP-43 and karyopherin-α in which DPR accumulation leads to mislocalisation of TDP-43, which in turn causes depletion of karyopherin-α in the nucleus, resulting in further mislocalisation of TDP-43.
Translation inhibition has been suggested to contribute to the toxicity conferred by GR and PR. Proteomic approaches have shown that GR(100) and PR(100) bind to large numbers of ribosomal proteins in the fly brain (Moens et al., 2019). This complements previous findings from interactome studies in human cells lines that suggest arginine-rich DPRs impair translation (Kanekura et al., 2016; Lee et al., 2016; Boeynaems et al., 2017; Hartmann et al., 2018). Furthermore, a genetic modifier eye screen in Drosophila identified translation factors eIF4B and eIF4H1, orthologs of eIF4H, as important for GR(50) expression (Goodman et al., 2019). These translation factors have been implicated in RAN translation and reported to bind G4C2 RNA through RNA recognition motifs (Cooper-Knock et al., 2014; Green et al., 2017). Depletion of these factors using RNAi mitigated toxicity conferred by GR expression, rescuing pigmentation defects, ommatidial disorganization and retinal loss (Goodman et al., 2019). Furthermore, eIF4B and eIF4H1 RNAi knockdown reduced the size and number of GR puncta per eye but did not alter G4C2(44) RNA transcript levels, suggesting they impact GR at a translational level (Goodman et al., 2019). Additionally, overexpression of eIF1A in PR(100)- and GR(100)-expressing flies partially rescued their shortened lifespan. Taken together, there is robust evidence from Drosophila that short-repeat GR and PR impair translation and that this could be a mechanism driving neurodegeneration in C9orf72-FTD/ALS.
Excitotoxicity is a common mechanism in neurodegenerative diseases, heavily implicated in Parkinson’s disease (Beal et al., 1993; Himmelberg et al., 2018) and Alzheimer’s disease (Mota et al., 2014; Pallo et al., 2016). Excitotoxicity, whereby neurons are damaged and killed by overactivation of glutamate receptors such as NMDA and AMPA, is nevertheless relatively understudied in C9orf72-FTD/ALS. One study indicated, through a combination of RNA-Sequencing and electrophysiological studies on induced pluripotent stem cell (iPSC)-derived motor neurons, that C9orf72 mutations cause increased expression of the GluA1 AMPA receptor subunit, leading to increased permeability of motor neurons to calcium ions, and thus vulnerability to excitotoxicity (Selvaraj et al., 2018). The potential for excitotoxicity to be a key mechanism in DPR-mediated toxicity was tested in flies expressing 36-repeat GR and PR in glutamatergic neurons. It was discovered that PR(36) and GR(36) induced an increase in extracellular glutamate and intracellular calcium, and an increase in synaptic boutons and active zones in the larval neuromuscular junction (Xu and Xu, 2018). Inhibition of glutamate transport or NMDA receptors could rescue motor deficits and shortened lifespan caused by motor-neuronal expression of GR(36) and PR(36) (Xu and Xu, 2018). Furthermore, selective inhibition of GR(36) and PR(36) in glutamatergic neurons rescued the phenotypes observed when the DPRs were expressed pan-neuronally (Xu and Xu, 2018). The authors suggest that this points to a selective susceptibility of glutamatergic neurons to PR and GR toxicity mediated through excitotoxicity. In contrast, however, larvae pan-neuronally expressing 1,000-repeat GR and PR, showed no significant changes in bouton number, although PR(1000) flies did show an increase in the number of bruchpilot positive active zones at the larval neuromuscular junction (West et al., 2020a). However, a capacity for excitotoxicity to potentiate neurodegeneration in a cell-autonomous capacity is an area of interest that remains relatively unexplored.
Recently, a novel mechanism and potential therapeutic target has been proposed based on RNA sequencing data from GR(100) brains and subsequent experiments showing that enhancement of the insulin pathway in neurons partially rescued the toxicity of GR (Atilano et al., 2021). Increased insulin signaling caused by either insulin treatment or over-expression of the fly insulin receptor reduced the level of GR, suggesting that insulin treatment may be a potential therapeutic strategy (Atilano et al., 2021). Another novel mechanism for GR toxicity was posited by Yang et al. (2015), using a novel GR(80) model. Consistent with other short-repeat GR models, it displayed extremely severe phenotypes. GMR-Gal4 expression induced drastic eye deformation and lethality in pupae (Yang et al., 2015). Expression in a range of neuronal and non-neuronal cell types in vivo resulted in a predominantly lethal phenotype (Yang et al., 2015). When driven by Vg-Gal4 in the wing imaginal disks, 90% of adult flies exhibited wing margin defects, which resembled those shown by flies with a partial loss of Notch activity (Yang et al., 2015). Indeed, ectopic expression of Notch partially alleviated GR(80) toxicity (Yang et al., 2015). However, the wing is not a pathologically relevant tissue in which to study FTD/ALS and the authors urge cautious interpretation. The Notch signaling pathway has broad activity in humans and is subject to regulation at multiple steps. It has yet to be further investigated and without further validation it is impossible to confidently evaluate the involvement of Notch in C9orf72-FTD/ALS. Subsequent studies using these GR(80) models have shown that when expressed in Drosophila muscles GR enters the mitochondria and interacts with components of the Mitochondrial Contact Site and Cristae Organizing System (MICOS) machinery, altering mitochondrial dynamics (Li et al., 2020a,b). This results in perturbations to the mitochondrial inner membrane, impairments to ion homeostasis and metabolism, and ultimately reduced muscle integrity. This was rescued by feeding flies nigericin, restoring ion homeostasis (Li et al., 2020a).
DNA damage is also a proposed mechanism underpinning toxicity of the arginine-rich DPRs (Farg et al., 2017; Lopez-Gonzalez et al., 2019; Andrade et al., 2020). In vitro studies have implicated different forms of DNA repair as disrupted by GA, GR, and PR (Farg et al., 2017; Andrade et al., 2020). It is theorized that nucleolar dysfunction can lead to DNA damage, and activation of the DNA damage response as a consequence. If DNA repair is prevented, apoptosis is triggered. A Drosophila GR(80) model (Yang et al., 2015) was used to complement work in iPSC-derived C9orf72 motor neurons that implicated DNA damage-induced p53 activation in GR toxicity (Lopez-Gonzalez et al., 2016). Further work from the same group identified an essential DNA repair protein as a genetic modifier of GR(80) toxicity (Lopez-Gonzalez et al., 2019). GR(80) expression in Drosophila neuronal cells induced a greatly increased levels of Ku80, a critical component of DNA repair pathways, compared to controls (Lopez-Gonzalez et al., 2019). Partial suppression of Ku80 suppressed retinal degradation in flies expressing GR(80) under the control of the eye specific driver GMR-Gal4 (Lopez-Gonzalez et al., 2019). However, induction of the temperature-sensitive Gal80, a negative regulator of Gal4, was required to reduce GR expression to allow screening, because it was semi-lethal and produced a severe phenotype when expressed in the absence of Gal80.
Co-expression of Dipeptide Repeat Proteins
In patients it has been observed that multiple DPRs can be present within the same cell (Ash et al., 2013; Mori et al., 2013a; Zu et al., 2013). As such, exacerbation of individual DPR toxicity and interaction between DPRs may contribute to C9orf72-related FTD/ALS disease progression and provide one explanation for the heterogeneity of symptoms seen in C9orf72 repeat expansion carriers. It is important to consider this when making inferences from DPR models, because the relative abundance of different DPRs in cells may influence the cytotoxic output and change the pathological mechanisms involved.
Whilst in vitro studies have provided early indications of DPR-DPR interactions, the effects of DPR co-expression remain relatively under-studied. This is likely due to a historic lack of appropriate models for research, owing to the often-lethal toxicity of expressing high levels of DPR in most existing models. Much of the existing research has centered around GA’s interactions with the other DPRs, especially the arginine-containing DPRs, and GA’s propensity to aggregate (Zhang et al., 2014; Chang et al., 2016; Nonaka et al., 2018). GA(50) has been found to sequester PR(50) from the nucleus into cytoplasmic inclusions, when co-expressed in NSC34 cells and mouse primary neurons (Darling et al., 2019). In NSC34 cells PR-induced cytotoxicity was ablated by co-expression with GA (Darling et al., 2019). This was proposed to be due to the morphological changes that occur when the two DPRs interact, preventing the associated toxic interactions of either individual DPR (Darling et al., 2019). Indeed, in an in vitro cell-free environment, concomitant expression of 20 repeats of GA and PR results in GA losing its β-sheet structure in favor of PR’s disordered structure, leading to GA/PR co-aggregation (Darling et al., 2019).
To date there remains a lack of in vivo studies exploring co-expression of DPRs. Despite its genetic tractability, only two studies have been published using Drosophila to explore DPR co-expression. Both studies use the UAS-Gal4 system to co-express DPRs in a tissue-specific manner. The first co-expressed GR(80) and GA(80) in Drosophila eyes, wing disks and salivary glands (Yang et al., 2015). Co-expression of 80 repeats of both GA and GR in Drosophila salivary glands reveals GA recruits GR to cytoplasmic inclusions, recapitulating observations made in both HeLa cells and iPSC-derived human neurons (Yang et al., 2015). Recruitment of GR by GA also suppressed GR-induced toxicity in these models. Co-expression of 1,000 repeat DPRs in the eye produced different results. When expressed individually, GR1000, AP1000, and PR1000 showed a dose-dependent increase in toxicity, only producing an eye phenotype when expressed homozygously (West et al., 2020a). GA(1000) showed no toxicity when expressed in the eye. Co-expression of any pairs of DPRs did not produce as severe a phenotype as doubling the dose of GR1000, AP(1000), or PR(1000) (West et al., 2020a). Co-expression of GR(1000) with each of the other DPRs proved the most toxic of the combinations, with the most severe phenotypes observed with GR(1000)/PR(1000) (West et al., 2020a). Co-expression of GA(1000) with GR(1000) produced a mostly wild type eye, but in a small proportion of flies mild perturbations were observed (West et al., 2020a). However, it is important to note that salivary glands, eyes, and wing disks, whilst useful systems, are not necessarily the most physiologically relevant models for an age-related neurodegenerative disease.
Co-expression of 1,000 repeat DPRs in the nervous system using a pan-neuronal driver (nSyb-Gal4) revealed age- and combination- specific motor phenotypes (West et al., 2020a). Combining alanine- and arginine- rich DPRs produced a significant decline in climbing speed between young (7 days old) and old (28 days old); for example, at 7 days post-eclosion, AP(1000)/PR(1000) expressing flies showed no significant climbing deficits, but by 28 days post-eclosion, they had a significantly slow speed compared to age-matched controls (West et al., 2020a). In fact, the only combination not to show an age-dependent decline in climbing speed was the alanine-positive DPR combination AP(1000)/GA(1000), suggesting that arginine-rich DPRs may be responsible for the age-related toxicity in this model (West et al., 2020a). This is supported by data from expression of each DPR individually, where both AP(1000) and GA(1000) had a consistent speed across lifespan, whereas GR(1000) and PR(1000) showed a significant decline (West et al., 2020a). The most severe combination was AP(1000)/GR(1000), which proved lethal before reaching 28 days-post eclosion (West et al., 2020a). Indeed, co-expression with GR(1000) exacerbated existing toxicity in all combinations. Given the interactions between DPRs in vivo is as yet relatively unexplored, one can only speculate as to the reasons behind these combination-specific phenotypes. However, it does highlight the importance of studying DPRs in a system that can be aged.
In addition to potentiating phenotypes previously observed in single DPR models, West et al. (2020a) also observed that concomitant expression of DPRs led to a novel, previously unreported, phenotype – seizures (West et al., 2020a). Seizure phenotypes are in keeping with epileptiform-like seizures observed in some C9orf72 patients (Capasso et al., 2017) and may highlight important mechanisms underpinning neuronal hyperexcitability, implicated in ALS. The observation that combinations of alanine and arginine DPRs, as well as concomitant expression of PR and GR, seem to increase bang-sensitive seizure susceptibility as well as motor dysfunction casts some doubt on that the previously stated hypothesis that GA sequestration of arginine-rich DPRs may prevent cellular dysfunction. It also highlights clear discrepancies between models, with both length-dependent and cell-type specific effects observed. It therefore remains unclear whether different DPRs are acting synergistically through the same toxic mechanisms, or via different mechanisms which may exacerbate or ameliorate phenotypes. Existing studies are also somewhat limited in that, so far, only two DPRs have been co-expressed at any given time, whereas all five could be present in patient cells. In C9orf72 patient frontal cortex, GA, GP, and AP have all been identified in large cytoplasmic inclusions (Lee et al., 2017). These inclusions often have a GA ‘core’ surrounded by either GP or AP, suggesting that GP and AP may be recruited to these inclusions by GA. The idea that GA is required for aggregate formation is supported by a study in HEK-293 cells, where 125-repeats of AP and GP would only aggregate when co-transfected with GA (Lee et al., 2017). The effects of co-expression on aggregation and morphological properties of each DPR remains to be investigated in vivo. It also remains to be answered what, if any, are the implications of these interactions on C9orf72-mediated disease.
In summary, it is clear from existing models that interactions between DPRs are possible and, knowing that multiple DPRs are expressed in patient cells, it is entirely likely that these interactions do occur in patients. To fully develop our understanding of DPR-DPR interactions, we need to investigate DPR co-expression under consistent conditions in in vivo and in vitro models. Drosophila models offer a powerful system with which we can further dissect the contribution of DPR-DPR interactions to molecular mechanisms underpinning disease progression.
Conclusion
Since the identification that C9orf72 hexanucleotide expansion mutations are the most common genetic cause of both FTD and ALS, Drosophila have proven to be an invaluable model to elucidate the mechanism contributing to neurodegeneration downstream of the expansion. Despite this, our understanding of how gain of function mechanisms, RNA and DPRs, underpin disease and how interplay between these mechanisms contributes to neurodegenerative cascades remains in its infancy. However, the unrivaled genetic tractability, ability to study age-dependent effects in an in vivo, whole organism context and establishment of longer repeat length models makes Drosophila a powerful model to further explore mechanisms underpinning FTD/ALS spectrum disorders.
Author Contributions
JS and RW wrote the manuscript with contributions from NH and DG. JS produced and formatted all figures and tables with input from RW, NH, and DG. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by an Alzheimer’s Society Fellowship awarded to RW [AS-JF-16b-004 (510)], and a Medical Research Council funded Ph.D. studentship undertaken by JS.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank Kate Whittaker for her comments on the manuscript.
References
Abramzon, Y. A., Fratta, P., Traynor, B. J., and Chia, R. (2020). The overlapping genetics of amyotrophic lateral sclerosis and frontotemporal dementia. Front. Neurosci. 14:42. doi: 10.3389/fnins.2020.00042
Al-Chalabi, A., Jones, A., Troakes, C., King, A., Al-Sarraj, S., and van den Berg, L. H. (2012). The genetics and neuropathology of amyotrophic lateral sclerosis. Acta Neuropathol. 124, 339–352. doi: 10.1007/s00401-012-1022-4
Al-Sarraj, S., King, A., Troakes, C., Smith, B., Maekawa, S., Bodi, I., et al. (2011). p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathol. 122, 691–702. doi: 10.1007/s00401-011-0911-2
Andrade, N. S., Ramic, M., Esanov, R., Liu, W. J., Rybin, M. J., Gaidosh, G., et al. (2020). Dipeptide repeat proteins inhibit homology-directed DNA double strand break repair in C9ORF72 ALS/FTD. Mol. Neurodegen. 15:18. doi: 10.1186/s13024-020-00365-9
Ash, P. E. A., Bieniek, K. F., Gendron, T. F., Caulfield, T., Lin, W. L., DeJesus-Hernandez, M., et al. (2013). Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77, 639–646. doi: 10.1016/j.neuron.2013.02.004
Atilano, M. L., Gronke, S., Niccoli, T., Kempthorne, L., Hahn, O., Moron-Oset, J., et al. (2021). Enhanced insulin signalling ameliorates C9orf72 hexanucleotide repeat expansion toxicity in Drosophila. Elife 10:21. doi: 10.7554/eLife.58565
Bakthavachalu, B., Huelsmeier, J., Sudhakaran, I. P., Hillebrand, J., Singh, A., Petrauskas, A., et al. (2018). RNP-granule assembly via ataxin-2 disordered domains is required for long-term memory and neurodegeneration. Neuron 98, 754–766. doi: 10.1016/j.neuron.2018.04.032
Batra, R., and Lee, C. W. (2017). Mouse models of C9orf72 hexanucleotide repeat expansion in amyotrophic lateral sclerosis/frontotemporal dementia. Front. Cell. Neurosci. 11:196. doi: 10.3389/fncel.2017.00196
Beal, M. F., Brouillet, E., Jenkins, B. G., Ferrante, R. J., Kowall, N. W., Miller, J. M., et al. (1993). Neurochemical and histologic characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J. Neurosci. 13, 4181–4192.
Beck, J., Poulter, M., Hensman, D., Rohrer, J. D., Mahoney, C. J., Adamson, G., et al. (2013). Large C9orf72 hexanucleotide repeat expansions are seen in multiple neurodegenerative syndromes and are more frequent than expected in the UK population. Am. J. Hum. Genet. 92, 345–353. doi: 10.1016/j.ajhg.2013.01.011
Becker, L. A., Huang, B., Bieri, G., Ma, R., Knowles, D. A., Jafar-Nejad, P., et al. (2017). Therapeutic reduction of ataxin-2 extends lifespan and reduces pathology in TDP-43 mice. Nature 544, 367–371. doi: 10.1038/nature22038
Belzil, V. V., Bauer, P. O., Prudencio, M., Gendron, T. F., Stetler, C. T., Yan, I. K., et al. (2013). Reduced C9orf72 gene expression in c9FTD/ALS is caused by histone trimethylation, an epigenetic event detectable in blood. Acta Neuropathol. 126, 895–905. doi: 10.1007/s00401-013-1199-1
Boeynaems, S., Bogaert, E., Kovacs, D., Konijnenberg, A., Timmerman, E., Volkov, A., et al. (2017). Phase separation of C9orf72 dipeptide repeats perturbs stress granule dynamics. Mol. Cell. 65, 1044–1055. doi: 10.1016/j.molcel.2017.02.013
Boeynaems, S., Bogaertl, E., Michiels, E., Gijselinck, I., Sieben, A., Jovicic, A., et al. (2016). Drosophila screen connects nuclear transport genes to DPR pathology in c9ALS/FTD. Sci. Rep. 6:8. doi: 10.1038/srep20877
Brooks, B. R., Miller, R. G., Swash, M., Munsat, T. L., and World Federation of Neurology Research Group on Motor Neuron Diseases. (2000). El Escorial revisited: Revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 1, 293–299. doi: 10.1080/146608200300079536
Burberry, A., Suzuki, N., Wang, J. Y., Moccia, R., Mordes, D. A., Stewart, M. H., et al. (2016). Loss-of-function mutations in the C9ORF72 mouse ortholog cause fatal autoimmune disease. Sci. Transl. Med. 8:12. doi: 10.1126/scitranslmed.aaf6038
Burguete, A. S., Almeida, S., Gao, F. B., Kalb, R., Akins, M. R., and Bonini, N. M. (2015). GGGGCC microsatellite RNA is neuritically localized, induces branching defects, and perturbs transport granule function. Elife 4:23. doi: 10.7554/eLife.0881
Callister, J. B., Ryan, S., Sim, J., Rollinson, S., and Pickering-Brown, S. M. (2016). Modelling C9orf72 dipeptide repeat proteins of a physiologically relevant size. Hum. Mol. Genet. 25, 5069–5082. doi: 10.1093/hmg/ddw327
Cao, W., Song, H. J., Gangi, T., Kelkar, A., Antani, I., Garza, D., et al. (2008). Identification of novel genes that modify phenotypes induced by Alzheimer’s beta-amyloid overexpression in Drosophila. Genetics 178, 1457–1471. doi: 10.1534/genetics.107.078394
Capasso, M., Anzellotti, F., Di Giacomo, R., and Onofrj, M. (2017). Epilepsy and electroencephalographic abnormalities in C9orf72 repeat expansion. Amyotroph. Lateral Scleros. Frontotemporal Degen. 18, 140–141. doi: 10.1080/21678421.2016.1231825
Chakraborty, R., Vepuri, V., Mhatre, S. D., Paddock, B. E., Miller, S., Michelson, S. J., et al. (2011). Characterization of a drosophila Alzheimer’s disease model: pharmacological rescue of cognitive defects. Plos One 6:e20799. doi: 10.1371/journal.pone.0020799
Chang, Y. J., Jeng, U. S., Chiang, Y. L., Hwang, I. S., and Chen, Y. R. (2016). The glycine-alanine dipeptide repeat from C9orf72 hexanucleotide expansions forms toxic amyloids possessing cell-to-cell transmission properties. J. Biol. Chem. 291, 4903–4911. doi: 10.1074/jbc.M115.694273
Chew, J., Gendron, T. F., Prudencio, M., Sasaguri, H., Zhang, Y. J., Castanedes-Casey, M., et al. (2015). C9ORF72 repeat expansions in mice cause TDP-43 pathology, neuronal loss, and behavioral deficits. Science 348, 1151–1154. doi: 10.1126/science.aaa9344
Chio, A., Borghero, G., Restagno, G., Mora, G., Drepper, C., Traynor, B. J., et al. (2012). Clinical characteristics of patients with familial amyotrophic lateral sclerosis carrying the pathogenic GGGGCC hexanucleotide repeat expansion of C9ORF72. Brain 135, 784–793. doi: 10.1093/brain/awr366
Chio, A., Logroscino, G., Hardiman, O., Swingler, R., Mitchell, D., Beghi, E., et al. (2009). Prognostic factors in ALS: A critical review. Amyotroph. Lateral Scleros. 10, 310–323. doi: 10.3109/17482960802566824
Chio, A., Logroscino, G., Traynor, B. J., Collins, J., Simeone, J. C., Goldstein, L. A., et al. (2013). Global epidemiology of amyotrophic lateral sclerosis: A systematic review of the published literature. Neuroepidemiology 41, 118–130. doi: 10.1159/000351153
Choi, M. L., and Gandhi, S. (2018). Crucial role of protein oligomerization in the pathogenesis of Alzheimer’s and Parkinson’s diseases. FEBS J. 285, 3631–3644. doi: 10.1111/febs.14587
Cooper-Knock, J., Hewitt, C., Highley, J. R., Brockington, A., Milano, A., Man, S., et al. (2012). Clinico-pathological features in amyotrophic lateral sclerosis with expansions in C9ORF72. Brain 135, 751–764. doi: 10.1093/brain/awr365
Cooper-Knock, J., Higginbottom, A., Stopford, M. J., Highley, J. R., Ince, P. G., Wharton, S. B., et al. (2015). Antisense RNA foci in the motor neurons of C9ORF72-ALS patients are associated with TDP-43 proteinopathy. Acta Neuropathol. 130, 63–75. doi: 10.1007/s00401-015-1429-9
Cooper-Knock, J., Walsh, M. J., Higginbottom, A., Highley, J. R., Dickman, M. J., Edbauer, D., et al. (2014). Sequestration of multiple RNA recognition motif-containing proteins by C9orf72 repeat expansions. Brain 137, 2040–2051. doi: 10.1093/brain/awu120
Cykowski, M. D., Dickson, D. W., Powell, S. Z., Arumanayagam, A. S., Rivera, A. L., and Appel, S. H. (2019). Dipeptide repeat (DPR) pathology in the skeletal muscle of ALS patients with C9ORF72 repeat expansion. Acta Neuropathol. 138, 667–670. doi: 10.1007/s00401-019-02050-8
Darling, A. L., Breydo, L., Rivas, E. G., Gebru, N. T., Zheng, D. L., Baker, J. D., et al. (2019). Repeated repeat problems: Combinatorial effect of C9orf72-derived dipeptide repeat proteins. Int. J. Biol. Macromol. 127, 136–145. doi: 10.1016/j.ijbiomac.2019.01.035
Davidson, Y. S., Barker, H., Robinson, A. C., Thompson, J. C., Harris, J., Troakes, C., et al. (2014). Brain distribution of dipeptide repeat proteins in frontotemporal lobar degeneration and motor neurone disease associated with expansions in C9ORF72. Acta Neuropathol. Commun. 2:13. doi: 10.1186/2051-5960-2-70
Deal, S. L., and Yamamoto, S. (2019). Unraveling novel mechanisms of neurodegeneration through a large-scale forward genetic screen in drosophila. Front. Genet. 9:700. doi: 10.3389/fgene.2018.00700
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., et al. (2011). Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron 72, 245–256. doi: 10.1016/j.neuron.2011.09.011
Doig, A. J. (2017). Frozen, but no accident - why the 20 standard amino acids were selected. FEBS J. 284, 1296–1305. doi: 10.1111/febs.13982
Edbauer, D., and Haass, C. (2016). An amyloid-like cascade hypothesis for C9orf72 ALS/FTD. Curr. Opin. Neurobiol. 36, 99–106. doi: 10.1016/j.conb.2015.10.009
Elden, A. C., Kim, H. J., Hart, M. P., Chen-Plotkin, A. S., Johnson, B. S., Fang, X. D., et al. (2010). Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 466, 1069–1077. doi: 10.1038/nature09320
Estevez-Fraga, C., Magrinelli, F., Moss, D. H., Mulroy, E., Di Lazzaro, G., Latorre, A., et al. (2021). Expanding the spectrum of movement disorders associated with C9orf72 hexanucleotide expansions. Neurol. Genet. 7:e575. doi: 10.1212/nxg.0000000000000575
Farg, M. A., Konopka, A., Soo, K. Y., Ito, D., and Atkin, J. D. (2017). The DNA damage response (DDR) is induced by the C9orf72 repeat expansion in amyotrophic lateral sclerosis. Hum. Mol. Genet. 26, 2882–2896. doi: 10.1093/hmg/ddx170
Foster, L. A., and Salajegheh, M. K. (2019). Motor neuron disease: pathophysiology, diagnosis, and management. Am. J. Med. 132, 32–37. doi: 10.1016/j.amjmed.2018.07.012
Fratta, P., Mizielinska, S., Nicoll, A. J., Zloh, M., Fisher, E. M. C., Parkinson, G., et al. (2012). C9orf72 hexanucleotide repeat associated with amyotrophic lateral sclerosis and frontotemporal dementia forms RNA G-quadruplexes. Sci. Rep. 2:6. doi: 10.1038/srep01016
Freibaum, B. D., Lu, Y. B., Lopez-Gonzalez, R., Kim, N. C., Almeida, S., Lee, K. H., et al. (2015). GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 525, 129–133. doi: 10.1038/nature14974
Freibaum, B. D., and Taylor, J. P. (2017). The role of dipeptide repeats in C9ORF72-Related ALS-FTD. Front. Mol. Neurosci. 10:35. doi: 10.3389/fnmol.2017.00035
Garcia-Redondo, A., Dols-Icardo, O., Rojas-Garcia, R., Esteban-Perez, J., Cordero-Vazquez, P., Munoz-Blanco, J. L., et al. (2013). Analysis of the C9orf72 gene in patients with amyotrophic lateral sclerosis in spain and different populations worldwide. Hum. Mutation 34, 79–82. doi: 10.1002/humu.22211
Gijselinck, I., Van Langenhove, T., van der Zee, J., Sleegers, K., Philtjens, S., Kleinberger, G., et al. (2012). A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol. 11, 54–65. doi: 10.1016/s1474-4422(11)70261-7
Gijselinck, I., Van Mossevelde, S., van der Zee, J., Sieben, A., Engelborghs, S., De Bleecker, J., et al. (2016). The C9orf72 repeat size correlates with onset age of disease, DNA methylation and transcriptional downregulation of the promoter. Mol. Psychiatry 21, 1112–1124. doi: 10.1038/mp.2015.159
Gomez-Tortosa, E., Gallego, J., Guerrero-Lopez, R., Marcos, A., Gil-Neciga, E., Sainz, M. J., et al. (2013). C9ORF72 hexanucleotide expansions of 20-22 repeats are associated with frontotemporal deterioration. Neurology 80, 366–370. doi: 10.1212/WNL.0b013e31827f08ea
Goodman, L. D., Prudencio, M., Kramer, N. J., Martinez-Ramirez, L. F., Srinivasan, A. R., Lan, M., et al. (2019). Toxic expanded GGGGCC repeat transcription is mediated by the PAF1 complex in C9orf72-associated FTD. Nat. Neurosci. 22, 863–874. doi: 10.1038/s41593-019-0396-1
Green, K. M., Glineburg, M. R., Kearse, M. G., Flores, B. N., Linsalata, A. E., Fedak, S. J., et al. (2017). RAN translation at C9orf72-associated repeat expansions is selectively enhanced by the integrated stress response. Nat. Commun. 8:13. doi: 10.1038/s41467-017-02200-0
Guo, Q., Lehmer, C., Martinez-Sanchez, A., Rudack, T., Beck, F., Hartmann, H., et al. (2018). In situ structure of neuronal C9orf72 Poly-GA aggregates reveals proteasome recruitment. Cell 172, 696–705. doi: 10.1016/j.cell.2017.12.030
Hardiman, O., Al-Chalabi, A., Chio, A., Corr, E. M., Logroscino, G., Robberecht, W., et al. (2017). Amyotrophic lateral sclerosis. Nat. Rev. Dis. Primers 3:18. doi: 10.1038/nrdp.2017.71
Hartmann, H., Hornburg, D., Czuppa, M., Bader, J., Michaelsen, M., Farny, D., et al. (2018). Proteomics and C9orf72 neuropathology identify ribosomes as poly-GR/PR interactors driving toxicity. Life Sci. Alliance 1:13. doi: 10.26508/lsa.201800070
Harvey, R. J., Skelton-Robinson, M., and Rossor, M. N. (2003). The prevalence and causes of dementia in people under the age of 65 years. J. Neurol. Neurosur. Psychiatry 74, 1206–1209. doi: 10.1136/jnnp.74.9.1206
Hayes, L. R., Duan, L., Bowen, K., Kalab, P., and Rothstein, J. D. (2020). C9orf72 arginine-rich dipeptide repeat proteins disrupt karyopherin-mediated nuclear import. Elife 9:29. doi: 10.7554/eLife.51685
Herranz-Martin, S., Chandran, J., Lewis, K., Mulcahy, P., Higginbottom, A., Walker, C., et al. (2017). Viral delivery of C9orf72 hexanucleotide repeat expansions in mice leads to repeat-length-dependent neuropathology and behavioural deficits. Dis. Models Mech. 10, 859–868. doi: 10.1242/dmm.029892
Himmelberg, M. M., West, R. J. H., Elliott, C. J. H., and Wade, A. R. (2018). Abnormal visual gain control and excitotoxicity in early-onset Parkinson’s disease Drosophila models. J. Neurophysiol. 119, 957–970. doi: 10.1152/jn.00681.2017
Holm, I. E., Isaacs, A. M., and Mackenzie, I. R. A. (2009). Absence of FUS-immunoreactive pathology in frontotemporal dementia linked to chromosome 3 (FTD-3) caused by mutation in the CHMP2B gene. Acta Neuropathol. 118, 719–720. doi: 10.1007/s00401-009-0593-1
Hsiung, G. Y. R., DeJesus-Hernandez, M., Feldman, H. H., Sengdy, P., Bouchard-Kerr, P., Dwosh, E., et al. (2012). Clinical and pathological features of familial frontotemporal dementia caused by C9ORF72 mutation on chromosome 9p. Brain 135, 709–722. doi: 10.1093/brain/awr354
Jiang, J., Zhu, Q., Gendron, T. F., Saberi, S., McAlonis-Downes, M., Seelman, A., et al. (2016). Gain of toxicity from ALS/FTD-linked repeat expansions in C9ORF72 is alleviated by antisense oligonucleotides targeting GGGGCC-containing RNAs. Neuron 90, 535–550. doi: 10.1016/j.neuron.2016.04.006
Jovicic, A., Mertens, J., Boeynaems, S., Bogaert, E., Chai, N., Yamada, S. B., et al. (2015). Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat. Neurosci. 18, 1226–1229. doi: 10.1038/nn.4085
Kanekura, K., Yagi, T., Cammack, A. J., Mahadevan, J., Kuroda, M., Harms, M. B., et al. (2016). Poly-dipeptides encoded by the C9ORF72 repeats block global protein translation. Hum. Mol. Genet. 25, 1803–1813. doi: 10.1093/hmg/ddw052
Kharel, P., Balaratnam, S., Beals, N., and Basu, S. (2020). The role of RNA G-quadruplexes in human diseases and therapeutic strategies. Wiley Interdis. Reviews-RNA 11:20. doi: 10.1002/wrna.1568
Koppers, M., Blokhuis, A. M., Westeneng, H. J., Terpstra, M. L., Zundel, C. A. C., de Sa, R. V., et al. (2015). C9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits. Ann. Neurol. 78, 426–438. doi: 10.1002/ana.24453
Kovanda, A., Zalar, M., Sket, P., Plavec, J., and Rogelj, B. (2015). Anti-sense DNA d(GGCCCC)(n) expansions in C9ORF72 form i-motifs and protonated hairpins. Sci. Rep. 5:7. doi: 10.1038/srep17944
Kramer, N. J., Carlomagno, Y., Zhang, Y. J., Almeida, S., Cook, C. N., Gendron, T. F., et al. (2016). Spt4 selectively regulates the expression of C9orf72 sense and antisense mutant transcripts. Science 353, 708–712. doi: 10.1126/science.aaf7791
Kwon, I., Xiang, S. H., Kato, M., Wu, L., Theodoropoulos, P., Wang, T., et al. (2014). Poly-dipeptides encoded by the C9orf72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science 345, 1139–1145. doi: 10.1126/science.1254917
Lee, K. H., Zhang, P. P., Kim, H. J., Mitrea, D. M., Sarkar, M., Freibaum, B. D., et al. (2016). C9orf72 dipeptide repeats impair the assembly, dynamics, and function of membrane-less organelles. Cell 167, 774–788. doi: 10.1016/j.cell.2016.10.002
Lee, Y. B., Baskaran, P., Gomez-Deza, J., Chen, H. J., Nishimura, A. L., Smith, B. N., et al. (2017). C9orf72 poly GA RAN-translated protein plays a key role in amyotrophic lateral sclerosis via aggregation and toxicity. Hum. Mol. Genet. 26, 4765–4777. doi: 10.1093/hmg/ddx350
Lee, Y. B., Chen, H. J., Peres, J. N., Gomez-Deza, J., Attig, J., Stalekar, M., et al. (2013). Hexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell. Rep. 5, 1178–1186. doi: 10.1016/j.celrep.2013.10.049
Lesage, S., Le Ber, I., Condroyer, C., Broussolle, E., Gabelle, A., Thobois, S., et al. (2013). C9orf72 repeat expansions are a rare genetic cause of parkinsonism. Brain 136, 385–391. doi: 10.1093/brain/aws357
Levine, T. P., Daniels, R. D., Gatta, A. T., Wong, L. H., and Hayes, M. J. (2013). The product of C9orf72, a gene strongly implicated in neurodegeneration, is structurally related to DENN Rab-GEFs. Bioinformatics 29, 499–503. doi: 10.1093/bioinformatics/bts725
Li, S. X., Wu, Z. H., Li, Y., Tantray, I., De Stefani, D., Mattarei, A., et al. (2020a). Altered MICOS morphology and mitochondrial ion homeostasis contribute to Poly(GR) toxicity associated with C9-ALS/FTD. Cell. Rep. 32:18. doi: 10.1016/j.celrep.2020.107989
Li, S. X., Wu, Z. H., Tantray, I., Li, Y., Chen, S. J., Dong, J., et al. (2020b). Quality-control mechanisms targeting translationally stalled and C-terminally extended poly(GR) associated with ALS/FTD. Proc. Natl. Acad. Sci. U S A. 117, 25104–25115. doi: 10.1073/pnas.2005506117
Lin, Y., Mori, E., Kato, M., Xiang, S. H., Wu, L. J., Kwon, I., et al. (2016). Toxic PR Poly-dipeptides encoded by the C9orf72 repeat expansion target LC domain polymers. Cell 167, 789–802. doi: 10.1016/j.cell.2016.10.003
Liu, Y. J., Pattamatta, A., Zu, T., Reid, T., Bardhi, O., Borchelt, D. R., et al. (2016). C9orf72 BAC mouse model with motor deficits and neurodegenerative features of ALS/FTD. Neuron 90, 521–534. doi: 10.1016/j.neuron.2016.04.005
Lopez-Gonzalez, R., Lu, Y. B., Gendron, T. F., Karydas, A., Tran, H., Yang, D. J., et al. (2016). Poly(GR) in C9ORF72-related ALS/FTD compromises mitochondrial function and increases oxidative stress and DNA damage in iPSC-derived motor neurons. Neuron 92, 383–391. doi: 10.1016/j.neuron.2016.09.015
Lopez-Gonzalez, R., Yang, D., Pribadi, M., Kim, T. S., Krishnan, G., Choi, S. Y., et al. (2019). Partial inhibition of the overactivated Ku80-dependent DNA repair pathway rescues neurodegeneration in C9ORF72-ALS/FTD. Proc. Natl. Acad. Sci. U S A. 116, 9628–9633. doi: 10.1073/pnas.1901313116
Mackenzie, I. R. A., Frick, P., Graesser, F. A., Gendron, T. F., Petrucelli, L., Cashman, N. R., et al. (2015). Quantitative analysis and clinico-pathological correlations of different dipeptide repeat protein pathologies in C9ORF72 mutation carriers. Acta Neuropathol. 130, 845–861. doi: 10.1007/s00401-015-1476-2
Mackenzie, I. R. A., Neumann, M., Bigio, E. H., Cairns, N. J., Alafuzoff, I., Kril, J., et al. (2010). Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol. 119, 1–4. doi: 10.1007/s00401-009-0612-2
Mackenzie, I.R., Arzberger, T., Kremmer, E., Troost, D., Lorenzl, S., Mori, K., et al. (2013). Dipeptide repeat protein pathology in C9ORF72 mutation cases: clinico-pathological correlations. Acta Neuropathol. 126, 859–879. doi: 10.1007/s00401-013-1181-y
Mahoney, C. J., Downey, L. E., Ridgway, G. R., Beck, J., Clegg, S., Blair, M., et al. (2012). Longitudinal neuroimaging and neuropsychological profiles of frontotemporal dementia with C9ORF72 expansions. Alzheimers Res. Ther. 4:10. doi: 10.1186/alzrt144
Majounie, E., Abramzon, Y., Renton, A. E., Perry, R., Bassett, S. S., Pletnikova, O., et al. (2012). Repeat expansion in C9ORF72 in Alzheimer’s Disease. New Eng. J. Med. 366, 283–284. doi: 10.1056/NEJMc1113592
Mann, D. M. A., Rollinson, S., Robinson, A., Callister, J. B., Thompson, J. C., Snowden, J. S., et al. (2013). Dipeptide repeat proteins are present in the p62 positive inclusions in patients with frontotemporal lobar degeneration and motor neurone disease associated with expansions in C9ORF72. Acta Neuropathol. Commun. 1:13. doi: 10.1186/2051-5960-1-68
May, S., Hornburg, D., Schludi, M. H., Arzberger, T., Rentzsch, K., Schwenk, B. M., et al. (2014). C9orf72 FTLD/ALS-associated Gly-Ala dipeptide repeat proteins cause neuronal toxicity and Unc119 sequestration. Acta Neuropathol. 128, 485–503. doi: 10.1007/s00401-014-1329-4
Mizielinska, S., Gronke, S., Niccoli, T., Ridler, C. E., Clayton, E. L., Devoy, A., et al. (2014). C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science 345, 1192–1194. doi: 10.1126/science.1256800
Mizielinska, S., and Isaacs, A. M. (2014). C9orf72 amyotrophic lateral sclerosis and frontotemporal dementia: gain or loss of function? Curr. Opin. Neurol. 27, 515–523. doi: 10.1097/wco.0000000000000130
Mizielinska, S., Lashley, T., Norona, F. E., Clayton, E. L., Ridler, C. E., Fratta, P., et al. (2013). C9orf72 frontotemporal lobar degeneration is characterised by frequent neuronal sense and antisense RNA foci. Acta Neuropathol. 126, 845–857. doi: 10.1007/s00401-013-1200-z
Mizielinska, S., Ridler, C. E., Balendra, R., Thoeng, A., Woodling, N. S., Grasser, F. A., et al. (2017). Bidirectional nucleolar dysfunction in C9orf72 frontotemporal lobar degeneration. Acta Neuropathol. Commun. 5:11. doi: 10.1186/s40478-017-0432-x
Moens, T. G., Mizielinska, S., Niccoli, T., Mitchell, J. S., Thoeng, A., Ridler, C. E., et al. (2018). Sense and antisense RNA are not toxic in Drosophila models of C9orf72-associated ALS/FTD. Acta Neuropathol. 135, 445–457. doi: 10.1007/s00401-017-1798-3
Moens, T. G., Niccoli, T., Wilson, K. M., Atilano, M. L., Birsa, N., Gittings, L. M., et al. (2019). C9orf72 arginine-rich dipeptide proteins interact with ribosomal proteins in vivo to induce a toxic translational arrest that is rescued by eIF1A. Acta Neuropathol. 137, 487–500. doi: 10.1007/s00401-018-1946-4
Moens, T. G., Partridge, L., and Isaacs, A. M. (2017). Genetic models of C9orf72: what is toxic? Curr. Opin. Genet. Dev. 44, 92–101. doi: 10.1016/j.gde.2017.01.006
Mori, K., Arzberger, T., Grasser, F. A., Gijselinck, I., May, S., Rentzsch, K., et al. (2013a). Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol. 126, 881–893. doi: 10.1007/s00401-013-1189-3
Mori, K., Weng, S. M., Arzberger, T., May, S., Rentzsch, K., Kremmer, E., et al. (2013b). The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339, 1335–1338. doi: 10.1126/science.1232927
Moron-Oset, J., Super, T., Esser, J., Isaacs, A. M., Gronke, S., and Partridge, L. (2019). Glycine-alanine dipeptide repeats spread rapidly in a repeat length- and age-dependent manner in the fly brain. Acta Neuropathol. Commun. 7:14. doi: 10.1186/s40478-019-0860-x
Moss, D. J. H., Poulter, M., Beck, J., Hehir, J., Polke, J. M., Campbell, T., et al. (2014). C9orf72 expansions are the most common genetic cause of Huntington disease phenocopies. Neurology 82, 292–299. doi: 10.1212/wnl.0000000000000061
Mota, S. I., Ferreira, I. L., and Rego, A. C. (2014). Dysfunctional synapse in Alzheimer’s disease - A focus on NMDA receptors. Neuropharmacology 76, 16–26. doi: 10.1016/j.neuropharm.2013.08.013
Murphy, N. A., Arthur, K. C., Tienari, P. J., Houlden, H., Chio, A., and Traynor, B. J. (2017). Age-related penetrance of the C9orf72 repeat expansion. Sci. Rep. 7:2116. doi: 10.1038/s41598-017-02364-1
Neary, D., Snowden, J. S., Gustafson, L., Passant, U., Stuss, D., Black, S., et al. (1998). Frontotemporal lobar degeneration - A consensus on clinical diagnostic criteria. Neurology 51, 1546–1554.
Neumann, M., Rademakers, R., Roeber, S., Baker, M., Kretzschmar, H. A., and Mackenzie, I. R. A. (2009). A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain 132, 2922–2931. doi: 10.1093/brain/awp214
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133. doi: 10.1126/science.1134108
Nonaka, T., Masuda-Suzukake, M., Hosokawa, M., Shimozawa, A., Hirai, S., Okado, H., et al. (2018). C9ORF72 dipeptide repeat poly-GA inclusions promote intracellular aggregation of phosphorylated TDP-43. Hum. Mol. Genet. 27, 2658–2670. doi: 10.1093/hmg/ddy174
Nordin, A., Akimoto, C., Wuolikainen, A., Alstermark, H., Jonsson, P., Birve, A., et al. (2015). Extensive size variability of the GGGGCC expansion in C9orf72 in both neuronal and non-neuronal tissues in 18 patients with ALS or FTD. Hum. Mol. Genet. 24, 3133–3142. doi: 10.1093/hmg/ddv064
O’Rourke, J. G., Bogdanik, L., Muhammad, A., Gendron, T. F., Kim, K. J., Austin, A., et al. (2015). C9orf72 BAC transgenic mice display typical pathologic features of ALS/FTD. Neuron 88, 892–901. doi: 10.1016/j.neuron.2015.10.027
Osborne, R. J., Lin, X. Y., Welle, S., Sobczak, K., O’Rourke, J. R., Swanson, M. S., et al. (2009). Transcriptional and post-transcriptional impact of toxic RNA in myotonic dystrophy. Hum. Mol. Genet. 18, 1471–1481. doi: 10.1093/hmg/ddp058
Pallo, S. P., DiMaio, J., Cook, A., Nilsson, B., and Johnson, G. V. W. (2016). Mechanisms of tau and A beta-induced excitotoxicity. Brain Res. 1634, 119–131. doi: 10.1016/j.brainres.2015.12.048
Peters, O. M., Cabrera, G. T., Tran, H., Gendron, T. F., McKeon, J. E., Metterville, J., et al. (2015). Human C9ORF72 hexanucleotide expansion reproduces RNA foci and dipeptide repeat proteins but not neurodegeneration in BAC transgenic mice. Neuron 88, 902–909. doi: 10.1016/j.neuron.2015.11.018
Quaegebeur, A., Glaria, I., Lashley, T., and Isaacs, A. M. (2020). Soluble and insoluble dipeptide repeat protein measurements in C9orf72-frontotemporal dementia brains show regional differential solubility and correlation of poly-GR with clinical severity. Acta Neuropathol. Commun. 8:13. doi: 10.1186/s40478-020-01036-y
Rademakers, R., Neumann, M., and Mackenzie, I. R. (2012). Advances in understanding the molecular basis of frontotemporal dementia. Nat. Rev. Neurol. 8, 423–434. doi: 10.1038/nrneurol.2012.117
Renton, A. E., Majounie, E., Waite, A., Simon-Sanchez, J., Rollinson, S., Gibbs, J. R., et al. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268. doi: 10.1016/j.neuron.2011.09.010
Ringholz, G. M., Appel, S. H., Bradshaw, M., Cooke, N. A., Mosnik, D. M., and Schulz, P. E. (2005). Prevalence and patterns of cognitive impairment in sporadic ALS. Neurology 65, 586–590. doi: 10.1212/01.wnl.0000172911.39167.b6
Rohrer, J. D., Guerreiro, R., Vandrovcova, J., Uphill, J., Reiman, D., Beck, J., et al. (2009). The heritability and genetics of frontotemporal lobar degeneration. Neurology 73, 1451–1456.
Saberi, S., Stauffer, J., Jiang, J., Garcia, S., Schulte, D., Ohkubo, T., et al. (2018). Sense-encoded poly-GR dipeptide repeat proteins correlate to neurodegeneration and uniquely co-localize with TDP-43 in dendrites of repeat expanded C9orf72 amyotrophic lateral sclerosis. Neurology 90:3.
Sareen, D., O’Rourke, J. G., Meera, P., Muhammad, A., Grant, S., Simpkinson, M., et al. (2013). Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci. Transl. Med. 5:13. doi: 10.1126/scitranslmed.3007529
Schludi, M. H., May, S., Grasser, F. A., Rentzsch, K., Kremmer, E., Kupper, C., et al. (2015). Distribution of dipeptide repeat proteins in cellular models and C9orf72 mutation cases suggests link to transcriptional silencing. Acta Neuropathol. 130, 537–555. doi: 10.1007/s00401-015-1450-z
Seelaar, H., Rohrer, J. D., Pijnenburg, Y. A. L., Fox, N. C., and van Swieten, J. C. (2011). Clinical, genetic and pathological heterogeneity of frontotemporal dementia: a review. J. Neurol. Neurosur. Psychiatry 82, 476–486. doi: 10.1136/jnnp.2010.212225
Selvaraj, B. T., Livesey, M. R., Zhao, C., Gregory, J. M., James, O. T., Cleary, E. M., et al. (2018). C9ORF72 repeat expansion causes vulnerability of motor neurons to Ca2+-permeable AMPA receptor-mediated excitotoxicity. Nat. Commun. 9:347. doi: 10.1038/s41467-017-02729-0
Snowden, J. S., Neary, D., and Mann, D. M. A. (2002). Frontotemporal dementia. Br. J. Psychiatry 180, 140–143. doi: 10.1192/bjp.180.2.140
Solomon, D. A., Stepto, A., Au, W. H., Adachi, Y., Diaper, D. C., Hall, R., et al. (2018). A feedback loop between dipeptide-repeat protein, TDP-43 and karyopherin-alpha mediates C9orf72-re ated neurodegeneration. Brain 141, 2908–2924. doi: 10.1093/brain/awy241
St Johnston, D. (2002). The art and design of genetic screens: Drosophila melanogaster. Nat. Rev. Genet. 3, 176–188. doi: 10.1038/nrg751
Tao, Z. T., Wang, H. F., Xia, Q., Li, K., Jiang, X. G., Xu, G. Q., et al. (2015). Nucleolar stress and impaired stress granule formation contribute to C9orf72 RAN translation-induced cytotoxicity. Hum. Mol. Genet. 24, 2426–2441. doi: 10.1093/hmg/ddv005
Tran, H., Almeida, S., Moore, J., Gendron, T. F., Chalasani, U., Lu, Y. B., et al. (2015). Differential toxicity of nuclear RNA foci versus dipeptide repeat proteins in a drosophila model of C9ORF72 FTD/ALS. Neuron 87, 1207–1214. doi: 10.1016/j.neuron.2015.09.015
Urwin, H., Josephs, K. A., Rohrer, J. D., Mackenzie, I. R., Neumann, M., Authier, A., et al. (2010). FUS pathology defines the majority of tau- and TDP-43-negative frontotemporal lobar degeneration. Acta Neuropathol. 120, 33–41. doi: 10.1007/s00401-010-0698-6
van Blitterswijk, M., Dejesus-Hernandez, M., Niemantsverdriet, E., Murray, M. E., Heckman, M. G., Diehl, N. N., et al. (2013). Association between repeat sizes and clinical and pathological characteristics in carriers of C90RF72 repeat expansions (Xpansize-72): a cross-sectional cohort study. Lancet Neurol. 12, 978–988. doi: 10.1016/s1474-4422(13)70210-2
van den Ameele, J., Jedlickova, I., Pristoupilova, A., Sieben, A., Van Mossevelde, S., Ceuterick-de Groote, C., et al. (2018). Teenage-onset progressive myoclonic epilepsy due to a familial C9orf72 repeat expansion. Neurology 90, E658–E663. doi: 10.1212/wnl.0000000000004999
Vilidaite, G., Norcia, A. M., West, R. J. H., Elliott, C. J. H., Pei, F., Wade, A. R., et al. (2018). Autism sensory dysfunction in an evolutionarily conserved system. Proc. R. Soc. B-Biol. Sci. 285:20182255. doi: 10.1098/rspb.2018.2255
Waite, A. J., Baumer, D., East, S., Neal, J., Morris, H. R., Ansorge, O., et al. (2014). Reduced C9orf72 protein levels in frontal cortex of amyotrophic lateral sclerosis and frontotemporal degeneration brain with the C9ORF72 hexanucleotide repeat expansion. Neurobiol. Aging 35:9. doi: 10.1016/j.neurobiolaging.2014.01.016
Wen, X. M., Tan, W. Z., Westergard, T., Krishnamurthy, K., Markandaiah, S. S., Shi, Y. X., et al. (2014). Antisense Proline-Arginine RAN dipeptides linked to C9ORF72-ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron 84, 1213–1225. doi: 10.1016/j.neuron.2014.12.010
West, R. J. H., Briggs, L., Fjeldstad, M. P., Ribchester, R. R., and Sweeney, S. T. (2018). Sphingolipids regulate neuromuscular synapse structure and function in Drosophila. J. Compar. Neurol. 526, 1995–2009. doi: 10.1002/cne.24466
West, R. J. H., Elliott, C. J. H., and Wade, A. R. (2015). Classification of Parkinson’s disease genotypes in drosophila using spatiotemporal profiling of vision. Sci. Rep. 5, 1–13. doi: 10.1038/srep16933
West, R. J. H., Sharpe, J. L., Voelzmann, A., Munro, A. L., Hahn, I., Baines, R. A., et al. (2020a). Co-expression of C9orf72 related dipeptide-repeats over 1000 repeat units reveals age- and combination-specific phenotypic profiles in Drosophila. Acta Neuropathol. Commun. 8:19. doi: 10.1186/s40478-020-01028-y
West, R. J. H., Ugbode, C., Fort-Aznar, L., and Sweeney, S. T. (2020b). Neuroprotective activity of ursodeoxycholic acid in CHMP2B(Intron5) models of frontotemporal dementia. Neurobiol. Dis. 144:105047. doi: 10.1016/j.nbd.2020.105047
White, M. R., Mitrea, D. M., Zhang, P. P., Stanley, C. B., Cassidy, D. E., Nourse, A., et al. (2019). C9orf72 Poly(PR) dipeptide repeats disturb biomolecular phase separation and disrupt nucleolar function. Mol. Cell. 74, 713–728. doi: 10.1016/j.molcel.2019.03.019
Wilke, C., Pomper, J. K., Biskup, S., Puskas, C., Berg, D., and Synofzik, M. (2016). Atypical parkinsonism in C9orf72 expansions: A case report and systematic review of 45 cases from the literature. Movement Disord. 31, S62–S62.
Xu, W. C., and Xu, J. (2018). C9orf72 dipeptide repeats cause selective neurodegeneration and cell-autonomous excitotoxicity in drosophila glutamatergic neurons. J. Neurosci. 38, 7741–7752. doi: 10.1523/jneurosci.0908-18.2018
Xu, Z. H., Poidevin, M., Li, X. K., Li, Y. J., Shu, L. Q., Nelson, D. L., et al. (2013). Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. Proc. Natl. Acad. Sci. U S A. 110, 7778–7783. doi: 10.1073/pnas.1219643110
Yang, D. J., Abdallah, A., Li, Z. D., Lu, Y. B., Almeida, S., and Gao, F. B. (2015). FTD/ALS-associated poly(GR) protein impairs the Notch pathway and is recruited by poly(GA) into cytoplasmic inclusions. Acta Neuropathol. 130, 525–535. doi: 10.1007/s00401-015-1448-6
Zhang, Y. J., Gendron, T. F., Ebbert, M. T. W., O’Raw, A. D., Yue, M., Jansen-West, K., et al. (2018). Poly(GR) impairs protein translation and stress granule dynamics in C9orf72-associated frontotemporal dementia and amyotrophic lateral sclerosis. Nat. Med. 24, 1136–1142. doi: 10.1038/s41591-018-0071-1
Zhang, Y. J., Gendron, T. F., Grima, J. C., Sasaguri, H., Jansen-West, K., Xu, Y. F., et al. (2016). C9ORF72 poly(GA) aggregates sequester and impair HR23 and nucleocytoplasmic transport proteins. Nat. Neurosci. 19, 668–677. doi: 10.1038/nn.4272
Zhang, Y. J., Jansen-West, K., Xu, Y. F., Gendron, T., Bieniek, K., Lin, W. L., et al. (2014). Aggregation-prone c9FTD/ALS poly(GA) RAN-translated proteins cause neurotoxicity by inducing ER stress. Acta Neuropathol. 128, 505–524. doi: 10.1007/s00401-014-1336-5
Zu, T., Gibbens, B., Doty, N. S., Gomes-Pereira, M., Huguet, A., Stone, M. D., et al. (2011). Non-ATG-initiated translation directed by microsatellite expansions. Proc. Natl. Acad. Sci. U S A. 108, 260–265. doi: 10.1073/pnas.1013343108
Keywords: Drosophila, C9orf72, dipeptide repeat proteins (DPRs), ALS (amyotrophic lateral sclerosis), FTD (frontotemporal dementia), MND (motor neurone disease)
Citation: Sharpe JL, Harper NS, Garner DR and West RJH (2021) Modeling C9orf72-Related Frontotemporal Dementia and Amyotrophic Lateral Sclerosis in Drosophila. Front. Cell. Neurosci. 15:770937. doi: 10.3389/fncel.2021.770937
Received: 05 September 2021; Accepted: 27 September 2021;
Published: 21 October 2021.
Edited by:
Agnes Lumi Nishimura, Queen Mary University of London, United KingdomReviewed by:
Daniel Solomon, King’s College London, United KingdomNam Chul Kim, University of Minnesota Twin Cities, United States
Copyright © 2021 Sharpe, Harper, Garner and West. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ryan J. H. West, ci5qLndlc3RAc2hlZmZpZWxkLmFjLnVr