 Rim Diab
Rim Diab Federica Pilotto1,2†
Federica Pilotto1,2† Smita Saxena
Smita Saxena- 1Department of Neurology, Center for Experimental Neurology, Inselspital University Hospital, Bern, Switzerland
- 2Department for BioMedical Research (DBMR), University of Bern, Bern, Switzerland
The proper functioning of the cell clearance machinery is critical for neuronal health within the central nervous system (CNS). In normal physiological conditions, the cell clearance machinery is actively involved in the elimination of misfolded and toxic proteins throughout the lifetime of an organism. The highly conserved and regulated pathway of autophagy is one of the important processes involved in preventing and neutralizing pathogenic buildup of toxic proteins that could eventually lead to the development of neurodegenerative diseases (NDs) such as Alzheimer’s disease or Amyotrophic lateral sclerosis (ALS). The most common genetic cause of ALS and frontotemporal dementia (FTD) is a hexanucleotide expansion consisting of GGGGCC (G4C2) repeats in the chromosome 9 open reading frame 72 gene (C9ORF72). These abnormally expanded repeats have been implicated in leading to three main modes of disease pathology: loss of function of the C9ORF72 protein, the generation of RNA foci, and the production of dipeptide repeat proteins (DPRs). In this review, we discuss the normal physiological role of C9ORF72 in the autophagy-lysosome pathway (ALP), and present recent research deciphering how dysfunction of the ALP synergizes with C9ORF72 haploinsufficiency, which together with the gain of toxic mechanisms involving hexanucleotide repeat expansions and DPRs, drive the disease process. This review delves further into the interactions of C9ORF72 with RAB proteins involved in endosomal/lysosomal trafficking, and their role in regulating various steps in autophagy and lysosomal pathways. Lastly, the review aims to provide a framework for further investigations of neuronal autophagy in C9ORF72-linked ALS-FTD as well as other neurodegenerative diseases.
Introduction
The most common attribute of a plethora of neurodegenerative diseases (NDs) is the misfolding and aggregation of proteins, leading to the loss of certain populations of neurons within defined regions of the central nervous system (CNS). A hallmark of NDs is the formation of inter- and intracellular protein accumulations or aggregates in neuronal and glial cells, due to protein misfolding or unfolding (Meyer and Kaspar, 2017; Rahman et al., 2022). Autophagy is regarded as a major clearance pathway within cells, whereby damaged organelles and aggregated proteins are degraded. Autophagy involves the degradation of cytoplasmic contents, enclosed within double-membrane vesicles called autophagosomes, that are delivered to the lysosomes for destruction. It is very likely that small alterations in protein turnover or impaired protein homeostasis during one’s lifetime have cumulative effects that manifest NDs in aged individuals. This may be due to failure of cellular clearance machinery. Thus, autophagy has been systematically linked to the occurrence of pathological changes in several NDs such as in Alzheimer’s, Parkinson’s, and Huntington’s disease (Yue et al., 2009). In this review, we expand the relationship between impaired autophagy and NDs. Further, we provide deeper insights into the frequent genetic cause of amyotrophic lateral sclerosis (ALS) and frontal temporal dementia (FTD) due to the hexanucleotide repeat expansions (HRE) in the C9ORF72 gene. Additionally, we discuss the impairments in the autophagy-lysosome pathway (ALP) that were recently identified in C9ORF72-ALS and FTD cases and in preclinical model systems. Moreover, we also highlight the physiological function of C9ORF72 protein in regulating and modulating the ALP machinery and cellular trafficking. Lastly, we touch upon some strategies to restore functional ALP, to ameliorate and restore the cell clearance machinery and to rescue the degeneration of neurons in ALS-FTD.
Functional autophagy
Four forms of autophagy have been identified: macroautophagy, microautophagy, chaperone-mediated autophagy, and crinophagy (Csizmadia and Juhász, 2020). However, macroautophagy is the most well studied process and is generally referred to as autophagy. Autophagy is tightly regulated by metabolic signals, by reducing equivalents, and by nutrient status, and it is particularly sensitive to energy levels. (Ryter et al., 2019). Unc-51-like Kinase 1 (ULK1) is a crucial autophagy initiator. Mammalian target-of-rapamycin mTORC1, together with adenosine monophosphate activated protein kinase (AMPK), are responsible for ULK1 phosphorylation, where ULK1 and AMPK interact in a nutrient-dependent way (Shang et al., 2011). Nevertheless, in conditions of nutrient starvation, ULK1 is dephosphorylated specifically at Ser638 and Ser758 (Shang et al., 2011). ULK1 must be properly phosphorylated to associate with AMPK, as reported by Shang et al. (2011). The interaction with AMPK can be compromised by a single serine-to-alanine mutation (S758A) at ULK1. This mutant ULK1-S758A initiates starvation-induced autophagy at a faster rate than the wild-type ULK1, but has no effect on the maximal autophagic capacity as starvation lengthens. Autophagy commences with the formation of autophagosomes that sequester the cytoplasmic organelles/inclusions targeted for destruction. The initiation of autophagy, leads to the assembly of ATG proteins at a specialized site, known as the pre-autophagosomal structure (PAS) (Mizushima et al., 2011; Rubinsztein et al., 2012). PAS subsequently undergo synchronized fusion of membranes, derived from endosome, Golgi and plasma membranes, to form the phagophore (Rubinsztein et al., 2012). Autolysosomes are formed due to the fusion of autophagosomes to free lysosomes, leading to the breakdown of the inner autophagosomal membrane and the ingestion of cargo by lysosomal hydrolytic enzymes. The lysosomes may export the catabolites produced during autophagosome breakdown to the cytoplasm, where they can be recycled to create new macromolecules (Mahapatra et al., 2021).
Autophagic lysosome reformation (ALR)
Following the breakdown of autophagic substrates, the levels of amino acids in the cytoplasm rise and mTORC1 is activated (Chen X. et al., 2021). In addition to negatively regulating autophagy and preventing its overactivation, mTORC1 reactivation aids in the regeneration of lysosomes from autolysosomes (Figure 1). When this process, known as ALR, is compromised, the cells become far more vulnerable to starvation-induced cell death (Chen X. et al., 2021; Nanayakkara et al., 2022). Among all the different cells of our body, neurons are particularly vulnerable to compromised ALR, which predominantly impacts correct neuronal functioning. Unsurprisingly, ALR impairment is linked to an ever-increasing number of diseases, and classified as a subgroup of lysosomal disorders, involving an imbalance in lysosome homeostasis, leading to autophagy inhibition (Nanayakkara et al., 2022). Mutations in different genes, such as spastizin and spatacsin are associated with autosomal recessive hereditary spastic paraplegia. The loss of function of spatacsin in mice causes hereditary spastic paraplegia-like phenotypes with degeneration of cerebellar Purkinje cells and cortical neurons (Varga et al., 2015). Both spastizin and spatacsin are physiologically important components of the ALR. Loss of spastizin or spatacsin resulted in the compete loss of free lysosomes that are required to fuse with autophagosomes, leading to an accumulation of autolysosomes and the subsequent failure of ALR. Defects in the regulation of ALR might ultimately result in the decline of functional lysosomes, leading to reduced autophagic clearance. This decline results in the accumulation of non-degraded toxic proteinaceous and membranous material and, finally, neuronal death (Chang et al., 2014).
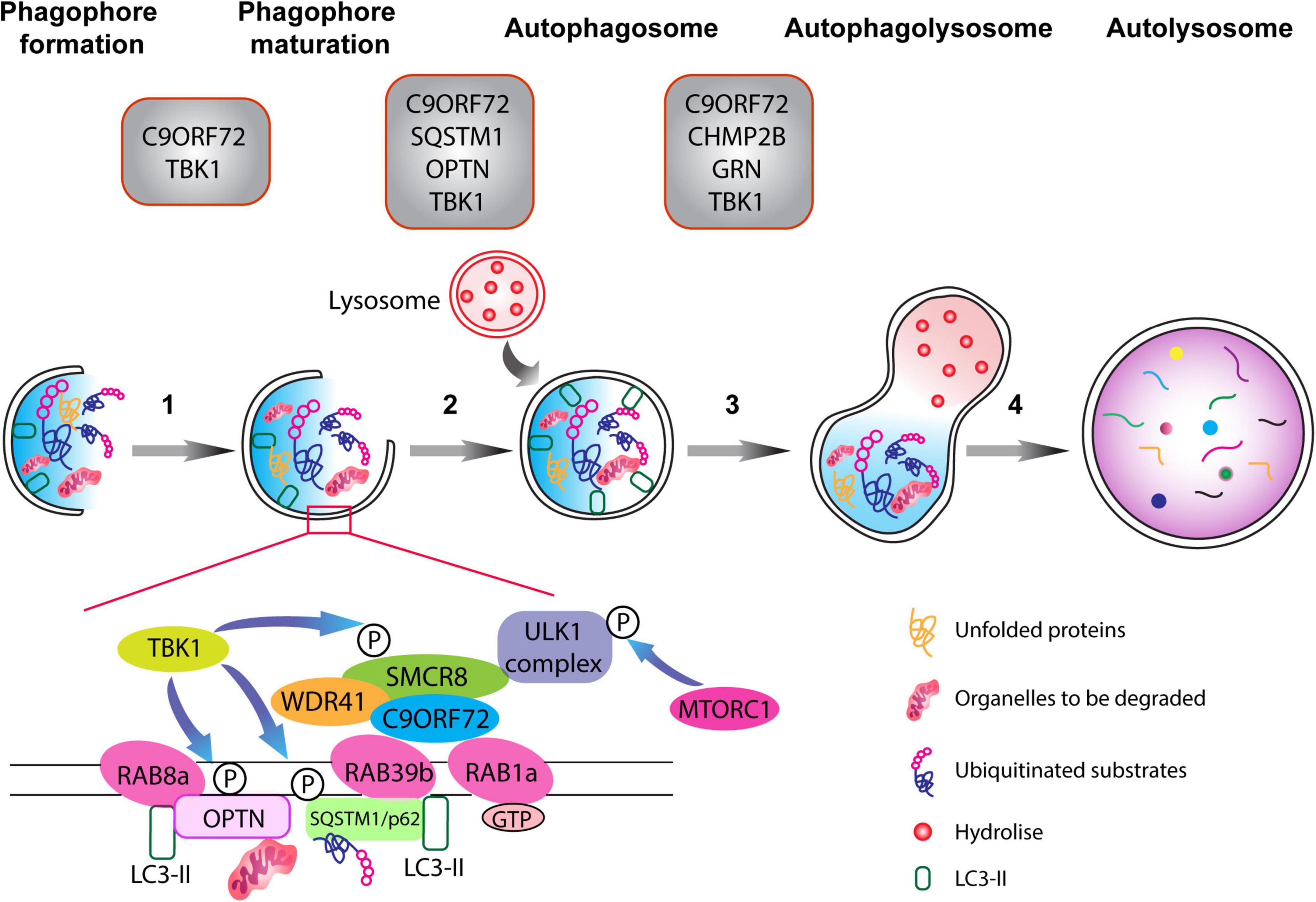
Figure 1. The autophagy pathway. Schematic representation of different phases of the autophagy pathway from phagophore formation to the degradation of dysfunctional organelles and ubiquitinated proteins (the autophagic cargo) within autolysosomes. Top: gray boxes depict the various proteins associated with ALS-FTD that normally function at defined stages of the autophagolysosome formation. Autophagy is composed of multiple steps (1) phagophore formation, where double membrane phagophore forms around damaged molecules, unfolded proteins, and compromised organelles, (2) phagophore maturation and elongation, (3) the formation of the mature phagophore carrying the autophagic cargo, (4) the fusion of the phagosome with lysosome containing hydrolytic enzymes, which degrade the content of the autophagic cargo. The C9ORF72-WDR1-SMCR8 complex regulates autophagy at the initiation and progression phases (steps 1–3). RAB1a GTPase can recruit the C9ORF72 complex to engage the ULK1 complex during the initial phase of the autophagy pathway. Importantly, RAB8a and RAB39b, respectively, bind to OPTN and SQSTM1/p62, and also interact with the C9ORF72-WDR1-SMCR8 complex. Finally, phosphorylation of SCMR8 by TBK1 is crucial for the initiation of the autophagy pathway.
Likewise, ALR dysfunction is causally implicated in autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS), a fatal, neurodegenerative disorder, wherein cerebellar Purkinje cells selectively degenerate, leading to spasticity and ataxia (Bagaria et al., 2022; Francis et al., 2022). ARSACS is caused by mutations in the gene of sacsin molecular chaperone (SACS), leading to a loss of function phenotype. Normally, SACS binds to the motor-protein KIF5B, which is then involved in the extrusion of autolysosome membrane buds along microtubules to create reformation tubules during ALR (Du et al., 2016; Francis et al., 2022). Loss of SACS function inhibits the generation of reformation tubules at autolysosomes during ALR, (Francis et al., 2022). In conclusion, ALR plays an important role in maintaining cellular health, and the impairment of this process may contribute to the development of NDs. However, more research is needed to fully understand the exact mechanisms involved and how they could be targeted for the development of new therapies for these debilitating diseases.
The cross talk between autophagy and UPS
Besides autophagy, it is vital to consider the impact of the ubiquitin-proteasome system (UPS) as one of the major quality control mechanisms involved in maintaining cellular homeostasis in eukaryotes. Both systems use ubiquitination as a degradation signal for their targets, but distinct processes are at work. Short-lived proteins and soluble misfolded proteins are degraded by UPS, whereas long-lived proteins, insoluble protein aggregates, whole organelles (such as mitochondria, peroxisomes), and intracellular parasites are destroyed by autophagy (Kocaturk and Gozuacik, 2018). There are links and reciprocal control mechanisms between both degradation pathways, in addition to an indirect connection between the two systems via ubiquitinated proteins (Nedelsky et al., 2008). The two degradative pathways play compensatory roles toward each other: when the function and ability of one process is mitigated or inhibited, the other’s activity levels rise dramatically to compensate for the deficits in the other pathway (Kocaturk and Gozuacik, 2018). The inhibition of the UPS using proteasome inhibitors (Wu et al., 2008; Selimovic et al., 2013; Fan et al., 2018) or by genetic manipulations (Demishtein et al., 2017) led to the induction of autophagy in cells as observed by the increased expression of autophagy-associated genes ATG5 and ATG7. This upregulation required the ER stress-dependent phosphorylation of eukaryotic translation initiation factor-2 alpha (eIF2α) (Zhu et al., 2010). Proteasome inhibition led to an increase in autophagy which correlated with the activation of two major regulators of autophagy, namely AMPK and mTORC1 (Xu et al., 2012; Jiang et al., 2015). Proteasome inhibition was also linked to an increase in sequestosome 1 (SQSTM1/p62) and GABARAPL1 expression by both Nrf1-dependent and independent pathways (Sha et al., 2018). Conversely, impairment in autophagy paralleled the activation of the UPS. Suppression of autophagy via chemical compounds, or via shRNA-mediated knock down of ATG genes, led to elevated activity of the UPS, mainly due to the increase in expression of proteasomal subunits (Lü and Wang, 2013). In another study, 3-MA-mediated autophagy inhibition in cultured neonatal rat ventricular myocytes increased chymotrypsin-like activity of proteasomes (Tannous et al., 2008).
The chaperone machinery, which oversees the quality control for proper protein folding, detects soluble portions of proteins with a folding issue and directs them to the UPS for destruction (Saibil, 2013). One of the E3 ligases that adds a K48-linked ubiquitin chain to unfolded or misfolded proteins has been identified as CHIP, a Hsp70 and Hsp90 chaperone interactor (Wang T. et al., 2020). The Hsp70 complex interacts with the BAG family of proteins, particularly BAG1, to initiate the proteasomal degradation of client proteins (Chen Y. et al., 2021). On the other side, aggresome production is necessary for the removal of insoluble aggregate-prone proteins. Ubiquitination via E3 ligases such as Parkin, CHIP, TRIM50, and HRD1 is the first step to priming insoluble protein aggregates (Olzmann et al., 2007; Olzmann and Chin, 2008; Mishra et al., 2009; Zhang and Qian, 2011). The process of aggresome formation is primarily achieved by HDAC6, serving as a linker protein between K63-ubiquitinated aggregates and dynein (Matthias et al., 2008). The aggregates are then transported toward microtubule organizing centers (MTOCs), resulting in their stacking up within the cytoplasm as aggresomes (Kopito, 2000). The direct interaction of SQSTM1/p62 and NBR1 with the ubiquitinated condensates further promotes their aggregation via their polymerization domain, followed by the delivery of ubiquitinated cargo to the autophagosomes (Ichimura et al., 2008; Lamark and Johansen, 2012). Alternatively, an autophagy-related protein known as ALFY has been identified as the main component in the onset of selective autophagy and the degradation of the aggresomal units (Clausen et al., 2010; Filimonenko et al., 2010).
Relationship between autophagy and neurodegeneration
Protein aggregates or inclusion bodies are common hallmarks of age-related neurodegenerative disorders (Dugger and Dickson, 2017). The disruptive nature of these aggregates is characterized by their ability to confer toxicity, and consequently impair physiological neuronal functions, including axonal transport, synaptic integrity, mitochondrial bioenergetic machinery, and the regulation of transcription (Higuchi et al., 2002; Ruegsegger and Saxena, 2016; Dugger and Dickson, 2017; Batool et al., 2019). Several studies have shown how deregulation of autophagy associates with a high correlation to NDs like Alzheimer’s disease (AD) and Parkinson’s disease (PD). Moreover, recent studies have established a significant relationship between impaired autophagy and the onset of psychiatric disorders, such as schizophrenia and bipolar disorders (Merenlender-Wagner et al., 2014; Vanderplow et al., 2021). Current research has also provided novel insights into the intricate roles of autophagy in the regulation of lysosomal degradation, and the subsequent recycling of the lysosomal components, enabling the maintenance of an equilibrium between pro-survival and cell death responses (Bar-Yosef et al., 2019).
Due to impairments in the ALP, protein aggregation is often complementary to the decline in physiological degradative function of neurons. In the case of NDs, ALP dysfunction can be attributed to multiple factors, for example loss of function mutations that directly impact ALP functionality. Frequently, loss of function mutations are coupled with gain of toxic function of the genes that promote toxic protein aggregation, thereby contributing to the disruption of physiological cellular function and leading to neuronal death (Stavoe and Holzbaur, 2019; Monaco and Fraldi, 2020). CNS-specific knockdown of autophagy genes, Atg5, Atg7, and FIP200, led to mice exhibiting behavioral and motor deficits and dramatically reduced lifespan due to impaired autophagolysosome clearance, highlighting the crucial role of autophagy in maintaining neuronal homeostasis (Hara et al., 2006; Komatsu et al., 2006; Liang et al., 2020). Several misfolded proteins that are linked to NDs directly target the ALP, however, the question of whether ALP itself may be considered protective or destructive, or even as an active contributor to neuronal death remains unclear. Although ALP is highly complex in nature, nevertheless, it is well acknowledged that boosting it might have neuroprotective effects against neurodegeneration (Bossy et al., 2008). For example, boosting the ALP via the mTORC1 inhibitor rapamycin was shown to be neuroprotective against protein aggregates in in vivo models of Huntington’s disease (HD) (Ravikumar et al., 2004).
Some studies identified autophagy as a facilitator of apoptotic pathways, for instance in vitro assays on astrocytes have shown that autophagy can trigger the activation of caspase 8 via the degradation of caveolin-1 (Chen et al., 2018; Peng et al., 2019). A new form of cell death involving the autophagy pathway, called karyoptosis, has been proposed for the CNS. Karyoptosis is induced by the chronic inhibition of autophagy and characterized by cytoplasmic relocation of Lamin B1, together with LC3 and the SQSTM1/p62 receptor (Dou et al., 2015; Baron et al., 2017; Baron and Fanto, 2018). Karyoptosis, has been observed in models of AD and polyglutamine expansion (PolyQ) diseases where caspase-3 independent cell death has been detected (Turmaine et al., 2000; Yang et al., 2008). The mechanisms linking neuroprotective effects of autophagy, or those mediating cell death, are still debated. This highlights a more complex role for autophagy, not only in sustaining neuronal physiology, but also as an ever-present challenge for the treatment of NDs.
Autophagy and autophagic lysosome reformation (ALR) impairments in NDs
Increasing evidence suggests that autophagy is implicated in neuronal disorders, ranging from AD and FTD, to movement disorders such as PD, ALS, multiple sclerosis and polyglutamine repeat diseases such as HD (Nah et al., 2015). As described in recent literature, dysfunctional autophagy is primarily implicated in the development of AD in human patients. The occurrence of impaired maturation or fusion of lysosomes with autophagosomes, or their movement toward the body of the neuronal cell, is commonly observed in AD neurons. Further, the presence of endosomal-lysosomal dysfunction has also been observed. In post-mortem human brain and in a mouse model of AD, this dysfunction is evident via the accumulation of double membrane autophagic vesicles (AVs) containing Aβ within dystrophic neurons and dendrites (Nixon et al., 2005; Boland et al., 2008). At the level of neuropathology, striking ultrastructural changes observed in AD involve the accumulation of AVs inside affected neurons (Terry et al., 1964; Nixon et al., 2005). Indeed, the extent of AVs accumulation in AD is so prominent that it often resembles AVs accumulation seen in lysosomal storage disorders (Nixon et al., 2008).
Recently, it was shown by Lee et al. (2022), that in AD, AVs are strongly immunoreactive to Aβ and are packed into large membrane blebs forming flower-like perikaryal rosettes. This unusual pattern of AV accumulation has been named as PANTHOS (poisonous anthos flower), and is widely observed within human post-mortem AD brains (Lee et al., 2022). Impairments in autophagic flux are known to occur in Aβ metabolism not only in the clearance and secretion process, but also in its generation (Turmaine et al., 2000). For instance, amyloid precursor protein (APP) and subunits of the γ-secretase complex are localized to autophagosomes, thus confirming a link between Aβ metabolism and autophagic flux (Di Meco et al., 2020). Rapamycin treatment of animal models of AD significantly improved cognitive skills. Additionally, reduced intracellular Aβ and extracellular amyloid deposition was observed, thus strongly linking AD with autophagy dysfunctions (Caccamo et al., 2010; Chandarlapaty et al., 2011). Moreover, brain specimens of AD patients were characterized by the presence of hyperphosphorylated tau colocalizing with LC3, an autophagosome marker, together with SQSTM1 (Piras et al., 2016).
In HD, a PolyQ expansion disorder, the gradual and progressive neuronal loss causes death of the striatal neurons. Compelling research points to autophagy as being crucial to the degradation of mutant Huntingtin (mHTT) protein (Martin et al., 2015). Indeed, research on HD animal models and human HD cell lines revealed a distinct autophagy-related deficit, whereby the AVs’ capacity to identify and capture cytosolic cargo is compromised (Martinez-Vicente et al., 2010). Of note, different post-translational events, such as SUMOylation, are known to play a role in autophagy in the context of HD. SUMOylation of the mHTT makes it more toxic to nerve cells. SUMO1 deletion in a humanized mouse model of HD, and in human fibroblasts, has been shown to boost and promote autophagic flux by increasing the association of mHTT with LAMP1 and LC3. This interaction also decreases the solubility of the toxic mHTT protein and its association with SQSTM1 (Ramírez-Jarquín et al., 2022). Additionally, a recent finding from Oh et al. (2022) reported that human fibroblasts directly reprogrammed to medium spiny neurons from HD patients show impairments in functional autophagy. Specifically, the authors identified an aging-associated upregulation of a microRNA (miR-29b-3p) that contributes to degeneration by targeting the human STAT3 3’ untranslated region, which is relevant for functional autophagy (You et al., 2015).
Parkinson’s disease is characterized by tremors, muscular rigidity, and bradykinesia caused by progressive loss of dopaminergic neurons in the substantia nigra pars compacta, followed by the accumulation of insoluble proteins in Lewy bodies (Gelb et al., 1999). It is interesting to note that various PD mutations can influence different autophagy phases. The pathophysiology of familial PD is caused by an accumulation of α-synuclein in Lewy bodies (Polymeropoulos et al., 1997; Krüger et al., 1998). It is assumed that wild-type α-synuclein, a protein with unidentified function, regulates dopamine neurotransmission at the pre-synaptic level and is routinely transported into lysosomes for chaperone-mediated autophagy (Winslow and Rubinsztein, 2011; Bar-Yosef et al., 2019). α-synuclein compromises ALP by directly inhibiting RAB1a, thereby impairing autophagy initiation. In addition, α-synuclein promotes the retention of TFEB, a key transcription factor regulating lysosomal biogenesis (Winslow et al., 2010; Settembre et al., 2011; Decressac et al., 2013). Interestingly, the process of ALR was impaired in PD neurons, leading to a deficit in functional lysosomes required to maintain prolonged autophagic clearance of α-synuclein. Notably, deficiency in glucosylceramidase beta 1 (GBA1) and cyclin G associated kinase (GAK) are established as genetic risk factors in 5–10% of cases of PD (Latourelle et al., 2009; Tysnes and Storstein, 2017; Avenali et al., 2020; Miyazaki et al., 2021). GBA1 deficiency specifically reduces lysosome function, leading to autophagy inhibition (Schöndorf et al., 2014; Brooker and Krainc, 2021; Kuo et al., 2022). GBA1 is required for ALR, and its deficiency leads to impaired lysosome homeostasis (Awad et al., 2015; Magalhaes et al., 2016). The neuronal accumulation of α-synuclein has been observed to coincide with ALR suppression, as during normal circumstances α-synuclein is cleared by autophagy (Magalhaes et al., 2016). Thus, ALR impairments might be causally associated with autophagy deficits and the accumulation of toxic α-synuclein species in the brain (Winslow et al., 2010; Magalhaes et al., 2016).
Relationship between autophagy and ALS-FTD mutations
In the case of ALS and FTD a peculiar relationship exists, as ALS and FTD represent two extremes of an overlapping clinico-pathological disease spectrum. They not only manifest different intermediate phenotypes, but also exhibit several shared pathological aspects between the two diseases (Andersen, 2013). C9ORF72 mutations are the most common genetic cause of ALS and, notably, most ALS-FTD causing monogenetic mutations overlap and mainly associate with endosomal-lysosomal and/or ALP. Point mutations in charged multivesicular body protein 2B (CHMP2B), valosin containing protein (VCP), ubiquilin 2 (UBQLN2), optineurin (OPTN), cyclin F (CCNF), TANK binding kinase 1 (TBK1), and sequestosome 1 (SQSTM1/p62), have been causally implicated in ALS-FTD pathology (Renton et al., 2014; Oakes et al., 2017; Rayner et al., 2021; Chua et al., 2022). Notably, systematic neuropathological analyses have revealed that 95–97% of patients with ALS and approximately 50% of patients with FTD share the presence of cytoplasmic TARDBP inclusions (Neumann et al., 2006; Hasegawa et al., 2008). In addition, there is co-accumulation of TARDB together with SQSTM1/p62, suggestive of impairments/disturbance in autophagy (Chua et al., 2022).
The fact that different mutations in proteins, which take part in the ALP, are causally liked to ALS-FTD clearly suggests that autophagy dysfunction is a key player in ALS-FTD disease progression. For instance, mutations in genes involved in the autophagic flux specifically target key molecular players in the ALP, altering their physiological function. For example, CHMP2B is part of the ESCRT-III complex that is involved in regulating endosomal sorting of transmembrane proteins as well as regulating the fusion of the lysosomal vesicles with the autophagosome (Krasniak and Ahmad, 2016). Numerous mutations in CHMP2B are associated with NDs, mainly involving the ALS-FTD spectrum. Within the ALS-FTD spectrum, a C to A substitution causing a T104N substitution, an A to G substitution resulting in a I29V substitution, and an A to G substitution leading to a Q206H substitution are all linked to primary muscular atrophy (Cox et al., 2010; Isaacs et al., 2011). Also, a G to T substitution leading to a D148Y mutation is associated with semantic dementia (Skibinski et al., 2005). Another ND, related to but not a part of the ALS-FTD spectrum, and caused by a CHMP2B mutation, is involved in corticobasal degeneration. This pathology is mainly due to an A to G substitution, resulting in a N143S mutation (van der Zee et al., 2008). Moreover, a dominant mutation in intron 5 of CHMP2B (CHMP2BIntron5) is linked to a fraction of heritable FTD associated with chromosome 3 (FTD-3) (Skibinski et al., 2005). Mouse models of CHMP2BIntron5 have revealed a characteristic autophagy phenotype, with increased SQSTM1/p62 levels as well as prominent ubiquitin deposits throughout the brain (Ghazi-Noori et al., 2012; Gascon et al., 2014; Clayton et al., 2015). Notably, mutant neurons exhibited axonal swellings caused by the accumulation of membrane bound vessels arising from endolysosomal and autolysosomal phenotype (Ghazi-Noori et al., 2012).
It has recently been shown that ALS-FTD-linked mutations of TBK1 and SQSTM1 diminish the phosphorylation of SQSTM1, thereby compromising the binding and clearance of ubiquitinated cargo (Deng et al., 2020). During periods of oxidative stress, SQSTM1 mutation G427R inhibits SQSTM1 phosphorylation at Ser351, thereby impairing KEAP1-SQSTM1 association. This impairment leads to reduced NFE2L2/Nrf2-mediated gene expression and substantially enhanced stress granule formation (Deng et al., 2020; Lee D. H. et al., 2020). Importantly, in physiological conditions, SQSTM1/p62 plays a key role in directing the ubiquitinated substrates to the degradation machinery, either via autophagy or UPS. This involves the activation of ULK1, which phosphorylates SQSTM1/p62 enabling its increase in affinity for ubiquitinated substrates (Lim et al., 2015). In addition, SQSTM1/p62 directly interacts with the LC3, an autophagosome membrane protein leading to the sequestration of targets that need to be degraded via ALP (Liu et al., 2016). Another important interacting partner of SQSTM1/p62 is optineurin (OPTN), and together they form an autophagy complex receptor for the degradation of damaged mitochondria. Of note, different causative mutations of familial and sporadic ALS involve mutations and pathological variants of OPTN (Maruyama et al., 2010). Finally, another important player recently linked to ALS pathogenesis is TBK1, which phosphorylates SQSTM1/p62 at Ser403, thereby enabling its binding to ubiquitinated cargos. In pathological cases, TBK1 mutation causes impairments in the clearance of ubiquitinated substrates, therefore impairing autophagic flux (Matsumoto et al., 2015). Interestingly, preclinical studies have shown how the TBK1- SQSTM1/p62 axis is important for the clearance of TDP43 aggregates (Lee S. et al., 2020). The finding that most patients suffering from ALS-FTD do not harbor mutations in TARDBP gene, yet present cytoplasmic TARDBP inclusions, is indicative of disturbed TARDBP function, likely due to disturbances in the ALP or endosomal-lysosomal trafficking in ALS-FTD (Wood et al., 2021). Furthermore, the conditional knock out mouse model of TBK1 exhibits disrupted autophagy (Duan et al., 2019).
Point mutations in VCP are found in a fraction of patients suffering from either familial form of PD or ALS (Johnson et al., 2010). VCP plays an important role in autophagosome maturation and autolysosome formation under basal conditions, and in cells where proteasome is compromised, but not in cells undergoing starvation responses (Tresse et al., 2010). RNAi-mediated knockdown or overexpression of dominant-negative VCP led to the prominent accumulation of immature and abnormal autophagic vesicles. The expression of disease-related VCP mutants (R155H and A232E) cause autophagy deficits (Ju et al., 2009). These findings strongly associate deficits in ALP with the pathophysiology of ALS-FTD, highlighting specific neuronal vulnerability to dysregulated ALP in ALS-FTD.
Physiological and pathological functions of C9ORF2 in autophagy pathways
Hexanucleotide repeat expansions (G4C2) in the 5’ non-coding sequence of the C9ORF72 gene was identified in 2011 as the most common inherited cause of ALS, FTD, and ALS-FTD (DeJesus-Hernandez et al., 2011; Renton et al., 2011). In contrast to healthy individuals which usually have 2 to 30 hexanucleotide repeats, ALS-FTD patients harbor 100 to even 1600 repeats (Buchman et al., 2013; Dols-Icardo et al., 2014). The pathogenic mechanisms by which HRE in the C9ORF72 gene cause ALS are not fully understood, however, three distinct but mutually non-exclusive disease mechanisms have been proposed (Balendra and Isaacs, 2018; Schmitz et al., 2021): firstly, haploinsufficiency of the C9ORF72 gene, leading to the loss of function associated with the reduced function of C9ORF72 protein (Shi et al., 2018); secondly, sequestration of RNA-binding proteins by RNA foci containing the C9ORF72 HRE RNA (Haeusler et al., 2014, 2016); and lastly, repeat-associated non-AUG (RAN) translation of the repeat expansion, leading to the generation of dipeptide repeat proteins (DPRs) cause toxic gain of function in C9ORF72 ALS-FTD (Mori et al., 2013; Figure 2).
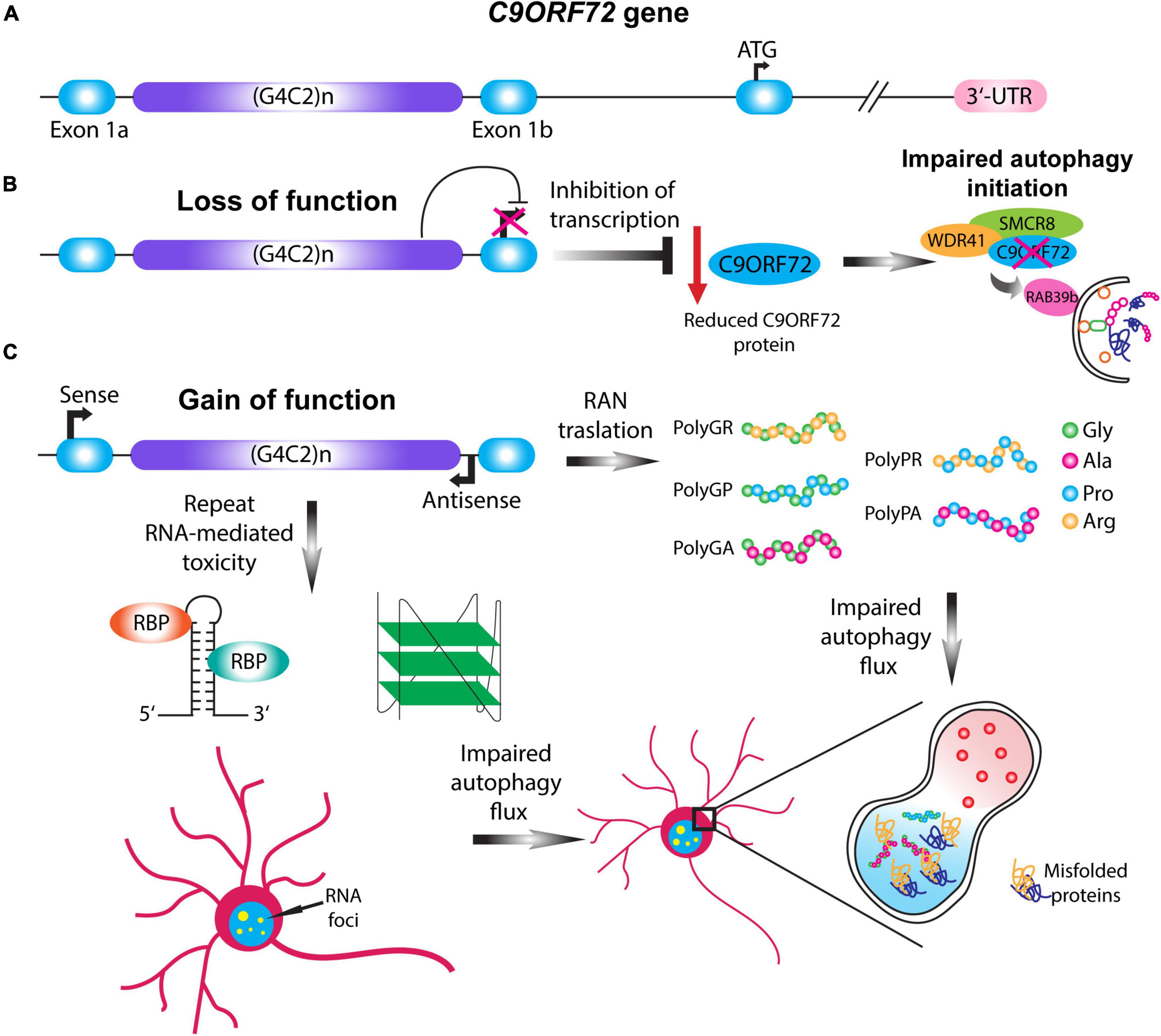
Figure 2. C9ORF72 loss and gain of function mechanisms impair autophagy. (A) Schematic illustration of the C9ORF72 gene. C9ORF72-ALS-FTD is characterized by a G4C2 hexanucleotide repeat expansion between exon 1a and exon 1b, which in patients can range up to thousands of repeats. (B) Haploinsufficiency of C9ORF72 gene causes the loss of function of the C9ORF72 protein by transcription inhibition. Given its importance in the initial steps of phagophore formation, reduction in the C9ORF72 protein impairs autophagy initiation, destabilizing WDR41 and SMCR8 complex. (C) Two pathological mechanisms involving the gain of function of the HRE gives rise to RAN translation and repeat-linked RNA mediated toxicity. In the case of RAN translation, the HRE can be translated in a sense or antisense manner giving rise to five different dipeptide repeats proteins (DPRs): PolyGA, PolyGP, PolyGR, PolyPR, and PolyPA. In the case of RNA-mediated toxicity the translated HRE can form higher order structures that sequester different RNA binding proteins, and thus forming RNA foci. The gain of function mechanisms can impair the autophagic flux and the formation of the autolysosome, leading to the accumulation of misfolded proteins.
Although not much is known about the physiological functions of normal C9ORF72 protein, recent research has implicated an important role for C9ORF72 protein in autophagy. Structure-based modeling suggested that C9ORF72 protein, together with its binding partner SMCR8, belong to the Differentially Expressed in Normal and Neoplastic cells (DENN) domain subfamily of proteins. The DENN domain-containing protein family is characterized by a conserved N-terminal Longin domain and a C-terminal DENN domain (Zhang et al., 2012; Levine et al., 2013a,b; Jiang et al., 2022). The autophagic function of C9ORF72 was deduced from C9ORF72, or SMCR8 knockout mice, which exhibited dysregulation of mTORC1 or TFEB signaling (Ugolino et al., 2016; Shao et al., 2020). Three independent studies in 2016 established that the C9ORF72 protein forms a tripartite complex with SMCR8 and WDR41, and harbors guanine nucleotide exchange factor (GEF) activity, which it exerts upon small GTPases (Amick et al., 2016; Sellier et al., 2016; Sullivan et al., 2016; Yang et al., 2016). Notably, the tight interaction between C9ORF72 and SMCR8 is achieved via the DENN domain within the C9ORF72 protein (Yang et al., 2016). Importantly, C9ORF72 protein levels diminished when the expression of SMCR8 was inhibited (Zhang et al., 2018), while neither C9ORF72 nor SMCR8 knockout negatively influenced WDR41 protein levels (Ugolino et al., 2016). Similarly, loss of SMCR8 and WDR41 significantly reduced C9ORF72 protein levels and, conversely, SMCR8 levels were also decreased after C9ORF72 and WDR41 loss (Zhang et al., 2018; Lan et al., 2019). These studies indicate that a direct relationship exists between SMCR8 and C9ORF72 expression, likely via the stabilization of the two proteins, whereas WDR41 association might be involved in stabilizing the C9ORF72-SMCR8 heterodimers.
Furthermore, a systematic proteomic analysis of the human autophagy system, together with the functional analysis of selected genes within the autophagy pathway, identified SMCR8 as an interacting protein of FIP200. The latter is a protein involved in the ULK1 complex, and serves as the upstream component of the autophagy machinery (Behrends et al., 2010; Wong et al., 2013; Lin and Hurley, 2016). Moreover, the tripartite C9ORF72 complex interacts with the ULK1 complex via the C9ORF72 DENN domain, which also associates with SMCR8 and ATG13 (Tang, 2016; Webster et al., 2016; Cali et al., 2019; Ho et al., 2019). C9ORF72 localizes to lysosomal membranes and, during conditions of nutrient starvation, association between C9ORF72 and ULK1 complex is enhanced via an elevated interaction between SLC66A1/PQLC2 and WDR41 (Aoki et al., 2017; Beckers et al., 2021). The mechanistic aspects associated with the regulation of ULK1-linked autophagy initiation via C9ORF72 complex remain unclear. Both augmented transcriptional and post-transcriptional modifications have been implicated in this regulation (Yang et al., 2016; Amick et al., 2020). Nevertheless, several studies have conclusively established that the main effect of the partial loss of C9ORF72 function is reduced ULK1 activity and the subsequent reduction in autophagy (Sellier et al., 2016; Webster et al., 2016; Yang et al., 2016; Jung et al., 2017).
Further, based on these previous observations, the reduction in C9ORF72 protein level has been specifically linked to a detrimental effect on autophagy initiation (Ugolino et al., 2016; Shi et al., 2018; Wang M. et al., 2020). In neurons from C9ORF72 depleted mice, both suppressed autophagy and reduced ULK1 protein levels were observed, suggesting that C9ORF72 and SMCR8 probably act against each other to regulate the autophagy initiation complex (Ho et al., 2019). Interestingly, ATXN2 with intermediate PolyQ repeats is associated not only with spinocerebellar ataxia type 2 (SCA2), but also with ALS (Elden et al., 2010), serving as a risk factor for C9ORF72-linked ALS (Zhang et al., 2017). Sellier et al. (2016) reported the existence of a distinct synergistic toxicity effect of ATXN2 intermediate PolyQ repeats, that induced motor neuron degeneration when coupled to C9ORF72 haploinsufficiency. Importantly, C9ORF72 reduction partially affects neuronal survival by impairing initial steps in the autophagy pathway and therefore allowing the accumulation of ATXN2 aggregates. This results in neuronal death in vitro and in zebrafish models, where this synergistic toxic effect is associated with swimming impairments and compromised spinal axonal projections (Ciura et al., 2016; Sellier et al., 2016).
As the three pathological mechanisms of C9ORF72-ALS-FTD are mutually non-exclusive, several studies have investigated the synergy between these mechanisms in the disease pathology. Zhu et al. (2020) have shown that reducing or completely abolishing C9ORF72 protein expression inhibited the HRE-mediated increase in the induction of autophagy. This suppression of autophagy accelerated disease pathology by triggering the premature accumulation of DPRs, activating the glial cells and, ultimately, initiating hippocampal neuronal degeneration (Zhu et al., 2020). Yet another study has highlighted a similar synergy between the loss of C9ORF72 expression and DPRs accumulation. Bonvin et al. have shown that a double hit mechanism confers DPR toxicity via reduced autophagy due to diminished C9ORF72 protein levels. This reduction directly impairs functional autophagy promoting the toxic accumulation of DPRs such as PolyGA, PolyGR, and PolyGP. Furthermore, siRNA−mediated depletion of C9ORF72 negatively impacted the recruitment of the SQSTM1/p62 autophagy adaptor protein to PolyGA aggregates, suggesting that C9ORF72 plays an important role at the first step of autophagosome formation (Boivin et al., 2020). In C9orf72-knockout mice, premature mortality is linked to dysfunctional immune responses, and they do not exhibit an overt neurodegenerative phenotype. However, upon crossing these knockout mice with mice carrying a repeat expansion transgene, the resulting toxic DPRs production attenuated autophagy induction. This attenuation led to the enhanced accumulation of DPRs, thus accelerating the pathology (Zhu et al., 2020). Based on these observations, one can envisage a toxic gain of function due to accumulations of DPRs, TARDBP, and HRE RNA; and, in parallel, the loss of function due to C9ORF72 haploinsufficiency might lead to a vicious exacerbation of the disease pathology.
Physiological and pathological functions of C9ORF72 in the regulation of RABs
The RAB family of proteins belongs to the RAS superfamily of proteins and, being enriched within distinct cellular organelles and membranes, plays a pivotal role in regulating membrane trafficking (Pang et al., 1988; Kiral et al., 2018). More than 60 RAB GTPases are present within the human genome, of which 24 are specific or highly enriched in expression within the CNS (Brighouse et al., 2010; D’Adamo et al., 2014). RAB proteins cycle between GTP-bound active and GDP-bound inactive states. The active vs. inactive state of RAB GTPases is tightly controlled by GDP-GTP exchange factors (GEFs) and GTPase-activating protein (GAPs). GAPs catalyze the hydrolysis of GTP to GDP, and GEFs promote the conversion of an inactive GDP-bound RAB to an active GTP-bound RAB (Barr and Lambright, 2010). Active RAB GTPases recruit downstream effector proteins in order to transmit signals involved in membrane trafficking and signaling. The effector proteins include tether proteins that facilitate vesicle docking and fusion, as well as GEFs and GAPs for other RAB GTPases (Grosshans et al., 2006). The active state and membrane association of RAB GTPases are further controlled by auxiliary proteins (Barr and Lambright, 2010; Chaineau et al., 2013; Ishida et al., 2016; Müller and Goody, 2018). The selective and highly defined membrane trafficking processes mediated by RAB GTPases rely on efficient interactions with other effector proteins such as motor proteins (kinesins and dyneins), tethering complexes (EEA1, Golgins, and the exocyst complex), coatamer complex (COPI, COPII, and clathrin), and SNAREs (Grosshans et al., 2006). Notably, the interaction of RABs with Guanosine nucleotide dissociation inhibitor (GDI), GEFs, GAPs and other effector proteins is regulated via the post-translational modifications of RABs such as phosphorylation (Pfeffer, 2013, 2017; Lamber et al., 2019).
RABs are mainly located within different endosomes, where they systematically regulate different steps of endocytic trafficking independently or cooperatively. RAB4, RAB5, RAB7, RAB9, RAB10, RAB11, and RAB35 have been primarily associated with intracellular endocytic trafficking (Tsubokawa and Katayama, 1985; Zhang et al., 2022). RAB4 is predominantly located on early endosomes, and regulates the recycling of receptors from early endosomes directly back to the plasma membrane (fast recycling pathway) (Van Der Sluijs et al., 1991; Daro et al., 1996). In contrast, RAB11 is associated with the slow recycling pathway, and is localized in the recycling endosome, promoting the redistribution of receptors from the recycling endosome to the plasma membrane (Ullrich et al., 1996; Ren et al., 1998). RAB5 plays an important role in regulating vesicle transport from the plasma membrane to the early endosome by fostering the fusion of newly formed endocytic vesicles with early endosomes, as well as promoting the fusion between early endosomes (Hoffenberg et al., 2000; Woodman, 2000). RAB7 is a late endosome/lysosome-associated small GTPase that participates in crucial endocytic processes (Langemeyer et al., 2018). It is involved in several regulatory mechanisms linked to sorting of endosomes, biogenesis of lysosome, and in the formation of phagosomes, autophagosomes, and other lysosome-associated organelles (Gutierrez et al., 2004; Guerra and Bucci, 2016). RAB7 modulates lysosomal biogenesis by associating with the RAB-interacting lysosomal protein (RILP) and affects the morphology and spatial distribution of lysosomes by regulating the cytoskeleton (Cantalupo et al., 2001; Wang et al., 2004; Figure 3).
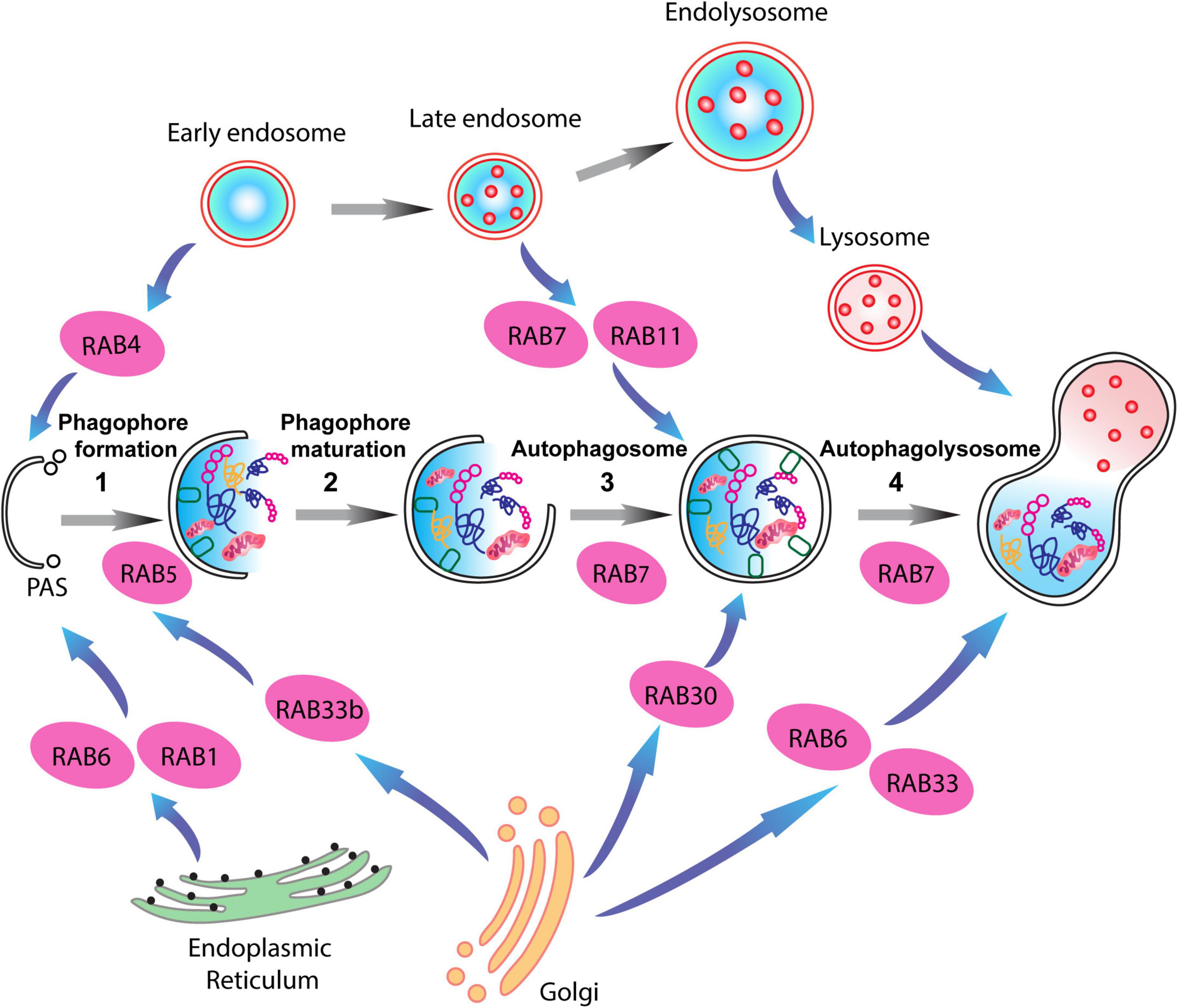
Figure 3. RAB proteins involved in different steps of the autophagy-lysosome pathway (ALP). Schematic representation of RAB GTPases and their binding partners in the ALP. RAB1, RAB6, and RAB4 are important for the formation of the pre-autophagosomal structure (PAS) providing membrane sources for the initiation of the autophagy pathway, while RAB5, RAB30, RAB33 are involved in autophagosome maturation together with RAB7, which is also involved in the fusion of the autophagosomes with late endosomes.
Notably, multiple RAB proteins are involved in the various stages of autophagic activity. RAB1, RAB4, and RAB11 regulate membrane trafficking originating in different organelles and direct them to the phagophore assembly site (Zoppino et al., 2010; Longatti and Tooze, 2012; Talaber et al., 2014). While, RAB5 interactions with the BECN1/PI3KC3 complex regulates the nucleation of the phagophore membranes (Morris et al., 2015; Pavlinov et al., 2020). RAB33 associates with Atg16L1, thus enlarging phagophore membrane area in a GTP-dependent manner (Itoh et al., 2008). Lastly, late endosome associated RAB proteins, RAB7, and RAB24, mediate the fusion of autophagosomes with lysosomes, and the clearance of autophagic compartments during basal conditions (Gutierrez et al., 2004; Ylä-Anttila et al., 2015; Ylä-Anttila and Eskelinen, 2018; Li et al., 2023). Importantly, a link between synaptic vesicle recycling and autophagy has been demonstrated via the interaction of synaptic vesicle-associated RAB26 with phagophore elongation factor Atg16L1. This leads to the targeting of synaptic vesicles to phagophores for bulk degradation (Binotti et al., 2015; Lüningschrör et al., 2017).
Differentially Expressed in Normal and Neoplastic cells-containing proteins primarily function as GEFs or GAPs for RAB GTPases (Yoshimura et al., 2010; Marat et al., 2011; Wu et al., 2011). They can also function as ADP-ribosylation factors (ARFs) (Su et al., 2020), and as Ras-related GTP binding proteins (RRAGs) (Müller and Goody, 2018; Lawrence et al., 2019). As C9ORF72 also belongs to the DENN family of proteins, it is likely that C9ORF72 serves as a GEF or GAP for multiple RAB GTPases (Levine et al., 2013a; Tang et al., 2020). Moreover, C9ORF72 not only interacts with several RAB molecules and key modulators of membrane trafficking (Tang, 2016; Boivin et al., 2020), but also because of its complex and multifaceted interactions, C9ORF2 protein harbors the potential to regulate autophagy at multiple steps. The identification of an unexpected interaction between C9ORF72 and RAB1A, which is involved in autophagy initiation, led to the discovery of a novel function for C9ORF72 protein, as an adaptor protein between RAB1A and the ULK1 complex. This interaction enables the translocation of the ULK1 complex to predetermined sites of autophagosome formation and links C9ORF72 in initial steps of autophagy (Webster et al., 2016). RAB5A is an important determinant in the process of autophagy initiation, and is mainly involved in delivering PtdIns3K complex and ATG7/ATG5/LC3 lipid complex to the newly formed phagophore (Li et al., 2013; Lin et al., 2016). An interaction between RAB5 and C9ORF72 has been identified in human motor neurons (Farg et al., 2017; Shi et al., 2018). Notably, a significant reduction in autophagosome numbers due to deficits in autophagosome formation was observed in C9ORF72-ALS patient-derived motor neurons (Shi et al., 2018). The elimination of C9ORF72 haploinsufficiency via the over expression of C9ORF72 protein, or the expression of a constitutively active RAB5A construct, could rescue the autophagosome formation deficits and enhance the survival of C9ORF72 neurons. These experiments convincingly implicate a critical role for this interaction in the cell clearance machinery as well as causally linking autophagosome formation deficits in the pathogenesis of C9ORF72-ALS-FTD (Shi et al., 2018).
Importantly, the C9ORF72-SMCR8-WDR41 complex confers GEF activity on RAB8A and RAB39B as well as GAP activity on RAB8A and RAB11A, thus playing a role in the activation of autophagy, and controlling autophagic flux (Yang et al., 2016). Since, RAB8A and RAB39B interact with other ALS-associated proteins such as TBK1, OPTN, and SQSTM1/p62, this suggests that autophagy dysfunctions are linked with the ALS pathogenesis (Corbier and Sellier, 2017). As expected, the knockdown of C9ORF72 in neurons led to impaired autophagy and concomitant accumulation of TARDBP and SQSTM1/p62 aggregates, recapitulating histopathological hallmarks of ALS-FTD (Sellier et al., 2016). It is now established that TBK1, SMCR8, and the C9ORF72 complex together with RAB39B belong to a common pathway regulating autophagy. This finding was inferred from experiments showing that TBK1 phosphorylates SMCR8, and the reduction in TBK1 expression levels can be normalized either by employing phosphomimetic mutants of SMCR8 or by over expressing the constitutively active version of RAB39B (Sellier et al., 2016). An additional role of the complex can be deduced from studies showing that the C9ORF72 complex regulates ARF GTPases and RAG GTPases. This regulation is mainly through SMCR8 which possesses GAP activity toward ARF1 and ARF6 GTPases, thereby regulating endocytic and Golgi trafficking (Sivadasan et al., 2016; Wang M. et al., 2020; Su et al., 2021). RAB8A can actively recruit TBK1, a protein kinase, which is then involved in the dual regulation of antiviral innate immunity and autophagy. Although TBK1 does not exhibit any kinase activity for C9ORF72, TBK1 phosphorylates SMCR8, ULK1, OPTN, and SQSTM1/p62. This in turn promotes the interaction with LC3, thereby facilitating the autophagic clearance of pathogenic proteins (Freischmidt et al., 2015; Heo et al., 2015; Yang et al., 2016; Harding et al., 2021). Recently, it was identified that TBK1 is hyperphosphorylated and sequestered within PolyGA aggregates, leading to the loss of TBK1 activity, enhancing TARDBP aggregation and triggering neurodegeneration in C9ORF72 ALS-FTD (Shao et al., 2022). See Table 1 describing the involvement of RAB proteins in ALP and the association of C9ORF72 proteins with specific RAB proteins. C9ORF72 haploinsufficiency leads to dysfunctional ALP, which accelerates the accumulation of toxic DPRs (Zhu et al., 2020; Beckers et al., 2021). Notably, DPRs such as PolyGA compromise neuronal proteostasis by accumulating within 26S proteasome and stalling cellular degradation (Guo et al., 2018). These observations indicate that, within the context of C9ORF72 ALS-FTD, the cross talk between ALP and UPS might be impaired, and that targeting both processes simultaneously could be a beneficial approach in treating C9ORF72 ALS-FTD.
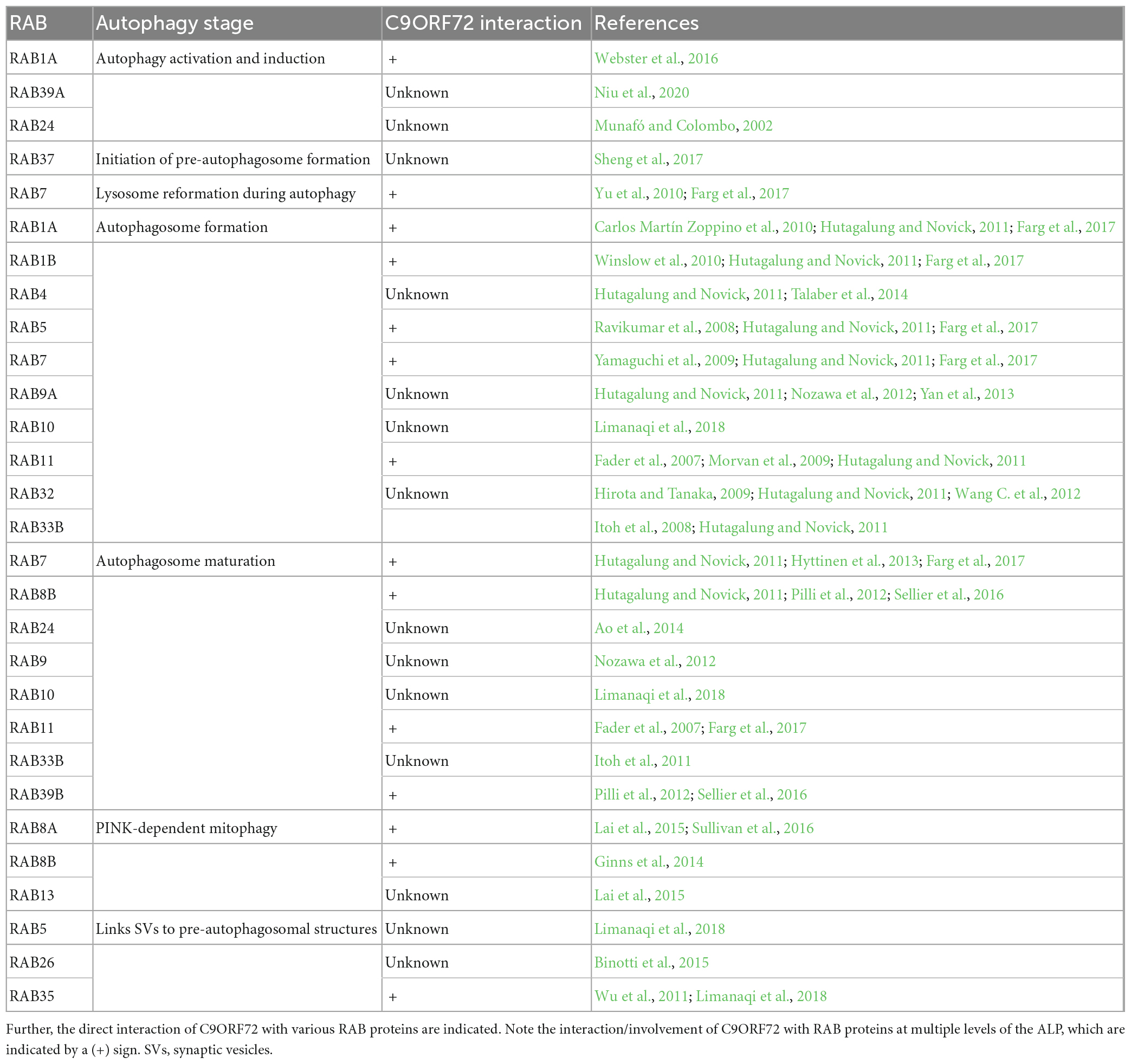
Table 1. List of RAB proteins involved in the autosomal-lysosomal pathway (ALP).
In the context of mTORC1 signaling, a reduction in the phosphorylation of RPS6KB1/p70S6K has been established in C9ORF72 knockdown models, implicating reduced mTORC1 activation (Wang M. et al., 2020). Any reduction in mTORC1 signaling leads to the nuclear translocation of transcription factors TFEB and TFE3. This translocation enhances transcription of genes associated with autophagy and lysosomal pathways, thus causing an excessive autophagic flux (Tang, 2016; Ji et al., 2017). Overall, loss of C9ORF72 function can synergistically exert dual responses on the regulation of autophagy. Firstly, by reducing mTORC1 signaling, thereby over-activating autophagy. Secondly, by reduced interaction with RAB GTPases due to C9ORF72 haploinsufficiency, thus causing impairment in vesicular trafficking. This impairment in trafficking hinders the proper functioning of the endosomal lysosomal pathways.
Physiological and pathological roles of C9ORF72 in lysosomal pathways
It is well established that deficits in lysosomal function and lysosomal biogenesis can invariably affect autophagic turnover and enhance cellular accumulation of autophagosomes. Considering that C9ORF72 has been reported to display GEF activity toward GTPases, a novel interaction of C9ORF72 via its DENN domain with inactive RRAG GTPases was identified, suggesting a novel role for C9ORF72 in nutrient sensing and lysosomal biogenesis (Martina and Puertollano, 2013; Corbier and Sellier, 2017; Fukuda and Shiozaki, 2018). During episodes of cellular starvation, the inactivation of mTORC1, results in the loss of the inhibitory effect of mTORC1 on ULK1. This causes the release of the ULK1 complex and the recruitment of the C9ORF72 complex to the lysosomes by SLC66A1. The movement of C9ORF72 to the lysosome is mediated via the interaction between SLC66A1 and WDR41 (Amick and Ferguson, 2017). Notably, an accumulation of enlarged lysosomes together with elevated expression of SQSTM1/p62 and LC3 has been observed in rodent C9ORF72 knock out macrophages, microglia and patient-derived motor neurons, suggestive of an increase in autophagy and/or a deficit in the clearance of autophagosomes by lysosomes (O’Rourke et al., 2016; Sullivan et al., 2016; Ugolino et al., 2016). Moreover, mice lacking C9ORF72 also exhibited an increased number and size of lysosomes within neurons, indicating increased lysosomal biogenesis as well as the accumulation of dysfunctional lysosomes (Shao et al., 2020). The well characterized accumulation of enlarged autophagosomes in C9ORF72 loss of function models has now been linked to M6PR vesicle trafficking (Saftig and Klumperman, 2009; Banik et al., 2020; Beckers et al., 2021). These vesicles bud from the trans-Golgi network and are actively involved in the transport of M6P-tagged lysosomal proteins to endosomes, thereby sustaining lysosomal biogenesis (Saftig and Klumperman, 2009). Intriguingly, in C9ORF72 knock out cells, M6PR is mislocalized within the cytoplasm in comparison to its correct perinuclear localization, suggesting that altered M6PR function within cells accounts for the observed changes in lysosomal biogenesis.
C9ORF72 and SMCR8 have been shown to play a role in regulating lysosomal exocytosis, involving the fusion of the lysosome with the plasma membrane, followed by the release of lysosomal contents (Medina et al., 2011). Likewise, the level of LAMP1 is increased in C9ORF72 or SMCR8-deficient macrophages, suggesting an increase in lysosomal exocytosis (Zhang et al., 2018; Shao et al., 2020). This enhanced lysosomal exocytosis is further consolidated by the presence of elevated extracellular levels of lysosome enzymes: cathepsin B and cathepsin D in C9ORF72-deficient cells (Shao et al., 2020). Moreover, C9ORF72 is implicated in the process of lysosomal targeting of CARM1, a histone arginine methyltransferase known to work as an epigenetic regulator of autophagy. During conditions of starvation, C9ORF72 interacts with CARM1, and facilitates its transport to the lysosome for degradation. C9ORF72 deficiency or reduction leads to cytoplasmic accumulation of CARM1, which translocates to the nucleus and functions as a transcriptional co-activator of autophagic and lysosomal genes such as Atg14 and Map1lc3b. CARM1 has been shown to be highly enriched on the promoters regulating autophagy and lysosomal genes in C9ORF72-deficient cells. This observation suggests that C9ORF72 haploinsufficiency leads to the dysregulated expression of genes involved in autophagy and lysosomal pathways (Liu and Wang, 2019). Importantly, several studies indicate a role for C9ORF72 in nutrient-sensing within the lysosome, independent of mTORC1 signaling (Ugolino et al., 2016; Amick et al., 2020; Pang and Hu, 2021). Thus, the recruitment of the C9ORF72-SMCR8-WDR41 complex to the lysosomes might be an active component of the lysosome-associated pathway, regulating the cellular response to nutrient variation (Amick et al., 2018). Additionally, the recruitment of C9ORF72 complex to the lysosome also inadvertently facilitates the interplay with mTORC1 (Wang M. et al., 2020). Notably, these two processes involving the C9ORF72-related network may reflect functions that parallel each other.
Therapeutic strategies for targeting ALP in C9ORF72-ALS-FTD
The lack of preventive or curative therapy for ALS-FTD, makes the targeting of ALP an attractive option. Given the strong evidence for disrupted ALP in all major forms of NDs, therapeutic strategies aimed at restoring autophagy deficits represent a common hub that would be beneficial to all NDs. One likely direction involves the early activation of autophagy, thereby enhancing the clearance of aggregated proteins. Notably, the role of rapamycin, one of the most well-known autophagy activators, is still debatable as it acts on multiple systems including the immune system. Different studies have reported both neuroprotective and detrimental effects when tested in vivo on various ALS animal models (Zhang et al., 2011; Wang I et al., 2012). Several studies have employed small-molecule autophagy activators such as trehalose, lithium, berberine, promethazine, and carbamazepine to achieve neuroprotection in ALS-FTD (Boivin et al., 2020; Lu et al., 2022). Other well-known autophagy-activating compounds, such as lithium, valproic acid, or carbamazepine, are already being employed in the clinics due to their positive effect on mood-stabilization. This class of compounds mainly act on inositol triphosphate (IP3) metabolism via the inhibition of PI3K and GSK-3β. IP3 acts on its receptor IP3R located at the ER thus promoting calcium release and ATP production that counteracts autophagic flux. Therefore the usage of mood stabilizing drugs, such as lithium, depletes IP3 viability and thus effectively boosts the autophagy process (Sarkar et al., 2005; Harris and Rubinsztein, 2012).
Furthermore, histone deacetylase 6 (HDAC6) has a relevant role in the process of fusion between autophagosomes and lysosomes and its therapeutical effect has been tested in animal models of superoxide dismutase 1 (SOD1)-linked ALS, where its genetic inhibition prolonged the lifespan of the animals (Taes et al., 2013). HDAC6 inhibitory effect in ameliorating ALS-FTD pathology has been tested in C9ORF72 mutation, where its inhibition reduced the accumulation of PolyGA in vivo (del Rosso et al., 2022). Interestingly, Metformin, an anti-hypertensive drug has been found to induce autophagy via both AMPK-dependent and SIRT1-dependent pathways and was employed in alleviating C9ORF72-linked DPR pathology in C9-500 BAC mouse model (Song et al., 2015; Zu et al., 2020). As Polito et al. (2014) have demonstrated, the overexpression of TFEB in AD rodent models inhibited neurofibrillary tangle pathology and improved neuronal survival and function. Yet another approach would be to enhance nuclear TFEB levels by reducing its nuclear export by selective inhibitors of nuclear export (SINEs) (McDermott, 2019). Several compounds that regulate the nuclear translocation of TFEB, such as 3,4-dimethoxychalcone (3,4-DC), (Chen et al., 2019) 2-Hydroxypropyl-β-cyclodextrin (HPβCD), (Song et al., 2014), Digoxin, proscillaridin A, and digoxigenin (Wang et al., 2017) have been employed. However, most of these compounds also modulate calcium signaling or mTORC1 or AKT, or PKC signaling pathways, thus it remains unclear whether the effects are directly TFEB specific (McMillan and Dwyer, 1989; Tong and Song, 2015).
Another approach would be to reduce DPRs burden within motor neurons. One method involves the overexpression of heat shock protein B8 (HSPB8), which led to the increase in the autophagy-mediated degradation of DPRs, and enabled the clearance of aggregation prone proteins before they could convert into inclusions (Liu et al., 2022). Furthermore, an interesting natural compound called berberine has been shown to influence TDP-43 protein aggregations boosting its degradation via autophagy (Chang et al., 2016). It is crucial to take into consideration the precise stage of the autophagy pathway on which the active compounds and drugs are acting. It has been reported that different active molecules like tamoxifen, progesterone, bosutinib, and lithium all exhibited beneficial effect in ameliorating ALS disease progression in vivo, because of increased autophagic flux (Figure 4). Nevertheless, other molecules (i.e., rapamycin and pimozide), despite increasing autophagy concomitantly, suppressed autophagy-mediated degradation. This resulted in the accumulation of autophagosomes and autophagy-related substrates such as misfolded protein aggregates together with SQSTM1/p62 (Fornai et al., 2008; Zhang et al., 2011; Wang I et al., 2012; Kim et al., 2013; Pozzi et al., 2018). Several studies have established a beneficial role for the overexpression of Parkin on α-synuclein toxicity (Shimura et al., 2001; Petrucelli et al., 2002; Rana et al., 2013; Meng et al., 2020). Notably, Parkin prevented the accumulation of SOD1 aggregates in ALS. In particular, Parkin fostered the Lys63-linked polyubiquitination of misfolded SOD1, promoting the clearance of aggregation-prone SOD1 through elevated autophagy (Yung et al., 2016). Given the beneficial effect of Parkin, several small molecules serving as activators of Parkin have been discovered which could be therapeutically employed for the clearance of toxic aggregates via autophagy (Miller and Muqit, 2019; Clark et al., 2021).
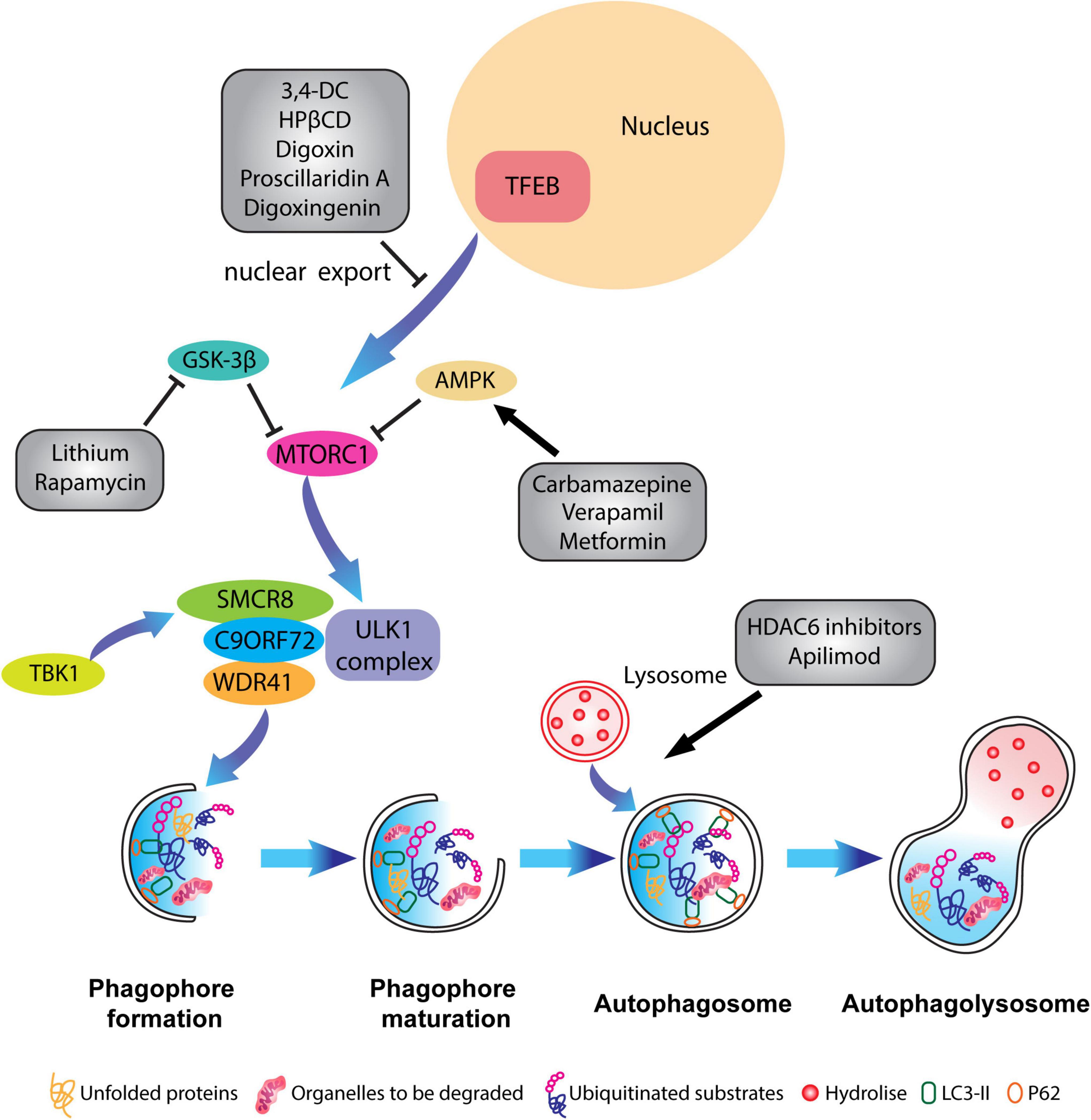
Figure 4. Pharmacological targeting of the autophagy pathway in neurons. Schematic representation of different compounds that have been used to alleviate ALP impairments and to restore autophagic flux. Some therapeutical agents promote the activation or inhibition of specific kinases which block mTORC1 activity such as Carbamazepine, Verapamil and Metformin, or inhibit the translocation of TFEB from the nucleus to the cytoplasm like in the case of 3,4-dimethoxychalcone (3,4-DC), 2-Hydroxypropyl-β-cyclodextrin (HPβCD), Digoxin, proscillaridin A. Other molecules such as HADC6 inhibitors and apilimod promote the fusion of lysosomes with the autophagosome.
Lastly, within the context of C9ORF72-ALS-FTD, targeting RAB GTPases could serve as a potential therapeutic strategy, as RAB GTPases are extensively involved in the biogenesis and function of endosomal-lysosomal systems (Langemeyer et al., 2018). PIKFYVE is a kinase that converts PtdIns3P to PtdIns (3, 5) P2, thereby inhibiting the fusion between autophagosomes and lysosomes. Inhibition of PIKFYVE kinase by Apilimod counteracts and promotes the fusion of lysosomes and autophagosomes, thereby normalizing lysosomal and autophagosomal deficits in C9ORF72 patient-derived motor neurons (Shi et al., 2018, 2019). RAB7 receptor antagonist CID1067700 inhibits the excessive transportation of cathepsin B from late endosomes to lysosomes, sustaining lysosomal membrane permeability and thereby reducing reactive astrogliosis in brain injury models (Qin et al., 2019). Although, lack of specificity toward RABs has been the biggest hinderance in the development of RAB-selective antagonists, viral-mediated expression of defined dominant negative/constitutively active RABs might enable the restoration of dysfunctional endosomal-lysosomal pathways.
Conclusion
It is important to understand the precise steps and function of ALP before attempting to implement it as a therapeutic procedure that potentially may lead to breakthroughs in tackling NDs. In the case of C9ORF72-ALS-FTD, the complex and multi-layered function of the protein in regulating ALP as well as endosomal trafficking has just begun to emerge, and further studies are required to dissect its precise function. The presence of C9ORF72 haploinsufficiency coupled together with the gain of function mechanisms involving both toxic DPRs and HRE further complicates the understanding of these events in the pathophysiology of C9ORF72-ALS-FTD. Lastly, the detailed understanding of processes such as phosphorylation, which regulates the initiation as well as the completion of the ALP, is required for future modulation of autophagy. Despite, this tangled scenario, it has become well established that ALP signaling is disrupted in NDs and especially in C9ORF72-ALS-FTD, and thus modulating or enhancing ALP might exert a beneficial outcome on the disease pathogenesis. Lastly such approaches might serve as an attractive entry point for future druggable targets.
Author contributions
SS wrote the review with equal contributions from FP and RD. FP designed all figures. All authors read and approved the submitted version.
Funding
This work was supported by the Marie Skłodowska-Curie actions (MSCA) Innovative, Training Network (ITN)- SAND funded through the H2020 program of the European Union, European Research Council (ERC) under the European Union’s Horizon 2020 Research and Innovation Program (grant agreement #725825), and Swiss National Science Foundation. Open access funding was provided by the University of Bern.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Amick, J., and Ferguson, S. M. (2017). C9orf72: At the intersection of lysosome cell biology and neurodegenerative disease. Traffic 18, 267–276. doi: 10.1111/tra.12477
Amick, J., Roczniak-Ferguson, A., and Ferguson, S. M. (2016). C9orf72 binds SMCR8, localizes to lysosomes, and regulates mTORC1 signaling. Mol. Biol. Cell 27, 3040–3051. doi: 10.1091/mbc.E16-01-0003
Amick, J., Tharkeshwar, A. K., Amaya, C., and Ferguson, S. M. (2018). WDR41 supports lysosomal response to changes in amino acid availability. Mol. Biol. Cell 29, 2213–2227. doi: 10.1091/mbc.E17-12-0703
Amick, J., Tharkeshwar, A. K., Talaia, G., and Ferguson, S. M. (2020). PQLC2 recruits the C9orf72 complex to lysosomes in response to cationic amino acid starvation. J. Cell Biol. 219:e201906076. doi: 10.1083/jcb.201906076
Andersen, P. M. (2013). ALS and FTD: Two sides of the same coin? Lancet Neurol. 12, 937–938. doi: 10.1016/S1474-4422(13)70218-7
Ao, X., Zou, L., and Wu, Y. (2014). Regulation of autophagy by the Rab GTPase network. Cell Death Differ. 21, 348–358. doi: 10.1038/cdd.2013.187
Aoki, Y., Manzano, R., Lee, Y., Dafinca, R., Aoki, M., Douglas, A. G. L., et al. (2017). C9orf72 and RAB7L1 regulate vesicle trafficking in amyotrophic lateral sclerosis and frontotemporal dementia. Brain 140, 887–897. doi: 10.1093/brain/awx024
Avenali, M., Blandini, F., and Cerri, S. (2020). Glucocerebrosidase defects as a major risk factor for Parkinson’s disease. Front. Aging Neurosci. 12:97. doi: 10.3389/fnagi.2020.00097
Awad, O., Sarkar, C., Panicker, L. M., Miller, D., Zeng, X., Sgambato, J. A., et al. (2015). Altered TFEB-mediated lysosomal biogenesis in Gaucher disease iPSC-derived neuronal cells. Hum. Mol. Genet. 24, 5775–5788. doi: 10.1093/hmg/ddv297
Bagaria, J., Bagyinszky, E., and An, S. S. A. (2022). Genetics of autosomal recessive spastic ataxia of charlevoix-saguenay (ARSACS) and role of sacsin in neurodegeneration. Int. J. Mol. Sci. 23:552. doi: 10.3390/ijms23010552
Balendra, R., and Isaacs, A. M. (2018). C9orf72-mediated ALS and FTD: Multiple pathways to disease. Nat. Rev. Neurol. 14, 544–558. doi: 10.1038/s41582-018-0047-2
Banik, S. M., Pedram, K., Wisnovsky, S., Ahn, G., Riley, N. M., and Bertozzi, C. R. (2020). Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature 584, 291–297. doi: 10.1038/s41586-020-2545-9
Baron, O., and Fanto, M. (2018). Karyoptosis: A novel type of cell death caused by chronic autophagy inhibition. Autophagy 14, 722–723. doi: 10.1080/15548627.2018.1434372
Baron, O., Boudi, A., Dias, C., Schilling, M., Nölle, A., Vizcay-Barrena, G., et al. (2017). Stall in canonical autophagy-lysosome pathways prompts nucleophagy-based nuclear breakdown in neurodegeneration. Curr. Biol. 27, 3626–3642.e6. doi: 10.1016/j.cub.2017.10.054
Barr, F., and Lambright, D. G. (2010). Rab GEFs and GAPs. Curr. Opin. Cell Biol. 22, 461–470. doi: 10.1016/j.ceb.2010.04.007
Bar-Yosef, T., Damri, O., and Agam, G. (2019). Dual role of autophagy in diseases of the central nervous system. Front. Cell Neurosci. 13:196. doi: 10.3389/fncel.2019.00196
Batool, S., Raza, H., Zaidi, J., Riaz, S., Hasan, S., and Syed, N. I. (2019). Synapse formation: From cellular and molecular mechanisms to neurodevelopmental and neurodegenerative disorders. J. Neurophysiol. 121, 1381–1397. doi: 10.1152/jn.00833.2018
Beckers, J., Tharkeshwar, A. K., and Van Damme, P. (2021). C9orf72 ALS-FTD: Recent evidence for dysregulation of the autophagy-lysosome pathway at multiple levels. Autophagy 17, 3306–3322. doi: 10.1080/15548627.2021.1872189
Behrends, C., Sowa, M. E., Gygi, S. P., and Harper, J. W. (2010). Network organization of the human autophagy system. Nature 466, 68–76. doi: 10.1038/nature09204
Binotti, B., Pavlos, N. J., Riedel, D., Wenzel, D., Vorbrüggen, G., Schalk, A. M., et al. (2015). The GTPase Rab26 links synaptic vesicles to the autophagy pathway. eLife 4:e05597. doi: 10.7554/eLife.05597
Boivin, M., Pfister, V., Gaucherot, A., Ruffenach, F., Negroni, L., Sellier, C., et al. (2020). Reduced autophagy upon C9ORF72 loss synergizes with dipeptide repeat protein toxicity in G4C2 repeat expansion disorders. EMBO J. 39:e100574. doi: 10.15252/embj.2018100574
Boland, B., Kumar, A., Lee, S., Platt, F. M., Wegiel, J., Yu, W. H., et al. (2008). Autophagy induction and autophagosome clearance in neurons: Relationship to autophagic pathology in Alzheimer’s disease. J. Neurosci. 28, 6926–6937. doi: 10.1523/JNEUROSCI.0800-08.2008
Bossy, B., Perkins, G., and Bossy-Wetzel, E. (2008). Clearing the brains cobwebs: The role of autophagy in neuroprotection. Curr. Neuropharmacol. 6, 97–101. doi: 10.2174/157015908784533897
Brighouse, A., Dacks, J. B., and Field, M. C. (2010). Rab protein evolution and the history of the eukaryotic endomembrane system. Cell Mol. Life Sci. 67, 3449–3465. doi: 10.1007/s00018-010-0436-1
Brooker, S. M., and Krainc, D. (2021). Glucocerebrosidase dysfunction in neurodegenerative disease. Essays Biochem. 65, 873–883. doi: 10.1042/EBC20210018
Buchman, V. L., Cooper-Knock, J., Connor-Robson, N., Higginbottom, A., Kirby, J., Razinskaya, O. D., et al. (2013). Simultaneous and independent detection of C9ORF72 alleles with low and high number of GGGGCC repeats using an optimised protocol of Southern blot hybridisation. Mol. Neurodegener. 8:12. doi: 10.1186/1750-1326-8-12
Caccamo, A., Majumder, S., Richardson, A., Strong, R., and Oddo, S. (2010). Molecular interplay between mammalian target of rapamycin (mTOR), Amyloid-β, and Tau. J. Biol. Chem. 285, 13107–13120. doi: 10.1074/jbc.M110.100420
Cali, C. P., Patino, M., Tai, Y. K., Ho, W. Y., McLean, C. A., Morris, C. M., et al. (2019). C9orf72 intermediate repeats are associated with corticobasal degeneration, increased C9orf72 expression and disruption of autophagy. Acta Neuropathol. 138, 795–811. doi: 10.1007/s00401-019-02045-5
Cantalupo, G., Alifano, P., Roberti, V., Bruni, C. B., and Bucci, C. (2001). Rab-interacting lysosomal protein (RILP): The Rab7 effector required for transport to lysosomes. EMBO J. 20, 683–693. doi: 10.1093/emboj/20.4.683
Carlos Martín Zoppino, F., Damián Militello, R., Slavin, I., Álvarez, C., and Colombo, M. I. (2010). Autophagosome formation depends on the small GTPase Rab1 and functional ER exit sites. Traffic 11, 1246–1261. doi: 10.1111/j.1600-0854.2010.01086.x
Chaineau, M., Ioannou, M. S., and McPherson, P. S. (2013). Rab35: GEFs, GAPs and effectors. Traffic 14, 1109–1117. doi: 10.1111/tra.12096
Chandarlapaty, S., Sawai, A., Scaltriti, M., Rodrik-Outmezguine, V., Grbovic-Huezo, O., Serra, V., et al. (2011). AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell 19, 58–71. doi: 10.1016/j.ccr.2010.10.031
Chang, C.-F., Lee, Y.-C., Lee, K.-H., Lin, H.-C., Chen, C.-L., Shen, C.-K. J., et al. (2016). Therapeutic effect of berberine on TDP-43-related pathogenesis in FTLD and ALS. J. Biomed. Sci. 23:72. doi: 10.1186/s12929-016-0290-z
Chang, J., Lee, S., and Blackstone, C. (2014). Spastic paraplegia proteins spastizin and spatacsin mediate autophagic lysosome reformation. J. Clin. Invest. 124, 5249–5262. doi: 10.1172/JCI77598
Chen, G., Xie, W., Nah, J., Sauvat, A., Liu, P., Pietrocola, F., et al. (2019). 3,4-Dimethoxychalcone induces autophagy through activation of the transcription factors TFE3 and TFEB. EMBO Mol. Med. 11:e10469. doi: 10.15252/emmm.201910469
Chen, X., Li, Z., Yang, C., Tang, J., Lan, H., and Liu, H. (2021). Lysosome depletion-triggered autophagy impairment in progressive kidney injury. Kidney Dis. 7, 254–267. doi: 10.1159/000515035
Chen, Y., Wang, K., Di, J., Guan, C., Wang, S., Li, Q., et al. (2021). Mutation of the BAG-1 domain decreases its protective effect against hypoxia/reoxygenation by regulating HSP70 and the PI3K/AKT signalling pathway in SY-SH5Y cells. Brain Res. 1751:147192. doi: 10.1016/j.brainres.2020.147192
Chen, Z., Nie, S.-D., Qu, M.-L., Zhou, D., Wu, L.-Y., Shi, X.-J., et al. (2018). The autophagic degradation of Cav-1 contributes to PA-induced apoptosis and inflammation of astrocytes. Cell Death Dis. 9:771. doi: 10.1038/s41419-018-0795-3
Chua, J. P., De Calbiac, H., Kabashi, E., and Barmada, S. J. (2022). Autophagy and ALS: Mechanistic insights and therapeutic implications. Autophagy 18, 254–282. doi: 10.1080/15548627.2021.1926656
Ciura, S., Sellier, C., Campanari, M.-L., Charlet-Berguerand, N., and Kabashi, E. (2016). The most prevalent genetic cause of ALS-FTD, C9orf72 synergizes the toxicity of ATXN2 intermediate polyglutamine repeats through the autophagy pathway. Autophagy 12, 1406–1408. doi: 10.1080/15548627.2016.1189070
Clark, E. H., Vázquez de la Torre, A., Hoshikawa, T., and Briston, T. (2021). Targeting mitophagy in Parkinson’s disease. J. Biol. Chem. 296:100209. doi: 10.1074/jbc.REV120.014294
Clausen, T. H., Lamark, T., Isakson, P., Finley, K., Larsen, K. B., Brech, A., et al. (2010). p62/SQSTM1 and ALFY interact to facilitate the formation of p62 bodies/ALIS and their degradation by autophagy. Autophagy 3, 330–344. doi: 10.4161/auto.6.3.11226
Clayton, E. L., Mizielinska, S., Edgar, J. R., Nielsen, T. T., Marshall, S., Norona, F. E., et al. (2015). Frontotemporal dementia caused by CHMP2B mutation is characterised by neuronal lysosomal storage pathology. Acta Neuropathol. 130, 511–523. doi: 10.1007/s00401-015-1475-3
Corbier, C., and Sellier, C. (2017). C9ORF72 is a GDP/GTP exchange factor for Rab8 and Rab39 and regulates autophagy. Small GTPases 8, 181–186. doi: 10.1080/21541248.2016.1212688
Cox, L. E., Ferraiuolo, L., Goodall, E. F., Heath, P. R., Higginbottom, A., Mortiboys, H., et al. (2010). Mutations in CHMP2B in lower motor neuron predominant amyotrophic lateral sclerosis (ALS). PLoS One 5:e9872. doi: 10.1371/journal.pone.0009872
Csizmadia, T., and Juhász, G. (2020). Crinophagy mechanisms and its potential role in human health and disease. Prog. Mol. Biol. Transl. Sci. 172, 239–255. doi: 10.1016/bs.pmbts.2020.02.002
D’Adamo, P., Masetti, M., Bianchi, V., Morè, L., Mignogna, M. L., Giannandrea, M., et al. (2014). RAB GTPases and RAB-interacting proteins and their role in the control of cognitive functions. Neurosci. Biobehav. Rev. 46(Pt 2), 302–314. doi: 10.1016/j.neubiorev.2013.12.009
Daro, E., van der Sluijs, P., Galli, T., and Mellman, I. (1996). Rab4 and cellubrevin define different early endosome populations on the pathway of transferrin receptor recycling. Proc. Natl. Acad. Sci. U.S.A. 93, 9559–9564. doi: 10.1073/pnas.93.18.9559
Decressac, M., Mattsson, B., Weikop, P., Lundblad, M., Jakobsson, J., and Björklund, A. (2013). TFEB-mediated autophagy rescues midbrain dopamine neurons from α-synuclein toxicity. Proc. Natl. Acad. Sci. U.S.A. 110, E1817–E1826. doi: 10.1073/pnas.1305623110
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-Linked FTD and ALS. Neuron 72, 245–256. doi: 10.1016/j.neuron.2011.09.011
del Rosso, G., Carlomagno, Y., Todd, T. W., Jones, C. Y., Prudencio, M., Daughrity, L. M., et al. (2022). HDAC6 interacts with poly (GA) and modulates its accumulation in c9FTD/ALS. Front. Cell Dev. Biol. 9:809942. doi: 10.3389/fcell.2021.809942
Demishtein, A., Fraiberg, M., Berko, D., Tirosh, B., Elazar, Z., and Navon, A. (2017). SQSTM1/p62-mediated autophagy compensates for loss of proteasome polyubiquitin recruiting capacity. Autophagy 13, 1697–1708. doi: 10.1080/15548627.2017.1356549
Deng, Z., Lim, J., Wang, Q., Purtell, K., Wu, S., Palomo, G. M., et al. (2020). ALS-FTLD-linked mutations of SQSTM1/p62 disrupt selective autophagy and NFE2L2/NRF2 anti-oxidative stress pathway. Autophagy 16, 917–931. doi: 10.1080/15548627.2019.1644076
Di Meco, A., Curtis, M. E., Lauretti, E., and Praticò, D. (2020). Autophagy dysfunction in Alzheimer’s disease: Mechanistic insights and new therapeutic opportunities. Biol. Psychiatry 87, 797–807. doi: 10.1016/j.biopsych.2019.05.008
Dols-Icardo, O., Garcia-Redondo, A., Rojas-Garcia, R., Sanchez-Valle, R., Noguera, A., Gomez-Tortosa, E., et al. (2014). Characterization of the repeat expansion size in C9orf72 in amyotrophic lateral sclerosis and frontotemporal dementia. Hum. Mol. Genet. 23, 749–754. doi: 10.1093/hmg/ddt460
Dou, Z., Xu, C., Donahue, G., Shimi, T., Pan, J.-A., Zhu, J., et al. (2015). Autophagy mediates degradation of nuclear lamina. Nature 527, 105–109. doi: 10.1038/nature15548
Du, W., Su, Q. P., Chen, Y., Zhu, Y., Jiang, D., Rong, Y., et al. (2016). Kinesin 1 drives autolysosome tubulation. Dev. Cell 37, 326–336. doi: 10.1016/j.devcel.2016.04.014
Duan, W., Guo, M., Yi, L., Zhang, J., Bi, Y., Liu, Y., et al. (2019). Deletion of Tbk1 disrupts autophagy and reproduces behavioral and locomotor symptoms of FTD-ALS in mice. Aging 11, 2457–2476. doi: 10.18632/aging.101936
Dugger, B. N., and Dickson, D. W. (2017). Pathology of neurodegenerative diseases. Cold Spring Harb. Perspect. Biol. 9:a028035. doi: 10.1101/cshperspect.a028035
Elden, A. C., Kim, H.-J., Hart, M. P., Chen-Plotkin, A. S., Johnson, B. S., Fang, X., et al. (2010). Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 466, 1069–1075. doi: 10.1038/nature09320
Fader, C. M., Sánchez, D., Furlán, M., and Colombo, M. I. (2007). Induction of autophagy promotes fusion of multivesicular bodies with autophagic vacuoles in K562 Cells. Traffic 9, 230–250. doi: 10.1111/j.1600-0854.2007.00677.x
Fan, T., Huang, Z., Wang, W., Zhang, B., Xu, Y., Mao, Z., et al. (2018). Proteasome inhibition promotes autophagy and protects from endoplasmic reticulum stress in rat alveolar macrophages exposed to hypoxia-reoxygenation injury. J. Cell Physiol. 233, 6748–6758. doi: 10.1002/jcp.26516
Farg, M. A., Sundaramoorthy, V., Sultana, J. M., Yang, S., Atkinson, R. A. K., Levina, V., et al. (2017). C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum. Mol. Genet. 26, 4093–4094. doi: 10.1093/hmg/ddx309
Filimonenko, M., Isakson, P., Finley, K. D., Anderson, M., Jeong, H., Melia, T. J., et al. (2010). The selective macroautophagic degradation of aggregated proteins requires the PI3P-binding protein Alfy. Mol. Cell. 38, 265–279. doi: 10.1016/j.molcel.2010.04.007
Fornai, F., Longone, P., Cafaro, L., Kastsiuchenka, O., Ferrucci, M., Manca, M. L., et al. (2008). Lithium delays progression of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U.S.A. 105, 2052–2057. doi: 10.1073/pnas.0708022105
Francis, V., Alshafie, W., Kumar, R., Girard, M., Brais, B., and McPherson, P. S. (2022). The ARSACS disease protein sacsin controls lysosomal positioning and reformation by regulating microtubule dynamics. J. Biol. Chem. 298:102320. doi: 10.1016/j.jbc.2022.102320
Freischmidt, A., Wieland, T., Richter, B., Ruf, W., Schaeffer, V., Müller, K., et al. (2015). Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci. 18, 631–636. doi: 10.1038/nn.4000
Fukuda, T., and Shiozaki, K. (2018). The Rag GTPase-Ragulator complex attenuates TOR complex 1 signaling in fission yeast. Autophagy 14, 1105–1106. doi: 10.1080/15548627.2018.1444313
Gascon, E., Lynch, K., Ruan, H., Almeida, S., Verheyden, J. M., Seeley, W. W., et al. (2014). Alterations in microRNA-124 and AMPA receptors contribute to social behavioral deficits in frontotemporal dementia. Nat. Med. 20, 1444–1451. doi: 10.1038/nm.3717
Gelb, D. J., Oliver, E., and Gilman, S. (1999). Diagnostic criteria for Parkinson disease. Arch. Neurol. 56:33. doi: 10.1001/archneur.56.1.33
Ghazi-Noori, S., Froud, K. E., Mizielinska, S., Powell, C., Smidak, M., Fernandez de Marco, M., et al. (2012). Progressive neuronal inclusion formation and axonal degeneration in CHMP2B mutant transgenic mice. Brain 135, 819–832. doi: 10.1093/brain/aws006
Ginns, E. I., Mak, S. K.-K., Ko, N., Karlgren, J., Akbarian, S., Chou, V. P., et al. (2014). Neuroinflammation and α-synuclein accumulation in response to glucocerebrosidase deficiency are accompanied by synaptic dysfunction. Mol. Genet. Metab. 111, 152–162. doi: 10.1016/j.ymgme.2013.12.003
Grosshans, B. L., Ortiz, D., and Novick, P. (2006). Rabs and their effectors: Achieving specificity in membrane traffic. Proc. Natl. Acad. Sci. U.S.A. 103, 11821–11827. doi: 10.1073/pnas.0601617103
Guerra, F., and Bucci, C. (2016). Multiple roles of the small GTPase Rab7. Cells 5:34. doi: 10.3390/cells5030034
Guo, Q., Lehmer, C., Martínez-Sánchez, A., Rudack, T., Beck, F., Hartmann, H., et al. (2018). In situ structure of neuronal C9orf72 Poly-GA aggregates reveals proteasome recruitment. Cell 172, 696–705.e12. doi: 10.1016/j.cell.2017.12.030
Gutierrez, M. G., Munafó, D. B., Berón, W., and Colombo, M. I. (2004). Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J. Cell Sci. 117, 2687–2697. doi: 10.1242/jcs.01114
Haeusler, A. R., Donnelly, C. J., and Rothstein, J. D. (2016). The expanding biology of the C9orf72 nucleotide repeat expansion in neurodegenerative disease. Nat. Rev. Neurosci. 17, 383–395. doi: 10.1038/nrn.2016.38
Haeusler, A. R., Donnelly, C. J., Periz, G., Simko, E. A. J., Shaw, P. G., Kim, M.-S., et al. (2014). C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature 507, 195–200. doi: 10.1038/nature13124
Hara, T., Nakamura, K., Matsui, M., Yamamoto, A., Nakahara, Y., Suzuki-Migishima, R., et al. (2006). Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441, 885–889. doi: 10.1038/nature04724
Harding, O., Evans, C. S., Ye, J., Cheung, J., Maniatis, T., and Holzbaur, E. L. F. (2021). ALS- and FTD-associated missense mutations in TBK1 differentially disrupt mitophagy. Proc. Natl. Acad. Sci. U.S.A. 118:e2025053118. doi: 10.1073/pnas.2025053118
Harris, H., and Rubinsztein, D. C. (2012). Control of autophagy as a therapy for neurodegenerative disease. Nat. Rev. Neurol. 8, 108–117. doi: 10.1038/nrneurol.2011.200
Hasegawa, M., Arai, T., Nonaka, T., Kametani, F., Yoshida, M., Hashizume, Y., et al. (2008). Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann. Neurol. 64, 60–70. doi: 10.1002/ana.21425
Heo, J.-M., Ordureau, A., Paulo, J. A., Rinehart, J., and Harper, J. W. (2015). The PINK1-PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol. Cell 60, 7–20. doi: 10.1016/j.molcel.2015.08.016
Higuchi, M., Lee, V. M.-Y., and Trojanowski, J. Q. (2002). Tau and axonopathy in neurodegenerative disorders. Neuromol. Med. 2, 131–150.
Hirota, Y., and Tanaka, Y. (2009). A small GTPase, human Rab32, is required for the formation of autophagic vacuoles under basal conditions. Cell Mol. Life Sci. 66, 2913–2932. doi: 10.1007/s00018-009-0080-9
Ho, W. Y., Tai, Y. K., Chang, J.-C., Liang, J., Tyan, S.-H., Chen, S., et al. (2019). The ALS-FTD-linked gene product, C9orf72, regulates neuronal morphogenesis via autophagy. Autophagy 15, 827–842. doi: 10.1080/15548627.2019.1569441
Hoffenberg, S., Liu, X., Nikolova, L., Hall, H. S., Dai, W., Baughn, R. E., et al. (2000). A novel membrane-anchored Rab5 interacting protein required for homotypic endosome fusion. J. Biol. Chem. 275, 24661–24669. doi: 10.1074/jbc.M909600199
Hutagalung, A. H., and Novick, P. J. (2011). Role of Rab GTPases in membrane traffic and cell physiology. Physiol. Rev. 91, 119–149. doi: 10.1152/physrev.00059.2009
Hyttinen, J. M. T., Niittykoski, M., Salminen, A., and Kaarniranta, K. (2013). Maturation of autophagosomes and endosomes: A key role for Rab7. Biochim. Biophys. Acta Mol. Cell Res. 1833, 503–510. doi: 10.1016/j.bbamcr.2012.11.018
Ichimura, Y., Kumanomidou, T., Sou, Y., Mizushima, T., Ezaki, J., Ueno, T., et al. (2008). Structural basis for sorting mechanism of p62 in selective autophagy. J. Biol. Chem. 283, 22847–22857. doi: 10.1074/jbc.M802182200
Isaacs, A. M., Johannsen, P., Holm, I., Nielsen, J. E., and FReJA consortium (2011). Frontotemporal dementia caused by CHMP2B mutations. Curr. Alzheimer Res. 8, 246–251. doi: 10.2174/156720511795563764
Ishida, M., Oguchi, M. E., and Fukuda, M. (2016). Multiple types of guanine nucleotide exchange factors (GEFs) for Rab small GTPases. Cell Struct. Funct. 41, 61–79. doi: 10.1247/csf.16008
Itoh, T., Fujita, N., Kanno, E., Yamamoto, A., Yoshimori, T., and Fukuda, M. (2008). Golgi-resident small GTPase Rab33B interacts with Atg16L and modulates autophagosome formation. Mol. Biol. Cell 19, 2916–2925. doi: 10.1091/mbc.e07-12-1231
Itoh, T., Kanno, E., Uemura, T., Waguri, S., and Fukuda, M. (2011). OATL1, a novel autophagosome-resident Rab33B-GAP, regulates autophagosomal maturation. J. Cell Biol. 192, 839–853. doi: 10.1083/jcb.201008107
Ji, Y. J., Ugolino, J., Brady, N. R., Hamacher-Brady, A., and Wang, J. (2017). Systemic deregulation of autophagy upon loss of ALS- and FTD-linked C9orf72. Autophagy 13, 1254–1255. doi: 10.1080/15548627.2017.1299312
Jiang, L., Zhang, T., Lu, K., and Qi, S. (2022). The progress in C9orf72 research: ALS/FTD pathogenesis, functions and structure. Small GTPases 13, 56–76. doi: 10.1080/21541248.2021.1892443
Jiang, S., Park, D. W., Gao, Y., Ravi, S., Darley-Usmar, V., Abraham, E., et al. (2015). Participation of proteasome-ubiquitin protein degradation in autophagy and the activation of AMP-activated protein kinase. Cell Signal 27, 1186–1197. doi: 10.1016/j.cellsig.2015.02.024
Johnson, J. O., Mandrioli, J., Benatar, M., Abramzon, Y., Van Deerlin, V. M., Trojanowski, J. Q., et al. (2010). Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 68, 857–864. doi: 10.1016/j.neuron.2010.11.036
Ju, J.-S., Fuentealba, R. A., Miller, S. E., Jackson, E., Piwnica-Worms, D., Baloh, R. H., et al. (2009). Valosin-containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J. Cell Biol. 187, 875–888. doi: 10.1083/jcb.200908115
Jung, J., Nayak, A., Schaeffer, V., Starzetz, T., Kirsch, A. K., Müller, S., et al. (2017). Multiplex image-based autophagy RNAi screening identifies SMCR8 as ULK1 kinase activity and gene expression regulator. eLife 6: e23063. doi: 10.7554/eLife.23063
Kim, J., Kim, T.-Y., Cho, K.-S., Kim, H. N., and Koh, J.-Y. (2013). Autophagy activation and neuroprotection by progesterone in the G93A-SOD1 transgenic mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 59, 80–85. doi: 10.1016/j.nbd.2013.07.011
Kiral, F. R., Kohrs, F. E., Jin, E. J., and Hiesinger, P. R. (2018). Rab GTPases and membrane trafficking in neurodegeneration. Curr. Biol. 28, R471–R486. doi: 10.1016/j.cub.2018.02.010
Kocaturk, N. M., and Gozuacik, D. (2018). Crosstalk Between mammalian autophagy and the ubiquitin-proteasome system. Front. Cell Dev. Biol. 6:128. doi: 10.3389/fcell.2018.00128
Komatsu, M., Waguri, S., Chiba, T., Murata, S., Iwata, J., Tanida, I., et al. (2006). Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441, 880–884. doi: 10.1038/nature04723
Kopito, R. R. (2000). Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 10, 524–530. doi: 10.1016/s0962-8924(00)01852-3
Krasniak, C. S., and Ahmad, S. T. (2016). The role of CHMP2BIntron5 in autophagy and frontotemporal dementia. Brain Res. 1649, 151–157. doi: 10.1016/j.brainres.2016.02.051
Krüger, R., Kuhn, W., Müller, T., Woitalla, D., Graeber, M., Kösel, S., et al. (1998). Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat. Genet. 18, 106–108. doi: 10.1038/ng0298-106
Kuo, S.-H., Tasset, I., Cheng, M. M., Diaz, A., Pan, M.-K., Lieberman, O. J., et al. (2022). Mutant glucocerebrosidase impairs α-synuclein degradation by blockade of chaperone-mediated autophagy. Sci. Adv. 8:eabm6393. doi: 10.1126/sciadv.abm6393
Lai, Y., Kondapalli, C., Lehneck, R., Procter, J. B., Dill, B. D., Woodroof, H. I., et al. (2015). Phosphoproteomic screening identifies Rab GTPases as novel downstream targets of PINK1. EMBO J. 34, 2840–2861. doi: 10.15252/embj.201591593
Lamark, T., and Johansen, T. (2012). Aggrephagy: Selective disposal of protein aggregates by macroautophagy. Int. J. Cell Biol. 2012:736905. doi: 10.1155/2012/736905
Lamber, E. P., Siedenburg, A.-C., and Barr, F. A. (2019). Rab regulation by GEFs and GAPs during membrane traffic. Curr. Opin. Cell Biol. 59, 34–39. doi: 10.1016/j.ceb.2019.03.004
Lan, Y., Sullivan, P. M., and Hu, F. (2019). SMCR8 negatively regulates AKT and MTORC1 signaling to modulate lysosome biogenesis and tissue homeostasis. Autophagy 15, 871–885. doi: 10.1080/15548627.2019.1569914
Langemeyer, L., Fröhlich, F., and Ungermann, C. (2018). Rab GTPase function in endosome and lysosome biogenesis. Trends Cell Biol. 28, 957–970. doi: 10.1016/j.tcb.2018.06.007
Latourelle, J. C., Pankratz, N., Dumitriu, A., Wilk, J. B., Goldwurm, S., Pezzoli, G., et al. (2009). Genomewide association study for onset age in Parkinson disease. BMC Med. Genet. 10:98. doi: 10.1186/1471-2350-10-98
Lawrence, R. E., Fromm, S. A., Fu, Y., Yokom, A. L., Kim, D. J., Thelen, A. M., et al. (2019). Structural mechanism of a Rag GTPase activation checkpoint by the lysosomal folliculin complex. Science 366, 971–977. doi: 10.1126/science.aax0364
Lee, D. H., Park, J. S., Lee, Y. S., Han, J., Lee, D.-K., Kwon, S. W., et al. (2020). SQSTM1/p62 activates NFE2L2/NRF2 via ULK1-mediated autophagic KEAP1 degradation and protects mouse liver from lipotoxicity. Autophagy 16, 1949–1973. doi: 10.1080/15548627.2020.1712108
Lee, S., Jeon, Y.-M., Cha, S. J., Kim, S., Kwon, Y., Jo, M., et al. (2020). PTK2/FAK regulates UPS impairment via SQSTM1/p62 phosphorylation in TARDBP/TDP-43 proteinopathies. Autophagy 16, 1396–1412. doi: 10.1080/15548627.2019.1686729
Lee, J.-H., Yang, D.-S., Goulbourne, C. N., Im, E., Stavrides, P., Pensalfini, A., et al. (2022). Faulty autolysosome acidification in Alzheimer’s disease mouse models induces autophagic build-up of Aβ in neurons, yielding senile plaques. Nat. Neurosci. 25, 688–701. doi: 10.1038/s41593-022-01084-8
Levine, T. P., Daniels, R. D., Wong, L. H., Gatta, A. T., Gerondopoulos, A., and Barr, F. A. (2013b). Discovery of new Longin and Roadblock domains that form platforms for small GTPases in Ragulator and TRAPP-II. Small GTPases 4, 62–69. doi: 10.4161/sgtp.24262
Levine, T. P., Daniels, R. D., Gatta, A. T., Wong, L. H., and Hayes, M. J. (2013a). The product of C9orf72, a gene strongly implicated in neurodegeneration, is structurally related to DENN Rab-GEFs. Bioinformatics 29, 499–503. doi: 10.1093/bioinformatics/bts725
Li, Y., Zhao, Y., Hu, J., Xiao, J., Qu, L., Wang, Z., et al. (2013). A novel ER-localized transmembrane protein, EMC6, interacts with RAB5A and regulates cell autophagy. Autophagy 9, 150–163. doi: 10.4161/auto.22742
Li, Z., Lai, M., Li, J., Yang, D., Zhao, M., Wang, D., et al. (2023). RAB7A GTPase is involved in mitophagosome formation and autophagosome-lysosome fusion in N2a cells treated with the prion protein fragment 106-126. Mol. Neurobiol. 60, 1391–1407. doi: 10.1007/s12035-022-03118-5
Liang, C. C., Wang, C., Peng, X., Gan, B., and Guan, J. L. (2020). Neural-specific deletion of FIP200 leads to cerebellar degeneration caused by increased neuronal death and axon degeneration. J. Biol. Chem. 285, 3499–3509. doi: 10.1074/jbc.M109.072389
Lim, J., Lachenmayer, M. L., Wu, S., Liu, W., Kundu, M., Wang, R., et al. (2015). Proteotoxic stress induces phosphorylation of p62/SQSTM1 by ULK1 to regulate selective autophagic clearance of protein aggregates. PLoS Genet. 11:e1004987. doi: 10.1371/journal.pgen.1004987
Limanaqi, F., Biagioni, F., Gambardella, S., Ryskalin, L., and Fornai, F. (2018). Interdependency between autophagy and synaptic vesicle trafficking: Implications for dopamine release. Front. Mol. Neurosci. 11:299. doi: 10.3389/fnmol.2018.00299
Lin, M. G., and Hurley, J. H. (2016). Structure and function of the ULK1 complex in autophagy. Curr. Opin. Cell Biol. 39, 61–68. doi: 10.1016/j.ceb.2016.02.010
Lin, M., Liu, H., Xiong, Q., Niu, H., Cheng, Z., Yamamoto, A., et al. (2016). Ehrlichia secretes Etf-1 to induce autophagy and capture nutrients for its growth through RAB5 and class III phosphatidylinositol 3-kinase. Autophagy 12, 2145–2166. doi: 10.1080/15548627.2016.1217369
Liu, F., Morderer, D., Wren, M. C., Vettleson-Trutza, S. A., Wang, Y., Rabichow, B. E., et al. (2022). Proximity proteomics of C9orf72 dipeptide repeat proteins identifies molecular chaperones as modifiers of poly-GA aggregation. Acta Neuropathol. Commun. 10:22. doi: 10.1186/s40478-022-01322-x
Liu, W. J., Ye, L., Huang, W. F., Guo, L. J., Xu, Z. G., Wu, H. L., et al. (2016). p62 links the autophagy pathway and the ubiqutin–proteasome system upon ubiquitinated protein degradation. Cell Mol. Biol. Lett. 21:29. doi: 10.1186/s11658-016-0031-z
Liu, Y., and Wang, J. (2019). C9orf72-dependent lysosomal functions regulate epigenetic control of autophagy and lipid metabolism. Autophagy 15, 913–914. doi: 10.1080/15548627.2019.1580106
Longatti, A., and Tooze, S. A. (2012). Recycling endosomes contribute to autophagosome formation. Autophagy 8, 1682–1683. doi: 10.4161/auto.21486
Lu, G., Wang, Y., Shi, Y., Zhang, Z., Huang, C., He, W., et al. (2022). Autophagy in health and disease: From molecular mechanisms to therapeutic target. MedComm (2020) 3:e150. doi: 10.1002/mco2.150
Lü, S., and Wang, J. (2013). The resistance mechanisms of proteasome inhibitor bortezomib. Biomark. Res. 1:13. doi: 10.1186/2050-7771-1-13
Lüningschrör, P., Binotti, B., Dombert, B., Heimann, P., Perez-Lara, A., Slotta, C., et al. (2017). Plekhg5-regulated autophagy of synaptic vesicles reveals a pathogenic mechanism in motoneuron disease. Nat. Commun. 8:678. doi: 10.1038/s41467-017-00689-z
Magalhaes, J., Gegg, M. E., Migdalska-Richards, A., Doherty, M. K., Whitfield, P. D., and Schapira, A. H. V. (2016). Autophagic lysosome reformation dysfunction in glucocerebrosidase deficient cells: Relevance to Parkinson disease. Hum. Mol. Genet. 25, 3432–3445. doi: 10.1093/hmg/ddw185
Mahapatra, K. K., Mishra, S. R., Behera, B. P., Patil, S., Gewirtz, D. A., and Bhutia, S. K. (2021). The lysosome as an imperative regulator of autophagy and cell death. Cell Mol. Life Sci. 78, 7435–7449. doi: 10.1007/s00018-021-03988-3
Marat, A. L., Dokainish, H., and McPherson, P. S. (2011). DENN domain proteins: Regulators of Rab GTPases. J. Biol. Chem. 286, 13791–13800. doi: 10.1074/jbc.R110.217067
Martin, D. D. O., Ladha, S., Ehrnhoefer, D. E., and Hayden, M. R. (2015). Autophagy in huntington disease and huntingtin in autophagy. Trends Neurosci. 38, 26–35. doi: 10.1016/j.tins.2014.09.003
Martina, J. A., and Puertollano, R. (2013). RRAG GTPases link nutrient availability to gene expression, autophagy and lysosomal biogenesis. Autophagy 9, 928–930. doi: 10.4161/auto.24371
Martinez-Vicente, M., Talloczy, Z., Wong, E., Tang, G., Koga, H., Kaushik, S., et al. (2010). Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat. Neurosci. 13, 567–576. doi: 10.1038/nn.2528
Maruyama, H., Morino, H., Ito, H., Izumi, Y., Kato, H., Watanabe, Y., et al. (2010). Mutations of optineurin in amyotrophic lateral sclerosis. Nature 465, 223–226. doi: 10.1038/nature08971
Matsumoto, G., Shimogori, T., Hattori, N., and Nukina, N. (2015). TBK1 controls autophagosomal engulfment of polyubiquitinated mitochondria through p62/SQSTM1 phosphorylation. Hum. Mol. Genet. 24, 4429–4442. doi: 10.1093/hmg/ddv179
Matthias, P., Yoshida, M., and Khochbin, S. (2008). HDAC6 a new cellular stress surveillance factor. Cell Cycle 7, 7–10. doi: 10.4161/cc.7.1.5186
McDermott, C. J. (2019). Clinical trials in amyotrophic lateral sclerosis. Curr. Opin. Neurol. 32, 758–763. doi: 10.1097/WCO.0000000000000731
McMillan, M. A., and Dwyer, J. (1989). Changing times changing paradigm (1): From hospital training and college education in Australia. Nurse Educ. Today 9, 13–18. doi: 10.1016/0260-6917(89)90004-x
Medina, D. L., Fraldi, A., Bouche, V., Annunziata, F., Mansueto, G., Spampanato, C., et al. (2011). Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev. Cell 21, 421–430. doi: 10.1016/j.devcel.2011.07.016
Meng, Y., Ding, J., Li, C., Fan, H., He, Y., and Qiu, P. (2020). Transfer of pathological α-synuclein from neurons to astrocytes via exosomes causes inflammatory responses after METH exposure. Toxicol. Lett. 331, 188–199. doi: 10.1016/j.toxlet.2020.06.016
Merenlender-Wagner, A., Shemer, Z., Touloumi, O., Lagoudaki, R., Giladi, E., Andrieux, A., et al. (2014). New horizons in schizophrenia treatment: Autophagy protection is coupled with behavioral improvements in a mouse model of schizophrenia. Autophagy 10, 2324–2332. doi: 10.4161/15548627.2014.984274
Meyer, K., and Kaspar, B. K. (2017). Glia–neuron interactions in neurological diseases: Testing non-cell autonomy in a dish. Brain Res. 1656, 27–39. doi: 10.1016/j.brainres.2015.12.051
Miller, S., and Muqit, M. M. K. (2019). Therapeutic approaches to enhance PINK1/Parkin mediated mitophagy for the treatment of Parkinson’s disease. Neurosci. Lett. 705, 7–13. doi: 10.1016/j.neulet.2019.04.029
Mishra, A., Godavarthi, S. K., Maheshwari, M., Goswami, A., and Jana, N. R. (2009). The ubiquitin ligase E6-AP is induced and recruited to aggresomes in response to proteasome inhibition and may be involved in the ubiquitination of Hsp70-bound misfolded proteins. J. Biol. Chem. 284, 10537–10545. doi: 10.1074/jbc.M806804200
Miyazaki, M., Hiramoto, M., Takano, N., Kokuba, H., Takemura, J., Tokuhisa, M., et al. (2021). Targeted disruption of GAK stagnates autophagic flux by disturbing lysosomal dynamics. Int. J. Mol. Med. 48:195. doi: 10.3892/ijmm.2021.5028
Mizushima, N., Yoshimori, T., and Ohsumi, Y. (2011). The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 27, 107–132. doi: 10.1146/annurev-cellbio-092910-154005
Monaco, A., and Fraldi, A. (2020). Protein aggregation and dysfunction of autophagy-lysosomal pathway: A vicious cycle in lysosomal storage diseases. Front. Mol. Neurosci. 13:37. doi: 10.3389/fnmol.2020.00037
Mori, K., Weng, S.-M., Arzberger, T., May, S., Rentzsch, K., Kremmer, E., et al. (2013). The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339, 1335–1338. doi: 10.1126/science.1232927
Morris, D. H., Yip, C. K., Shi, Y., Chait, B. T., and Wang, Q. J. (2015). BECLIN 1-VPS34 complex architecture: Understanding the nuts and bolts of therapeutic targets. Front. Biol. 10:398–426. doi: 10.1007/s11515-015-1374-y
Morvan, J., Köchl, R., Watson, R., Collinson, L. M., Jefferies, H. B. J., and Tooze, S. A. (2009). In vitro reconstitution of fusion between immature autophagosomes and endosomes. Autophagy 5, 676–689. doi: 10.4161/auto.5.5.8378
Müller, M. P., and Goody, R. S. (2018). Molecular control of Rab activity by GEFs, GAPs and GDI. Small GTPases 9, 5–21. doi: 10.1080/21541248.2016.1276999
Munafó, D. B., and Colombo, M. I. (2002). Induction of autophagy causes dramatic changes in the subcellular distribution of GFP-Rab24. Traffic 3, 472–482. doi: 10.1034/j.1600-0854.2002.30704.x
Nah, J., Yuan, J., and Jung, Y.-K. (2015). Autophagy in neurodegenerative diseases: From mechanism to therapeutic approach. Mol. Cells 38, 381–389. doi: 10.14348/molcells.2015.0034
Nanayakkara, R., Gurung, R., Rodgers, S. J., Eramo, M. J., Ramm, G., Mitchell, C. A., et al. (2022). Autophagic lysosome reformation in health and disease. Autophagy 1–18. doi: 10.1080/15548627.2022.2128019
Nedelsky, N. B., Todd, P. K., and Taylor, J. P. (2008). Autophagy and the ubiquitin-proteasome system: Collaborators in neuroprotection. Biochim. Biophys. Acta Mol. Basis Dis. 1782, 691–699. doi: 10.1016/j.bbadis.2008.10.002
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133. doi: 10.1126/science.1134108
Niu, M., Zheng, N., Wang, Z., Gao, Y., Luo, X., Chen, Z., et al. (2020). RAB39B deficiency impairs learning and memory partially through compromising autophagy. Front. Cell Dev. Biol. 8:598622. doi: 10.3389/fcell.2020.598622
Nixon, R. A., Wegiel, J., Kumar, A., Yu, W. H., Peterhoff, C., Cataldo, A., et al. (2005). Extensive involvement of autophagy in alzheimer disease: An immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 64, 113–122. doi: 10.1093/jnen/64.2.113
Nixon, R. A., Yang, D.-S., and Lee, J.-H. (2008). Neurodegenerative lysosomal disorders: A continuum from development to late age. Autophagy 4, 590–599. doi: 10.4161/auto.6259
Nozawa, T., Aikawa, C., Goda, A., Maruyama, F., Hamada, S., and Nakagawa, I. (2012). The small GTPases Rab9A and Rab23 function at distinct steps in autophagy during Group A Streptococcus infection. Cell Microbiol. 14, 1149–1165. doi: 10.1111/j.1462-5822.2012.01792.x
O’Rourke, J. G., Bogdanik, L., Yáñez, A., Lall, D., Wolf, A. J., Muhammad, A. K. M. G., et al. (2016). C9orf72 is required for proper macrophage and microglial function in mice. Science 351, 1324–1329. doi: 10.1126/science.aaf1064
Oakes, J. A., Davies, M. C., and Collins, M. O. (2017). TBK1: A new player in ALS linking autophagy and neuroinflammation. Mol. Brain 10:5. doi: 10.1186/s13041-017-0287-x
Oh, Y. M., Lee, S. W., Kim, W. K., Chen, S., Church, V. A., Cates, K., et al. (2022). Age-related Huntington’s disease progression modeled in directly reprogrammed patient-derived striatal neurons highlights impaired autophagy. Nat. Neurosci. 25, 1420–1433. doi: 10.1038/s41593-022-01185-4
Olzmann, J. A., and Chin, L.-S. (2008). Parkin-mediated K63-linked polyubiquitination: A signal for targeting misfolded proteins to the aggresome-autophagy pathway. Autophagy 4, 85–87. doi: 10.4161/auto.5172
Olzmann, J. A., Li, L., Chudaev, M. V., Chen, J., Perez, F. A., Palmiter, R. D., et al. (2007). Parkin-mediated K63-linked polyubiquitination targets misfolded DJ-1 to aggresomes via binding to HDAC6. J. Cell Biol. 178, 1025–1038. doi: 10.1083/jcb.200611128
Pang, W., and Hu, F. (2021). Cellular and physiological functions of C9ORF72 and implications for ALS/FTD. J. Neurochem. 157, 334–350. doi: 10.1111/jnc.15255
Pang, Y. J., Du, N. Z., and Liu, J. L. (1988). [The clinical analysis of malignant tumors in eyelid and postoperative reconstruction of the eyelid]. Yan Ke Xue Bao 4, 246–249.
Pavlinov, I., Salkovski, M., and Aldrich, L. N. (2020). Beclin 1-ATG14L Protein-protein interaction inhibitor selectively inhibits autophagy through disruption of VPS34 Complex I. J. Am. Chem. Soc. 142, 8174–8182. doi: 10.1021/jacs.9b12705
Peng, W., Minakaki, G., Nguyen, M., and Krainc, D. (2019). Preserving lysosomal function in the aging brain: Insights from neurodegeneration. Neurotherapeutics 16, 611–634. doi: 10.1007/s13311-019-00742-3
Petrucelli, L., O’Farrell, C., Lockhart, P. J., Baptista, M., Kehoe, K., Vink, L., et al. (2002). Parkin protects against the toxicity associated with Mutant α-Synuclein. Neuron 36, 1007–1019. doi: 10.1016/S0896-6273(02)01125-X
Pfeffer, S. R. (2013). Rab GTPase regulation of membrane identity. Curr. Opin. Cell Biol. 25, 414–419. doi: 10.1016/j.ceb.2013.04.002
Pfeffer, S. R. (2017). Rab GTPases: Master regulators that establish the secretory and endocytic pathways. Mol. Biol. Cell 28, 712–715. doi: 10.1091/mbc.E16-10-0737
Pilli, M., Arko-Mensah, J., Ponpuak, M., Roberts, E., Master, S., Mandell, M. A., et al. (2012). TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity 37, 223–234. doi: 10.1016/j.immuni.2012.04.015
Piras, A., Collin, L., Grüninger, F., Graff, C., and Rönnbäck, A. (2016). Autophagic and lysosomal defects in human tauopathies: Analysis of post-mortem brain from patients with familial Alzheimer disease, corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol. Commun. 4:22. doi: 10.1186/s40478-016-0292-9
Polito, V. A., Li, H., Martini-Stoica, H., Wang, B., Yang, L., Xu, Y., et al. (2014). Selective clearance of aberrant tau proteins and rescue of neurotoxicity by transcription factor EB. EMBO Mol. Med. 6, 1142–1160. doi: 10.15252/emmm.201303671
Polymeropoulos, M. H., Lavedan, C., Leroy, E., Ide, S. E., Dehejia, A., Dutra, A., et al. (1997). Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276, 2045–2047. doi: 10.1126/science.276.5321.2045
Pozzi, S., Thammisetty, S. S., and Julien, J.-P. (2018). Chronic administration of pimozide fails to attenuate motor and pathological deficits in two mouse models of amyotrophic lateral sclerosis. Neurotherapeutics 15, 715–727. doi: 10.1007/s13311-018-0634-3
Qin, Y., He, Y., Zhu, Y., Li, M., Ni, Y., Liu, J., et al. (2019). CID1067700, a late endosome GTPase Rab7 receptor antagonist, attenuates brain atrophy, improves neurologic deficits and inhibits reactive astrogliosis in rat ischemic stroke. Acta Pharmacol. Sin. 40, 724–736. doi: 10.1038/s41401-018-0166-8
Rahman, A., Saikia, B., Gogoi, C. R., and Baruah, A. (2022). Advances in the understanding of protein misfolding and aggregation through molecular dynamics simulation. Prog. Biophys. Mol. Biol. 175, 31–48. doi: 10.1016/j.pbiomolbio.2022.08.007
Ramírez-Jarquín, U. N., Sharma, M., Zhou, W., Shahani, N., and Subramaniam, S. (2022). Deletion of SUMO1 attenuates behavioral and anatomical deficits by regulating autophagic activities in Huntington disease. Proc. Natl. Acad. Sci. U.S.A. 119:e2107187119. doi: 10.1073/pnas.2107187119
Rana, A., Rera, M., and Walker, D. W. (2013). Parkin overexpression during aging reduces proteotoxicity, alters mitochondrial dynamics, and extends lifespan. Proc. Natl. Acad. Sci. U.S.A. 110, 8638–8643. doi: 10.1073/pnas.1216197110
Ravikumar, B., Imarisio, S., Sarkar, S., O’Kane, C. J., and Rubinsztein, D. C. (2008). Rab5 modulates aggregation and toxicity of mutant huntingtin through macroautophagy in cell and fly models of Huntington disease. J. Cell Sci. 121, 1649–1660. doi: 10.1242/jcs.025726
Ravikumar, B., Vacher, C., Berger, Z., Davies, J. E., Luo, S., Oroz, L. G., et al. (2004). Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 36, 585–595. doi: 10.1038/ng1362
Rayner, S. L., Cheng, F., Hogan, A. L., Grima, N., Yang, S., Ke, Y. D., et al. (2021). ALS/FTD-causing mutation in cyclin F causes the dysregulation of SFPQ. Hum. Mol. Genet. 30, 971–984. doi: 10.1093/hmg/ddab073
Ren, M., Xu, G., Zeng, J., De Lemos-Chiarandini, C., Adesnik, M., and Sabatini, D. D. (1998). Hydrolysis of GTP on rab11 is required for the direct delivery of transferrin from the pericentriolar recycling compartment to the cell surface but not from sorting endosomes. Proc. Natl. Acad. Sci. U.S.A. 95, 6187–6192. doi: 10.1073/pnas.95.11.6187
Renton, A. E., Chiò, A., and Traynor, B. J. (2014). State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 17, 17–23. doi: 10.1038/nn.3584
Renton, A. E., Majounie, E., Waite, A., Simón-Sánchez, J., Rollinson, S., Gibbs, J. R., et al. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-Linked ALS-FTD. Neuron 72, 257–268. doi: 10.1016/j.neuron.2011.09.010
Rubinsztein, D. C., Shpilka, T., and Elazar, Z. (2012). Mechanisms of autophagosome biogenesis. Curr. Biol. 22, R29–R34. doi: 10.1016/j.cub.2011.11.034
Ruegsegger, C., and Saxena, S. (2016). Proteostasis impairment in ALS. Brain Res. 1648, 571–579. doi: 10.1016/j.brainres.2016.03.032
Ryter, S. W., Bhatia, D., and Choi, M. E. (2019). Autophagy: A lysosome-dependent process with implications in cellular redox homeostasis and human disease. Antioxid. Redox Signal. 30, 138–159. doi: 10.1089/ars.2018.7518
Saftig, P., and Klumperman, J. (2009). Lysosome biogenesis and lysosomal membrane proteins: Trafficking meets function. Nat. Rev. Mol. Cell Biol. 10, 623–635. doi: 10.1038/nrm2745
Saibil, H. (2013). Chaperone machines for protein folding, unfolding and disaggregation. Nat. Rev. Mol. Cell Biol. 14, 630–642. doi: 10.1038/nrm3658
Sarkar, S., Floto, R. A., Berger, Z., Imarisio, S., Cordenier, A., Pasco, M., et al. (2005). Lithium induces autophagy by inhibiting inositol monophosphatase. J. Cell Biol. 170, 1101–1111. doi: 10.1083/jcb.200504035
Schmitz, A., Pinheiro Marques, J., Oertig, I., Maharjan, N., and Saxena, S. (2021). Emerging perspectives on dipeptide repeat proteins in C9ORF72 ALS/FTD. Front. Cell Neurosci. 15:637548. doi: 10.3389/fncel.2021.637548
Schöndorf, D. C., Aureli, M., McAllister, F. E., Hindley, C. J., Mayer, F., Schmid, B., et al. (2014). iPSC-derived neurons from GBA1-associated Parkinson’s disease patients show autophagic defects and impaired calcium homeostasis. Nat. Commun. 5:4028. doi: 10.1038/ncomms5028
Selimovic, D., Porzig, B. B. O. W., El-Khattouti, A., Badura, H. E., Ahmad, M., Ghanjati, F., et al. (2013). Bortezomib/proteasome inhibitor triggers both apoptosis and autophagy-dependent pathways in melanoma cells. Cell Signal 25, 308–318. doi: 10.1016/j.cellsig.2012.10.004
Sellier, C., Campanari, M.-L., Julie Corbier, C., Gaucherot, A., Kolb-Cheynel, I., Oulad-Abdelghani, M., et al. (2016). Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neuron dysfunction and cell death. EMBO J. 35, 1276–1297. doi: 10.15252/embj.201593350
Settembre, C., Di Malta, C., Polito, V. A., Arencibia, M. G., Vetrini, F., Erdin, S., et al. (2011). TFEB links autophagy to lysosomal biogenesis. Science 332, 1429–1433. doi: 10.1126/science.1204592
Sha, Z., Schnell, H. M., Ruoff, K., and Goldberg, A. (2018). Rapid induction of p62 and GABARAPL1 upon proteasome inhibition promotes survival before autophagy activation. J. Cell Biol. 217, 1757–1776. doi: 10.1083/jcb.201708168
Shang, L., Chen, S., Du, F., Li, S., Zhao, L., and Wang, X. (2011). Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc. Natl. Acad. Sci. U.S.A. 108, 4788–4793. doi: 10.1073/pnas.1100844108
Shao, Q., Yang, M., Liang, C., Ma, L., Zhang, W., Jiang, Z., et al. (2020). C9orf72 and smcr8 mutant mice reveal MTORC1 activation due to impaired lysosomal degradation and exocytosis. Autophagy 16, 1635–1650. doi: 10.1080/15548627.2019.1703353
Shao, W., Todd, T. W., Wu, Y., Jones, C. Y., Tong, J., Jansen-West, K., et al. (2022). Two FTD-ALS genes converge on the endosomal pathway to induce TDP-43 pathology and degeneration. Science 378, 94–99. doi: 10.1126/science.abq7860
Sheng, Y., Song, Y., Li, Z., Wang, Y., Lin, H., Cheng, H., et al. (2017). RAB37 interacts directly with ATG5 and promotes autophagosome formation via regulating ATG5-12-16 complex assembly. Cell Death Differ. 25, 918–934. doi: 10.1038/s41418-017-0023-1
Shi, Y., Hung, S.-T., Rocha, G., Lin, S., Linares, G. R., Staats, K. A., et al. (2019). Identification and therapeutic rescue of autophagosome and glutamate receptor defects in C9ORF72 and sporadic ALS neurons. JCI Insight 5:e127736. doi: 10.1172/jci.insight.127736
Shi, Y., Lin, S., Staats, K. A., Li, Y., Chang, W.-H., Hung, S.-T., et al. (2018). Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat. Med. 24, 313–325. doi: 10.1038/nm.4490
Shimura, H., Schlossmacher, M. G., Hattori, N., Frosch, M. P., Trockenbacher, A., Schneider, R., et al. (2001). Ubiquitination of a new form of α-Synuclein by parkin from human brain: Implications for Parkinson’s disease. Science 293, 263–269. doi: 10.1126/science.1060627
Sivadasan, R., Hornburg, D., Drepper, C., Frank, N., Jablonka, S., Hansel, A., et al. (2016). C9ORF72 interaction with cofilin modulates actin dynamics in motor neurons. Nat. Neurosci. 19, 1610–1618. doi: 10.1038/nn.4407
Skibinski, G., Parkinson, N. J., Brown, J. M., Chakrabarti, L., Lloyd, S. L., Hummerich, H., et al. (2005). Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat. Genet. 37, 806–808. doi: 10.1038/ng1609
Song, W., Wang, F., Lotfi, P., Sardiello, M., and Segatori, L. (2014). 2-Hydroxypropyl-β-cyclodextrin promotes transcription factor EB-mediated activation of autophagy: Implications for therapy. J. Biol. Chem. 289, 10211–10222. doi: 10.1074/jbc.M113.506246
Song, Y. M., Lee, Y., Kim, J.-W., Ham, D.-S., Kang, E.-S., Cha, B. S., et al. (2015). Metformin alleviates hepatosteatosis by restoring SIRT1-mediated autophagy induction via an AMP-activated protein kinase-independent pathway. Autophagy 11, 46–59. doi: 10.4161/15548627.2014.984271
Stavoe, A. K. H., and Holzbaur, E. L. F. (2019). Autophagy in neurons. Annu. Rev. Cell Dev. Biol. 35, 477–500. doi: 10.1146/annurev-cellbio-100818-125242
Su, M.-Y., Fromm, S. A., Remis, J., Toso, D. B., and Hurley, J. H. (2021). Structural basis for the ARF GAP activity and specificity of the C9orf72 complex. Nat. Commun. 12:3786. doi: 10.1038/s41467-021-24081-0
Su, M.-Y., Fromm, S. A., Zoncu, R., and Hurley, J. H. (2020). Structure of the C9orf72 ARF GAP complex that is haploinsufficient in ALS and FTD. Nature 585, 251–255. doi: 10.1038/s41586-020-2633-x
Sullivan, P. M., Zhou, X., Robins, A. M., Paushter, D. H., Kim, D., Smolka, M. B., et al. (2016). The ALS/FTLD associated protein C9orf72 associates with SMCR8 and WDR41 to regulate the autophagy-lysosome pathway. Acta Neuropathol. Commun. 4:51. doi: 10.1186/s40478-016-0324-5
Taes, I., Timmers, M., Hersmus, N., Bento-Abreu, A., Van Den Bosch, L., Van Damme, P., et al. (2013). Hdac6 deletion delays disease progression in the SOD1G93A mouse model of ALS. Hum. Mol. Genet. 22, 1783–1790. doi: 10.1093/hmg/ddt028
Talaber, G., Miklossy, G., Oaks, Z., Liu, Y., Tooze, S. A., Chudakov, D. M., et al. (2014). HRES-1/Rab4 promotes the formation of LC3(+) autophagosomes and the accumulation of mitochondria during autophagy. PLoS One 9:e84392. doi: 10.1371/journal.pone.0084392
Tang, B. L. (2016). C9orf72’s interaction with Rab GTPases—modulation of membrane traffic and autophagy. Front. Cell Neurosci. 10:228. doi: 10.3389/fncel.2016.00228
Tang, D., Sheng, J., Xu, L., Zhan, X., Liu, J., Jiang, H., et al. (2020). Cryo-EM structure of C9ORF72-SMCR8-WDR41 reveals the role as a GAP for Rab8a and Rab11a. Proc. Natl. Acad. Sci. U.S.A. 117, 9876–9883. doi: 10.1073/pnas.2002110117
Tannous, P., Zhu, H., Nemchenko, A., Berry, J. M., Johnstone, J. L., Shelton, J. M., et al. (2008). Intracellular protein aggregation is a proximal trigger of cardiomyocyte autophagy. Circulation 117, 3070–3078. doi: 10.1161/CIRCULATIONAHA.107.763870
Terry, R. D., Gonatas, N. K., and Weiss, M. (1964). The ultrastructure of the cerebral cortex in Alzheimer’s disease. Trans. Am. Neurol. Assoc. 89:12.
Tong, Y., and Song, F. (2015). Intracellular calcium signaling regulates autophagy via calcineurin-mediated TFEB dephosphorylation. Autophagy 11, 1192–1195. doi: 10.1080/15548627.2015.1054594
Tresse, E., Salomons, F. A., Vesa, J., Bott, L. C., Kimonis, V., Yao, T.-P., et al. (2010). VCP/p97 is essential for maturation of ubiquitin-containing autophagosomes and this function is impaired by mutations that cause IBMPFD. Autophagy 6, 217–227. doi: 10.4161/auto.6.2.11014
Tsubokawa, T., and Katayama, Y. (1985). Active neural processes within the brain stem in production of coma–Araki’s coma-puncture revisited. Neurol. Med. Chir. 25, 503–514. doi: 10.2176/nmc.25.503
Turmaine, M., Raza, A., Mahal, A., Mangiarini, L., Bates, G. P., and Davies, S. W. (2000). Nonapoptotic neurodegeneration in a transgenic mouse model of Huntington’s disease. Proc. Natl. Acad. Sci. U.S.A. 97, 8093–8097. doi: 10.1073/pnas.110078997
Tysnes, O.-B., and Storstein, A. (2017). Epidemiology of Parkinson’s disease. J. Neural Transm. 124, 901–905. doi: 10.1007/s00702-017-1686-y
Ugolino, J., Ji, Y. J., Conchina, K., Chu, J., Nirujogi, R. S., Pandey, A., et al. (2016). Loss of C9orf72 Enhances Autophagic Activity via Deregulated mTOR and TFEB Signaling. PLoS Genet. 12:e1006443. doi: 10.1371/journal.pgen.1006443
Ullrich, O., Reinsch, S., Urbé, S., Zerial, M., and Parton, R. G. (1996). Rab11 regulates recycling through the pericentriolar recycling endosome. J. Cell Biol. 135, 913–924. doi: 10.1083/jcb.135.4.913
Van Der Sluijs, P., Hull, M., Zahraoui, A., Tavitian, A., Goud, B., and Mellman, I. (1991). The small GTP-binding protein rab4 is associated with early endosomes. Proc. Natl. Acad. Sci. U.S.A. 88, 6313–6317. doi: 10.1073/pnas.88.14.6313
van der Zee, J., Urwin, H., Engelborghs, S., Bruyland, M., Vandenberghe, R., Dermaut, B., et al. (2008). CHMP2B C-truncating mutations in frontotemporal lobar degeneration are associated with an aberrant endosomal phenotype in vitro. Hum. Mol. Genet. 17, 313–322. doi: 10.1093/hmg/ddm309
Vanderplow, A. M., Eagle, A. L., Kermath, B. A., Bjornson, K. J., Robison, A. J., and Cahill, M. E. (2021). Akt-mTOR hypoactivity in bipolar disorder gives rise to cognitive impairments associated with altered neuronal structure and function. Neuron 109, 1479–1496.e6. doi: 10.1016/j.neuron.2021.03.008
Varga, R.-E., Khundadze, M., Damme, M., Nietzsche, S., Hoffmann, B., Stauber, T., et al. (2015). In vivo evidence for lysosome depletion and impaired autophagic clearance in hereditary spastic paraplegia Type SPG11. PLoS Genet. 11:e1005454. doi: 10.1371/journal.pgen.1005454
Wang, I., Guo, B.-S., Liu, Y.-C., Wu, C.-C., Yang, C.-H., Tsai, K.-J., et al. (2012). Autophagy activators rescue and alleviate pathogenesis of a mouse model with proteinopathies of the TAR DNA-binding protein 43. Proc. Natl. Acad. Sci. U.S.A. 109, 15024–15029. doi: 10.1073/pnas.1206362109
Wang, C., Liu, Z., and Huang, X. (2012). Rab32 is important for autophagy and lipid storage in Drosophila. PLoS One 7:e32086. doi: 10.1371/journal.pone.0032086
Wang, C., Niederstrasser, H., Douglas, P. M., Lin, R., Jaramillo, J., Li, Y., et al. (2017). Small-molecule TFEB pathway agonists that ameliorate metabolic syndrome in mice and extend C. elegans lifespan. Nat. Commun. 8:2270. doi: 10.1038/s41467-017-02332-3
Wang, T., Wang, W., Wang, Q., Xie, R., Landay, A., and Chen, D. (2020). The E3 ubiquitin ligase CHIP in normal cell function and in disease conditions. Ann. N. Y. Acad. Sci. 1460, 3–10. doi: 10.1111/nyas.14206
Wang, M., Wang, H., Tao, Z., Xia, Q., Hao, Z., Prehn, J. H. M., et al. (2020). C9orf72 associates with inactive Rag GTPases and regulates mTORC1-mediated autophagosomal and lysosomal biogenesis. Aging Cell 19:e13126. doi: 10.1111/acel.13126
Wang, T., Wong, K. K., and Hong, W. (2004). A unique region of RILP distinguishes it from its related proteins in its regulation of lysosomal morphology and interaction with Rab7 and Rab34. Mol. Biol. Cell 15, 815–826. doi: 10.1091/mbc.e03-06-0413
Webster, C. P., Smith, E. F., Bauer, C. S., Moller, A., Hautbergue, G. M., Ferraiuolo, L., et al. (2016). The C9orf72 protein interacts with Rab1a and the ULK 1 complex to regulate initiation of autophagy. EMBO J. 35, 1656–1676. doi: 10.15252/embj.201694401
Winslow, A. R., and Rubinsztein, D. C. (2011). The Parkinson disease protein α-synuclein inhibits autophagy. Autophagy 7, 429–431. doi: 10.4161/auto.7.4.14393
Winslow, A. R., Chen, C.-W., Corrochano, S., Acevedo-Arozena, A., Gordon, D. E., Peden, A. A., et al. (2010). α-Synuclein impairs macroautophagy: Implications for Parkinson’s disease. J. Cell Biol. 190, 1023–1037. doi: 10.1083/jcb.201003122
Wong, P.-M., Puente, C., Ganley, I. G., and Jiang, X. (2013). The ULK1 complex: Sensing nutrient signals for autophagy activation. Autophagy 9, 124–137. doi: 10.4161/auto.23323
Wood, A., Gurfinkel, Y., Polain, N., Lamont, W., and Lyn Rea, S. (2021). Molecular mechanisms underlying TDP-43 pathology in cellular and animal models of ALS and FTLD. Int. J. Mol. Sci. 22:4705. doi: 10.3390/ijms22094705
Woodman, P. G. (2000). Biogenesis of the sorting endosome: The role of Rab5. Traffic 1, 695–701. doi: 10.1034/j.1600-0854.2000.010902.x
Wu, W. K. K., Wu, Y. C., Yu, L., Li, Z. J., Sung, J. J. Y., and Cho, C. H. (2008). Induction of autophagy by proteasome inhibitor is associated with proliferative arrest in colon cancer cells. Biochem. Biophys. Res. Commun. 374, 258–263. doi: 10.1016/j.bbrc.2008.07.031
Wu, X., Bradley, M. J., Cai, Y., Kümmel, D., De La Cruz, E. M., Barr, F. A., et al. (2011). Insights regarding guanine nucleotide exchange from the structure of a DENN-domain protein complexed with its Rab GTPase substrate. Proc. Natl. Acad. Sci. U.S.A. 108, 18672–18677. doi: 10.1073/pnas.1110415108
Xu, J., Wang, S., Viollet, B., and Zou, M.-H. (2012). Regulation of the proteasome by AMPK in endothelial cells: The role of O-GlcNAc transferase (OGT). PLoS One 7:e36717. doi: 10.1371/journal.pone.0036717
Yamaguchi, H., Nakagawa, I., Yamamoto, A., Amano, A., Noda, T., and Yoshimori, T. (2009). An initial step of GAS-containing autophagosome-like vacuoles formation requires Rab7. PLoS Pathog. 5:e1000670. doi: 10.1371/journal.ppat.1000670
Yan, H., Li, W., Xu, J., Zhu, S., Long, X., and Che, J. (2013). D2 dopamine receptor antagonist raclopride induces non-canonical autophagy in cardiac myocytes. J. Cell Biochem. 114, 103–110. doi: 10.1002/jcb.24306
Yang, D.-S., Kumar, A., Stavrides, P., Peterson, J., Peterhoff, C. M., Pawlik, M., et al. (2008). Neuronal apoptosis and autophagy cross talk in aging PS/APP mice, a model of Alzheimer’s disease. Am. J. Pathol. 173, 665–681. doi: 10.2353/ajpath.2008.071176
Yang, M., Liang, C., Swaminathan, K., Herrlinger, S., Lai, F., Shiekhattar, R., et al. (2016). A C9ORF72/SMCR8-containing complex regulates ULK1 and plays a dual role in autophagy. Sci. Adv. 2:e1601167. doi: 10.1126/sciadv.1601167
Ylä-Anttila, P., and Eskelinen, E.-L. (2018). Roles for RAB24 in autophagy and disease. Small GTPases 9, 57–65. doi: 10.1080/21541248.2017.1317699
Ylä-Anttila, P., Mikkonen, E., Happonen, K. E., Holland, P., Ueno, T., Simonsen, A., et al. (2015). RAB24 facilitates clearance of autophagic compartments during basal conditions. Autophagy 11, 1833–1848. doi: 10.1080/15548627.2015.1086522
Yoshimura, S., Gerondopoulos, A., Linford, A., Rigden, D. J., and Barr, F. A. (2010). Family-wide characterization of the DENN domain Rab GDP-GTP exchange factors. J. Cell Biol. 191, 367–381. doi: 10.1083/jcb.201008051
You, L., Wang, Z., Li, H., Shou, J., Jing, Z., Xie, J., et al. (2015). The role of STAT3 in autophagy. Autophagy 11, 729–739. doi: 10.1080/15548627.2015.1017192
Yu, L., McPhee, C. K., Zheng, L., Mardones, G. A., Rong, Y., Peng, J., et al. (2010). Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 465, 942–946. doi: 10.1038/nature09076
Yue, Z., Friedman, L., Komatsu, M., and Tanaka, K. (2009). The cellular pathways of neuronal autophagy and their implication in neurodegenerative diseases. Biochim. Biophys. Acta Mol. Cell Res. 1793, 1496–1507. doi: 10.1016/j.bbamcr.2009.01.016
Yung, C., Sha, D., Li, L., and Chin, L.-S. (2016). Parkin protects against Misfolded SOD1 toxicity by promoting its aggresome formation and autophagic clearance. Mol. Neurobiol. 53, 6270–6287. doi: 10.1007/s12035-015-9537-z
Zhang, D., Iyer, L. M., He, F., and Aravind, L. (2012). Discovery of novel DENN proteins: Implications for the evolution of eukaryotic intracellular membrane structures and human disease. Front. Genet. 3:283. doi: 10.3389/fgene.2012.00283
Zhang, J., Jiang, Z., and Shi, A. (2022). Rab GTPases: The principal players in crafting the regulatory landscape of endosomal trafficking. Comput. Struct. Biotechnol. J. 20, 4464–4472. doi: 10.1016/j.csbj.2022.08.016
Zhang, M., Xi, Z., Misquitta, K., Sato, C., Moreno, D., Liang, Y., et al. (2017). C9orf72 and ATXN2 repeat expansions coexist in a family with ataxia, dementia, and parkinsonism. Mov. Disord. 32, 158–162. doi: 10.1002/mds.26841
Zhang, X., and Qian, S.-B. (2011). Chaperone-mediated hierarchical control in targeting misfolded proteins to aggresomes. Mol. Biol. Cell 22, 3277–3288. doi: 10.1091/mbc.E11-05-0388
Zhang, X., Li, L., Chen, S., Yang, D., Wang, Y., Zhang, X., et al. (2011). Rapamycin treatment augments motor neuron degeneration in SOD1 G93A mouse model of amyotrophic lateral sclerosis. Autophagy 7, 412–425. doi: 10.4161/auto.7.4.14541
Zhang, Y., Burberry, A., Wang, J.-Y., Sandoe, J., Ghosh, S., Udeshi, N. D., et al. (2018). The C9orf72-interacting protein Smcr8 is a negative regulator of autoimmunity and lysosomal exocytosis. Genes Dev. 32, 929–943. doi: 10.1101/gad.313932.118
Zhu, K., Dunner, K., and McConkey, D. J. (2010). Proteasome inhibitors activate autophagy as a cytoprotective response in human prostate cancer cells. Oncogene 29, 451–462. doi: 10.1038/onc.2009.343
Zhu, Q., Jiang, J., Gendron, T. F., McAlonis-Downes, M., Jiang, L., Taylor, A., et al. (2020). Reduced C9ORF72 function exacerbates gain of toxicity from ALS/FTD-causing repeat expansion in C9orf72. Nat. Neurosci. 23, 615–624. doi: 10.1038/s41593-020-0619-5
Zoppino, F. C. M., Militello, R. D., Slavin, I., Alvarez, C., and Colombo, M. I. (2010). Autophagosome formation depends on the small GTPase Rab1 and functional ER exit sites. Traffic 11, 1246–1261.
Keywords: C9ORF72, ALS-FTD, autophagy, autophagy-lysosomal pathway, RAB proteins, endosomal trafficking, neurodegeneration
Citation: Diab R, Pilotto F and Saxena S (2023) Autophagy and neurodegeneration: Unraveling the role of C9ORF72 in the regulation of autophagy and its relationship to ALS-FTD pathology. Front. Cell. Neurosci. 17:1086895. doi: 10.3389/fncel.2023.1086895
Received: 01 November 2022; Accepted: 01 March 2023;
Published: 16 March 2023.
Edited by:
Annakaisa Haapasalo, University of Eastern Finland, FinlandReviewed by:
Sheng Zhang, The University of Texas Health Science Center at Houston, United StatesMorwena Latouche, Université Paris Sciences et Lettres, France
Copyright © 2023 Diab, Pilotto and Saxena. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Smita Saxena, c21pdGEuc2F4ZW5hQGRibXIudW5pYmUuY2g=
†These authors have contributed equally to this work