




- 1 Department of Signal Processing, Tampere University of Technology, Tampere, Finland
- 2 School of Computer Science and Communication, Royal Institute of Technology, Stockholm, Sweden
- 3 Stockholm Brain Institute, Karolinska Institutet, Stockholm, Sweden
- 4 Krasnow Institute for Advanced Study, George Mason University, Fairfax, VA, USA
More than a hundred biochemical species, activated by neurotransmitters binding to transmembrane receptors, are important in long-term potentiation (LTP) and long-term depression (LTD). To investigate which species and interactions are critical for synaptic plasticity, many computational postsynaptic signal transduction models have been developed. The models range from simple models with a single reversible reaction to detailed models with several hundred kinetic reactions. In this study, more than a hundred models are reviewed, and their features are compared and contrasted so that similarities and differences are more readily apparent. The models are classified according to the type of synaptic plasticity that is modeled (LTP or LTD) and whether they include diffusion or electrophysiological phenomena. Other characteristics that discriminate the models include the phase of synaptic plasticity modeled (induction, expression, or maintenance) and the simulation method used (deterministic or stochastic). We find that models are becoming increasingly sophisticated, by including stochastic properties, integrating with electrophysiological properties of entire neurons, or incorporating diffusion of signaling molecules. Simpler models continue to be developed because they are computationally efficient and allow theoretical analysis. The more complex models permit investigation of mechanisms underlying specific properties and experimental verification of model predictions. Nonetheless, it is difficult to fully comprehend the evolution of these models because (1) several models are not described in detail in the publications, (2) only a few models are provided in existing model databases, and (3) comparison to previous models is lacking. We conclude that the value of these models for understanding molecular mechanisms of synaptic plasticity is increasing and will be enhanced further with more complete descriptions and sharing of the published models.
1. Introduction
Synaptic plasticity is an activity-dependent change in the strength or efficacy of the synaptic connection between a pre- and postsynaptic neuron. It is induced with brief periods of synaptic activity, for example, using tetanic, high-frequency neuronal activity. Changes in synapses, in general, can last from milliseconds into years. These long-lasting changes, which require protein synthesis and gene transcription, are suggested to lead to learning and formation of memories.
The long-term activity-dependent strengthening and weakening of synapses are known as long-term potentiation (LTP; Bliss and Gardner-Medwin, 1973; Bliss and Lømo, 1973) and long-term depression (LTD; Ito et al., 1982; Ito, 1989; Dudek and Bear, 1992), respectively. Frequency-dependent LTP and LTD in the cornu ammonis 1 (CA1) region of the hippocampus, triggered by activation of N-methyl-d-aspartate (NMDA) receptors (NMDARs), are the most studied forms of long-term plasticity (see, e.g., Malenka and Bear, 2004; Citri and Malenka, 2008). In addition to hippocampal NMDAR-dependent LTP and LTD, diverse forms of LTP and LTD have been discovered in different brain regions. One example of non-NMDAR-dependent plasticity is cerebellar LTD. Some forms of LTP require neither the NMDA nor the non-NMDA ionotropic glutamate receptors (non-NMDARs include kainate receptors and α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors, AMPARs), but do require activation of metabotropic glutamate receptors (mGluRs). This form is found, for example, in the CA1 region of the hippocampus (Lanté et al., 2006). Despite the variation in NMDAR dependence, all forms of synaptic plasticity are calcium ion (Ca2+)-dependent; only the mechanisms for Ca2+ elevation vary.
Two broad types of computational models, phenomenological and biophysical models, have been developed to understand the pre- and postsynaptic events in LTP and LTD. Phenomenological models use abstract equations to describe a relationship between neuronal activity and synaptic plasticity. Biophysical models include electrophysiological models, biochemical models, and models that include both electrophysiological properties and biochemical reactions (signaling pathways) underlying the relationship between neuronal activity and synaptic plasticity, though even these include simplifications because all the mechanisms cannot be modeled in detail. The focus of the present study is on biophysical models which concentrate on postsynaptic biochemical reactions.
This review presents an overview of 117 postsynaptic signal transduction models, categorizes them so that similarities and differences are more readily apparent, and explains how these models can be used to identify key molecules and address questions related to mechanisms underlying LTP and LTD. Section 2 presents the biological background of synaptic plasticity, Section 3 classifies the computational postsynaptic signal transduction models, and Section 4 summarizes the directions and trends of this field.
2. Synaptic Plasticity
Many different classification schemes for synaptic plasticity exist. Synaptic potentiation can be classified into three main types: short-term potentiation (STP), which lasts as long as 30–45 min; early phase LTP (E-LTP), which lasts for 1–2 h; and late phase LTP (L-LTP), which persists for considerably more than 2 h (Sweatt, 1999; Soderling and Derkach, 2000; Citri and Malenka, 2008). Synaptic depression, on the other hand, is typically classified into two types: short-term depression (STD) and LTD (Ito, 2001); though there appears to be an early and late phase LTD (E-LTD, L-LTD) also (Kauderer and Kandel, 2000). In addition, all types of plasticity involve three processes: induction, in which the mechanisms leading to plasticity are engaged; expression, which involves mechanisms allowing the plasticity to be exhibited and measured; and maintenance, which involves processes occurring after the induction phase is complete and allowing the plasticity to persist for long periods of time (Sweatt, 1999).
2.1. Mechanisms to Trigger Synaptic Plasticity
Many different plasticity induction protocols have been developed. In general, potentiation is induced by a high-frequency stimulation and depression by a low-frequency stimulation of a chemical synapse, but there are variations in the experimental procedures depending on the cell type. Short-term plasticity is triggered typically by short trains of stimulation (Citri and Malenka, 2008). LTP is typically triggered with longer 1 s trains of high-frequency (100 Hz) stimulation (Citri and Malenka, 2008). One train triggers only E-LTP, whereas repetitive trains trigger L-LTP (Citri and Malenka, 2008). L-LTD is typically triggered with prolonged repetitive low-frequency (1 Hz) stimulation (Citri and Malenka, 2008). Theta stimulation consists of short bursts of trains repeated with 200 ms intervals and produces L-LTP, even though the number of pulses is more similar to that producing E-LTP. Spike-timing-dependent plasticity (STDP) is another protocol to trigger LTP as well as LTD. In STDP, pre- and postsynaptic neurons are stimulated independently and the timing between pre- and postsynaptic spikes determines whether potentiation or depression occurs (Markram et al., 1997; Bi and Poo, 1998; Bi and Rubin, 2005; Dan and Poo, 2006).
2.2. Molecular Mechanisms of Synaptic Plasticity
There are various mechanisms, both pre- and postsynaptic, that lead to changes in synaptic strength, for example changes in neurotransmitter release, conductance of receptors, numbers of receptors, numbers of active synapses, and structure of synapses (Hayer and Bhalla, 2005). Several reviews about the molecular mechanisms underlying synaptic plasticity have been published (see, e.g., Bliss and Collingridge, 1993; Malenka and Nicoll, 1999; Sweatt, 1999; Soderling and Derkach, 2000; Ito, 2002; Lisman et al., 2002; Malenka and Bear, 2004; Blitzer et al., 2005; Cooke and Bliss, 2006; Wang et al., 2006; Bruel-Jungerman et al., 2007; Citri and Malenka, 2008; Santos et al., 2009). Cytosolic Ca2+ is inarguably the most critical factor: chemical buffering of Ca2+ or pharmacological blocking of Ca2+ influx prevents both potentiation and depression. There are several sources of Ca2+, depending on the brain region and the cell type. Influx through NMDARs is the most common source for LTP; influx through Ca2+-permeable AMPARs, voltage-gated Ca2+ channels, or release from intracellular stores (triggered by mGluRs which are G protein-coupled receptors) are important in many cell types. Ca2+ can activate, both directly and indirectly, protein kinases and phosphatases leading to phosphorylation–dephosphorylation cycles and, ultimately, to LTP and LTD. The next paragraphs focus on the molecular mechanisms behind NMDAR-dependent LTP and LTD, as well as cerebellar LTD, because these forms of plasticity have been studied the most both experimentally and computationally.
NMDAR-dependent potentiation is triggered by release of the neurotransmitter glutamate from the presynaptic neuron and subsequent binding to NMDARs on the postsynaptic neuron (Bliss and Collingridge, 1993; Malenka and Nicoll, 1999; Sweatt, 1999; Malenka and Bear, 2004; Citri and Malenka, 2008). After NMDARs are activated, Ca2+ can flow into the cell if the postsynaptic membrane is sufficiently depolarized to relieve the magnesium ion block from NMDAR. NMDAR-dependent LTP requires a large increase in postsynaptic Ca2+ concentration which triggers several events inside the cell. One of the most important events is Ca2+ binding to calmodulin, which then activates Ca2+/calmodulin-dependent protein kinase II (CaMKII), leading to phosphorylation of AMPARs, increase in single-channel conductance of AMPARs, and incorporation of additional AMPARs into the postsynaptic density (Citri and Malenka, 2008). Ca2+ also binds to protein kinase C (PKC) which is involved in E-LTP in some cell types (Malinow et al., 1989; Klann et al., 1993). In the hippocampus, the calmodulin-4Ca2+ complex (CaMCa4) further activates adenylyl cyclase, leading to activation of cyclic adenosine monophosphate (cAMP)-dependent protein kinase (PKA) which is required for some forms of L-LTP (Woo et al., 2003).
Transcription and also somatic and dendritic protein synthesis are required for induction of L-LTP (Bradshaw et al., 2003b), but it is unclear whether protein synthesis is required for induction of E-LTP. These nuclear and somatic events involve Ca2+/calmodulin-dependent protein kinase IV (CaMKIV), mitogen-activated protein kinase (MAPK, ERK), and PKA. For maintenance of L-LTP, the atypical PKC isozyme (PKMζ), which is an autonomously active form of PKC, is required in addition to local dendritic protein synthesis (Serrano et al., 2005).
NMDAR-dependent LTD needs only a modest increase in Ca2+ concentration (instead of the large Ca2+ increase for LTP). This modest increase in Ca2+ concentration leads to preferential activation of protein phosphatase 2B also known as calcineurin, because it has a much higher affinity for CaMCa4 than CaMKII has. Activation of protein phosphatases leads to dephosphorylation and endocytosis of AMPARs located on the plasma membrane (Citri and Malenka, 2008), and thereby the expression of LTD. Protein translation may be needed for expression and maintenance of L-LTD (Citri and Malenka, 2008), but otherwise mechanisms behind maintenance of NMDAR-dependent LTD have not been studied extensively. Some forms of LTD also require Ca2+-dependent production of endocannabinoids which travel retrogradely to produce changes in presynaptic release of neurotransmitters (Gerdeman and Lovinger, 2003).
Cerebellar LTD, the best studied form of non-NMDAR-dependent LTD, is observed at the parallel fiber to Purkinje cell synapse. Purkinje cells form synapses with several thousand parallel fibers and also receive many synaptic contacts from a single climbing fiber (Ito, 2002; Citri and Malenka, 2008). Cerebellar LTD is induced when parallel fibers and a climbing fiber are activated simultaneously. Glutamate released by parallel fibers activates mGluRs which in turn activate phospholipase C (Ito, 2002). Phospholipase C catalyzes the reaction producing diacylglycerol and inositol trisphosphate (IP3). Diacylglycerol activates PKC, and IP3 causes the release of Ca2+ from endoplasmic reticulum through IP3 receptors (IP3Rs). Phospholipase A2, which is activated by an elevation in Ca2+ concentration, produces arachidonic acid which more persistently activates PKC that is transiently activated by diacylglycerol. PKC phosphorylates AMPARs and this leads to endocytosis of AMPARs from the plasma membrane. As in hippocampal LTP, protein synthesis is needed for L-LTD (Ito, 2001).
Given that Ca2+ activates multiple processes and enzymes, such as endocannabinoid production, calcineurin, and CaMKII, it is still not clear why some stimulation protocols produce depression and some produce potentiation. Non-linear interactions between multiple pathways make a quantitative understanding difficult solely from experiments. Computer modeling synthesizes information from myriad studies ranging from plasma membrane level phenomena to intracellular phenomena. Simulations therefore provide deeper insight into mechanisms underlying plasticity and this is why modeling studies have become more and more popular during the last 10 years.
3. Computational Models
Many computational models have been developed to understand pre- and postsynaptic events in LTP and LTD. Several focused reviews that include models of a specific neural system or type of plasticity have appeared during the last 20 years (Brown et al., 1990; Neher, 1998; Hudmon and Schulman, 2002a,b; Bi and Rubin, 2005; Holmes, 2005; Wörgötter and Porr, 2005; Ajay and Bhalla, 2006; Klipp and Liebermeister, 2006; Zou and Destexhe, 2007; Morrison et al., 2008; Ogasawara et al., 2008; Bhalla, 2009; Ogasawara and Kawato, 2009; Tanaka and Augustine, 2009; Urakubo et al., 2009; Castellani and Zironi, 2010; Gerkin et al., 2010; Graupner and Brunel, 2010; Hellgren Kotaleski and Blackwell, 2010; Shouval et al., 2010); however, a comprehensive review on postsynaptic signal transduction models for LTP and LTD is lacking.
In this study, an analysis of altogether 117 postsynaptic signal transduction models published through the year 2009 is presented (see Table 1). We limit the present analysis to models of postsynaptic signal transduction pathways that are defined using several characteristics. First, the output of the model needs to be a postsynaptic aspect of the neuron. Second, some part of intracellular signaling is explicitly modeled. Thus, models in this review are required to include at least mechanisms for postsynaptic Ca2+ dynamics, Ca2+ buffers, phosphorylation–dephosphorylation cycles, LTP and LTD related enzymes, retrograde signals, or synaptic strength that depends on Ca2+ concentration. Alternatively, models that explicitly include the kinases and phosphatases underlying changes in AMPAR phosphorylation or synthesis of plasticity-related proteins are included. Models which have intracellular signaling pathways in neurons but do not address plasticity are excluded. Models of AMPAR and NMDAR activation alone, or models including only anchoring and scaffolding proteins as intracellular molecules are excluded. Lastly, purely phenomenological models of plasticity are excluded. These strict criteria are needed because of the large number of models. In addition, a few models published during 2010 are excluded (see, e.g., Clopath et al., 2010; Kim et al., 2010; Kubota and Kitajima, 2010; Nakano et al., 2010; Pepke et al., 2010; Qi et al., 2010; Rackham et al., 2010; Santamaria et al., 2010; Tolle and Le Novère, 2010a).

Table 1. List of postsynaptic signal transduction models published each year.
3.1. Main Characteristics of Models
The lists of LTP models (Table 2), LTD models (Table 3), and dual LTP and LTD models (Table 4) order the models alphabetically by the first author and by the publication month and year. Dual LTP and LTD models are able to simulate both forms of plasticity. Characteristics listed under the methods include the computational techniques: either deterministic ordinary and partial differential equations (Det.) or stochastic techniques (Stoch.) which include, for example, reaction algorithms such as the Gillespie stochastic simulation algorithm (Gillespie, 1976, 1977) and diffusion algorithms such as Brownian dynamics. A few studies also use so-called hybrid methods where different techniques are combined. The models are further classified according to the biochemical phenomena that are modeled: some models only describe reactions between chemical species (Reac.) and some also take into account the diffusion of at least some chemical species (Diff.). In addition to biochemical models, there are models which not only describe intracellular events associated with synaptic plasticity, but also take into account the associated plasma membrane and ion channel level phenomena by modeling the membrane voltage; these models are referred to as electrophysiological (Elect.). Tables 2–4 indicate the simulation tool or programing language used when known, but this piece of information is not always given in the publications. Other characteristics included in Tables 2–4 are the cell type of the model, which process of synaptic plasticity is modeled [induction (Ind.), expression (Expr.), or maintenance (Maint.)] according to the publications, time required for the dynamics of the model to reach a steady state, the model outputs used to demonstrate the change in synaptic strength, and the size of the model [less than 20 different chemical species or other model variables is defined as small (S), between 20 and 50 is medium (M), and more than 50 is large (L)]. If several different types of models are used in one publication, the size of the largest model is given. The time required for the dynamics of the model to reach a steady state is suggestive and it is not possible to compare all the models according to the time because different models use, for example, different inputs.
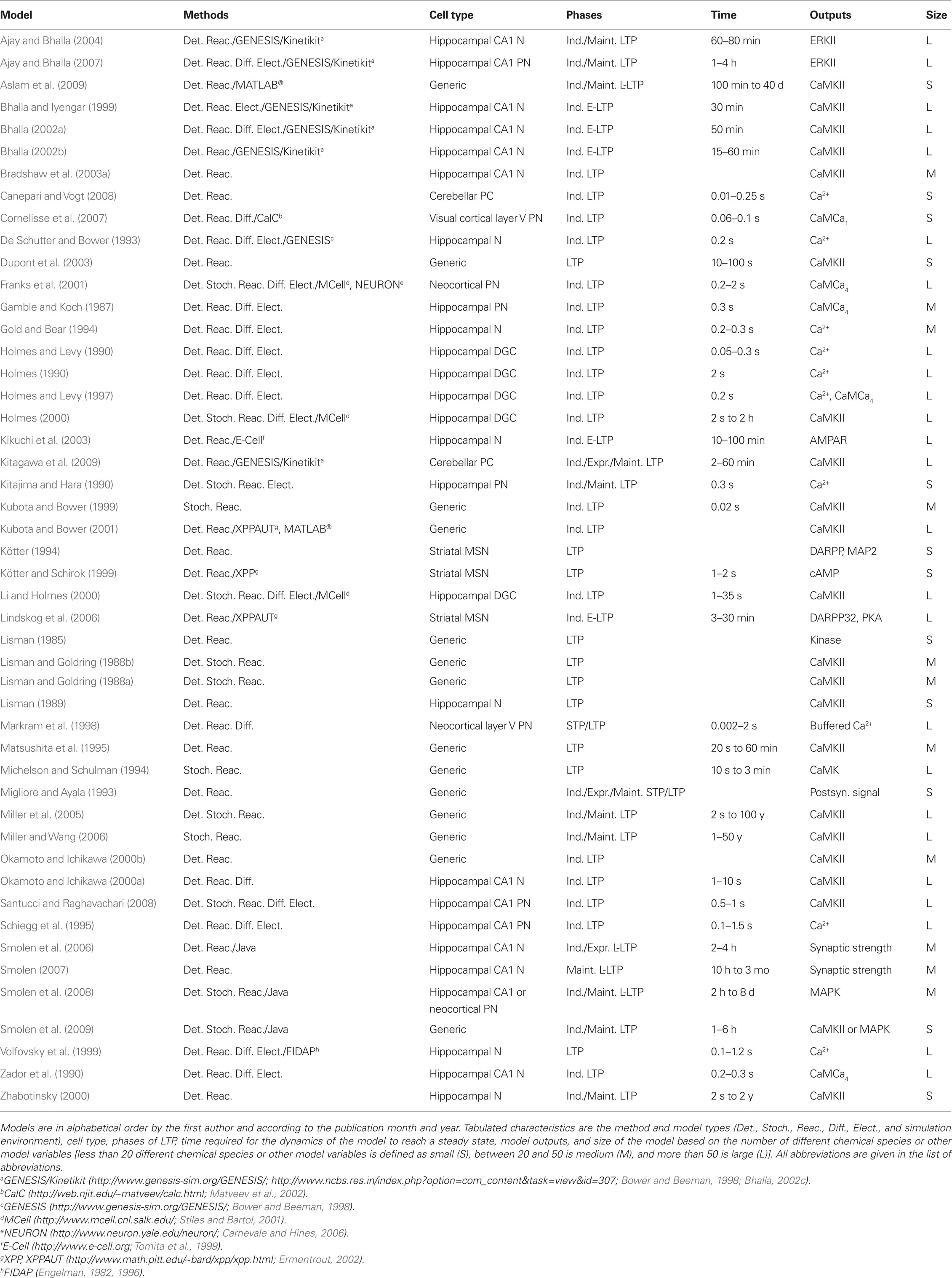
Table 2. List of LTP models.
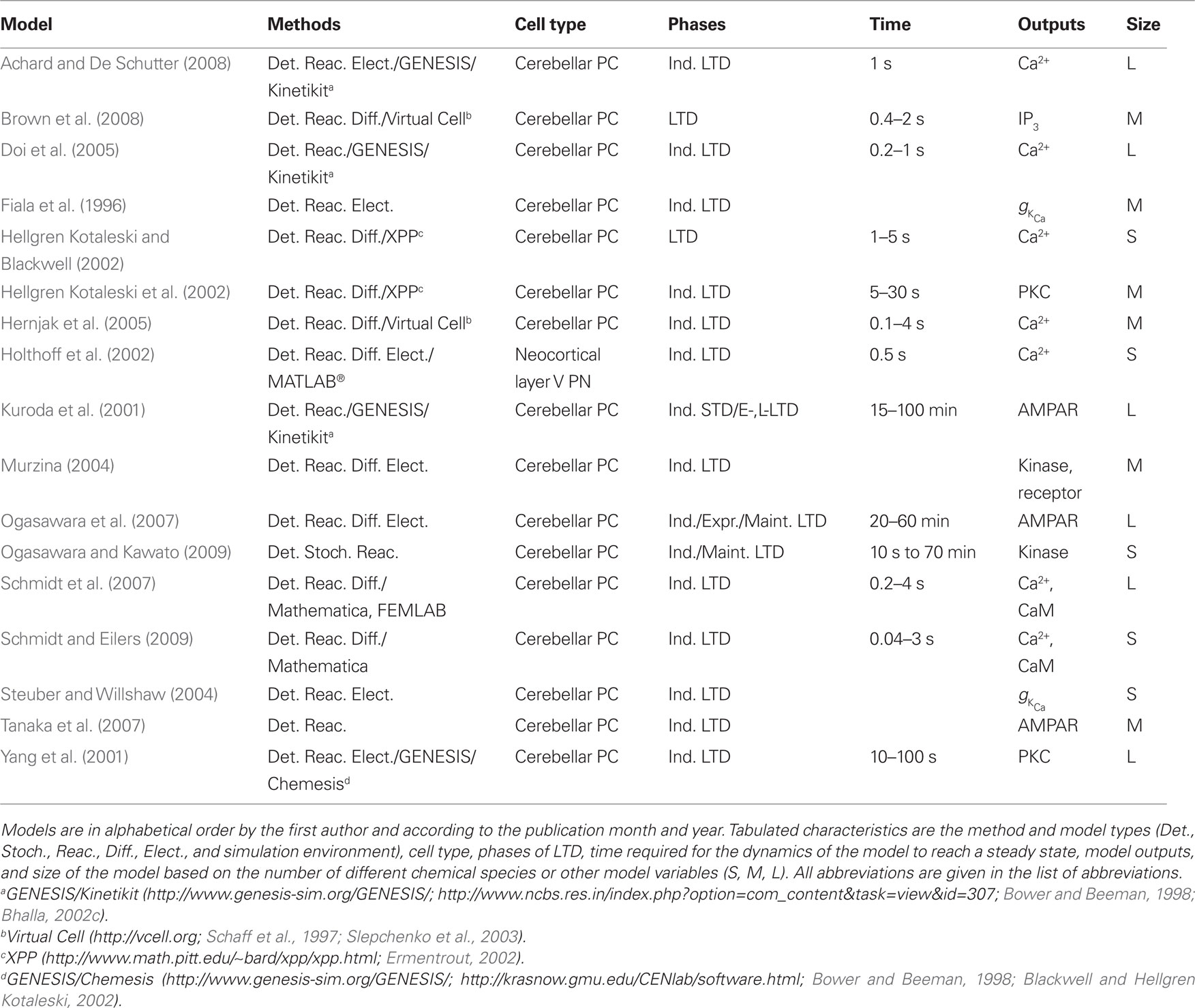
Table 3. List of LTD models.
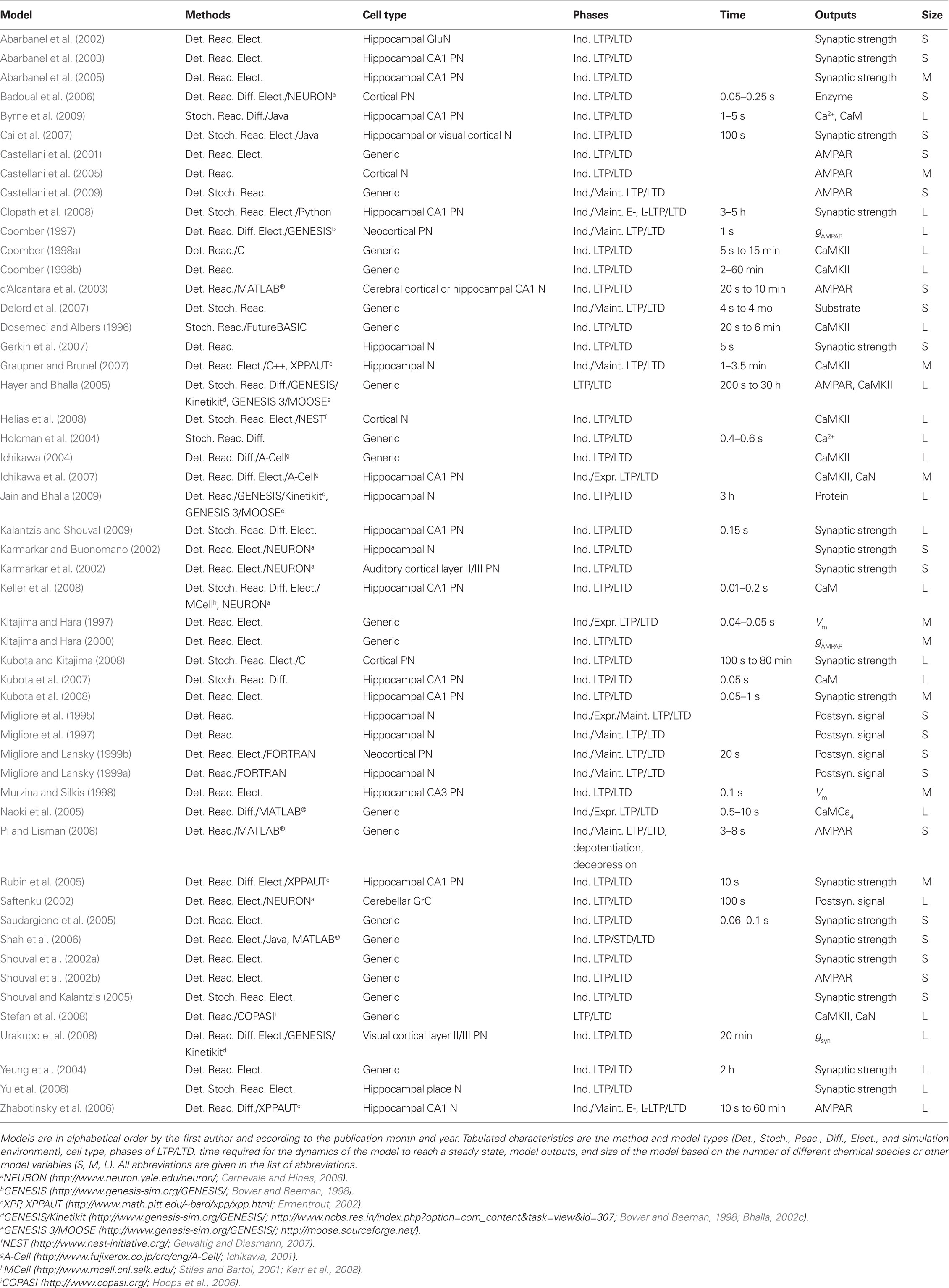
Table 4. List of dual LTP and LTD models.
3.2. Categorization of Models
In this study, models are further categorized (Figure 1) into models for single pathways (Table 5), models for calcium mechanisms or simplified intracellular processes (Table 6), and models for signaling networks (Table 7). Models for single pathways involve at most one kinase as a model variable and do not include any receptors, ion channels, or pumps on the plasma membrane. Typically single pathways contain a pathway involving calmodulin and CaMKII and sometimes also phosphatases. Models for calcium mechanisms or simplified intracellular processes include postsynaptic Ca2+ buffers together with ion channels, receptors, or pumps, or simplified intracellular processes. The last group of models, consisting of signaling networks, takes into account interactions between at least two pathways and thus often have several protein kinases and phosphatases. These models can also include ion channels, receptors, and pumps. Several characteristics, such as model inputs, number and types of morphological compartments, molecules, ion channels, and receptors, are described for the models in the following sections. In some cases it is difficult to determine the model inputs based on the information given in the publications. For detailed biophysical models, the input is typically coupled with the plasma membrane level phenomena, such as membrane voltage. In these cases, we have indicated the change in membrane current (ΔIm) or membrane voltage (ΔVm) as the input. For more simplified models, a variety of mathematical equations are used to describe the model and the input. In these cases, we have indicated which physical property the input equation represents, such as synaptic stimulus (causing elevation in Ca2+ concentration). See also Section 4 for further comments on the presentation of input for models.
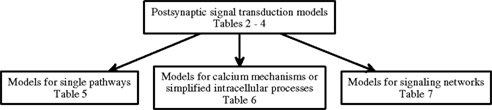
Figure 1. Categorization of postsynaptic signal transduction models.
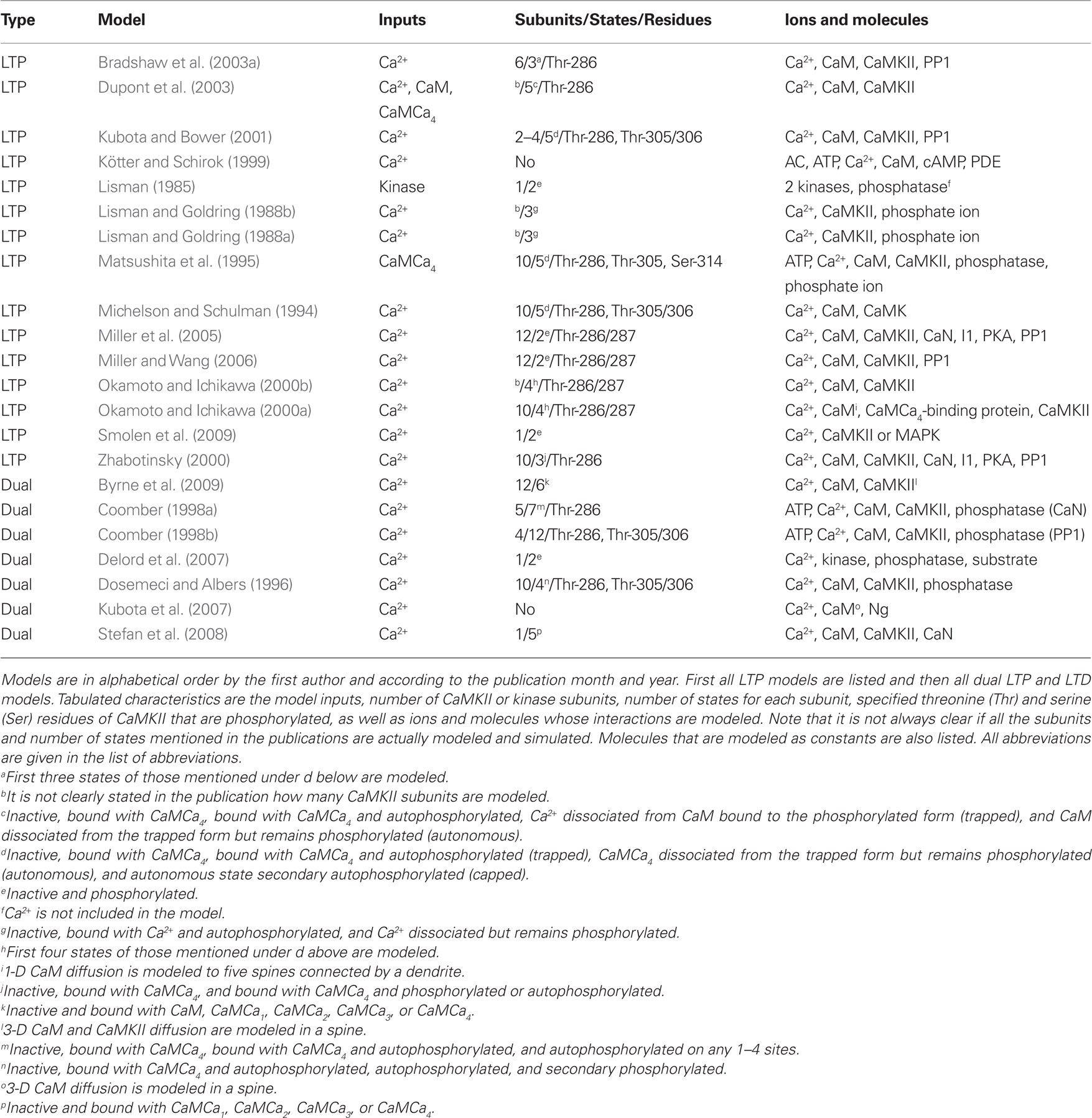
Table 5. Characteristics of models for single pathways.
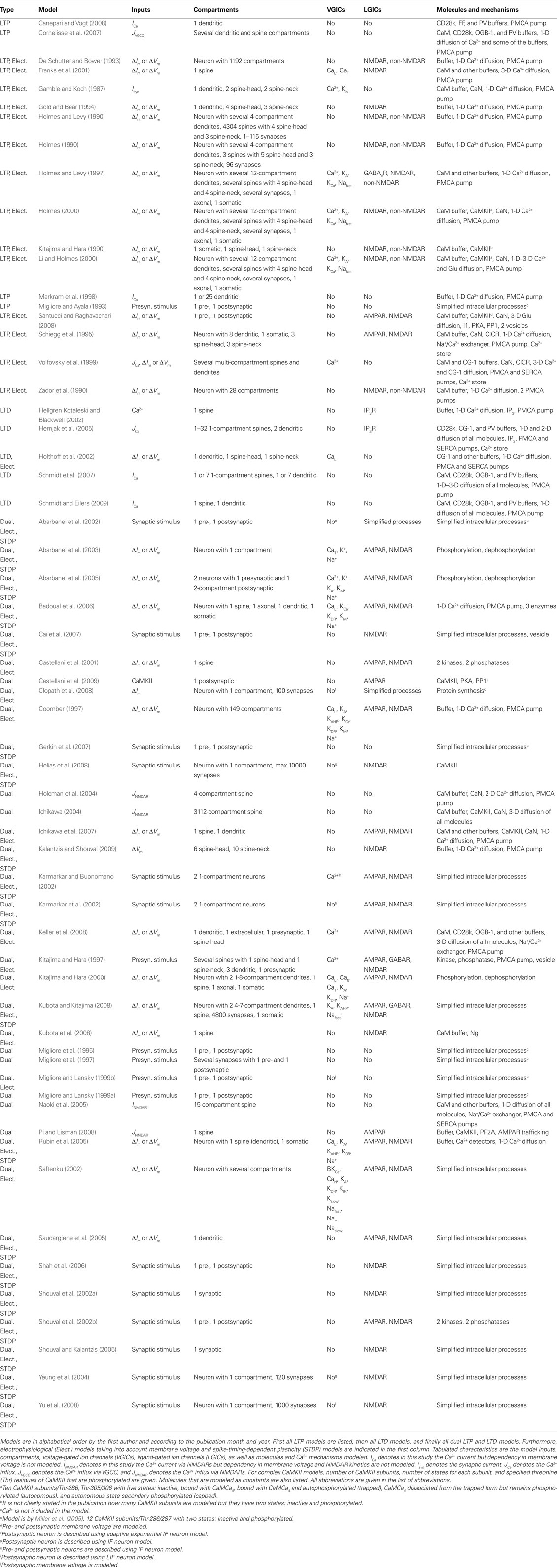
Table 6. Characteristics of models for calcium mechanisms or simplified intracellular processes.
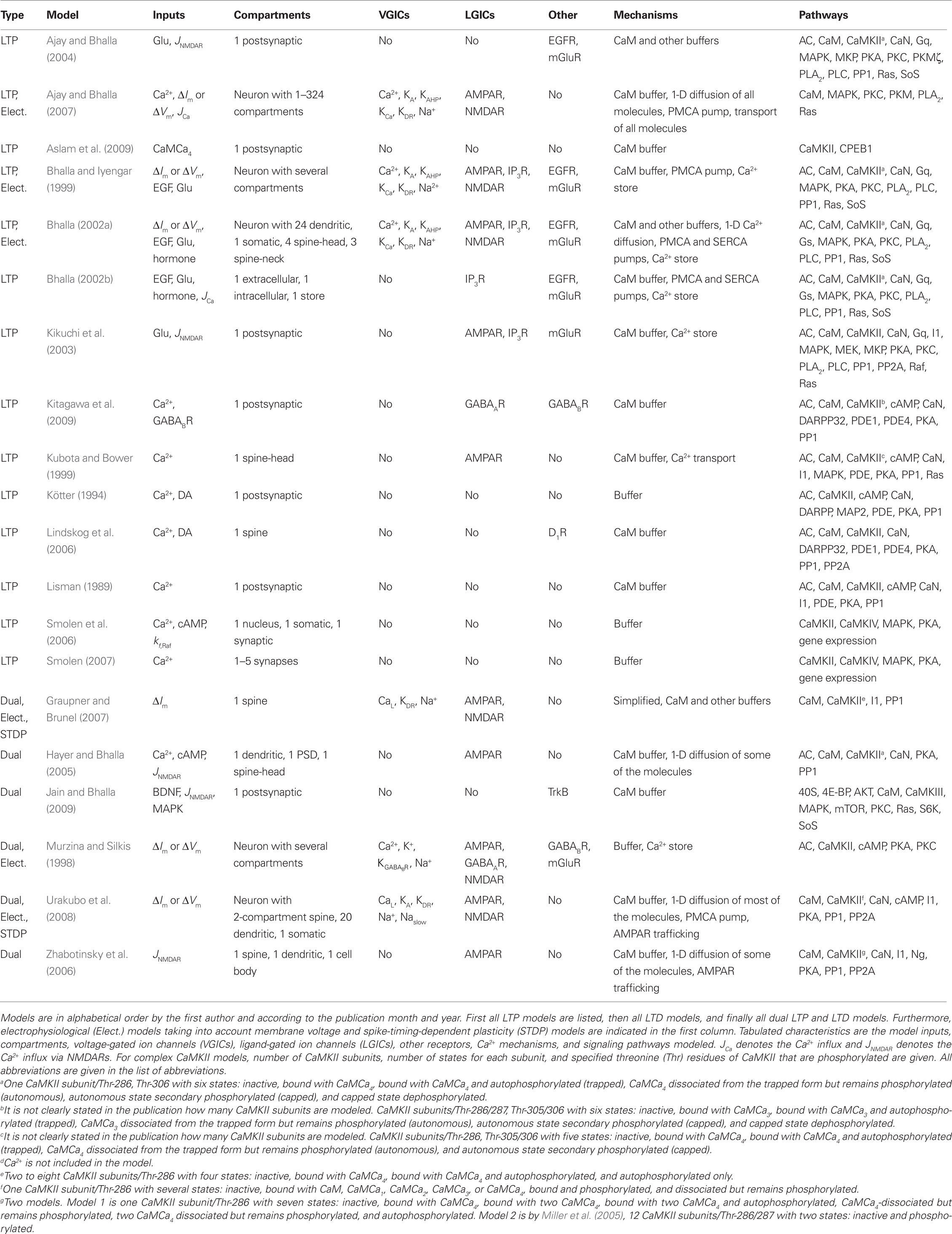
Table 7. Characteristics of models for signaling networks.
3.2.1. Models for single pathways
The models for single pathways typically focus on CaMKII (e.g., Dosemeci and Albers, 1996; Okamoto and Ichikawa, 2000a; Smolen et al., 2009), though one model for cAMP production (Kötter and Schirok, 1999) exists and several models are focused on calmodulin activation (e.g., Kubota et al., 2007; Stefan et al., 2008). Most of these models use Ca2+ concentration as the input and include reaction kinetics of CaMCa4 binding and unbinding to CaMKII subunits. Many of the models do not take into account the dodecameric structure of the CaMKII holoenzyme nor the spatial aspect of CaMCa4-dependent autophosphorylation of CaMKII between adjacent subunits. Because of the importance of CaMKII in LTP, most of these single pathway models address the same issues of amplitude and frequency dependence of Ca2+-bound calmodulin or CaMKII activation; subsequent models usually build on previous models and then advance the simulation technique (e.g., stochastic instead of deterministic simulations), or incorporate new experimental details on the CaMKII molecule.
Lisman (1985) presents one of the first models for LTP, which shows that a simple switch model has two stable states, one in which the kinase is dephosphorylated and the other in which it is almost completely phosphorylated. Switch-like behavior, important for memory formation, can be created even when reactions occur stochastically (Smolen et al., 2009), using fast and slow feedback loops. Another stochastic model (Miller et al., 2005) shows that the highly phosphorylated state of CaMKII can remain stable for years, another property which could be important for memory storage.
Okamoto and Ichikawa (2000a) demonstrate the crucial role of competition for calmodulin between spines by modeling several morphological compartments. They model CaMKII in a set of five spines connected to a dendrite and show that after autophosphorylation of CaMKII in a spine, calmodulin in the dendrite can diffuse into that spine for CaMCa4 trapping, which leads to competition since there is a limited concentration of calmodulin. Most of calmodulin is taken by those spines that experience relatively large increases in Ca2+ concentration.
A few of the models contribute to understanding of CaMKII activation though they do not explicitly model CaMKII. Delord et al. (2007) use simple models for Ca2+-controlled phosphorylation–dephosphorylation cycles with non-specific phosphoprotein substrates. Despite the simplicity of these models, the fraction of phosphorylated protein remains elevated for prolonged time periods after Ca2+ concentration returns to its basal level, representing a form of memory storage. Furthermore, the substrate phosphorylation persists in the presence of substrate turnover. Kubota et al. (2007) demonstrate that neurogranin regulates the spatiotemporal pattern of Ca2+-bound calmodulin, which has important implications for CaMKII activation and spatial specificity, by modeling diffusion of single molecules in a spine using 3-D Brownian dynamics.
Several studies show the importance of phosphatases for persistence of synaptic plasticity. Kubota and Bower (2001) show that asymptotic Ca2+ frequency sensitivity of CaMKII depends on both CaMKII and protein phosphatase 1 (PP1). Matsushita et al. (1995) show that phosphatase concentration not only controls whether CaMKII remains phosphorylated, but also controls the intensity of the input required to switch on the persistently phosphorylated state. Lisman and Zhabotinsky (2001) revisit this issue, and show that the CaMKII and PP1 bistable switch activated during the induction of LTP remains active despite the protein turnover. The bistable switch allows CaMKII autophosphorylation to be maintained at low Ca2+ concentrations, even after considering the effect of phosphatases and protein turnover. On the other hand, Bradshaw et al. (2003a) show that the presence of PP1 transforms the CaMKII bistable switch into a reversible (ultrasensitive) switch because PP1 dephosphorylates CaMKII when Ca2+ concentration is lowered to a basal level. Coomber (1998a) studies autophosphorylation and dephosphorylation of CaMKII and includes autophosphorylation of an inhibitory site caused by low-frequency stimulation. In this manner, either LTP or LTD can occur. Though using different mechanisms, both Dosemeci and Albers (1996) and Coomber (1998a,b) show that the phosphorylation of CaMKII can be sensitive to the temporal pattern of Ca2+ pulses, and this may allow CaMKII in the postsynaptic density to act as synaptic frequency detectors. The large allosteric model for calmodulin activation in the postsynaptic density by Stefan et al. (2008) explains how different Ca2+ concentrations can trigger the activation of either CaMKII or calcineurin.
3.2.2. Models for calcium mechanisms or simplified intracellular processes
Models for calcium mechanisms or simplified intracellular processes are a diverse group of models which typically address the role of Ca2+ in producing changes in synaptic strength. Most of these models focus on mechanisms controlling Ca2+ dynamics, such as Ca2+ buffers, pumps, glutamate receptors, or Ca2+-permeable ion channels. Another set of these models use more abstract equations representing intracellular processes and include an equation describing the Ca2+-dependent change in synaptic strength, in order to evaluate whether LTP or LTD occurs with repeated patterns of stimulation.
One of the most compelling questions in the field of LTP is whether high-frequency stimulation increases the spine Ca2+ concentration more than low-frequency stimulation. This has been addressed using models of Ca2+ dynamics in spines alone (see, e.g., Gamble and Koch, 1987; Kitajima and Hara, 1990; Gold and Bear, 1994; Volfovsky et al., 1999; Franks et al., 2001) or spines that include NMDAR activation by electrical activity in models of an entire neuron (see, e.g., Holmes and Levy, 1990; Zador et al., 1990; Koch and Zador, 1993). Zador et al. (1990) further demonstrate that spines compartmentalize Ca2+ (i.e., the Ca2+ signal is limited to those spines that are stimulated), thus providing a mechanism for spatial specificity. Holmes and Levy (1990) show that the frequency sensitivity of LTP requires Ca2+ buffers in addition to NMDAR properties.
A variation of this question is the effect of spine geometry on Ca2+ concentration and synaptic plasticity. Both Volfovsky et al. (1999) and Schmidt and Eilers (2009) test different spine-neck lengths and show that a long neck isolates Ca2+ signaling and calmodulin activation to the spine while stubby spines have a strong coupling between spines and the dendrite. Cornelisse et al. (2007) investigate the role of spine geometry compared to the dendrite. In particular, they demonstrate that the surface area to volume does not completely explain the difference in Ca2+ decay between a spine and dendrite. Instead, a lower buffer capacity of the spine is required to explain the experimental data.
Another important question is the role of various Ca2+ buffers in controlling Ca2+ dynamics. Many models of Ca2+ dynamics have only one or two Ca2+-binding proteins, instead of the many types found in real neurons. Markram et al. (1998) show that competition among Ca2+-binding proteins of various speeds and affinities influences the differential activation of intracellular targets. Models of Ca2+ dynamics permit tight coupling between experiments and models, but require the use of both intrinsic buffers, such as calbindin and parvalbumin, as well as Ca2+ indicators, such as Fura-FF, which themselves are fast, highly diffusible buffers. Other models have shown that buffer saturation is a crucial factor producing supralinear increases in Ca2+ concentration (Hellgren Kotaleski and Blackwell, 2002; Hernjak et al., 2005; Canepari and Vogt, 2008).
Improvements in Ca2+ imaging techniques have been accompanied by the development of sophisticated models that investigate mechanisms underlying Ca2+ microdomains. Naoki et al. (2005) take into account buffering by Ca2+-binding proteins and show that the diffusion coefficient of calmodulin has a strong effect on calmodulin activation in the microdomain near NMDARs. Kubota et al. (2008) investigate the Ca2+-binding protein neurogranin which increases Ca2+ dissociation from calmodulin. Their results show that with no Ca2+ extrusion mechanism, neurogranin increases the steady state concentration of Ca2+; however, in the presence of Ca2+ extrusion mechanisms, neurogranin instead enhances the decay rate of Ca2+. Keller et al. (2008) use MCell (Stiles and Bartol, 2001; Kerr et al., 2008) to develop one of the most advanced models of Ca2+ dynamics in a spine, including Ca2+ pumps, and both voltage-gated Ca2+ channels and NMDA-type of glutamate receptors. The voltage-dependent activation of the channels is coupled to a NEURON (Carnevale and Hines, 2006) simulation of membrane voltage. Keller et al. (2008) show that the Ca2+ gradient and calmodulin activation in the postsynaptic density depend on the order of glutamate release and action potential, and thus may explain the results of STDP experiments.
Just as recent models of Ca2+ dynamics include additional biophysical details, other models explore how biophysical processes related to, for example, glutamate receptors modulate LTP induction. Santucci and Raghavachari (2008) study the role of different types of NMDAR NR2 subunits on subsequent CaMKII activation. They show that though NR2B subunits have a more prolonged time course, the higher open probability of NR2A subunits leads to greater Ca2+ influx and CaMKII activation. The model of Li and Holmes (2000) shows that the variability in NMDAR opening, the spine-head Ca2+ concentration, and levels of CaMKII activation can play an important role in LTP induction. The spine model by Schiegg et al. (1995) includes calcineurin and Ca2+ release from stores, for example through IP3Rs, in the spine head. This study shows that the inclusion of calcineurin alone, which is a Ca2+ sensitive protein phosphatase important for synaptic depression, eliminates LTP; further inclusion of Ca2+ release from stores is required to restore LTP induction. Pi and Lisman (2008) study the role of AMPAR trafficking, modeled by inserting and removing AMPARs in the postsynaptic membrane with a rate that depends on phosphorylated CaMKII and dephosphorylated protein phosphatase 2A (PP2A). Pi and Lisman (2008) show that CaMKII activity is high during LTP, PP2A activity remains high during LTD, and neither activity is high during a basal state; thus, LTD is not a reversal of previous LTP, rather a distinct phenomenon. Clopath et al. (2008) focus on synaptic tagging, an experimental concept important for synaptic specificity of protein synthesis-dependent LTP. The model includes production of plasticity-related proteins which can be captured by tagged synapses. Non-tagged synapses can be tagged stochastically in either a high or low state. They show that synapses share protein synthesis processes which have an effect on the stabilization of potentiated synapses during the transition from E-LTP to L-LTP.
As with all computational models, verification by direct comparison with experimental data strengthens the ability to make experimental predictions and resolve conflicting experimental evidence. The study by Santucci and Raghavachari (2008) is an excellent example on developing a computationally realistic model from good quality data, using the model to resolve conflicting experimental evidence, and then making further experimental predictions. Other examples of direct comparison with experiments include studies by Markram et al. (1998), Volfovsky et al. (1999), Cornelisse et al. (2007), and Schmidt and Eilers (2009). In addition, the prediction that PP2A is critical for LTD induction has been confirmed experimentally (Nicholls et al., 2008). Cai et al. (2007) demonstrate that including the stochastic properties of synaptic transmission significantly affects the form of STDP curves, and indeed is required to explain the experimental data.
3.2.3. Models for signaling networks
Many LTP models for signaling networks are extensions of the single pathway CaMKII models. The model by Lisman (1989) is a landmark because it is one of the first to show that synaptic strength stored by CaMKII could be bidirectionally modified by physiological activity according to the postsynaptic Ca2+ concentration. Kubota and Bower (1999) predict that the CaMKII activity can be sensitive to small changes in the timing of presynaptic signal to the spine head and that CaMKII can exhibit temporal sensitivity even in the presence of PP1. Kitagawa et al. (2009) evaluate the effect of inhibitory G protein-coupled gamma-aminobutyric acid (GABA) B receptor (GABABR) activation on LTP. They show that a transient increase in Ca2+ concentration induces long-term activation of CaMKII, which is attenuated by GABABR activation due to inhibition of PKA. They further show a role for a novel positive feedback loop – one involving CaMKII-mediated downregulation of phosphodiesterase type 1.
Bhalla and Iyengar (1999), Bhalla (2002a,b), Ajay and Bhalla (2004, 2007), and Hayer and Bhalla (2005) have modeled pathways for several protein kinases and phosphatases to investigate information processing. The first study (Bhalla and Iyengar, 1999) uses synaptic stimulation of a compartmental neuron model (Holmes and Levy, 1990; Traub et al., 1991; De Schutter and Bower, 1993) to determine the Ca2+ concentration that is the input to signaling network models. Simulations show that several properties not present in individual pathways, such as feedback loops, thresholds, and sensitivity to signal strength and duration, can emerge from the interaction of pathways. Feedback loops and thresholds can give rise to bistability, offering the possibility that information can be stored within biochemical reactions in the signaling network. The role of temporal sensitivity is further explored (Bhalla, 2002a). This study shows that different input patterns are processed differently by the signaling network, thus giving rise to different outputs (input pattern discrimination). The role of the feedback loop involving MAPK and PKC is further explored in additional studies that integrate experiments and modeling (Bhalla, 2002b). The signaling network models are further refined to include PKMζ (Ajay and Bhalla, 2004, 2007), diffusional processes (Ajay and Bhalla, 2007), and electrical activity (Ajay and Bhalla, 2007) to explore mechanisms underlying MAPK activation in LTP. Ajay and Bhalla (2007) show that extracellular signal-regulated kinase (ERK, MAPK) type II (ERKII) activation after an LTP-inducing stimuli is not explained with reaction–diffusion alone but requires a distributed synaptic input and activation of voltage-gated Ca2+ channels. The model by Hayer and Bhalla (2005) shows that CaMKII and AMPAR phosphorylation form distinct bistable switches, allowing for multiple stable states of the system.
The models of striatal medium spiny neurons (Kötter, 1994; Lindskog et al., 2006) focus on integration of dopamine and glutamate signals, and explore mechanisms which are important for striatal learning. The model by Kötter (1994) is the first to investigate signaling pathways underlying plasticity in the striatum, and shows that, with Ca2+-activated adenylyl cyclase, dopamine and Ca2+ synergistically activate PKA. The model by Lindskog et al. (2006) includes the striatal adenylyl cyclase type 5, which is inhibited by Ca2+, and shows that separate transient dopamine or Ca2+ elevations each may increase the phosphorylation of cAMP-regulated phosphoprotein (DARPP32), due to Ca2+ activation of PP2A. Through this mechanism, paired stimuli yield increased PKA activation and DARPP32 phosphorylation compared to dopamine alone, in contrast to the effect of prolonged stimuli in which Ca2+ decreases DARPP32 phosphorylation. Fernandez et al. (2006) study the functions of DARPP32 with a detailed signaling network model but they do not address plasticity, thus this study is not included in Table 7. However, their study may be used as a valuable model to build on for future modeling efforts studying plasticity.
More recently models have been constructed to investigate mechanisms underlying L-LTP, by incorporating molecules such as CaMKIV, transcription factors, or the translation factor cytoplasmic polyadenylation element binding protein (CPEB1). Smolen (2007) shows that long periods of decreased activity reset synaptic strength to a low value, whereas episodic activity with short inactive periods maintains strong synapses. Smolen et al. (2008) implement a stochastic model to show that the feedback loop from MAPK to MAPK kinase kinase (Raf) increases the robustness of both stable states of MAPK activity to stochastic fluctuations. Aslam et al. (2009) show that the positive feedback loop between CaMKII and CPEB1 forms a bistable switch accounting for the protein synthesis dependence of L-LTP. In addition, Jain and Bhalla (2009) are interested in protein synthesis dependence of L-LTP, and thus investigate how the synaptic input pattern affects dendritic protein synthesis. These types of models are likely to increase because behavioral memories require protein synthesis.
Long-term depression is predominant for synapses in the cerebellum; thus, most models of LTD describe signaling networks in cerebellar Purkinje cells. Kuroda et al. (2001) investigate the mechanism producing persistent phosphorylation of AMPARs, required for LTD. Simulations show that the initial phase of phosphorylation of AMPARs depends on the activation of PKC by arachidonic acid, Ca2+, and diacylglycerol, whereas a later phase depends on the activation of a positive feedback loop and especially phospholipase A2 and arachidonic acid. Tanaka et al. (2007) further demonstrate that disrupting the positive feedback loop between several protein kinases can affect Ca2+ triggering of LTD. Brown et al. (2008) present an elaborate three-dimensional model of a Purkinje cell dendrite with spines to investigate the issue of whether sufficient phosphatidylinositol biphosphate (PIP2) is available in a single spine to achieve the experimentally estimated concentrations of IP3 required for Ca2+ release and subsequent LTD. They elegantly show that a relatively novel mechanism, namely stimulated synthesis of PIP2, is required to account for experimental results. Three of the LTD models (Yang et al., 2001; Ogasawara et al., 2007; Achard and De Schutter, 2008) use the multi-compartment, multi-channel Purkinje cell model by De Schutter and Bower (1994a,b) to simulate electrical activity leading to Ca2+ influx through synaptic and voltage-gated ion channels. Ogasawara et al. (2007) show that the nitric oxide concentration is critical for induction of LTD and for its input specificity. Achard and De Schutter (2008) re-evaluate the importance of conjunctive parallel fiber and climbing fiber inputs. They show that both inputs are required to produce a sufficient Ca2+ elevation to trigger LTD.
Because of the role of the cerebellum in eyeblink classical conditioning, several signaling network models investigate whether temporal characteristics of classical conditioning can be explained by temporal characteristics of LTD in single Purkinje cells. Fiala et al. (1996) have developed the first model to explain adaptive timing of the eyeblink response in classical conditioning. They use a biochemical variant of spectral timing for their parallel fiber inputs, and also include the effect of Ca2+-gated potassium channel activation on membrane voltage. They show that the phosphorylation state of target proteins responsible for LTD depends on the timing between climbing fiber and parallel fiber stimulation. Hellgren Kotaleski et al. (2002) include production of PKC activators by parallel fiber and climbing fiber stimulation in order to evaluate the relationship between LTD and behavior. Both Hellgren Kotaleski et al. (2002) and Doi et al. (2005) show that IP3-dependent Ca2+ dynamics are sensitive to temporal interval between parallel fiber and climbing fiber stimulation. Hellgren Kotaleski et al. (2002) further demonstrate that PKC activation is sensitive to temporal interval between parallel fiber and climbing fiber inputs (which is analogous to classical conditioning being sensitive to temporal interval). The importance of conjunctive parallel fiber and climbing fiber inputs for Ca2+ elevation is confirmed using a multi-compartment, multi-channel Purkinje cell model by Ogasawara et al. (2007) which more accurately simulates Ca2+ influx through synaptic and voltage-gated ion channels. Steuber and Willshaw (2004) show that replacing the spectral timing mechanism with Ca2+-dependent phosphorylation of mGluRs allows a single Purkinje cell to learn the adaptive timing of the eyeblink response.
More recent dual LTP and LTD models evaluate signaling network activation using spike-timing-dependent protocols (Graupner and Brunel, 2007; Urakubo et al., 2008). Urakubo et al. (2008) show that Ca2+ influx through NMDARs does not vary with spike timing (contrary to expectations) without suppression of NMDARs by Ca2+-bound calmodulin. Graupner and Brunel (2007) have constructed models for Ca2+/CaM-dependent autophosphorylation of CaMKII and PP1-dependent dephosphorylation of CaMKII. Graupner and Brunel (2007) show that CaMKII plays a central role in LTD because it is dephosphorylated during induction of LTD. More importantly, their bistable model can reproduce plasticity in response to STDP and high-frequency stimulation, without requiring abnormally low Ca2+ concentrations for dephosphorylation.
4. Analysis and Discussion
This study provides an extensive overview of 117 computational models for postsynaptic signal transduction pathways in synaptic plasticity developed over the past 25 years through 2009. Our purpose is to categorize the models so that similarities and differences are more readily apparent. Due to the large number of models, many models, though valuable, are excluded since they do not reach our criteria given in the beginning of Section 3. Some of the models included in this study are very simplified biochemical models meaning that a specific phenomenon is expressed using only a couple of reactions (see, e.g., Delord et al., 2007; Pi and Lisman, 2008). In the other extreme are the complex biophysical models that include detailed reaction–diffusion systems coupled to neuronal electrical activity (see, e.g., Bhalla, 2002a; Urakubo et al., 2008). Though model complexity has been increasing (Figures 2 and 3), the simpler biochemical models remain a valuable approach. They are relatively easy to construct, and the number of parameters to be fine-tuned is small. Not only are they computationally efficient, but they allow theoretical analysis and identification of which pathway, or combination of pathways, produces which property. On the other hand, models with detailed mechanisms are ideal for investigating which of several candidate molecules and mechanisms control or modulate a particular response. Furthermore, the direct correspondence between a detailed model and real neuron allows specific model predictions to be tested experimentally.
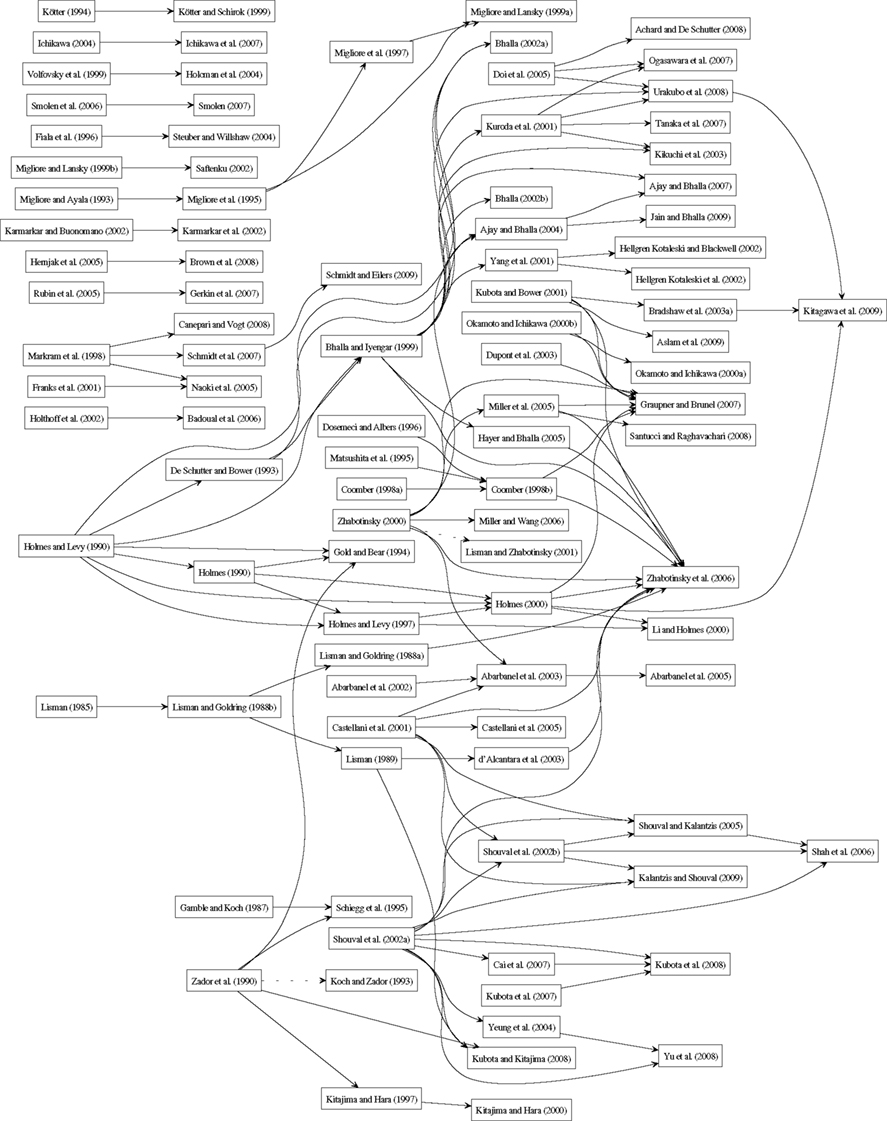
Figure 2. Evolution of postsynaptic signal transduction models from 1985 to 2009. The starting point of an arrow represents the model which is used by the latter model indicated as the arrowhead. A dotted line in the arrow means that the two studies use exactly the same model (the latter study is not presented in Tables 1–9).
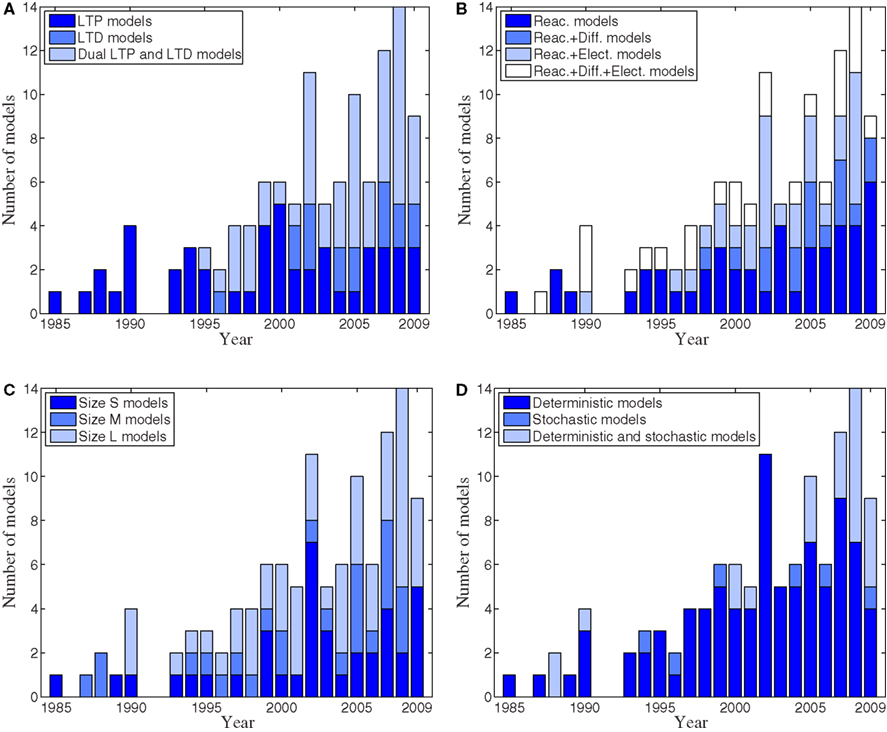
Figure 3. Numbers of published postsynaptic signal transduction models per year from 1985 to 2009. (A) Numbers of LTP, LTD, and dual LTP and LTD models. (B) Numbers of reaction, reaction and diffusion, reaction and electrophysiological, as well as reaction, diffusion, and electrophysiological models. (C) Numbers of different size (S, M, and L) models. (D) Numbers of deterministic, stochastic, and deterministic and stochastic models.
In our study, the emphasis is more on evaluating the model components and on the significance of the models rather than on comparison of the actual model responses. The comparison of model responses is not trivial because all models would need to be implemented and simulated before a comparative analysis could be performed (see also Pettinen et al., 2005). Indeed, this is not only time consuming, but impossible since many of the models are neither described in sufficient detail nor provided in model databases or by other open-access means (see Table 8). Even qualitative comparison is difficult since only a few publications provide a graphical illustration of the model components and in many cases it is difficult to interpret the model input or stimulus. These observations serve also as guidelines for reviewers evaluating future publications and models: (1) all models should be described in sufficient detail including equations, inputs, outputs, compartments, variables, constants, parameters, and initial conditions; (2) graphical illustration of the model should include only those model components that actually participate in simulations; (3) the simulation tool or programing language should be specified; and (4) the model should be provided in a model database. Nordlie et al. (2009) propose a good model description practice for neuronal network models. A similar description practice is needed for signal transduction models and our study is one step toward this, as is the BioModels Database project (Le Novère et al., 2006).

Table 8. Models provided in databases or by other open-access means.
Every computational model needs to be stimulated to study evoked activity even though this aspect is not always clearly indicated in the publications. In other words, an input similar to the one given in experimental wet-lab studies or as in the physiological in vivo state is required. In many cases, however, it is a challenge to mimic the input used in experiments. The construction of input stimulus is quite straightforward in cases where biophysically detailed models and a high-frequency stimulation protocol are used. In the other extreme are the models which use some function mimicking synaptic stimulus. This input type is not adequately described in many of the publications analyzed in the present study. This makes the reproduction of simulation results and the comparison of the models impossible. Therefore, the description of input stimuli should be taken into account when developing specific description language solutions for computational neuroscience and neuroinformatics.
Testing sensitivity to changes in parameter values is very important because many of the model parameters are not sufficiently constrained by experimental data. Table 9 highlights the models that evaluate whether the simulation results are sensitive to changes in parameter values. In this study, small-scale testing means that values for 10 parameters or less (for example rate constants) are varied, and large-scale testing means that values for greater than 10 parameters are varied. Table 9 shows that only a few models employ the large-scale testing of sensitivity to changes in parameter values. Publications that only test sensitivity to changes in input parameter values or do parameter estimation to fit experimental data, without analyzing the different model responses, are not included in Table 9.

Table 9. Models testing sensitivity to changes in parameter values.
In order to predict the future direction of the field, trends regarding the development of models of postsynaptic signal transduction pathways underlying LTP and LTD are illustrated (Figures 2 and 3). Figure 2 shows how different models reviewed in this study have evolved from each other. Two models are connected in Figure 2 if the publication either states directly that other models are used or the publication uses a subset of the exact same equations appearing in the older publications by the same authors. Models are excluded from Figure 2 if there is no clear evidence that they have used some other model as the basis, or if they are only based on models not reviewed in this study. Figure 2 shows that the models by Holmes and Levy (1990), Bhalla and Iyengar (1999), and Shouval et al. (2002a) are most often used as a starting point when developing new models. Zhabotinsky et al. (2006) and Graupner and Brunel (2007) cite the largest number of models when developing their models, but, on the other hand, they do not clearly state which parts of their model are taken from which other models.
Though LTP models appeared first, most of the new models are dual LTP and LTD models (Figure 3A), suggesting that these are being developed to investigate which characteristics of synaptic input patterns lead to LTP versus LTD. Despite limiting the review to models of signaling pathways, the models are extremely diverse in scope, with less than half including only reactions. Other models combine reactions and diffusion, or reactions and electrophysiological phenomena; about one-fifth have all three (Figure 3B). About one-third of the models are size small, meaning that there are less than 20 different chemical species or other model variables, and about half of the models are size large meaning that there are more than 50 different chemical species or other model variables (Figure 3C). The trend is toward increasing numbers of large models, reflecting both the increase in computational power and increasing knowledge of the biochemical pathways. Nonetheless, the continued development of small models reflects their utility in theoretical analysis. Most of the models are still deterministic even though stochastic methods have been developed more and more recently (Figure 3D). The scarcity of stochastic models compared to large models may reflect the availability of software modeling tools and analytic tools. However, several stochastic reaction–diffusion simulation tools have appeared recently (see, e.g., Kerr et al., 2008; Wils and De Schutter, 2009; Andrews et al., 2010; Byrne et al., 2010; Oliveira et al., 2010; Tolle and Le Novère, 2010b). Stochastic methods are important because very small numbers of molecules can have a dramatic effect on either strengthening or weakening the synapses and these effects should be taken into account. Another possibility is to develop and use so-called hybrid simulation methods where specific events are modeled as stochastic and others as deterministic. Though not illustrated graphically, only about one-fourth of the reviewed publications specify the simulation tool or programing language used. Most often the simulation tool used is GENESIS/Kinetikit (Bower and Beeman, 1998; Bhalla, 2002c), XPPAUT (Ermentrout, 2002), and NEURON (Carnevale and Hines, 2006). Programing languages most often used are Java and MATLAB®.
The trends in Figure 3 lead to several predictions about the future of signaling pathway modeling. The first prediction is that both the number of large models and the size of the largest model will continue to increase. Thus, existing models will be expanded to include additional signaling pathways, in parallel with the increase in experimental data of additional molecular mechanisms. Second, the trend in Figure 3D suggests that increasing number of models will be implemented stochastically or using hybrid deterministic–stochastic methods. The stochastic part of the models in particular may focus on events in the postsynaptic density and other multi-protein complexes. The third prediction is that the scope of the models will expand, with more models of dual LTP and LTD phenomena, in part because both phenomena have been measured in most cell types, and in part because the increase in size of the models is expanding to include signaling pathways for both phenomena. Related to the increase in scope of the models, more will blend reactions with diffusion or electrophysiological phenomena in order to study spatial aspects of signaling and also to better relate to experiments. In particular, modeling reactions alone is not sufficient for understanding synaptic plasticity but also electrophysiological phenomena needs to be taken into account by modeling neuronal networks (Hellgren Kotaleski and Blackwell, 2010). Further development of simulation tools (Pettinen et al., 2005; Alves et al., 2006) together with improvements in parallel computing should help in this endeavor.
Though the trend is toward larger and more complex models, this does not imply that all larger models are better than simpler models. As explained above, the quality of a model depends on many factors. Probably the most important criteria is whether the model can address a question of general scientific interest. For this reason, we have tried to organize our description of the models in order to highlight the questions addressed. Another related criteria is whether a model can make verifiable, i.e. falsifiable, predictions. Using these two criteria, models incorporating more biochemical details often appear superior, but only if the parameters can be adequately constrained. However, models which simplify the equations describing intracellular signaling pathways are more easily integrated with whole neuron electrophysiological models or able to simulate longer time frames. From this perspective they may excel for investigating whether different stimulation patterns change synaptic strength differently. It is important to note that earlier models may have been groundbreaking at the time of publication, yet their perceived quality decreases as more is learned about the interactions of intracellular molecules. Only a couple of studies reduce complex models to simpler ones and show comparative simulation results between the models (see, e.g., Hayer and Bhalla, 2005; Smolen, 2007). The reduction of model complexity will be an important research area in the future because simplified models that can capture relevant aspects of dynamics could be embedded, for example, into biologically-inspired neuronal network models when the activity of individual neurons is modeled in more detail.
To fully understand synaptic plasticity, many different characteristics of signaling pathways need to be considered. Temporal and spatial aspects of signaling are crucially important because they relate the cellular phenomenon of plasticity to the behavioral phenomenon of learning. Not only do theoreticians and modelers need to incorporate experimental findings, but also experimental progress can be enhanced by using model simulations to select the most promising experiments. Careful attention to these issues should improve the utility of modeling approaches for investigating molecular mechanisms of synaptic plasticity. The ultimate future goal of LTP and LTD modeling is to find such models for different brain regions and cells that can explain all the phases of synaptic plasticity, and then use these models to explain the differences in plasticity between brain regions or cell types. Many of the modeling studies have so far concentrated on only one type of synaptic plasticity. We believe that an analysis like the one provided by us will help in this endeavor to make more predictive models for synaptic plasticity in the future.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was partly supported by research project grants from Academy of Finland [106030 and 124615 (Marja-Leena Linne), 126556 (Tiina Manninen), and 129657 (Finnish Programme for Centres of Excellence in Research 2006–2011)], Swedish Research Council (Jeanette Hellgren Kotaleski), the Parkinson’s Foundation (Jeanette Hellgren Kotaleski), HFSP programme (Kim T. Blackwell), and the joint NSF-NIH CRCNS programme through NIH grant R01 AA16022 and R01 AA18060 (Kim T. Blackwell). Additional support was obtained from Finnish Foundation for Economic and Technology Sciences – KAUTE (Tiina Manninen), Otto A. Malm Foundation (Tiina Manninen and Katri Hituri), Emil Aaltonen Foundation (Katri Hituri), Finnish Foundation for Technology Promotion (Katri Hituri), and two graduate schools (Tampere University of Technology Graduate School and Tampere Doctoral Programme in Information Science and Engineering) (Katri Hituri).
References
Abarbanel, H. D. I., Gibb, L., Huerta, R., and Rabinovich, M. I. (2003). Biophysical model of synaptic plasticity dynamics. Biol. Cybern. 89, 214–226.
Abarbanel, H. D. I., Huerta, R., and Rabinovich, M. I. (2002). Dynamical model of long-term synaptic plasticity. Proc. Natl. Acad. Sci. U.S.A. 99, 10132–10137.
Abarbanel, H. D. I., Talathi, S. S., Gibb, L., and Rabinovich, M. I. (2005). Synaptic plasticity with discrete state synapses. Phys. Rev. E 72, 031914.
Achard, P., and De Schutter, E. (2008). Calcium, synaptic plasticity and intrinsic homeostasis in Purkinje neuron models. Front. Comput. Neurosci. 2:8. doi: 10.3389/neuro.10.008.2008
Ajay, S. M., and Bhalla, U. S. (2004). A role for ERKII in synaptic pattern selectivity on the time-scale of minutes. Eur. J. Neurosci. 20, 2671–2680.
Ajay, S. M., and Bhalla, U. S. (2006). Synaptic plasticity in vitro and in silico: insights into an intracellular signaling maze. Physiology 21, 289–296.
Ajay, S. M., and Bhalla, U. S. (2007). A propagating ERKII switch forms zones of elevated dendritic activation correlated with plasticity. HFSP J. 1, 49–66.
Alves, R., Antunes, F., and Salvador, A. (2006). Tools for kinetic modeling of biochemical networks. Nat. Biotechnol. 24, 667–672.
Andrews, S. S., Addy, N. J., Brent, R., and Arkin, A. P. (2010). Detailed simulations of cell biology with Smoldyn 2.1. PLoS Comput. Biol. 6, e1000705. doi: 10.1371/journal.pcbi.1000705
Aslam, N., Kubota, Y., Wells, D., and Shouval, H. Z. (2009). Translational switch for long-term maintenance of synaptic plasticity. Mol. Syst. Biol. 5, 284.
Badoual, M., Zou, Q., Davison, A. P., Rudolph, M., Bal, T., Fregnac, Y., and Destexhe, A. (2006). Biophysical and phenomenological models of multiple spike interactions in spike-timing dependent plasticity. Int. J. Neural Syst. 16, 79–97.
Bhalla, U. S. (2002a). Biochemical signaling networks decode temporal patterns of synaptic input. J. Comput. Neurosci. 13, 49–62.
Bhalla, U. S. (2002b). Mechanisms for temporal tuning and filtering by postsynaptic signaling pathways. Biophys. J. 83, 740–752.
Bhalla, U. S. (2002c). “Use of Kinetikit and GENESIS for modeling signaling pathways,” in Methods in Enzymology, Vol. 345, eds J. D. Hildebrandt and R. Iyengar (San Diego: Academic Press), 3–23.
Bhalla, U. S. (2009). “Molecules, networks, and memory,” in Systems Biology: The Challenge of Complexity, 1st Edn., eds S. Nakanishi, R. Kageyama, and D. Watanabe (Tokyo: Springer), 151–158.
Bhalla, U. S., and Iyengar, R. (1999). Emergent properties of networks of biological signaling pathways. Science 283, 381–387.
Bi, G.-Q., and Poo, M.-M. (1998). Synaptic modifications in cultured hippocampal neurons: dependence on spike timing, synaptic strength, and postsynaptic cell type. J. Neurosci. 18, 10464–10472.
Bi, G.-Q., and Rubin, J. (2005). Timing in synaptic plasticity: from detection to integration. Trends Neurosci. 28, 222–228.
Blackwell, K. T., and Hellgren Kotaleski, J. (2002). “Modeling the dynamics of second messenger pathways,” in Neuroscience Databases: A Practical Guide, ed. R. Kötter (Norwell, MA: Kluwer Academic Publishers), 63–80.
Bliss, T. V. P., and Collingridge, G. L. (1993). A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361, 31–39.
Bliss, T. V. P., and Gardner-Medwin, A. R. (1973). Long-lasting potentiation of synaptic transmission in the dentate area of the unanaesthetized rabbit following stimulation of the perforant path. J. Physiol. 232, 357–374.
Bliss, T. V. P., and Lømo, T. (1973). Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J. Physiol. 232, 331–356.
Blitzer, R. D., Iyengar, R., and Landau, E. M. (2005). Postsynaptic signaling networks: cellular cogwheels underlying long-term plasticity. Biol. Psychiatry 57, 113–119.
Bower, J. M., and Beeman, D. (1998). The Book of GENESIS: Exploring Realistic Neural Models with the GEneral NEural SImulation System, 2nd Edn. New York: Telos/Springer-Verlag.
Bradshaw, J. M., Kubota, Y., Meyer, T., and Schulman, H. (2003a). An ultrasensitive Ca2+/calmodulin-dependent protein kinase II-protein phosphatase 1 switch facilitates specificity in postsynaptic calcium signaling. Proc. Natl. Acad. Sci. U.S.A. 100, 10512–10517.
Bradshaw, K. D., Emptage, N. J., and Bliss, T. V. P. (2003b). A role for dendritic protein synthesis in hippocampal late LTP. Eur. J. Neurosci. 18, 3150–3152.
Brown, S.-A., Morgan, F., Watras, J., and Loew, L. M. (2008). Analysis of phosphatidylinositol-4,5-bisphosphate signaling in cerebellar Purkinje spines. Biophys. J. 95, 1795–1812.
Brown, T. H., Kairiss, E. W., and Keenan, C. L. (1990). Hebbian synapses: biophysical mechanisms and algorithms. Annu. Rev. Neurosci. 13, 475–511.
Bruel-Jungerman, E., Davis, S., and Laroche, S. (2007). Brain plasticity mechanisms and memory: a party of four. Neuroscientist 13, 492–505.
Byrne, M. J., Putkey, J. A., Waxham, M. N., and Kubota, Y. (2009). Dissecting cooperative calmodulin binding to CaM kinase II: a detailed stochastic model. J. Comput. Neurosci. 27, 621–638.
Byrne, M. J., Waxham, M. N., and Kubota, Y. (2010). Cellular dynamic simulator: an event driven molecular simulation environment for cellular physiology. Neuroinformatics 8, 63–82.
Cai, Y., Gavornik, J. P., Cooper, L. N., Yeung, L. C., and Shouval, H. Z. (2007). Effect of stochastic synaptic and dendritic dynamics on synaptic plasticity in visual cortex and hippocampus. J. Neurophysiol. 97, 375–386.
Canepari, M., and Vogt, K. E. (2008). Dendritic spike saturation of endogenous calcium buffer and induction of postsynaptic cerebellar LTP. PLoS ONE 3, e4011. doi: 10.1371/journal.pone.0004011
Carnevale, T., and Hines, M. (2006). The NEURON Book, 1st Edn. Cambridge, UK: Cambridge University Press.
Castellani, G. C., Bazzani, A., and Cooper, L. N. (2009). Toward a microscopic model of bidirectional synaptic plasticity. Proc. Natl. Acad. Sci. U.S.A. 106, 14091–14095.
Castellani, G. C., Quinlan, E. M., Bersani, F., Cooper, L. N., and Shouval, H. Z. (2005). A model of bidirectional synaptic plasticity: from signaling network to channel conductance. Learn. Mem. 12, 423–432.
Castellani, G. C., Quinlan, E. M., Cooper, L. N., and Shouval, H. Z. (2001). A biophysical model of bidirectional synaptic plasticity: dependence on AMPA and NMDA receptors. Proc. Natl. Acad. Sci. U.S.A. 98, 12772–12777.
Castellani, G. C., and Zironi, I. (2010). “Biophysics-based models of LTP/LTD,” in Hippocampal Microcircuits: A Computational Modeler’s Resource Book, eds V. Cutsuridis, B. Graham, S. Cobb, and I. Vida (New York: Springer), 555–570.
Citri, A., and Malenka, R. C. (2008). Synaptic plasticity: multiple forms, functions, and mechanisms. Neuropsychopharmacology 33, 18–41.
Clopath, C., Büsing, L., Vasilaki, E., and Gerstner, W. (2010). Connectivity reflects coding: a model of voltage-based STDP with homeostasis. Nat. Neurosci. 13, 344–352.
Clopath, C., Ziegler, L., Vasilaki, E., Büsing, L., and Gerstner, W. (2008). Tag-trigger-consolidation: a model of early and late long-term-potentiation and depression. PLoS Comput. Biol. 4, e1000248. doi: 10.1371/journal.pcbi.1000248
Cooke, S. F., and Bliss, T. V. P. (2006). Plasticity in the human central nervous system. Brain 129, 1659–1673.
Coomber, C. (1997). A model of associative long-term potentiation and long-term depression in a compartmental reconstruction of a neuron. Neurocomputing 16, 189–205.
Coomber, C. (1998a). Current theories of neuronal information processing performed by Ca2+/calmodulin-dependent protein kinase II with support and insights from computer modelling and simulation. Comput. Chem. 22, 251–263.
Coomber, C. J. (1998b). Site-selective autophosphorylation of Ca2+/calmodulin-dependent protein kinase II as a synaptic encoding mechanism. Neural Comput. 10, 1653–1678.
Cornelisse, L. N., van Elburg, R. A. J., Meredith, R. M., Yuste, R., and Mansvelder, H. D. (2007). High speed two-photon imaging of calcium dynamics in dendritic spines: consequences for spine calcium kinetics and buffer capacity. PLoS ONE 2, e1073. doi: 10.1371/journal.pone.0001073
d’Alcantara, P., Schiffmann, S. N., and Swillens, S. (2003). Bidirectional synaptic plasticity as a consequence of interdependent Ca2+-controlled phosphorylation and dephosphorylation pathways. Eur. J. Neurosci. 17, 2521–2528.
Dan, Y., and Poo, M.-M. (2006). Spike timing-dependent plasticity: from synapse to perception. Physiol. Rev. 86, 1033–1048.
Delord, B., Berry, H., Guigon, E., and Genet, S. (2007). A new principle for information storage in an enzymatic pathway model. PLoS Comput. Biol. 3, e124. doi: 10.1371/journal.pcbi.0030124
De Schutter, E., and Bower, J. M. (1993). Sensitivity of synaptic plasticity to the Ca2+ permeability of NMDA channels: a model of long-term potentiation in hippocampal neurons. Neural Comput. 5, 681–694.
De Schutter, E., and Bower, J. M. (1994a). An active membrane model of the cerebellar Purkinje cell. I. Simulation of current clamps in slice. J. Neurophysiol. 71, 375–400.
De Schutter, E., and Bower, J. M. (1994b). An active membrane model of the cerebellar Purkinje Cell. II. Simulation of synaptic responses. J. Neurophysiol. 71, 401–419.
Doi, T., Kuroda, S., Michikawa, T., and Kawato, M. (2005). Inositol 1,4,5-trisphosphate-dependent Ca2+ threshold dynamics detect spike timing in cerebellar Purkinje cells. J. Neurosci. 25, 950–961.
Dosemeci, A., and Albers, R. W. (1996). A mechanism for synaptic frequency detection through autophosphorylation of CaM kinase II. Biophys. J. 70, 2493–2501.
Dudek, S. M., and Bear, M. F. (1992). Homosynaptic long-term depression in area CA1 of hippocampus and effects of N-methyl-d-aspartate receptor blockade. Proc. Natl. Acad. Sci. U.S.A. 89, 4363–4367.
Dupont, G., Houart, G., and De Koninck, P. (2003). Sensitivity of CaM kinase II to the frequency of Ca2+ oscillations: a simple model. Cell Calcium 34, 485–497.
Engelman, M. S. (1982). FIDAP (A Fluid Dynamics Analysis Program). Adv. Eng. Softw. (1978) 4, 163–166.
Ermentrout, B. (2002). Simulating, Analyzing, and Animating Dynamical Systems: A Guide to XPPAUT for Researchers and Students, 1st Edn. Philadelphia: Society for Industrial and Applied Mathematics (SIAM).
Fernandez, é., Schiappa, R., Girault, J.-A., and Le Novère, N. (2006). DARPP-32 is a robust integrator of dopamine and glutamate signals. PLoS Comput. Biol. 2, e176. doi: 10.1371/journal.pcbi.0020176
Fiala, J. C., Grossberg, S., and Bullock, D. (1996). Metabotropic glutamate receptor activation in cerebellar Purkinje cells as substrate for adaptive timing of the classically conditioned eye-blink response. J. Neurosci. 16, 3760–3774.
Franks, K. M., Bartol, T. M., and Sejnowski, T. J. (2001). An MCell model of calcium dynamics and frequency-dependence of calmodulin activation in dendritic spines. Neurocomputing 38–40, 9–16.
Gamble, E., and Koch, C. (1987). The dynamics of free calcium in dendritic spines in response to repetitive synaptic input. Science 236, 1311–1315.
Gerdeman, G. L., and Lovinger, D. M. (2003). Emerging roles for endocannabinoids in long-term synaptic plasticity. Br. J. Pharmacol. 140, 781–789.
Gerkin, R. C., Bi, G.-Q., and Rubin, J. E. (2010). “A phenomenological calcium-based model of STDP,” in Hippocampal Microcircuits: A Computational Modeler’s Resource Book, eds V. Cutsuridis, B. Graham, S. Cobb, and I. Vida (New York: Springer), 571–591.
Gerkin, R. C., Lau, P.-M., Nauen, D. W., Wang, Y. T., and Bi, G.-Q. (2007). Modular competition driven by NMDA receptor subtypes in spike-timing-dependent plasticity. J. Neurophysiol. 97, 2851–2862.
Gillespie, D. T. (1976). A general method for numerically simulating the stochastic time evolution of coupled chemical reactions. J. Comput. Phys. 22, 403–434.
Gillespie, D. T. (1977). Exact stochastic simulation of coupled chemical reactions. J. Phys. Chem. 81, 2340–2361.
Gold, J. I., and Bear, M. F. (1994). A model of dendritic spine Ca2+ concentration exploring possible bases for a sliding synaptic modification threshold. Proc. Natl. Acad. Sci. U.S.A. 91, 3941–3945.
Graupner, M., and Brunel, N. (2007). STDP in a bistable synapse model based on CaMKII and associated signaling pathways. PLoS Comput. Biol. 3, e221. doi: 10.1371/journal.pcbi.0030221
Graupner, M., and Brunel, N. (2010). Mechanisms of induction and maintenance of spike-timing dependent plasticity in biophysical synapse models. Front. Comput. Neurosci. 4:136. doi:10.3389/fncom.2010.00136
Hayer, A., and Bhalla, U. S. (2005). Molecular switches at the synapse emerge from receptor and kinase traffic. PLoS Comput. Biol. 1, e20. doi: 10.1371/journal.pcbi.0010020
Helias, M., Rotter, S., Gewaltig, M.-O., and Diesmann, M. (2008). Structural plasticity controlled by calcium based correlation detection. Front. Comput. Neurosci. 2:7. doi: 10.3389/neuro.10.007.2008
Hellgren Kotaleski, J., and Blackwell, K. T. (2002). Sensitivity to interstimulus interval due to calcium interactions in the Purkinje cell spines. Neurocomputing 44–46, 13–18.
Hellgren Kotaleski, J., and Blackwell, K. T. (2010). Modelling the molecular mechanisms of synaptic plasticity using systems biology approaches. Nat. Rev. Neurosci. 11, 239–251.
Hellgren Kotaleski, J., Lester, D., and Blackwell, K. T. (2002). Subcellular interactions between parallel fibre and climbing fibre signals in Purkinje cells predict sensitivity of classical conditioning to interstimulus interval. Integr. Physiol. Behav. Sci. 37, 265–292.
Hernjak, N., Slepchenko, B. M., Fernald, K., Fink, C. C., Fortin, D., Moraru, I. I., Watras, J., and Loew, L. M. (2005). Modeling and analysis of calcium signaling events leading to long-term depression in cerebellar Purkinje cells. Biophys. J. 89, 3790–3806.
Hines, M. L., Morse, T., Migliore, M., Carnevale, N. T., and Shepherd, G. M. (2004). ModelDB: a database to support computational neuroscience. J. Comput. Neurosci. 17, 7–11.
Holcman, D., Schuss, Z., and Korkotian, E. (2004). Calcium dynamics in dendritic spines and spine motility. Biophys. J. 87, 81–91.
Holmes, W. R. (1990). Is the function of dendritic spines to concentrate calcium? Brain Res. 519, 338–342.
Holmes, W. R. (2000). Models of calmodulin trapping and CaM kinase II activation in a dendritic spine. J. Comput. Neurosci. 8, 65–86.
Holmes, W. R. (2005). “Calcium signaling in dendritic spines,” in Modeling in the Neurosciences: From Biological Systems to Neuromimetic Robotics, 2nd Edn., eds G. N. Reeke, R. R. Poznanski, K. A. Lindsay, J. R. Rosenberg, and O. Sporns (Boca Raton: CRC Press), 25–60.
Holmes, W. R., and Levy, W. B. (1990). Insights into associative long-term potentiation from computational models of NMDA receptor-mediated calcium influx and intracellular calcium concentration changes. J. Neurophysiol. 63, 1148–1168.
Holmes, W. R., and Levy, W. B. (1997). Quantifying the role of inhibition in associative long-term potentiation in dentate granule cells with computational models. J. Neurophysiol. 78, 103–116.
Holthoff, K., Tsay, D., and Yuste, R. (2002). Calcium dynamics of spines depend on their dendritic location. Neuron 33, 425–437.
Hoops, S., Sahle, S., Gauges, R., Lee, C., Pahle, J., Simus, N., Singhai, M., Xu, L., Mendes, P., and Kummer, U. (2006). COPASI – a complex pathway simulator. Bioinformatics 22, 3067–3074.
Hudmon, A., and Schulman, H. (2002a). Neuronal Ca2+/calmodulin-dependent protein kinase II: the role of structure and autoregulation in cellular function. Annu. Rev. Biochem. 71, 473–510.
Hudmon, A., and Schulman, H. (2002b). Structure-function of the multifunctional Ca2+/calmodulin-dependent protein kinase II. Biochem. J. 364(Pt 3), 593–611.
Ichikawa, K. (2001). A-Cell: graphical user interface for the construction of biochemical reaction models. Bioinformatics 17, 483–484.
Ichikawa, K. (2004). Localization of activated Ca2+/calmodulin-dependent protein kinase II within a spine: modeling and computer simulation. Neurocomputing 58–60, 443–448.
Ichikawa, K. (2005). A modeling environment with three-dimensional morphology, A-Cell-3D, and Ca2+ dynamics in a spine. Neuroinformatics 3, 49–63.
Ichikawa, K., Hoshino, A., and Kato, K. (2007). Induction of synaptic depression by high-frequency stimulation in area CA1 of the rat hippocampus: modeling and experimental studies. Neurocomputing 70, 2055–2059.
Ito, M. (2001). Cerebellar long-term depression: characterization, signal transduction, and functional roles. Physiol. Rev. 81, 1143–1195.
Ito, M. (2002). The molecular organization of cerebellar long-term depression. Nat. Rev. Neurosci. 3, 896–902.
Ito, M., Sakurai, M., and Tongroach, P. (1982). Climbing fiber induced depression of both mossy fiber responsiveness and glutamate sensitivity of cerebellar Purkinje cells. J. Physiol. 324, 113–124.
Jain, P., and Bhalla, U. S. (2009). Signaling logic of activity-triggered dendritic protein synthesis: an mTOR gate but not a feedback switch. PLoS Comput. Biol. 5, e1000287. doi: 10.1371/journal.pcbi.1000287
Kalantzis, G., and Shouval, H. Z. (2009). Structural plasticity can produce metaplasticity. PLoS ONE 4, e8062. doi: 10.1371/journal.pone.0008062
Karmarkar, U. R., and Buonomano, D. V. (2002). A model of spike-timing dependent plasticity: one or two coincidence detectors? J. Neurophysiol. 88, 507–513.
Karmarkar, U. R., Najarian, M. T., and Buonomano, D. V. (2002). Mechanisms and significance of spike-timing dependent plasticity. Biol. Cybern. 87, 373–382.
Kauderer, B. S., and Kandel, E. R. (2000). Capture of a protein synthesis-dependent component of long-term depression. Proc. Natl. Acad. Sci. U.S.A. 97, 13342–13347.
Keller, D. X., Franks, K. M., Bartol, Jr., T. M., and Sejnowski, T. J. (2008). Calmodulin activation by calcium transients in the postsynaptic density of dendritic spines. PLoS ONE 3, e2045. doi: 10.1371/journal.pone.0002045
Kerr, R. A., Bartol, T. M., Kaminsky, B., Dittrich, M., Chang, J.-C. J., Baden, S. B., Sejnowski, T. J., and Stiles, J. R. (2008). Fast Monte Carlo simulation methods for biological reaction–diffusion systems in solution and on surfaces. SIAM J. Sci. Comput. 30, 3126–3149.
Kikuchi, S., Fujimoto, K., Kitagawa, N., Fuchikawa, T., Abe, M., Oka, K., Takei, K., and Tomita, M. (2003). Kinetic simulation of signal transduction system in hippocampal long-term potentiation with dynamic modeling of protein phosphatase 2A. Neural Netw. 16, 1389–1398.
Kim, M. S., Huang, T., Abel, T., and Blackwell, K. T. (2010). Temporal sensitivity of protein kinase A activation in late-phase long term potentiation. PLoS Comput. Biol. 6, e1000691. doi: 10.1371/journal.pcbi.1000691
Kitagawa, Y., Hirano, T., and Kawaguchi, S.-Y. (2009). Prediction and validation of a mechanism to control the threshold for inhibitory synaptic plasticity. Mol. Syst. Biol. 5, 280.
Kitajima, T., and Hara, K. (1990). A model of the mechanisms of long-term potentiation in the hippocampus. Biol. Cybern. 64, 33–39.
Kitajima, T., and Hara, K. (2000). A generalized Hebbian rule for activity-dependent synaptic modifications. Neural Netw. 13, 445–454.
Kitajima, T., and Hara, K.-I. (1997). An integrated model for activity-dependent synaptic modifications. Neural Netw. 10, 413–421.
Klann, E., Chen, S. J., and Sweatt, J. D. (1993). Mechanism of protein kinase C activation during the induction and maintenance of long-term potentiation probed using a selective peptide substrate. Proc. Natl. Acad. Sci. U.S.A. 90, 8337–8341.
Klipp, E., and Liebermeister, W. (2006). Mathematical modeling of intracellular signaling pathways. BMC Neurosci. 7(Suppl. 1), S10.
Koch, C., and Zador, A. (1993). The function of dendritic spines: devices subserving biochemical rather than electrical compartmentalization. J. Neurosci. 13, 413–422.
Kötter, R. (1994). Postsynaptic integration of glutamatergic and dopaminergic signals in the striatum. Prog. Neurobiol. 44, 163–196.
Kötter, R., and Schirok, D. (1999). Towards an integration of biochemical and biophysical models of neuronal information processing: a case study in the nigro-striatal system. Rev. Neurosci. 10, 247–266.
Kubota, S., and Kitajima, T. (2008). A model for synaptic development regulated by NMDA receptor subunit expression. J. Comput. Neurosci. 24, 1–20.
Kubota, S., and Kitajima, T. (2010). Possible role of cooperative action of NMDA receptor and GABA function in developmental plasticity. J. Comput. Neurosci. 28, 347–359.
Kubota, Y., and Bower, J. M. (1999). Decoding time-varying calcium signals by the postsynaptic biochemical network: computer simulations of molecular kinetics. Neurocomputing 26–27, 29–38.
Kubota, Y., and Bower, J. M. (2001). Transient versus asymptotic dynamics of CaM kinase II: possible roles of phosphatase. J. Comput. Neurosci. 11, 263–279.
Kubota, Y., Putkey, J. A., Shouval, H. Z., and Waxham, M. N. (2008). IQ-motif proteins influence intracellular free Ca2+ in hippocampal neurons through their interactions with calmodulin. J. Neurophysiol. 99, 264–276.
Kubota, Y., Putkey, J. A., and Waxham, M. N. (2007). Neurogranin controls the spatiotemporal pattern of postsynaptic Ca2+/CaM signaling. Biophys. J. 93, 3848–3859.
Kuroda, S., Schweighofer, N., and Kawato, M. (2001). Exploration of signal transduction pathways in cerebellar long-term depression by kinetic simulation. J. Neurosci. 21, 5693–5702.
Lanté, F., de Jésus Ferreira, M.-C., Guiramand, J., Récasens, M., and Vignes, M. (2006). Low-frequency stimulation induces a new form of LTP, metabotropic glutamate (mGlu5) receptor- and PKA-dependent, in the CA1 area of the rat hippocampus. Hippocampus 16, 345–360.
Le Novère, N., Bornstein, B., Broicher, A., Courtot, M., Donizelli, M., Dharuri, H., Li, L., Sauro, H., Schilstra, M., Shapiro, B., Snoep, J. L., and Hucka, M. (2006). BioModels Database: a free, centralized database of curated, published, quantitative kinetic models of biochemical and cellular systems. Nucleic Acids Res. 34, D689–D691.
Li, Y., and Holmes, W. R. (2000). Comparison of CaMKinase II activation in a dendritic spine computed with deterministic and stochastic models of the NMDA synaptic conductance. Neurocomputing 32–33, 1–7.
Lindskog, M., Kim, M., Wikström, M. A., Blackwell, K. T., and Hellgren Kotaleski, J. (2006). Transient calcium and dopamine increase PKA activity and DARPP-32 phosphorylation. PLoS Comput. Biol. 2, e119. doi: 10.1371/journal.pcbi.0020119
Lisman, J. (1989). A mechanism for the Hebb and the anti-Hebb processes underlying learning and memory. Proc. Natl. Acad. Sci. U.S.A. 86, 9574–9578.
Lisman, J., and Goldring, M. A. (1988a). Evaluation of a model of long-term memory based on the properties of the Ca2+/calmodulin-dependent protein kinase. J. Physiol. (Paris) 83, 187–197.
Lisman, J. E., and Goldring, M. A. (1988b). Feasibility of long-term storage of graded information by the Ca2+/calmodulin-dependent protein kinase molecules of the postsynaptic density. Proc. Natl. Acad. Sci. U.S.A. 85, 5320–5324.
Lisman, J., Schulman, H., and Cline, H. (2002). The molecular basis of CaMKII function in synaptic and behavioural memory. Nat. Rev. Neurosci. 3, 175–190.
Lisman, J. E. (1985). A mechanism for memory storage insensitive to molecular turnover: a bistable autophosphorylating kinase. Proc. Natl. Acad. Sci. U.S.A. 82, 3055–3057.
Lisman, J. E., and Zhabotinsky, A. M. (2001). A model of synaptic memory: a CaMKII/PP1 switch that potentiates transmission by organizing an AMPA receptor anchoring assembly. Neuron 31, 191–201.
Lloyd, C. M., Lawson, J. R., Hunter, P. J., and Nielsen, P. F. (2008). The CellML model repository. Bioinformatics 24, 2122–2123.
Luciano, J. S., and Stevens, R. D. (2007). e-Science and biological pathway semantics. BMC Bioinformatics 8(Suppl. 3), S3.
Malenka, R. C., and Nicoll, R. A. (1999). Long-term potentiation – a decade of progress? Science 285, 1870–1874.
Malinow, R., Schulman, H., and Tsien, R. W. (1989). Inhibition of postsynaptic PKC or CaMKII blocks induction but not expression of LTP. Science 245, 862–866.
Markram, H., Lübke, J., Frotscher, M., and Sakmann, B. (1997). Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science 275, 213–215.
Markram, H., Roth, A., and Helmchen, F. (1998). Competitive calcium binding: implications for dendritic calcium signaling. J. Comput. Neurosci. 5, 331–348.
Matsushita, T., Moriyama, S., and Fukai, T. (1995). Switching dynamics and the transient memory storage in a model enzyme network involving Ca2+/calmodulin-dependent protein kinase II in synapses. Biol. Cybern. 72, 497–509.
Matveev, V., Sherman, A., and Zucker, R. S. (2002). New and corrected simulations of synaptic facilitation. Biophys. J. 83, 1368–1373.
Michelson, S., and Schulman, H. (1994). CaM kinase: a model for its activation and dynamics. J. Theor. Biol. 171, 281–290.
Migliore, M., Alicata, F., and Ayala, G. F. (1995). A model for long-term potentiation and depression. J. Comput. Neurosci. 2, 335–343.
Migliore, M., Alicata, F., and Ayala, G. F. (1997). Possible roles of retrograde messengers on LTP, LTD, and associative memory. Biosystems 40, 127–132.
Migliore, M., and Ayala, G. F. (1993). A kinetic model of short- and long-term potentiation. Neural Comput. 5, 636–647.
Migliore, M., and Lansky, P. (1999a). Computational model of the effects of stochastic conditioning on the induction of long-term potentiation and depression. Biol. Cybern. 81, 291–298.
Migliore, M., and Lansky, P. (1999b). Long-term potentiation and depression induced by a stochastic conditioning of a model synapse. Biophys. J. 77, 1234–1243.
Migliore, M., Morse, T. M., Davison, A. P., Marenco, L., Shepherd, G. M., and Hines, M. L. (2003). ModelDB: making models publicly accessible to support computational neuroscience. Neuroinformatics 1, 135–139.
Miller, P., and Wang, X.-J. (2006). Stability of discrete memory states to stochastic fluctuations in neuronal systems. Chaos 16, 026109.
Miller, P., Zhabotinsky, A. M., Lisman, J. E., and Wang, X.-J. (2005). The stability of a stochastic CaMKII switch: dependence on the number of enzyme molecules and protein turnover. PLoS Biol. 3, e107. doi: 10.1371/journal.pbio.0030107
Morrison, A., Diesmann, M., and Gerstner, W. (2008). Phenomenological models of synaptic plasticity based on spike timing. Biol. Cybern. 98, 459–478.
Murzina, G. B. (2004). Mathematical simulation of the induction of long-term depression in cerebellar Purkinje cells. Neurosci. Behav. Physiol. 34, 115–121.
Murzina, G. B., and Silkis, I. G. (1998). Studies of long-term potentiation and depression of inhibitory transmission by mathematical modeling of post-synaptic processes. Neurosci. Behav. Physiol. 28, 121–129.
Nakano, T., Doi, T., Yoshimoto, J., and Doya, K. (2010). A kinetic model of dopamine- and calcium-dependent striatal synaptic plasticity. PLoS Comput. Biol. 6, e1000670. doi: 10.1371/journal.pcbi.1000670
Naoki, H., Sakumura, Y., and Ishii, S. (2005). Local signaling with molecular diffusion as a decoder of Ca2+ signals in synaptic plasticity. Mol. Syst. Biol. 1, 2005.0027.
Neher, E. (1998). Usefulness and limitations of linear approximations to the understanding of Ca2+ signals. Cell Calcium 24, 345–357.
Nicholls, R. E., Alarcon, J. M., Malleret, G., Carroll, R. C., Grody, M., Vronskaya, S., and Kandel, E. R. (2008). Transgenic mice lacking NMDAR-dependent LTD exhibit deficits in behavioral flexibility. Neuron 58, 104–117.
Nordlie, E., Gewaltig, M.-O., and Plesser, H. E. (2009). Towards reproducible descriptions of neuronal network models. PLoS Comput. Biol. 5, e1000456. doi: 10.1371/journal.pcbi.1000456
Ogasawara, H., Doi, T., Doya, K., and Kawato, M. (2007). Nitric oxide regulates input specificity of long-term depression and context dependence of cerebellar learning. PLoS Comput. Biol. 3, e179. doi: 10.1371/journal.pcbi.0020179
Ogasawara, H., Doi, T., and Kawato, M. (2008). Systems biology perspectives on cerebellar long-term depression. Neurosignals 16, 300–317.
Ogasawara, H., and Kawato, M. (2009). “Computational models of cerebellar long-term memory,” in Systems Biology: The Challenge of Complexity, 1st Edn., eds S. Nakanishi, R. Kageyama, and D. Watanabe (Tokyo: Springer), 169–182.
Okamoto, H., and Ichikawa, K. (2000a). A model for molecular mechanisms of synaptic competition for a finite resource. Biosystems 55, 65–71.
Okamoto, H., and Ichikawa, K. (2000b). Switching characteristics of a model for biochemical-reaction networks describing autophosphorylation versus dephosphorylation of Ca2+/calmodulin-dependent protein kinase II. Biol. Cybern. 82, 35–47.
Oliveira, R. F., Terrin, A., Di Benedetto, G., Cannon, R. C., Koh, W., Kim, M., Zaccolo, M., and Blackwell, K. T. (2010). The role of type 4 phosphodiesterases in generating microdomains of cAMP: large scale stochastic simulations. PLoS ONE 5, e11725. doi: 10.1371/journal.pone.0011725
Pepke, S., Kinzer-Ursem, T., Mihalas, S., and Kennedy, M. B. (2010). A dynamic model of interactions of Ca2+, calmodulin, and catalytic subunits of Ca2+/calmodulin-dependent protein kinase II. PLoS Comput. Biol. 6, e1000675. doi: 10.1371/journal.pcbi.1000675
Pettinen, A., Aho, T., Smolander, O.-P., Manninen, T., Saarinen, A., Taattola, K.-L., Yli-Harja, O., and Linne, M.-L. (2005). Simulation tools for biochemical networks: evaluation of performance and usability. Bioinformatics 21, 357–363.
Pi, H. J., and Lisman, J. E. (2008). Coupled phosphatase and kinase switches produce the tristability required for long-term potentiation and long-term depression. J. Neurosci. 28, 13132–13138.
Qi, Z., Miller, G. W., and Voit, E. O. (2010). The internal state of medium spiny neurons varies in response to different input signals. BMC Syst. Biol. 4, 26.
Rackham, O. J. L., Tsaneva-Atanasova, K., Ganesh, A., and Mellor, J. R. (2010). A Ca2+-based computational model for NMDA receptor-dependent synaptic plasticity at individual post-synaptic spines in the hippocampus. Front. Syn. Neurosci. 2:31. doi: 10.3389/fnsyn.2010.00031
Rubin, J. E., Gerkin, R. C., Bi, G.-Q., and Chow, C. C. (2005). Calcium time course as a signal for spike-timing-dependent plasticity. J. Neurophysiol. 93, 2600–2613.
Saftenku, E. E. (2002). A simplified model of long-term plasticity in cerebellar mossy fiber-granule cell synapses. Neurophysiology 34, 216–218.
Santamaria, F., Gonzalez, J., Augustine, G. J., and Raghavachari, S. (2010). Quantifying the effects of elastic collisions and non-covalent binding on glutamate receptor trafficking in the post-synaptic density. PLoS Comput. Biol. 6, e1000780. doi: 10.1371/journal.pcbi.1000780
Santos, S. D., Carvalho, A. L., Caldeira, M. V., and Duarte, C. B. (2009). Regulation of AMPA receptors and synaptic plasticity. Neuroscience 158, 105–125.
Santucci, D. M., and Raghavachari, S. (2008). The effects of NR2 subunit-dependent NMDA receptor kinetics on synaptic transmission and CaMKII activation. PLoS Comput. Biol. 4, e1000208. doi: 10.1371/journal.pcbi.1000208
Saudargiene, A., Porr, B., and Wörgötter, F. (2005). Synaptic modifications depend on synapse location and activity: a biophysical model of STDP. Biosystems 79, 3–10.
Schaff, J., Fink, C. C., Slepchenko, B., Carson, J. H., and Loew, L. M. (1997). A general computational framework for modeling cellular structure and function. Biophys. J. 73, 1135–1146.
Schiegg, A., Gerstner, W., Ritz, R., and van Hemmen, J. L. (1995). Intracellular Ca2+ stores can account for the time course of LTP induction: a model of Ca2+ dynamics in dendritic spines. J. Neurophysiol. 74, 1046–1055.
Schmidt, H., and Eilers, J. (2009). Spine neck geometry determines spino-dendritic cross-talk in the presence of mobile endogenous calcium binding proteins. J. Comput. Neurosci. 27, 229–243.
Schmidt, H., Kunerth, S., Wilms, C., Strotmann, R., and Eilers, J. (2007). Spino-dendritic cross-talk in rodent Purkinje neurons mediated by endogenous Ca2+-binding proteins. J. Physiol. 581, 619–629.
Serrano, P., Yao, Y., and Sacktor, T. C. (2005). Persistent phosphorylation by protein kinase Mζ maintains late-phase long-term potentiation. J. Neurosci. 25, 1979–1984.
Shah, N. T., Yeung, L. C., Cooper, L. N., Cai, Y., and Shouval, H. Z. (2006). A biophysical basis for the inter-spike interaction of spike-timing-dependent plasticity. Biol. Cybern. 95, 113–121.
Shouval, H. Z., Bear, M. F., and Cooper, L. N. (2002a). A unified model of NMDA receptor-dependent bidirectional synaptic plasticity. Proc. Natl. Acad. Sci. U.S.A. 99, 10831–10836.
Shouval, H. Z., Castellani, G. C., Blais, B. S., Yeung, L. C., and Cooper, L. N. (2002b). Converging evidence for a simplified biophysical model of synaptic plasticity. Biol. Cybern. 87, 383–391.
Shouval, H. Z., and Kalantzis, G. (2005). Stochastic properties of synaptic transmission affect the shape of spike time-dependent plasticity curves. J. Neurophysiol. 93, 1069–1073.
Shouval, H. Z., Wang, S. S. H., and Wittenberg, G. M. (2010). Spike timing dependent plasticity: a consequence of more fundamental learning rules. Front. Comput. Neurosci. 4:19. doi: 10.3389/fncom.2010.00019
Sivakumaran, S., Hariharaputran, S., Mishra, J., and Bhalla, U. S. (2003). The Database of Quantitative Cellular Signaling: management and analysis of chemical kinetic models of signaling networks. Bioinformatics 19, 408–415.
Slepchenko, B. M., Schaff, J. C., Macara, I., and Loew, L. M. (2003). Quantitative cell biology with the Virtual Cell. Trends Cell Biol. 13, 570–576.
Smolen, P. (2007). A model of late long-term potentiation simulates aspects of memory maintenance. PLoS ONE 2, e445. doi: 10.1371/journal.pone.0000445
Smolen, P., Baxter, D. A., and Byrne, J. H. (2006). A model of the roles of essential kinases in the induction and expression of late long-term potentiation. Biophys. J. 90, 2760–2775.
Smolen, P., Baxter, D. A., and Byrne, J. H. (2008). Bistable MAP kinase activity: a plausible mechanism contributing to maintenance of late long-term potentiation. Am. J. Physiol. Cell Physiol. 294, C503–C515.
Smolen, P., Baxter, D. A., and Byrne, J. H. (2009). Interlinked dual-time feedback loops can enhance robustness to stochasticity and persistence of memory. Phys. Rev. E 79, 031902.
Soderling, T. R., and Derkach, V. A. (2000). Postsynaptic protein phosphorylation and LTP. Trends Neurosci. 23, 75–80.
Stefan, M. I., Edelstein, S. J., and Le Novère, N. (2008). An allosteric model of calmodulin explains differential activation of PP2B and CaMKII. Proc. Natl. Acad. Sci. U.S.A. 105, 10768–10773.
Steuber, V., and Willshaw, D. J. (2004). A biophysical model of synaptic delay learning and temporal pattern recognition in a cerebellar Purkinje cell. J. Comput. Neurosci. 17, 149–164.
Stiles, J. R., and Bartol, T. M. (2001). “Monte Carlo methods for simulating realistic synaptic microphysiology using MCell,” in Computational Neuroscience: Realistic Modeling for Experimentalists, ed. E. De Schutter (Boca Raton: CRC Press), 87–127.
Sweatt, J. D. (1999). Toward a molecular explanation for long-term potentiation. Learn. Mem. 6, 399–416.
Tanaka, K., and Augustine, G. J. (2009). “Systems biology meets single-cell physiology: role of a positive-feedback signal transduction network in cerebellar long-term synaptic depression,” in Systems Biology: The Challenge of Complexity, 1st Edn., eds S. Nakanishi, R. Kageyama, and D. Watanabe (Tokyo: Springer), 159–168.
Tanaka, K., Khiroug, L., Santamaria, F., Doi, T., Ogasawara, H., Ellis-Davies, G. C. R., Kawato, M., and Augustine, G. J. (2007). Ca2+ requirements for cerebellar long-term synaptic depression: role for a postsynaptic leaky integrator. Neuron 54, 787–800.
Tolle, D. P., and Le Novère, N. (2010a). Brownian diffusion of AMPA receptors is sufficient to explain fast onset of LTP. BMC Syst. Biol. 4, 25.
Tolle, D. P., and Le Novère, N. (2010b). Meredys, a multi-compartment reaction–diffusion simulator using multistate realistic molecular complexes. BMC Syst. Biol. 4, 24.
Tomita, M., Hashimoto, K., Takahashi, K., Shimizu, T. S., Matsuzaki, Y., Miyoshi, F., Saito, K., Tanida, S., Yugi, K., Venter, J. C., and Hutchison III, C. A. (1999). E-CELL: software environment for whole-cell simulation. Bioinformatics 15, 72–84.
Traub, R. D., Wong, R. K., Miles, R., and Michelson, H. (1991). A model of a CA3 hippocampal pyramidal neuron incorporating voltage-clamp data on intrinsic conductances. J. Neurophysiol. 66, 635–650.
Urakubo, H., Honda, M., Froemke, R. C., and Kuroda, S. (2008). Requirement of an allosteric kinetics of NMDA receptors for spike timing-dependent plasticity. J. Neurosci. 28, 3310–3323.
Urakubo, H., Honda, M., Tanaka, K., and Kuroda, S. (2009). Experimental and computational aspects of signaling mechanisms of spike-timing-dependent plasticity. HFSP J. 3, 240–254.
Volfovsky, N., Parnas, H., Segal, M., and Korkotian, E. (1999). Geometry of dendritic spines affects calcium dynamics in hippocampal neurons: theory and experiments. J. Neurophysiol. 82, 450–462.
Wang, H., Hu, Y., and Tsien, J. Z. (2006). Molecular and systems mechanisms of memory consolidation and storage. Prog. Neurobiol. 79, 123–135.
Wils, S., and De Schutter, E. (2009). STEPS: modeling and simulating complex reaction–diffusion systems with Python. Front. Neuroinform. 3:15. doi: 10.3389/neuro.11.015.2009
Woo, N. H., Duffy, S. N., Abel, T., and Nguyen, P. V. (2003). Temporal spacing of synaptic stimulation critically modulates the dependence of LTP on cyclic AMP-dependent protein kinase. Hippocampus 13, 293–300.
Wörgötter, F., and Porr, B. (2005). Temporal sequence learning, prediction, and control: a review of different models and their relation to biological mechanisms. Neural Comput. 17, 245–319.
Yang, K.-H., Hellgren Kotaleski, J., and Blackwell, K. T. (2001). The role of protein kinase C in the biochemical pathways of classical conditioning. Neurocomputing 38–40, 79–85.
Yeung, L. C., Shouval, H. Z., Blais, B. S., and Cooper, L. N. (2004). Synaptic homeostasis and input selectivity follow from a calcium-dependent plasticity model. Proc. Natl. Acad. Sci. U.S.A. 101, 14943–14948.
Yu, X., Shouval, H. Z., and Knierim, J. J. (2008). A biophysical model of synaptic plasticity and metaplasticity can account for the dynamics of the backward shift of hippocampal place fields. J. Neurophysiol. 100, 983–992.
Zador, A., Koch, C., and Brown, T. H. (1990). Biophysical model of a Hebbian synapse. Proc. Natl. Acad. Sci. U.S.A. 87, 6718–6722.
Zhabotinsky, A. M. (2000). Bistability in the Ca2+/calmodulin-dependent protein kinase-phosphatase system. Biophys. J. 79, 2211–2221.
Zhabotinsky, A. M., Camp, R. N., Epstein, I. R., and Lisman, J. E. (2006). Role of the neurogranin concentrated in spines in the induction of long-term potentiation. J. Neurosci. 26, 7337–7347.
Keywords: computational model, kinetic model, long-term depression, long-term potentiation, plasticity, postsynaptic signal transduction model
Citation: Manninen T, Hituri K, Hellgren Kotaleski J, Blackwell KT and Linne M-L (2010) Postsynaptic signal transduction models for long-term potentiation and depression. Front. Comput. Neurosci. 4:152. doi: 10.3389/fncom.2010.00152
Received: 15 July 2010;
Paper pending published: 26 August 2010;
Accepted: 22 November 2010;
Published online: 13 December 2010.
Edited by:
Nicolas Brunel, Centre National de la Recherche Scientifique, FranceReviewed by:
Harel Z. Shouval, University of Texas Medical School at Houston, USAPaul Miller, Brandeis University, USA
Copyright: © 2010 Manninen, Hituri, Hellgren Kotaleski, Blackwell and Linne. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Tiina Manninen, Computational Neuroscience Laboratory, Department of Signal Processing, Tampere University of Technology, P.O. Box 553, FI-33101 Tampere, Finland. e-mail:dGlpbmEubWFubmluZW5AdHV0LmZp