 Hongye Zhao1,2Tiezheng Zheng1Xiaohan Yang1Ming Fan3Lingling Zhu3Shuhong Liu3Liying Wu3
Hongye Zhao1,2Tiezheng Zheng1Xiaohan Yang1Ming Fan3Lingling Zhu3Shuhong Liu3Liying Wu3 Changkai Sun1,4*
Changkai Sun1,4*- 1Department of Physiology and Key Laboratory of Brain Diseases of Liaoning Province, School of Basic Medical Sciences, Dalian Medical University, Dalian, China
- 2Department of Physiology, School of Basic Medical Sciences, Qiqihar Medical University, Qiqihar, China
- 3Department of Brain Protection and Plasticity, Institute of Basic Medical Sciences, Academy of Military Medical Sciences, Beijing, China
- 4Department of Biomedical Engineering, Faculty of Electronic Information and Electrical Engineering & Research Center for the Control Engineering of Translational Precision Medicine, Dalian University of Technology, Dalian, China
Cryptotanshinone (CTs), an active component isolated from the root of Salvia miltiorrhiza (SM), has been shown to exert potent neuroprotective property. We here established an oxygen-glucose deprivation/recovery (OGD/R)-injured Neurovascular Unit (NVU) model in vitro to observe the neuroprotective effects of CTs on cerebral ischemia/reperfusion injury (CIRI), and explore the underlying mechanisms. CTs was observed to significantly inhibit the OGD/R-induced neuronal apoptosis, and decease the activation of Caspase-3 and the degradation of poly-ADP-ribose polymerase (PARP), as well as the increase of Bax/Bcl-2 ratio in neurons under OGD/R condition. The inhibitory effects of CTs on neuron apoptosis were associated with the blocking of mitogen-activated protein kinase (MAPK) signaling pathway. CTs also remarkably ameliorated OGD/R-induced reduction of transepithelial electrical resistance (TEER) values and the increase of transendothelial permeability coefficient (Pe) of sodium fluorescein (SF) by upregulating the expression of ZO-1, Claudin-5, and Occludin in brain microvascular endothelial cells (BMECs), which might be related to the down-regulation of matrix metalloproteinase (MMP)-9 expression. Based on these findings, CTs may play a neuroprotective role in OGD/R injure in NVU models in vitro by inhibiting cell apoptosis and alleviating the damage of blood-brain barrier (BBB).
Introduction
Stroke is the second leading cause of morbidity and mortality worldwide, with acute ischemic stroke (AIS) making up more than 80% of all the cases (1). AIS is caused by sudden interruption of artery supplying blood to the brain, usually resulting in a disorder of nervous system. Rapid restoration of blood supply is a critical therapeutic strategy for AIS, but it can bring secondary damage and further lead to more serious disturbance in the function of nervous system, called cerebral ischemia/reperfusion injury (CIRI). CIRI initiates a complex cascade of events, such as intracellular calcium overload, glutamate exitotoxicity, free radicals accumulation, excessive release of inflammatory mediators, DNA damage, blood-brain barrier (BBB) disruption, and apoptosis (2). Therefore, not only is CIRI involved in neurons but also in microvessels and gliacytes (3).
Hunting for an effective therapy for CIRI constitutes a challenging task in neuroscience for decades. Neuroprotection remains the central focus of CIRI treatment. Many mechanisms are involved in the origination and development of CIRI. Hence, neuroprotection targeting to a single therapeutic target is invalid. Based on this, LO EH and his colleagues proposed Neurovascular Unit (NVU) in 2003 (4). NVU is regarded as the basic structural and functional unit of brain, and mainly including neurons, astrocytes, microglia, microvascular endothelial cells, pericytes, even with basement membrane, and extracellular matrix. This concept not only shows the interactions among these types of cells, but also reflects their roles in the origination and development of brain diseases (5). So, NVU has become an important model for studying multi-target and multi-level therapy for brain diseases.
We here established an in vitro model of NVU, as described in previous reports (6). It is a co-culture system made up of three kinds of rat primary cells including brain microvascular endothelial cells (BMECs), astrocytes and neurons. This model can be used in brain research, potential drug targets screening and therapeutic drug discovery.
Salvia miltiorrhiza (SM) root has been commonly used to treat cardiovascular and cerebrovascular diseases in China and other Asian countries (7, 8). Cryptotanshinone (CTs), one of the major tanshinones isolated from the root of SM, is a kind of lipophilic compound and can pass through BBB (9, 10). CTs has various biological activities, such as anti-oxidation, anti-inflammation, anti-tumor, anti-apoptosis, anti-platelet aggregation activities, and so on (11–14). The previous studies demonstrated CTs possessed protective effects on the ischemic damage of multiple organs (15) and has the potential protective effects for CIRI (16). However, the protective effects of CTs on CIRI have not yet been confirmed, and its exact mechanism is unknown.
Oxygen-glucose deprivation/recovery (OGD/R) model is the most widely used in studies of CIRI in vitro (17), and many typical pathologic changes in CIRI were observed on OGD/R-injured NVU model (3). In the present study, we successfully established an OGD/R-injured NVU model in vitro to elucidate the potential protective effects of CTs on CIRI and explore its underlying mechanisms.
Materials and Methods
Animals
Sprague-Dawley (SD) rats were obtained from the Experimental Animal Center and housed in the Experimental Animal Center, Academy of Military Medical Sciences (Beijing, China). Newborn rats were sacrificed for isolating the primary cerebral astrocytes and neurons, and 120–150 g male rats were sacrificed for isolating the primary BMECs. All experiments followed an institutionally approved protocol in accordance with the China's Guidelines for Care and Use of Laboratory Animals.
Preparation of CTs
CTs (Mw: 296.35, purity ≥98%) was purchased from the National Institute for Food and Drug Control (Beijing, China). CTs was dissolved in dimethyl sulfoxide (DMSO, Sigma-Aldrich, USA) to prepare for the stock solution with a concentration of 100 mM. The final concentration of DMSO in the testing solution was <0.1% (v/v) to prevent possible cytotoxicity.
Reagents
MCDB 131 medium, DMEM, neurobasal A medium, microvascular growth supplement (MVGS) and B-27 were purchased from GIBCO (Thermo Fisher Scientific Inc., USA). DNase I, collagen type I and sodium fluorescein (Mw: 376.27, SF) were purchased Sigma (Sigma-Aldrich Co. LLC., USA). CCK-8 was purchased from Dojindo (Dojindo Laboratories, Japan). In situ cell death detection Kit (Fluorescein) and collagenase/dispase were purchased from Roche (Roche Applied Science, Germany). Clarity Western ECL Substrate kit were purchased from Bio-Rad (Bio-Rad Laboratories, Inc., USA). All the antibodies used in this research were purchased from CST (Cell Signaling Technology, Inc., USA), except the antibodies specific for ZO-1, Claudin-5, Occludin, MMP-2 and MMP-9 were from Abcam [Abcam (Shanghai) trading Co., Ltd., China].
Isolation and Purification of Three Types of Rat Cerebral Cells
Primary rat BMECs were obtained from the cerebral cortex of 120–150 g rats according to previous reports (6) with some improvements. Primary rat cerebral astrocytes and neurons were obtained from cerebral cortex of 24 h newborn rats as previous reports (18) (Appendix 1).
The purified BMECs, astrocytes and neurons were used to establish the in vitro NVU model (Appendix 2).
Establishment of OGD/R-Injured NVU Model in vitro
At first, we established a NVU model in vitro by referring to previous reports (6, 19) with a slight modification (Appendix 3).
The OGD/R treatment on NVU models in vitro were performed as previous descriptions (6). Briefly, the prepared NVU models in vitro were subsequently transferred into an anaerobic incubator (Coy laboratory, USA) with condition of 95% N2 and 5% CO2 at 37°C. Within the anaerobic incubator, the cell culture mediums were replaced with oxygen/glucose-free balanced DMEM without serum, which were previously saturated with 95% N2 and 5% CO2 at 37°C for 3 h. After OGD treatment for 2 h, the NVU models in vitro were switched to the normoxic incubator with high-glucose DMEM without serum for 24 h.
Experimental Groups and Treatment
NVU models in vitro were randomly divided into 4 groups of Control, OGD2h/R24h and two doses of CTs (2.5 and 5.0 μM). Except for the Control group, each group was exposed to OGD2h/R24h. Drug treatment groups were treated with CTs (2.5 and 5.0 μM) for 3 h before OGD and during the OGD period (CTs was added into the culture mediums of insert). The experimental condition was established by a preliminary study involving different concentrations of CTs (0.32–10.0 μM) at different (3 and 24 h) pre- and post-hypoxia incubation periods (Appendix 4).
Detection of Cell Viability
The cell viability of neurons was evaluated by cell counting kit-8 (CCK-8). The values were expressed as the percentage of living cells.
The cell viabilities of BMECs and astrocytes were tested by trypan blue stain after digestion with 0.25% trypsin-EDTA. The values were expressed as cells/cm2.
TUNEL Assay
TUNEL assay was performed to analyze neuron apoptosis according to the manufacturer's instructions using In situ Cell Death Detection Kit. Finally, images were captured on a fluorescence microscope at × 100 and × 400 magnification. The neurons with green fluorescence were described as apoptotic neurons. The number of apoptotic neurons per view was counted using microscopy at × 400 magnification.
Permeability Measurement of BBB by TEER and SF
The integrity of the BBB was measured via the transepithelial electrical resistance (TEER) assay using a Millicell ERS-2 Voltohmmeter (Millipore, USA) according to the protocol provided by the manufacturer. The TEER value of cell-free well was regarded as background TEER value. The final TEER value = (sample TEER value–background TEER value) × the area of insert membrane (4.52 cm2). The values were expressed as Ω × cm2.
Transendothelial permeability coefficient (Pe) of SF was performed as previously described (20). Briefly, the culture mediums in the inserts were switched to 1 ml Ringer-Hepes buffer containing a final concentration of 10 μg/ml SF. The inserts were transferred after 5, 15, 30, and 60 min to a new well with 2.5 ml Ringer-Hepes buffer, respectively. Hundred microliter culture mediums under the insert membrane were taken out at each observing time. The absorbance was measured by fluorospectrophotometer (Fluoroskan Ascent FL, Thermo Fisher Scientific Inc., USA, excitation wavelength: 485 nm; emission wavelength: 535 nm). The absorbance of cell-free well was regarded as background absorbance. Pe was calculated as previously described (21).
Western Blotting
BMECs and neurons from NVU models were collected and lysed by RIPA buffer containing protease inhibitor, respectively. Protein concentration was determined by the BCA protein assay kit. Equivalent amounts of proteins from each group were subjected to SDS-PAGE gel electrophoresis, and transferred onto polyvinylidene fluoride (PVDF) membranes (Millipore, USA). After being blocked with 5% non-fat milk in TBST buffer for 1 h, the PVDF membranes were incubated with primary antibody at 4°C overnight. The primary antibodies used in this study were rabbit anti-Caspase-3 (1:1,000), rabbit anti-PARP (1:1,000), rabbit anti-Bax (1:1,000), rabbit anti-Bcl-2 (1:1,000), rabbit anti-p-ERK1/2 (1:1,000), rabbit anti-ERK1/2 (1:1,000), rabbit anti-p-JNK (1:1,000), rabbit anti-JNK (1:1,000), rabbit anti-p-p38 (1:1,000), rabbit anti-p38 (1:1,000), rabbit anti-ZO-1 (1:1,000), rabbit anti-Occludin (1:1,000), mouse anti-Claudin-5(1:2,000), rabbit anti-MMP-2 (1:1,000), rabbit anti-MMP-9 (1:1,000), rabbit anti-β-actin (1:1,000). After three washes with TBST buffer, the membranes were incubated with goat anti-mouse HRP or goat anti-rabbit HRP-conjugated IgG secondary antibodies (1:3,000) for 1 h each at room temperature. Protein bands were visualized with an Clarity Western ECL Substrate kit. The density of the bands was quantified using Image J software (National Institutes of Health, USA).
Statistical Analysis
Data were expressed as the mean ± SD from at least three independent experiments. Data were analyzed by one-way analysis of variance (ANOVA) followed by Tukey's post hoc test using GraphPad Prism 6.0 software (GraphPad, La Jolla, CA, USA). Significant difference was accepted at P < 0.05.
Results
The Effects of CTs on Cell Viability and Cellular Apoptosis in Neurons in OGD/R-Injured NVU Model in vitro
Compared with the neurons in control group, cell viability was significantly reduced to 47.35 ± 3.80% after OGD/R treatment (P < 0.01). CTs pre-treatment was able to increase the cell viability in neurons. The cell viability in CTs 2.5 and 5.0 μM group neurons were respectively, 54.10 ± 4.54% and 60.56 ± 5.01% (P < 0.05 and P < 0.01, Figure 1A). Meanwhile, the neuronal apoptosis was detected by TUNEL assay after OGD/R. In Control group, only about 8.50 ± 1.30% neurons were of apoptotic phenotype. This phenotype of neurons was remarkably increased to 51.73 ± 3.99% in the OGD2h/R24h group (P < 0.01). Pre-treatment with CTs 2.5 and 5.0 μM could protect against OGD/R induced apoptosis. The apoptotic neurons were significantly decreased to 45.00 ± 4.44% and 39.33 ± 2.78% respectively (P < 0.05 and P < 0.01, Figures 1B,C).
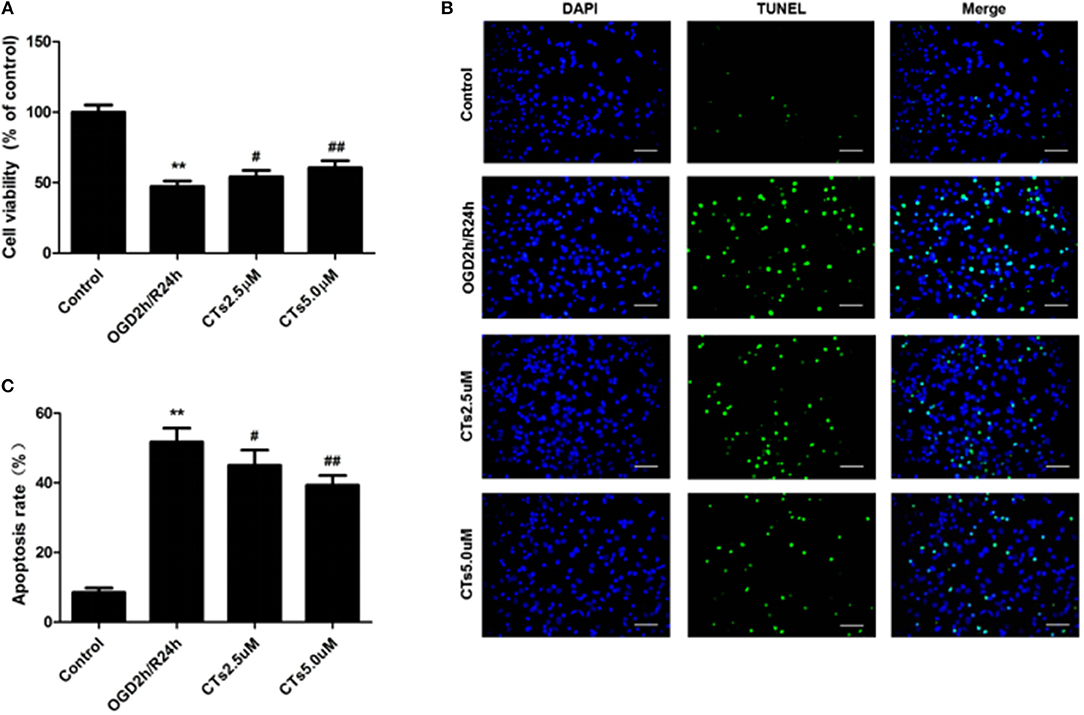
Figure 1. The effects of CTs on cell viability and cellular apoptosis in neurons in OGD/R-injured NVU model in vitro. (A) Cell viability was measured using the CCK-8 assay (n = 4). The cell viability of neurons was calculated by dividing the optical density of samples with the optical density of control. (B) Representative photomicrographs of apoptotic neurons were determined by TUNEL staining. Green fluorescence shows TUNEL-positive nuclei; blue fluorescence shows nuclei of total neurons. Scale bars: 25 μm. (C) The neuronal apoptosis rate was presented as the percentage of TUNEL-positive neurons. The percentage of TUNEL-positive neurons was calculated by dividing the number of TUNEL positive apoptotic neurons with the total number of neurons in 5 high-power fields. Data are presented as the mean ± SD. **P < 0.01 vs. Control group; #P < 0.05, ##P < 0.01 vs. OGD2h/R24h group. CTs, cryptotanshinone.
The Effects of CTs on Apoptosis-Related Proteins Expressions in Neurons in OGD/R-Injured NVU Model in vitro
The effect of CTs pre-treatment on apoptosis-related proteins expressions were confirmed by western blotting. As shown in Figure 2, it is clear that the Caspase-3 activation, PARP degradation and Bax/Bcl-2 ratio were significantly increased in neurons after OGD/R damage (all P < 0.01). However, the increased Caspase-3 activation, PARP degradation and Bax/Bcl-2 ratio were reduced by pre-treatment with CTs 2.5 and 5.0 μM (all P < 0.05 and P < 0.01).
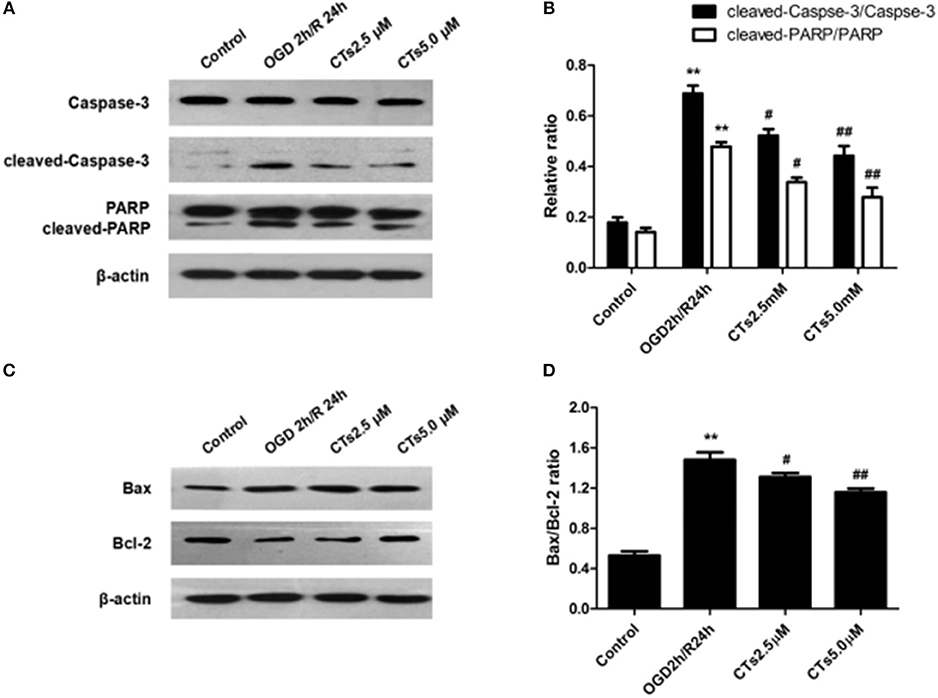
Figure 2. The effects of CTs on apoptosis-related proteins expressions in neurons in OGD/R-injured NVU model in vitro. (A) Representative Western blots showing the levels of Caspase-3, cleaved-Caspase-3(17, 19 kD), PARP and cleaved-PARP in neurons. (B) Relative density refers to the ratio of cleaved-Caspase-3(17, 19 kD) to Caspase-3 and cleaved-PARP to PARP (n = 4). (C) Representative Western blots showing the levels of Bax and Bcl-2 in neurons. (D) Relative density refers to the ratio of Bax to Bcl-2 (n = 4). The semiquantitative analyse results indicated that CTs protected neurons from apoptosis induced by OGD/R. Data are presented as the mean±SD. **P < 0.01 vs. Control group; #P < 0.05, ##P < 0.01 vs. OGD2h/R24h group. PARP, poly-ADP-ribose polymerase; Bcl-2, B-cell lymphoma 2; Bax, Bcl-2-associated X protein; CTs, cryptotanshinone.
CTs Regulated the MAPK Signaling Pathways in Neurons in OGD/R-Injured NVU Model in vitro
To assess whether CT pre-treatment would modulate the mitogen-activated protein kinase (MAPK) signaling pathways in vitro, we also examined the expression of total proteins [total extracellular regulated protein kinases1/2 (t-ERK1/2), total c-Jun N-terminal kinase (t-JNK), and total p38 mitogen-activated protein kinases (t-p38 MAPK)] and their phosphorylation (p-ERK1/2, p-JNK1/2, and p-p38 MAPK) on this pathway by western blotting. The results showed that there was a significant upregulation of p-ERK1/2, p-JNK, and p-p38 MAPK in neurons after OGD/R treatment (all P < 0.01). Pre-treatment with CTs 2.5 and 5.0 μM significantly downregulated p-ERK1/2, p-JNK, and p-p38 MAPK (all P < 0.05 and P < 0.01, Figures 3A,B).
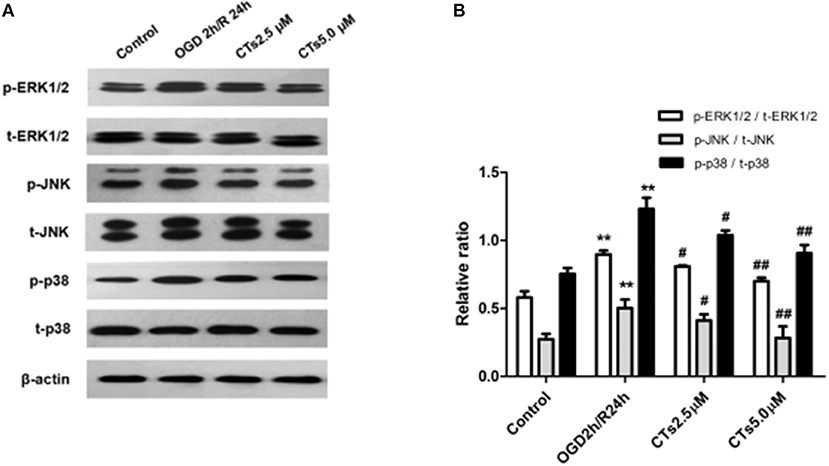
Figure 3. The effects of CTs on the MAPK signaling pathways in neurons in OGD/R-injured NVU model. (A) Representative Western blots showing the levels of p-ERK1/2, p-JNK, p-p38, t-ERK1/2, t-JNK, and t-p38 in neurons. (B) Relative density refers to the ratio of p-ERK1/2 to t-ERK1/2, p-JNK to t-JNK and p-p38 to t-p38 (n = 4). The semiquantitative analyses suggested that CTs pre-treatment obviously reduced the phosphorylation of ERK1/2, JNK and p38 in OGD/R injury neurons, thus regulated cell apoptosis. Data are presented as the mean ± SD. **P < 0.01 vs. Control group; #P < 0.05, ##P < 0.01 vs. OGD2h/R24h group. ERK1/2, extracellular regulated protein kinases1/2; JNK, c-Jun N-terminal kinase; p38, p38 mitogen-activated protein kinases; CTs, cryptotanshinone.
CTs Improved the BBB Function in OGD/R-Injured NVU Model in vitro
BMECs and astrocytes are major cells that that comprise the blood-brain barrier. The TEER values and the Pe of SF can reflect paracellular permeability of BMECs. After OGD/R treatment, compared with Control group, the numbers of survival BMECs and astrocytes (both P < 0.01, Figures 4A,B) and the TEER value (P < 0.01, Figure 4C) significantly decreased, while Pe of SF (P < 0.01, Figure 4D) increased significantly, demonstrating the BBB integrity was destroyed. The above changes could be markedly reversed by pre-treatmet with CTs 2.5 and 5.0 μM (all P < 0.05 and P < 0.01).
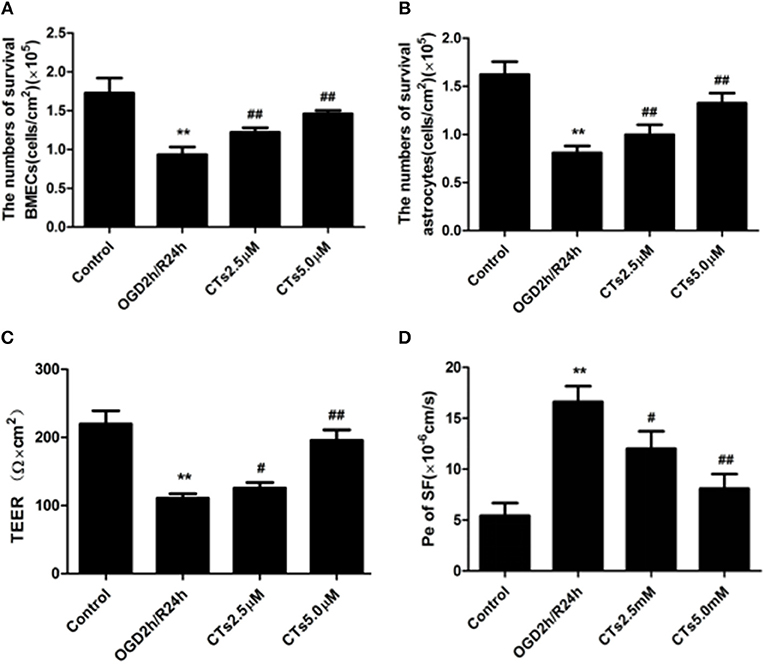
Figure 4. CTs improved the BBB function in OGD/R-injured NVU model. (A,B) The numbers of survival BMECs and astrocytes were detected by trypan blue stain.The values were expressed as cells/cm2 (n = 4). (C) TEER value was measured using a Millicell ERS-2 Voltohmmeter. The values were calculated according to the corresponding formula, and were expressed as Ω × cm2 (n = 4). (D) Pe of SF was calculated according to the corresponding methods. Pe were expressed as × 10−6cm/s (n = 4). The detection results showed CTs had an effect of protecting BBB function from OGD/R injury. Data are presented as the mean ± SD. **P < 0.01 vs. Control group; #P < 0.05, ##P < 0.01 vs. OGD2h/R24h group. BMECs, brain microvascular endothelial cells; TEER, transepithelial electrical resistance; Pe, permeability coefficient; SF, sodium fluorescein; CTs, cryptotanshinone.
The Effects of CTs on Tight Junction Proteins and Matrix Metalloproteinases in BMECs in OGD/R-Injured NVU Model in vitro
To determine the effects of CTs pre-treatment on the tight junction proteins (TJPs) between endothelial cells, the levels of tight junction proteins (TJPs), ZO-1, Claudin-5 and Occludin, in BMECs were determined by Western blotting. As shown in Figures 5A,B, the protein expression levels of ZO-1, Claudin-5 and Occludin were significantly decreased after OGD/R treatment (all P < 0.01). Pre-treatmet with CTs 2.5 and 5.0 μM up-regulated obviously all of the expression levels of those proteins (all P < 0.05 and P < 0.01). Moreover, to explore the potential mechanisms of CTs on the protection of BBB function, the expression of matrix metalloproteinase-2 and -9 (MMP-2 and -9) were also examined by western blotting in the BMECs. The western blotting results exhibited that the expression levels of MMP-2 and -9 obviously increased after OGD/R injury (both P < 0.01). The expression levels of MMP-9 significantly decreased in CTs 2.5 and 5.0 μM group (P < 0.05 and P < 0.01, Figures 5C,D). The expression of MMP-2 decreased in CTs 2.5 and 5.0 μM group although the difference was not significant (P >0.05).

Figure 5. The effects of CTs on tight junction proteins and matrix metalloproteinase in BMECs in OGD/R-injured NVU model. (A) Representative Western blots showing the levels of ZO-1, Claudin-5, and Occludin in BMECs. (B) Relative density refers to the ratio of ZO-1, Claudin-5, and Occludin to β-actin. (C) Representative Western blots showing the levels of MMP-2 and MMP-9 in BMECs. (D) Relative density refers to the ratio of MMP-2 and MMP-9 to β-actin (n = 4). The semiquantitative analyses suggested CTs upregulated the expression of tight junction proteins by downregulating the expression of MMP-9 in OGD/R injury BMECs. Data are presented as the mean ± SD. **P < 0.01 vs. Control group; #P < 0.05, ##P < 0.01 vs. OGD2h/R24h group. BMECs, brain microvascular endothelial cells; ZO-1, Zonula occludens-1; MMP-2, matrix metalloproteinase-2; MMP-9, matrix metalloproteinase-9; CTs, cryptotanshinone.
Discussion
AIS is induced by temporary occlusion of cerebral artery supplying blood. Reperfusion remains the critical therapeutic strategy for limiting brain injury following AIS, but the rapid restoration of blood flow is frequently associated with a serious secondary brain injury, called CIRI (22). AIS, particularly CIRI, triggers multiple cell signaling pathways in the brain, which may lead to neuron survival or damage (23, 24). However, the mechanisms involving neuronal fate following CIRI are complex and not fully clear. There is increasing evidence to show that cell apoptosis and disruption of the BBB play vital roles in neuron damage after CIRI (25–27).
CTs, one of the main active components of SM root, has already been shown to exert potent neuroprotective and antiapoptotic properties (28, 29). CTs could attenuates CIRI through inhibiting thrombosis formation, platelet aggregation and activation of PLC/PKC signaling pathway (8). CTs protected primary cortical neurons from glutamate-induced neurotoxicity through the activation of PI3K/Akt signaling pathway (9). Recent evidence has further shown that CTs exhibits a protective effect against cerebral stroke through suppressing the PI3K/AKT-eNOS signaling pathway (16). Taken together, these studies clearly show that CTs protects against CIRI and suggest that modulation of anti-apoptotic signaling cascades might be one of the molecular mechanisms of its neuroprotective effect.
In this study, we used OGD/R-injured NVU model in vitro to further investigate the neuroprotective effects of CTs on CIRI as well as the underlying mechanisms by focusing on MAPK signaling pathways, as these pathways were closely related to cell survival and apoptosis (Figure 6). In line with previous studies, our results indicated that CTs had significant neuroprotective effects against CIRI.
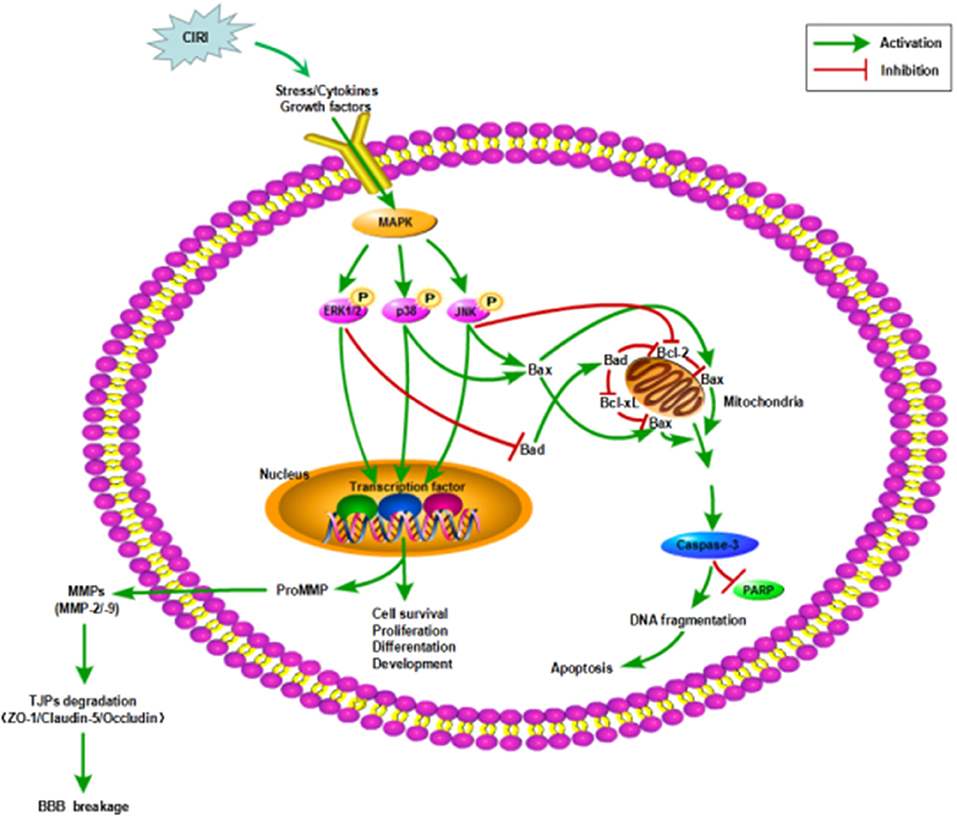
Figure 6. Mechanisms in CIRI inducing neuronal apoptosis and BBB disruption by activiating MAPK signaling pathways. AIS may lead to CIRI causing the release of proinflammatory cytokines and free radicals at the neurovascular unit activating MAPK signaling pathways. MAPK signaling pathways, including ERK1/2, JNK and p38 MAPK, participate in the regulation of neuronal survival or apoptosis and the activation of MMPs. Then MMPs activation disrupts the BBB integrity by degrading TJPs. ZO-1, Claudin-5 and Occludin are three of the important TJPs associated proteins. AIS, acute ischemic stroke; CIRI, cerebral ischemia/reperfusion injury; BBB, blood-brain barrier; MAPK, mitogen-activated protein kinase signaling pathways; ERK1/2, extracellular regulated protein kinases1/2; JNK, c-Jun N-terminal kinase; p38, p38 mitogen-activated protein kinases; Bcl-2, B-cell lymphoma 2; Bcl-xL, B-cell lymphoma-extra large; Bax, Bcl-2-associated X protein; Bad, Bcl-2-associated death protein; PARP, poly-ADP-ribose polymerase; TJPs, tight junctions proteins; ZO-1, Zonula occludens-1; MMP-2/-9, matrix metalloproteinase-2/-9.
Cell apoptosis is a major characteristic of CIRI. Reducing neuronal apoptosis could minimize or even prevent the occurrence of CIRI (25). In this study, we found that CTs pre-treatment could decrease the apoptotic rate in the neurons of NVU model in vitro exposed to OGD/R. Meanwhile, we further investigated the Bax/Bcl-2 ratio. The Bcl-2 family member Bax was markedly up-regulated after OGD/R, which resulted in the release of cytochrome c to the cytosol. Bcl-2 exerts the anti-apoptosis efficiency through inhibiting the function of Bax (30, 31). We found that the OGD/R-induced up-regulation of Bax/Bcl-2 ratio could be reversed by CTs pre-treatment. Moreover, we analyzed the activation of Caspase-3 and the degradation of PARP. Cytochrome c release triggers the cleavage of Caspase-3 to activate the Caspase-3 protein, which results in DNA fragmentation and cell apoptosis by cleaving PARP (31, 32). Our data revealed that pre-treatment with CTs prevented the OGD/R-induced increase in cleaved-Caspase-3 and cleaved-PARP. Our findings suggested that CTs could protect neurons from apoptosis induced by OGD/R.
Neuronal damage after CIRI is usually caused by oxidative stress, inflammation response, or mitochondrial dysfunction, and activates ultimately an apoptotic cascade. MAPK signaling pathways, as both targets and mediators of CIRI, are involved in neuronal survival or apoptosis regulation after stroke (33–35). Thus, inhibiting or regulating the expression and activity of MAPK signalings may constitute novel therapeutic strategies for CIRI (36, 37). Recent studies have found that several drugs might play protective roles in CIRI by inhibition of MAPK signaling pathways (38, 39). The MAPK family includes extracellular signal-regulated kinase 1/2 (ERK1/2), c-Jun amino terminal kinase (JNK) and p38 MAPK. These pathways are activated through phosphorylation by upstream kinases recruited through diverse extracellular signaling events. Our results demonstrated that the phosphorylation of ERK1/2, JNK, and p38 MAPK was significantly increased in the neurons of NVU model in vitro exposed to OGD/R. Pre-treatment with CTs resulted in decreased phosphorylation of all the three molecules. These results suggested that MAPK signaling pathways could be involved in the neuroprotective effects of CTs. It is known that, the activated ERK1/2, JNK and p38 MAPK mainly mediate the cellular stress in CIRI by phosphorylating intracellular enzymes, transcription factors, and cytosolic proteins involved in cell survival and apoptosis (40–42). Our results indicated that the protective effects of CTs against neuronal apoptosis induced by OGD/R could associate with suppressing the activation of MAPK signalings.
In addition, the activation of MAPK signaling pathways is associated with BBB damage (Figure 6) and aggravates CIRI by promoting the production of inflammatory cytokines (43, 44). BBB disruption is a hallmark of stroke and a mediator of CIRI and stroke progression (45). Restoration of the BBB can relieve neuronal damage caused by CIRI (46). Our findings demonstated that CTs could suppress the activation of MAPK signalings in neurons. Therefore, we hypothesized that CTs might have a beneficial effect on CIRI-induced BBB dysfunction. As we know, BBB, is mainly made up of BMECs and astrocytes, plays a critical role in maintaining the microenvironment of brain (18). Accumulating evidence has indicated that BBB dysfunction is a key event during the progression of CIRI (47). Therefore, protecting BBB from disruption may be a promising strategy for prevention and treatment of CIRI (48). In the current study, we found that the cell viabilities of BMECs and astrocytes of NVU model in vitro were significantly reduced after OGD/R. Pre-treatment with CTs increased notably the cell viabilities of BMECs and astrocytes. Moreover, the permeability measurement of BBB by TEER and SF showed that CTs pre-treatment remarkably reversed the leakage of BBB in the OGD/R-injured NVU model in vitro. These results suggested that CTs could effectively improve BBB integrity during OGD/R.
Tight junctions proteins(TJPs) between BMECs participate in forming the BBB and are composed of Zonula Occludens (ZO), Claudins and Occludin, all of which play important roles in regulating BBB permeability and function (49, 50). And there is growing evidence that the disruption of ZO-1, Claudin-5, and Occludin led to the functional changes of TJs (51–53). In this study, we found that the expression of ZO-1, Claudin-5 and Occludin was significantly down-regulated in BMECs of NVU model in vitro after OGD/R. Pre-treatment with CTs could maintain the expression of these three kinds of TJPs. These data confirmed that CTs could attenuate the BBB damage during CIRI by maintaining the TJPs expression in BMECs. Furthermore, alterations of MMPs also affect the function of the BBB (54). MMPs, a family of zinc- and calcium-dependent enzymes, disrupt the BBB integrity by degrading TJPs (55–57). The expression of MMPs is only low level in normal brain tissue, but many MMPs are activated and their levels increase after ischemic stroke (58). Members of the MMPs family, specifically MMP-2 and MMP-9 are involved in the breakdown of the BBB and increased levels of these MMPs have been observed during CIRI in stroke (59, 60). Consistent with these observations, we found the increase of MMP-2 and -9 protein levels in BMECs of NVU model in vitro exposed to OGD/R. CTs pre-treatment down-regulated the expression of MMP-9, but not MMP-2. These data further supported that CTs maintained the expression of ZO-1, Claudin-5 and Occludin, probably during OGD/R via inhibiting protein expression of MMP-9. MMPs expression is tightly regulated at the transcriptional and post-translationa levels. However, the exact mechanism is unclear. At present, it is believed that multiple signaling pathways are involved in this complex regulatory process, such as MAPK, NF-κB, and PI3K/Akt signaling pathway (61–63), which may be the reason why CTs only down-regulated the expression of MMP-9. Further study is required to explore the functional relationship between the regulation of MMP-2 expression and BBB integrity and health.
Limitations
Several limitations should be acknowledged for the present study. Again further studies are needed to reveal the effects of CTs on oxidative stress, inflammation and the other two types of cells apoptosis in the NVU model during OGD/R injury, BMECs and astrocytes for instance.
Conclusions
Despite the above limitations, we indicate that the protective mechanism of CTs against OGD/R damage might exert via inhibiting neuron apoptosis and attenuating BBB disruption. Furthermore, we also clarified that CTs inhibited neuronal apoptosis possibly by blocking the activation of MAPK signaling pathways, and CTs alleviating BBB disruption may associated with the regulation of TJPs and MMP-9 in our experiment. Accordingly, CTs will represent a novel and potent candidate for the treatment of CIRI in the future.
Ethics Statement
This study was carried out in accordance with the recommendations of China's Guidelines for Care and Use of Laboratory Animals, Medical Ethics Committee of Dalian Medical University. The protocol was approved by the Medical Ethics Committee of Dalian Medical University.
Author Contributions
CS, LZ, and MF were involved in designing the study, interpreting the data and writing the manuscript. HZ, TZ, and XY performed the majority of the experiments. SL and LW contributed to the analysis of the data.
Funding
This work was supported by funding from the National Science and Technology Major Special Project on Major New Drug Innovation of China (No. 2012ZX09503-001-003).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2019.00381/full#supplementary-material
References
1. Benjamin EJ, Virani SS, Callaway CW, Chamberlain AM, Chang AR, Cheng S, et al. Heart disease and stroke statistics-2018 update: a report from the American heart association. Circulation. (2018) 137:e67–492. doi: 10.1161/CIR.0000000000000558
2. Doyle KP, Simon RP, Stenzel-Poore MP. Mechanisms of ischemic brain damage. Neuropharmacology. (2008) 55:310–8. doi: 10.1016/j.neuropharm.2008.01.005
3. Xue Q, Liu Y, He R, Yang S, Tong J, Li X, et al. Lyophilized powder of catalpol and puerarin protects neurovascular unit from stroke. Int J Biol Sci. (2016) 12:367–80. doi: 10.7150/ijbs.14059
4. Lo EH, Dalkara T, Moskowitz MA. Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci. (2003) 4:399–415. doi: 10.1038/nrn1106
5. Cai W, Zhang K, Li P, Zhu L, Xu J, Yang B, et al. Dysfunction of the neurovascular unit in ischemic stroke and neurodegenerative diseases: an aging effect. Ageing Res Rev. (2017) 34:77–87. doi: 10.1016/j.arr.2016.09.006
6. Xue Q, Liu Y, Qi H, Ma Q, Xu L, Chen W, et al. A novel brain neurovascular unit model with neurons, astrocytes and microvascular endothelial cells of rat. Int J Biol Sci. (2013) 9:174–89. doi: 10.7150/ijbs.5115
7. Li ZM, Xu SW, Liu PQ. Salvia miltiorrhizaBurge (Danshen): a golden herbal medicine in cardiovascular therapeutics. Acta Pharmacol Sin. (2018) 39:802–24. doi: 10.1038/aps.2017.193
8. Fei YX, Wang SQ, Yang LJ, Qiu YY, Li YZ, Liu WY, et al. Salvia miltiorrhiza Bunge (Danshen) extract attenuates permanent cerebral ischemia through inhibiting platelet activation in rats. J Ethnopharmacol. (2017) 207:57–66. doi: 10.1016/j.jep.2017.06.023
9. Zhang F, Zheng W, Pi R, Mei Z, Bao Y, Gao J, et al. Cryptotanshinone protects primary rat cortical neurons from glutamate-induced neurotoxicity via the activation of the phosphatidylinositol 3-kinase/Akt signaling pathway. Exp Brain Res. (2009) 193:109–18. doi: 10.1007/s00221-008-1600-9
10. Yu XY, Lin SG, Chen X, Zhou ZW, Liang J, Duan W, et al. Transport of cryptotanshinone, a major active triterpenoid in Salvia miltiorrhiza Bunge widely used in the treatment of stroke and Alzheimer's disease, across the blood-brain barrier. Curr Drug Metab. (2007) 8:365–78. doi: 10.2174/138920007780655441
11. Wang W, Wang X, Zhang XS, Liang CZ. Cryptotanshinone attenuates oxidative stress and inflammation through the regulation of Nrf-2 and NF-kappaB in mice with unilateral ureteral obstruction. Basic Clin Pharmacol Toxicol. (2018) 123:714–20. doi: 10.1111/bcpt.13091
12. Yang Y, Cao Y, Chen L, Liu F, Qi Z, Cheng X, et al. Cryptotanshinone suppresses cell proliferation and glucose metabolism via STAT3/SIRT3 signaling pathway in ovarian cancer cells. Cancer Med. (2018) 7:4610–8. doi: 10.1002/cam4.1691
13. Sun PP, Yuan F, Xu J, Sai K, Chen J, Guan S. Cryptotanshinone ameliorates hepatic normothermic ischemia and reperfusion injury in rats by anti-mitochondrial apoptosis. Biol Pharm Bull. (2014) 37:1758–65. doi: 10.1248/bpb.b14-00389
14. Zhang Y, Chen L, Li F, Wang H, Yao Y, Shu J, et al. Cryptotanshinone protects against adriamycin-induced mitochondrial dysfunction in cardiomyocytes. Pharm Biol. (2016) 54:237–42. doi: 10.3109/13880209.2015.1029052
15. Han JY, Fan JY, Horie Y, Miura S, Cui DH, Ishii H, et al. Ameliorating effects of compounds derived from Salvia miltiorrhiza root extract on microcirculatory disturbance and target organ injury by ischemia and reperfusion. Pharmacol Ther. (2008) 117:280–95. doi: 10.1016/j.pharmthera.2007.09.008
16. Zhu W, Qiu W, Lu A. Cryptotanshinone exhibits therapeutical effects on cerebral stroke through the PI3K/AKTeNOS signaling pathway. Mol Med Rep. (2017) 16:9361–6. doi: 10.3892/mmr.2017.7824
17. Alluri H, Anasooya Shaji C, Davis ML, Tharakan B. Oxygen-glucose deprivation and reoxygenation as an in vitro ischemia-reperfusion injury model for studying blood-brain barrier dysfunction. J Vis Exp. (2015) 99: e52699. doi: 10.3791/52699
18. Khatri R, McKinney AM, Swenson B, Janardhan V. Blood-brain barrier, reperfusion injury, and hemorrhagic transformation in acute ischemic stroke. Neurology. (2012) 79(13 Suppl. 1): S52–7. doi: 10.1212/WNL.0b013e3182697e70
19. Adriani G, Ma D, Pavesi A, Kamm RD, Goh EL. A 3D neurovascular microfluidic model consisting of neurons, astrocytes and cerebral endothelial cells as a blood-brain barrier. Lab Chip. (2017) 17:448–59. doi: 10.1039/C6LC00638H
20. Deli MA, Abraham CS, Kataoka Y, Niwa M. Permeability studies on in vitro blood-brain barrier models: physiology, pathology, and pharmacology. Cell Mol Neurobiol. (2005) 25:59–127. doi: 10.1007/s10571-004-1377-8
21. Czupalla CJ, Liebner S, Devraj K. In vitro models of the blood-brain barrier. Methods Mol Biol. (2014) 1135:415–37. doi: 10.1007/978-1-4939-0320-7_34
22. Wang HJ, Wei JY, Liu DX, Zhuang SF, Li Y, Liu H, et al. Endothelial Atg7 deficiency ameliorates acute cerebral injury induced by ischemia/reperfusion. Front Neurol. (2018) 9:998. doi: 10.3389/fneur.2018.00998
23. Mehta SL, Manhas N, Raghubir R. Molecular targets in cerebral ischemia for developing novel therapeutics. Brain Res Rev. (2007) 54:34–66. doi: 10.1016/j.brainresrev.2006.11.003
24. Nakka VP, Gusain A, Mehta SL, Raghubir R. Molecular mechanisms of apoptosis in cerebral ischemia: multiple neuroprotective opportunities. Mol Neurobiol. (2008) 37:7–38. doi: 10.1007/s12035-007-8013-9
25. Li K, Ding D, Zhang M. Neuroprotection of osthole against cerebral ischemia/reperfusion injury through an anti-apoptotic pathway in rats. Biol Pharm Bull. (2016) 39:336–42. doi: 10.1248/bpb.b15-00699
26. Li F, Geng X, Yip J, Ding Y. Therapeutic target and cell-signal communication of chlorpromazine and promethazine in attenuating blood-brain barrier disruption after ischemic stroke. Cell Transplant. (2019) 28:145–56. doi: 10.1177/0963689718819443
27. Zhang QY, Wang ZJ, Sun DM, Wang Y, Xu P, Wu WJ, et al. Novel therapeutic effects of leonurine on ischemic stroke: new mechanisms of BBB integrity. Oxid Med Cell Longev. (2017) 2017:7150376. doi: 10.1155/2017/7150376
28. Park OK, Choi JH, Park JH, Kim IH, Yan BC, Ahn JH, et al. Comparison of neuroprotective effects of five major lipophilic diterpenoids from Danshen extract against experimentally induced transient cerebral ischemic damage. Fitoterapia. (2012) 83:1666–74. doi: 10.1016/j.fitote.2012.09.020
29. Adams JD, Wall M, Garcia C. Salvia columbariae contains tanshinones. Evid Based Complement Alternat Med. (2005) 2:107–10. doi: 10.1093/ecam/neh067
30. Kiraz Y, Adan A, Kartal Yandim M, Baran Y. Major apoptotic mechanisms and genes involved in apoptosis. Tumour Biol. (2016) 37:8471–86. doi: 10.1007/s13277-016-5035-9
31. Li T, Wang L, Hu Q, Liu S, Bai X, Xie Y, et al. Neuroprotective roles of l-cysteine in attenuating early brain injury and improving synaptic density via the CBS/H2S pathway following subarachnoid hemorrhage in rats. Front Neurol. (2017) 8:176. doi: 10.3389/fneur.2017.00176
32. van Wijk SJ, Hageman GJ. Poly(ADP-ribose) polymerase-1 mediated caspase-independent cell death after ischemia/reperfusion. Free Radic Biol Med. (2005) 39:81–90. doi: 10.1016/j.freeradbiomed.2005.03.021
33. Lawrence MC, Naziruddin B, Levy MF, Jackson A, McGlynn K. Calcineurin/nuclear factor of activated T cells and MAPK signaling induce TNF-{alpha} gene expression in pancreatic islet endocrine cells. J Biol Chem. (2011) 286:1025–36. doi: 10.1074/jbc.M110.158675
34. Nozaki K, Nishimura M, Hashimoto N. Mitogen-activated protein kinases and cerebral ischemia. Mol Neurobiol. (2001) 23:1–19. doi: 10.1385/MN:23:1:01
35. Lennmyr F, Karlsson S, Gerwins P, Ata KA, Terent A. Activation of mitogen-activated protein kinases in experimental cerebral ischemia. Acta Neurol Scand. (2002) 106:333–40. doi: 10.1034/j.1600-0404.2002.01313.x
36. Kovalska M, Kovalska L, Pavlikova M, Janickova M, Mikuskova K, Adamkov M, et al. Intracellular signaling MAPK pathway after cerebral ischemia-reperfusion injury. Neurochem Res. (2012) 37:1568–77. doi: 10.1007/s11064-012-0752-y
37. Zeng Z, Zhang Y, Liang X, Wang F, Zhao J, Xu Z, et al. Qingnao dripping pills mediate immune-inflammatory response and MAPK signaling pathway after acute ischemic stroke in rats. J Pharmacol Sci. (2019) 139:143–50. doi: 10.1016/j.jphs.2018.12.009
38. Che N, Ma Y, Xin Y. Protective role of fucoidan in cerebral ischemia-reperfusion injury through inhibition of MAPK signaling pathway. Biomol Ther. (2017) 25:272–8. doi: 10.4062/biomolther.2016.098
39. Xin L, Junhua W, Long L, Jun Y, Yang X. Exogenous hydrogen sulfide protects SH-SY5Y cells from OGD/R induced injury. Curr Mol Med. (2017) 17:563–7. doi: 10.2174/1566524018666180222121643
40. Irving EA, Bamford M. Role of mitogen- and stress-activated kinases in ischemic injury. J Cereb Blood Flow Metab. (2002) 22:631–47. doi: 10.1097/00004647-200206000-00001
41. Cheng YL, Choi Y, Seow WL, Manzanero S, Sobey CG, Jo DG, et al. Evidence that neuronal Notch-1 promotes JNK/c-Jun activation and cell death following ischemic stress. Brain Res. (2014) 1586:193–202. doi: 10.1016/j.brainres.2014.08.054
42. Gladbach A, van Eersel J, Bi M, Ke YD, Ittner LM. ERK inhibition with PD184161 mitigates brain damage in a mouse model of stroke. J Neural Transm. (2014) 121:543–7. doi: 10.1007/s00702-013-1138-2
43. Yao X, Wang Y, Zhang D. microRNA-21 confers neuroprotection against cerebral ischemia-reperfusion injury and alleviates blood-brain barrier disruption in rats via the MAPK signaling pathway. J Mol Neurosci. (2018) 65:43–53. doi: 10.1007/s12031-018-1067-5
44. Lu D, Mai HC, Liang YB, Xu BD, Xu AD, Zhang YS. Beneficial role of rosuvastatin in blood-brain barrier damage following experimental ischemic stroke. Front Pharmacol. (2018) 9:926. doi: 10.3389/fphar.2018.00926
45. Yang Y, Rosenberg GA. Blood-brain barrier breakdown in acute and chronic cerebrovascular disease. Stroke. (2011) 42:3323–8. doi: 10.1161/STROKEAHA.110.608257
46. Gong P, Zhang Z, Zou C, Tian Q, Chen X, Hong M, et al. Hippo/YAP signaling pathway mitigates blood-brain barrier disruption after cerebral ischemia/reperfusion injury. Behav Brain Res. (2019) 356:8–17. doi: 10.1016/j.bbr.2018.08.003
47. Moretti R, Pansiot J, Bettati D, Strazielle N, Ghersi-Egea JF, Damante G, et al. Blood-brain barrier dysfunction in disorders of the developing brain. Front Neurosci. (2015) 9:40. doi: 10.3389/fnins.2015.00040
48. Sifat AE, Vaidya B, Abbruscato TJ. Blood-brain barrier protection as a therapeutic strategy for acute ischemic stroke. AAPS J. (2017) 19:957–72. doi: 10.1208/s12248-017-0091-7
49. Hainsworth AH, Fisher MJ. A dysfunctional blood-brain barrier and cerebral small vessel disease. Neurology. (2017) 88:420–1. doi: 10.1212/WNL.0000000000003561
50. Abdullahi W, Tripathi D, Ronaldson PT. Blood-brain barrier dysfunction in ischemic stroke: targeting tight junctions and transporters for vascular protection. Am J Physiol Cell Physiol. (2018) 315:C343–56. doi: 10.1152/ajpcell.00095.2018
51. Hu S, Wu Y, Zhao B, Hu H, Zhu B, Sun Z, et al. Panax notoginseng saponins protect cerebral microvascular endothelial cells against oxygen-glucose deprivation/reperfusion-induced barrier dysfunction via activation of PI3K/Akt/Nrf2 antioxidant signaling pathway. Molecules. (2018) 23:11. doi: 10.3390/molecules23112781
52. Song J, Kang SM, Lee WT, Park KA, Lee KM, Lee JE. The beneficial effect of melatonin in brain endothelial cells against oxygen-glucose deprivation followed by reperfusion-induced injury. Oxid Med Cell Longev. (2014) 2014:639531. doi: 10.1155/2014/639531
53. Jiao H, Wang Z, Liu Y, Wang P, Xue Y. Specific role of tight junction proteins claudin-5, occludin, and ZO-1 of the blood-brain barrier in a focal cerebral ischemic insult. J Mol Neurosci. (2011) 44:130–9. doi: 10.1007/s12031-011-9496-4
54. Zhang J, Zhang Z, Zhang W, Li X, Wu T, Li T, et al. Jia-Jian-Di-Huang-Yin-Zi decoction exerts neuroprotective effects on dopaminergic neurons and their microenvironment. Sci Rep. (2018) 8:9886. doi: 10.1038/s41598-018-27852-w
55. Woessner JF, Jr. The family of matrix metalloproteinases. Ann N Y Acad Sci. (1994) 732:11–21. doi: 10.1111/j.1749-6632.1994.tb24720.x
56. Cheng S, Pollock AS, Mahimkar R, Olson JL, Lovett DH. Matrix metalloproteinase 2 and basement membrane integrity: a unifying mechanism for progressive renal injury. FASEB J. (2006) 20:1898–900. doi: 10.1096/fj.06-5898fje
58. McColl BW, Rothwell NJ, Allan SM. Systemic inflammation alters the kinetics of cerebrovascular tight junction disruption after experimental stroke in mice. J Neurosci. (2008) 28:9451–62. doi: 10.1523/JNEUROSCI.2674-08.2008
59. Rempe RG, Hartz AMS, Soldner ELB, Sokola BS, Alluri SR, Abner EL, et al. Matrix metalloproteinase-mediated blood-brain barrier dysfunction in epilepsy. J Neurosci. (2018) 38:4301–15. doi: 10.1523/JNEUROSCI.2751-17.2018
60. Yang Y, Estrada EY, Thompson JF, Liu W, Rosenberg GA. Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J Cereb Blood Flow Metab. (2007) 27:697–709. doi: 10.1038/sj.jcbfm.9600375
61. Zhu H, Dai R, Zhou Y, Fu H, Meng Q. TLR2 ligand Pam3CSK4 regulates MMP-2/9 expression by MAPK/NF-kappaB signaling pathways in primary brain microvascular endothelial cells. Neurochem Res. (2018) 43:1897–904. doi: 10.1007/s11064-018-2607-7
62. Zhang HT, Zhang P, Gao Y, Li CL, Wang HJ, Chen LC, et al. Early VEGF inhibition attenuates blood-brain barrier disruption in ischemic rat brains by regulating the expression of MMPs. Mol Med Rep. (2017) 15:57–64. doi: 10.3892/mmr.2016.5974
Keywords: cryptotanshinone, oxygen-glucose deprivation/recovery, cerebral protection, neurovascular unit, apoptosis, BBB disruption
Citation: Zhao H, Zheng T, Yang X, Fan M, Zhu L, Liu S, Wu L and Sun C (2019) Cryptotanshinone Attenuates Oxygen-Glucose Deprivation/ Recovery-Induced Injury in an in vitro Model of Neurovascular Unit. Front. Neurol. 10:381. doi: 10.3389/fneur.2019.00381
Received: 07 December 2018; Accepted: 28 March 2019;
Published: 18 April 2019.
Edited by:
Anna Rita Bilia, University of Florence, ItalyReviewed by:
Shekher Mohan, Manchester University, United StatesHarshini Sarojini, University of Louisville, United States
Copyright © 2019 Zhao, Zheng, Yang, Fan, Zhu, Liu, Wu and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Changkai Sun, cksun110@yeah.net