 Bert M. Verheijen
Bert M. Verheijen Kiyomitsu Oyanagi
Kiyomitsu Oyanagi Fred W. van Leeuwen
Fred W. van Leeuwen- 1Department of Translational Neuroscience, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht University, Utrecht, Netherlands
- 2Department of Neurology and Neurosurgery, Brain Center Rudolf Magnus, University Medical Center Utrecht, Utrecht University, Utrecht, Netherlands
- 3Division of Neuropathology, Department of Brain Disease Research, Shinshu University School of Medicine, Nagano, Japan
- 4Brain Research Laboratory, Hatsuishi Hospital, Chiba, Japan
- 5Department of Neuroscience, Faculty of Health, Medicine and Life Sciences, Maastricht University, Maastricht, Netherlands
Guam parkinsonism–dementia complex (G-PDC) is an enigmatic neurodegenerative disease that is endemic to the Pacific island of Guam. G-PDC patients are clinically characterized by progressive cognitive impairment and parkinsonism. Neuropathologically, G-PDC is characterized by abundant neurofibrillary tangles, which are composed of hyperphosphorylated tau, marked deposition of 43-kDa TAR DNA-binding protein, and neuronal loss. Although both genetic and environmental factors have been implicated, the etiology and pathogenesis of G-PDC remain unknown. Recent neuropathological studies have provided new clues about the pathomechanisms involved in G-PDC. For example, deposition of abnormal components of the protein quality control system in brains of G-PDC patients indicates a role for proteostasis imbalance in the disease. This opens up promising avenues for new research on G-PDC and could have important implications for the study of other neurodegenerative disorders.
Introduction
Guam parkinsonism–dementia complex (G-PDC) is a mysterious neurodegenerative disorder that afflicts the indigenous Chamorro people of Guam (Mariana Islands). It was first described in clinicopathological studies by Hirano et al. in 1961 (1, 2). Mental deterioration and extrapyramidal signs (rigidity, tremors, and bradykinesia) characterize G-PDC clinically (3). Neuropathologically, G-PDC is characterized by widespread neurofibrillary tangles (NFTs), consisting of highly phosphorylated tau, marked deposition of 43-kDa TAR DNA-binding protein (TDP-43) and neuronal loss (4).
A remarkably high incidence of amyotrophic lateral sclerosis (ALS) was previously found in the same Western Pacific focus of neurological disease (5–12). Because of co-occurrence and overlapping pathology, displaying features of classical ALS in combination with NFTs, it has been proposed that these cases of ALS and G-PDC are variations of the same disorder, i.e., ALS/PDC of Guam (known in Guam as lytico-bodig) (13, 14). However, ALS and G-PDC might be completely separate disease entities: NFTs have been reported to be a frequent background feature in the affected population, and, therefore, occurrence of ALS in Guamanians often results in mixed pathology (15–20).
Although several causes have been suggested for G-PDC, including genetics, infectious agents, mineral deficiencies, and environmental toxins, the etiology and pathogenesis of this disease remain unknown (21–26). Determining the cause(s) of G-PDC and the pathways that lead to neurodegeneration in G-PDC is of great interest, as this could lead to new insights into more common neurodegenerative diseases, such as Alzheimer’s disease (AD) and other tauopathies. Similarly, insights derived from studies on other neurodegenerative disorders may be helpful for understanding and revealing unique aspects of G-PDC. Here, we highlight some recent observations that open up new opportunities to investigate disease mechanisms in G-PDC.
Neuropathological Features of G-PDC
The search for the cause of G-PDC has become increasingly challenging due to a notable decline in neurological disease incidence on the island in the last decades (27–31). The rapid Westernization of Guam may have contributed to this decrease. However, neuropathology has been found to be unaltered during this time (although the severity and distribution of pathological lesions may have changed to some extent) (32).
Neuropathologically, G-PDC is characterized by severe neuronal loss and abundant NFTs in different brain regions, including the temporal and frontal cortex, basal ganglia, thalamus, and brainstem (2, 17, 32, 33). NFTs (Figures 1A,B) are biochemically and ultrastructurally similar to those found in AD brains (34) and closely resemble those seen in frontotemporal dementia (FTD) tauopathies (35).
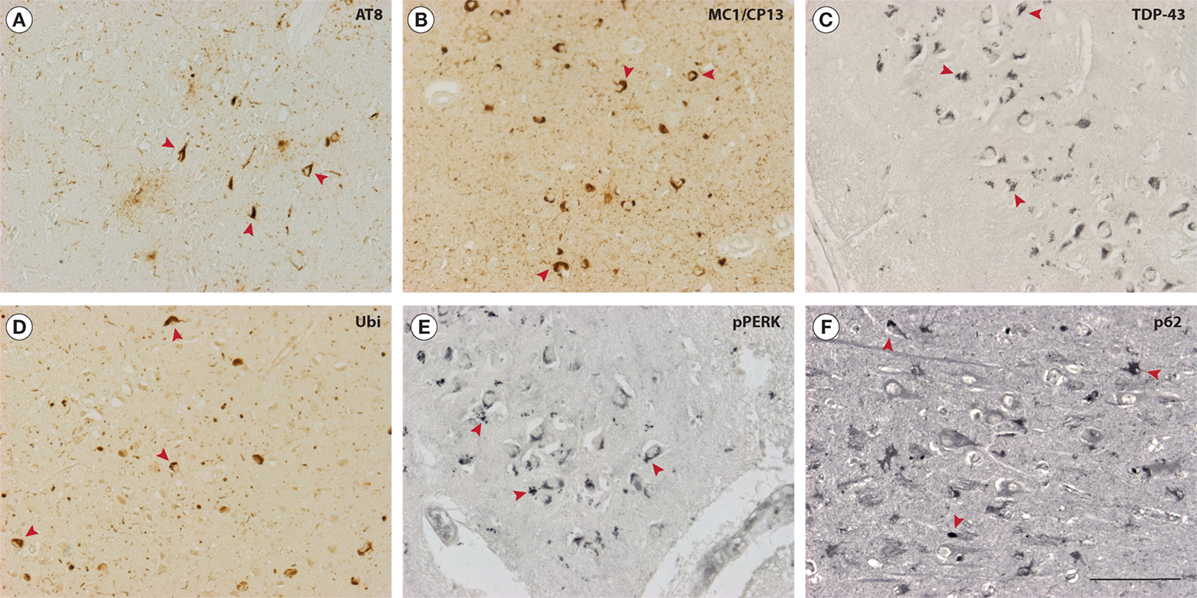
Figure 1. Neuropathological features of Guam parkinsonism–dementia complex (G-PDC). In the brains of G-PDC patients, various pathological protein aggregates can be found, e.g., neurofibrillary tangles (NFTs) (mouse anti-phosphorylated tau, AT8, 1:3,000, Innogenetics; mouse anti-MC1/CP13, 1:200, gift from Dr. P. Davies) (A,B) and 43-kDa TAR DNA-binding protein (TDP-43)-positive inclusions (mouse anti-TDP-43, 1:1,000, Abnova) (C). Abnormal protein deposits are decorated with ubiquitin (rabbit anti-ubiquitin, Z0458, 1:3,000, DAKO) (D). Immunoreactivity for phosphorylated pancreatic ER kinase (pPERK) indicates activation of the unfolded protein response and is associated with granulovacuolar degeneration (rabbit anti-pPERK, sc-32577, 1:400, Santa Cruz Biotechnology) (E). Furthermore, the ubiquitin-binding protein p62 is deposited in G-PDC brains (rabbit anti-p62, 1:500, Biomol) (F). Accumulation of p62 is a marker for the inhibition of autophagic flux. The photomicrographs in panels (A–F) show representative hippocampal sections from G-PDC brains. Arrowheads indicate distinct immunoreactive structures. Scale bar: 100 µm (Verheijen et al., unpublished data).
Besides frequent tangles, other pathological protein deposits have been identified in neurons of G-PDC brains. These include cytoplasmic TDP-43-containing aggregates (Figure 1C) and focal α-synuclein pathology (32, 36–40). This could hint at a role for these particular proteins in specific disease processes in G-PDC. For example, abnormal aggregation of the RNA-binding protein TDP-43 may be linked to defective RNA processing (e.g., pre-mRNA splicing) and several other cellular perturbations, like disrupted nucleocytoplasmic transport, mitochondrial dysfunction, and inhibition of endocytosis (41–46). Amyloid-β (Aβ) deposits were initially reported to be rare or absent in G-PDC (47) but were later found to be present by several authors (32, 48–51). Intracellular inclusions are often ubiquitinated (32) (Figure 1D).
It is interesting to point out that glial cells, i.e., astrocytes and oligodendroglia, also contain pathological inclusions (tau, TDP-43) in G-PDC and that these cells may play a significant role in G-PDC pathogenesis (32, 52). Extracellular NFTs are associated with reactive microglia (53, 54).
Importantly, findings related to multiple proteinopathy have been complemented by the detection of molecular markers that link G-PDC to certain protein homeostasis (proteostasis) pathways. We have recently found some evidence for activation of the unfolded protein response (UPR) in G-PDC brain (Verheijen et al., unpublished data). The UPR is an adaptive signaling cascade that is triggered by endoplasmic reticulum (ER) stress and is associated with abnormal protein aggregation and neurodegeneration (55, 56). Phosphorylation of pancreatic ER kinase, an ER transmembrane protein and ER stress sensor, plays an important role in the initiation and regulation of the UPR (Figure 1E). The primary goal of the UPR is to restore proteostasis in the ER via translational block and activation of ER stress responsive genes, but extensive or prolonged ER stress and UPR activation in disease can turn the UPR maladaptive. In addition, the autophagy substrate p62/SQSTM1 accumulates in G-PDC brains (Figure 1F), which could indicate compromised autophagy-mediated degradation (57, 58).
Together, some of these pathological characteristics suggest proteostasis network dysfunction in G-PDC. Disruption of the intracellular protein degradation machinery and proteostasis collapse has been associated with many neurodegenerative diseases, and it will be interesting to see if, and to what extent, protein degradation pathways are affected in G-PDC (59, 60). Failure of protein quality control (PQC) mechanisms to maintain proteostasis may represent a key pathogenic mechanism in G-PDC.
Mutant Ubiquitin and Impaired Proteolysis in G-PDC
Multiple neuropathological observations imply impaired PQC in G-PDC. In a recent study, abnormal components of the ubiquitin-proteasome pathway (UPP), i.e., frameshift mutants of ubiquitin-B (UBB+1), have been found to accumulate in G-PDC brains (61).
The UPP is a major mechanism for the clearance of (abnormal) proteins in cells and impairment of the UPP has been reported to occur during neurodegeneration (62–65). UBB+1 is a dose-dependent inhibitor of the UPP that is thought to be generated through “molecular misreading,” a poorly understood process that introduces mutations (e.g., ΔGA or ΔGU dinucleotide deletions) not present in DNA into mRNA, resulting in the generation of aberrant proteins (66–68) (Figure 2A). The mutant protein lacks a C-terminal glycine residue (G76, which is replaced by a 20-amino acid extension in UBB+1) that is necessary to ubiquitinate other proteins, but can still be ubiquitinated itself. The abnormal C-terminal domain of UBB+1 can be recognized by UBB+1 antibodies (69) (Figures 2B,C).
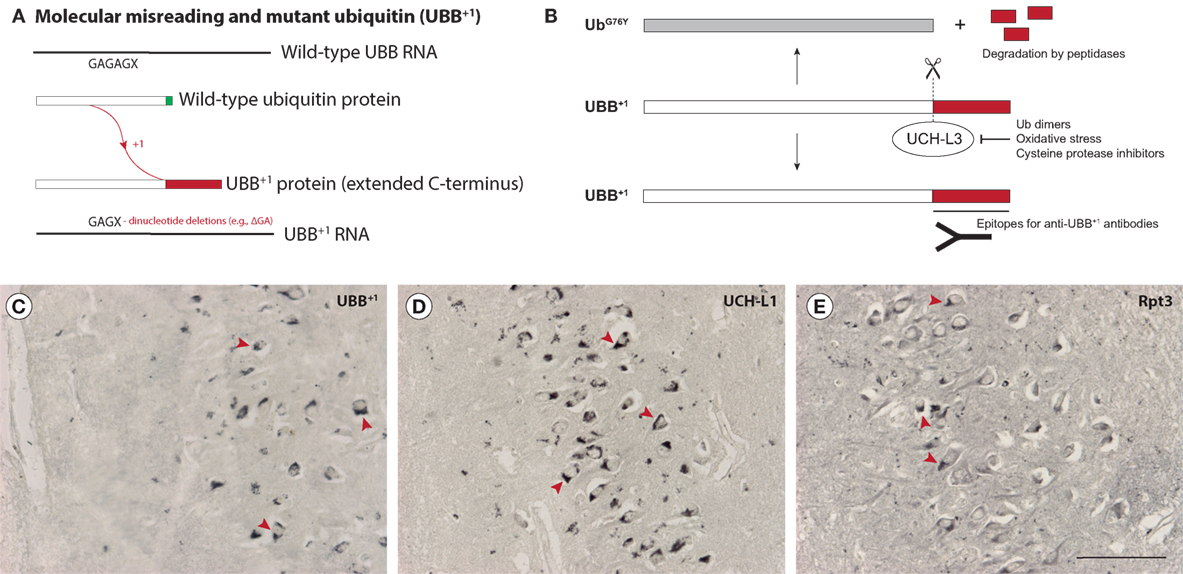
Figure 2. Accumulation of mutant ubiquitin (UBB+1) in Guam parkinsonism-dementia complex (G-PDC). UBB+1 is a frameshift mutant of ubiquitin that is generated through “molecular misreading,” a form of transcriptional mutagenesis that introduces dinucleotide deletions (; ΔGA or ΔGU) in or near GAGAG motifs in mRNA (A). UBB+1 contains an extended C-terminal tail that can be recognized by anti-UBB+1 antibodies. Deubiquitinating enzymes (DUBs), i.e., ubiquitin C-terminal hydrolase L3 (UCH-L3), can cleave this abnormal C-terminal domain, destroying the epitope. However, inhibition of DUBs, e.g., by oxidative stress conditions, prevents this cleavage (B). Accumulation of UBB+1 (rabbit anti-UBB+1, Ubi2A, 1:400, Dr. F. W. van Leeuwen) (C) and specific ubiquitin-proteasome pathway components, i.e., the DUB ubiquitin C-terminal hydrolase L1 (UCH-L1) (rabbit anti-UCH-L1, 1:500, Biomol) (D) and the proteasomal ATPase subunit Rpt3 (rabbit anti-Rpt3, 1:400, Biomol) (E) can be observed in G-PDC patient brains (hippocampal sections), which strongly suggests proteostasis breakdown. UBB+1 is not present in young control brains (non-Guamanian cases). Arrowheads indicate various immunoreactive structures. Scale bar: 100 µm (Verheijen et al., unpublished data).
UBB+1 accumulates in several neurodegenerative diseases other than G-PDC, including tauopathies (e.g., AD) and polyglutamine (polyQ) diseases (e.g., Huntington’s disease) and has been shown to be detrimental to neurons (66, 70–73). The mechanisms by which UBB+1 exerts its effects on the UPP and neuronal function are not known exactly, but it has been demonstrated that UBB+1 is an inhibitor of deubiquitinating enzymes (DUBs) (74), and that it can affect mitochondrial function (75). UBB+1 expression in primary neurons causes neuritic beading of mitochondria in association with neuronal degeneration, presumably due to impaired axonal transport (76). Transgenic expression of UBB+1 in mice results in contextual memory deficits and central breathing dysfunction, which are consistent with neurodegenerative disease (77, 78). Also, UBB+1 was shown to increase mutant protein load in a mouse model for familial encephalopathy with neuroserpin inclusion bodies (FENIB), by impairing ER-associated degradation (79). UBB+1 might act as a modifier of the aggregation and cytotoxicity of other aggregation-prone proteins, such as prion protein and disease-associated huntingtin (80).
Dissecting the precise role of UBB+1 in neurodegeneration is complicated by its dual role as both substrate and inhibitor of the UPP: at low concentrations, UBB+1 is degraded via the ubiquitin-fusion degradation pathway, but at high concentrations it is a potent inhibitor of the UPP (81–85). In addition, UBB+1 can induce cytoprotective programs, like chaperone expression (86), which may compensate for diminished UPP function. Overexpression of UBB+1 in AD transgenic mice resulted in an unexpected decrease in Aβ plaque load (87), which could suggest that accumulation of UBB+1 is actually part of a cellular protective response. It is tempting to speculate that UBB+1 can interfere with non-degradation-based PQC processes like misfolding-associated protein secretion (MAPS) as well, because MAPS enables PQC when conventional proteasomal degradation is impaired (88). Recent progress in identifying and understanding the roles of different ubiquitin chain topologies potentially adds another layer of complexity to the study of UBB+1, because different ubiquitin chain linkages on UBB+1 could change its properties (89–91). The E3 ubiquitin ligases and DUBs that regulate UBB+1 ubiquitination in neurons also remain to be identified (73).
In G-PDC, UBB+1 deposits are not exclusively located in neurons, but are also present in glial cells (61). Such glial deposits have previously been observed in progressive supranuclear palsy (71), a disease that shares some similarities with G-PDC (23, 92, 93). The role of the UPP in glia with regard to neurodegenerative disease has been somewhat neglected (94), and it would be interesting to find out how UBB+1 impacts neuroglia. In cultured astrocytes, UBB+1 expression seems to confer protective effects via various mechanisms, ranging from regulation of pro-inflammatory signaling to altering mitochondrial dynamics (95, 96). Thus, UBB+1 and UPP inhibition may elicit cell type-specific responses in the context of disease.
In addition to UBB+1, several other proteins related to the UPP were found to be present in G-PDC aggregates, i.e., the DUB ubiquitin C-terminal hydrolase L1 (UCH-L1) (Figure 2D) and the proteasomal ATPase subunit Rpt3 (Figure 2E) (61). Accumulation of these UPP components has been associated with neuropathology in earlier studies (97, 98). Mutations in UCH-L1 lead to motor dysfunction in patients (99, 100) and mice lacking functional UCH-L1 show neurodegeneration (101). Strikingly, motor neuron-specific knockout of Rpt3 results in an ALS-like phenotype in mice (102).
Based on these findings, we conclude that the UPP is disturbed in G-PDC. The definite roles of UBB+1, different UPP components, and PQC mechanisms in G-PDC remain to be determined.
Proteostasis Imbalance and Neurodegeneration in G-PDC
Accumulation of (abnormal) PQC components in G-PDC brains could be an important clue as to why disease-associated proteins aggregate and neurons degenerate in G-PDC. Impaired PQC and several neurodegenerative diseases have been causally linked through genetic analyses of familial cases [e.g., in familial Parkinson’s disease (PD) (103)], demonstrating that PQC dysfunction can be a disease-initiating factor. Moreover, pharmacological inhibition of PQC via injection of proteasome inhibitors causes parkinsonian features in rats (104). Age-related decline in PQC function might (in part) explain the loss of proteome integrity associated with age-related neurodegenerative disorders, including G-PDC (105, 106).
Detailed examination of the PQC system and UBB+1 in the context of the multiproteinopathy that characterizes G-PDC, in different experimental models (e.g., in vitro neuronal cell culture and experimental animal models), will likely increase our knowledge of the timing (early vs. late) and relative importance of different disease processes. We take the view that mechanistic studies in such experimental model systems, and validating new findings against the reality of human tissues (neuropathology), will be a powerful approach for making discoveries. It is well recognized that abnormal PQC is implicated in AD, ALS(-FTD), PD, and other neurodegenerative disorders. Because this is a quite recent finding for G-PDC, insights derived from studies on other neurodegenerative diseases will be valuable to help understanding G-PDC, including its unique characteristics.
The cellular mechanisms that control proteostasis are not limited to protein degradation by the UPP, but also involve protein synthesis, folding, trafficking, and other (extracellular) clearance routes (107). PQC critically depends on additional cellular machinery, such as molecular chaperones (108, 109), and is often associated with specific cellular compartments/organelles (e.g., ER, mitochondria). Crosstalk within the proteostasis network and interactions between disease-related proteins and various disease mechanisms (e.g., DNA damage, defective RNA processing, ER and mitochondrial dysfunction, nitrative and oxidative stress, cytoskeletal defects, Golgi fragmentation, abnormal stress granule formation, prion-like mechanisms, neuroinflammation, synaptic malfunction, disrupted membrane trafficking, and excitotoxicity) can determine whether a neuron will degrade (toxic protein aggregates) or degenerate. It should be mentioned that protein aggregation itself, even though it reflects failure of PQC to get rid of potentially harmful proteins, need not be pathogenic and in some cases may even be protective (110).
Ultimately, new mechanistic insights may result in the identification of therapeutic targets that prevent or slow down neurodegeneration. If G-PDC and other neurological disorders converge on the dysregulation of proteostasis as a common underlying mechanism of pathogenesis, exploring PQC pathways to restore proteostasis could be a promising strategy to find therapies for multiple neurodegenerative diseases. Current advances include the use of UPP activation to remove and stop propagation of pathogenic protein species (111–113). Other targets, such as chaperones and autophagy, are also being explored (58, 109, 114). However, different proteins, in different protein states, can be cleared via separate pathways. Ideally, a proteostasis “reset,” rebalancing the entire proteostasis network in proteotoxically stressed neurons, would be desirable, because this could counteract many defects at the same time. It is unknown whether such a reset switch exists in human neurons, but some cells appear to exhibit a remarkable capacity to reestablish proteostasis (e.g., via lysosome acidification and metabolic shift in C. elegans germ lineage, “exopher” formation in C. elegans neurons and during differentiation of mouse embryonic stem cells) (115–117). Combinatorial therapies are likely to be required to generate robust treatments that effectively restore homeostasis in human somatic cells in such a way. The diverse pathology of G-PDC provides an excellent paradigm for interrogating interactions between multiple pathogenic proteins, the proteostasis network and different mechanisms of neurodegeneration.
Future Directions
Novel neuropathological findings in G-PDC stress the importance of utilizing pathway-specific markers to unravel disease mechanisms. While determining the (possibly multifactorial) cause of G-PDC, perhaps through new genetic and/or exposome (i.e., the totality of all environmental exposures) studies in the geographical isolate, remains a major outstanding problem, more studies into the pathomechanisms underlying neurodegeneration in G-PDC are warranted. Applying insights derived from studies on other neurological diseases that share molecular neuropathology, including work on postmortem human brains and various experimental model systems, will help to identify critical disease mechanisms. Investigating the interplay between G-PDC-associated pathological proteins and specific cellular/molecular pathways (e.g., PQC pathways), using relevant experimental models (e.g., patient induced pluripotent stem cell-derived neurons), will be an exciting and challenging task for the future. Such studies have also been initiated on the Kii peninsula of Japan, another hyperendemic focus of ALS/PDC (118–120) (Verheijen et al., under investigation). The resulting insights could improve our understanding of other neurodegenerative diseases and might result in the identification of new biomarkers or therapeutic targets. It will be important for future studies to replicate findings in other populations with increased prevalence of ALS/PDC (121, 122).
Some Open Questions
• Is impairment of the PQC system an important pathogenic mechanism in G-PDC?
• Does UBB+1 modify the cytotoxicity of aggregation-prone proteins, like tau and TDP-43?
• What is the role of glia in G-PDC? What are the effects of UBB+1 on glial cells?
• Are UBB+1 and PQC proteins also deposited in diseases that are similar to G-PDC, e.g., Kii ALS/PDC?
• What are the roles of the UPR and other adaptive stress response pathways in G-PDC?
• Is accumulation of UBB+1 and PQC dysfunction an early or late event in G-PDC? Is it a cause or consequence (or both) of disease?
• How can new insights into pathogenic mechanisms of G-PDC be used to advance the understanding and treatment of other neurodegenerative diseases? E.g., can UBB+1 and particular PQC components be used as therapeutic targets in neurodegenerative disorders?
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Drs. J.-M. Graïc, J. J. van Heerikhuize, and D. F. Swaab (Netherlands Institute for Neuroscience, Amsterdam, The Netherlands), Dr. R. J. Braun (Institute of Cell Biology, University of Bayreuth, Bayreuth, Germany), and Professor M. Yamada (Department of Brain Disease Research, Shinshu University School of Medicine, Nagano, Japan) for assistance. We would like to thank Dr. P. Davies (Albert Einstein College of Medicine, Bronx, NY, USA) for his generous gift of anti-MC1 and anti-CP13. This study was supported in part by the JSPS KAKENHI (Grant-in-Aid for Scientific Research [C] No. 15K06754) and by the Collaborative Research Project (2017-#2907) of Brain Research Institute, Niigata University.
References
1. Hirano A, Kurland LT, Krooth RS, Lessell S. Parkinsonism-dementia complex, an endemic disease on the island of Guam. I. Clinical features. Brain (1961) 84:642–61. doi:10.1093/brain/84.4.642
2. Hirano A, Malamud N, Kurland LT. Parkinsonism-dementia complex, an endemic disease on the island of Guam. II. Pathological features. Brain (1961) 84:662–79. doi:10.1093/brain/84.4.662
3. Murakami N. Parkinsonism-dementia complex on Guam – overview of clinical aspects. J Neurol (1999) 246(Suppl 2):II16–8. doi:10.1007/BF03161077
4. Oyanagi K, Hashimoto T, Yamazaki M. Parkinsonism–dementia complex of Guam. In: Dickson DW, Weller RO, editors. Neurodegeneration: The Molecular Pathology of Dementia and Movement Disorders. Oxford, UK: Wiley-Blackwell (2011). p. 171–8.
5. Zimmerman H. Progress Report of Work in the Laboratory of Pathology during May 1945. Guam. Washington, DC: US Naval Medical Research (1945). Unit Number 2, 1 June.
6. Koerner DR. Amyotrophic lateral sclerosis on Guam: a clinical study and review of the literature. Ann Intern Med (1952) 37:1204–20. doi:10.7326/0003-4819-37-6-1204
7. Arnold A, Edgren DC, Palladino VS. Amyotrophic lateral sclerosis; fifty cases observed on Guam. J Nerv Ment Dis (1953) 117:135–9. doi:10.1097/00005053-195302000-00005
8. Tillema S, Wijnberg CJ. Endemic amyotrophic lateral sclerosis on Guam; epidemiological data; a preliminary report. Doc Med Geogr Trop (1953) 5:366–70.
9. Kurland LT, Mulder DW. Epidemiologic investigations of amyotrophic lateral sclerosis. 1. Preliminary report on geographic distribution, with special reference to the Mariana Islands, including clinical and pathologic observations. Neurology (1954) 4:355–78. doi:10.1212/WNL.4.5.355
10. Mulder DW, Kurland LT, Iriarte LL. Neurologic diseases on the island of Guam. U S Armed Forces Med J (1954) 5:1724–39.
11. Kurland LT, Mulder DW. Epidemiologic investigations of amyotrophic lateral sclerosis. 2. Familial aggregations indicative of dominant inheritance. I. Neurology (1955) 5:182–96. doi:10.1212/WNL.5.3.182
12. Kurland LT, Mulder DW. Epidemiologic investigations of amyotrophic lateral sclerosis. 2. Familial aggregations indicative of dominant inheritance. II. Neurology (1955) 5:249–68. doi:10.1212/WNL.5.3.182
13. Elizan TS, Hirano A, Abrams BM, Need RL, Van Nuis C, Kurland LT. Amyotrophic lateral sclerosis and parkinsonism-dementia complex of Guam. Neurological reevaluation. Arch Neurol (1966) 14:356–68. doi:10.1001/archneur.1966.00470100012002
14. Hirano A, Malamud N, Elizan TS, Kurland LT. Amyotrophic lateral sclerosis and Parkinsonism-dementia complex on Guam. Further pathologic studies. Arch Neurol (1966) 15:35–51. doi:10.1001/archneur.1966.00470130039004
15. Anderson FH, Richardson EP, Okazaki H, Brody JA. Neurofibrillary degeneration on Guam: frequency in Chamorros and non Chamorros with no known neurological disease. Brain (1979) 102:65–77. doi:10.1093/brain/102.1.65
16. Chen L. Neurofibrillary change on Guam. Arch Neurol (1981) 38:16–8. doi:10.1001/archneur.1981.00510010042006
17. Oyanagi K, Makifuchi T, Ohtoh T, Chen KM, van der Schaaf T, Gajdusek DC, et al. Amyotrophic lateral sclerosis of Guam: the nature of the neuropathological findings. Acta Neuropathol (1994) 88:405–12. doi:10.1007/BF00389491
18. Oyanagi K, Wada M. Neuropathology of parkinsonism-dementia complex and amyotrophic lateral sclerosis of Guam: an update. J Neurol (1999) 246(Suppl 2):II19–27. doi:10.1007/BF03161078
19. Morris HR, Al-Sarraj S, Schwab C, Gwinn-Hardy K, Perez-Tur J, Wood NW, et al. A clinical and pathological study of motor neurone disease on Guam. Brain (2001) 124:2215–22. doi:10.1093/brain/124.11.2215
20. Perl DP, Hof PR, Purohit DP, Loerzel AJ, Kakulas BA. Hippocampal and entorhinal cortex neurofibrillary tangle formation in Guamanian Chamorros free of overt neurologic dysfunction. J Neuropathol Exp Neurol (2003) 62:381–8. doi:10.1093/jnen/62.4.381
21. Perl DP, Gajdusek DC, Garruto RM, Yanagihara RT, Gibbs CJ. Intraneuronal aluminum accumulation in amyotrophic lateral sclerosis and Parkinsonism-dementia of Guam. Science (1982) 217:1053–5. doi:10.1126/science.7112111
22. Spencer PS, Nunn PB, Hugon J, Ludolph AC, Ross SM, Roy DN, et al. Guam amyotrophic lateral sclerosis-parkinsonism-dementia linked to a plant excitant neurotoxin. Science (1987) 237:517–22. doi:10.1126/science.3603037
23. Steele JC. Parkinsonism-dementia complex of Guam. Mov Disord (2005) 20(Suppl 12):S99–107. doi:10.1002/mds.20547
24. Steele JC, Guella I, Szu-Tu C, Lin MK, Thompson C, Evans DM, et al. Defining neurodegeneration on Guam by targeted genomic sequencing. Ann Neurol (2015) 77:458–68. doi:10.1002/ana.24346
25. Steele JC, Wresch R, Hanlon SD, Keystone J, Ben-Shlomo Y. A unique retinal epitheliopathy is associated with amyotrophic lateral sclerosis/Parkinsonism-Dementia complex of Guam. Mov Disord (2015) 30:1271–5. doi:10.1002/mds.26264
26. Cox PA, Davis DA, Mash DC, Metcalf JS, Banack SA. Dietary exposure to an environmental toxin triggers neurofibrillary tangles and amyloid deposits in the brain. Proc Biol Sci (2016) 283:20152397. doi:10.1098/rspb.2015.2397
27. Garruto RM, Yanagihara R, Gajdusek DC. Disappearance of high-incidence amyotrophic lateral sclerosis and parkinsonism-dementia on Guam. Neurology (1985) 35:193–8. doi:10.1212/WNL.35.2.193
28. Wiederholt WC. Neuroepidemiologic research initiatives on Guam: past and present. Neuroepidemiology (1999) 18:279–91. doi:10.1159/000026223
29. Galasko D, Salmon DP, Craig U-K, Thal LJ, Schellenberg G, Wiederholt W. Clinical features and changing patterns of neurodegenerative disorders on Guam, 1997–2000. Neurology (2002) 58:90–7. doi:10.1212/WNL.58.1.90
30. Plato CC, Galasko D, Garruto RM, Plato M, Gamst A, Craig U-K, et al. ALS and PDC of Guam: forty-year follow-up. Neurology (2002) 58:765–73. doi:10.1212/WNL.58.5.765
31. Plato CC, Garruto RM, Galasko D, Craig U-K, Plato M, Gamst A, et al. Amyotrophic lateral sclerosis and parkinsonism-dementia complex of Guam: changing incidence rates during the past 60 years. Am J Epidemiol (2003) 157:149–57. doi:10.1093/aje/kwf175
32. Miklossy J, Steele JC, Yu S, McCall S, Sandberg G, McGeer EG, et al. Enduring involvement of tau, beta-amyloid, alpha-synuclein, ubiquitin and TDP-43 pathology in the amyotrophic lateral sclerosis/parkinsonism-dementia complex of Guam (ALS/PDC). Acta Neuropathol (2008) 116:625–37. doi:10.1007/s00401-008-0439-2
33. Nakano I, Hirano A. Neuron loss in the nucleus basalis of Meynert in parkinsonism-dementia complex of Guam. Ann Neurol (1983) 13:87–91. doi:10.1002/ana.410130118
34. Buée-Scherrer V, Buée L, Hof PR, Leveugle B, Gilles C, Loerzel AJ, et al. Neurofibrillary degeneration in amyotrophic lateral sclerosis/parkinsonism-dementia complex of Guam. Immunochemical characterization of tau proteins. Am J Pathol (1995) 146:924–32.
35. Winton MJ, Joyce S, Zhukareva V, Practico D, Perl DP, Galasko D, et al. Characterization of tau pathologies in gray and white matter of Guam parkinsonism-dementia complex. Acta Neuropathol (2006) 111:401–12. doi:10.1007/s00401-006-0053-0
36. Yamazaki M, Arai Y, Baba M, Iwatsubo T, Mori O, Katayama Y, et al. Alpha-synuclein inclusions in amygdala in the brains of patients with the parkinsonism-dementia complex of Guam. J Neuropathol Exp Neurol (2000) 59:585–91. doi:10.1093/jnen/59.7.585
37. Forman MS, Schmidt ML, Kasturi S, Perl DP, Lee VM-Y, Trojanowski JQ. Tau and alpha-synuclein pathology in amygdala of Parkinsonism-dementia complex patients of Guam. Am J Pathol (2002) 160:1725–31. doi:10.1016/S0002-9440(10)61119-4
38. Sebeo J, Hof PR, Perl DP. Occurrence of alpha-synuclein pathology in the cerebellum of Guamanian patients with parkinsonism-dementia complex. Acta Neuropathol (2004) 107:497–503. doi:10.1007/s00401-004-0840-4
39. Hasegawa M, Arai T, Akiyama H, Nonaka T, Mori H, Hashimoto T, et al. TDP-43 is deposited in the Guam parkinsonism-dementia complex brains. Brain (2007) 130:1386–94. doi:10.1093/brain/awm065
40. Geser F, Winton MJ, Kwong LK, Xu Y, Xie SX, Igaz LM, et al. Pathological TDP-43 in parkinsonism-dementia complex and amyotrophic lateral sclerosis of Guam. Acta Neuropathol (2008) 115:133–45. doi:10.1007/s00401-007-0257-y
41. Lagier-Tourenne C, Polymenidou M, Cleveland DW. TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum Mol Genet (2010) 19:R46–64. doi:10.1093/hmg/ddq137
42. Braun RJ, Sommer C, Carmona-Gutierrez D, Khoury CM, Ring J, Büttner S, et al. Neurotoxic 43-kDa TAR DNA-binding protein (TDP-43) triggers mitochondrion-dependent programmed cell death in yeast. J Biol Chem (2011) 286:19958–72. doi:10.1074/jbc.M110.194852
43. Scotti MM, Swanson MS. RNA mis-splicing in disease. Nat Rev Genet (2016) 17:19–32. doi:10.1038/nrg.2015.3
44. Liu G, Coyne AN, Pei F, Vaughan S, Chaung M, Zarnescu DC, et al. Endocytosis regulates TDP-43 toxicity and turnover. Nat Commun (2017) 8:2092. doi:10.1038/s41467-017-02017-x
45. Chou C-C, Zhang Y, Umoh ME, Vaughan SW, Lorenzini I, Liu F, et al. TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat Neurosci (2018) 79:416. doi:10.1038/s41593-017-0047-3
46. Leibiger C, Deisel J, Aufschnaiter A, Ambros S, Tereshchenko M, Verheijen BM, et al. TDP-43 controls lysosomal pathways thereby determining its own clearance and cytotoxicity. Hum Mol Genet (2018). doi:10.1093/hmg/ddy066
47. Gentleman SM, Perl D, Allsop D, Clinton J, Royston MC, Roberts GW. Beta (A4)-amyloid protein and parkinsonian dementia complex of Guam. Lancet (1991) 337:55–6. doi:10.1016/0140-6736(91)93378-M
48. Ito H, Hirano H, Yen SH, Kato S. Demonstration of beta amyloid protein-containing neurofibrillary tangles in parkinsonism-dementia complex on Guam. Neuropathol Appl Neurobiol (1991) 17:365–73. doi:10.1111/j.1365-2990.1991.tb00736.x
49. Guiroy DC, Mellini M, Miyazaki M, Hilbich C, Safar J, Garruto RM, et al. Neurofibrillary tangles of Guamanian amyotrophic lateral sclerosis, parkinsonism-dementia and neurologically normal Guamanians contain a 4- to 4.5-kilodalton protein which is immunoreactive to anti-amyloid beta/A4-protein antibodies. Acta Neuropathol (1993) 86:265–74. doi:10.1007/BF00304141
50. Schwab C, Steele JC, Akiyama H, McGeer EG, McGeer PL. Relationship of amyloid beta/A4 protein to the neurofibrillary tangles in Guamanian parkinsonism-dementia. Acta Neuropathol (1995) 90:287–98. doi:10.1007/BF00296513
51. Schmidt ML, Lee VM, Saido T, Perl D, Schuck T, Iwatsubo T, et al. Amyloid plaques in Guam amyotrophic lateral sclerosis/parkinsonism-dementia complex contain species of A beta similar to those found in the amyloid plaques of Alzheimer’s disease and pathological aging. Acta Neuropathol (1998) 95:117–22. doi:10.1007/s004010050774
52. Oyanagi K, Makifuchi T, Ohtoh T, Chen K-M, Gajdusek DC, Chase TN. Distinct pathological features of the Gallyas- and tau-positive glia in the parkinsonism-dementia complex and amyotrophic lateral sclerosis of Guam. J Neuropathol Exp Neurol (1997) 56:308–16. doi:10.1097/00005072-199703000-00010
53. Schwab C, Steele JC, McGeer PL. Neurofibrillary tangles of Guam parkinson-dementia are associated with reactive microglia and complement proteins. Brain Res (1996) 707:196–205. doi:10.1016/0006-8993(95)01257-5
54. McGeer PL, Schwab C, McGeer EG, Haddock RL, Steele JC. Familial nature and continuing morbidity of the amyotrophic lateral sclerosis-parkinsonism dementia complex of Guam. Neurology (1997) 49:400–9. doi:10.1212/WNL.49.2.400
55. Scheper W, Hoozemans JJM. The unfolded protein response in neurodegenerative diseases: a neuropathological perspective. Acta Neuropathol (2015) 130:315–31. doi:10.1007/s00401-015-1462-8
56. Hetz C, Saxena S. ER stress and the unfolded protein response in neurodegeneration. Nat Rev Neurol (2017) 13:477–91. doi:10.1038/nrneurol.2017.99
57. Menzies FM, Fleming A, Rubinsztein DC. Compromised autophagy and neurodegenerative diseases. Nat Rev Neurosci (2015) 16:345–57. doi:10.1038/nrn3961
58. Menzies FM, Fleming A, Caricasole A, Bento CF, Andrews SP, Ashkenazi A, et al. Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron (2017) 93:1015–34. doi:10.1016/j.neuron.2017.01.022
59. Rubinsztein DC. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature (2006) 443:780–6. doi:10.1038/nature05291
60. Hipp MS, Park S-H, Hartl FU. Proteostasis impairment in protein-misfolding and -aggregation diseases. Trends Cell Biol (2014) 24:506–14. doi:10.1016/j.tcb.2014.05.003
61. Verheijen BM, Hashimoto T, Oyanagi K, van Leeuwen FW. Deposition of mutant ubiquitin in parkinsonism-dementia complex of Guam. Acta Neuropathol Commun (2017) 5:82. doi:10.1186/s40478-017-0490-0
62. Ciechanover A, Brundin P. The ubiquitin proteasome system in neurodegenerative diseases: sometimes the chicken, sometimes the egg. Neuron (2003) 40:427–46. doi:10.1016/S0896-6273(03)00606-8
63. Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature (2003) 426:895–9. doi:10.1038/nature02263
64. Lehman NL. The ubiquitin proteasome system in neuropathology. Acta Neuropathol (2009) 118:329–47. doi:10.1007/s00401-009-0560-x
65. Dennissen FJA, Kholod N, van Leeuwen FW. The ubiquitin proteasome system in neurodegenerative diseases: culprit, accomplice or victim? Prog Neurobiol (2012) 96:190–207. doi:10.1016/j.pneurobio.2012.01.003
66. van Leeuwen FW, de Kleijn DPV, van den Hurk HH, Neubauer A, Sonnemans MAF, Sluijs JA, et al. Frameshift mutants of beta amyloid precursor protein and ubiquitin-B in Alzheimer’s and down patients. Science (1998) 279:242–7. doi:10.1126/science.279.5348.242
67. van Leeuwen FW, Burbach JP, Hol EM. Mutations in RNA: a first example of molecular misreading in Alzheimer’s disease. Trends Neurosci (1998) 21:331–5. doi:10.1016/S0166-2236(98)01280-6
68. van Leeuwen FW, Fischer DF, Kamel D, Sluijs JA, Sonnemans MAF, Benne R, et al. Molecular misreading: a new type of transcript mutation expressed during aging. Neurobiol Aging (2000) 21:879–91. doi:10.1016/S0197-4580(00)00151-2
69. Dennissen FJA, Kholod N, Hermes DJHP, Kemmerling N, Steinbusch HWM, Dantuma NP, et al. Mutant ubiquitin (UBB+1) associated with neurodegenerative disorders is hydrolyzed by ubiquitin C-terminal hydrolase L3 (UCH-L3). FEBS Lett (2011) 585:2568–74. doi:10.1016/j.febslet.2011.06.037
70. de Vrij FM, Sluijs JA, Gregori L, Fischer DF, Hermens WT, Goldgaber D, et al. Mutant ubiquitin expressed in Alzheimer’s disease causes neuronal death. FASEB J (2001) 15:2680–8. doi:10.1096/fj.01-0438com
71. Fischer DF, de Vos RAI, van Dijk R, De Vrij FMS, Proper EA, Sonnemans MAF, et al. Disease-specific accumulation of mutant ubiquitin as a marker for proteasomal dysfunction in the brain. FASEB J (2003) 17:2014–24. doi:10.1096/fj.03-0205com
72. de Pril R, Fischer DF, Maat-Schieman MLC, Hobo B, de Vos RAI, Brunt ER, et al. Accumulation of aberrant ubiquitin induces aggregate formation and cell death in polyglutamine diseases. Hum Mol Genet (2004) 13:1803–13. doi:10.1093/hmg/ddh188
73. Gentier RJ, van Leeuwen FW. Misframed ubiquitin and impaired protein quality control: an early event in Alzheimer’s disease. Front Mol Neurosci (2015) 8:47. doi:10.3389/fnmol.2015.00047
74. Krutauz D, Reis N, Nakasone MA, Siman P, Zhang D, Kirkpatrick DS, et al. Extended ubiquitin species are protein-based DUB inhibitors. Nat Chem Biol (2014) 10:664–70. doi:10.1038/nchembio.1574
75. Braun RJ, Sommer C, Leibiger C, Gentier RJG, Dumit VI, Paduch K, et al. Accumulation of basic amino acids at mitochondria dictates the cytotoxicity of aberrant ubiquitin. Cell Rep (2015) 10:1557–71. doi:10.1016/j.celrep.2015.02.009
76. Tan Z, Sun X, Hou F-S, Oh H-W, Hilgenberg LGW, Hol EM, et al. Mutant ubiquitin found in Alzheimer’s disease causes neuritic beading of mitochondria in association with neuronal degeneration. Cell Death Differ (2007) 14:1721–32. doi:10.1038/sj.cdd.4402180
77. Fischer DF, van Dijk R, van Tijn P, Hobo B, Verhage MC, van der Schors RC, et al. Long-term proteasome dysfunction in the mouse brain by expression of aberrant ubiquitin. Neurobiol Aging (2009) 30:847–63. doi:10.1016/j.neurobiolaging.2008.06.009
78. Irmler M, Gentier RJG, Dennissen FJA, Schulz H, Bolle I, Hölter SM, et al. Long-term proteasomal inhibition in transgenic mice by UBB+1 expression results in dysfunction of central respiration control reminiscent of brainstem neuropathology in Alzheimer patients. Acta Neuropathol (2012) 124:187–97. doi:10.1007/s00401-012-1003-7
79. Schipanski A, Oberhauser F, Neumann M, Lange S, Szalay B, Krasemann S, et al. Lectin OS-9 delivers mutant neuroserpin to endoplasmic reticulum associated degradation in familial encephalopathy with neuroserpin inclusion bodies. Neurobiol Aging (2014) 35:2394–403. doi:10.1016/j.neurobiolaging.2014.04.002
80. Tank EMH, True HL. Disease-associated mutant ubiquitin causes proteasomal impairment and enhances the toxicity of protein aggregates. PLoS Genet (2009) 5:e1000382. doi:10.1371/journal.pgen.1000382
81. Lam YA, Pickart CM, Alban A, Landon M, Jamieson C, Ramage R, et al. Inhibition of the ubiquitin-proteasome system in Alzheimer’s disease. Proc Natl Acad Sci U S A (2000) 97:9902–6. doi:10.1073/pnas.170173897
82. Lindsten K, De Vrij FMS, Verhoef LGGC, Fischer DF, van Leeuwen FW, Hol EM, et al. Mutant ubiquitin found in neurodegenerative disorders is a ubiquitin fusion degradation substrate that blocks proteasomal degradation. J Cell Biol (2002) 157:417–27. doi:10.1083/jcb.200111034
83. van Tijn P, De Vrij FMS, Schuurman KG, Dantuma NP, Fischer DF, van Leeuwen FW, et al. Dose-dependent inhibition of proteasome activity by a mutant ubiquitin associated with neurodegenerative disease. J Cell Sci (2007) 120:1615–23. doi:10.1242/jcs.03438
84. Verhoef LGGC, Heinen C, Selivanova A, Halff EF, Salomons FA, Dantuma NP. Minimal length requirement for proteasomal degradation of ubiquitin-dependent substrates. FASEB J (2009) 23:123–33. doi:10.1096/fj.08-115055
85. van Tijn P, Verhage MC, Hobo B, van Leeuwen FW, Fischer DF. Low levels of mutant ubiquitin are degraded by the proteasome in vivo. J Neurosci Res (2010) 88:2325–37. doi:10.1002/jnr.22396
86. Hope AD, de Silva R, Fischer DF, Hol EM, van Leeuwen FW, Lees AJ. Alzheimer’s associated variant ubiquitin causes inhibition of the 26S proteasome and chaperone expression. J Neurochem (2003) 86:394–404. doi:10.1046/j.1471-4159.2003.01844.x
87. van Tijn P, Dennissen FJA, Gentier RJG, Hobo B, Hermes D, Steinbusch HWM, et al. Mutant ubiquitin decreases amyloid β plaque formation in a transgenic mouse model of Alzheimer’s disease. Neurochem Int (2012) 61:739–48. doi:10.1016/j.neuint.2012.07.007
88. Lee J-G, Takahama S, Zhang G, Tomarev SI, Ye Y. Unconventional secretion of misfolded proteins promotes adaptation to proteasome dysfunction in mammalian cells. Nat Cell Biol (2016) 18:765–76. doi:10.1038/ncb3372
89. Komander D, Rape M. The ubiquitin code. Annu Rev Biochem (2012) 81:203–29. doi:10.1146/annurev-biochem-060310-170328
90. Yau R, Rape M. The increasing complexity of the ubiquitin code. Nat Cell Biol (2016) 18:579–86. doi:10.1038/ncb3358
91. Kwon YT, Ciechanover A. The ubiquitin code in the ubiquitin-proteasome system and autophagy. Trends Biochem Sci (2017) 42:873–86. doi:10.1016/j.tibs.2017.09.002
92. Steele JC, Caparros-Lefebvre D, Lees AJ, Sacks OW. Progressive supranuclear palsy and its relation to pacific foci of the parkinsonism-dementia complex and Guadeloupean parkinsonism. Parkinsonism Relat Disord (2002) 9:39–54. doi:10.1016/S1353-8020(02)00043-3
93. Dickson DW. Parkinson’s disease and parkinsonism: neuropathology. Cold Spring Harb Perspect Med (2012) 2:a009258. doi:10.1101/cshperspect.a009258
94. Jansen AHP, Reits EAJ, Hol EM. The ubiquitin proteasome system in glia and its role in neurodegenerative diseases. Front Mol Neurosci (2014) 7:73. doi:10.3389/fnmol.2014.00073
95. Choi K, Park J, Lee J, Han EC, Choi C. Mutant ubiquitin attenuates interleukin-1β- and tumor necrosis factor-α-induced pro-inflammatory signaling in human astrocytic cells. PLoS One (2013) 8:e67891. doi:10.1371/journal.pone.0067891
96. Yim N, Ryu S-W, Han EC, Yoon J, Choi K, Choi C. Mutant ubiquitin UBB+1 induces mitochondrial fusion by destabilizing mitochondrial fission-specific proteins and confers resistance to oxidative stress-induced cell death in astrocytic cells. PLoS One (2014) 9:e99937. doi:10.1371/journal.pone.0099937
97. Lowe J, McDermott H, Landon M, Mayer RJ, Wilkinson KD. Ubiquitin carboxyl-terminal hydrolase (PGP 9.5) is selectively present in ubiquitinated inclusion bodies characteristic of human neurodegenerative diseases. J Pathol (1990) 161:153–60. doi:10.1002/path.1711610210
98. Zouambia M, Fischer DF, Hobo B, de Vos RAI, Hol EM, Varndell IM, et al. Proteasome subunit proteins and neuropathology in tauopathies and synucleinopathies: consequences for proteomic analyses. Proteomics (2008) 8:1221–36. doi:10.1002/pmic.200700679
99. Leroy E, Boyer R, Auburger G, Leube B, Ulm G, Mezey E, et al. The ubiquitin pathway in Parkinson’s disease. Nature (1998) 395:451–2. doi:10.1038/26652
100. Bilguvar K, Tyagi NK, Ozkara C, Tuysuz B, Bakircioglu M, Choi M, et al. Recessive loss of function of the neuronal ubiquitin hydrolase UCHL1 leads to early-onset progressive neurodegeneration. Proc Natl Acad Sci U S A (2013) 110:3489–94. doi:10.1073/pnas.1222732110
101. Saigoh K, Wang YL, Suh JG, Yamanishi T, Sakai Y, Kiyosawa H, et al. Intragenic deletion in the gene encoding ubiquitin carboxy-terminal hydrolase in gad mice. Nat Genet (1999) 23:47–51. doi:10.1038/12647
102. Tashiro Y, Urushitani M, Inoue H, Koike M, Uchiyama Y, Komatsu M, et al. Motor neuron-specific disruption of proteasomes, but not autophagy, replicates amyotrophic lateral sclerosis. J Biol Chem (2012) 287:42984–94. doi:10.1074/jbc.M112.417600
103. Przedborski S. The two-century journey of Parkinson disease research. Nat Rev Neurosci (2017) 18:251–9. doi:10.1038/nrn.2017.25
104. McNaught KSP, Perl DP, Brownell A-L, Olanow CW. Systemic exposure to proteasome inhibitors causes a progressive model of Parkinson’s disease. Ann Neurol (2004) 56:149–62. doi:10.1002/ana.20186
106. Labbadia J, Morimoto RI. The biology of proteostasis in aging and disease. Annu Rev Biochem (2015) 84:435–64. doi:10.1146/annurev-biochem-060614-033955
107. Klaips CL, Jayaraj GG, Hartl FU. Pathways of cellular proteostasis in aging and disease. J Cell Biol (2018) 217:51–63. doi:10.1083/jcb.201709072
108. Hartl FU, Bracher A, Hayer-Hartl M. Molecular chaperones in protein folding and proteostasis. Nature (2011) 475:324–32. doi:10.1038/nature10317
109. Ciechanover A, Kwon YT. Protein quality control by molecular chaperones in neurodegeneration. Front Neurosci (2017) 11:2491. doi:10.3389/fnins.2017.00185
110. Kopito RR. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol (2000) 10:524–30. doi:10.1016/S0962-8924(00)01852-3
111. Lee B-H, Lee MJ, Park S, Oh D-C, Elsasser S, Chen P-C, et al. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature (2010) 467:179–84. doi:10.1038/nature09299
112. Myeku N, Clelland CL, Emrani S, Kukushkin NV, Yu WH, Goldberg AL, et al. Tau-driven 26S proteasome impairment and cognitive dysfunction can be prevented early in disease by activating cAMP-PKA signaling. Nat Med (2016) 22:46–53. doi:10.1038/nm.4011
113. Myeku N, Duff KE. Targeting the 26S proteasome to protect against proteotoxic diseases. Trends Mol Med (2018) 24:18–29. doi:10.1016/j.molmed.2017.11.006
114. Ciechanover A, Kwon YT. Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. Exp Mol Med (2015) 47:e147. doi:10.1038/emm.2014.117
115. Hernebring M, Brolén G, Aguilaniu H, Semb H, Nyström T. Elimination of damaged proteins during differentiation of embryonic stem cells. Proc Natl Acad Sci U S A (2006) 103:7700–5. doi:10.1073/pnas.0510944103
116. Bohnert KA, Kenyon C. A lysosomal switch triggers proteostasis renewal in the immortal C. elegans germ lineage. Nature (2017) 551:629–33. doi:10.1038/nature24620
117. Melentijevic I, Toth ML, Arnold ML, Guasp RJ, Harinath G, Nguyen KC, et al. C. elegans neurons jettison protein aggregates and mitochondria under neurotoxic stress. Nature (2017) 542:367–71. doi:10.1038/nature21362
118. Kuzuhara S, Kokubo Y, Sasaki R, Narita Y, Yabana T, Hasegawa M, et al. Familial amyotrophic lateral sclerosis and parkinsonism-dementia complex of the Kii Peninsula of Japan: clinical and neuropathological study and tau analysis. Ann Neurol (2001) 49:501–11. doi:10.1002/ana.100
119. Kuzuhara S, Kokubo Y. Atypical parkinsonism of Japan: amyotrophic lateral sclerosis-parkinsonism-dementia complex of the Kii peninsula of Japan (Muro disease): an update. Mov Disord (2005) 20(Suppl 12):S108–13. doi:10.1002/mds.20548
120. Mimuro M, Yoshida M, Kuzuhara S, Kokubo Y. Amyotrophic lateral sclerosis and parkinsonism-dementia complex of the Hohara focus of the Kii Peninsula: a multiple proteinopathy? Neuropathology (2018) 38:98–107. doi:10.1111/neup.12434
121. Gajdusek DC, Salazar AM. Amyotrophic lateral sclerosis and parkinsonian syndromes in high incidence among the Auyu and Jakai people of West New Guinea. Neurology (1982) 32:107–26. doi:10.1212/WNL.32.8.919-b
Keywords: Guam parkinsonism–dementia complex, mutant ubiquitin, protein quality control, protein aggregation, tau, neurofibrillary tangles, 43-kDa TAR DNA-binding protein, neuropathology
Citation: Verheijen BM, Oyanagi K and van Leeuwen FW (2018) Dysfunction of Protein Quality Control in Parkinsonism–Dementia Complex of Guam. Front. Neurol. 9:173. doi: 10.3389/fneur.2018.00173
Received: 30 December 2017; Accepted: 06 March 2018;
Published: 20 March 2018
Edited by:
Cintia Roodveldt, Centro Andaluz de Biología Molecular y Medicina Regenerativa (CABIMER), SpainReviewed by:
Arianna Bellucci, University of Brescia, ItalyCarlos M. Opazo, University of Melbourne, Australia
Copyright: © 2018 Verheijen, Oyanagi and van Leeuwen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bert M. Verheijen, l.m.verheijen-3@umcutrecht.nl