 Yujun Wang
Yujun Wang Yuan Jin
Yuan Jin Qiong Zhang2
Qiong Zhang2- 1Department of Intensive Care Unit, The Central Hospital of Wuhan, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 2Department of Otorhinolaryngology–Head and Neck Surgery, The Central Hospital of Wuhan, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
Hearing loss is the most common congenital sensory impairment. Mutations or deficiencies of the GJB2 gene are the most common genetic cause of congenital non-syndromic deafness. Pathological changes such as decreased potential in the cochlea, active cochlear amplification disorders, cochlear developmental disorders and macrophage activation have been observed in various GJB2 transgenic mouse models. In the past, researchers generally believed that the pathological mechanisms underlying GJB2-related hearing loss comprised a K+ circulation defect and abnormal ATP-Ca2+ signals. However, recent studies have shown that K+ circulation is rarely associated with the pathological process of GJB2-related hearing loss, while cochlear developmental disorders and oxidative stress play an important, even critical, role in the occurrence of GJB2-related hearing loss. Nevertheless, these research has not been systematically summarized. In this review, we summarize the pathological mechanisms of GJB2-related hearing loss, including aspects of K+ circulation, developmental disorders of the organ of Corti, nutrition delivery, oxidative stress and ATP-Ca2+ signals. Clarifying the pathological mechanism of GJB2-related hearing loss can help develop new prevention and treatment strategies.
Introduction
Changes in the genes encoding gap junction proteins (connexins, Cx) are the most common genetic causes of hearing loss (He et al., 2017; Chai et al., 2022). Thus far, 21 human gap junction proteins have been identified. These are transmembrane proteins consisting of six identical or different gap junction protein monomers that form a hemichannel, which called connexon; the two connexons then form the gap junction channel (GJCs) between adjacent cell. GJCs allow for material exchange between adjacent cells, and small molecules (<1.4 kDa) such as ions, adenosine triphosphate (ATP) and miRNA can pass through them. Cx26 encoded by the GJB2 gene and Cx30 encoded by the GJB6 gene, the most abundant gap junction proteins in the inner ear, are expressed in the support cells of the organ of Corti (OC), the basal and intermediate cells of the stria vascularis, and fibroblasts in the spiral ligament. GJCs junctions formed by Cx26 plays an important role in the development, formation and maintenance of auditory function, and material exchange in inner ear cells. Up to now, more than 100 mutations associated with human hearing loss have been identified in the region of GJB2 gene. GJB2 mutation has been observed to be involved in the pathogenesis of hereditary deafness, delayed deafness and presbycusis. Although many valuable pathological phenomena have been elucidated using Gjb2 gene knockdown (KD) and Gjb2 mutant transgenic mice, the main pathological mechanism of hearing loss caused by GJB2 mutation is still unclear. This paper summarizes recent progress in research on GJB2-related hearing loss, attempting to identify the pathological mechanism of hearing loss caused by Cx26 mutation.
K+ circulation
The cochlea, a coiled structure located in the ventral region of the inner ear, acts as the primary structure for the perception of sound. The cochlea consists of three internal cavities filled with lymph fluid: from top to bottom, the scala vestibuli (SV), scala media (SM), and scala tympani (ST). The SV and ST contain perilymph fluid and traffic with each other through the cochlear aperture at the top of the cochlea (Jagger and Forge, 2015), while the SM contains endolymph fluid. Endolymph fluid and perilymph fluid constitute unique ionic environments (Zhang et al., 2023). Perilymph is similar to extracellular fluid, with a high concentration of Na+ and a low concentration of K+, and endolymph is similar to intracellular fluid, with a low concentration of Na+ and a high concentration of K+; this leads to a difference in potential between different parts of the cochlea in the resting state (Zhao, 2017). As endolymph contains up to 150 mM K+, 2 mM Na+, and 20 mM Ca2+, it exhibits a potential of approximately + 80 mV relative to the neighboring extracellular spaces. The cell bodies of hair cells are immersed in endolymph. When sound is transmitted to the inner ear, the mechanical vibration of the basilar membrane makes the hair cell bundle deflect and open the mechanically sensitive channel at the top of the stereocilia (Chai et al., 2018). Driven by endocochlear positive potential, K+ in the endolymph passes through the mechano-transduction channels in the hair cells’ hair bundles, thus generating auditory receptor current and potential. The K+ entering the hair cells passes through their basal lateral membrane via the K+ channel and is discharged. This K+ reaches the lateral cochlear wall through the GJCs network composed of Cx26 and Cx30 between Deiters cells and epithelial cells in the outer lymph or basement membrane and is finally discharged into the endolymph (Figure 1; Nin et al., 2012). Cx26 mutation is thought to damage the inner ear GJCs function and destroy the cochlear GJCs-mediated K+ circulation, resulting in toxic K+ accumulation around hair cells and eventually leading to hair cell degeneration and hearing loss. This process was once considered the main pathological mechanism of GJB2-related hearing loss. However, this model lacks sufficient experimental evidence and its theory is based on indirect evidence related to the expression of ion channels and connexins. This model also cannot explain why either mutated Cx26 or Cx30 cannot be compensated for by another kind of connexin; for example, Cx30 still exists in the cochlea of Gjb2 KD mice and should be located in the same region. Additionally, GJCs in the inner ear of Gjb2 gene KD mice retains permeability, which suggests that it theoretically still has K+ circulation ability. More importantly, hearing loss was detected in a Gjb2 R75W mouse model, but its endolymphatic potential was still within the normal range, which showed that impaired K+ circulation was not the main pathological mechanism of deafness caused by Gjb2 mutation but only a concomitant phenomenon (Inoshita et al., 2008). Moreover, an important aspect of the K+ circulation hypothesis is the degeneration of hair cells caused by the accumulation of K+ around them following Cx26 mutation. However, some studies have shown that the hearing loss caused by Gjb2 mutation occurs before the degeneration of hair cells, which also indicates that the K+ circulation is not the pathogenic mechanism of GJB2-related hearing loss (Liang et al., 2012).
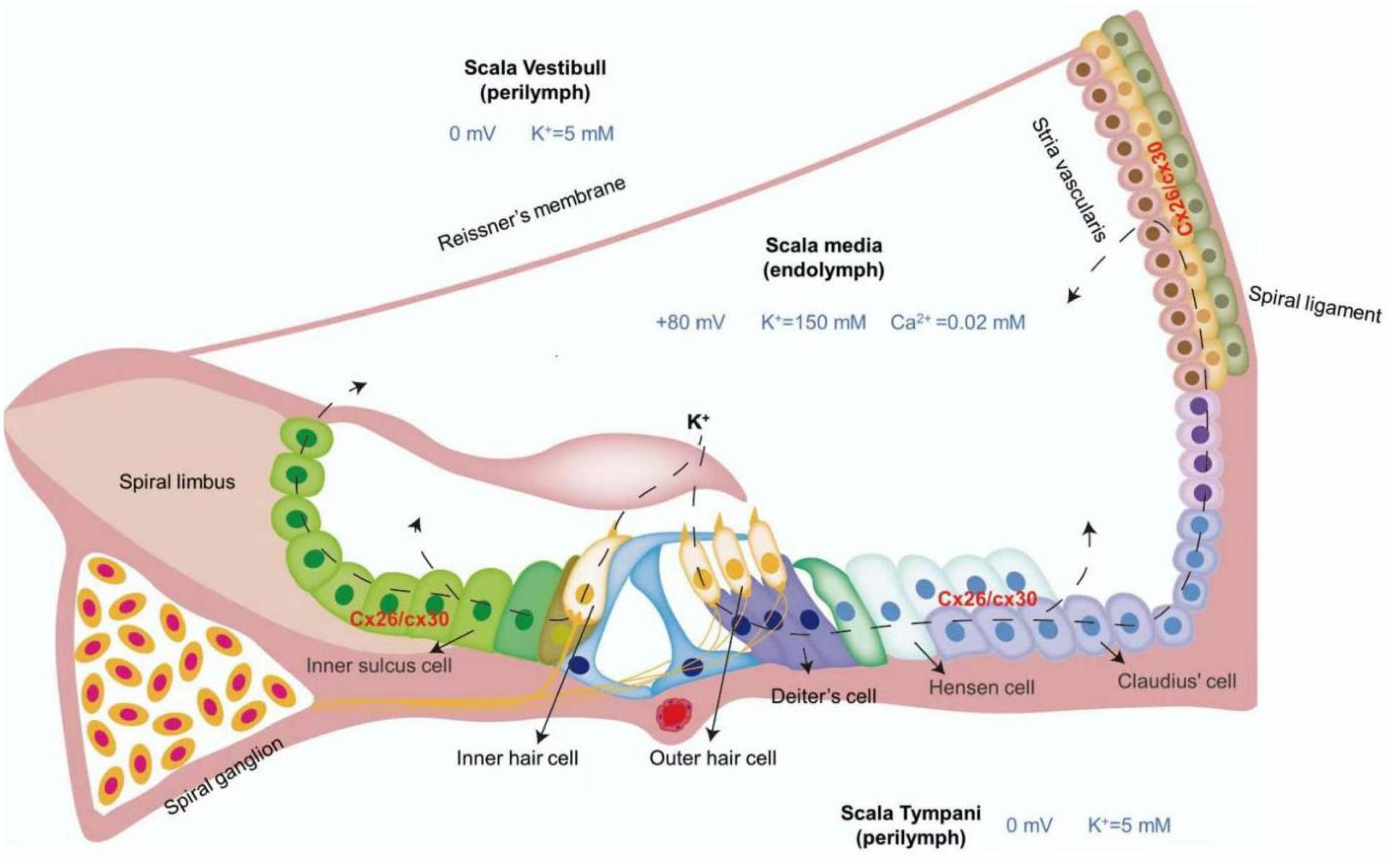
Figure 1. Diagram of K+ circulation.
Development of the organ of Corti
In independent lines of Cx26 null mice, researchers observed that the tunnel of Corti (TC) and Nuel’s space (NS) never opened and the postnatal development of OC was blocked (Minekawa et al., 2009). Morphological abnormalities in TC and NS were also observed in a Gjb2 R75W transgenic mouse model (Zhang et al., 2005). Thus, in four different Gjb2 transgenic mouse models, the postnatal development of OC was observed to stop before cell death in this organ. The opening of the TC and the formation of Nuel’s space are important events for the facilitation of hearing. The delayed or impaired development of normal physiological structures can cause serious hearing loss (Forrest et al., 2002). Dysplastic OC were observed in patients with keratitis-ichthyosis-deafness (KID) syndrome with a heterozygous GJB2 G45E variant (Griffith et al., 2006). Additionally, unopened TC and abnormal NS were observed in both Gjb2 variant carriers and mice. This cross-species phenomenon suggests that abnormalities in OC caused by the absence of Cx26 with normal physiological function may form the pathological basis for hearing defects.
The mechanism by which GJB2 mutation causes malformation of OC has not been fully clarified. Current research suggests that the opening of TC is related to the development of inner and outer pillar cells. Inoshita et al. observed that pillar cell bodies were differentiated from the surrounding cells during the expansion of TC at postnatal day (P)8 in both non-transgenic mice and Gjb2 R75W mice (Inoshita et al., 2008). However, there was a significant decrease in the number of microtubules in the inner column cells of the deformed OC in Gjb2 R75W mutant mice, while abundant microtubules were formed parallelly in non-transgenic mice. Chen et al. observed pathological changes in the cochlea of transgenic mice through the conditional KD of cochlear Cx26 at P0 and P8 (Chen et al., 2018). In the P8 KD group, Gjb2 KD did not significantly affect the morphology of OC. In the P0 KD group, TC and NS did not form during P5–P7 and the phalangeal process of Deiters cells did not develop into finger-like structures. The formation of microtubules in the pillar cells was significantly reduced, as was the amount of acetylated α-tubulin in these cells. These results suggest that the KD of the Gjb2 gene during the early postnatal period in mice may cause disorders in the cytoskeletal development of pillar cells. Liu X. et al. (2021) also observed that the KD of the Gjb2 gene during the early postnatal period in mice resulted in the reduction of F-actin levels in pillar cells. These studies suggest that the malformation of OC caused by Cx26 KD may be related to the disordered development of the cytoskeleton of pillar cells. Qiu et al. (2022) showed that the KD of cochlear Cx26 led to the malformed assembly of non-centrosomal microtubule-organizing centers far from the centrosome, accompanied by a decrease in the microtubule arrays emitted by abnormal non-centrosomal microtubule-organizing centers. The malformation of non-central microtubule-organizing centers in Cx26-deleted mice may lead to problems in the capture and anchoring of the microtubules of column cells, resulting in the malformation of OC.
The opening of TC may be related to the reduction of cell connections between the inner and outer pillar cells. The reduction of adhesion via E-cadherin between pillar cells is considered a key event (Whitlon, 1993; Johnen et al., 2012). Defourny et al. (2019) showed that Eph receptor A4 (EphA4) could bind with A Disintegrin And Metalloproteinase 10 (ADAM10) to promote the destruction of adhesion between inner and outer pillar cells based on E-cadherin. In the presence of defective EphA4 or an ADAM10 inhibitor, the adhesion junctions between pillar cells based on E-cadherin cannot be degraded normally. The cutting effect of the molecular scissors ADAM10 on cadherin was also found in other tissues and cells (Maretzky et al., 2005, 2008; Guerra et al., 2021). Zhang et al. (2022) observed that OC opened earlier in mice treated with triiodothyronine (T3) than in normal mice, accompanied by the formation of acetylated microtubules and a decrease of the adhesion junction molecule P-cadherin but not E-cadherin in pillar cells. This is consistent with the conclusions of other researchers. In general, the current research supports the notion that the development of the cytoskeleton of the pillar cells supports the height and deformation of the columnar cells, and the reduction of the adhesive connections formed by E-cadherin or P-cadherin between the columnar cells also participates in the opening of OC during its development and maturation (Figure 2). In addition, the abnormal development of the cytoskeleton may lead to other problems, such as affecting the transport of membrane proteins and thus affecting other cell functions. The Gjb2 variant or Gjb2 gene KD may affect the degradation of adhesion junctions between pillar cells through unknown pathways The Tspan protein family reported in other systems has a regulatory effect on ADAM10 splicing E-cadherin. Thus, further research on the relationship between Cx26 and Tspan may be valuable (Reyat et al., 2017; Seipold et al., 2018).
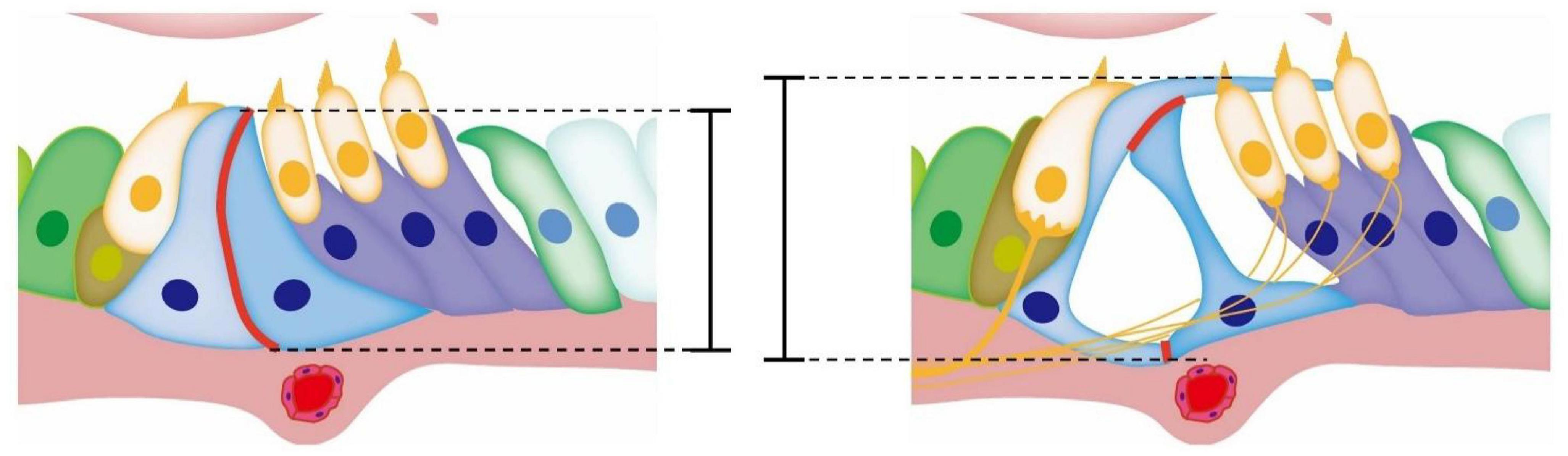
Figure 2. Diagram of the development of OC. The red mark denotes the adhesive connection between pillar cells. During the opening of TC, the adhesive junctions between pillar cell bodies gradually degrade and gaps appear in the originally tight cell membrane. With the development and growth of the cytoskeleton of inner and outer pillar cells, the height of OC increases, while TC appears between the inner and outer pillar cells.
Nutrient delivery
The spiral vessel exists at the bottom of the avascular sensor epiphyllum in the mammalian cochlea, but it is generally believed that blood vessel networks in the stria vascularis, spiral ligaments and spiral limbus also transport glucose and other metabolites to the sensory epithelium and sensory hair cells through GJCs between supporting cells (Santos-Sacchi and Dallos, 1983). Studies have shown that mice carrying Gjb2 homozygous defects die in the womb during the short period from early to middle pregnancy (Gabriel et al., 1998). This is because the placenta of Cx26-deficient mouse embryos is severely impaired in its ability to absorb glucose and other nutrients from maternal blood, hence inhibiting rapid organogenesis during the second trimester of pregnancy. Intercellular coupling via supporting cells’ GJCs provides an activity-dependent intercellular pathway for the delivery of nutrients from blood vessels to distal neurons. Fetoni et al. (2018) assessed the glucose uptake of Gjb2+/– mice with an insufficient Cx26 level using a fluorescent non-metabolizable D-glucose analyzer, 2-NBDG, and found that the 2-NBDG fluorescence emission in the stria vascularis of Gjb2+/– mice was significantly reduced compared with Gjb2+/– mice. Like Cx26, Cx30, which is most abundantly expressed in the cochlea and co-located with Cx26, has also been proven to play an indispensable role in the energy supply of the cochlear sensory epithelium. Nutritional deficiency affects ATP production and normal function and causes oxidative stress and other damage.
Oxidative stress
Reactive oxygen species (ROS) play an important role in cell biology (He et al., 2017, 2020; Liu W. et al., 2021). ROS are indispensable in physiological processes such as protein phosphorylation, ion channels and the redox regulation of transcription factors (Zhong et al., 2020). However, prolonged exposure to high ROS concentrations may cause non-specific damage to proteins, lipids and nucleic acids, leading to the loss or destruction of cell function (Wang et al., 2022). Like the plasma membrane half-channel formed by Cx43 through which glutathione (GSH) is released by brain astrocytes to help neurons resist oxidation, the intercellular connection formed by supporting cells in the cochlea also has positive significance for GSH release and oxidation. Fetoni et al. (2018) compared the mRNA expression profile of Gjb2–/– mice at P5 with age-matched Gjb2 loxP/loxP mice. The results showed the activation of metabolic signaling pathways such as oxidative metabolism, the antioxidant defense system and glutathione metabolism (Fetoni et al., 2018). The amount of GSH released from cochlear explants from P5 Gjb2–/– mice was significantly lower than that from Gjb2 loxP/loxP mice. The total amount of glutathionylated proteins in the cochlea and their expression in the spiral ganglion neurons (SGNs), OC and stria vascularis were significantly reduced in 6-month-old Gjb2+/– mice compared with Gjb2 loxP/loxP mice. These findings indicate that the function of the cochlear antioxidant defense system in Cx26-deficient mice is impaired. Dihydroethidium (DHE, a lipophilic cell-permeable dye that is rapidly oxidized to ethidium in the presence of superoxide free radicals) was used to compare ROS content in the cochlea of Gjb2–/– mice at P5 and age-matched Gjb2 loxP/loxP mice. The results showed that the DHE signal in the cochlea of Cx26-deficient mice was significantly higher. Similarly, in the cochlea of Gjb2+/– mice aged 2, 6, and 12 months, the staining signals of DHE and 4-hydroxy-2-nonenal (4-HNE, a lipid peroxidation product) were also observed to be significantly higher than those in age-matched Gjb2 loxP/loxP mice, while the 2-month-old Gjb2+/– mice showed no significant hearing loss compared with Gjb2 loxP/loxP mice. Moreover, the antioxidant stress signaling pathway Nrf2/ARE and its response product, the HO-1 enzyme, are also activated in the cochlea of Gjb2+/– mice. These findings indicate that oxidative metabolism and ROS play an important role in hearing loss caused by Cx26 KD. A significantly higher ROS level was also observed in the cochlea of Cx30 null mice (Teubner et al., 2003). A genome-wide association studies (GWAS) carried out in a large cohort of 4,091 individuals originating from Europe, Caucasus and Central Asia, with hearing phenotype (including 1,076 presbycusis patients and 1,290 healthy matched controls), showed a statistically significant association with two genes (PRKCE and TGFB1) related to the Nrf2 pathway, further supporting the hypothesis that elements of the Nrf2 pathway are essential for hearing maintenance (Fetoni et al., 2018).
ATP release and Ca2+ signals
ATP release and the associated calcium signal transduction play an important role in inner ear development and hearing (Bobbin and Thompson, 1978; Kujawa et al., 1994; Muñoz et al., 1995; Housley et al., 1999). ATP can be used as a neurotransmitter, and its receptors can be divided into two major classes of purinergic receptors (P1 receptors and P2 receptors), each with multiple subtypes (Huang et al., 2010). P2 receptors are classified into two major groups, P2X and P2Y. P2X receptors are non-selective ATP-gated ion channels with high permeability for Na+, K+, and Ca2+, while P2Y receptors are G protein-coupled metabotropic receptors that activate phospholipase C (PLC), resulting in the activation of the second messengers diacylglycerol and inositol trisphosphate (IP3). The P2Y1, P2Y2, P2Y4, P2Y6, and P2Y12 receptors have been reported to be expressed in the sensory and non-sensory cells of OC and the spiral ganglion neurons of developing rat cochlea (Huang et al., 2010), while P2X2, P2Y4, and P2X7 are distributed on the OHC surface and correlated with the electrical activity of the OHC (Nikolic et al., 2003; Tritsch et al., 2007, 2010; Dayaratne et al., 2014). When the purinergic receptor P2Y of inner ear cells is activated by ATP, PLC-dependent IP3 is released and activates the endoplasmic reticulum IP3 receptor, which promotes the release of endoplasmic reticulum Ca2+ and raises the cytosolic free Ca2+ concentration, thus inducing Ca2+ signal transduction. ATP has been observed to trigger Ca2+ waves in the Kolliker’s organ of the immature OC as well as Ca2+ APs in IHCs (Tritsch et al., 2007, 2010; Anselmi et al., 2008; Majumder et al., 2010). ATP-induced IP3-dependent intercellular Ca2+ signals propagate radially across this cochlear cellular network at a uniform speed of 10 to 15 μm/s (Gale et al., 2004; Piazza et al., 2007). In the developing cochlea, the waves of ATP released from non-sensory cells of the greater epithelial ridge cause Ca2+ action potentials of immature inner hair cells to occur in brief bursts during episodes of spontaneous depolarization, which promotes the maturation of hair cells and SGNs and refines axonal projections from the cochlea to the central nervous system (Tritsch et al., 2010). In addition, exogenous ATP can depolarize type II neurons both directly and by evoking glutamate synaptic inputs from external hair cells, but the effect of this type of ATP decreases with age at birth (Greenwood et al., 2007).
Stout et al. (2002) reported that the level of extracellular ATP in a culture of Cx43-transfected cells was higher than that in a culture of the parent cells, which suggested that ATP release in astrocytes and glioma cells might be mediated by the Cx43 half-channel. Zhao and Santos-Sacchi (1999) showed that connexin hemichannels in cochlear supporting cells could release ATP under physiological conditions, with the level corresponding with physiological ATP concentrations in the cochlea, thus accounting for the submicromolar concentrations measured in cochlear fluids in vivo (Inoshita et al., 2008). Extracellular ATP could alter OHC electromotility through the activation of P2 purinergic receptors. Therefore, Cx26 dysfunction affects OHC electromotility and hearing function. A cochlear organotypic culture study showed that connexins make up the channels responsible for ATP-induced Ca2+ waves in the outer sulcus over Panx1 and P2X7 (Zhao and Santos-Sacchi, 1999). Additionally, the infusion of ATP into the cochlea can reduce and inhibit the otoacoustic emission of cochlear microsounds, complex action potential and distortion products, which can reduce active cochlear mechanics and hearing sensitivity. These studies suggest that GJc plays an important role in the ATP release and intercellular Ca2+ signaling transmission of inner ear Sertoli cells. In addition, a decrease in Ca2+ concentration increases the opening of chemical channels, which promotes ATP release from the cytoplasm to the endolymph through connexin hemichannels. Mechanical stimulation also induces hemichannels to release ATP. As sound-induced mechanical vibration increases, hemichannel ATP release increases to reduce OHC electromotility as a possible protective mechanism, which can enhance auditory sensitivity when the sound intensity is low (Brownell et al., 1985; Ashmore, 1987; Dallos, 1992; Anselmi et al., 2008; Tritsch et al., 2010).
The study of Ca2+ signals and ATP release requires drugs such as Cx channel blockers and Ca2+ indicators. The pharmacological compounds used in most studies involving connexin are not specific and have been proven to affect the activities of various other channels, while the putative conduits expressing ATP in inner ear cells may include connexin channels, P2X7 receptors (P2X7Rs), pannexin channels, anime channels, vessels and transports (Spray et al., 2006). Thus, rigorous standards must be applied for the detection of hemichannel expression and function.
Summary and outlook
Gap junction channels composed of Cx26 allow for the exchange of small molecules such as glucose, second messengers, ions and miRNA. Previous studies have suggested that Cx26 plays a role in the K+ circulation of the inner ear, maintaining the high potential of the inner ear and the electrical activity of hair cells. Mutant Cx26 is unable to support K+ circulation, thus impairing the amplification function of the cochlea. However, recent studies have shown that K+ circulation disorder is not the main pathological mechanism of Cx26 mutation-related deafness. Researchers believe that Cx26 mutation causes changes in Ca2+ signaling and ATP release as well as columnar cell cytoskeletal developmental disorders, all of which contribute to the occurrence of hearing loss. Recent research also found that cochlear macrophages participated in the process of outer hair cell loss in Cx26 mutant mice. The systemic application of dexamethasone can prevent the loss of outer hair cells and improve hearing. The study of Cx26 mutation-related hearing loss should be based on a deeper understanding of the physiological functions and regulatory networks of the Cx26 protein. There is still a long way to go in developing treatments based on the pathological processes of Cx26-related deafness to completely restore hearing.
Cochlear implantation is an effective treatment strategy for patients with hearing loss caused by GJB2 variants. Gene therapy has also made significant progress in restoring the hearing of patients with hereditary deafness. Adeno-associated virus (AAV)-mediated gene therapy has proved to be helpful in restoring the hearing of Gjb2 mutant mice. Using an AAV vector, the Gjb2 gene was successfully expressed in the cochlear Sertoli cells of newborn mice through the round window membrane, leading to significant improvements in the auditory responses and development of the cochlear structure (Yu et al., 2014). With the continuous improvement of AAV serotypes and capsid modifications, this technology will likely contribute to the development of clinical treatments for hereditary deafness.
Author contributions
YJ and YW wrote the manuscript. WC designed the manuscript. QZ, YX, XG, and SZ provided the reference materials. All authors read and approved the final manuscript.
Funding
This work was supported by the project funded by the Wuhan Municipal Health Commission (WX21Q51).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Anselmi, F., Hernandez, V. H., Crispino, G., Seydel, A., Ortolano, S., Roper, S., et al. (2008). ATP release through connexin hemichannels and gap junction transfer of second messengers propagate Ca2+ signals across the inner ear. Proc. Natl. Acad. Sci. U.S.A. 105, 18770–18775. doi: 10.1073/pnas.0800793105
Ashmore, J. F. (1987). A fast motile response in guinea-pig outer hair cells: the cellular basis of the cochlear amplifier. J. Physiol. 388, 323–347.
Bobbin, R. P., and Thompson, M. H. (1978). Effects of putative transmitters on afferent cochlear transmission. Ann. Otol. Rhinol. Laryngol. 87(2 Pt 1), 185–190.
Brownell, W. E., Bader, C. R., Bertrand, D., and Ribaupierre, Y d (1985). Evoked mechanical responses of isolated cochlear outer hair cells. Science 227, 194–196.
Chai, R., Li, G., Wang, J., and Zou, J. (2018). hearing loss: reestablish the neural plasticity in regenerated spiral ganglion neurons and sensory hair cells 2018. Neural Plast. 2018:4759135. doi: 10.1155/2018/4759135
Chai, R., Li, H., Yang, T., Sun, S., and Yuan, Y. (2022). Editorial: hearing loss: mechanisms and prevention. Front. Cell Dev. Biol. 10:838271. doi: 10.3389/fcell.2022.838271
Chen, S., Xie, L., Xu, K., Cao, H., Wu, X., Xu, X., et al. (2018). Developmental abnormalities in supporting cell phalangeal processes and cytoskeleton in the GJB2GJB2 knockdown mouse model. Dis. Model Mech. 11:dmm033019. doi: 10.1242/dmm.033019
Dayaratne, M. W., Vlajkovic, S., Lipski, J., and Thorne, P. R. (2014). Kolliker’s organ and the development of spontaneous activity in the auditory system: implications for hearing dysfunction. Biomed. Res. Int. 2014:367939. doi: 10.1155/2014/367939
Defourny, J., Peuckert, C., Kullander, K., and Malgrange, B. (2019). EphA4-ADAM10 interplay patterns the cochlear sensory epithelium through local disruption of Adherens junctions. iScience 11, 246–257. doi: 10.1016/j.isci.2018.12.017
Fetoni, A. R., Zorzi, V., Paciello, F., Ziraldo, G., Peres, C., Raspa, M., et al. (2018). Cx26 partial loss causes accelerated presbycusis by redox imbalance and dysregulation of Nfr2 pathway. Redox Biol. 19, 301–317. doi: 10.1016/j.redox.2018.08.002
Forrest, D., Reh, T. A., and Rusch, A. (2002). Neurodevelopmental control by thyroid hormone receptors. Curr. Opin. Neurobiol. 12, 49–56.
Gabriel, H. D., Jung, D., Bützler, C., Temme, A., Traub, O., Winterhager, E., et al. (1998). Transplacental uptake of glucose is decreased in embryonic lethal connexin26-deficient mice. J. Cell Biol. 140, 1453–1461. doi: 10.1083/jcb.140.6.1453
Gale, J. E., Piazza, V., Ciubotaru, C. D., and Mammano, F. (2004). A mechanism for sensing noise damage in the inner ear. Curr. Biol. 14, 526–529.
Greenwood, D., Jagger, D., Huang, L., Hoya, N., Thorne, P., and Wildman, S. (2007). P2X receptor signaling inhibits BDNF-mediated spiral ganglion neuron development in the neonatal rat cochlea. Development 134, 1407–1417. doi: 10.1242/dev.002279
Griffith, A., Yang, Y., Pryor, S., Park, H., Jabs, E., and Joseph, B. Jr., et al. (2006). Cochleosaccular dysplasia associated with a connexin 26 mutation in keratitis-ichthyosis-deafness syndrome. Laryngoscope 116, 1404–1408. doi: 10.1097/01.mlg.0000224549.75161.ca
Guerra, E., Trerotola, M., Relli, V., Lattanzio, R., Tripaldi, R., Vacca, G., et al. (2021). Trop-2 induces ADAM10-mediated cleavage of E-cadherin and drives EMT-less metastasis in colon cancer. Neoplasia 23, 898–911. doi: 10.1016/j.neo.2021.07.002
He, Z., Guo, L., Shu, Y., Fang, Q., Zhou, H., Liu, Y., et al. (2017). Autophagy protects auditory hair cells against neomycin-induced damage. Autophagy 13, 1884–1904. doi: 10.1080/15548627.2017.1359449
He, Z., Zou, S., Li, M., Liao, F., Wu, X., Sun, H., et al. (2020). The nuclear transcription factor FoxG1 affects the sensitivity of mimetic aging hair cells to inflammation by regulating autophagy pathways. Redox Biol. 28:101364. doi: 10.1016/j.redox.2019.101364
Housley, G. D., Kanjhan, R., Raybould, N. P., Greenwood, D., Salih, S. G., Järlebark, L., et al. (1999). Expression of the P2X(2) receptor subunit of the ATP-gated ion channel in the cochlea: implications for sound transduction and auditory neurotransmission. J. Neurosci. 19, 8377–8388. doi: 10.1523/JNEUROSCI.19-19-08377.1999
Huang, L., Thorne, P., Vlajkovic, S., and Housley, G. (2010). Differential expression of P2Y receptors in the rat cochlea during development. Purinergic Signal. 6, 231–248.
Inoshita, A., Iizuka, T., Okamura, H., Minekawa, A., Kojima, K., Furukawa, M., et al. (2008). Postnatal development of the organ of Corti in dominant-negative GJB2GJB2 transgenic mice. Neuroscience 156, 1039–1047. doi: 10.1016/j.neuroscience.2008.08.027
Jagger, D. J., and Forge, A. (2015). Connexins and gap junctions in the inner ear–it’s not just about K(+) recycling. Cell Tissue Res. 360, 633–644.
Johnen, N., Francart, M., Thelen, N., Cloes, M., and Thiry, M. (2012). Evidence for a partial epithelial-mesenchymal transition in postnatal stages of rat auditory organ morphogenesis. Histochem. Cell Biol. 138, 477–488. doi: 10.1007/s00418-012-0969-5
Kujawa, S. G., Erostegui, C., Fallon, M., Crist, J., and Bobbin, R. P. (1994). Effects of adenosine 5’-triphosphate and related agonists on cochlear function. Hear Res. 76, 87–100.
Liang, C., Zhu, Y., Zong, L., Lu, G., and Zhao, H. (2012). Cell degeneration is not a primary causer for Connexin26 (GJB2) deficiency associated hearing loss. Neurosci. Lett. 528, 36–41.
Liu, W., Xu, L., Wang, X., Zhang, D., Sun, G., Wang, M., et al. (2021). PRDX1 activates autophagy via the PTEN-AKT signaling pathway to protect against cisplatin-induced spiral ganglion neuron damage. Autophagy 17, 4159–4181. doi: 10.1080/15548627.2021.1905466
Liu, X., Jin, Y., Chen, S., Xu, K., Xie, L., Qiu, Y., et al. (2021). F-actin dysplasia involved in organ of corti deformity in GJB2 knockdown mouse model. Front. Mol. Neurosci. 14:808553. doi: 10.3389/fnmol.2021.808553
Majumder, P., Crispino, G., Rodriguez, L., Ciubotaru, C., Anselmi, F., and Piazza, V. (2010). ATP-mediated cell-cell signaling in the organ of Corti: the role of connexin channels. Purinergic Signal 6, 167–187. doi: 10.1007/s11302-010-9192-9
Maretzky, T., Reiss, K., Ludwig, A., Buchholz, J., Scholz, F., and Proksch, E. (2005). ADAM10 mediates E-cadherin shedding and regulates epithelial cell-cell adhesion, migration, and beta-catenin translocation. Proc. Natl. Acad. Sci. U.S.A. 102, 9182–9187. doi: 10.1073/pnas.0500918102
Maretzky, T., Scholz, F., Köten, B., Proksch, E., Saftig, P., and Reiss, K. (2008). ADAM10-mediated E-cadherin release is regulated by proinflammatory cytokines and modulates keratinocyte cohesion in eczematous dermatitis. J. Invest. Dermatol. 128, 1737–1746. doi: 10.1038/sj.jid.5701242
Minekawa, A., Abe, T., Inoshita, A., Iizuka, T., Kakehata, S., Narui, Y., et al. (2009). Cochlear outer hair cells in a dominant-negative connexin26 mutant mouse preserve non-linear capacitance in spite of impaired distortion product otoacoustic emission. Neuroscience 164, 1312–1319. doi: 10.1016/j.neuroscience.2009.08.043
Muñoz, D. J., Thorne, P. R., Housley, G. D., Billett, T. E., and Battersby, J. M. (1995). Extracellular adenosine 5’-triphosphate (ATP) in the endolymphatic compartment influences cochlear function. Hear Res. 90, 106–118.
Nikolic, P., Housley, G., and Thorne, P. (2003). Expression of the P2X7 receptor subunit of the adenosine 5’-triphosphate-gated ion channel in the developing and adult rat cochlea. Audiol. Neurootol. 8, 28–37. doi: 10.1159/000067891
Nin, F., Hibino, H., Murakami, S., Suzuki, T., Hisa, Y., and Kurachi, Y. (2012). Computational model of a circulation current that controls electrochemical properties in the mammalian cochlea. Proc. Natl. Acad. Sci. U.S.A. 109, 9191–9196. doi: 10.1073/pnas.1120067109
Piazza, V., Ciubotaru, C. D., Gale, J., and Mammano, F. (2007). Purinergic signalling and intercellular Ca2+ wave propagation in the organ of Corti. Cell Calcium 41, 77–86.
Qiu, Y., Xu, K., Xie, L., Chen, S., and Sun, Y. (2022). The reduction in microtubule arrays caused by the dysplasia of the non-centrosomal microtubule-organizing center leads to a malformed organ of Corti in the Cx26-null mouse. Biomedicines 10:1364. doi: 10.3390/biomedicines10061364
Reyat, J. S., Chimen, M., Noy, P., Szyroka, J., Rainger, G., and Tomlinson, M. (2017). ADAM10-interacting tetraspanins Tspan5 and Tspan17 regulate VE-cadherin expression and promote T lymphocyte transmigration. J. Immunol. 199, 666–676. doi: 10.4049/jimmunol.1600713
Santos-Sacchi, J., and Dallos, P. (1983). Intercellular communication in the supporting cells of the organ of Corti. Hear Res. 9, 317–326.
Seipold, L., Altmeppen, H., Koudelka, T., Tholey, A., Kasparek, P., Sedlacek, R., et al. (2018). In vivo regulation of the A disintegrin and metalloproteinase 10 (ADAM10) by the tetraspanin 15. Cell Mol. Life Sci. 75, 3251–3267. doi: 10.1007/s00018-018-2791-2
Spray, D. C., Ye, Z. C., and Ransom, B. R. (2006). Functional connexin “hemichannels”: a critical appraisal. Glia 54, 758–773. doi: 10.1002/glia.20429
Stout, C., Costantin, J., Naus, C., and Charles, A. C. (2002). Intercellular calcium signaling in astrocytes via ATP release through connexin hemichannels. J. Biol. Chem. 277, 10482–10488.
Teubner, B., Michel, V., Pesch, J., Lautermann, J., Cohen-Salmon, M., Söhl, G., et al. (2003). Connexin30 (Gjb6)-deficiency causes severe hearing impairment and lack of endocochlear potential. Hum. Mol. Genet. 12, 13–21. doi: 10.1093/hmg/ddg001
Tritsch, N. X., Rodríguez-Contreras, A., Crins, T. H., Wang, H., Borst, J. G., and Bergles, D. E. (2010). Calcium action potentials in hair cells pattern auditory neuron activity before hearing onset. Nat. Neurosci. 13, 1050–1052.
Tritsch, N. X., Yi, E., Gale, J. E., Glowatzki, E., and Bergles, D. E. (2007). The origin of spontaneous activity in the developing auditory system. Nature 450, 50–55.
Wang, M., Dong, Y., Gao, S., Zhong, Z., Cheng, C., Qiang, R., et al. (2022). Hippo/YAP signaling pathway protects against neomycin-induced hair cell damage in the mouse cochlea. Cell Mol. Life Sci. 79:79. doi: 10.1007/s00018-021-04029-9
Whitlon, D. S. (1993). E-cadherin in the mature and developing organ of Corti of the mouse. J. Neurocytol. 22, 1030–1038.
Yu, Q., Wang, Y., Chang, Q., Wang, J., Gong, S., Li, H., et al. (2014). Virally expressed connexin26 restores gap junction function in the cochlea of conditional GJB2GJB2 knockout mice. Gene Ther. 21, 71–80. doi: 10.1038/gt.2013.59
Zhang, H., Xie, L., Chen, S., Qiu, Y., Sun, Y., and Kong, W. (2022). Thyroxine regulates the opening of the organ of corti through affecting P-cadherin and acetylated microtubule. Int. J. Mol. Sci. 23:13339. doi: 10.3390/ijms232113339
Zhang, Y., Fang, Q., Wang, H., Qi, J., Sun, S., Liao, M., et al. (2023). Increased mitophagy protects cochlear hair cells from aminoglycoside-induced damage. Autophagy 19, 75–91.
Zhang, Y., Tang, W., Ahmad, S., Sipp, J., Chen, P., and Lin, X. (2005). Gap junction-mediated intercellular biochemical coupling in cochlear supporting cells is required for normal cochlear functions. Proc. Natl. Acad. Sci. U.S.A. 102, 15201–15206. doi: 10.1073/pnas.0501859102
Zhao, H. B. (2017). Hypothesis of K(+)-recycling defect is not a primary deafness mechanism for Cx26 (GJB2) deficiency. Front. Mol. Neurosci. 10:162.
Zhao, H. B., and Santos-Sacchi, J. (1999). Auditory collusion and a coupled couple of outer hair cells. Nature 399, 359–362. doi: 10.1038/20686
Keywords: GJB2 gene, gap junction protein, hearing loss, K+ circulation, cochlear
Citation: Wang Y, Jin Y, Zhang Q, Xiong Y, Gu X, Zeng S and Chen W (2023) Research progress in delineating the pathological mechanisms of GJB2-related hearing loss. Front. Cell. Neurosci. 17:1208406. doi: 10.3389/fncel.2023.1208406
Received: 19 April 2023; Accepted: 15 May 2023;
Published: 02 June 2023.
Edited by:
Zuhong He, Wuhan University, ChinaReviewed by:
Ao Li, Nanjing Drum Tower Hospital, ChinaZheng-De Du, Capital Medical University, China
Copyright © 2023 Wang, Jin, Zhang, Xiong, Gu, Zeng and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Chen, Y3c5OTk5MEAxMjYuY29t
†These authors have contributed equally to this work