

- 1 Center for Neural Science, New York University, New York City, NY, USA
- 2 Laboratoire de Neurophysique et Physiologie, Université Paris Descartes, Paris, France
- 3 Centre National de la Recherche Scientifique, Paris, France
We review biophysical models of synaptic plasticity, with a focus on spike-timing dependent plasticity (STDP). The common property of the discussed models is that synaptic changes depend on the dynamics of the intracellular calcium concentration, which itself depends on pre- and postsynaptic activity. We start by discussing simple models in which plasticity changes are based directly on calcium amplitude and dynamics. We then consider models in which dynamic intracellular signaling cascades form the link between the calcium dynamics and the plasticity changes. Both mechanisms of induction of STDP (through the ability of pre/postsynaptic spikes to evoke changes in the state of the synapse) and of maintenance of the evoked changes (through bistability) are discussed.
Introduction
Long-term synaptic modifications have long been postulated to occur in response to the simultaneous activation of both pre- and postsynaptic neurons (Hebb, 1949). Recent experimental techniques allow precise control over pre- and postsynaptic spiking activity. Such experiments provide evidence at the single-cell level that coincidence between afferent input with postsynaptic spiking evokes long-term modifications. In general, presynaptic input (onset of the excitatory postsynaptic potential – EPSP) occurring closely before or after a postsynaptic action potential results in maximal synaptic modification, while no plasticity occurs if the temporal difference between both is large.
In the hippocampus (Levy and Steward, 1983; Gustafsson et al., 1987; Magee and Johnston, 1997; Bi and Poo, 1998), the Xenopus tectum (Zhang et al., 1998), the visual cortex (Markram et al., 1997; Sjöström et al., 2001), and the somatosensory cortex (Feldman, 2000) an EPSP occurring prior to the backpropagating action potential (pre–post pairing) evokes long-term potentiation (LTP), and the anti-causal order, i.e., the EPSP occurs after the postsynaptic neuron spiked (post–pre pairing), leads to long-term depression (LTD). Such a temporal order of potentiation and depression occurrence is generally referred to as the “classical” spike-timing dependent plasticity (STDP) rule. Since the early STDP experiments, numerous studies in different brain regions and under varying experimental conditions have revealed a plethora of STDP shapes (see Abbott and Nelson, 2000 for a review). Further studies investigating plasticity results in response to triplets and quadruplets of spikes have highlighted the non-linearity of plasticity results (Bi and Wang, 2002; Froemke and Dan, 2002; Wang et al., 2005). In this review we will mostly focus on biophysical models accounting for the “classical” form of STDP observed in hippocampal and neocortical pyramidal cells as well as in retinotectal connections.
What are the biochemical mechanisms operating at the synapse leading to the observed plasticity outcomes? Enormous experimental effort has been devoted to the identification of the molecular players both mediating and modulating synaptic plasticity (Malenka and Bear, 2004). Tremendous advances have been made in identifying which constituents take part in the induction of LTP for pre–post pairs or LTD for post–pre pairs, for example. The biological mechanisms underlying spike-timing synaptic plasticity have furthermore inspired mathematical models that strive to reproduce aspects of STDP results.
Other less well studied questions are: What is the nature of the synaptic change (continuous or discrete)? What biological machinery stably maintains the evoked synaptic state over time scales of minutes to hours or more? Only a few experimental studies have investigated synaptic changes on putative single synaptic connections. These studies consistently find all-or-none switch like events (Petersen et al., 1998; Bagal et al., 2005; O’Connor et al., 2005b). These experiments suggest that the synapse exists in only two states of high- and low transmission strength respectively, and that transitions between these states can be evoked by specific stimulation protocols.
Synapse models including protein signaling cascades often exhibit bistability due to positive feedback loops in the modeled pathways. In such models, synaptic changes correspond to transitions between two states, and the stable maintenance of synaptic changes is accounted for by bistability (we discuss briefly in section “Bi/multistable models based on alternative mechanisms” recent models that have more than two stable states). In the absence of bistability, changes evoked by a given stimulation protocol are expected to decay in time. Therefore, models lacking bistability are not expected to possess long-term memory properties at the level of a single synapse (but see Delord et al., 2007). Alternatively, stable memory retention has been proposed to rely on reinforcing the LTP/LTD of a synapse through network dynamics, thus stabilizing otherwise unstable synapses (Abraham and Robins, 2005; Billings and van Rossum, 2009).
Here, we review “biophysical” models of synaptic plasticity whose aim is to reproduce spike-timing dependent plasticity experiments but also to understand the mechanisms that convert a given firing pattern of pre- and postsynaptic cells into a specific synaptic change. Such approaches are to be distinguished from purely phenomenological models of STDP which directly link the time difference between pre- and postsynaptic spikes to a particular synaptic change (see Morrison et al., 2008 for a review). In particular, we focus on models which try to link the calcium dynamics evoked by pre- and postsynaptic activity to observed plasticity outcomes. We discuss in particular how specific biophysical features give rise to specific components of the STDP outcome. We start from phenomenological models of synaptic plasticity based purely on the dynamics of calcium concentration in dendritic spines (section ‘Phenomenological models based on calcium dynamics’). We then turn to models that explicitly describe the protein signaling cascades that have been shown experimentally to be involved in synaptic plasticity, with a special emphasis on calcium/calmodulin-dependent protein kinase II (CaMKII)-based models (section ‘Models including biochemical signaling cascades beyond calcium’). We point out that, unlike the more phenomenological models based on calcium only, the detailed models exhibit bistable behavior, which allows them to maintain synaptic changes for (in principle) arbitrary amounts of time. Last, we discuss how such bistable synapse models can account for spike-timing dependent plasticity.
Phenomenological Models Based on Calcium Dynamics
An increase in postsynaptic calcium concentration is a necessary (Lynch et al., 1983; Zucker, 1999; Mizuno et al., 2001; Ismailov et al., 2004; Nevian and Sakmann, 2006) and sufficient (Malenka et al., 1988; Neveu and Zucker, 1996; Yang et al., 1999) signal to induce synaptic changes. We start here by briefly reviewing the experimental evidence supporting this statement. We then turn to discuss phenomenological models reproducing STDP plasticity results based on the calcium concentration dynamics.
Calcium is a Key Signal for Plasticity: Experimental Data
In most synapses, synaptic activation leads to calcium entry in the postsynaptic terminal through N-methyl-D-aspartic acid receptor (NMDA-R)-channels (Koester and Sakmann, 1998; Yuste et al., 1999; Kovalchuk et al., 2000). Backpropagating action potentials (BPAPs) produce calcium influx through voltage-dependent calcium channels (VDCCs) (Jaffe et al., 1992; Yuste and Denk, 1995; Majewska et al., 2000; Sabatini and Svoboda, 2000). The induction of LTP at the hippocampal Schaffer collateral – CA1 neuron synapse necessitates activation of NMDA receptors (Collingridge et al., 1983; Bliss and Collingridge, 1993), while basal synaptic transmission and the maintenance of the potentiated state are not affected by NMDA blockade (Morris et al., 1986). The requirement of NMDA activation for LTP induction has also been identified between thick, tufted layer V pyramidal neurons in rat visual cortex (Artola and Singer, 1987; Bear et al., 1992; Markram et al., 1997; Sjöström et al., 2001), in layer IV to layer II/III pyramidal cell synapses in the somatosensory cortex (Castro-Alamancos et al., 1995; Feldman, 2000; Nevian and Sakmann, 2006), and in the lateral geniculate nucleus (Hahm et al., 1991; Mooney et al., 1993).
Long-term potentiation induction evoked by STDP protocols also depends on the large calcium influx through NMDA-Rs in the hippocampus (Magee and Johnston, 1997) and the somatosensory cortex (Nevian and Sakmann, 2006). The induction of spike-timing dependent LTD, however, is mediated by the activation of presynaptic NMDA-Rs (Sjöström et al., 2003; Bender et al., 2006; Nevian and Sakmann, 2006). Nevian and Sakmann (2006) show in the somatosensory cortex that burst-pairing induced LTD is independent of postsynaptic activation of NMDA-Rs, while the postsynaptic calcium influx through VDCCs is necessary for the induction of LTD. On the other hand, VDCC antagonists (nimodipine for L-type channels; or Ni2+ for T-type channels) block spike-timing evoked LTP without any effect on baseline EPSPs in hippocampal slices (Magee and Johnston, 1997). In hippocampal cultures, Bi and Poo (1998) report that blocking L-type Ca2+ channels (by nimodipine) does not affect LTP induction by pre–post pairings but prevents LTD induction in response to post–pre pairings.
Long-term potentiation and LTD rely on calcium influx through different channels but both require postsynaptic calcium elevations (Lynch et al., 1983; Malenka et al., 1988; Neveu and Zucker, 1996; Yang et al., 1999; Zucker, 1999; Mizuno et al., 2001; Ismailov et al., 2004; Nevian and Sakmann, 2006). One of the main conclusions from those studies is that LTP is triggered by a brief increase of calcium with relatively high magnitude, whereas a prolonged modest rise of calcium reliably induces LTD. Neveu and Zucker (1996) show that the release of caged-calcium by photolysis in hippocampal CA1 pyramidal cells is sufficient to evoke LTP and LTD, and that concurrent presynaptic activity is not required. Nevian and Sakmann (2006) demonstrate that LTP and LTD are equally sensitive to fast (1,2-bis(o-aminophenoxy)ethane-N,N,N’,N’,-tetraacetic acid – BAPTA) and slow (ethylene glycol tetraacetic acid – EGTA) Ca2+ buffers loaded in the postsynaptic cell. They conclude that the calcium sensors that trigger the long-lasting synaptic changes respond to the global, volume-averaged increase in intracellular calcium concentration rather than to local calcium concentrations in microdomains. Note that cortical LTD involving the activation of metabotropic glutamate receptors (mGluRs) and retrograde signaling (see below) also requires postsynaptic calcium elevations (Nevian and Sakmann, 2006).
Models Based Exclusively on the Dynamics of Calcium Concentration
How do synaptic modifications emerge from specific patterns of pre- and postsynaptic spiking? We discuss in this section a first series of biophysical models that have tried to reproduce STDP results from the postsynaptic calcium dynamics induced by pre- and postsynaptic activity. While such models readily account for LTD induction in response to post–pre pairs and for LTP in response to pre–post pairs, they consistently observe a second LTD window for pre–post pairs with large time differences, Δt, between pre- and postsynaptic spikes.
We start by discussing the properties of postsynaptic calcium transients evoked by spike-pairs with different Δts. An isolated postsynaptic spike generates a short-lasting calcium transient due to opening of VDCCs induced by the depolarization through the BPAP (see Δt = −100 ms case in Figure 1A). Likewise, an isolated presynaptic spike generates a long-lasting calcium transient due to NMDA channel opening (Figure 1A). When the presynaptic spike is immediately followed by a postsynaptic spike, the strong depolarization by the BPAP increases drastically the voltage-dependent NMDA-R mediated calcium current due to removal of the magnesium block (Nowak et al., 1984; Jahr and Stevens, 1990; magenta line in Figure 1A). This supralinear superposition of the two contributions at positive Δts is particularly apparent in the maximal amplitude and the integral of the calcium transients (Figure 1B). Note that the dependence of calcium dynamics on Δt is not fully captured by amplitude or integral alone. In fact, varying Δt changes in a pronounced fashion the amplitude histogram of the calcium transient even in ranges where the maximal amplitude or the integral depend very weakly on Δt (compare for example protocols with Δt = −100 and −20 ms in Figure 1C).
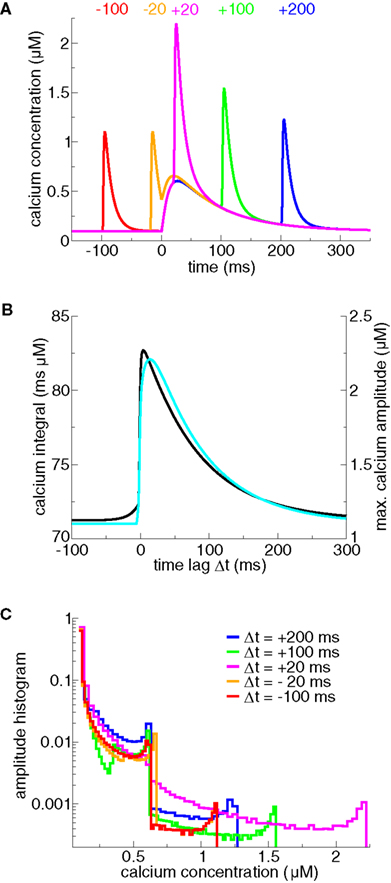
Figure 1. Calcium dynamics in response to spike-pairs for different Δts. (A) Calcium transients for five different time differences between pre- and postsynaptic spikes, Δts (marked in the panel in ms). The transients are generated using the model for postsynaptic calcium and postsynaptic membrane potential dynamics presented in Graupner and Brunel (2007). There, the postsynaptic membrane potential is modeled using the Hodgkin–Huxley formalism in a single compartment. In the model, calcium influx is mediated by VDCCs (high-voltage activated L-type current) and voltage-dependent NMDA-Rs. The presynaptic spike is occurring at t = 0 ms. The presynaptically evoked calcium amplitude is 0.5 μM and the postsynaptic Ca2+ amplitude is 1 μM (Sabatini et al., 2002). Note that the calcium amplitudes are the only parameters that are changed compared to Graupner and Brunel (2007). (B) Maximal calcium amplitude (cyan line, right-hand y axis) and integral of the calcium transient (black line, left-hand y axis) as a function of Δt. (C) Calcium amplitude distributions of calcium transients evoked by spike-pairs for five different Δts. The five calcium transients shown in (A) give rise to the amplitude distributions shown in the same color. The histograms are calculated by binning the calcium concentration (bins size ΔCa = 0.02 μM for Δt = −100, −20, + 100 ms, and 0.035 μM for Δt = + 20, + 200 ms) and counting the number of data points whose amplitude falls in each bin. The amplitude histograms illustrate the fraction of time spent at a certain calcium level by the calcium trace for a given Δt.
In calcium-based models, the induced synaptic weight change is determined by the time course of the calcium transients triggered by pre- and postsynaptic spikes (Shouval et al., 2002; Cai et al., 2007). The magnitude and sign of the resulting synaptic changes are based on the calcium control hypothesis (Figure 2A) which is derived from experimental evidence showing that different calcium levels trigger different forms of synaptic plasticity (Yang et al., 1999; Zucker, 1999; Mizuno et al., 2001; Ismailov et al., 2004; Nevian and Sakmann, 2006). According to this hypothesis, no modification occurs when the calcium level is below a threshold θd which is larger than the resting concentration. If calcium resides in an intermediate calcium range, between θd and a second threshold θp > θd, the synaptic weight is decreased. Finally, if calcium increases above the second threshold, θp, the synaptic strength is potentiated (Figure 2A).
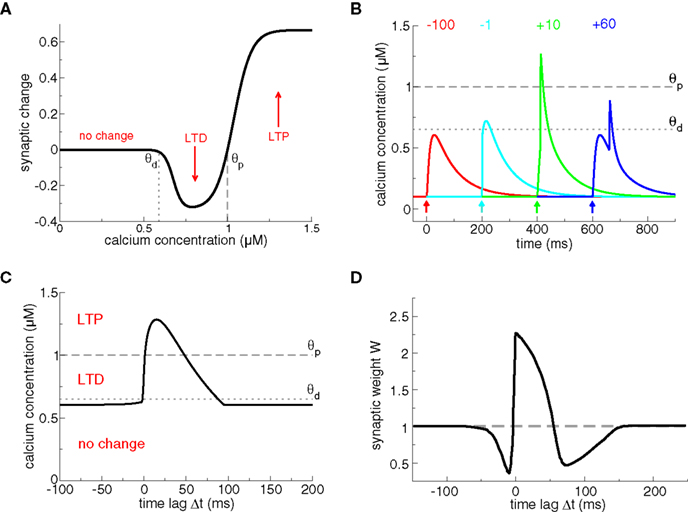
Figure 2. Plasticity results based on calcium control hypothesis. (A) Calcium control hypothesis. The calcium control hypothesis implies that low calcium levels do not evoke any changes, intermediate calcium levels (between θd and θp) depress the synapse (corresponding to an LTD event) and high calcium transients (above θp) potentiate the synapse (corresponding to an LTP event). Note that depression and potentiation are not sudden events but occur with a calcium-dependent time constant, such that LTP induction is faster than LTD induction (see Shouval et al., 2002 for more details). (B) Calcium transients evoked by spike-pairs and mediated exclusively by NMDA-Rs. In this plot, we use the model of Graupner and Brunel (2007), except that calcium influx occurs through NMDA-Rs only, i.e., there is no Ca2+ current mediated by VDCCs, as in Shouval et al. (2002). Otherwise, we use the same parameters as in Figure 1, i.e., the presynaptically evoked Ca2+ amplitude is 0.5 μM. Calcium transients are shown for four different Δts (values given in the panel in ms). The timing of the presynaptic spike is indicated by an arrow for each particular Δt. The thresholds θd (dotted line) and θp (dashed line) from the calcium control hypothesis (see A) are chosen appropriately, that is, large Δt transients do not cross any threshold, short negative Δt transients cross θd, and short positive Δt transients cross θp. (C) Maximal calcium amplitude as a function of Δt, plotted together with the thresholds θd (dotted line) and θp (dashed line). (D) Plasticity outcomes in response to spike-pairs. Spike-pairs with short positive Δts evoke LTP. Spike-pairs with short negative and with large positive Δts lead to LTD. Figure reproduced from Shouval et al. (2002). Note the large extent of the LTD range for short negative Δts as compared to (C). The difference is due to the slow after-depolarizing tail of the BPAP used in Shouval et al. (2002). See text for more details.
Models based on the calcium control hypothesis explains to a large extent the spike-timing dependence of plasticity, as shown in Figure 2, provided the maximal amplitude of the calcium transient for pre–post pairings at short Δt is larger than the potentiating threshold θp. Post–pre pairings evoke calcium transients which linearly superimpose and therefore yield moderate calcium elevations promoting LTD. Pre–post pairings result in supralinear superpositions of the calcium transients which attain high calcium levels required to evoke LTP. If Δt grows larger, the calcium transients pass again through a region of moderate levels inducing LTD (see Figure 2C). Note that Shouval et al. (2002) assume the dominant source of calcium influx to be NMDA-Rs (compare Figures 1A and 2B). They furthermore model the BPAPs with a slow after-depolarizing tail which increases the range of interaction between the postsynaptic spike and NMDA activation by the presynaptic action potential for Δt < 0. That interaction range defines the width of the LTD window in their model (compare LTD range in Figure 2C without after-depolarizing tail, and the LTD range obtained in Shouval et al. (2002), reproduced in Figure 2D).
Most STDP spike-pair experiments however have not found a “second LTD window” at large positive Δt (but see Nishiyama et al., 2000; Wittenberg and Wang, 2006). Shouval and Kalantzis (2005) show that stochastic properties of synaptic transmission can markedly reduce the LTD magnitude at positive time lags. The main idea is that the NMDA-mediated calcium transients at large positive Δts show a high level of relative fluctuations (high coefficient of variation) since the effective number of activated NMDA receptors is small. It is shown that a low number of NMDA-Rs (∼10) gives rise to a sufficient amount of variability to average out the second LTD window (Shouval and Kalantzis, 2005).
Adding features such as short-term depression, stochastic transmitter release, and BPAP depression/facilitation to calcium-based models allows to reproduce spike-triplet data of hippocampal and visual cortex neurons (Cai et al., 2007). The non-linearity of plasticity results between pre–post–pre and post–pre–post triplets is attributed in this model to the consecutive occurrence of either two presynaptic- or two postsynaptic spikes, respectively. Depending on the recovery dynamics of neurotransmitter release, release probability and the depression/facilitation dynamics of BPAPs, two successive presynaptic spikes (in pre–post–pre triplets) and two successive postsynaptic spikes (in post–pre–post triplets) can generate markedly different calcium dynamics leading to different plasticity results.
The class of models described in this section has been surprisingly successful in reproducing experimental results about spike-timing dependent plasticity, given the simplicity of the models. However, these models leave open the question of the mechanisms that translate a given calcium level into a particular synaptic change.
Models Based on Calcium Dynamics and Abstract Readout Systems
We now turn to models that include additional dynamical variables driven by the calcium concentration. These phenomenological variables can be seen as calcium-sensitive “detectors” mediating LTP and LTD (Karmarkar et al., 2002; Abarbanel et al., 2003; Rubin et al., 2005; Badoual et al., 2006). Such phenomenological detectors are assumed to represent biological signaling pathways in an abstract fashion.
Both Karmarkar et al. (2002) and Badoual et al. (2006) account for STDP using distinct but converging dynamical variables modeling calcium- and mGluR (see section ‘Models including biochemical signaling cascades beyond calcium’)-activated pathways. In Badoual et al. (2006), an “LTP-mediating” enzyme is activated by large calcium transients (see Figures 3A,B). In contrast, LTD is evoked by the coincident activation of two enzymes, one activated by calcium and the other briefly activated by the presence of glutamate (see black dashed line in Figure 3A), potentially describing a mGluR-mediated signaling cascade. In turn, LTD occurs only when calcium is present at the time of the occurrence of the presynaptic spike, which is the case if the presynaptic spike is preceded by a BPAP (see Figures 3A,B). The model also accounts for plasticity results in response to pre–post–pre triplets in the visual cortex (Froemke and Dan, 2002). Karmarkar and Buonomano (2002) implement the calcium- and the mGluR pathway by assuming two functionally distinct calcium pools. In that view, calcium influx through VDCCs modulates the mGluR-mediated pathway leading to LTD induction, while calcium from NMDA-Rs is involved in LTP induction.
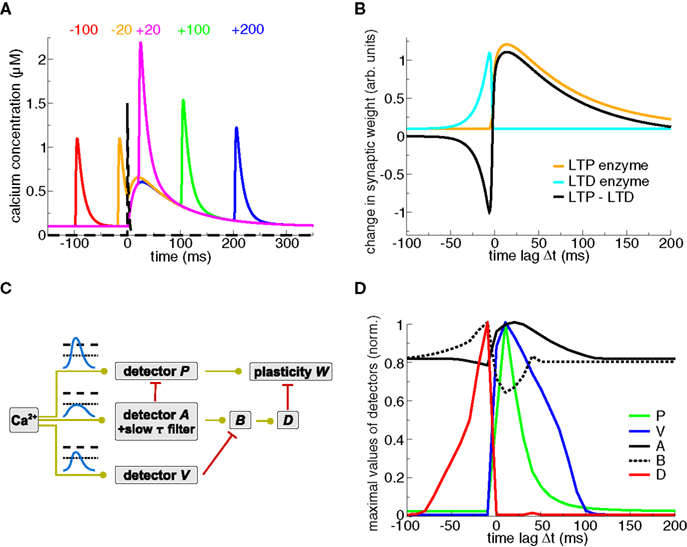
Figure 3. Plasticity results based on phenomenological readout systems. (A) Calcium- and glutamate transients evoked by spike-pairs for five different Δts. The calcium transients are identical to the ones in Figure 1A. The presynaptic spike occurs at t = 0 ms leading to the release of glutamate in the synaptic cleft (black dashed line, see Badoual et al., 2006 for details). (B) Schematic representation of the STDP curve from the biophysical models by Karmarkar et al. (2002) and Badoual et al. (2006). An “LTD enzyme” (cyan line) is activated by glutamate and calcium, e.g., it is represented here to be proportional to the calcium concentration at the occurrence of the presynaptic spike (t = 0 ms in panel A). An “LTP enzyme” (orange line) is activated by high calcium concentrations, e.g., it is represented here to be proportional to the maximal calcium concentration of spike-pair evoked transients. The total change in synaptic weight (black line) is the difference between both. See Karmarkar et al. (2002) and Badoual et al. (2006) for more details. (C) Phenomenological calcium detector system (Rubin et al., 2005). The three different detector systems respond to different calcium signals (illustrated in the panel, see text). The interactions of the detector cascades drive the evolution of the readout variable, the synaptic weight W. Green lines with circles denote activation of the target activity, and red lines with bars signify inhibition of the target. Adapted from Rubin et al. (2005). (D) Maximal detector levels with respect to Δt. The maximal values of the detector variables (shown in C) over a spike-pair cycle is depicted. Note the resemblance of the “D” and “P” activation with the LTP and LTD enzyme activation, respectively, in panel (B). Figure kindly provided by Jonathan Rubin (see Rubin et al., 2005 for more details).
Abarbanel et al. (2003) propose a non-linear competition between two calcium-sensitive detectors to evoke LTP/LTD, that is, phosphorylation and dephosphorylation processes which relate to the α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate receptor (AMPA-R, see section ‘Models including biochemical signaling cascades beyond calcium’) conductance. The half activation concentrations of the two opposing processes (described by Hill functions) are chosen well above the calcium amplitudes of single pre- or postsynaptic transients (Abarbanel et al., 2003). In consequence and similar to the results of Shouval et al. (2002), plasticity results in response to spike-pair stimulation yield LTD for short negative Δts, LTP for short positive Δts, and a further LTD window for large positive Δts (compare with Figure 2D, Abarbanel et al., 2003).
Rubin et al. (2005) propose a “detector” system based on pathways resembling the CaMKII kinase-phosphatase system (see below), implementing three calcium-sensitive detectors (“P”, “A”, and “V”, see Figure 3C). In that model, high, short-lasting calcium levels evoke LTP by activating a detector promoting the increase of synaptic weight (“P” in their model, see Figures 3C,D). Another detector builds up in response to low and prolonged calcium elevations (agent “A” and in turn “B”) evoking LTD above a certain threshold. Importantly, intermediate calcium levels activate a “Veto” agent (“V”) with a fast time constant providing fast tracking of the calcium transient. This veto mechanism suppresses the LTD induction pathway (Figures 3C,D). The dynamics of the “veto” mechanism prevents in particular the appearance of LTD for large positive Δts in response to spike-pair stimulation (see Figure 3D and Gerkin et al., 2010 for an in-depth review of the model).
The models discussed here indicate that synaptic changes combine in a highly non-linear fashion in between spike-pairs and are most likely not a result of piecewise, linear additions of changes evoked by single spike-pairs. Attention should be drawn to the fact that in all the models discussed so far, the time constant of the synaptic variable has to become essentially infinite at resting calcium concentration for the evoked synaptic changes not to decay after the presentation of the stimulation protocol. In the presence of noise and/or finite time constants, such models cannot maintain the evoked synaptic changes in a stable manner. This is in contrast to the models described in the next section, in which bistability leads naturally to maintenance of the evoked synaptic state.
Models Including Biochemical Signaling Cascades Beyond Calcium
Several specific biochemical pathways have been shown to be involved in induction and maintenance of long-term synaptic modifications. We briefly list experimental evidence emphasizing the role of the CaMKII kinase-phosphatase system in synaptic plasticity. We also discuss another line of experimental studies suggesting that synaptic changes are binary all-or-none transitions. We then turn to review biochemical models investigating the dynamics of the CaMKII system. We point out that such models generally exhibit bistability suggesting the CaMKII system to be involved in both induction and maintenance of synaptic changes. See Kotaleski and Blackwell (2010) for a more general review of modeling approaches of the molecular mechanisms underlying LTP and LTD.
Protein Signaling Cascades Linking Calcium Transients to Synaptic Changes: Experimental Data
The postsynaptic calcium signal activates a multitude of calcium-responsive signaling cascades that have been identified to mediate or modulate LTP/LTD induction and expression as well as learning and memory (see review by Malenka and Bear, 2004). Here, we review three key pathways mediating long-term changes in hippocampal CA3–CA1 synapses: (i) the CaMKII-dependent cascade, (ii) the cyclic adenosine monophosphate (cAMP)-dependent protein kinase A (PKA) cascade, (iii) and the calcineurin cascade. See Figure 4 for a schematic depiction of the biochemical pathways. Note that we limit the discussion to the biochemistry involved in spike-timing dependent plasticity at the Schaffer collateral – CA1 neuron synapse. Although synaptic plasticity in other systems rely on different induction pathways and expression mechanisms (e.g., in the cerebellum, Hansel et al., 2006), the CA3–CA1 synapse exhibits characteristics which are shared by other glutamatergic excitatory synapses throughout the mammalian brain, including the cerebral cortex (Kirkwood et al., 1993).
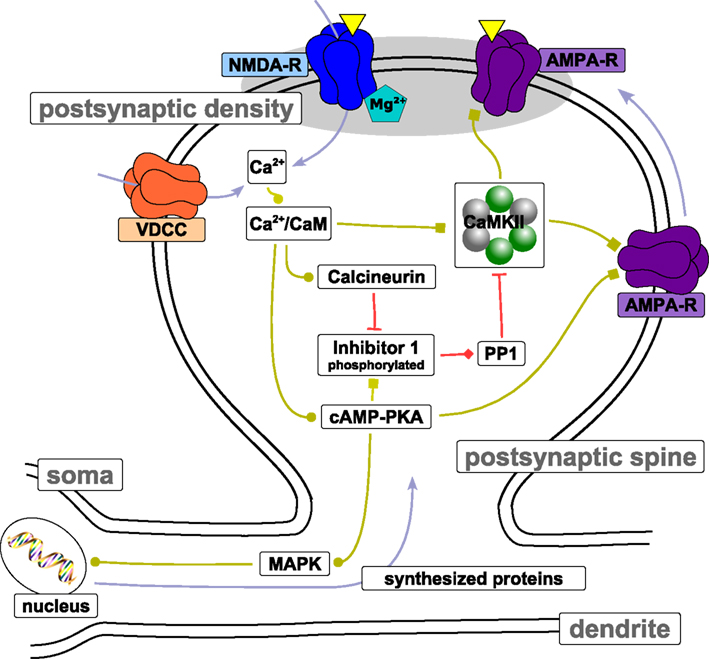
Figure 4. Protein signaling cascades involved in LTP/LTD. The figure shows biochemical pathways that have been identified to be involved in LTP/LTD induction and maintenance at the Schaffer collateral – CA3 neuron synapse. Light blue arrows indicate transport of the corresponding entity. Light green connections indicate stimulation of target activity. Squares at the end of light green connections indicate that the stimulation is due to phosphorylation of the target. Red connections depict inhibition of target activity through dephosphorylation (indicated by a bar at the end of the connection) or binding (indicated by the diamond at the end of the connection). The yellow triangles illustrate neurotransmitter binding to receptors located in the postsynaptic density. Note that the spatial proportions between spine, dendrite, and soma are not preserved. See text for more details.
Calcium/calmodulin-dependent protein kinase II
In its basal state, the enzymatic activity of the CaMKII towards target proteins is extremely low. Regulation of intracellular calcium levels allows the neuron to link neural activity with the phosphorylation level of CaMKII. CaMKII activation is governed by calcium/calmodulin (Ca2+/CaM) binding and is prolonged beyond fast-decaying calcium transients by its autophosphorylation (Fink and Meyer, 2002). Autophosphorylation of CaMKII at the autoregulatory domain occurs after calcium/calmodulin binding to two neighboring subunits in the CaMKII holoenzyme ring and enables the enzyme to remain autonomously active after dissociation of calcium/calmodulin (Hanson and Schulman, 1992). See reviews by Hudmon and Schulman (2002) and Griffith (2004) for more details of the regulation of CaMKII activity.
In its activated state, CaMKII is reversibly translocated to a postsynaptic density (PSD)-bound state where it interacts with multiple LTP related partners structurally organizing protein anchoring assemblies (Shen and Meyer, 1999; Hayashi et al., 2000; Fink and Meyer, 2002; Lisman et al., 2002; Colbran, 2004). The direct phosphorylation of the AMPA receptor GluR1 subunit by CaMKII enhances AMPA channel function (Mammen et al., 1997; Derkach et al., 1999), and drives AMPA receptors into synapses (Hayashi et al., 2000; see also review article by Lisman et al., 2002). Mutated mice lacking the ability of CaMKII autophosphorylation exhibit profound deficits in hippocampus-dependent learning and memory and also completely fail to exhibit LTP induction in the hippocampal CA1 subfield under standard stimulation protocols (Giese et al., 1998). Furthermore, LTP induction in the hippocampus via spike-timing stimulation protocols is blocked in the presence of KN-62, a specific blocker of CaMKII which binds to the enzyme and blocks the activation by calcium/calmodulin (Wang et al., 2005).
The role of CaMKII beyond induction of synaptic long-term modifications remains controversial. Enzymatic activity of CaMKII decreases to baseline within ∼15 min after LTP induction (Lengyel et al., 2004). This is in agreement with recent findings indicating that autonomous CaMKII activity is not required for LTP maintenance or for memory storage/retrieval in vivo (Buard et al., 2010). In contrast, Sanhueza et al. (2007) show that a non-competitive inhibitor of CaMKII can reverse LTP suggesting that a component of synaptic memory maintenance is attributable to CaMKII in CA1 synapses.
Cyclic adenosine monophosphate-dependent protein kinase A
The cAMP-dependent PKA cascade is thought to mediate synapse to nucleus signaling and seems to initiate synthesis of proteins and RNA during the late phase of LTP induction in the hippocampal area CA1 (on time scales > 1 h; Abel et al., 1997; Nguyen and Kandel, 1997). These studies suggest that the early phase of LTP induction and basal synaptic transmission are not affected by cAMP–PKA inactivation. In hippocampus to prefrontal cortex connections however, LTP induction is accompanied by a rapid increase in PKA activity during the early phase (Jay et al., 1998). Also for the CA3–CA1 pathway, LTP induction by high-frequency stimulations can be blocked by inhibiting postsynaptic cAMP–PKA in contrast to the experimental results above (Blitzer et al., 1995, 1998). The requirement of PKA for LTP induction can be overcome by direct inhibition of postsynaptic phosphatases (Blitzer et al., 1995), suggesting that PKA gates LTP by blocking/or competing with protein phosphatases (see below).
The calcium-sensitivity of the PKA pathway relies upon calcium/calmodulin-initiated conversion of adenosine triphosphate into cAMP by adenylyl cyclase (Cooper et al., 1995). Elevation of cAMP, in turn, activates the cAMP-dependent PKA (Carr et al., 1992; Glantz et al., 1992). Stimulating this pathway by increasing the adenylyl cyclase activity is shown to induce LTP in hippocampal slices without the requirement for any electrical stimulation, an effect that can be blocked with PKA inhibitors (Frey et al., 1993). Similarly, overexpression of adenylyl cyclase in transgenic mice enhances LTP and learning (Wang, 2004). Though PKA directly phosphorylates the AMPA receptor GluR4 subunit, both PKA activity and CaMKII activity are necessary to incorporate AMPA-Rs into the cell membrane (Esteban et al., 2003).
The signaling cascade continues towards the nucleus through the mitogen-activated protein kinase (MAPK). PKA activates this enzyme after hippocampus-dependent learning in mice. Furthermore, MAPK inhibitors block the maintenance of LTP (Waltereit and Weller, 2003; Sweatt, 2004). This cascade targets the cAMP-responsive element-binding protein (CREB) in the nucleus and therefore governs the expression of LTP/memory effector proteins (Bozon et al., 2003; Chen et al., 2003). These results indicate that this branch of the cAMP-dependent signaling cascade plays a key role during the late phase of LTP most likely accompanied by altered gene expression (Goelet et al., 1986; Alberini et al., 1995).
Calcineurin
Experimental results indicate that the sign of hippocampal synaptic plasticity is regulated by the balance between protein phosphorylation and dephosphorylation mediated by PKA and calcineurin, respectively. Consistent with this idea, overexpression of calcium/calmodulin-dependent calcineurin in the forebrain of transgenic mice is found to impair an intermediate and PKA-dependent phase of LTP, as well as the transition from short- to long-term memory and memory retrieval (Mansuy et al., 1998; Winder et al., 1998). On the other hand, inhibition of calcineurin activity facilitates LTP in vitro and in vivo in a PKA-dependent manner (Malleret et al., 2001). Consistent with these findings, LTD evoked during STDP stimulation by post–pre spike-pairs is blocked in the presence of calcineurin inhibitors while the same blockade unmasks potentiation for spike-triplets (Wang et al., 2005). Similar results are found for presynaptic stimulation protocols of varying frequencies inducing LTD at low (1 −10 Hz) and LTP at high frequencies (10 −100 Hz) in control conditions (O’Connor et al., 2005a). A kinase inhibitor (inhibiting CaMKII and protein kinase C) blocks LTP and reveals LTD for 1 −100 Hz stimulation protocols. On the other hand, a phosphatase inhibitor (blocking protein phosphatase 1, PP1, and protein phosphatase 2A) prevents LTD for intermediate stimulation frequencies (1 −10 Hz) but leaves LTP induction unchanged at high stimulation frequencies ( > 10 Hz; O’Connor et al., 2005a).
The results discussed so far suggest that the kinase and the phosphatase pathways interact at one or several points in the signaling cascade. A possible converging point of the cAMP–PKA and the calcineurin pathways is inhibitor 1 (I1, see Figure 4). Evidence for the role of I1 as a point of convergence is: (i) Hippocampal LTD induction involves calcium/calmodulin-dependent calcineurin dephosphorylating I1 (Mulkey et al., 1994), (ii) Synaptic stimulation that induces cAMP-dependent LTP raises the amount of phosphorylated I1 in the CA1 region (Blitzer et al., 1998). This increase is dependent on PKA activity since it is blocked by PKA inhibitors. Phosphorylated I1 is a specific blocker of PP1 (Ingebritsen and Cohen, 1983). Hence, the differential calcium-dependent activation of the calcineurin and the cAMP–PKA pathway is expressed in the phosphorylation level of I1 which in turn inhibits PP1 in its phosphorylated state. During the induction of hippocampal LTD, the inactivation of I1 through dephosphorylation increases PP1 activity (Mulkey et al., 1994). Disruption of PP1 binding to synaptic targeting proteins is reported to block synaptically evoked LTD but does not affect basal synaptic transmission in CA1 pyramidal cells. PP1 has no direct access to synaptic AMPA-Rs, but it is the only phosphatase able to dephosphorylate CaMKII in the PSD (Strack et al., 1997).
Which molecular pathways underlie spike-timing dependent plasticity in other brain areas? LTD seems to involve retrograde signaling to the presynaptic terminal in the visual and the somatosensory cortex (Sjöström et al., 2003; Nevian and Sakmann, 2006). Both postsynaptic calcium elevations mediated by VDCCs as well as the activation of mGluRs are necessary for such LTD induction. An application of a mGluR antagonist results in a complete block of LTD but has no effect on the calcium transients (Nevian and Sakmann, 2006). This block of LTD is attributable to the disruption of the G-protein coupled cascade involving retrograde endocannabinoid signaling (Piomelli, 2003; Sjöström et al., 2003, 2004; Nevian and Sakmann, 2006). Calcium in turn modulates the efficiency of the G-protein coupled phospholipase C-dependent pathway which synthesizes endocannabinoids (Hashimotodani et al., 2005; Maejima et al., 2005). Note that a mGluR- and postsynaptic calcium-dependent form of LTD has also been found at the Schaffer collateral – CA1 synapse (Stanton et al., 1991; Bolshakov and Siegelbaum, 1994; Otani and Connor, 1998). The simultaneous presence of two seemingly independent LTD-inducing pathways, that is, a mGluR- and a PP1-dependent cascade, at Schaffer collateral – CA1 synapses sparks ongoing debates (Lisman, 2009). Some of the controversy might be settled in light of a recent study showing that in contrast to PP1-dependent LTD (Debanne et al., 1996), multiple converging Schaffer collateral inputs are required for the induction of mGluR-dependent LTD in a CA1 pyramidal cell (Fan et al., 2010).
More proteins have been suggested to be related to LTP/LTD, such as protein kinase C, phosphatidylinostiol 3-kinase, tyrosine kinase Src to name just a few of them (see reviews by Bliss and Collingridge, 1993; Malenka and Bear, 2004). Apart for the pathways involving CaMKII and associated proteins, the signal transduction pathways involved in triggering long-term synaptic changes remain elusive with many potential players but few definite answers about specific roles in induction and maintenance mechanisms.
Nature of Synaptic Changes – Experimental Evidence for Bistable Synapses in the Hippocampus
We now turn to experiments which address the nature of synaptic changes and suggest that these changes are switch-like all-or-none transitions, consistent with a bistable system.
In experiments on long-term synaptic modifications, LTP (resp. LTD) refers to a long-lasting increase (resp. decrease) of the EPSP recorded at the soma of the postsynaptic neuron after the stimulation protocol. LTP/LTD protocols typically involve the stimulation of a large number of afferents – the recorded signals and changes in EPSP size stem therefore from an ensemble of synapses and reflect properties of a compound signal. For this reason, most of the plasticity experiments provide no insights into the nature of synaptic changes at the single-synapse level. A few plasticity experiments at the Schaffer collateral – CA1 synapse address the question whether synaptic strength changes occur in an analog or a digital manner (Petersen et al., 1998; O’Connor et al., 2005b). In other words, is the size of the EPSP at the level of a single synapse changing continuously or can it take specific values only (despite the variability due to neurotransmitter release, diffusion, and channel opening)? If the latter is true, can the synapse take two, three, or more states? In the most simple case of two stable states, the synaptic efficacy can be considered a binary variable in the long-term.
Experiments by Petersen et al. (1998) and O’Connor et al. (2005b) address this question using a minimal stimulation technique on Schaffer collateral – CA1 connections, whose aim is to evoke single-synapse responses (Raastad, 1995; Bolshakov and Siegelbaum, 1995; Stevens and Wang, 1995; Isaac et al., 1996). The excitatory postsynaptic current (EPSC) size increases abruptly during the LTP stimulation protocol and decreases abruptly during the LTD protocol (Figure 5A, O’Connor et al., 2005b). Applying statistical tests to this change in EPSC size leads the authors to the conclusion that the changes are all-or-none, sudden switch-like events, taking place on the time scale of seconds (O’Connor et al., 2005b). They show furthermore that these events saturate synapses to full potentiation or depression. That means that once a synapse got potentiated it cannot be potentiated a second time, but the potentiation can be reversed by a subsequent LTD induction protocol. Accordingly, a second LTD induction protocol cannot decrease EPSC size further suggesting that the investigated synapse has two stable states, that is, the synapse is binary. Results obtained by Petersen et al. (1998) on potentiation of putative single-synapses reach the same conclusion, but LTD induction has not been considered in this study. Bagal et al. (2005) use glutamate uncaging paired with brief postsynaptic depolarization and report long-lasting potentiation of single dendritic spines in hippocampal CA1 cells. This potentiation shares many features with conventional LTP such as a dependence on NMDA activation and the ability to be reversed, or depotentiated, in a NMDA-R-dependent manner by low-frequency stimulus trains. Again, potentiation was expressed in a stepwise, all-or-none manner.
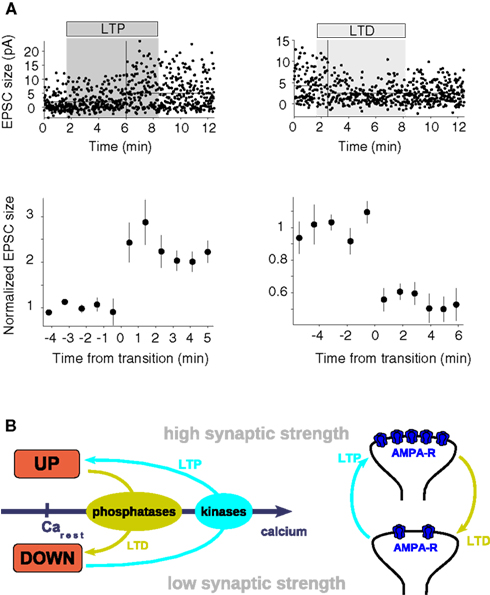
Figure 5. The bistable synapse. (A) Unitary plasticity events take place in single, reversible steps. The depicted synaptic changes are measured in hippocampal CA1 neurons in response to minimal stimulation of the Schaffer collateral pathway. Putative single-synapse responses (EPSCs) recorded before, during (shaded region) and after the LTP (above, left-hand side) and the LTD (above, right-hand side) stimulation protocol are shown as a function of time. To illustrate the immediate step-like transitions 10-response bins are grouped and aligned with respect to the point of transition at t = 0 min for the LTP (below, left-hand side) and the LTD (below, right-hand side) event (figure adapted from O’Connor et al., 2005b). (B) Scheme of a bistable synapse. A bistable synapse exhibits two stable states – DOWN and UP – at resting calcium levels, i.e., transitions between both states are not possible (or happen with vanishingly low probability) at resting calcium levels. The UP and the DOWN states are characterized by high and low synaptic strength, respectively. Since intracellular calcium elevations are a necessary and sufficient signal to induce LTP and LTD, transitions from the DOWN to the UP state (LTP, cyan arrows) supposedly occur during high amplitude calcium transients, while transitions from the UP to the DOWN state (LTD, light green arrows) occur if the system is exposed to moderate calcium concentrations for a long time (left panel). Low and high calcium transients activate protein phosphatases and protein kinases, respectively, whose activation switch the system between the UP and the DOWN states. The two discrete states are likely expressed by different numbers of functional AMPA receptors in the membrane in the hippocampus (right panel). See text for more details.
These experimental results suggest that synapses can be described as occupying two states of low- or high transmission efficacy (see Figure 5B). These states can be termed DOWN and UP states, respectively. In that framework, LTP corresponds to a transition from the DOWN to the UP state, while LTD corresponds to a transition from the UP to the DOWN state. This implies that potentiation or depression observed in experiments involving stimulation of ensembles of synapses are a combination of multiple step-like events of single synapses. LTP (resp. LTD) experiments on such an ensemble start from a mixture of states and can either partially or maximally potentiate (resp. depress) all connections (O’Connor et al., 2005b). Therefore, smooth plasticity curves (such as STDP curves) can be obtained through averaging over multiple synapses of otherwise discrete single synaptic changes (Appleby and Elliott, 2005).
A key mechanism proposed for the expression of LTP involves an increase in the number of functional AMPA-Rs in the plasma membrane (see reviews by Malenka and Nicoll, 1999; Malinow and Malenka, 2002) or the phosphorylation state of AMPA-Rs (Benke et al., 1998; Lee et al., 2000, 2003; see also review by Soderling and Derkach, 2000) or both. Consequently, binary changes would imply that synaptic changes always involve a cluster of AMPA receptors, inserted all at once in the membrane (see scheme in Figure 5B and review by Lisman, 2003). Note that the number of AMPA receptors in the PSD of a spine is believed to vary from none for silent synapses to around 50 (Kennedy, 2000).
As a means of information storage, graded synaptic changes (Bienenstock et al., 1982; Oja, 1982) are more susceptible to drift due to biochemical noise (e.g., protein turnover) and ongoing neural activity than all-or-none binary changes (see, e.g., discussion in Petersen et al., 1998). Thus synaptic discreteness might help to make information storage in a neural network robust. However, we should emphasize that the experiments described in this section are limited to time scales on the order of minutes. On longer time scales, step-like changes of synaptic strength might give way to events like altered gene expression that may take a more continuous character.
Bistable Models Based on the CaMKII Kinase – Phosphatase System
We now turn to review a line of modeling research addressing the issue of maintenance of the evoked synaptic state during the early phase of LTP/LTD. The mathematical studies presented here show that detailed biochemical models of protein networks often exhibit bistability and therefore behave as bistable switches. Positive feedback loops are at the origin of such switches which express in the simplest form two stable states. Those stable states are proposed to maintain evoked synaptic states beyond stimulation protocols.
As outlined above, the expression of plasticity involves multiple molecular players. Molecules, however, have a short lifetime of the order of minutes to days, whereas some memories can be retained for years. Despite the fact that long-term modifications involve structural reorganization, altered gene transcription and new protein synthesis in the late phase (see review by Malenka and Bear, 2004), the question remains how the induced state can be preserved by a machinery involving a limited number of proteins in the presence of protein turnover. Here, we also discuss models which show that bistable switches formed by an ensemble of proteins can recruit newly synthesized proteins to adopt a particular “stored” state and thus retain state information despite molecular turnover.
The problem of synaptic stability is first noted by Crick (1984) who suggests that cooperative interactions among proteins can overcome the problem of molecular turnover for long-term memory. The idea of a molecular switch storing information beyond protein lifetime is further developed by Lisman (1985). By using known enzymatic reactions, he shows in a simple mathematical model that a kinase can exist in two stable states: a unphosphorylated “off” state and a phosphorylated “on” state. Transitions from “off” to “on” can be induced by another phosphorylating kinase and the evoked state acts autocatalytically to sustain the phosphorylation level and to phosphorylate downstream kinases. Reverse transitions could be induced by phosphatase activity. The first candidate protein proposed to be at the origin for such a switch is the CaMKII. Theoretical studies in the late 1980s show that the complex holoenzyme composed of 12 subunits can exhibit a switch-like behavior due to calcium-independent autophosphorylation even in the presence of phosphatase activity and of protein turnover (Lisman and Goldring, 1988).
We now turn to the description of a specific mathematical model of CaMKII autophosphorylation/dephosphorylation behavior (Zhabotinsky, 2000, see also Okamoto and Ichikawa, 2000 for a closely related model). This model shows that the CaMKII protein can exhibit a bistable phosphorylation behavior in a range of calcium concentrations. It includes crucial biochemical details of calcium-triggered autophosphorylation and dephosphorylation of the CaMKII protein: (i) initial and subsequent phosphorylation steps are calcium–calmodulin dependent; (ii) subsequent phosphorylation steps are facilitated due to the increase in calcium/calmodulin affinity of a phosphorylated subunit and the fact that a phosphorylated subunit stays active as catalyst for the autophosphorylation reaction (see section ‘Protein signaling cascades linking calcium transients to synaptic changes: experimental data’ and Hudmon and Schulman, 2002); and (iii) the dephosphorylation of CaMKII subunits by PP1 is implemented according to the Michaelis–Menten scheme (Michaelis and Menten, 1913). These features lead to three steady-states of the CaMKII phosphorylation level in a range of calcium concentrations: a stable weakly phosphorylated steady-state (i.e., a DOWN state), a stable highly phosphorylated state (i.e., an UP state) and an intermediate unstable fixed-point (see Figure 6B). This unstable steady-state separates the basins of attraction of the two stable steady-states. The range of bistability is shown to potentially include the resting calcium concentration, providing stability of UP and DOWN states at resting conditions.
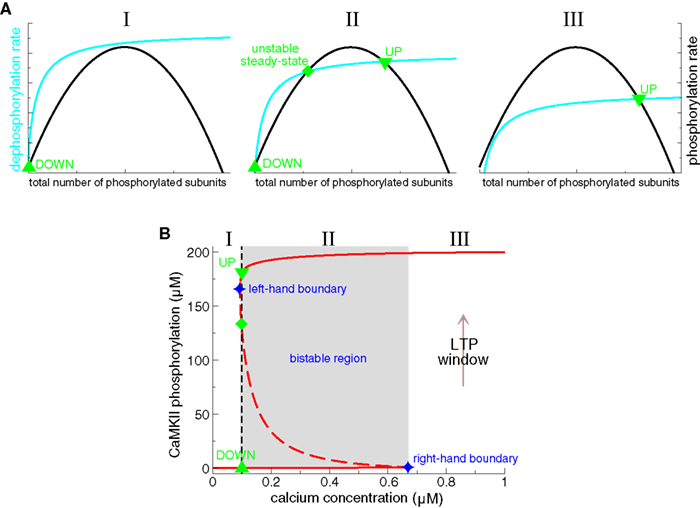
Figure 6. Bistability of the CaMKII phosphorylation level. (A) Total phosphorylation (black lines) and dephosphorylation rates (cyan lines) illustrating the steady-states of the system. The overall phosphorylation and dephosphorylation rates are shown as functions of the total number of phosphorylated subunits of CaMKII holoenzymes. The intersection points of both rates characterize balanced phosphorylation and dephosphorylation and mark the steady-states of the system. Depending on the relative position of both curves, there can be either a single stable steady-state present (I and III), or three steady-states (II). When three steady-states are present, two of them are stable (an “UP” state with a high phosphorylation level, and a “DOWN” state with low phosphorylation level) and the intermediate steady-state is unstable. (I) only the DOWN state is stable when dephosphorylation is strong, and/or phosphorylation is weak (left-hand panel), (II) bistability occurs if phosphorylation and dephosphorylation are balanced (middle panel), (III) the UP state is the only stable state if dephosphorylation is weaker than phosphorylation (right-hand panel). Note that the dephosphorylation rate saturates and the phosphorylation rate vanishes at high total phosphorylation levels. See text for more details. (B) Concentration of phosphorylated CaMKII subunits as a function of calcium in detailed biochemical models (Okamoto and Ichikawa, 2000; Zhabotinsky, 2000). In such models, the phosphorylation level of the CaMKII exhibits a bistable behavior. That is, two stable phosphorylation states – a weakly and a highly phosphorylated state – exist in a range of calcium (region II, middle panel in A, Ca∈[0.094,0.67] μM, gray area, adapted from Zhabotinsky, 2000). Stable steady-states are depicted by full red lines and unstable fixed points by the dashed line. UP to DOWN transitions occur below the left-hand boundary of this bistable region (region I), and DOWN to UP transition are evoked if the system is exposed during a sufficiently long interval to calcium concentrations higher than the right-hand boundary of the bistable region (“LTP window”, region III). In Zhabotinsky (2000) the bistable region includes the calcium resting concentration Ca0 = 0.1 μM. Reproduced from Zhabotinsky (2000).
What is the mechanism of the bistability in the phosphorylation behavior of CaMKII subunits? This question can be answered by inspecting the total rates of autophosphorylation and dephosphorylation of CaMKII subunits. Figure 6A shows schematically how both rates vary as a function of the total number of phosphorylated subunits. The intersection points, the points where the total autophosphorylation rate balances the total dephosphorylation rate, mark the three steady-states of the system (Figure 6A, middle panel). The left- and the right-hand steady-states are stable and the middle one is unstable. Two criteria are necessary for these three intersection points to emerge: (i) The total dephosphorylation rate has to saturate at high phosphorylation levels (see cyan lines in Figure 6A). Such a saturation naturally occurs if dephosphorylation is described according to the Michaelis–Menten scheme, which is valid if the enzyme (PP1) is present in small amounts compared to the substrate (phosphorylated subunits). (ii) The cooperativity of autophosphorylation is at the origin of the bump-like behavior of the total autophosphorylation rate. Subsequent phosphorylation in the ring is faster than the initial autophosphorylation rate since only a single calcium/calmodulin complex is required as compared to two for the initiation step (Hudmon and Schulman, 2002). Without this facilitation of autophosphorylation, the total rate would stay constant with increasing number of phosphorylated CaMKII subunits and the three intersection points with the dephosphorylation rate could not be realized. In summary, the saturation of the phosphatase activity dephosphorylating CaMKII combined with the cooperativity of the autophosphorylation rate yields the bistability of the CaMKII phosphorylation level.
These mathematical models have a number of limitations. First, they are strictly speaking valid only in the limit of a large number of interacting molecules. Only a relatively small number of CaMKII proteins are however present in a typical PSD – of the order 50 −100 holoenzymes (Harris and Stevens, 1989; Hanson and Schulman, 1992; McNeill and Colbran, 1995; Doi et al., 2005). Furthermore, the average lifetime of a single CaMKII protein is about 30 h independently of its phosphorylation state (Ehlers, 2003). This raises the question of the stability of the CaMKII switch with respect to stochastic fluctuations induced by protein turnover. Miller et al. (2005) investigate this question using the model of Zhabotinsky (2000), and show that the CaMKII switch composed of a realistic number of CaMKII protein is stable for years even in the presence of protein turnover, phosphatase as well as free calcium fluctuations.
Second, the localization of CaMKII is not restricted to the PSD, but translocation and diffusive exchange between the PSD and the cytosol create an ongoing flux (Shen and Meyer, 1999; Shen et al., 2000; Sharma et al., 2006). This raises the question of how these exchanges affect the stability of the CaMKII switch. Hayer and Bhalla (2005) investigate both the stability of the CaMKII switch and the insertion of AMPA receptors in the presence of protein trafficking and turnover. Besides the bistability of the CaMKII phosphorylation level, they identify an independent AMPA receptor switch based on self-recruitment of receptors into the synapse (Ehlers, 2000; Esteban et al., 2003). Depending on whether both switches function completely independently, or tightly coupled determines if three or two stable states exist in such a system, respectively. The average lifetime of such switches depends on the coupling and ranges from 24 h to more than a year (Hayer and Bhalla, 2005). The existence of a second switch besides CaMKII ensuring the maintenance of AMPA receptors in the membrane could be a means to maintain the evoked synaptic state on longer time scales. This is a crucial issue since CaMKII enzymatic activity decreases to baseline within ∼15 min after LTP induction (Lengyel et al., 2004; see above).
Finally, several experimental studies have shown that in some cases LTD is not a simple reversal of previously evoked LTP (termed depotentiation) but rather involves separate biochemical mechanisms (Zhuo et al., 1999, see also section ‘Protein signaling cascades linking calcium transients to synaptic changes: experimental data’). To account for these experimental results, there should be at least three states available to the synapse: a “basal state”, a potentiated state, and a depressed state. Depotentiation would correspond to the transition from potentiated to basal, while LTD would be the transition from basal to depressed. Pi and Lisman (2008) propose a model which accounts for bidirectional changes starting from a basal state of the synapse. They demonstrate that the coupling of a kinase (e.g., CaMKII) and a phosphatase switch (e.g., protein phosphatase 2 A, PP2A) could give rise to tristability of the synapse. Both switches stably maintain the induction of LTP and LTD through respective autocatalytic reactions (compare with Lisman, 1985). The phosphatase switch is proposed to be based on PP2A since LTD induction results in persistent activation of the PP2A (Thiels et al., 1998).
STDP in CaMKII-Based Bistable Models
Using the knowledge about biochemical pathways involved in the induction of synaptic changes and the existence of bistability in such networks, two recent studies have investigated STDP in CaMKII-based models (Graupner and Brunel, 2007; Urakubo et al., 2008). These two studies describe known protein signaling cascades providing the link between the calcium level and the phosphorylation level of the CaMKII protein (see Figure 4), whose phosphorylation level exhibits bistability. In addition, Urakubo et al. (2008) describe AMPA receptor trafficking which translates CaMKII bistability into bistable synaptic conductance since AMPA-Rs are clustered in the PSD through phosphorylation by CaMKII.
The calcium control hypothesis (Figure 2A) implies no synaptic changes for resting and low calcium levels, LTD at intermediate calcium levels and LTP induction at high calcium elevations. These three functionally different calcium regions translate for a bistable system into the following three criteria: (i) UP and DOWN states should exist at resting and low levels of calcium. This requirement assures the stability of the evoked synaptic state under resting conditions and activity which does not lead to considerable calcium accumulations. (ii) Prolonged, intermediate calcium elevations should move the system from the UP to the DOWN state. Such transitions would take place in the range of calcium levels typically present in response to LTD stimulation protocols. Starting from the DOWN state, no transition should occur. Note that such a “LTD region” does not exist in Zhabotinsky (2000) and Okamoto and Ichikawa (2000) (compare Figure 6B). (iii) Repetitive exposures to high calcium levels should move the system from the DOWN to the UP state. Such a transition conforms to a LTP event occurring in response to fast and high calcium transients (“LTP window” in Figure 6B). In contrast, no transition should occur in such conditions if the system resides initially in the UP state.
Realistic calcium transients evoked by spike-pairs with short positive Δts move the CaMKII system from the weakly to the highly phosphorylated state, that is, LTP occurs corresponding to criterion (iii) above (Figures 8B,C) (Graupner and Brunel, 2007; Urakubo et al., 2008). In both models, such high calcium transients boost CaMKII autophosphorylation and inhibit PP1 activity dephosphorylating the CaMKII protein (Figure 8A). High calcium concentrations have previously been shown to move the CaMKII system from the DOWN to the UP state (Okamoto and Ichikawa, 2000; Zhabotinsky, 2000, see “LTP window” Figure 6B).
In addition, both Graupner and Brunel (2007) and Urakubo et al. (2008) show that the parameters of the protein signaling cascade can be set such that there is a region at intermediate calcium levels within which the CaMKII moves from the UP to the DOWN state (see Figure 7C, region II). That region, called “LTD-window”, emerges from an elevation in active PP1 which in turn stems from the calcium-dependent activation of the calcineurin pathway (see section ‘Protein signaling cascades linking calcium transients to synaptic changes: experimental data and Figure 7). Importantly, calcium transients in response to spike-pairs with short negative Δts are shown to amplify PP1 activity leading to LTD transitions, which corresponds to criterion (ii) above (Figures 8A,B) (Graupner and Brunel, 2007).
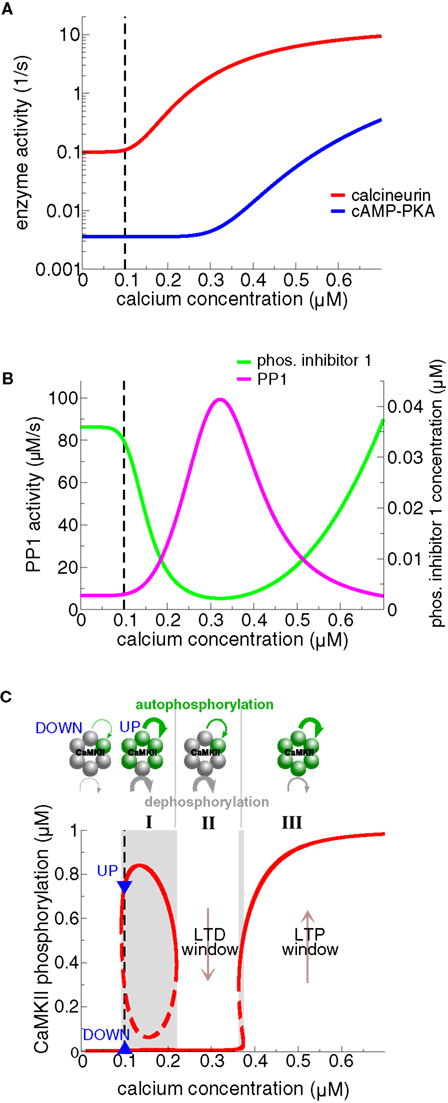
Figure 7. Steady-states of the protein signaling cascade and the CaMKII phosphorylation level exhibiting bistability and an “LTD window”. (A) Calcineurin and cAMP–PKA activities as a function of calcium. The model assumes that the calcineurin pathway activates at moderate calcium levels, while the cAMP–PKA pathway activates at high calcium levels. See Figure 4 for a depiction of the signaling cascades. (B) Phosphorylated I1 and PP1 activities as a function of calcium. The activation of the calcineurin pathway at intermediate calcium levels promotes I1 dephosphorylation (green line) and in turn activation of PP1 activity (magenta line). Activation of the cAMP–PKA pathway at high calcium levels promotes I1 phosphorylation and thereby PP1 inhibition. The differential activation of calcineurin vs. cAMP–PKA gives therefore rise to a peak of PP1 activity at intermediate calcium levels. (C) Steady-states of the phosphorylated CaMKII subunit concentration and the autophosphorylation – dephosphorylation balance as functions of calcium. The upper row illustrates rings of six functionally coupled subunits of the CaMKII holoenzyme. A gray subunit stands for dephosphorylated and a green one for phosphorylated. The green and the gray curved lines indicate calcium-dependent autophosphorylation and PP1-mediated dephosphorylation, respectively. Their width corresponds to the strength of the respective process in the three different calcium regions (I, II, and III). At low calcium levels, including the calcium resting level (region I), autophosphorylation and dephosphorylation balance each other at two different CaMKII phosphorylation levels, giving rise to bistability at resting calcium (Ca0 = 0.1 μM, lower panel). The PP1 activity dephosphorylating CaMKII has a peak at intermediate calcium levels (magenta line in panel B). As a result, the UP state loses stability, leaving the weakly phosphorylated state as the only stable steady-state in region II (“LTD window”). The PP1 dephosphorylation activity is suppressed and autophosphorylation is strong at high calcium levels (“LTP window”, region III). Consequently, the highly phosphorylated state is the only stable state of the CaMKII system in region III. The resting calcium concentration is indicated by the dashed black line in all panels. Figures are adapted from Graupner and Brunel (2007).
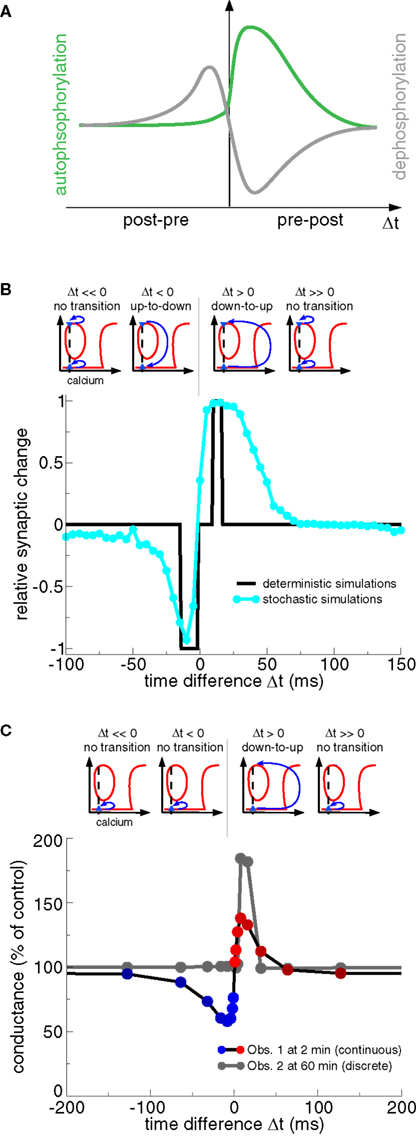
Figure 8. Transition dynamics of the CaMKII phosphorylation level in response to the STDP protocol as a function of Δt. (A) The total level of PP1 activity after the presentation of a pre- and postsynaptic spike-pair as a function of Δt (gray line), and the maximal rate of phosphorylation of a CaMKII subunit for the same protocol (green line). Note the resemblance of the PP1 activity and the phosphorylation rate as a function Δt to the activity of the “LTD” and the “LTP” enzymes in Figure 3B, and to the “P” and “D” activation in Figure 3D, respectively. Adapted from Graupner and Brunel (2007). (B) CaMKII transition behavior in response to the STDP stimulation protocol (Graupner and Brunel, 2007). Bidirectional transitions between the DOWN and the UP states in response to calcium transients evoked by the STDP stimulation protocol are illustrated in the upper row. Up-to-down transitions occur when PP1 activity is high and autophoshorylation reaches moderate levels (see panel A) which is the case when Δt < 0 (lower panel). Δt > 0 stimulation protocols yield large calcium elevations which strongly activate autophosphorylation and suppress PP1 activity (see panel A) evoking down-to-up transitions (lower panel). The CaMKII transition results are summarized in the lower panel for stimulation with deterministic calcium transients (black line) and noisy calcium transients (cyan line). Results adapted from Graupner and Brunel (2007). (C) CaMKII transition behavior in response to the STDP stimulation protocol (Urakubo et al., 2008). As in (B) transitions between the DOWN and the UP state in response to STDP stimulation are illustrated in the upper row. Note that all CaMKII proteins are initially in the DOWN state (blue triangle). As a result, only down-to-up transitions are observed for short positive Δts, despite the fact that the steady-states of the phosphorylated CaMKII also exhibit a “LTD window” (compare with Figure 7C). Spike-timing dependent synaptic conductance changes are shown at 2 min (Obs. 1) and at 60 min (Obs. 2) after the onset of stimulation (lower panel). Synaptic changes show a continuous character right after the deterministic stimulation protocols (Obs. 1), while the changes are discrete on the long run due to bistability (Obs. 2). Results adapted from Urakubo et al. (2008).
Both studies show that STDP plasticity results can be accounted for (at least on short time scales, see Obs. 1 in Figure 8C) if the balance between calcineurin- and cAMP–PKA activation results in high PP1 activity for post–pre pairs and low PP1 activity together with strong autophosphorylation for pre–post pairs (Figure 8A) (Graupner and Brunel, 2007; Urakubo et al., 2008). The studies differ however in the way how that differential response is obtained. Graupner and Brunel (2007) demonstrate that the activation of the calcineurin pathway at intermediate calcium concentrations and of the cAMP–PKA pathway at high calcium concentrations is sufficient to obtain the STDP curve (Figures 7A and 8B). They show in particular that the stronger cAMP–PKA pathway activation due to higher calcium elevations for large positive Δt protocols acts like a realistic veto preventing LTD transitions to occur in this range (compare Figures 3C,D, and Rubin et al., 2005). In contrast, Urakubo et al. (2008) suggest time-difference sensitive allosteric kinetics of the NMDA receptor to be at the origin of STDP results. Based on experimental data, they include the suppression of NMDA-R-mediated currents by calcium/calmodulin binding in their model (Ehlers et al., 1996; Rycroft and Gibb, 2004). In order to achieve a timing-dependent suppression of NMDA-Rs, they assume that calcium/calmodulin suppresses rapidly glutamate-unbound NMDA-Rs but suppresses slowly the glutamate-bound NMDA-R. This allosteric model leads to inhibition of calcium influx for post–pre pairs activating PP1, and boosts calcium influx for pre–post pairs resulting in PKA activation in conjunction with CaMKII autophosphorylation (Figure 8A). Urakubo et al. (2008) show furthermore that their allosteric model predicts correctly the direction of synaptic plasticity in response to spike-triplet and -quadruplet stimulation as obtained in the visual cortex (Froemke and Dan, 2002).
The shape of the STDP results in response to spike-pair stimulation in bistable models depends on the initial distribution of synapses across UP and DOWN states. All synapses are initially in the DOWN state and no noise is present in the model by Urakubo et al. (2008). Their long-term plasticity results are therefore discrete showing deterministic DOWN to UP transitions for pre–post pairs with short Δts (Figure 8C, Obs. 2). Graupner and Brunel (2007) assume an equal initial occupation of UP and DOWN states, that is, 50% of the synapses are initially in the UP and 50% in the DOWN state. Together with noisy calcium transients, they obtain smooth STDP results reflecting stochastic transitions for pre–post and post–pre pairs with short time differences (Figure 8B). Note that O’Connor et al. (2005b) find that 71 ± 11% of Schaffer collateral-CA1 synapses could potentiate or were unable to depress, and 29 ± 11% of the synapses could depress or were unable to potentiate, suggesting that synapses initially occupy both states but with unequal probabilities.
In summary, the CaMKII system can reproduce experimentally observed transition behavior in response to the STDP spike-pair stimulation protocol. The bistability of the CaMKII kinase – phosphatase system allows furthermore to stably maintain the evoked state.
Bi/Multistable Models Based on Alternative Mechanisms
The models discussed so far generate bistability through the properties of the CaMKII kinase – phosphatase system. However, other potential mechanisms giving rise to bistability have been described in recent years, and we discuss them shortly in this section. We also mention models with more than two stable states – three in practice. In such models, multistability is generated through coupling of several bistable switches.
The CaMKII kinase – phosphatase system discussed so far is only a part of the extensive protein signaling network at the synapse (Bhalla and Iyengar, 1999). Bhalla and Iyengar (1999) account for the convergence of mGluR- and NMDA-R-activated pathways based on known interactions involving a multiplicity of proteins (such as protein kinase C and MAPK, for example). It is shown that positive feedback loops in such protein networks give rise to bistability. In contrast to the studies outlined in the previous section, the intrinsic activation and the intrinsic enzymatic properties of single proteins are not resolved in Bhalla and Iyengar (1999) (the intersubunit autophosphorylation of CaMKII is not described at length, for example). Instead, two types of signal transmission mechanisms are implemented: (i) protein–protein interactions as well as enzymatic reactions such as protein phosphorylation and dephosphorylation; (ii) and protein degradation or production of intracellular messengers.
Castellani et al. (2009) describe the two step phosphorylation cycle of AMPA receptors using enzymatic Michaelis–Menten equations. Phosphorylation steps occur through PKA and CaMKII activity and both dephosphorylation steps are mediated by PP1, the concentration of which are used as input variables of the system. The non-linearity of the Michaelis–Menten description endows the system with bistability depending on the CaMKII concentration. The receptor exists in the dephosphorylated state only at low CaMKII concentrations; in its phosphorylated state at high CaMKII concentrations; and can exist in both states at intermediate CaMKII levels. Castellani et al. (2009) use furthermore a stochastic formulation of their phosphorylation scheme to investigate the stability of the states at low receptor numbers and in the presence of noise.
Similarly, Delord et al. (2007) propose a one-step phosphorylation cycle of a substrate “S” (e.g., synaptic AMPA-Rs). Phosphorylation and dephosphorylation are mediated by kinases and phosphatases, respectively, and their respective rates are calcium-dependent. The maintenance of plastic modifications relies on negligible reaction rates in basal conditions, that is, the de- and phosphorylation rates are on the order of 1/month at resting calcium concentrations. Moreover, Delord et al. (2007) show that information coding and memory maintenance are robust to stochastic fluctuations in their model.
The late phase of LTP involves the synthesis of new proteins (Frey et al., 1988; Kang and Schuman, 1996). Aslam et al. (2009) demonstrate that self-sustained regulation of translation can form a bistable synaptic switch that persistently regulates the onsite synthesis of plasticity-related proteins. In particular, they model the CaMKII – cytoplasmic polyadenylation element-binding protein (CPEB1) molecular loop which stably increases the local CaMKII concentration at the potentiated synapse. Protein–protein interactions are implemented based on standard Michaelis–Menten-type kinetics in that approach.
The phosphorylation of the AMPA receptor by CaMKII enhances synaptic AMPA channel function (Mammen et al., 1997; Derkach et al., 1999; Hayashi et al., 2000). Hayer and Bhalla (2005) show that the combination of a CaMKII- and a AMPA receptor switch can lead to tri- or bistability, depending on whether both switches function completely independently, or tightly coupled, respectively. The independent AMPA receptor switch in that model is based on self-recruitment of receptors into the synapse (see discussion of the Hayer and Bhalla, 2005 model at the end of section ‘Bistable models based on the CaMKII kinase – phosphatase system’). Similarly, Pi and Lisman (2008) obtain tristability through coupling of a bistable kinase- and a bistable phosphatase switch (see discussion of Pi and Lisman, 2008 at the end of section ‘Bistable models based on the CaMKII kinase – phosphatase system’).
Another possibility for bistability to arise is the modulation of trafficking rates due to local clustering of receptors in the synaptic membrane (Shouval, 2005). Contrary to the approaches described above, the stability of synaptic efficacies stems from local interactions between individual receptors within a single synapse in that model. This leads to metastable states that can outlast the lifetime of individual receptors, thus providing a mechanism for long-term maintenance of bidirectional synaptic changes.
Besides the existence of bi- and multistability, the question whether experimental stimulation protocols known to induce LTP/LTD evoke transitions between the stable steady-states has not been tested in these models.
Discussion
We reviewed here biophysical models describing how pre- and postsynaptic activity patterns can translate into changes of synaptic efficacy and how those changes can be maintained persistently. We compared several classes of models that incorporate an increasing amount of biological details. A first class of models are based on calcium dynamics, with the possible addition of “readout” variables that translate information contained in the local calcium transients into experimentally observed plasticity results. Those models account for induction of STDP, but leave open the question of the maintenance of synaptic changes over long time scales. A second class of models include explicitly specific protein signaling cascades present at the synapse. These models typically feature bistability, which allows them to stably maintain evoked synaptic changes over long-time scales. Furthermore, several models belonging to this class have been shown recently to be able to reproduce experimentally observed STDP results.
The studies discussed here share a number of common features. Pathways decreasing synaptic strength (e.g., protein phosphatases) activate at intermediate calcium levels, while pathways increasing synaptic strength (e.g., protein kinases) activate at high calcium levels. This is embodied in the calcium control hypothesis (Figure 2A) but it is not sufficient to account for STDP experiments that do not see a second LTD range at positive Δts (see Figure 2). Models with effectively three calcium-triggered pathways (“P”, “A”, and “V” detectors in Rubin et al., 2005; calcineurin, cAMP–PKA, direct CaMKII phosphorylation pathways in Graupner and Brunel, 2007) or with the inclusion of converging pathways other than calcium (mGluR-mediated pathways in Karmarkar and Buonomano, 2002 and Badoual et al., 2006; allosteric kinetics of NMDA receptors in Urakubo et al., 2008) have been proposed to yield LTD at short negative Δts and LTP at short positive Δts only.
The detailed biophysical models discussed here predict that synapses should have a small number of stable states (two in the simplest case). Whether synapses are bistable or not is a controversial issue. At first sight, it seems difficult to accept the idea of a binary system on the basis of recorded synaptic changes, which show a continuous character in most experiments (Dudek and Bear, 1992; Bi and Poo, 1998; Ngezahayo et al., 2000). However, continuous changes can be reconciled with binary individual synapses, if one takes into account stochasticity inherent in synaptic processes and the fact that stimulation protocols typically comprise ensembles of synapses (Appleby and Elliott, 2005; Graupner and Brunel, 2007). The all-or-none potentiation behavior, which one would expect from a bistable synapse, can therefore only be revealed during stimulation of single synapses (Petersen et al., 1998; Bagal et al., 2005; O’Connor et al., 2005b). Bistability has been observed experimentally in a number of distinct biochemical systems (Degn, 1968; Naparstek et al., 1974; Eschrich et al., 1980; Frenzel et al., 1995). It allows to durably store information in a noisy environment in which a continuous variable would progressively deteriorate due to ongoing perturbations.
One implication of bistability at the level of a single synapse is that LTP is the reversal of LTD and vice versa. Experimentalists have however reported several types of decrease in synaptic strength. LTD (decrease of efficacy from “basal” strength) and depotentiation (decrease of efficacy after potentiation) have been considered to be two distinct processes. Some of the differences between the two can be reconciled in bistable models by considering that a “basal” condition is likely to be a mix of synapses in the UP and the DOWN state. In CA3–CA1 hippocampal synapses, 71% of synapses seem to be initially in the DOWN state, and the remaining 29% in the UP state (O’Connor et al., 2005b). This difference in UP and DOWN state occupations gives in principle rise to a higher probability to evoke LTP than LTD. Furthermore, a LTD protocol will decrease synaptic strength by provoking up-to-down transitions in some synapses that were initially in the UP state. On the other hand, the initial conditions are different in depotentiation protocols because a larger fraction of the synapses are in the UP state. However, some studies indicate that depotentiation and LTD might operate through different molecular mechanisms (Zhuo et al., 1999; Lee et al., 2000; Jouvenceau et al., 2003). More complex models than the ones discussed here would be necessary to account for these experimental data with respect to induction and maintenance of synaptic changes (see discussion of Pi and Lisman, 2008 at the end of section ‘Bistable models based on the CaMKII kinase – phosphatase system’).
The CaMKII-based models discussed here suggest that the signaling cascades involved in the early phase of LTP/LTD (<60 min) also maintain the evoked synaptic changes on the long run via bistability. Another possibility for stable memory storage could be that the protein synthesis mechanisms recruited during the late phase of LTP/LTD (Krug et al., 1984; Frey et al., 1988) are bistable (see Aslam et al., 2009), while the early mechanisms triggering those changes are not. Such a possibility has been investigated in models of synaptic tagging and capture (Clopath et al., 2008; Barrett et al., 2009). These models account for the synapse-specific capturing of plasticity-related proteins that are synthesized in the cell body and globally available. In both models, the consolidation mechanism involved in the late phase of synaptic plasticity exhibits bistability (or quasi-bistability through very slow time scales in Barrett et al., 2009).
All the models presented here focus on STDP outcomes evoked with spike-pairs. Few of these studies address plasticity results in response to spike-triplets or -quadruplets (Rubin et al., 2005; Badoual et al., 2006; Urakubo et al., 2008), and none of those investigate the rate dependence of STDP results, i.e., how do plasticity results change if the frequency of the spike-pair presentation is varied (see Sjöström et al., 2001; Wittenberg and Wang, 2006). It remains to be shown that the proposed signaling cascades account for experimental plasticity results obtained with all these distinct stimulation protocols. Furthermore, it remains to be elucidated how differences in the underlying biochemical machinery at a synapse give rise to different plasticity results in different brain areas in response to a given stimulation protocol (e.g. compare STDP results obtained in the hippocampus: Magee and Johnston, 1997; Bi and Poo, 1998; the cortex: Markram et al., 1997; Sjöström et al., 2001; or a cerebellum-like structure Bell et al., 1997, for example). Biophysically detailed models can help to obtain insights into such differences by elucidating which component of the models is related to which property of the plasticity outcome. Finally, one future challenge for such synapse models remains to investigate synaptic plasticity in response to naturalistic activity patterns occurring in vivo.
All the studies discussed here focus on the direction and amplitude of synaptic changes. To our knowledge, none of the studies has investigated the underlying temporal dynamics of synaptic changes in detail. O’Connor et al. (2005b) report that synaptic changes are sudden switch-like events, taking place on the time scale of seconds. LTP transitions occur on average 1.3 min after the onset of the stimulation protocol and LTD transitions on average after 3.1 min (O’Connor et al., 2005b). In the experiments of Bagal et al. (2005), the step-like potentiation event occurs ≈38 s after the pairing pulse and when it occurs it is expressed in less than 10 s. How can the underlying protein signaling cascades give rise to such temporal dynamics? What defines how many presentations of a certain stimulation pattern are required to evoke a transition? That also relates to the question of how synaptic changes are summed up across individual stimulation patterns? Continuing effort from experimentalists and modelers is required to answer those questions, which relate to the underlying time course of learning in the intact brain.
The models reviewed here account for protein signaling cascades local to one synapse. However, CaMKII, for example, is actively translocated to the PSD upon synaptic activation (Strack et al., 1997; Schulman, 2004; Merrill et al., 2005). Experiments show furthermore that spines are not biochemically isolated compartments but interact with each other in an activity-dependent manner (Harvey et al., 2008). As a result, LTP at individual synapses reduces the threshold for potentiation at neighboring synapses (Harvey and Svoboda, 2007). How competition or positive feedback between synapses can lead to sparse or clustered plasticity models of memory storage on single dendritic branches remains to be addressed in the models outlined here.
We reviewed here biophysical models of single synapses. A challenge for future modeling studies is to insert such synaptic dynamics in networks, in order to understand how they affect network dynamics and how they allow networks to durably store information provided by external inputs. A prerequisite for such an endeavor would be to derive simplified models (i.e., described by the smallest possible number of variables) that keep the main qualitative features of the biophysically detailed models (i.e., bistability, ability to reproduce plasticity outcomes seen in experiment). Work in this direction is in progress (Graupner and Brunel, in preparation). An interesting question is whether and how the emerging synaptic connectivity patterns in such models will be different from studies implementing phenomenological spike-based learning rules based on spike-pairs or -triplets (reviewed in Morrison et al., 2008). Such investigations should further advance our understanding of the role of synaptic plasticity in shaping the spatio-temporal dynamics of recurrent networks and thereby memory formation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Wulfram Gerstner for encouraging us to write a review paper. MG was partly supported by grants from the National Institutes of Health (DC005787-01A1), the Deutscher Akademischer Austausch Dienst (DAAD), the Ministère des Affaires étrangères du Gouvernement Français, the Agence Nationale de la Recherche (ANR) Neurosciences, and the Centre National de la Recherche Scientifique (CNRS). NB was supported by ANR grants ANR-05-NEUR-030 and ANR-08-SYSC-005.
References
Abarbanel, H. D. I., Gibb, L., Huerta, R., and Rabinovich, M. (2003). Biophysical model of synaptic plasticity dynamics. Biol. Cybern. 89, 214–226.
Abbott, L., and Nelson, S. (2000). Synaptic plasticity: taming the beast. Nat. Neurosci. 3(Suppl.) 1178–1183.
Abel, T., Nguyen, P. V., Barad, M., Deuel, T. A., Kandel, E. R., and Bourtchouladze, R. (1997). Genetic demonstration of a role for PKA in the late phase of LTP and in hippocampus-based long-term memory. Cell 88, 615–626.
Abraham, W. C., and Robins, A. (2005). Memory retention–the synaptic stability versus plasticity dilemma. Trends Neurosci. 28, 73–78.
Alberini, C. M., Ghirardi, M., Huang, Y. Y., Nguyen, P. V., and Kandel, E. R. (1995). A molecular switch for the consolidation of long-term memory: cAMP-inducible gene expression. Ann. N.Y. Acad. Sci. 758, 261–286.
Appleby, P. A., and Elliott, T. (2005). Synaptic and temporal ensemble interpretation of spike-timing-dependent plasticity. Neural Comput. 17, 2316–2336.
Artola, A., and Singer, W. (1987). Long-term potentiation and NMDA receptors in rat visual cortex. Nature 330, 649–652.
Aslam, N., Kubota, Y., Wells, D., and Shouval, H. Z. (2009). Translational switch for long-term maintenance of synaptic plasticity. Mol. Syst. Biol. 5, 284.
Badoual, M., Zou, Q., Davison, A. P., Rudolph, M., Bal, T., Frégnac, Y., and Destexhe, A. (2006). Biophysical and phenomenological models of multiple spike interactions in spike-timing dependent plasticity. Int. J. Neural Syst. 16, 79–97.
Bagal, A. A., Kao, J., P. Y., Tang, C.-M., and Thompson, S. M. (2005). Long-term potentiation of exogenous glutamate responses at single dendritic spines. Proc. Natl. Acad. Sci. U.S.A. 102, 14434–14439.
Barrett, A. B., Billings, G. O., R. Morris, G. M., and van Rossum, M. C. W. (2009). State based model of long-term potentiation and synaptic tagging and capture. PLoS Comput. Biol. 5:1, e1000259. doi:10.1371/journal.pcbi.1000259.
Bear, M. F., Press, W. A., and Connors, B. W. (1992). Long-term potentiation in slices of kitten visual cortex and the effects of NMDA receptor blockade. J. Neurophysiol. 67, 841–851.
Bell, C., Han, V., Sugawara, Y., and Grant, K. (1997). Synaptic plasticity in a cerebellum-like structure depends on temporal order. Nature 387, 278–281.
Bender, V. A., Bender, K. J., Brasier, D. J., and Feldman, D. E. (2006). Two coincidence detectors for spike timing-dependent plasticity in somatosensory cortex. J. Neurosci. 26, 4166–4177.
Benke, T. A., Lüthi, A., Isaac, J. T., and Collingridge, G. L. (1998). Modulation of AMPA receptor unitary conductance by synaptic activity. Nature 393, 793–797.
Bhalla, U., and Iyengar, R. (1999). Emergent properties of networks of biological signaling pathways. Science 283, 381–387.
Bi, G., and Poo, M. (1998). Synaptic modifications in cultured hippocampal neurons: dependence on spike timing, synaptic strength, and postsynaptic cell type. J. Neurosci. 18, 10464–10472.
Bi, G. Q., and Wang, H. X. (2002). Temporal asymmetry in spike timing-dependent synaptic plasticity. Physiol. Behav. 77, 551–555.
Bienenstock, E. L., Cooper, L. N., and Munro, P. W. (1982). Theory for the development of neuron selectivity: orientation specificity and binocular interaction in visual cortex. J. Neurosci. 2, 32–48.
Billings, G., and van Rossum, M. C. W. (2009). Memory retention and spike-timing-dependent plasticity. J. Neurophysiol. 101, 2775–2788.
Bliss, T., and Collingridge, G. (1993). A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361, 31–39.
Blitzer, R. D., Connor, J. H., Brown, G. P., Wong, T., Shenolikar, S., Iyengar, R., and Landau, E. M. (1998). Gating of CaMKII by cAMP-regulated protein phosphatase activity during LTP. Science 280, 1940–1942.
Blitzer, R. D., Wong, T., Nouranifar, R., Iyengar, R., and Landau, E. M. (1995). Postsynaptic cAMP pathway gates early LTP in hippocampal CA1 region. Neuron 15, 1403–1414.
Bolshakov, V. Y., and Siegelbaum, S. A. (1994). Postsynaptic induction and presynaptic expression of hippocampal long-term depression. Science 264, 1148–1152.
Bolshakov, V. Y., and Siegelbaum, S. A. (1995). Regulation of hippocampal transmitter release during development and long-term potentiation. Science 269, 1730–1734.
Bozon, B., Kelly, A., Josselyn, S. A., Silva, A. J., Davis, S., and Laroche, S. (2003). MAPK, CREB and Zif268 are all required for the consolidation of recognition memory. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 358, 805–814.
Buard, I., Coultrap, S. J., Freund, R. K., Lee, Y.-S., Dell’Acqua, M. L., Silva, A. J., and Bayer, K. U. (2010). CaMKII “autonomy” is required for initiating but not for maintaining neuronal long-term information storage. J. Neurosci. 30, 8214–8220.
Cai, Y., Gavornik, J. P., Cooper, L. N., Yeung, L. C., and Shouval, H. Z. (2007). Effect of stochastic synaptic and dendritic dynamics on synaptic plasticity in visual cortex and hippocampus. J. Neurophysiol. 97, 375–386.
Carr, D. W., Stofko-Hahn, R. E., Fraser, I. D., Cone, R. D., and Scott, J. D. (1992). Localization of the cAMP-dependent protein kinase to the postsynaptic densities by A-kinase anchoring proteins. Characterization of AKAP 79. J. Biol. Chem. 267, 16816–16823.
Castellani, G. C., Bazzani, A., and Cooper, L. N. (2009). Toward a microscopic model of bidirectional synaptic plasticity. Proc. Natl. Acad. Sci. U.S.A. 106, 14091–14095.
Castro-Alamancos, M. A., Donoghue, J. P., and Connors, B. W. (1995). Different forms of synaptic plasticity in somatosensory and motor areas of the neocortex. J. Neurosci. 15, 5324–5333.
Chen, A., Muzzio, I. A., Malleret, G., Bartsch, D., Verbitsky, M., Pavlidis, P., Yonan, A. L., Vronskaya, S., Grody, M. B., Cepeda, I., Gilliam, T. C., and Kandel, E. R. (2003). Inducible enhancement of memory storage and synaptic plasticity in transgenic mice expressing an inhibitor of ATF4 (CREB-2) and C/EBP proteins. Neuron 39, 655–669.
Clopath, C., Ziegler, L., Vasilaki, E., Büsing, L., and Gerstner, W. (2008). Tag-trigger-consolidation: a model of early and late long-term-potentiation and depression. PLoS Comput. Biol. 4:12, e1000248. doi:10.1371/journal.pcbi.1000248.
Colbran, R. J. (2004). Targeting of calcium/calmodulin-dependent protein kinase II. Biochem. J. 378, 1–16.
Collingridge, G. L., Kehl, S. J., and McLennan, H. (1983). Excitatory amino acids in synaptic transmission in the schaffer collateral-commissural pathway of the rat hippocampus. J. Physiol. 334, 33–46.
Cooper, D., Mons, N., and Karpen, J. (1995). Adenylyl cyclases and the interaction between calcium and cAMP signalling. Nature 374, 421–424.
Debanne, D., Gähwiler, B. H., and Thompson, S. M. (1996). Cooperative interactions in the induction of long-term potentiation and depression of synaptic excitation between hippocampal CA3-CA1 cell pairs in vitro. Proc. Natl. Acad. Sci. U.S.A. 93, 11225–11230.
Degn, H. (1968). Bistability caused by substrate inhibition of peroxidase in an open reaction system. Nature 217, 1047–1050.
Delord, B., Berry, H., Guigon, E., and Genet, S. (2007). A new principle for information storage in an enzymatic pathway model. PLoS Comput. Biol. 3: 6, e124. doi:10.1371/journal.pcbi.0030124.
Derkach, V., Barria, A., and Soderling, T. (1999). Ca2+/calmodulin-kinase II enhances channel conductance of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate type glutamate receptors. Proc. Natl. Acad. Sci. U.S.A. 96, 3269–3274.
Doi, T., Kuroda, S., Michikawa, T., and Kawato, M. (2005). Inositol 1,4,5-trisphosphate-dependent Ca2+ threshold dynamics detect spike timing in cerebellar purkinje cells. J. Neurosci. 25, 950–961.
Dudek, S., and Bear, M. (1992). Homosynaptic long-term depression in area CA1 of hippocampus and effects of N-methyl-D-aspartate receptor blockade. Proc. Natl. Acad. Sci. U.S.A. 89, 4363–4367.
Ehlers, M. D. (2000). Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron 28, 511–525.
Ehlers, M. D. (2003). Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat. Neurosci. 6, 231–242.
Ehlers, M. D., Zhang, S., Bernhadt, J. P., and Huganir, R. L. (1996). Inactivation of NMDA receptors by direct interaction of calmodulin with the NR1 subunit. Cell 84, 745–755.
Eschrich, K., Schellenberger, W., and Hofmann, E. (1980). In vitro demonstration of alternate stationary states in an open enzyme system containing phosphofructokinase. Arch. Biochem. Biophys. 205, 114–121.
Esteban, J. A., Shi, S.-H., Wilson, C., Nuriya, M., Huganir, R. L., and Malinow, R. (2003). PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat. Neurosci. 6, 136–143.
Fan, W., Ster, J., and Gerber, U. (2010). Activation conditions for the induction of metabotropic glutamate receptor-dependent long-term depression in hippocampal CA1 pyramidal cells. J. Neurosci. 30, 1471–1475.
Feldman, D. E. (2000). Timing-based LTP and LTD at vertical inputs to layer II/III pyramidal cells in rat barrel cortex. Neuron 27, 45–56.
Fink, C. C., and Meyer, T. (2002). Molecular mechanisms of CaMKII activation in neuronal plasticity. Curr. Opin. Neurobiol. 12, 293–299.
Frenzel, J., Schellenberger, W., and Eschrich, K. (1995). Bistability and damped oscillations in the fructose 6-phosphate/fructose 1,6-bisphosphate cycle in cell-free extracts from rat liver. Biol. Chem. Hoppe Seyler 376, 17–24.
Frey, U., Huang, Y. Y., and Kandel, E. R. (1993). Effects of cAMP simulate a late stage of LTP in hippocampal CA1 neurons. Science 260, 1661–1664.
Frey, U., Krug, M., Reymann, K. G., and Matthies, H. (1988). Anisomycin, an inhibitor of protein synthesis, blocks late phases of LTP phenomena in the hippocampal CA1 region in vitro. Brain Res. 452, 57–65.
Froemke, R. C., and Dan, Y. (2002). Spike-timing-dependent synaptic modification induced by natural spike trains. Nature 416, 433–438.
Gerkin, R. C., Bi, G.-Q., and Rubin, J. E. (2010). Hippocampal Microcircuits: A Computational Modeler’s Resource Book, Vol. 5. Springer Series in Computational Neuroscience.
Giese, K., Fedorov, N., Filipkowski, R., and Silva, A. (1998). Autophosphorylation at Thr286 of the alpha calcium-calmodulin kinase II in LTP and learning. Science 279, 870–873.
Glantz, S. B., Amat, J. A., and Rubin, C. S. (1992). cAMP signaling in neurons: patterns of neuronal expression and intracellular localization for a novel protein, AKAP 150, that anchors the regulatory subunit of cAMP-dependent protein kinase II beta. Mol. Biol. Cell 3, 1215–1228.
Goelet, P., Castellucci, V. F., Schacher, S., and Kandel, E. R. (1986). The long and the short of long-term memory–a molecular framework. Nature 322, 419–422.
Graupner, M., and Brunel, N. (2007). STDP in a bistable synapse model based on CaMKII and associated signaling pathways. PLoS Comput. Biol. 3, 2299–2323. doi:10.1371/journal.pcbi.0030221.
Griffith, L. C. (2004). Regulation of calcium/calmodulin-dependent protein kinase II activation by intramolecular and intermolecular interactions. J. Neurosci. 24, 8394–8398.
Gustafsson, B., Wigström, H., Abraham, W. C., and Huang, Y. Y. (1987). Long-term potentiation in the hippocampus using depolarizing current pulses as the conditioning stimulus to single volley synaptic potentials. J. Neurosci. 7, 774–780.
Hahm, J. O., Langdon, R. B., and Sur, M. (1991). Disruption of retinogeniculate afferent segregation by antagonists to NMDA receptors. Nature 351, 568–570.
Hansel, C., de Jeu, M., Belmeguenai, A., Houtman, S. H., G. Buitendijk, H. S., Andreev, D., C. Zeeuw, I. D., and Elgersma, Y. (2006). alphaCaMKII is essential for cerebellar LTD and motor learning. Neuron 51, 835–843.
Hanson, P. I., and Schulman, H. (1992). Neuronal Ca2+/calmodulin-dependent protein kinase. Annu. Rev. Biochem. 61, 559–601.
Harris, K. M., and Stevens, J. K. (1989). Dendritic spines of CA1 pyramidal cells in the rat hippocampus: serial electron microscopy with reference to their biophysical characteristics. J. Neurosci. 9, 2982–2997.
Harvey, C. D., and Svoboda, K. (2007). Locally dynamic synaptic learning rules in pyramidal neuron dendrites. Nature 450, 1195–1200.
Harvey, C. D., Yasuda, R., Zhong, H., and Svoboda, K. (2008). The spread of Ras activity triggered by activation of a single dendritic spine. Science 321, 136–140.
Hashimotodani, Y., Ohno-Shosaku, T., Tsubokawa, H., Ogata, H., Emoto, K., Maejima, T., Araishi, K., Shin, H.-S., and Kano, M. (2005). Phospholipase Cbeta serves as a coincidence detector through its Ca2+ dependency for triggering retrograde endocannabinoid signal. Neuron 45, 257–268.
Hayashi, Y., Shi, S. H., Esteban, J. A., Piccini, A., Poncer, J. C., and Malinow, R. (2000). Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science 287, 2262–2267.
Hayer, A., and Bhalla, U. S. (2005). Molecular switches at the synapse emerge from receptor and kinase traffic. PLoS Comput. Biol. 1:2, 137–154. doi:10.1371/journal.pcbi.0010020.
Hudmon, A., and Schulman, H. (2002). Neuronal Ca2+/calmodulin-dependent protein kinase II: The role of structure and autoregulation in cellular function. Annu. Rev. Biochem. 71, 473–510.
Ingebritsen, T. S., and Cohen, P. (1983). Protein phosphatases: properties and role in cellular regulation. Science 221, 331–338.
Isaac, J. T., Hjelmstad, G. O., Nicoll, R. A., and Malenka, R. C. (1996). Long-term potentiation at single fiber inputs to hippocampal CA1 pyramidal cells. Proc. Natl. Acad. Sci. U.S.A. 93, 8710–8715.
Ismailov, I., Kalikulov, D., Inoue, T., and Friedlander, M. J. (2004). The kinetic profile of intracellular calcium predicts long-term potentiation and long-term depression. J. Neurosci. 24, 9847–9861.
Jaffe, D. B., Johnston, D., N. Lasser-Ross, Lisman, J. E., Miyakawa, H., and Ross, W. N. (1992). The spread of Na+ spikes determines the pattern of dendritic Ca2+ entry into hippocampal neurons. Nature 357, 244–246.
Jahr, C., and Stevens, C. (1990). A quantitative description of NMDA receptor-channel kinetic behavior. J. Neurosci. 10, 1830–1837.
Jay, T., Gurden, H., and Yamaguchi, T. (1998). Rapid increase in PKA activity during long-term potentiation in the hippocampal afferent fibre system to the prefrontal cortex in vivo. Eur. J. Neurosci. 10, 3302–3306.
Jouvenceau, A., Billard, J.-M., Haditsch, U., Mansuy, I. M., and Dutar, P. (2003). Different phosphatase-dependent mechanisms mediate long-term depression and depotentiation of long-term potentiation in mouse hippocampal CA1 area. Eur. J. Neurosci. 18, 1279–1285.
Kang, H., and Schuman, E. M. (1996). A requirement for local protein synthesis in neurotrophin-induced hippocampal synaptic plasticity. Science 273, 1402–1406.
Karmarkar, U. R., and Buonomano, D. V. (2002). A model of spike-timing dependent plasticity: one or two coincidence detectors? J. Neurophysiol. 88, 507–513.
Karmarkar, U. R., Najarian, M. T., and Buonomano, D. V. (2002). Mechanisms and significance of spike-timing dependent plasticity. Biol. Cybern. 87, 373–382.
Kirkwood, A., Dudek, S. M., Gold, J. T., Aizenman, C. D., and Bear, M. F. (1993). Common forms of synaptic plasticity in the hippocampus and neocortex in vitro. Science 260, 1518–1521.
Koester, H. J., and Sakmann, B. (1998). Calcium dynamics in single spines during coincident pre- and postsynaptic activity depend on relative timing of back-propagating action potentials and subthreshold excitatory postsynaptic potentials. Proc. Natl. Acad. Sci. U.S.A. 95, 9596–9601.
Kotaleski, J. H., and Blackwell, K. T. (2010). Modelling the molecular mechanisms of synaptic plasticity using systems biology approaches. Nat. Rev. Neurosci. 11, 239–251.
Kovalchuk, Y., Eilers, J., Lisman, J., and Konnerth, A. (2000). NMDA receptor-mediated subthreshold Ca2+ signals in spines of hippocampal neurons. J. Neurosci. 20, 1791–1799.
Krug, M., Lössner, B., and Ott, T. (1984). Anisomycin blocks the late phase of long-term potentiation in the dentate gyrus of freely moving rats. Brain Res. Bull. 13, 39–42.
Lee, H., Barbarosie, M., Kameyama, K., Bear, M., and Huganir, R. (2000). Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature 405, 955–959.
Lee, H.-K., Takamiya, K., Han, J.-S., Man, H., Kim, C.-H., Rumbaugh, G., Yu, S., Ding, L., He, C., Petralia, R. S., Wenthold, R. J., Gallagher, M., and Huganir, R. L. (2003). Phosphorylation of the AMPA receptor GluR1 subunit is required for synaptic plasticity and retention of spatial memory. Cell 112, 631–643.
Lengyel, I., Voss, K., Cammarota, M., Bradshaw, K., Brent, V., Murphy, K. P., Giese, S. J., Rostas, J. A., and Bliss, T. V. (2004). Autonomous activity of CaMKII is only transiently increased following the induction of long-term potentiation in the rat hippocampus. Eur. J. Neurosci. 20, 3063–3072.
Levy, W. B., and Steward, O. (1983). Temporal contiguity requirements for long-term associative potentiation/depression in the hippocampus. Neuroscience 8, 791–797.
Lisman, J. (1985). A mechanism for memory storage insensitive to molecular turnover: a bistable autophosphorylating kinase. Proc. Natl. Acad. Sci. U.S.A. 82, 3055–3057.
Lisman, J. (2003). Long-term potentiation: outstanding questions and attempted synthesis. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 358, 829–842.
Lisman, J., Schulman, H., and Cline, H. (2002). The molecular basis of CaMKII function in synaptic and behavioural memory. Nat. Rev. Neurosci. 3, 175–190.
Lisman, J. E., and Goldring, M. A. (1988). Feasibility of long-term storage of graded information by the Ca2+/calmodulin-dependent protein kinase molecules of the postsynaptic density. Proc. Natl. Acad. Sci. U.S.A. 85, 5320–5324.
Lynch, G., Larson, J., Kelso, S., Barrionuevo, G., and Schottler, F. (1983). Intracellular injections of EGTA block induction of hippocampal long-term potentiation. Nature 305, 719–721.
Maejima, T., Oka, S., Hashimotodani, Y., T. Ohno-Shosaku, Aiba, A., Wu, D., Waku, K., Sugiura, T., and Kano, M. (2005). Synaptically driven endocannabinoid release requires Ca2+-assisted metabotropic glutamate receptor subtype 1 to phospholipase Cbeta4 signaling cascade in the cerebellum. J. Neurosci. 25, 6826–6835.
Magee, J., and Johnston, D. (1997). A synaptically controlled, associative signal for Hebbian plasticity in hippocampal neurons. Science 275, 209–213.
Majewska, A., Brown, E., Ross, J., and Yuste, R. (2000). Mechanisms of calcium decay kinetics in hippocampal spines: role of spine calcium pumps and calcium diffusion through the spine neck in biochemical compartmentalization. J. Neurosci. 20, 1722–1734.
Malenka, R., and Nicoll, R. (1999). Long-term potentiation–a decade of progress? Science 285, 1870–1874.
Malenka, R. C., Kauer, J. A., Zucker, R. S., and Nicoll, R. A. (1988). Postsynaptic calcium is sufficient for potentiation of hippocampal synaptic transmission. Science 242, 81–84.
Malinow, R., and Malenka, R. C. (2002). AMPA receptor trafficking and synaptic plasticity. Annu. Rev. Neurosci. 25, 103–126.
Malleret, G., Haditsch, U., Genoux, D., Jones, M., Bliss, T., Vanhoose, A., Weitlauf, C., Kandel, E., Winder, D., and Mansuy, I. (2001). Inducible and reversible enhancement of learning, memory, and long-term potentiation by genetic inhibition of calcineurin. Cell 104, 675–686.
Mammen, A. L., Kameyama, K., Roche, K. W., and Huganir, R. L. (1997). Phosphorylation of the alpha-amino-3-hydroxy-5-methylisoxazole4-propionic acid receptor GluR1 subunit by calcium/calmodulin-dependent kinase II. J. Biol. Chem. 272, 32528–32533.
Mansuy, I. M., Mayford, M., Jacob, B., Kandel, E. R., and Bach, M. E. (1998). Restricted and regulated overexpression reveals calcineurin as a key component in the transition from short-term to long-term memory. Cell 92, 39–49.
Markram, H., Lübke, J., Frotscher, M., and Sakmann, B. (1997). Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science 275, 213–215.
McNeill, R. B., and Colbran, R. J. (1995). Interaction of autophosphorylated Ca2+/calmodulin-dependent protein kinase II with neuronal cytoskeletal proteins. characterization of binding to a 190-kDa postsynaptic density protein. J. Biol. Chem. 270, 10043–10049.
Merrill, M. A., Chen, Y., Strack, S., and Hell, J. W. (2005). Activity-driven postsynaptic translocation of CaMKII. Trends Pharmacol. Sci. 26, 645–653.
Miller, P., Zhabotinsky, A. M., Lisman, J. E., and Wang, X.-J. (2005). The stability of a stochastic CaMKII switch: dependence on the number of enzyme molecules and protein turnover. PLoS Biol. 3:4, e107. doi:10.1371/journal.pbio.0030107.
Mizuno, T., Kanazawa, I., and Sakurai, M. (2001). Differential induction of LTP and LTD is not determined solely by instantaneous calcium concentration: an essential involvement of a temporal factor. Eur. J. Neurosci. 14, 701–708.
Mooney, R., Madison, D. V., and Shatz, C. J. (1993). Enhancement of transmission at the developing retinogeniculate synapse. Neuron 10, 815–825.
Morris, R. G., Anderson, E., Lynch, G. S., and Baudry, M. (1986). Selective impairment of learning and blockade of long-term potentiation by an N-methyl-D-aspartate receptor antagonist, AP5. Nature 319, 774–776.
Morrison, A., Diesmann, M., and Gerstner, W. (2008). Phenomenological models of synaptic plasticity based on spike timing. Biol. Cybern. 98, 459–478.
Mulkey, R., Endo, S., Shenolikar, S., and Malenka, R. (1994). Involvement of a calcineurin/inhibitor-1 phosphatase cascade in hippocampal long-term depression. Nature 369, 486–488.
Naparstek, A., Romette, J. L., Kernevez, J. P., and Thomas, D. (1974). Memory in enzyme membranes. Nature 249, 490–491.
Neveu, D., and Zucker, R. S. (1996). Long-lasting potentiation and depression without presynaptic activity. J. Neurophysiol. 75, 2157–2160.
Nevian, T., and Sakmann, B. (2006). Spine Ca2+ signaling in spike-timing-dependent plasticity. J. Neurosci. 26, 11001–11013.
Ngezahayo, A., Schachner, M., and Artola, A. (2000). Synaptic activity modulates the induction of bidirectional synaptic changes in adult mouse hippocampus. J. Neurosci. 20, 2451–2458.
Nguyen, P. V., and Kandel, E. R. (1997). Brief theta-burst stimulation induces a transcription-dependent late phase of LTP requiring cAMP in area CA1 of the mouse hippocampus. Learn. Mem. 4, 230–243.
Nishiyama, M., Hong, K., Mikoshiba, K., Poo, M. M., and Kato, K. (2000). Calcium stores regulate the polarity and input specificity of synaptic modification. Nature 408, 584–588.
Nowak, L., Bregestovski, P., Ascher, P., Herbet, A., and Prochiantz, A. (1984). Magnesium gates glutamate-activated channels in mouse central neurones. Nature 307, 462–465.
O’Connor, D. H., Wittenberg, G. M., and Wang, S. S.-H. (2005a). Dissection of bidirectional synaptic plasticity into saturable unidirectional processes. J. Neurophysiol. 94, 1565–1573.
O’Connor, D. H., Wittenberg, G. M., and Wang, S. S.-H. (2005b). Graded bidirectional synaptic plasticity is composed of switch-like unitary events. Proc. Natl. Acad. Sci. U.S.A. 102, 9679–9684.
Oja, E. (1982). A simplified neuron model as a principal component analyzer. J. Math. Biol. 15, 267–273.
Okamoto, H., and Ichikawa, K. (2000). Switching characteristics of a model for biochemical-reaction networks describing autophosphorylation versus dephosphorylation of Ca2+/calmodulin-dependent protein kinase II. Biol. Cybern. 82, 35–47.
Otani, S., and Connor, J. A. (1998). Requirement of rapid Ca2+ entry and synaptic activation of metabotropic glutamate receptors for the induction of long-term depression in adult rat hippocampus. J. Physiol. 511, 761–770.
Petersen, C., Malenka, R., Nicoll, R., and Hopfield, J. (1998). All-or-none potentiation at CA3-CA1 synapses. Proc. Natl. Acad. Sci. U.S.A. 95, 4732–4737.
Pi, H. J., and Lisman, J. E. (2008). Coupled phosphatase and kinase switches produce the tristability required for long-term potentiation and long-term depression. J. Neurosci. 28, 13132–13138.
Piomelli, D. (2003). The molecular logic of endocannabinoid signalling. Nat. Rev. Neurosci. 4, 873–884.
Raastad, M. (1995). Extracellular activation of unitary excitatory synapses between hippocampal CA3 and CA1 pyramidal cells. Eur. J. Neurosci. 7, 1882–1888.
Rubin, J. E., Gerkin, R. C., Bi, G.-Q., and Chow, C. C. (2005). Calcium time course as a signal for spike-timing-dependent plasticity. J. Neurophysiol. 93, 2600–2613.
Rycroft, B. K., and Gibb, A. J. (2004). Regulation of single NMDA receptor channel activity by alpha-actinin and calmodulin in rat hippocampal granule cells. J. Physiol. 557(Pt 3), 795–808.
Sabatini, B. L., Oertner, T. G., and Svoboda, K. (2002). The life cycle of Ca2+ ions in dendritic spines. Neuron 33, 439–452.
Sabatini, B. L., and Svoboda, K. (2000). Analysis of calcium channels in single spines using optical fluctuation analysis. Nature 408, 589–593.
Sanhueza, M., McIntyre, C. C., and Lisman, J. E. (2007). Reversal of synaptic memory by Ca2+ /calmodulin-dependent protein kinase II inhibitor. J. Neurosci. 27, 5190–5199.
Schulman, H. (2004). Activity-dependent regulation of calcium/calmodulin-dependent protein kinase II localization. J. Neurosci. 24, 8399–8403.
Sharma, K., Fong, D. K., and Craig, A. M. (2006). Postsynaptic protein mobility in dendritic spines: long-term regulation by synaptic NMDA receptor activation. Mol. Cell. Neurosci. 31, 702–712.
Shen, K., and Meyer, T. (1999). Dynamic control of CaMKII translocation and localization in hippocampal neurons by NMDA receptor stimulation. Science 284, 162–166.
Shen, K., Teruel, M. N., Connor, J. H., Shenolikar, S., and Meyer, T. (2000). Molecular memory by reversible translocation of calcium/calmodulin-dependent protein kinase I. Nat. Neurosci. 3, 881–886.
Shouval, H. Z. (2005). Clusters of interacting receptors can stabilize synaptic efficacies. Proc. Natl. Acad. Sci. U.S.A. 102, 14440–14445.
Shouval, H. Z., Bear, M. F., and Cooper, L. N. (2002). A unified model of NMDA receptor-dependent bidirectional synaptic plasticity. Proc. Natl. Acad. Sci. U.S.A. 99, 10831–10836.
Shouval, H. Z., and Kalantzis, G. (2005). Stochastic properties of synaptic transmission affect the shape of spike time-dependent plasticity curves. J. Neurophysiol. 93, 1069–1073.
Sjöström, P. J., Turrigiano, G. G., and Nelson, S. (2001). Rate, timing, and cooperativity jointly determine cortical synaptic plasticity. Neuron 32, 1149–1164.
Sjöström, P. J., Turrigiano, G. G., and Nelson, S. B. (2003). Neocortical LTD via coincident activation of presynaptic NMDA and cannabinoid receptors. Neuron 39, 641–654.
Sjöström, P. J., Turrigiano, G. G., and Nelson, S. B. (2004). Endocannabinoid-dependent neocortical layer-5 LTD in the absence of postsynaptic spiking. J. Neurophysiol. 92, 3338–3343.
Soderling, T. R., and Derkach, V. A. (2000). Postsynaptic protein phosphorylation and LTP. Trends Neurosci. 23, 75–80.
Stanton, P. K., Chattarji, S., and Sejnowski, T. J. (1991). 2-amino-3-phosphonopropionic acid, an inhibitor of glutamate-stimulated phosphoinositide turnover, blocks induction of homosynaptic long-term depression, but not potentiation, in rat hippocampus. Neurosci. Lett. 127, 61–66.
Stevens, C. F., and Wang, Y. (1995). Facilitation and depression at single central synapses. Neuron 14, 795–802.
Strack, S., Choi, S., Lovinger, D. M., and Colbran, R. J. (1997). Translocation of autophosphorylated calcium/calmodulin-dependent protein kinase II to the postsynaptic density. J. Biol. Chem. 272, 13467–13470.
Sweatt, J. D. (2004). Mitogen-activated protein kinases in synaptic plasticity and memory. Curr. Opin. Neurobiol. 14, 311–317.
Thiels, E., Norman, E. D., Barrionuevo, G., and Klann, E. (1998). Transient and persistent increases in protein phosphatase activity during long-term depression in the adult hippocampus in vivo. Neuroscience 86, 1023–1029.
Urakubo, H., Honda, M., Froemke, R. C., and Kuroda, S. (2008). Requirement of an allosteric kinetics of NMDA receptors for spike timing-dependent plasticity. J. Neurosci. 28, 3310–3323.
Waltereit, R., and Weller, M. (2003). Signaling from cAMP/PKA to MAPK and synaptic plasticity. Mol. Neurobiol. 27, 99–106.
Wang, H.-X., Gerkin, R. C., Nauen, D. W., and Bi, G.-Q. (2005). Coactivation and timing-dependent integration of synaptic potentiation and depression. Nat. Neurosci. 8, 187–193.
Winder, D., Mansuy, I., Osman, M., Moallem, T., and Kandel, E. (1998). Genetic and pharmacological evidence for a novel, intermediate phase of long-term potentiation suppressed by calcineurin. Cell 92, 25–37.
Wittenberg, G. M., and Wang, S. S.-H. (2006). Malleability of spike-timing-dependent plasticity at the CA3-CA1 synapse. J. Neurosci. 26, 6610–6617.
Yang, S., Tang, Y., and Zucker, R. (1999). Selective induction of LTP and LTD by postsynaptic [Ca2+]i elevation. J. Neurophysiol. 81, 781–787.
Yuste, R., and Denk, W. (1995). Dendritic spines as basic functional units of neuronal integration. Nature 375, 682–684.
Yuste, R., Majewska, A., Cash, S. S., and Denk, W. (1999). Mechanisms of calcium influx into hippocampal spines: heterogeneity among spines, coincidence detection by NMDA receptors, and optical quantal analysis. J. Neurosci. 19, 1976–1987.
Zhabotinsky, A. M. (2000). Bistability in the Ca2+/calmodulin-dependent protein kinase-phosphatase system. Biophys. J. 79, 2211–2221.
Zhang, L. I., Tao, H. W., Holt, C. E., Harris, W. A., and Poo, M. (1998). A critical window for cooperation and competition among developing retinotectal synapses. Nature 395, 37–44.
Zhuo, M., Zhang, W., Son, H., Mansuy, I., Sobel, R. A., Seidman, J., and Kandel, E. R. (1999). A selective role of calcineurin A-alpha in synaptic depotentiation in hippocampus. Proc. Natl. Acad. Sci. U.S.A. 96, 4650–4655.
Keywords: STDP, biophysical models, bistability, induction, maintenance, protein signaling cascade, calcium control hypothesis, CaMKII
Citation: Graupner M and Brunel N (2010) Mechanisms of induction and maintenance of spike-timing dependent plasticity in biophysical synapse models. Front. Comput. Neurosci. 4:136. doi:10.3389/fncom.2010.00136
Received: 04 February 2010;
Paper pending published: 18 May 2010;
Accepted: 25 August 2010;
Published online: 17 September 2010
Edited by:
Wulfram Gerstner, Ecole Polytechnique Fédérale de Lausanne, SwitzerlandReviewed by:
Mark C. W. van Rossum, University of Edinburgh, UKLorric Ziegler, Ecole Polytechnique Fédérale de Lausanne, Switzerland
Copyright: © 2010 Graupner and Brunel. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Michael Graupner, Center for Neural Science, New York University, 4 Washington Place, New York, NY 10003-6603, USA. e-mail:bWljaGFlbC5ncmF1cG5lckBueXUuZWR1
Abbreviations: EPSP/C, excitatory postsynaptic potential/current; LTP, long-term potentiation; LTD, long-term depression; STDP, spike-timing dependent plasticity; CA, Cornu Ammonis area; CaMKII, calcium/calmodulin-dependent protein kinase II; BPAP, backpropagating action potential; VDCC, voltage-dependent calcium channel; NMDA-R, N-methyl-d-aspartic acid receptor; mGluR, metabotropic glutamate receptor; AMPA-R, α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate receptor; CaM, calmodulin; PP1, protein phosphatase 1; PKA, protein kinase A; I1, inhibitor 1; cAMP, cyclic adenosine monophosphate; MAPK, mitogen-activated protein kinase.