Abstract
Autism covers a wide spectrum of disorders for which there are many views, hypotheses and theories. Here we propose a unifying theory of autism, the Intense World Theory. The proposed neuropathology is hyper-functioning of local neural microcircuits, best characterized by hyper-reactivity and hyper-plasticity. Such hyper-functional microcircuits are speculated to become autonomous and memory trapped leading to the core cognitive consequences of hyper-perception, hyper-attention, hyper-memory and hyper-emotionality. The theory is centered on the neocortex and the amygdala, but could potentially be applied to all brain regions. The severity on each axis depends on the severity of the molecular syndrome expressed in different brain regions, which could uniquely shape the repertoire of symptoms of an autistic child. The progression of the disorder is proposed to be driven by overly strong reactions to experiences that drive the brain to a hyper-preference and overly selective state, which becomes more extreme with each new experience and may be particularly accelerated by emotionally charged experiences and trauma. This may lead to obsessively detailed information processing of fragments of the world and an involuntarily and systematic decoupling of the autist from what becomes a painfully intense world. The autistic is proposed to become trapped in a limited, but highly secure internal world with minimal extremes and surprises. We present the key studies that support this theory of autism, show how this theory can better explain past findings, and how it could resolve apparently conflicting data and interpretations. The theory also makes further predictions from the molecular to the behavioral levels, provides a treatment strategy and presents its own falsifying hypothesis.
Introduction
The neurobiology of autism has been researched extensively with growing urgency and major strides and insights over the past 30 years (Rubenstein and Merzenich, 2003; Belmonte et al., 2004b; Courchesne, 2004; Casanova, 2007; Minshew and Williams, 2007; Amaral et al., 2008), yet no coherent neurobiologically based theory of autism has yet emerged to explain its entire heterogeneity. A wide range of interpretations, hypotheses, and theories has been put forward, each casting a different light on an important but specific aspect of autism. The central question is whether the spectrum of autism is due to a spectrum of neuropathologies or whether a single common pathology can explain the spectrum. Recently, we put forth a bottom-up hypothesis for autism that is neurobiologically grounded and works its way up from the molecular, cellular, and circuit levels toward the potential cognitive consequences, called the Intense World Syndrome (for extensive review see Markram et al., 2007b). The Intense World Syndrome hypothesis was grounded in original experiments using the valproic acid (VPA) rat model of autism to explore possible alterations across molecular, cellular, synaptic, circuit, and behavioral levels. Such experiments can only be performed using an animal model. This animal model was chosen because VPA intake during pregnancy was linked to an increased risk of giving birth to an autistic child (Moore et al., 2000; Rasalam et al., 2005) and VPA exposure in rats has remarkably similar effects as in humans. The multi-level approach from molecules to behavior, possible only in an animal model, allowed piecing together the different levels of the brain's organization and up toward its emergent behavior, which revealed a common and coherent theme of alterations and suggested that autism could be explained as an Intense World Syndrome (Markram et al., 2007b). Naturally, there is a vast gap between an animal model and autism that is currently only recognized for humans. In this paper, we therefore explored previous studies, results, hypotheses, and theories in the light of the Intense World hypothesis, and examined whether this hypothesis can stand as a formal and unifying theory of autism.
The original notion of an Intense WorldSyndrome in autism arose, because VPA-exposed animals exhibited amplified fear processing and memories (Markram et al., 2005, 2008), which indicated that fragments of the world could easily become emotionally aversive and be stored excessively. In strong support of this, we found that on the neural circuit level, VPA-exposed animals exhibited enhanced neuronal reactivity and plasticity across several brain regions, such as the amygdala and neocortex. This provided the potential cellular and circuit explanation for how an autistic brain could be easily trapped in a painfully intense world, potentially explaining a broad range of common autistic symptoms such as sensory sensitivity, withdrawal, repetitive behavior, idiosyncrasies, and even exceptional talents.
The experimentally based and common neuropathology proposed in the Intense World Theory is hyper-functioning of elementary brain modules, called local neural microcircuits, which are characterized by hyper-reactivity and hyper-plasticity, both of which seem to be caused by a tendency for excitatory neurons to dominate their neighbors. Such hyper-functional microcircuits are proposed to easily become autonomous, leading to runaway information processing, over-specialization in tasks and a hyper-preference syndrome. The proposed core cognitive consequences are hyper-perception, hyper-attention, hyper-memory, functions mediated by the neocortex, and hyper-emotionality, mediated by the hyper-functionality of the limbic system. These four dimensions could potentially explain the full spectrum of symptoms in autism, depending on the severity of the microcircuit pathology in different brain regions. The degree of hyper-functionality in different brain regions could vary in each child depending on genetic personality traits, on unique epigenetic conditions, and unique sequence of postnatal experiences.
This article begins by shortly reviewing the validity of the VPA rat model of autism as well as the experimental insights obtained from this model, before delving deeper into an a re-examination and re-interpretation of previous studies on human autism in the light of these experimental results from the animal model. We make the case for a unified Intense World Theory for autism that can potentially explain many of the varied past results and resolve many conflicting findings and views, and by making some falsifiable experimental predictions.
VPA and its Link to Autism
Valproic acid is widely used to treat epilepsy and bipolar disorder and is also a potent teratogen. It was first introduced in the 60s as an anticonvulsant and later as the mood-stabilizing drug for the treatment of bipolar disorder. Case reports started to appear in the 90s on children with Fetal Anticonvulsant Syndrome, which included autistic traits (Christianson et al., 1994; Williams and Hersh, 1997; Williams et al., 2001). Two independent follow-up population studies confirmed a strong link between VPA and autism with approximately 10% of exposed children exhibiting full blown autism and 80% with one or more autistic features (Moore et al., 2000; Rasalam et al., 2005). Overall, the autism prevalence in the prenatally VPA-exposed population is approximately 11–100 times higher than in the general population assuming prevalence rates of 10–91 cases per 10,000 in the general population (Fombonne, 2006; Autism Speaks).
The physical malformations caused by VPA, such as facial dysmorphy and ear abnormalities, indicate an early insult to the brainstem during embryogenesis and, more specifically, around the time of neural tube closure. Support for the so-called brain-stem hypothesis of autism (Stromland et al., 1994; Rodier et al., 1996, 1997; Arndt et al., 2005) originate from a study on the brain stem-related teratogenic effects of thalidomide (Stromland et al., 1994), another prescribed drug strongly associated with autism. This study was the first to reveal that the occurrence of autism is strikingly high (30%) and exclusively when thalidomide intake occurred during gestational days 20–24, which led to the conclusions that autism is associated with a brainstem injury at the time point of neural tube closure. This notion is supported by magnetic resonance imaging studies which revealed brainstem hypoplasia in autism (Hashimoto et al., 1995; Gaffney et al., 1988) as well as a post-mortem study on an autistic subject who exhibited severe morphological abnormalities and neuronal loss in the brain stem (Rodier et al., 1996).
While the early brainstem hypothesis of autism assumes that all other brain alterations observed in autism are a consequence of this “big bang” (Rodier et al., 1996) it is possible that traces of the impact of VPA exposure could be carried by progenitors into the whole brain (and body), and these effects would only manifest when these brain regions begin to develop and have to start performing their functions. Indeed, VPA is teratogenic at sub-lethal doses and enhances gene transcription induced by a variety of exogenous and endogenous promoters by inhibiting histone deacetylase (Phiel et al., 2001).
Valproic acid given continuously throughout pregnancy to rats, as for humans, has been known for some time to cause severe behavioral alterations (for review see Vorhees, 1987a,b; Wagner et al., 2006; Markram et al., 2007b). However, theses alterations could be confounded by the numerous other VPA-induced insults and cognitive and motor impairments, more associated with generalized Fetal Anticonvulsant Syndrome than with autism. To target the autism component of this anticonvulsant syndrome, Rodier et al. (1996) developed a rat model for autism by specifically administering VPA only during the time period of neural tube closure according to the neurological hypothesis that the brainstem is injured in autism (reviewed in Markram et al., 2007b). The administration of a single i.p. injection of VPA (350 mg/kg) administered to pregnant dams on embryonic days (ED) 12–13 results in a reduction of the trigeminal and hypoglossal motor nuclei, loss of neurons in the abducens nucleus and in the oculomotor nucleus (Rodier et al., 1996), which parallels losses found in the brainstem in autism (Hashimoto et al., 1995; Rodier et al., 1996; Gaffney et al., 1988). Follow-up anatomical studies in the rat showed that VPA exposure on ED12.5 also results in a loss of cerebellar neurons (Rodier et al., 1997; Ingram et al., 2000), one of the most prominent features in the autistic brain (Ritvo et al., 1986; Kemper and Bauman, 1998; Palmen et al., 2004). Abnormalities in the serotonergic system, one of the most indicative biochemical pathological markers in autism (Lam et al., 2006), were also found (Narita et al., 2002; Miyazaki et al., 2005; Tsujino et al., 2007). Behaviorally, prenatal exposure to VPA on ED12.5 produces the two cardinal symptoms of autism in the rat offspring: decreased social interactions and increased repetitive behaviors (Schneider and Przewlocki, 2005; Markram et al., 2008). In the emotional domain, the offspring also exhibit enhanced anxiety (Schneider and Przewlocki, 2005; Schneider et al., 2007; Markram et al., 2008), in the motor domain, locomotor hyperactivity (Schneider and Przewlocki, 2005), in the nociception domain, lower sensitivity to pain (Schneider et al., 2001; Schneider and Przewlocki, 2005; Markram et al., 2008), in the sensory domain, hyper-sensitivity to non-painful sensory stimulation and impaired pre-pulse inhibition (Schneider and Przewlocki, 2005; Markram et al., 2008), and in the memory domain, enhanced eye-blink conditioning (Stanton et al., 2007) – all of which are common features of autism described in the DSM-IV and/or in the autism literature (Sears et al., 1994; Muris et al., 1998; American Psychiatric Association, 2000; McAlonan et al., 2002; Perry et al., 2007).
It is often argued that “autism is a human disorder” which is based on the higher cognitive symptoms that are most commonly associated with autism such a theory of mind and language deficits as well as unusual human talents. Albeit in a far more rudimentary form, many of the high-level cognitive functions can also be observed and measured in much lower mammals such as rats and mice. The common thread is the neocortex, which is the source of mammalian higher brain functions. The microcircuitry of the mammalian neocortex is remarkably similar from mouse to man in terms of layering, types of neurons, interconnections, and long-range connectivity principles (Silberberg et al., 2002). It would be very difficult to argue that insults and predispositions are exclusive to human neocortex. One may also argue that such a model ignores the well-established heredity component of autism (for review see, e.g., Persico and Bourgeron, 2006), but then not all homozygote twins succumb to autism. A pure genetic argument also ignores the high incidence of autism reported with thalidomide and high doses of VPA in human offspring. The most parsimonious interpretation is that autism is a poly-genetically predisposed disorder that is triggered by an insult and that the pathology unfolds during development. The low incidence of VPA-linked autism today is another possible argument, but the doses used today (around 5–10 mg/kg) are about 5–10 times lower than doses used in the earlier times (40–50 mg/kg). The animal models however used even higher doses (300–500 mg/kg) primarily because it would not be possible to systematically study the alterations if only 5–10% of the offspring are affected as reported in the earlier times. The higher doses seem to bring the incidence more into the 70–80% range and doses above 800 mg/kg are lethal (unpublished data). It may also turn out that these doses could be significantly lowered if combined with animals genetically engineered with identified predisposing mutations. Nevertheless, high doses do limit the strength of conclusions drawn from this model and further validation in human autism is required.
The Neurobiological Basis of the Intense World Theory
The Intense World Theory is experimentally based on direct neuronal recordings and behavioral testing on rat offspring exposed prenatally to a single dose (500 mg/kg) of VPA on embryonic day 12.5. In the course of these studies, we focused on the neocortex and the amygdala for these reasons specified below.
The neocortex is fundamental for all higher-order cognitive functions such as perception, attention, and memory. The entire neocortical sheet can be viewed as a collection of functional columns or modules that process different features and spatial positions of the sensory environment and their relationships to the body. The remarkable property of these columns is that they are very similar from mouse to man and across all neocortical regions with a stereotypical template design and only subtle variations for different neocortical regions and species (Silberberg et al., 2002). These columns are therefore designed to be “general purpose processors” and they are interlinked via short and long-range connections to form brain areas, regions, and the neocortex as a whole. These columns react to input, and their activity must be carefully coordinated across the entire neocortex to orchestrate coherent higher brain function. We therefore examined the alterations in the somatosensory cortex and the medial prefrontal cortex (mPFC). The knowledge of the normal somatosensory circuit in the rat is the most extensive and somatosensory abnormalities, such as increased sensitivity to touch, are common in autism. The prefrontal cortex has received extensive attention in autism research due to its pivotal role in executive function, language, social cognition, and regulation of emotional behavior (Struss and Knight, 2002). Based on non-invasive imaging studies during task performance in autistic and control subjects, some studies initially suggested that the prefrontal cortex is not sufficiently activated in autism (Happé et al., 1996; Baron-Cohen et al., 1999; Ring et al., 1999; Castelli et al., 2002), but more recent studies show hyper-activation in this brain area (Gomot et al., 2008; Knaus et al., 2008; Dichter et al., 2009; Belmonte et al., 2010). As described further below, direct measurements from somatosensory and prefrontal neurons of VPA-treated offspring indeed suggest that these brain regions may be hyper-reactive and hyper-plastic (Rinaldi et al., 2007, 2008a,b).
The amygdala is a key part of the emotional and social brain circuits and has many functional roles such as detecting and interpreting signs of emotional and social significance in the environment, modulating memory storage across multiple brain sites, establishing fear memories, anxiety, and the regulation of autonomic and hormonal responses (reviewed in Davis and Whalen, 2001; LeDoux, 2003; Zald, 2003; McGaugh, 2004; Adolphs, 2006). Dysfunction of the amygdala has been related to disorders of fear processing, anxiety, and social behaviors (reviewed in Cottraux, 2005; Damsa et al., 2005; Hajek et al., 2005; Shayegan and Stahl, 2005; Blair et al., 2006). In autism research, the amygdala was studied primarily due to its role in the processing and interpretation of socio-emotional cues and its influence on social behaviors (Baron-Cohen et al., 2000; Sweeten et al., 2002; Amaral et al., 2003; Schultz, 2005; Bachevalier and Loveland, 2006; Schulkin, 2007). The very first animal model of autism was based on lesioning the amygdala and studying the effects on social behavior and hierarchy (Bachevalier, 1994), implying that the lack of amygdala activity may explain the lack of social interactions or social intelligence in autism. This view dominated the research performed on the role of the amygdala in autism. Parallels were drawn between amygdala lesioned patients and autistic subjects (Adolphs et al., 2001), functional magnet resonance imaging (fMRI) studies revealing an insufficiently activating amygdala in autistic subjects were associated with deficits in interpreting other people's state of minds and feelings (Baron-Cohen et al., 1999; Critchley et al., 2000; Pierce et al., 2001). However, the opposite could also be true and lead to similar symptoms: rather than being hypo-active or not sufficiently responding, the amygdala could be overly reactive in autism. Consequently, autistic people could be processing too much emotionally relevant information, including enhanced fear and anxiety processing. The outcome could be a similar one to a not sufficiently active amygdala: withdrawal and decreased social interaction due to an enhanced stress-response and socio-emotional overflow. Indeed, as described below our studies on VPA-treated rat offspring indicate that the amygdala is hyper-reactive, hyper-plastic, and generates enhanced anxiety and fear processing (Markram et al., 2008). In accordance with this, more recent fMRI studies as well reveal amygdaloid hyper-activation in autism (Dalton et al., 2005; Kleinhans et al., 2009; Monk et al., 2010).
Hyper-reactivity
Autistic children can be overtly sensitive to sensory stimulation, including light, sounds, and touch. VPA-exposed rat offspring exhibit impaired habituation to sensory stimulation as measured by the level of pre-pulse inhibition in vivo (Schneider and Przewlocki, 2005; Markram et al., 2008). To test neuronal reactivity to network stimulation in vitro single neuron reactivity was recorded in the somatosensory cortex, the prefrontal cortex, and the lateral amygdala, while simultaneously stimulating the brain slices with a multi-electrode array.
In both neocortical areas neuronal reactivity to network stimulation was nearly twice as strong in VPA slices than control slices, which could be observed across different neocortical layers (Rinaldi et al., 2008a,b). Neuronal responses in the VPA-exposed amygdala were also greatly amplified to network stimulation and in addition increased and prolonged episodes of bursting behavior were observed, which were greater in number, frequency, and duration (Markram et al., 2008). Thus, the slightest network stimulation compared to controls, triggers a run-away-like response in the amygdala in this animal model of autism.
In order to account for this massive increase in neuronal reactivity to network stimulation several possibilities were tested. To check if enhanced excitability of the individual neurons within the microcircuit could account for the hyper-reactivity to network stimulation, excitatory synaptic currents were studied in paired neuronal recordings and revealed that the α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) mediated synaptic responses of connections between neurons were actually weaker in the VPA-treated rat offspring, which was accounted for by fewer synapses deployed (around five synapses are normally used in these connections) in each synaptic connection (Rinaldi et al., 2008b). In addition, examination of the passive and active conductance properties of the excitatory pyramidal neurons revealed that pyramidal neurons required more current to drive the neuron to spiking threshold and that the number of spikes generated for a series of current injections was lower than in controls (Rinaldi et al., 2008b). Thus overall, when stimulated individually VPA-neurons were hypo-excitable, which could not account for hyper-reactivity to network stimulation. We then tested if reduced inhibition could account for the microcircuit hyper-reactivity. Inhibitory currents were increased proportionally to the increased excitation in the neocortex, indicating that the excitation was able to recruit a constant matching level of inhibition without an imbalance developing (Rinaldi et al., 2008b). In the amygdala however, we found that inhibition was greatly reduced (Markram et al., 2008), resulting in an excitation/inhibition imbalance. Thus, these results paint an ambiguous picture for the hypothesized imbalance of excitation and inhibition in autism (Casanova et al., 2003; Rubenstein and Merzenich, 2003). As far as the neocortex is concerned, excitation is balanced perfectly well with inhibition in VPA-treated offspring, while in the amygdala an imbalance toward increased reactivity due to a loss of inhibition can be observed. It remains to be examined if the loss of inhibition is the primary mechanism of producing hyper-reactive microcircuits in brain areas that have a high incidence of inhibitory neurons, such as the amygdala or the cerebellum.
We then examined if changes in morphology could account for the observed hyper-reactivity. Morphological examination of 3D reconstructions of somatosensory pyramidal neurons did not show any significant differences in the extent of axonal or dendritic arbors, in the spine or bouton densities, and in the size of pyramidal neuron somata. There was also no change in the number of pyramidal neurons or in the level of apoptosis (Rinaldi et al., 2008b).
In summary, the neuronal network response in VPA-treated offspring is greatly amplified, resulting in hyper-reactivity to network stimulation as a common denominator between several distinct brain regions (Figure 1). Several potential mechanisms can be excluded to underlie this hyper-reactivity, such as more excitable neurons or stronger excitatory synaptic connections, or changes in morphology. Quite to the contrary, VPA-neurons seem to be hypo-excitable, as if they were to compensate the strong network hyper-reactivity. A loss of inhibition can be a potential mechanism to account for hyper-reactive microcircuits in some, but not in all, brain regions. In the neocortex, inhibition matched excitation levels.
Figure 1
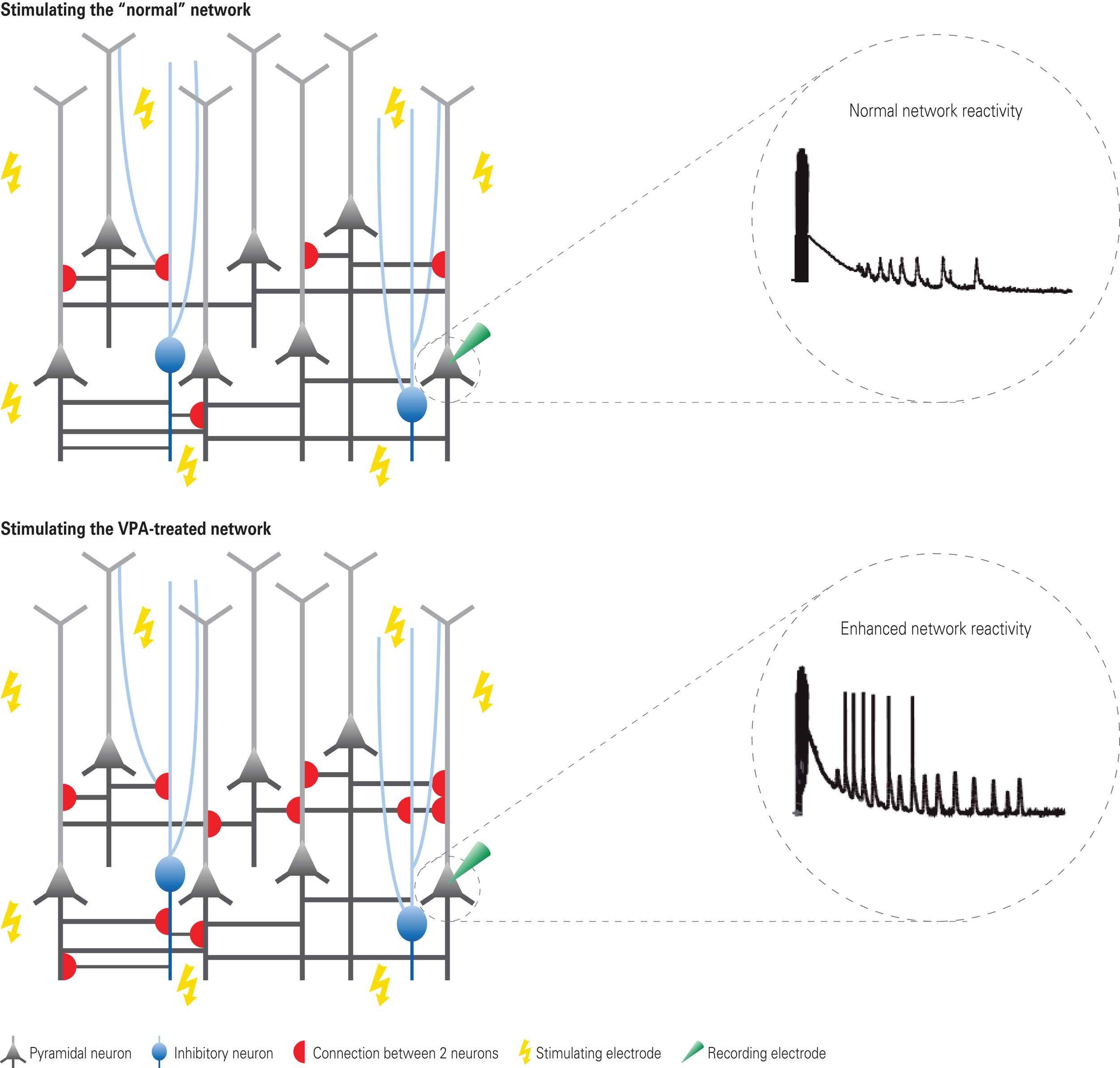
Hyper-reactivity and hyper-connectivity in VPA-treated offspring. Depicted are schematic neural microcircuits from control (top) and VPA-treated offspring (bottom). Brain slices were electrically stimulated through multiple stimulation electrodes underneath the slice – a multi-electrode array. The responsiveness to this network stimulation was recorded from individually patched cells. In comparison to controls, neurons from VPA-treated offspring were excessively reacting to the stimulation as depicted in the exemplary voltage traces. Further examination by recording from pairs of neurons revealed that this hyper-reactivity was due to the excessive connectivity in VPA-treated microcircuits (schematically depicted by the red half-circles). The connection probability was increased by approximately 50% in microcircuits from VPA-treated offspring.
After excluding all above parameters, we examined synaptic connectivity patterns between neurons as a potential mechanism for the observed hyper-reactivity.
Hyper-connectivity
Alterations in synaptic connectivity in autism were proposed previously (Belmonte et al., 2004a; Just et al., 2004; Courchesne and Pierce, 2005b). Several fMRI studies suggested that long-range connections between different brain areas are underdeveloped in autism (Horwitz et al., 1988; Castelli et al., 2002; Welchew et al., 2005; Just et al., 2007), and by extrapolation, that short-range connections may be overly developed (Casanova, 2004; Courchesne and Pierce, 2005a,b; Courchesne et al., 2005; Mottron et al., 2006). While in the human connectivity can be deduced only indirectly (e.g., through synchronization states between different brain regions), the animal model poses the advantage of a direct and quantitative assessment of neuronal connectivity. We examined the number of direct connections established between excitatory pyramidal neurons as well as between excitatory pyramidal neurons onto inhibitory interneurons within the microcircuitry of the somatosensory and prefrontal cortex using paired neuron recordings in brain slices of VPA treated and control offspring. We found an increase of around 50% of both neuronal connection types, excitatory, and inhibitory, in VPA-treated offspring (Rinaldi et al., 2008a,b; Silva et al., 2009; Figure 1). When examining the circuit in an unperturbed baseline state, this hyper-connectivity was only found for very close neighboring neurons confined to the typical dimensions of a neocortical minicolumn (less than 50 μm intersomatic distances). However, we also applied an advanced over-night stimulation protocol (for more details see the following section on hyper-plasticity) and found that hyper-connectivity could also emerge beyond the mini-columnar range as a result of enhanced micro-circuit plasticity in VPA-treated offspring (Silva et al., 2009; Figure 3).
In summary, VPA-treated neuronal microcircuits exhibited a 50% increase in connectivity between neurons. Hyper-connectivity between neurons and thus enhanced information flow could be the on the underlying mechanisms causing hyper-reactivity across different brain regions.
Hyper-plasticity
Memory processes are often altered in autism, but research in this area has produced quite controversial results with hypothesis postulating amnesia (Boucher and Warrington, 1976) normal (e.g., Renner et al., 2000; Toichi and Kamio, 2002) and enhanced memory functioning (Sears et al., 1994; Caron et al., 2004). Long-term potentiation (LTP) is the neuronal mechanism widely assumed to underlie memory formation (e.g., Muller et al., 2002). We therefore examined whether synaptic plasticity was affected in the VPA-treated somatosensory cortex, prefrontal cortex, and amygdala following a Hebbian pairing stimulation protocol. In all three brain regions, the amount of LTP was doubled in VPA-treated offspring as compared to controls (Rinaldi et al., 2007, 2008a; Markram et al., 2008; Figure 2). In the neocortex at the age of 2 weeks, LTP usually takes on a presynaptic form by increasing the release of glutamate (Markram and Tsodyks, 1996). This type of presynaptic LTP was normal in the VPA-exposed animals. However, postsynaptic LTP, normally not present at this age in the neocortex, contributed significantly to the enhanced responses. This additional and boosted form of postsynaptic LTP was found in both neocortical layers 2/3 and layer 5 pyramidal neurons. These results indicate that glutamatergic synapses are remarkably hyper-plastic in this animal model of autism.
Figure 2
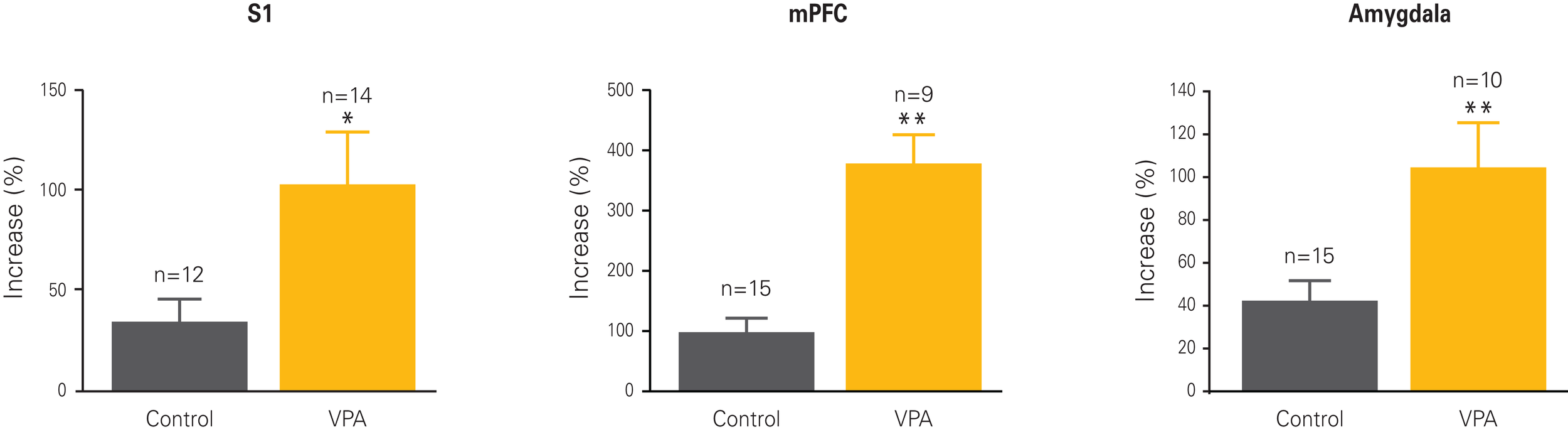
Cellular hyper-plasticity in VPA-treated offspring. Long-term potentiation (LTP) is increased in neurons from VPA-treated offspring as compared to controls. The graphs depict the percentual increase in response amplitude recorded from neurons before and after a strong electrical stimulation. Increases are at least two to four times higher in VPA-treated offspring than controls across all three recorded brain regions, the somatosensory cortex (S1), the medial prefrontal cortex (mPFC) and the lateral amygdala. Results rearranged from original research articles (Rinaldi et al., 2007, 2008a; Markram et al., 2008), with permission from PNAS, Neurospychopharmacology, and Frontiers in Neural Circuits.
Previously, we found that connectivity between neurons can also change as a result of each stimulation to reconfigure the network – termed “microcircuit plasticity,” which is a form of wiring plasticity (Le Be and Markram, 2006). To test the rewiring capacity of the micro-networks, brain slices from VPA-treated and control rats were perfused with glutamate overnight and neuronal connectivity patterns were established before and after the stimulation. Indeed, in brain slices from VPA-exposed rat offspring we found a striking increase in the rate at which neurons connect and disconnect to rewire the circuit (Figure 3). Interestingly, this increased rate of rewiring was particularly pronounced above the intersomatic distance of 50 μm, which lies beyond the mini-columnar range. Within the mini-columnar range (intersomatic distances below 50 μm) the VPA-treated circuit seemed to be saturated, since connectivity is already increased at baseline levels. These results indicate a remarkably increased capacity for rewiring microcircuits as a result to stimulation and learning experiences. This suggests that the microcircuits in this animal model of autism do not only react excessively and modify their existing synapses in a stronger manner, but that overall the microcircuits are also more adaptive to stimulation (Silva et al., 2009).
Figure 3
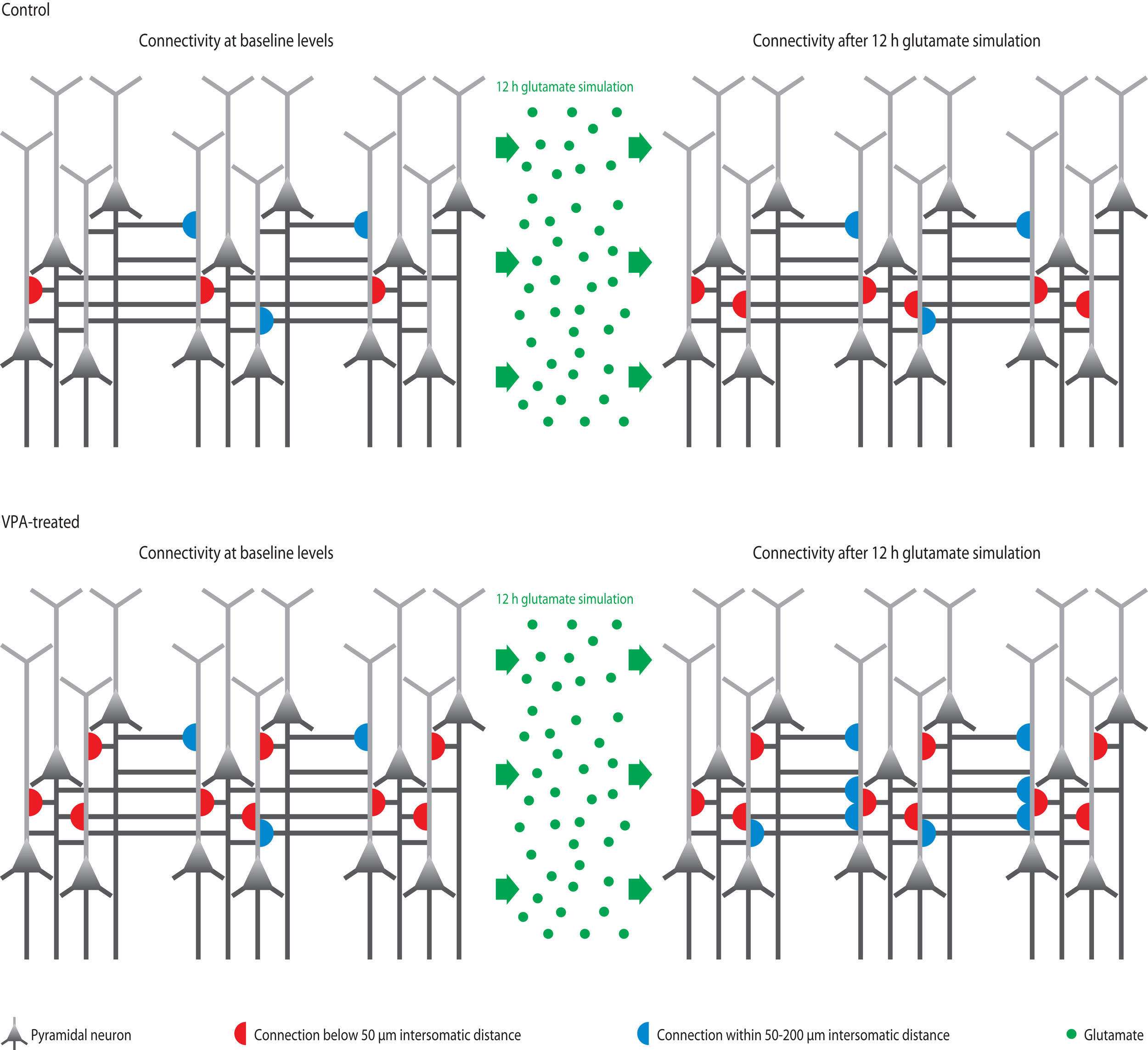
Microcircuit hyper-plasticity in VPA-treated offspring. Depicted are schematic neural microcircuits from control (top) and VPA-treated offspring (bottom). In this experiment, brain slices were perfused for 12 h with a glutamate solution in order to stimulate the circuits and induce rewiring of connections (red and blue half-circles). The connectivity probability between neurons was accessed before (left panels) and after (right panels) the glutamate stimulation. In controls, the connection probability increased significantly within a range of less than 50 μm (red half-circles), which is the mini-columnar range, but did not change in ranges higher than 50 μm (measured up to 200 μm, blue half-circles), which is a columnar range. In VPA-treated offspring, the connections probability was already increased within the short mini-columnar range (below 50 μm, red half-circles) before the glutamate stimulation and did not increase any further after the stimulation (right panel), because the connection capacity was already boosted and probably saturated to a maximum at baseline levels. Indeed, controls only reached a similarly high connectivity probability within the mini-columnar range as VPA-treated offspring already exhibited at baseline levels after the 12 h stimulation was applied. However, in VPA-treated offspring, the connectivity probability increased significantly at ranges above 50 μm (right panel, blue half-circles), revealing a further remarkable rewiring capacity at the columnar range due to stimulation – a feature “normal” control microcircuits did not exhibit.
Hyper-NMDA receptor expression
The glutamatergic neurotransmitter and receptor systems, particularly N-methyl-d-aspartate (NMDA), mediates synaptic plasticity (Nicoll and Malenka, 1999) and alterations in this system could contribute to the above observed hyper-plasticity. As compared to control, the VPA-treated neocortex did not exhibit any alterations in the AMPA receptor subunits GluR1, GluR2, and GluR3 and the obligatory subunit of the NMDA receptor, NR1. However, the NMDA receptor subunits NR2A and NR2B were more than twofold over-expressed (Rinaldi et al., 2007). Electrophysiological experiments that isolated the NMDA component of the synaptic currents confirmed a boosted NMDA receptor mediated current at these glutamatergic synapses. We further examined the various second-messenger systems and found an enhancement in CaMKII expression, which is known to mediate NMDA receptor plasticity induction (Silva et al., 1992; Liao et al., 1995; Giese et al., 1998; Lisman et al., 2002). We did not find any evidence at this age that enhanced NMDA receptor levels might render neurons more vulnerable to neurotoxicity, but have not examined whether this could cause damage later on in life.
Hyper-learning
If neocortical columns are hyper-reactive and hyper-plastic, there could be significant consequences for perception, attention, as well as for learning and memory. We therefore examined whether these changes had any impact on learning and memory tasks that depend on the neocortex. The rat whiskers are comparable to the human fingertips and each whisker is neatly represented in the barrel cortex (Woolsey and Van der Loos, 1970), a part of the somatosensory cortex. Rats use their whiskers to build spatial representations of their environment, locate objects, and perform fine-grain texture discriminations (Petersen, 2007). Rats can therefore be trained with a reward to discriminate between a wide and narrow aperture using their whiskers, a task which depends on the barrel cortex (Krupa et al., 2001). VPA-exposed offspring learned and memorized better to discriminate between apertures of different sizes than normal control rats (Markram et al., 2007a) consistent with the hyper-functionality.
Hyper-fear
It is widely established that the amygdala mediates the formation and probably also storage of fear memories (LeDoux, 2003) and enhances memory formation throughout other brain regions by acting as an emotional amplifier (Cahill and McGaugh, 1996). Since we observed enhanced plasticity in the amygdala in the VPA model of autism, we asked whether this would also manifest in enhanced fear memory formation. Indeed, VPA-exposed offspring exhibited greatly amplified conditioned cued and contextual fear memories when tested up to 3 months after conditioning (Figure 4). Moreover, in VPA-treated offspring, this fear generalized to another previously non-fear evocative tone and was more resistant to extinction than in controls (Markram et al., 2005, 2008). This indicated that VPA-treated animals store fear memories in an exaggerated and more persistent manner, generalize learned fear more easily to similar stimuli and once fear to a particular stimulus configuration is acquired it is difficult to erase.
Figure 4
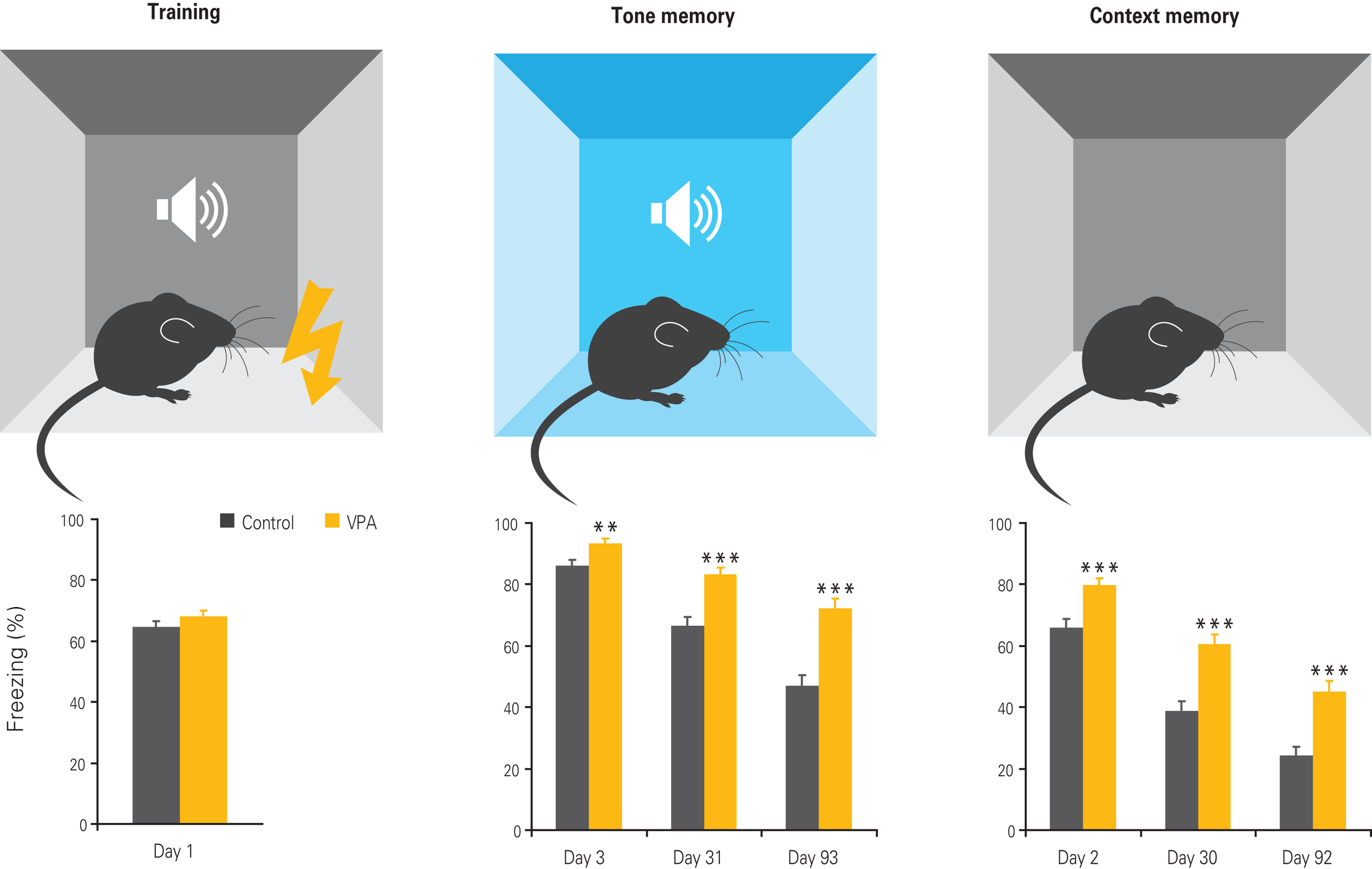
Enhanced fear memories in VPA-treated offspring. Offspring of VPA-treated dams exhibits normal fear conditioning during training, but enhanced fear memories to the tone and context 1, 30, and 90 days after training, with the differences becoming more pronounced with time. Results rearranged from original research article (Markram et al., 2008) with permission of Neurospychopharmacology.
Anxiety and phobias are known features of autism (Muris et al., 1998; Gillott et al., 2001; Evans et al., 2005) and were initially reported by Kanner (1943) himself and are furthermore widely claimed in anecdotal parental reports. Nevertheless, with the exception of two studies (Bernier et al., 2005; Gaigg and Bowler, 2007), fear processing has largely been overlooked in autism research and this was the first demonstration of enhanced fear processing in an animal model of autism suggesting that this may also be occurring in autism. Enhanced fear memory formation and a progressive generalization of fears could have major consequences on behavior and account for inappropriate reactions to the environment, sudden and apparently inexplicable anxiety attacks, loss of the finesse required in social interactions, and phobias. Over-generalization may also accelerate the progression in autism by more rapidly limiting the repertoire of safe stimuli, environments, and situations. While deficits in extinction were previously observed in autistic children (Mullins and Rincover, 1985; Sears et al., 1994; Coldren and Halloran, 2003) and may lead to preservation tendencies observed in autism, fear extinction was never studied in autism. If present, a deficit in extinguishing acquired fear in autism would make it more difficult to relinquish old fears that are no longer relevant or justifiable. This deficit combined with longer-lasting fear memories that are also over-generalized, could lead to a progressive and irreversible reduction in the repertoire of acceptable stimuli and drive a complete lock down and blanketing out of what would rapidly become a painfully intense world.
The Intense World Theory of Autism
The challenge for any unified theory of autism is to understand the common cause for the wide spectrum of autistic disorders and the autistic traits that are found in other disorders, if there is one. The Intense World Theory proposes that autistic traits could emerge if a molecular syndrome is activated that sensitizes gene expression pathways to respond excessively to environmental stimulation. Under normal conditions such pathways would enable enriched environments to nurture brain development, but if these pathways are sensitized, then environmental stimulation may cause exaggerated and accelerated development of the brain in general and the glutamatergic system of neural microcircuits in particular. Microcircuit glutamatergic hyper-functionality in the neocortex could cause hyper-perception, hyper-attention, and hyper-memory, which are proposed as the core triad of cognitive traits common to all autistic symptoms. Microcircuit hyper-functionality in the limbic system could cause hyper-emotionality adding a forth dimension that could scale the cognitive impact of the triad pathology. The severity on each of these four axes could perhaps account for autism on any part of the spectrum. The sensitivity to the environment could drive the brain to develop in a premature sequence and in a manner that enhances functionality. At its peak, the environment could become excessively intense and set in motion a systematic regression to where the brain is forced to take refuge in a highly specialized “cocoon” where extremes and surprises are actively avoided and blocked out.
The specific molecular cascade that drives hyper-functionality in brain microcircuits is not thought to be necessarily the same in different parts of the brain since there may be different ways to produce hyper-functionality in different regions and we therefore propose a common syndrome rather than a common specific molecular cascade, for all brain regions. The characteristics of the syndrome could be an exaggerated response to stimulation. In the brain, this common molecular syndrome is proposed to have a dual effect of causing hyper-reactivity and hyper-plasticity of microcircuits to produce hyper-functionality (Figure 5).
Figure 5
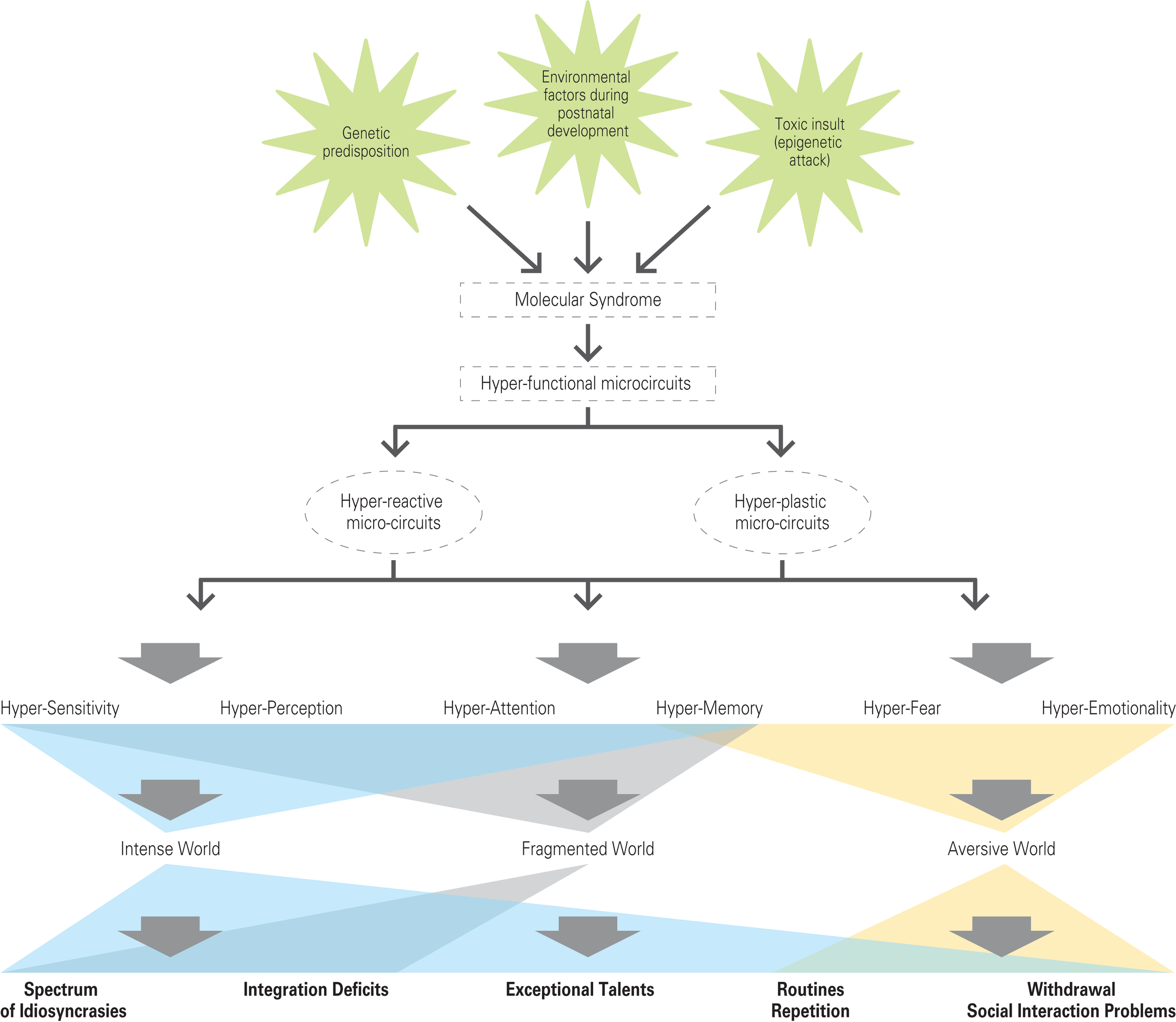
The hyper-functional circuits in autism. As suggested by the Intense World Theory three etiological factors (a genetic predisposition; an epigenetics attack in form of a toxic insult; environmental factors during postnatal development) cause autism by activating a molecular syndrome that may be different across different brain regions, but that leads to hyper-functional microcircuits (expressed as hyper-reactivity and hyper-plasticity) in all brain regions. Two regions known to be affected include the neocortex and amygdala and we hypothesize that other regions may be similarly affected. The consequences on cognitive processing include hyper-sensitivity, -perception, -attention, -memory, -fear, and -emotionality. We propose that this leads to an intense, fragmented, and aversive world syndrome for the autistic child, which could account for a spectrum of behavioral abnormalities.
In the neocortex, the consequences could be severe, because microcircuits lie within functional modules called neocortical columns and if these columns are hyper-functional, the delicate balance between intra- and inter-columnar processing would be upset. The approximately million neocortical columns in humans each need to be precisely excited and inhibited to coordinate higher brain functions and complex behavior. Excessive intra-columnar processing, particularly during development, could enhance the most elementary sensory, motor, and cognitive processes at the expense of being able to orchestrate “symphonies” of higher cognitive functions. With excessive learning and memory processes, sensory regions could consolidate into overspecialized modules and lead to hyper-preference processing. During early development, this may lead to excessive flow of information from sensory areas to the higher integration areas such as the association cortices and prefrontal lobe which may cause prematurely accelerated growth of these higher order brain areas, as indeed observed in autism (Courchesne et al., 2001; Carper et al., 2002), but this is only one out of several possible explanations for this phenomenon. Deficits in higher brain functions such as executive control and holistic processing may also be better explained by overly autonomous elementary modules rather than deficits in these areas or weak long-range connectivity. Indeed, structural MRI indicates higher levels of white matter in the cerebrum and cerebellum in young children with autism (Courchesne et al., 2001; Carper et al., 2002), which – in the light of the Intense World Theory – could be interpreted as a compensatory action to coordinate excessive activity in columns within and between different brain areas.
More specifically, each neocortical column is known to be involved in processing multiple features of stimuli. In the visual cortex for example, such features include the orientation, spatial frequency, contrast or color (Hubel and Wiesel, 1962; Tootell et al., 1981). The emphasis that each column must place on processing the different features is carefully crafted to allow the neocortex to simultaneously specialize in processing many different features locally and generalize in combining features globally. Hyper-reactivity and hyper-plasticity could act synergistically during experiences to enhance sensitivity to features and consolidate memories of features processed. Selective feature sensitivity would more easily allow specific features to trigger column processing, but could make it more difficult to activate other features (and tasks) and to interrupt any processing, once started. Overly strong consolidation of memories of the processing of features in development could also rapidly lead to dominance of the earliest features (“impressions”) and avoidance of processing of other features (manifesting in higher thresholds). Such hyper-autonomous and overly selective columns could make bottom-up control of the activation of columns from the thalamus and top-down control from higher associational areas more difficult leaving the neocortex fragmented into independent modules that are difficult to control and coordinate – an exaggerated and runaway response to stimulation.
Hyper-preference processing in the sensory domain could lead to exaggerated selectivity, sensitivity, and specialization of sensory features and hence hyper-perception, while hyper-preference processing in cognitive processing in general could lead to hyper-attention and in the memory domain, to hyper-memory. Therefore, the degree of hyper-preference processing in neocortical regions, areas and columns and the normal variation of individuals, could contribute a spectrum of autistic traits all manifesting as part of an Intense World Syndrome.
The molecular syndrome, however, also seems to extend beyond the neocortex. While in the neocortex this drives hyper-functionality by hyper-connecting neighboring neurons to produce excessive excitation and by hyper-expressing NMDA receptors to produce excessive plasticity, in the amygdala inhibition is also reduced. The amygdala contains relatively higher numbers of inhibitory interneurons than in the neocortex and reducing inhibition may be more effective in causing hyper-functionality than hyper-connecting through glutamatergic synapses. Hyper-functionality in the amygdala (and related emotion centers) could add a very important hyper-emotional dimension to the triad pathology to render the already intense world progressively more painful and aversive with each experience leading to a progressive lock down and social withdrawal.
While the Intense World Theory is primarily based on experimental data derived from the neocortex and the amygdala, we do not exclude that the same molecular syndrome driving hyper-reactivity and hyper-plasticity could be active in other structures of the forebrain or the mid- and hindbrain as well. For example, it is well established that the cerebellum is strongly affected in autism with decreased numbers of the inhibitory Purkinje cells (Ritvo et al., 1986; Bailey et al., 1998; Bauman and Kemper, 2005), which would predict hyper-reactivity. It is also known that there are increased white matter volumes (Courchesne et al., 2001) which would increase activation of brain regions from other regions. Indeed, others already postulated a disinhibition of the cerebellum that would lead to increased reactivity and could alter connectivity not only within the cerebellum, but also across the cerebello-thalamo-corticial circuits (Carper and Courchesne, 2000; Belmonte et al., 2004b; Boso et al., 2010).
In summary, autonomously acting hyper-functional microcircuits in the neocortex may cause exaggerated perception to fragments of a sensory world that must normally be holistically processed, and may cause hyper-focusing on fragments of the sensory world with exaggerated and persistent attention. This hyper-attention could become difficult to shift to new stimuli due to the difficulty for bottom-up and top-down mechanisms to coordinate the overly autonomous low-level neocortical columns. The hyper-plasticity component may also drive exaggerated memories to further amplify hyper-attention toward the same stimulus and drive over-generalization of attention to all related forms of the stimulus. The positive consequences are exceptional capabilities for elementary and specific tasks while the negative consequences are impairment of holistic processing, a rapid lock to a limited repertoire of behavioral routines, which are then repeated obsessively.
As a consequence the autistic person would remain with a fragmented and amplified perception of bits and pieces of the world. The intense world that the autistic person faces could also easily become aversive if the amygdala and related emotional areas are significantly affected with local hyper-functionality. The lack of social interaction in autism may therefore not be because of deficits in the ability to process social and emotional cues, but because a sub-set of cues are overly intense, compulsively attended to, excessively processed and remembered with frightening clarity and intensity. Typical autistic symptoms, such as averted eye gaze, social withdrawal, and lack of communication, may be explained by an initial over-awareness of sensory and social fragments of the environment, which may be so intense, that avoidance is the only refuge. This active avoidance strategy could be triggered at a very early stage in a child's development and could progress rapidly with each experience manifesting as a regression, which is striking in some cases. With such early over-specialization, many other important elementary certain skills may never be properly developed to enable normal navigation in a socially rich world with an appropriate understanding of social cues and communication. As already stated by other authors (for example Frith, 2003) “autism affects development, and in turn development affects autism” (p. 2). Compiling higher order functions such as abstract thought and language when the elementary alphabet of features is so overly attended to may become difficult if not impossible in severe cases.
Support, Contradictions, and Unification
The Intense World Theory proposes that core elementary cognitive consequences in any child on the autistic spectrum are hyper-perception, hyper-attention, hyper-memory, and hyper-emotionality. Hyper-capabilities are one positive aspect of such a brain, sensory overload, avoidance of stimulus-loaded situations and rapid lock-down into behavioral routines are the downside of it. The next four subsections will examine if there are indeed any indications for such postulates in the autism literature.
The remaining subsections will discuss support for the main neurophysiological postulates of the Intense World Theory, namely hyper-reactivity and hyper-plasticity.
In each section we propose support for these postulates, point out possible data contradictions and contradictory postulates of other neurological and/or psychological autism theories. Finally, the Intense World Theory proposes to be a unifying theory of autism and as such will attempt to resolve apparent conflicting data, interpretations, views and hypotheses, and reconcile debates.
Hyper-perception
Super-charged microcircuits in primary sensory areas could produce enhanced sensitivity to sensory stimulation and sensory overload. It is often reported that autistic children are hyper-sensitive to touch, noises and visual input and react with temper tantrums, extreme anxiety, and even panic when exposed to novel or stimulus-overloaded situations. Sensory stimuli that are bearable and normal to a typically developing child may be unbearable to an autistic child. A person who is suffering from sensory overload will naturally avoid situations that are unpredictable and filled with aversive stimuli, such as supermarket visits, social encounters etc. The hyper-sensory component of autism is for example epitomized in the personal account of the well-known autistic Temple Grandin(Grandin, 1996). Autistics also pay extreme attention to detail and can notice the smallest changes in their environment. These every-day experiences are supported by a number of rather recent quantitative scientific studies, which point to the superior performance of autistics in a variety of auditory and visual tasks.
In the auditory domain, autistics exhibit enhanced discrimination between auditory stimuli (Bonnel et al., 2003), more accurate local target detection of auditory stimuli (Mottron et al., 2000) and diminished global interference on auditory processing (Foxton et al., 2003). Moreover, autistic individuals also exhibit impaired pre-pulse inhibition (McAlonan et al., 2002; Perry et al., 2007). In this paradigm, an auditory softer pre-pulse reduces the reactivity to a second louder pulse, a well-known phenomenon which is called “pre-pulse inhibition” and represents a form of sensorimotor gating. Impaired pre-pulse inhibition in autistics suggests impaired inhibitory regulation of more complex forms of sensory processing and may be a contributing factor to sensory overload.
In the visual domain, autistics exhibit enhanced visual discrimination capabilities (Plaisted et al., 1998; O'Riordan and Plaisted, 2001; O'Riordan et al., 2001), faster target detection in feature and conjunctive visual search (O'Riordan et al., 2001; Jarrold et al., 2005), more accurate local target detection (Plaisted et al., 1999), diminished global interference on visual processing (Mottron et al., 2003), and enhanced orientation discrimination of first-order gratings (Bertone et al., 2005).
In general, autistics perform better than controls on tasks that favor the detail-oriented, “piece-meal” processing of stimuli, which gave rise to cognitive theories such the Weak Central Coherence Theory (Frith, 1989; Frith and Happé, 1994; Happé, 1999) and Enhanced Perceptual Functioning Theory (Mottron and Burack, 2001; Mottron et al., 2006). For example, autistic individuals perform better on the Wechsler Block Design task, which is due to a greater ability to segment the whole design into its component parts (Shah and Frith, 1993), the Embedded Figures Test, which requires to spot and reproduce a figure from a complex background (Shah and Frith, 1983; Jolliffe and Baron-Cohen, 1997) and graphical reproductions of impossible figures (Mottron et al., 1999). On the other hand, autistics seem to perform worse on tasks that require holistic and contextual processing of stimuli (Happé and Frith, 1997). These results were summarized in and emerged from the Weak Central Coherence Theory, which claims that autistic perception is characterized by a focus on details at the expense of feature integration and holistic Gestalt-processing. Actually, it is postulated that a deficit in holistic top-down processing of sensory information produces the advantage for local detail processing (Frith, 1989; Frith and Happé, 1994; Happé, 1999). Later it was suggested that enhanced detailed-focused perceptual processing per se, rather than a failure in central processing, is the root cause (Plaisted et al., 2003) and the theory was revised accordingly more recently (Happé and Frith, 2006). This is also the main hypothesis of the Enhanced Perceptual Functioning Theory, which claims that the main features of autistic perception are locally oriented perception and enhanced low-level discrimination (Mottron et al., 2006).
The Intense World Theory also predicts enhanced perception of sensory fragments, focus on details and piece-meal perception and a deficit in complex and more holistic processing and therefore encompasses these different theories. The Intense World Theory however explains integration deficits differently from the Weak Central Coherence Theory, namely as arising from autonomous hyper-functional neocortical columns that are more difficult to control and orchestrate by both top-down and stimulus entrainment by bottom-up mechanisms, rather than a deficit in top-down pathways or mechanisms. The consequences are however the same as those predicted by the Weak Central Coherence Theory and the Enhanced Perceptual Functioning Theory.
Hyper-attention
Attention is a conglomerate of different functionalities, including sustained attention, orienting attention, response inhibition, and set shifting (Posner and Petersen, 1990). Sustained attention is defined by the ability to maintain attention to repetitive stimuli over prolonged periods of times. Orienting refers to the capability to disengage attention, shift attention, and re-engage attention. Response inhibition refers to the capability to suppress irrelevant or interfering stimuli and finally, set shifting is thought to reflect cognitive flexibility and refers to the ability to shift to a different thought or action according to changes in a situation.
Autistics are well known to pay abnormal and obsessive attention to detail, and to note and record their environment with exquisite clarity. They can become hyper-focused and locked-in on apparently arbitrary subjects of interest and sustain their attention on these subjects for unusually long time periods. However, they are also known not to pay attention to things on demand, for example when pointed out by parents, being called by their name or when somebody enters the room. It is in fact notoriously difficult to engage their attention on demand. We suggest that this apparent attention deficit is the result of excessive on-going processing and excessive attention to endogenous domains where attention is fed back onto oneself. As a consequence of this internal hyper-focus, it would be more difficult for another person to command the autistic person's attention as well as it would be more difficult for the autistic person himself to command his own attention voluntarily. This form of exaggerated self-engrossment with internally on-going processes could perhaps also explain the apparent deficit in theory of mind so often reported in autism.
The scientific literature reports normal sustained attention in autism (Garretson et al., 1990; Buchsbaum et al., 1992; Siegel et al., 1995; Pascualvaca et al., 1998; Noterdaeme et al., 2001; Voelbel et al., 2006; Johnson et al., 2007), deficits in orienting attention (Casey et al., 1993; Wainwright-Sharp and Bryson, 1993; Courchesne et al., 1994; Townsend et al., 1996; Wainwright and Bryson, 1996; Rinehart et al., 2001; Belmonte and Yurgelun-Todd, 2003; Renner et al., 2006), deficits in disengaging attention (Landry and Bryson, 2004), impaired set-shifting response, such as on the Wisconsin Card Sorting test (Rumsey and Hamburger, 1988; Szatmari et al., 1989; Prior and Hoffmann, 1990; Ozonoff et al., 1991, 1994; Ozonoff, 1995; Shu et al., 2001) and other set-shifting tasks (Geurts et al., 2004; Ozonoff et al., 2004; Kenworthy et al., 2005; Verte et al., 2005). Conflicting results are reported concerning impaired response inhibition, which may be normal (Ozonoff and Strayer, 1997; Goldberg et al., 2005) or impaired (Johnson et al., 2007; Luna et al., 2007). The impairments are usually attributed to an executive function deficit in autism mediated by an under-performing prefrontal cortex (for review see Sanders et al., 2008).
The Intense World Theory suggests that hyper-functional microcircuits become autonomous processing modules that are difficult to control voluntarily. Once activated these columns may reverberate and not require continual external stimulus entrainment and can easily escape top-down control from areas such as the prefrontal cortex. It is understandable that this has been interpreted as a deficit in the prefrontal functioning, but in fact the prefrontal cortex may even be over-performing in its attempts to catch up with the runaway columns. In fact, we found the same hyper-functionality caused by hyper-connectivity and hyper-plasticity in the prefrontal cortex as in the somatosensory cortex in the animal model of autism (Rinaldi et al., 2008a). The prefrontal cortex may therefore actually be over-performing, but relative to other activity in the neocortex only appear as if it is under-performing. Indeed, autistic subjects exhibit hyper-activation in this brain area as revealed in recent fMRI studies (Gomot et al., 2008; Knaus et al., 2008; Dichter et al., 2009; Belmonte et al., 2010) and structural MRI studies most commonly report an overgrowth of ipsilateral cortico-cortical connections and this overgrowth of white matter is most pronounced in the prefrontal cortex (e.g., Herbert et al., 2003., 2004; Hardan et al., 2006; Craig et al., 2007). This is more consistent with a hyper-functional prefrontal cortex than weak long-range connections as usually interpreted from fMRI studies (Just et al., 2004, 2007; Koshino et al., 2005, 2008; Cherkassky et al., 2006; Kana et al., 2006, 2007).
Hyper-memory and intellect
Enhanced learning and memory as proposed by the Intense World Theory, could explain the astonishing savant talents, such as exceptional memory for music, extraordinary calendar and numerical calculations, the ability to draw complex scenes in exquisite detail from memory, or extraordinary factual memory (Pring, 2005; Treffert, 1999). However, the general tendency in autism research is to consider these exceptional cases almost as “aberrations of autism” only present in a small subsection of the autistic spectrum. Most (70–75% of the cases) autistics are usually classified as mentally retarded with diminished cognitive capabilities (American Psychiatric Association, 1980, 1987, 1994, 2000; Lord and Spence, 2006). Within this framework the most puzzling question is: can hyper-memory then be a core cognitive abnormality in all of autism? This chapter briefly discusses the state of memory research in autism and makes a statement regarding the general vista of low cognitive capabilities and mental retardation in autism. We argue that enhanced memory capabilities in autism may lie at the heart of many core symptoms of autism.
Initial scientific studies on autistic memory postulated a memory dysfunction rather than hyper-function behind the deficits in social interactions and language deficits. In particular, the prevailing opinion was to compare autism to amnesia (Boucher and Warrington, 1976). Later studies could not substantiate this claim and the current research vista states that basic perceptual based memory functionality in the visual, auditory, spatial, and even verbal domains is normal or even enhanced, but deteriorates with increasing complexity and contextual enrichment (Minshew et al., 1992, 1997; Rumsey and Hamburger, 1988; Minshew and Goldstein, 2001; Toichi and Kamio, 2002; Williams et al., 2005, 2006). The conclusions is that an underlying core deficit in Executive Function (Ozonoff et al., 1991; Bennetto et al., 1996) or a deficit in Complex Information Processing (Minshew and Goldstein, 1998) contribute to this type of memory dysfunctions.
The Complex Information Processing Disorder Theory of autism (Minshew and Goldstein, 1998) proposes an increasing impairment in integrating progressively more complex information due to a failure of neuronal integration mechanisms across different brain regions, the so-called functional Under-Connectivity Theory (Just et al., 2004) and thus explains memory deficits for complex and abstract material. While the Intense World Theory also predicts deficits in integration mechanisms, the latter theory suggests that the underlying neuronal mechanism is that of low-level hyper-functional and autonomous columns that excessively process and store simple features. Thus, the Intense World Theory predicts that simple classical and operant conditioning mechanisms as well as low-level and simple perceptual memory processing should be enhanced in autism. Indeed, such enhanced memory capabilities were already observed in classical conditioning paradigms (Sears et al., 1994) and some perceptual memory paradigms (Toichi and Kamio, 2002; Caron et al., 2004).
However, the general tone of the vast majority of studies is to find and define, with increasingly minuscule detail “deficits” and “impairments” in autistic memory and hypothesize about their contributions to the autistic syndrome. Normal and in particular enhanced memory capabilities (Sears et al., 1994; Toichi and Kamio, 2002; Caron et al., 2004) are usually ignored in neuropsychological theories of autism, with the notable exceptions of the Enhanced Perceptual Functioning Theory (Mottron et al., 2006; Mottron et al., 2010) and Extreme Male or Systemizing Theory (Baron-Cohen, 2002). As noted above the current research vista strongly associates autism with the stigma of mental retardation and low intelligence. However, is there really a foundation for this association? Taking into account the profound consequences of this type of stigmatization, it is a rather astonishing fact that typical diagnostic procedures for autism (such as the Diagnostic and Statistical Manual of Mental Disorders, the Autism Diagnostic Observation Schedule or Autism Diagnostic Interview) do not include a proper cognitive evaluation, but focus on the symptomatic triad of social impairments, communication deficits, and repetitive behaviors (American Psychiatric Association, 1980, 1987, 1994, 2000). Along this line, a meta-study evaluating 215 research articles published between 1937 and 2003 with claims about mental retardation found that 74% of these claims came from non-empirical sources, of which 53% were never traced back to empirical data. Astonishingly, most of the empirical evidence was published 25–45 years ago and based on measures of development rather than tests of intelligence or cognitive capabilities (Goldberg Edelson, 2006). But even when autistic intelligence is tested with approved intelligence tests it is necessary to interpret the results with caution. The most commonly used test to measure intelligence is the Wechsler Intelligence test and it was widely applied in autism research, yielding a characteristic profile of weak executive function, low working memory, and low abstracting skills (Happé, 1994). Autistic people would usually perform well only on one subscale, the Block Design test, which requires assembling a geometrical figure from memory. In general, however, the Wechsler Intelligence test is heavily based on verbal skills, command of language, and uses questions that require social and practical understanding. Based on this test, intelligence was generally classified as low in autism with exceptional islets of performance (Happé, 1994). However, this view is now being seriously challenged from a number of quarters. A recent study compared performance of autistic children and adults on the Wechsler and the Raven Progressive Matrices Intelligence Test. This test measures high-level analytical reasoning, such as inferring rules, managing hierarchical goals, and forming high-level abstractions in a presumably non-verbal way (Raven et al., 1998). In comparison to the Wechsler Test, autistic subjects had higher intelligence scores on the Raven Test and did not differ from control subjects, suggesting grossly under-estimated fluid intelligence and cognitive capabilities in autism (Dawson et al., 2007). Further support for under-estimated intelligence and abstracting skills stems from the Extreme Male Theory, which suggest that the prominent cognitive style is autism is prone toward analyzing the variables in a system, deriving the underlying rules that govern it and to construct, predict, and control systems (Baron-Cohen, 2002). This cognitive style is called “systemizing” and reads like a conglomerate of higher order cognitive abstracting capabilities that autistics were previously believed to be less capable of. Research by Baron-Cohen and colleagues suggests that autistics exhibit enhanced systemizing capabilities and evidence cited for this are savant talents, attention to detail, preference for rule-based, structured and factual information, higher scores on tests of intuitive physics, preference for toys such as cars, obsession with collecting items, obsession with closed controllable system, such as computers, and enhanced systemizing quotients (reviewed in Baron-Cohen, 2002).
Accumulation of this type of evidence poses a serious challenge to the current mostly deficit-oriented research approaches in autism. The Intense World Theory suggests a novel strategy for autism research of cognitive capabilities and proposes to study idiosyncratic excessive memory formation patterns in autism. Probably due to enhanced sensory processing and hyper-attention, the Intense World Theory explicitly predicts excessive and idiosyncratic memory formation in autism. While it needs to be clarified if this hyper-memorization is a consequence of hyper-perception and hyper-attention or a super-capacity on its own, learning and memorization patterns in autism should be clearly different from “normal” children and even vary substantially between autistic children, depending on their early life experiences and exposure to sensory material. While the case study literature and anecdotal accounts were always numerous and rich in documenting unconventional learning strategies and unexpected mnemonic capabilities in autistic children and adults – often discovered only accidentally by parents or care-takers (Baron-Cohen, 2003; Dawson et al., 2008), a recent large-scale survey of parents of over 144 autistic children revealed that 43% of these children exhibited exceptional memory capabilities for individually selective material. In up to 10% this pattern was even striking (Liss et al., 2006). It is easy to argue that hyper-plasticity at the columnar micro-circuit level may account for these exceptional memory achievements as well as the savant skills.
However, the Intense World Theory also draws attention to what may be a serious oversight in autism research and diagnosis: that is the downside of excessive memory processing. Excessive memory in low-level sensory and elementary cognitive regions could lead to an early over-specialization of feature processing and missed developmental opportunities to acquire a full spectrum of low-level processing strategies and to build higher order strategies. This might lead to a fragmented alphabet of feature processing capabilities in the vocabulary of sensory processing and impede the development of higher cognitive functions such as abstract thinking and language processing. The autistic person may also become locked into powerful memories that are difficult to correct or extinguish and that dominate every-day life. Quick and almost arbitrary association building based on enhanced perception of sensory features paired with excessive internal emotions – positive or negative – may rapidly lock the person down into behavioral routines. A failure to extinguish such associations may underlie the insistence on sameness and obsession with routines and may make rehabilitation difficult.
Finally, we would like to draw attention to fear memory formation in autism, a domain which has not yet received enough attention. While autism is clinically associated with enhanced anxiety and phobias (Muris et al., 1998; Gillott et al., 2001; Evans et al., 2005), the current scientific literature on fear memory formation in autism is sparse, consisting of only two recent publications on high-functioning autistic adults. Both of them report normal fear conditioning (Bernier et al., 2005; Gaigg and Bowler, 2007) and thus stand in striking contradiction to the clinically observed enhanced fears and anxiety. Based on the observed hyper-reactivity and hyper-plasticity in the amygdala and concomitant enhanced fear memory formation in the VPA model of autism (Markram et al., 2008), we argue that further research on fear memory formation in autism (such as studying younger age groups, using varied fear conditioning paradigms including controls for context) will provide insights into the underlying nature of withdrawal, social avoidance, and awkwardness.
Hyper-emotionality
The forth axis in the Intense World Theory is proposed to be hyper-emotionality as an inevitable consequence of limbic hyper-reactivity and hyper-plasticity. The amygdala, a key part of the limbic system, plays a pivotal role in modulating and regulating emotional responses (Davis and Whalen, 2001; LeDoux, 2003; Zald, 2003; McGaugh, 2004; Adolphs, 2006) and a malfunctioning in this particular brain region has been proposed to underlie the social deficits in autism (Baron-Cohen et al., 2000; Sweeten et al., 2002; Amaral et al., 2003; Bachevalier and Loveland, 2006; Schultz, 2005). For example, the Theory of Mind suggests that autistics are severely impaired in “reading other people's minds” and empathizing with other people (Baron-Cohen et al., 1985). This theory involves two elements: (1) the ability to attribute mental states to oneself and others, to be able to distinguish between oneself and others and realize that others have independent minds and may pursue different goals from oneself; (2) the ability to express an appropriate emotional reaction to the other person's mental state, thus to be able to empathize with the others’ mind. The proposed deficits in reading other people's feelings and thoughts and the lack in empathizing with other people have been commonly used to explain the impairments in social interactions and communication as well as inappropriate responses in social encounters even in high-functioning forms of autism such as in an Asperger. It was suggested that these deficits are mediated by a not sufficiently activated amygdala (Baron-Cohen et al., 1999; Critchley et al., 2000; Pierce et al., 2001). These and other data, such as post-mortem examinations of amygdaloid morphology (e.g., Kemper and Bauman, 1998; Schumann and Amaral, 2006), amygdala lesion studies in non-human primates (Bachevalier, 1994; but see also more recently, Emery et al., 2001; Prather et al., 2001), as well as comparison between amygdala-lesioned patients and autistics (Adolphs et al., 2001) have led to the Amygdala Theory of autism (Bachevalier, 1994; Baron-Cohen et al., 2000; Sweeten et al., 2002; Amaral et al., 2003; Bachevalier and Loveland, 2006). In its current version it implies that the amygdala is hypo-functioning, thus the autistic person does not “feel” enough or does not process socio-emotional cues sufficiently (reviewed in Markram et al., 2007b).
On the other hand there is evidence that the amygdala may be overly activated in autism. First, structurally the amygdala is enlarged in autism as early as 18 months of age and this enlargement persists throughout childhood until about 12 years of age (Sparks et al., 2002; Schumann et al., 2004; Mosconi et al., 2009). In adolescence the enlargement disappears (Schumann et al., 2004) and by early adulthood the amygdala may even end up smaller than in control subjects (Aylward et al., 1999; Rojas et al., 2004). These changes may reflect an over-activation of the amygdala in early childhood. Second, functional hyper-reactivity was demonstrated when autistic subjects are confronted with socially relevant stimuli, such as faces and eyes (Dalton et al., 2005; Kleinhans et al., 2009; Monk et al., 2010). For example, Kleinhans et al. (2009) showed that compared to controls the amygdala of autistic subjects exhibits attenuated habitation to facial stimuli and that increased amygdala-arousal in autistics was associated with increased social impairment. Monk et al. (2010) recently showed that right amygdala activation is enhanced in autistic subjects during face processing when controlling for attention, that is when the autistic subjects pay attention to the stimuli. Dalton et al. (2005) revealed that high-functioning autistics showed greater activation in the right amygdala when viewing familiar and unfamiliar faces and greater activation in the left amygdala and also in the left orbito-frontal cortex when viewing emotional faces. Both areas form part of the emotion circuit of the brain and increased reactivity to faces in these areas means a heightened emotional response to these stimuli. Autistics in this study also spent less time fixating the eyes region (deviant eye gaze is a core feature in autism – American Psychiatric Association, 2000). Moreover, in autistics, but not in controls, the amount of eye gaze fixation was strongly correlated with amygdala activation when viewing both, inexpressive or emotional faces (Dalton et al., 2005). This suggests that that eye gaze fixation is associated with emotional and possibly negative arousal in autistics and this could explain why autistics have “trouble looking other people in the eye.” Eye contact and watching the facial expressions are one of the first signs of cognitively healthy infants, are natural to people, and serve to build the basis for successful navigation through a social environment. For an autistic person however, these stimuli may be just too intense or even aversive to cope with and hence they are avoided. Obviously, continuous avoidance of a special class of cues will consolidate feature preference processing and prevent learning in this domain, thus some later developed social awkwardness and inappropriateness described in autism may be due to this lack of acquired knowledge. However, contrary to the deficit-oriented or disconnected Amygdala Theory and Theory of Mind of autism, we propose that the amygdala may be overtly active in autism, and hence autistic individuals may in principle be very well able to attend to social cues, feel emotions and even empathize with others or read their minds, but they avoid doing so, because it is emotionally too overwhelming, anxiety-inducing, and stressful.
The Intense World Theory proposes that amygdaloid hyper-reactivity and hyper-plasticity may in particular provoke a disproportional level of negative emotions and affect in autism, such as elevated stress responses and anxiety as well as enhanced fear memory formation. Enhanced phobias and anxiety levels were first noted by Kanner himself in his original case studies (Kanner, 1943) and later confirmed by population studies on children with autism (Muris et al., 1998; Gillott et al., 2001; Evans et al., 2005) and their relatives (Micali et al., 2004). A peek into the autistic world of increased anxiety, stress, and fear formation is delivered in the fascinating introspection of autistics Temple Grandin and Sean Barron, who vividly describe how anxiety and fear lead to social withdrawal and avoidance (Grandin and Barron, 2005).
In the research community, the idea of generally enhanced arousal levels in autism was brought forward in the mid sixties (Hutt and Hutt, 1965; Hutt et al., 1965) and since then enhanced autonomic activity, in terms of either enhanced reactivity to stimulation, diminished habituation to stimuli or enhanced baseline levels, was reported in autistics using skin conductance or cardio-vascular indicators (Palkovitz and Wiesenfeld, 1980; James and Barry, 1984; van Engeland, 1984; Barry and James, 1988; Hirstein et al., 2001; Ming et al., 2005) and increased stress responses were observed measuring stress hormone levels in the blood stream (Lake et al., 1977; Tordjman et al., 1997; Corbett et al., 2006). Interestingly, we have also observed increased levels of the stress hormone corticosterone in the VPA-exposed rat offspring in the blood stream (unpublished data). Thus, based on the human subject studies and our VPA rat model data the Intense World Theory suggests that the autistics perceive their surroundings not only as overwhelmingly intense due to hyper-reactivity of primary sensory areas, but also as aversive and highly stressful due to an overly reactive amygdala, which also makes quick and powerful fear associations with usually neutral stimuli – fear of a color for example. A natural coping strategy to deal with this kind of emotional overflow could be social avoidance and withdrawal. In further support of this view, decreased amygdala activation has been linked to genetic hyper-sociability (Meyer-Lindenberg et al., 2005), whereas increased activation is observed in social avoidance and phobia (Stein et al., 2002).
Hyper-connectivity
Autism was previously proposed as a disorder of brain connectivity (Belmonte et al., 2004a,b; Casanova, 2004; Just et al., 2004; Courchesne and Pierce, 2005a,b; Courchesne et al., 2005; Mottron et al., 2006; Geschwind and Levitt, 2007; Minshew and Williams, 2007). In human subjects, cortical connectivity can be studied either on the anatomical or the functional level. White matter volumes within or between brain regions indicate macro-anatomical connectivity. Functional connectivity between brain regions is usually assessed as the amount of temporal activity synchronization between these brain regions during task performance. By comparing synchronization states of autistic versus control subjects abnormal functional connectivity patterns are deduced.
Magnet resonance imaging studies show that white matter volumes are increased in young (2–3 years old) autistic children in the cerebrum (18%) and cerebellum (39%) (Courchesne et al., 2001). This increase is not uniform, but is most pronounced in the frontal, followed by the temporal and parietal lobes, whereas occipital lobes remain normal (Carper et al., 2002). In the same two studies, older autistic children did not differ from healthy children in terms of their white matter volumes, suggesting an initial overgrowth of connections, followed by an abnormally slow or arrested growth in later childhood and adolescence (Courchesne et al., 2001; Carper et al., 2002). Herbert and colleagues examined white matter in more detail in 6- to 11-year-old autistic children. They subdivided cerebral white matter into an outer radiate zone composed of intra-hemispheric cortico-cortical connections and an inner zone composed of sagittal and bridging compartments mainly containing the long-range ipsilateral association fibers, projection fibers to sub-cortical regions and inter-hemispheric commissural fibers. They found that while the long-range fibers in the inner zone were not affected in the autistic group, the more short-and middle-range fibers in the outer zone were increased in volume in the autistic children. Consequently, they concluded that short- and middle-range connections are overgrown in autistic children (Herbert et al., 2004). Post-mortem studies indicate a Minicolumnar Pathology in autism. Casanova et al. (2002) found that minicolumns are in general narrower in the frontal and temporal lobes of autistic brains than in “normal” brains. Both the core, holding the cell bodies, as well as the neurophil, holding unmyelinated axons from inhibitory interneurons as well as dendritic arborizations and synapses, were found to be reduced in size. Two important, albeit speculative, conclusions were drawn from these observations: (1) An increased number of processing units as well as loss of inhibition in the neurophil could lead to hyper-excitability (Casanova et al., 2002). (2) Increased numbers of minicolumns could contribute to increased short-range connectivity (Casanova, 2004).
While structural imaging and post-mortem studies suggest an overgrowth of short- and middle-range connections in autism, functional imaging studies suggest reduced functionallong-range connectivity during complex task processing between occipital and frontal or temporal lobes (Castelli et al., 2002), superior temporal and inferior frontal lobes (Just et al., 2004), parietal and frontal lobes (Horwitz et al., 1988; Just et al., 2007) as well as amygdala and parahippocampal gyrus (Welchew et al., 2005). This led to the functional Under-Connectivity Hypothesis of autism (Just et al., 2004), which states that long-range connections between brain regions are functionally impaired and thus complex information processing is not properly integrated across brain regions. Just et al. (2004) suggested that long-range under-connectivity may provide a useful framework to explain executive function deficits, theory of mind deficits, empathy deficits, complex information processing deficits, and weak central coherence in autism.
The Intense World Theory is in accordance with the observed anatomical short- and middle-range over-connectivity and it contributes to the connectivity debate the first direct evidence at the neurophysiological level that hyper-connectivity in the mini-columnar range could be a major pathology in autism (Rinaldi et al., 2008b). Furthermore, the Intense World Theory is based on empirical evidence demonstrating that hyper-connectivity within minicolumns produces hyper-reactivity and may render these basic processing units overtly sensitive, autonomous and more difficult to control. Thus, from this perspective, the observed functional under-connectivity data do not necessarily mean that functional long-range connectivity is weaker than in normal subjects, because the lack of activation and control of distant brain regions may be due to the difficulty to control and coordinate more autonomously active columns. Furthermore, the theory predicts that most tests conducted on autistic children would face a serious challenge of hyper-preference processing because the autistic person may actively shut down processing of selected features and tasks and prefer to perform different tasks, which could easily be confused with generalized hypo-functionality of a brain region or pathway. An illustration of this point is discussed in the next section regarding the fusiform face area and how conventional stimuli may fail while non-conventional stimuli may succeed to activate a brain region (Grelotti et al., 2005).
Hyper-reactivity
Autism was described as a disorder of chronic hyper-arousal already several decades ago (Hutt and Hutt, 1965). Several studies suggested enhanced central (Hutt et al., 1964, 1965; Hutt and Hutt, 1965) or autonomic (Palkovitz and Wiesenfeld, 1980; James and Barry, 1984; van Engeland, 1984; Barry and James, 1988; Hirstein et al., 2001; Ming et al., 2005) arousal levels in autism, which is indicative of increased neuronal reactivity toward stimulation and/or increased basal activity levels. Motivated by the need to explain the high tendency for seizures and the sensory hyper-sensitivity more recent hypotheses suggested autism to be characterized by an imbalance of excitation and inhibition in the neocortex (Hussman, 2001; Casanova et al., 2002, 2003; Rubenstein and Merzenich, 2003), with excitation winning over inhibition and leading to hyper-reactivity. While neuronal hyper-reactivity can be explained by reduced inhibition (Hussman, 2001; Casanova et al., 2003) and many other mechanisms (Rubenstein and Merzenich, 2003), the experiments on which the Intense World Theory is based rule out most of these mechanisms except a striking hyper-connectivity. These studies also rule out an imbalance in excitation and inhibition in the neocortex (Rinaldi et al., 2008b), but do find that inhibition is reduced in the amygdala instead (Markram et al., 2008). A loss of inhibition is speculated in brain regions where the inhibitory neurons are present in higher fractions than in the neocortex since glutamatergic hyper-connectivity may not be a sufficient driving factor for hyper-reactivity. This prediction is consistent with a significant loss of inhibitory Purkinje cells in the cerebellum (Ritvo et al., 1986; Rodier et al., 1996; Bailey et al., 1998; Kemper and Bauman, 1998).
How does the postulate of overall neocortical hyper-reactivity fit with functional neuroimaging and EEG studies that seem to suggest hypo-activation and hypo-functionality in higher order brain areas during task processing (Courchesne et al., 1984; Ciesielski et al., 1990; Muller et al., 1998, 1999; Baron-Cohen et al., 1999; Ring et al., 1999; Townsend et al., 1999; Critchley et al., 2000; Schultz et al., 2000; Pierce et al., 2001, 2004; Castelli et al., 2002; Luna et al., 2002; Belmonte and Yurgelun-Todd, 2003; Boddaert et al., 2003; Hubl et al., 2003; Gervais et al., 2004; Just et al., 2004)? The Intense World Theory proposes several possible explanations for this apparent contradiction: Firstly, hyper-functional columns in the primary areas could persistently interrupt high order processing with their runaway processing. Secondly, high order areas must face a major challenge to orchestrate hyper-reactive and hyper-plastic columns, which may be impossible in severe cases of autism. Thirdly, a lack of synchrony or coherence in low order areas, do not necessarily imply deficits within higher order areas. Fourthly, functional imaging is baseline-based and these relative measures do not accurately reflect absolute levels of activity. Thus, what appears as hypo-activation in comparison, may actually be higher activation on absolute terms. Fifthly, the Intense World Theory predicts that there could be extreme feature preference selection and active avoidance, which could severely limit the tasks that are valid comparisons for each autistic subject.
The Intense World Theory therefore predicts that there could be hypo-reactivity to many conventional stimuli that drive normal brains, but hyper-reactivity to highly selective sets of preferred stimuli depending on the unique interests and earliest impressions of the specific autistic individual. This could drive highly idiosyncratic activation patterns in autistics. An example for such idiosyncratic activation pattern stems from the fusiform face area. As suggested by its name, in normal subjects this area is highly reactive to faces (for example Haxby et al., 1994; Puce et al., 1995; Kanwisher, 2000). In autistic subjects, the fusiform face area is usually observed to be hypo-reactive (Critchley et al., 2000; Pierce and Courchesne, 2000; Hall et al., 2003; Hubl et al., 2003; Piggot et al., 2004; Wang et al., 2004), which has been taken as evidence for abnormal face perception and social development in autism. However, this observed hypo-functional area may activate to a different set of stimuli in autistics. For example, Grelotti et al. (2005) studied a boy with autism who was an expert at distinguishing cartoon characters known as “Digimon.” This boy exhibited reduced activation of the fusiform face area to faces, but actually enhanced activation to the Digimon images (Grelotti et al., 2005). The point is that conventional stimulus paradigms may reveal a specific task deficit, but not necessarily a brain deficit in general, that is reduced reactivity toward conventional stimulation may not be indicative of lower functionality of this brain area per se. The main problem here and the challenge in the study of autism in general is that conventional stimulus paradigms may be appropriate to reveal the effective functional deficits for a specific task, but not optimal and valid to dissect out the neuropathology of autism.
Why should the autistic brain exhibit such idiosyncratic stimulus patterns and is this not an argument that could hold for any individual's brain? The autistic brain is particular, due to the proposed hyper-reactivity and hyper-plasticity in circumscribed and autonomously acting microcircuits. Enhanced processing and memorization during early development may imprint the young autistic brain with very idiosyncratic memory pathways and lead to passive and active avoidance of certain classes of stimuli – in particular those with high arousal levels (faces, eyes, social situations, novel environments, etc) and at the same time peculiar, almost random preference for other classes of stimuli (certain food, certain routines, etc). These preferred stimuli and interests may activate even higher order regions at normal and even above normal levels. An additional factor that is brought up by the Intense World Theory is that the autistic brain may actively avoid certain stimulus patterns and tasks as part of the aversive component brought in by hyper-emotionality. Active avoidance implies powerful gating of activity that may not even be within the capability of normal subjects.
Hyper-glutamatergia
Studies on the VPA rat model of autism revealed an over-expression of the NMDA receptors in the neocortex, in particular the receptor subunits NR2A and NR2B, as well as the CAM-kinase-linked second-messenger pathway and these increases are correlated with a hyper-plasticity of synaptic connections (Rinaldi et al., 2007). The Intense World Theory therefore proposes that blocking NMDA receptors in particular may reduce the hyper-plasticity component of the autistic brains and therefore alleviate some autistic symptoms. Improvements should be best observed in behavioral routines and obsessive preferences and dislikes, which should be manifest consequences of increased absorption and storage of information. From the Intense World Theory perspective, lessening of the excessive processing and storage capability of the autistic brain should also lead to a general “opening-up” to the world and environment. However, it is not yet clear whether elevated NMDA receptor levels occur in other parts of the brain. Preliminary studies in our laboratory show that other glutamatergic receptors may be elevated at different developmental stages as well (unpublished data). Since it seems that a molecular syndrome is active rather than a specific molecular pathway, it may be that different brain regions achieve hyper-plasticity using different mechanisms.
Glutamate was introduced into the search for the neurotransmitter abnormality in autism by proposing autism as a hypo-glutamatergic disorder (Carlsson, 1998). This was not based on measurements of glutamate levels, receptor expression or activation of glutamatergic neurons, but on the observation that glutamatergic antagonists may lead to sensory distortions and hyper-sensitivity similar to those observed in autism. The glutamatergic hypothesis swung to the opposite end to propose that autism could be a hyper-glutamatergic disorder (Belsito et al., 2001). This was also not based on specific measurements, but on reports that glutamate is important for brain development, and excessive glutamate expression can cause toxicity and damage the brain, and since the autistic brain was considered severely damaged (the a priori assumption), it is important to block glutamate release. Based on this reasoning, clinical trials were begun to reduce glutamate transmission. At about the same time two studies indicated abnormal glutamate blood levels (see below). However, the drug lamotrigine, an anticonvulsant that attenuates glutamate release, but that also has numerous other effects including blocking Na+ channels (Coulter, 1997), was not successful in attenuating autistic symptoms (Belsito et al., 2001). On the other hand, treatment of autistic children with amantadine hydrochloride, a partial NMDA receptor antagonist (and also as an indirect dopamine agonist), usually used in young children for influenza prophylaxis, was able to ameliorate some autistic symptoms, such as hyperactivity and language problems (King et al., 2001a,b). Further support for the beneficial effects of NMDA receptor blockade in autism stems from a more recent clinical study with memantine, a moderate NMDA receptor antagonist (Chez et al., 2007). In an open-label add-on therapy memantine was administered to 151 patients with prior diagnoses of autism or Pervasive Developmental Disorder Not Otherwise Specified over a 21 month period. Improvements were observed in language function, social behavior, and self-stimulatory behaviors (Chez et al., 2007). However, the results with NMDA receptor agents are not yet straightforward and conclusive since another single-blind study on 10 autistic subjects with D-Cycloserine, a partial NMDA receptor agonist at the glycine-B site of the NR1 subunit, also produced improvements in social behavior (Posey et al., 2004). Within the framework of the Intense World Theory blocking excessive processing and memory formation would lead to an amelioration of the symptoms. Thus, we suggest that blockage of the glutamatergic system, possibly through NMDA receptors, may provide a promising treatment strategy for autism.
Amino acids such as glutamate and aspartic acid or glutamate receptor levels in the brain (Purcell et al., 2001; Page et al., 2006; DeVito et al., 2007) or in blood samples (Moreno-Fuenmayor et al., 1996; Aldred et al., 2003; Shinohe et al., 2006) were not measured until more recently. One post-mortem study compared glutamate receptor expression in autistic and control brains and revealed that the mRNA levels of the AMPA receptors GluR1, GluR2, and GluR3 were increased in the cerebellum of autistics. At the protein level GluR1 and the NMDA receptor subunit NR1 were also found to be increased in the cerebellum of autistic subjects. However contrary to the increase at the gene and protein level, determining the density of glutamate receptors by autoradiography in the cerebellum, prefrontal cortex, and caudate putamen revealed a significant density decrease of AMPA receptors in the cerebellum of autistics. NMDA receptor density was normal throughout these brain regions (Purcell et al., 2001). Measurement of NMDA receptor levels may however yield ambiguous results, because it possible that NMDA receptor expression is stimulus-dependent. This could explain high levels during brain development as observed in VPA-exposed rat offspring (Rinaldi et al., 2007). These high levels might relax with aging, but could be more readily induced.
Furthermore, both the mRNA and protein levels of EAAT 1 and EAAT 2 were found to be significantly increased in the autistic cerebellum (Purcell et al., 2001). EAAT 1 and EAAT 2 are predominantly located on astroglia and their main function is to remove glutamate from the extra-synaptic space (Danbolt, 1994). Since the protein level and activity of glutamate transporters are controlled by the extracellular glutamate concentration or by activity-dependent mechanisms (Levy et al., 1995), increased EAAT 1 and EAAT 2 protein levels may be due to increased extracellular glutamate concentrations in autism. Using proton magnetic resonance spectroscopy, higher concentrations of glutamate were also reported in the amygdala/hippocampal pathway, but not the parietal cortex (Page et al., 2006). Yet another recent study reported reduced composite levels of glutamate/glutamine widespread in cerebral and cerebellum gray matter (DeVito et al., 2007).
Glutamine, the precursor to glutamate, has been reported to be deregulated in autistic brain tissue and blood samples (Rolf et al., 1993; Moreno-Fuenmayor et al., 1996; Aldred et al., 2003; Page et al., 2006; DeVito et al., 2007; but see also Shinohe et al., 2006). Glutamate (Moreno-Fuenmayor et al., 1996; Aldred et al., 2003; Shinohe et al., 2006) and aspartic acid (Moreno-Fuenmayor et al., 1996) were also found to be increased in the blood of autistics (but see also Rolf et al., 1993).
Several genetic studies point to the involvement of the glutamatergic system in autism. Single nucleotide polymorphisms (SNPs) in the genes encoding ionotropic glutamate 6 receptors (GluR6; Jamain et al., 2002; Shuang et al., 2004) and the metabotropic glutamate receptor 8 (mGluR8; Serajee et al., 2003) were reported in autistic subjects. SNPs were also found in autistic subjects within SLC25A12 (Ramoz et al., 2004; Segurado et al., 2005), a gene encoding the mitochondrial aspartate/glutamate carrier (AGC1). Even though larger follow up-studies were not able to confirm the association of SLC25A12 with autism (Blasi et al., 2006; Rabionet et al., 2006), two more recent studies again suggest a link between SLC25A12 and autism: one study reported an increase of SLC25A12 expression in prefrontal and cerebellar post-mortem tissue of autistic brains (Lepagnol-Bestel et al., 2008) and another study suggested a link between SLC25A12 expression and in behavioral routines in autism (Silverman et al., 2008). A reason for the contradictory findings could be differences in sample composition regarding age and gender inclusion (the latter in particular for control samples).
Several glutamatergic synapse gene mutations on chromosome 22 were also associated with autism. These include neuroligin 3 and 4 (Jamain et al., 2003; Laumonnier et al., 2004), SHANK3 (Durand et al., 2007), and MAPK8IP2 (also termed IB2 or JIP2; Giza et al., 2010). Neuroligins are cell adhesion molecules expressed postsynaptically and bind to the presynaptically expressed neurexins. Interactions between neuroligin and β-neurexin increases the size and number of presynaptic terminals (Levinson et al., 2005) and triggers the recruitment of presynaptic (Scheiffele et al., 2000) and postsynaptic molecules (Graf et al., 2004; Nam and Chen, 2005). Neuroligins can also bind to SHANK3 (Meyer et al., 2004), a scaffolding protein found in the postsynaptic density (PSD) of excitatory synapses. Due to their ability to form multimeric complexes with postsynaptic receptors, signaling molecules and cytoskeletal proteins they are thought to function as organizing molecules (Naisbitt et al., 1999; Boeckers et al., 2002). IB2 (for Islet Brain-2) is a the protein expressed in neural and neuroendocrine cells throughout the brain (Negri et al., 2000) and its disruption has been associated with reduced AMPA-mediated transmission and enhanced NMDA-mediated transmission in the cerebellum (Giza et al., 2010). Thus, mutations in these glutamatergic pathways could lead to some form of hyper-excitability or hyper-plasticity in autism, a hypothesis which would require further testing.
The metabotropic glutamate receptor 5 (mGluR5) has also been linked to autism and hyper-plasticity (Huber et al., 2002) via the genetic mutation of fragile X mental retardation 1 (FMR1) gene (for a more detailed discussion see next section).
Driven by the toxicity reasoning, a more recent study links hypo-digoxinemia often found in autism, with excessive activation of NMDA receptors. Digoxin is a hormone released from the hypothalamus, which inhibits the Na+K+-ATPase pump with a concomitant effect of maintaining the extracellular levels of Mg2+. Since Mg2+ blocks NMDA receptors, this is proposed to cause excessive activation of NMDA and hence the feared toxicity and brain damage (Kurup and Kurup, 2003). While excessive activation of NMDA can cause toxicity in the adult, in the neonate it is more likely to accelerate the environment-driven nurturing of brain development and acquisition of new memories. Lowered Mg2+ will also affect synaptic transmission of all transmitters in the entire brain and alter the excitability of all neurons by lowering surface charges and hence voltage dependencies, and so it is neither possible nor valid to link any potential effects of hypo-digoxinemia to autism only through the potential effects on NMDA receptors.
In summary, anything seems possible when theories of the neuropathology of autism are generated by top-down inference from cognitive deficits to brain regions and cellular and molecular alterations. The important change that would temper much of the wild speculation is to begin at the bottom and work up toward the emergent systems, behavioral, and cognitive level deficits. A hyper-glutamatergia in general and hyper-NMDA receptor expression in particular needs to be confirmed in human subjects and the different brain regions affected need to be examined.
Hyper-plasticity
The Intense World Theory claims enhanced learning and memory process in autism, based on amplified synaptic plasticity (Rinaldi et al., 2007; Silva et al., 2009). In human subjects, cellular plasticity can be induced with theta-burst stimulation using trans-cranial magnetic stimulation (TMS) and recent studies demonstrate that long-term potentiation as well as long-term depression are greatly enhanced in autistic subject (Oberman et al., 2009, 2010), which is the first proof of hyper-plasticity proposed by the Intense World Theory in human autism.
More traditionally, cellular plasticity is studied with invasive techniques in animal brains and based on this approach autism research focuses mainly on measuring synaptic communication and plasticity in animal models of autism and relating the result to the human condition. However, currently there is a fundamental drawback in this research, which is not technical, but ideological in nature: most animal models of autism assume a priori a cognitive or emotional impairment and consequently neural under-functioning as the basis for autism and thus only such animal models are conceived and only such results seem to get published that are based on this deficit-focused conception of autism. Currently, to our knowledge, apart from the VPA rat model of autism, only two mouse mutant models were extensively characterized in terms of cellular dynamics – the Rett model and the Fragile-X model, both of which are oriented toward explaining the implicit postulation of learning impairments and mental retardation in autism.
Rett syndrome (RTT) is an X-linked dominant progressive neurodevelopmental disorder affecting girls and associated with severe mental retardation. It is caused by loss-of-function mutations in the gene encoding methyl CpG binding protein 2 (MeCP2; Amir et al., 1999) and a mutation is this gene is observed in at least 80% of all RTT cases (Buyse et al., 2000; Dragich et al., 2000; Huppke et al., 2000). Because RTT has resemblance with autism, such as initially normal development with sudden regression around the age of 3, stereotyped behavior and impaired social communication, but also because of the popular postulate that over 70% of autistic cases are also mentally retarded, a MeCP2 mutation has also been speculated to underlie the assumed learning disabilities and/or other symptoms in autism (Lam et al., 2000; Vourc'h et al., 2001; Beyer et al., 2002; Carney et al., 2003). Up to date only 3 out of 301 studied autistic patients have been identified with a MeCP2 mutation (Lam et al., 2000; Vourc'h et al., 2001; Beyer et al., 2002; Carney et al., 2003), which excludes a MeCP2 mutation as a common genetic cause of autism. Mice with a mutated MeCP2 gene have been generated (Guy et al., 2001) to study possible neuronal alterations underlying RTT and possibly autism (Dani et al., 2005; Asaka et al., 2006; Chang et al., 2006; Moretti et al., 2006; Medrihan et al., 2008). MeCP2 ko mice exhibited impaired LTP and LTD in the hippocampus (Asaka et al., 2006; Moretti et al., 2006) and additionally also impaired LTP in the primary motor and sensory cortices (Moretti et al., 2006), which is commonly interpreted as being in line with the severe mental retardation observed in RTT and extrapolated to autism as well.
Fragile X syndrome is a syndrome of X-linked mental retardation, caused by a mutation in the FMR1 gene that encodes the fragile X mental retardation protein (FMRP). Aside from the most prominent feature of intellectual disability, behavioral symptoms may include abnormal speech patterns, stereotypic movements (e.g., hand-flapping) and abnormal social behavior, in particular shyness and limited eye contact. The overall prevalence of Fragile X Syndrome in autism is on average 4% (reviewed in Dykens and Volkmar, 1997), which – even though higher than in the normal population – also excludes Fragile X as a common cause of autism. However, since the prevalence of autism in the Fragile X Syndrome population is high with rates of 15–33% or even more (for review see Hagerman et al., 1986; Dykens and Volkmar, 1997; Rogers et al., 2001; Hayashi et al., 2007), it has been suggested that a mutation in the FMR1 gene may also underlie autism (most recently for example Hayashi et al., 2007; Bear et al., 2008). To further study the neurobiological causes of Fragile X Syndrome a mutant mouse was created lacking the expression of the FMR1 gene (Consortium, 1994). FMR1 ko mice typically exhibit elongated dendritic spines and an increase in spine density (Irwin et al., 2002; Galvez and Greenough, 2005; McKinney et al., 2005; Hayashi et al., 2007), which indicates a hyper-connected and potentially hyper-reactive network. These findings are similar to those found in the VPA model and may suggest that hyper-reactivity of neural microcircuits is also a core feature of Fragile X and may explain the high degree of comorbidity with autism.
In terms of synaptic plasticity, FMR1 ko mice exhibit a pattern of impaired LTP in the neocortex (Li et al., 2002; Larson et al., 2005; Hayashi et al., 2007), but normal LTP in CA1 of the hippocampus (Godfraind et al., 1996; Paradee et al., 1999; Li et al., 2002; Larson et al., 2005) and finally increased LTD in hippocampal CA1 (Huber et al., 2002) and cerebellum (Koekkoek et al., 2005). There is indication that loss of FMRP leads to disinhibition of the mGluR5, which may trigger exaggerated synthesis of certain proteins (Zalfa et al., 2003) and account for the increased mGluR-dependent LTD in the CA1 region of the hippocampus (Huber et al., 2002), which has led to the mGluR Theory of Fragile X (Bear et al., 2004). While the theory is intriguing and opens up a potentially powerful treatment strategy for Fragile X, it remains to be established how much FMRP and mGluRs contribute to autism as well. These findings would indicate synapses are dampened down in Fragile X which is the opposite to the findings in the valproate model where LTP is enhanced both in the neocortex and amygdala. We have not tested whether LTD is also enhanced, but enhanced LTP supported by a massive over-expression of NMDA receptors at synapses indicates that individual synapses can become excessively strong in autism. The animal model of Fragile X is therefore not the same in this respect to the valproate animal model. It is possible therefore that Fragile X and autism share the core pathology of hyper-reactivity, but not hyper-plasticity that enhances synaptic strength.
The first, and perhaps most important, step toward a unifying theory of autism is to turn from the traditional view of impaired intellectual capabilities and the popularized stigma of mental retardation because this view excludes a large body of scientific, anecdotal data, and alternative interpretations of enhanced brain functions. This could be a crucial step since virtually all patented treatments and pharmaceutical therapies for autism aim to enhance cognitive capabilities (Nakamura, 2002; Yoo et al., 2007). However, in the light of the Intense World Theory these treatments strategies may have to be reversed and aimed at reducing the hyper-functional cognitive and emotive system.
Progression and Treatment According to the Intense World Theory
The central claims of the Intense World Theory are based on a synergy of data from the VPA rat model of autism as well as data and observations gathered from the human autism literature. As such, the central claims of hyper-reactivity and hyper-plasticity on the neurophysiological level as well as hyper-perception, hyper-attention, hyper-memory, and hyper-emotionality on the psychological level, have to be substantiated in systematic and controlled experiments on human subjects. Thus, it is only prudent to take the predictions on the progression of autism as well as how to best treat autism with extreme caution until such controlled experiments are conducted. However, if it turns out that some or even all of the central claims of the Intense World Theory turn out to be valid, the implications for the progression as well as the treatment of autism are quite far-reaching and in the case of treatment even counter-intuitive to best-intended parental or professional strategies and are thus worth-while of a discussion.
The Intense World Theory proposes a particular form of brain hypertrophy triggered by an epigenetic insult, which may render the brain excessively reactive to the environment. The excessive reactivity and rapid memory formation of experiences boosted by an amplified emotional component may trigger the acceleration of brain maturation until the environment becomes painfully intense. The intensity of lower-order sensory processing becomes so overwhelming that the autistic may actively avoid specific features that have become negatively associated and focus on elementary features that have become positively associated. Because of the powerful and persistent memories each experience is predicted to systematically drive the autistic child into a world that is secured by only letting in positively associated stimuli and actively avoiding any surprises. Since new information must necessarily be surprising, autistics could rapidly become resistant to rehabilitation. If the intensity peaks too early and the shutdown is triggered, the autistic may not learn to assemble elementary information into higher order concepts needed for abstract thinking and language. Driven by a painfully intense world, the autistic brain progresses functionally to become highly fragmented with islets of excessive processing which manifest in arbitrary preferences and tasks that may be exceptionally well processed. The plasticity that follows may lead to irreversible structural changes and fragmentation of the brain. The prognosis is proposed to become worse in a sensory enriched and dynamically changing world. It is also likely that providing an enriched environment and a directive teaching and aggressive rehabilitation program may in fact accelerate the progression of the disorder. The prognosis may be improved by filtering sensory and emotional extremes, preventing surprises, and pharmacologically by suppressing sensory reactivity and memory formation. The disorder might even be preventable if intervention begins before the “intense world” reaction is triggered, that is before critical periods of neurodevelopment. Critical periods are moments when circuit architectures consolidate and gradual exposure to a more normal world may not have such adverse effects after these critical periods. There may therefore be a preventative time window for autism. Reversing consolidated hyper-functional circuits after these critical periods will be more difficult, but due to the potential for learning and memory, an extinction-based rehabilitation program may be effective.
Behavioral treatment according to the Intense World Theory is proposed to focus on filtering the extremes in the intensity of all sensory and emotional exposure as well as relaxation and progressive systematic desensitization to stimuli presentation. The probably most counter-intuitive suggestion that emerges from the Intense World Theory is to surround the child with a highly predictable and calm environment protected from abrupt sensory and emotional transients and surprises for the first years of life to prevent excessive sensory and emotion driven brain development. The child should be introduced to new stimuli and tasks gently and with caution, retracting at any sign of distress. The adoption of a responsive rehabilitation program would ensure that the teacher works carefully to avoid triggering adverse reactions. Introduction to strangers should be controlled, brief, indirect, and as inert as possible.
Pharmacological treatment according to the Intense World Theory should focus on reducing brain reactivity in general, blocking memory formation, reducing stress responses, and enhancing memory extinction. Treatment should be applied as early as possible and should last until after the completion of the critical periods of brain development (probably beyond the age of 6). Such critical periods are often irreversible milestones and hence the disorder might be avoidable if these milestones can be crossed without triggering or further driving the autistic child to seek refuge in a limited albeit secure world. Treatment for elderly children and adults with autism would be more difficult to reverse but memory attenuation and extinction-based rehabilitation programs as well as reducing anxiety levels and stress responses may at least partially reverse or ameliorate the pathology. An important consideration in any rehabilitation program as predicted by the Intense World Theory is the complication of hyper-emotionality, which may be well masked from the observer and which would demand even greater care in how the autistic is handled. Punishments may be greatly amplified for the autistic and imprinted rigorously and indefinitely into the future.
Predictions of the Intense World Theory
The Intense World Theory is not only consistent with a large number of previous studies, and has the explanatory power to reconcile a large number of apparently contradictory data and interpretations, but also has significant predictive power because it is grounded at the molecular, cellular and circuit levels. The set of some testable predications derived from the Intense World Theory are listed below.
Circuit level:
Some level of hyper-reactive and hyper-plastic circuits in all brain regions. This can be tested by progressively increasing the intensity of multi-site stimulation of a brain region. The molecular, synaptic, and cellular causes may differ in different regions.
The loss of inhibition and hence hyper-reactivity also in sub-cortical regions where inhibitory neurons are more prominent such as in the cerebellum.
System level:
The variance, as opposed to the mean, of the pixel intensities in fMRI measures should be higher in autistic subjects with the upper and lower intensities being greater than in neurotypical subjects. Higher upper pixel intensities may even be present in under-activated brain regions. These predictions would best be tested using fMRI at the highest spatial resolution.
Enhanced low-level feature processing in primary sensory brain regions. This could manifest in greater peaks in some tuning curves for different features as well as lower detection thresholds.
Cognitive and emotional levels:
Highly idiosyncratic stimulus preference to activate brain areas, which should become more idiosyncratic with progression of the disorder.
Vulnerability to sensory overflow. Consequential behavior would be panic, aggression, and withdrawal as already suggested by anecdotal reports.
Associated Pathologies:
A lower threshold for stimulus-induced epilepsy (such as stroboscopically induced epilepsy).
Neglect syndrome.
Learning impairments correlated to the degree and variance of associations required.
Panic attacks and phobias triggered by uniquely personal situations (as originally noted by Leo Kanner).
Post-traumatic stress disorder.
Circuit level:
Abnormally high levels of persistent reverberant activity within circuits that is difficult to interrupt.
Systems level:
Amplified neocortical response during thalamic stimulation.
Abnormally high degree of synchronization between the mPFC with some brain regions and abnormally low synchronization, with other regions.
Abnormally high tendency for phase locking with smaller phase lags indicating a higher degree of oscillations and coherence across those brain areas engaged.
Cognitive level:
Enhanced sustained attention to material of interest (as anecdotally reported).
Once attention is captured, impaired shifting of attention to different features or tasks, because of internal hyper-processing (as also suggested by the difficulty to engage the attention of an autistic child).
Attention to arbitrary fragments related strongly to early life experiences.
Strongly enhanced focused on internal processes.
Associated Pathologies:
Attention deficit disorder (ADD) or Attention deficit hyperactivity disorder (ADHD).
Learning impairments in some cases and super-learning in others.
May seem distracted and disengaged, but are actually hyper-focused on internal processes.
Molecular:
Enhanced NMDA receptor subunit expression in the neocortex. Alterations in the adult, after critical periods, may be absent and increases may still be observable following stimulation.
Other markers associated with synaptic plasticity may follow a similar pattern as some NMDA receptor subunits.
Cellular:
NMDA mediated Ca2+ toxicity in adult and aged autistics. This will have consequences for the enhanced learning and memory postulate of the Intense World Theory, which may be less pronounced in aged autistics.
Cognitive level:
Strong imprint of early life experiences and learning material, which is highly resistant to extinction and dominates over new learning challenges. E.g., this could potentially show in high interference in learning semantic material.
Enhanced simple associate learning (conditioning), which is resistant to extinction. This should be particularly well observable early in life. Later in life strong early life memory traces will over-shadow further learning attempts.
Learning impairments are predicted for all negatively associated stimuli and resistance to rehabilitation is predicted because of fear of previous negative associations. The Intense World Theory predicts that all children with autism will display greater than average memory performance for the tasks that the autistic has chosen as non-threatening.
Enhanced learning and memory should be most observable in very young autistic children and less so in progressively older autistics. In elderly autistics – due to the neurotoxicity effects of enhanced NMDA signaling throughout in particular the early lifespan – learning and memory impairments may become more evident.
Related Pathologies:
Idiosyncratic, albeit exceptional memory capabilities.
Repetitive tendencies.
Obsession compulsive disorder.
Stronger and more persistent memories following conditioning.
Behavioral inflexibility.
Cellular:
Cell toxicity in brain areas that are easily excitable such as the amygdala and hippocampus due to over-excitation.
Circuit:
Hyper-reactive and hyper-plastic amygdala and other limbic structures.
Systems:
Enhanced amygdala activation when autistics are instructed by teachers or parents.
Enhanced amygdala activation when autistics view eyes, faces or social scenes.
Enhanced sympathetic responses to social content such as eyes, faces, social situations etc. This could be tested with measures of the autonomic system, such as skin conductance, heart measures or stress and anxiety hormone levels.
Enhanced sympathetic responses to novel and sensory rich situations.
Enhanced sympathetic responses to negatively associated stimuli.
Cognitive level:
Enhanced fear conditioning in autistics – most pronounced in young children.
Enhanced generalization of fear responses to similar content.
Resistance to fear extinction.
Enhanced active avoidance of fear-associated material.
Enhanced active avoidance of high-emotion content such as eyes, emotional faces, social encounters.
Enhanced active avoidance of novel environments is predicted due to fear of surprises that arise from over-generalization of previous negative associations.
At the behavioral level unpredictable, exaggerated, extreme, and inappropriate reactions to surprising situations.
Associated Pathologies:
Anxiety.
Panic attacks and phobias.
Post-traumatic stress disorder.
Paranoia.
Depression.
Hyper-emotionality.
Behavioral inflexibility.
Repetitive tendencies.
The Intense World Theory predicts higher variance in any set of scores from a large psychophysical test battery as compared to controls with the highest and lowest scores. In particular, since columns are hyper-reactive, but highly selective, the reaction times to a battery of tests should display a higher variance with the longest and shortest reaction times. The theory predicts that the key parameter to measure is the variance of performance on different scores rather than focusing on any one task, on absolute values, or on the average performance of many tasks. The theory predicts the same high variance characteristic for cognitive profiling.
oHigh measures of variance on cognitive battery tests should be independent of intelligence, but due to the proposed highly idiosyncratic feature over-selectivity of neural columns.
Falsifying the Intense World Theory
The Intense World Theory is merely a theory. The importance of a theory comes from its power to unify and account for past data and reconcile inconsistencies and the value of a theory comes from its testable predictions. A theory must also be falsifiable. A large number of studies will have to be carried out to validate or invalidate this theory. In particular experiments need to be performed that verify that the core neuropathologies found in the animal model are also valid in human autism. This will become possible with a molecular marker of hyper-reactivity and hyper-plasticity in post-mortem tissue or perhaps with high-resolution fMRI that is becoming possible with the 9.4 Tesla machines. The genetic vulnerability for insult-induced autism must also be tested. Typically genetic mutations are chased as causes of autisms, which have revealed dozens of candidates – we propose that they may rather be setting the threshold for epigenetic insult. The theory can be falsified if a single gene mutation is found to cause all variants of autism, if neural circuits are not found to be hyper-reactive and hyper-plastic, if glutamatergic synapses are hypo-functional in autism, if these core neuropathologies are found not to cause hyper-functionality of local microcircuits, if hyper-functionality of local neural microcircuits are found not to cause abnormal perception, attention, memory, and emotional processing and if these core cognitive-emotive functions are found not to be affected at least to some measurable extent, in all cases of autism.
Conclusions
Our earlier and on-going studies on the valproate rat model of autism suggested a coherent theme of alterations from the molecular to the behavioral level. We applied this single theme to previous studies, hypotheses, theories, and even anecdotal reports on human autism and found that this theme can potentially explain these ideas and reconcile contradictory evidences. The theme is therefore a potentially unifying theory for the neurobiological and affective-cognitive basis of autism – the Intense World Theory of autism. Different from previous theories, the Intense World Theory is driven from a neurobiological perspective where we attempt to reconstruct the disorder from fundamental molecular, cellular, and circuit changes. This theory is by far not a complete explanation of the cause of autism, but it provides a coherent multi-level framework for a complete explanation. On a neurobiological level, the Intense World Theory of autism proposes excessive functioning of neural microcircuits, with the main symptoms being hyper-reactivity and hyper-plasticity and together, hyper-functionality. On a perceptual and cognitive level this excessive functioning of local neuronal circuits may lead to an intensely perceived world, which may turn aversive and highly stressful if the amygdala and other parts of the limbic system are also affected.
In contrast to other deficit-oriented theories of autism, the Intense World Theory points out that enhanced brain functioning may lie at the heart of autism. In this light, autistic individuals may in general – and not only in exceptional cases – exhibit enhanced perception, attention, and memory capabilities and it is in fact these capabilities, which may turn the world too intense and even aversive and lead to many of the autistic symptoms including withdrawal and social avoidance. Thus, while enhanced brain functioning may be debilitating, the hopeful insight offered by the Intense World Theory is that the right treatment strategies of cocooning the autistic infant to protect from surprising situations, and dampening brain functioning in early development to prevent driving the brains circuits into an irreversible trajectory, may reveal truly capable and highly gifted individuals who can integrate with their social environment successfully.
Statements
Conflict of interest
The authors declare that this review was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
Adolphs R. (2006). How do we know the minds of others? Domain-specificity, simulation, and enactive social cognition. Brain Res.1079, 25–35.10.1016/j.brainres.2005.12.127
2
Adolphs R. Sears L. Piven J. (2001). Abnormal processing of social information from faces in autism. J. Cogn. Neurosci.13, 232–240.10.1162/089892901564289
3
Aldred S. Moore K. M. Fitzgerald M. Waring R. H. (2003). Plasma amino acid levels in children with autism and their families. J. Autism Dev. Disord.33, 93–97.10.1023/A:1022238706604
4
Amaral D. G. Bauman M. D. Schumann C. M. (2003). The amygdala and autism: implications from non-human primate studies. Genes Brain Behav.2, 295–302.10.1034/j.1601-183X.2003.00043.x
5
Amaral D. G. Schumann C. M. Nordahl C. W. (2008). Neuroanatomy of autism. Trends Neurosci.31, 137–145.10.1016/j.tins.2007.12.005
6
American Psychiatric Association. (1980). Diagnostic and Statistical Manual of Mental Disorders3rd Edn. Washington, DC: American Psychiatric Association.
7
American Psychiatric Association . (1987). Diagnostic and Statistical Manual of Mental Disorders, 3rd revised Edn.Washington, DC: American Psychiatric Association.
8
American Psychiatric Association . (1994). Diagnostic and Statistical Manual of Mental Disorders, 4th Edn. Washington, DC: American Psychiatric Association.
9
American Psychiatric Association . (2000). Diagnostic and Statistical Manual of Mental Disorders, 4th revised Edn.Washington, DC: American Psychiatric Association.
10
Amir R. E. Van den Veyver I. B. Wan M. Tran C. Q. Francke U. Zoghbi H. Y. (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet.23, 185–188.10.1038/13810
11
Arndt T. L. Stodgell C. J. Rodier P. M. (2005). The teratology of autism. Int. J. Dev. Neurosci.23, 189–199.10.1016/j.ijdevneu.2004.11.001
12
Asaka Y. Jugloff D. G. Zhang L. Eubanks J. H. Fitzsimonds R. M. (2006). Hippocampal synaptic plasticity is impaired in the Mecp2-null mouse model of Rett syndrome. Neurobiol. Dis.21, 217–227.10.1016/j.nbd.2005.07.005
13
Aylward E. H. Minshew N. J. Goldstein G. Honeycutt N. A. Augustine A. M. Yates K. O. Barta P. E. Pearlson G. D. (1999). MRI volumes of amygdala and hippocampus in non-mentally retarded autistic adolescents and adults. Neurology53, 2145–2150.
14
Bachevalier J. (1994). Medial temporal lobe structures and autism: a review of clinical and experimental findings. Neuropsychologia32, 627–648.10.1016/0028-3932(94)90025-6
15
Bachevalier J. Loveland K. A. (2006). The orbitofrontal-amygdala circuit and self-regulation of social-emotional behavior in autism. Neurosci. Biobehav. Rev.30, 97–117.10.1016/j.neubiorev.2005.07.002
16
Bailey A. Luthert P. Dean A. Harding B. Janota I. Montgomery M. Rutter M. Lantos P. (1998). A clinicopathological study of autism. Brain121 (Pt 5), 889–905.10.1093/brain/121.5.889
17
Baron-Cohen S. (2002). The extreme male brain theory of autism. Trends Cogn. Sci.6, 248–254.10.1016/S1364-6613(02)01904-6
18
Baron-Cohen S. (2003). The Essential Difference: Male and Female Brains and the Truth about Autism. New York: Basic.
19
Baron-Cohen S. Leslie A. M. Frith U. (1985). Does the autistic child have a “theory of mind”?Cognition21, 37–46.10.1016/0010-0277(85)90022-8
20
Baron-Cohen S. Ring H. A. Bullmore E. T. Wheelwright S. Ashwin C. Williams S. C. (2000). The amygdala theory of autism. Neurosci. Biobehav. Rev.24, 355–364.10.1016/S0149-7634(00)00011-7
21
Baron-Cohen S. Ring H. A. Wheelwright S. Bullmore E. T. Brammer M. J. Simmons A. Williams S. C. (1999). Social intelligence in the normal and autistic brain: an fMRI study. Eur. J. Neurosci.11, 1891–1898.10.1046/j.1460-9568.1999.00621.x
22
Barry R. J. James A. L. (1988). Coding of stimulus parameters in autistic, retarded, and normal children: evidence for a two-factor theory of autism. Int. J. Psychophysiol.6, 139–149.10.1016/0167-8760(88)90045-1
23
Bauman M. L. Kemper T. L. (2005). Neuroanatomic observations of the brain in autism: a review and future directions. Int. J. Dev. Neurosci.23, 183–187.10.1016/j.ijdevneu.2004.09.006
24
Bear M. F. Dolen G. Osterweil E. Nagarajan N. (2008). Fragile X: translation in action. Neuropsychopharmacology33, 84–87.10.1038/sj.npp.1301610
25
Bear M. F. Huber K. M. Warren S. T. (2004). The mGluR theory of fragile X mental retardation. Trends Neurosci.27, 370–377.10.1016/j.tins.2004.04.009
26
Belmonte M. K. Allen G. Beckel-Mitchener A. Boulanger L. M. Carper R. A. Webb S. J. (2004a). Autism and abnormal development of brain connectivity. J. Neurosci.24, 9228–9231.10.1523/JNEUROSCI.3340-04.2004
27
Belmonte M. K. Cook E. H. Jr. Anderson G. M. Rubenstein J. L. Greenough W. T. Beckel-Mitchener A. Courchesne E. Boulanger L. M. Powell S. B. Levitt P. R. Perry E. K. Jiang Y. H. DeLorey T. M. Tierney E. (2004b). Autism as a disorder of neural information processing: directions for research and targets for therapy. Mol. Psychiatry9, 646–663.
28
Belmonte M. K. Gomot M. Baron-Cohen S. (2010). Visual attention in autism families: “unaffected” sibs share atypical frontal activation. J. Child Psychol. Psychiatry51, 259–276.10.1111/j.1469-7610.2009.02153.x
29
Belmonte M. K. Yurgelun-Todd D. A. (2003). Functional anatomy of impaired selective attention and compensatory processing in autism. Brain Res. Cogn. Brain Res.17, 651–664.10.1016/S0926-6410(03)00189-7
30
Belsito K. M. Law P. A. Kirk K. S. Landa R. J. Zimmerman A. W. (2001). Lamotrigine therapy for autistic disorder: a randomized, double-blind, placebo-controlled trial. J. Autism Dev. Disord.31, 175–181.10.1023/A:1010799115457
31
Bennetto L. Pennington B. F. Rogers S. J. (1996). Intact and impaired memory functions in autism. Child Dev.67, 1816–1835.10.2307/1131734
32
Bernier R. Dawson G. Panagiotides H. Webb S. (2005). Individuals with autism spectrum disorder show normal responses to a fear potential startle paradigm. J. Autism Dev. Disord.35, 575–583.10.1007/s10803-005-0002-0
33
Bertone A. Mottron L. Jelenic P. Faubert J. (2005). Enhanced and diminished visuo-spatial information processing in autism depends on stimulus complexity. Brain128, 2430–2441.10.1093/brain/awh561
34
Beyer K. S. Blasi F. Bacchelli E. Klauck S. M. Maestrini E. Poustka A. (2002). Mutation analysis of the coding sequence of the MECP2 gene in infantile autism. Hum. Genet.111, 305–309.10.1007/s00439-002-0786-3
35
Blair R. J. Peschardt K. S. Budhani S. Mitchell D. G. Pine D. S. (2006). The development of psychopathy. J. Child Psychol. Psychiatry47, 262–276.10.1111/j.1469-7610.2006.01596.x
36
Blasi F. Bacchelli E. Carone S. Toma C. Monaco A. P. Bailey A. J. Maestrini E. (2006). SLC25A12 and CMYA3 gene variants are not associated with autism in the IMGSAC multiplex family sample. Eur. J. Hum. Genet.14, 123–126.
37
Boddaert N. Belin P. Chabane N. Poline J. B. Barthelemy C. Mouren-Simeoni M. C. Brunelle F. Samson Y. Zilbovicius M. (2003). Perception of complex sounds: abnormal pattern of cortical activation in autism. Am. J. Psychiatry160, 2057–2060.10.1176/appi.ajp.160.11.2057
38
Boeckers T. M. Bockmann J. Kreutz M. R. Gundelfinger E. D. (2002). ProSAP/Shank proteins – a family of higher order organizing molecules of the postsynaptic density with an emerging role in human neurological disease. J. Neurochem.81, 903–910.10.1046/j.1471-4159.2002.00931.x
39
Bonnel A. Mottron L. Peretz I. Trudel M. Gallun E. Bonnel A. M. (2003). Enhanced pitch sensitivity in individuals with autism: a signal detection analysis. J. Cogn. Neurosci.15, 226–235.10.1162/089892903321208169
40
Boso M. Emanuele E. Prestori F. Politi P. Barale F. D'Angelo E. (2010). Autism and genius: is there a link? The involvement of central brain loops and hypotheses for functional testing. Funct. Neurol.25, 15–20.
41
Boucher J. Warrington E. K. (1976). Memory deficits in early infantile autism: some similarities to the amnesic syndrome. Br. J. Psychol.67, 73–87.
42
Buchsbaum M. S. Siegel B. V. Jr. Wu J. C. Hazlett E. Sicotte N. Haier R. Tanguay P. Asarnow R. Cadorette T. Donoghue D. Lott I. Paek J. Sabalesky D. (1992). Brief report: attention performance in autism and regional brain metabolic rate assessed by positron emission tomography. J. Autism Dev. Disord.22, 115–125.10.1007/BF01046407
43
Buyse I. M. Fang P. Hoon K. T. Amir R. E. Zoghbi H. Y. Roa B. B. (2000). Diagnostic testing for Rett syndrome by DHPLC and direct sequencing analysis of the MECP2 gene: identification of several novel mutations and polymorphisms. Am. J. Hum. Genet.67, 1428–1436.10.1086/316913
44
Cahill L. McGaugh J. L. (1996). Modulation of memory storage. Curr. Opin. Neurobiol.6, 237–242.10.1016/S0959-4388(96)80078-X
45
Carlsson M. L. (1998). Hypothesis: is infantile autism a hypoglutamatergic disorder? Relevance of glutamate – serotonin interactions for pharmacotherapy. J. Neural Transm.105, 525–535.10.1007/s007020050076
46
Carney R. M. Wolpert C. M. Ravan S. A. Shahbazian M. Ashley-Koch A. Cuccaro M. L. Vance J. M. Pericak-Vance M. A. (2003). Identification of MeCP2 mutations in a series of females with autistic disorder. Pediatr. Neurol.28, 205–211.
47
Caron M. J. Mottron L. Rainville C. Chouinard S. (2004). Do high functioning persons with autism present superior spatial abilities?Neuropsychologia42, 467–481.10.1016/j.neuropsychologia.2003.08.015
48
Carper R. A. Courchesne E. (2000). Inverse correlation between frontal lobe and cerebellum sizes in children with autism. Brain123 (Pt 4), 836–844.10.1093/brain/123.4.836
49
Carper R. A. Moses P. Tigue Z. D. Courchesne E. (2002). Cerebral lobes in autism: early hyperplasia and abnormal age effects. Neuroimage16, 1038–1051.10.1006/nimg.2002.1099
50
Casanova M. F. (2004). White matter volume increase and minicolumns in autism. Ann. Neurol.56, 453; author reply 454.10.1002/ana.20196
51
Casanova M. F. (2007). The neuropathology of autism. Brain Pathol.17, 422–433.10.1111/j.1750-3639.2007.00100.x
52
Casanova M. F. Buxhoeveden D. Gomez J. (2003). Disruption in the inhibitory architecture of the cell minicolumn: implications for autisim. Neuroscientist9, 496–507.10.1177/1073858403253552
53
Casanova M. F. Buxhoeveden D. P. Switala A. E. Roy E. (2002). Minicolumnar pathology in autism. Neurology58, 428–432.
54
Casey B. J. Gordon C. T. Mannheim G. B. Rumsey J. M. (1993). Dysfunctional attention in autistic savants. J. Clin. Exp. Neuropsychol.15, 933–946.10.1080/01688639308402609
55
Castelli F. Frith C. Happé F. Frith U. (2002). Autism, Asperger syndrome and brain mechanisms for the attribution of mental states to animated shapes. Brain125, 1839–1849.10.1093/brain/awf189
56
Chang Q. Khare G. Dani V. Nelson S. Jaenisch R. (2006). The disease progression of Mecp2 mutant mice is affected by the level of BDNF expression. Neuron49, 341–348.10.1016/j.neuron.2005.12.027
57
Cherkassky V. L. Kana R. K. Keller T. A. Just M. A. (2006). Functional connectivity in a baseline resting-state network in autism. Neuroreport17, 1687–1690.10.1097/01.wnr.0000239956.45448.4c
58
Chez M. G. Burton Q. Dowling T. Chang M. Khanna P. Kramer C. (2007). Memantine as adjunctive therapy in children diagnosed with autistic spectrum disorders: an observation of initial clinical response and maintenance tolerability. J. Child Neurol.22, 574–579.10.1177/0883073807302611
59
Christianson A. L. Chesler N. Kromberg J. G. (1994). Fetal valproate syndrome: clinical and neuro-developmental features in two sibling pairs. Dev. Med. Child Neurol.36, 361–369.10.1111/j.1469-8749.1994.tb11858.x
60
Ciesielski K. T. Courchesne E. Elmasian R. (1990). Effects of focused selective attention tasks on event-related potentials in autistic and normal individuals. Electroencephalogr. Clin. Neurophysiol.75, 207–220.10.1016/0013-4694(90)90174-I
61
Coldren J. T. Halloran C. (2003). Spatial reversal as a measure of executive functioning in children with autism. J. Genet. Psychol.164, 29–41.10.1080/00221320309597501
62
Consortium D.-B. F. X. (1994). Fmr1 knockout mice: a model to study fragile X mental retardation. Cell. Mol. Neurobiol.78, 23–33.
63
Corbett B. A. Mendoza S. Abdullah M. Wegelin J. A. Levine S. (2006). Cortisol circadian rhythms and response to stress in children with autism. Psychoneuroendocrinology31, 59–68.10.1016/j.psyneuen.2005.05.011
64
Cottraux J. (2005). Recent developments in research and treatment for social phobia (social anxiety disorder). Curr. Opin. Psychiatry18, 51–54.
65
Coulter D. A. (1997). Antiepileptic drug cellular mechanisms of action: where does lamotrigine fit in?J. Child Neurol.12 (Suppl. 1), S2–S9.10.1177/0883073897012001031
66
Courchesne E. (2004). Brain development in autism: early overgrowth followed by premature arrest of growth. Ment. Retard. Dev. Disabil. Res. Rev.10, 106–111.10.1002/mrdd.20020
67
Courchesne E. Karns C. M. Davis H. R. Ziccardi R. Carper R. A. Tigue Z. D. Chisum H. J. Moses P. Pierce K. Lord C. Lincoln A. J. Pizzo S. Schreibman L. Haas R. H. Akshoomoff N. A. Courchesne R. Y. (2001). Unusual brain growth patterns in early life in patients with autistic disorder: an MRI study. Neurology57, 245–254.
68
Courchesne E. Kilman B. A. Galambos R. Lincoln A. J. (1984). Autism: processing of novel auditory information assessed by event-related brain potentials. Electroencephalogr. Clin. Neurophysiol.59, 238–248.10.1016/0168-5597(84)90063-7
69
Courchesne E. Pierce K. (2005a). Brain overgrowth in autism during a critical time in development: implications for frontal pyramidal neuron and interneuron development and connectivity. Int. J. Dev. Neurosci.23, 153–170.10.1016/j.ijdevneu.2005.01.003
70
Courchesne E. Pierce K. (2005b). Why the frontal cortex in autism might be talking only to itself: local over-connectivity but long-distance disconnection. Curr. Opin. Neurobiol.15, 225–230.10.1016/j.conb.2005.03.001
71
Courchesne E. Redcay E. Morgan J. T. Kennedy D. P. (2005). Autism at the beginning: microstructural and growth abnormalities underlying the cognitive and behavioral phenotype of autism. Dev. Psychopathol.17, 577–597.10.1017/S0954579405050285
72
Courchesne E. Townsend J. Akshoomoff N. A. Saitoh O. Yeung-Courchesne R. Lincoln A. J. James H. E. Haas R. H. Schreibman L. Lau L. (1994). Impairment in shifting attention in autistic and cerebellar patients. Behav. Neurosci.108, 848–865.10.1037/0735-7044.108.5.848
73
Craig M. C. Zaman S. H. Daly E. M. Cutter W. J. Robertson D. M. Hallahan B. Toal F. Reed S. Ambikapathy A. Brammer M. Murphy C. M. Murphy D. G. (2007). Women with autistic-spectrum disorder: magnetic resonance imaging study of brain anatomy. Br. J. Psychiatry191, 224–228.10.1192/bjp.bp.106.034603
74
Critchley H. D. Daly E. M. Bullmore E. T. Williams S. C. Van Amelsvoort T. Robertson D. M. Rowe A. Phillips M. McAlonan G. Howlin P. Murphy D. G. (2000). The functional neuroanatomy of social behaviour: changes in cerebral blood flow when people with autistic disorder process facial expressions. Brain123 (Pt 11), 2203–2212.10.1093/brain/123.11.2203
75
Dalton K. M. Nacewicz B. M. Johnstone T. Schaefer H. S. Gernsbacher M. A. Goldsmith H. H. Alexander A. L. Davidson R. J. (2005). Gaze fixation and the neural circuitry of face processing in autism. Nat. Neurosci.8, 519–526.
76
Damsa C. Maris S. Pull C. B. (2005). New fields of research in posttraumatic stress disorder: brain imaging. Curr. Opin. Psychiatry18, 55–64.
77
Danbolt N. C. (1994). The high affinity uptake system for excitatory amino acids in the brain. Prog. Neurobiol.44, 377–396.10.1016/0301-0082(94)90033-7
78
Dani V. S. Chang Q. Maffei A. Turrigiano G. G. Jaenisch R. Nelson S. B. (2005). Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. U.S.A.102, 12560–12565.10.1073/pnas.0506071102
79
Davis M. Whalen P. J. (2001). The amygdala: vigilance and emotion. Mol. Psychiatry6, 13–34.10.1038/sj.mp.4000812
80
Dawson M. Mottron L. Gernsbacher M. A. (2008). “Learning in autism,” in Learning and Memory: A Comprehensive Reference: Cognitive Psychology, eds ByrneJ.RoedigerH. L. I. (Oxford, UK: Elsevier), 759–772.10.1016/B978-012370509-9.00152-2
81
Dawson M. Soulieres I. Gernsbacher M. A. Mottron L. (2007). The level and nature of autistic intelligence. Psychol. Sci.18, 657–662.10.1111/j.1467-9280.2007.01954.x
82
DeVito T. J. Drost D. J. Neufeld R. W. Rajakumar N. Pavlosky W. Williamson P. Nicolson R. (2007). Evidence for cortical dysfunction in autism: a proton magnetic resonance spectroscopic imaging study. Biol. Psychiatry61, 465–473.10.1016/j.biopsych.2006.07.022
83
Dichter G. S. Felder J. N. Bodfish J. W. (2009). Autism is characterized by dorsal anterior cingulate hyperactivation during social target detection. Soc. Cogn. Affect. Neurosci.4, 215–226.10.1093/scan/nsp017
84
Dragich J. Houwink-Manville I. Schanen C. (2000). Rett syndrome: a surprising result of mutation in MECP2. Hum. Mol. Genet.9, 2365–2375.10.1093/hmg/9.16.2365
85
Durand C. M. Betancur C. Boeckers T. M. Bockmann J. Chaste P. Fauchereau F. Nygren G. Rastam M. Gillberg I. C. Anckarsater H. Sponheim E. Goubran-Botros H. Delorme R. Chabane N. Mouren-Simeoni M. C. de Mas P. Bieth E. Roge B. Heron D. Burglen L. Gillberg C. Leboyer M. Bourgeron T. (2007). Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet.39, 25–27.10.1038/ng1933
86
Dykens E. Volkmar F. (1997). “Medical conditions associated with autism,” in Handbook of Autism and Pervasive Developmental Disorders, eds CohenD.VolkmarF. (New York: Wiley), 388–410.
87
Emery N. J. Capitanio J. P. Mason W. A. Machado C. J. Mendoza S. P. Amaral D. G. (2001). The effects of bilateral lesions of the amygdala on dyadic social interactions in rhesus monkeys (Macaca mulatta). Behav. Neurosci.115, 515–544.10.1037/0735-7044.115.3.515
88
Evans D. W. Canavera K. Kleinpeter F. L. Maccubbin E. Taga K. (2005). The fears, phobias and anxieties of children with autism spectrum disorders and down syndrome: comparisons with developmentally and chronologically age matched children. Child Psychiatry Hum. Dev.36, 3–26.10.1007/s10578-004-3619-x
89
Fombonne E. (2006). “Past and future perspectives on autism epidemiology,” in Understanding Autism: From Basic Neuroscience to Treatment, eds MoldinS. O.RubensteinJ. R. L. (Boca Raton: CRC Press, Taylor and Francis Group), 25–48.10.1201/9781420004205.ch2
90
Foxton J. M. Stewart M. E. Barnard L. Rodgers J. Young A. H. O'Brien G. Griffiths T. D. (2003). Absence of auditory “global interference” in autism. Brain126, 2703–2709.10.1093/brain/awg274
91
Frith U. (1989). Autism: Explaining the Enigma. Oxford: Basil Blackwell.
92
Frith U. (2003). Autism: Explaining the Enigma, 2nd Edn. Oxford: Wiley-Blackwell.
93
Frith U. Happé F. (1994). Autism: beyond “theory of mind”. Cognition50, 115–132.10.1016/0010-0277(94)90024-8
94
Gaffney G. R. Kuperman S. Tsai L. Y. Minchin S. (1988). Morphological evidence for brainstem involvement in infantile autism. Biol. Psychiatry24, 578–586.10.1016/0006-3223(88)90168-0
95
Gaigg S. B. Bowler D. M. (2007). Differential fear conditioning in Asperger's syndrome: implications for an amygdala theory of autism. Neuropsychologia45, 2125–2134.10.1016/j.neuropsychologia.2007.01.012
96
Galvez R. Greenough W. T. (2005). Sequence of abnormal dendritic spine development in primary somatosensory cortex of a mouse model of the fragile X mental retardation syndrome. Am. J. Med. Genet.135, 155–160.10.1002/ajmg.a.30709
97
Garretson H. B. Fein D. Waterhouse L. (1990). Sustained attention in children with autism. J. Autism Dev. Disord.20, 101–114.10.1007/BF02206860
98
Gervais H. Belin P. Boddaert N. Leboyer M. Coez A. Sfaello I. Barthelemy C. Brunelle F. Samson Y. Zilbovicius M. (2004). Abnormal cortical voice processing in autism. Nat. Neurosci.7, 801–802.10.1038/nn1291
99
Geschwind D. H. Levitt P. (2007). Autism spectrum disorders: developmental disconnection syndromes. Curr. Opin. Neurobiol.17, 103–111.10.1016/j.conb.2007.01.009
100
Geurts H. M. Verte S. Oosterlaan J. Roeyers H. Sergeant J. A. (2004). How specific are executive functioning deficits in attention deficit hyperactivity disorder and autism?J. Child Psychol. Psychiatry45, 836–854.10.1111/j.1469-7610.2004.00276.x
101
Giese K. P. Fedorov N. B. Filipkowski R. K. Silva A. J. (1998). Autophosphorylation at Thr286 of the alpha calcium-calmodulin kinase II in LTP and learning. Science279, 870–873.10.1126/science.279.5352.870
102
Giza J. Urbanski M. J. Prestori F. Bandyopadhyay B. Yam A. Friedrich V. Kelley K. D'Angelo E. Goldfarb M. (2010). Behavioral and cerebellar transmission deficits in mice lacking the autism-linked gene islet brain-2. J. Neurosci.30, 14805–14816.10.1523/JNEUROSCI.1161-10.2010
103
Gillott A. Furniss F. Walter A. (2001). Anxiety in high-functioning children with autism. Autism5, 277–286.10.1177/1362361301005003005
104
Godfraind J. M. Reyniers E. De Boulle K. D'Hooge R. De Deyn P. P. Bakker C. E. Oostra B. A. Kooy R. F. Willems P. J. (1996). Long-term potentiation in the hippocampus of fragile X knockout mice. Am. J. Med. Genet.64, 246–251.10.1002/(SICI)1096-8628(19960809)64:2<246::AID-AJMG2>3.0.CO;2-S
105
Goldberg M. C. Mostofsky S. H. Cutting L. E. Mahone E. M. Astor B. C. Denckla M. B. Landa R. J. (2005). Subtle executive impairment in children with autism and children with ADHD. J. Autism Dev. Disord.35, 279–293.10.1007/s10803-005-3291-4
106
Goldberg Edelson M. (2006). Are the majority of children with autism mentally retarded? A systematic evaluation of the data. Focus Autism Other Dev. Disabl.21, 66–83.10.1177/10883576060210020301
107
Gomot M. Belmonte M. K. Bullmore E. T. Bernard F. A. Baron-Cohen S. (2008). Brain hyper-reactivity to auditory novel targets in children with high-functioning autism. Brain131, 2479–2488.10.1093/brain/awn172
108
Graf E. R. Zhang X. Jin S. X. Linhoff M. W. Craig A. M. (2004). Neurexins induce differentiation of GABA and glutamate postsynaptic specializations via neuroligins. Cell119, 1013–1026.
109
Grandin T. (1996). Thinking in Pictures. New York: Vintage.
110
Grandin T. Barron S. (2005). Unwritten Rules of Social Relationships: Decoding Social Mysteries Through the Unique Perspectives of Autism. Arlington, TX: Future Horizons, Inc.
111
Grelotti D. J. Klin A. J. Gauthier I. Skudlarski P. Cohen D. J. Gore J. C. Volkmar F. R. Schultz R. T. (2005). fMRI activation of the fusiform gyrus and amygdala to cartoon characters but not to faces in a boy with autism. Neuropsychologia43, 373–385.10.1016/j.neuropsychologia.2004.06.015
112
Guy J. Hendrich B. Holmes M. Martin J. E. Bird A. (2001). A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat. Genet.27, 322–326.10.1038/85899
113
Hagerman R. J. Jackson A. W. 3rd Levitas A. Rimland B. Braden M. (1986). An analysis of autism in fifty males with the fragile X syndrome. Am. J. Med. Genet.23, 359–374.10.1002/ajmg.1320230128
114
Hajek T. Carrey N. Alda M. (2005). Neuroanatomical abnormalities as risk factors for bipolar disorder. Bipolar Disord.7, 393–403.10.1111/j.1399-5618.2005.00238.x
115
Hall G. B. Szechtman H. Nahmias C. (2003). Enhanced salience and emotion recognition in autism: a PET study. Am. J. Psychiatry160, 1439–1441.10.1176/appi.ajp.160.8.1439
116
Happé F. (1999). Autism: cognitive deficit or cognitive style?Trends Cogn. Sci.3, 216–222.10.1016/S1364-6613(99)01318-2
117
Happé F. Ehlers S. Fletcher P. Frith U. Johansson M. Gillberg C. Dolan R. Frackowiak R. Frith C. (1996). “Theory of mind” in the brain. Evidence from a PET scan study of Asperger syndrome. Neuroreport8, 197–201.10.1097/00001756-199612200-00040
118
Happé F. Frith U. (1997). Central coherence and theory of mind in autism: reading homographs in context. Br. J. Dev. Psychol.15, 1–12.
119
Happé F. Frith U. (2006). The weak coherence account: detail-focused cognitive style in autism spectrum disorders. J. Autism Dev. Disord.36, 5–25.10.1007/s10803-005-0039-0
120
Happé F. G. (1994). Wechsler IQ profile and theory of mind in autism: a research note. J. Child Psychol. Psychiatry35, 1461–1471.10.1111/j.1469-7610.1994.tb01287.x
121
Hardan A. Y. Muddasani S. Vemulapalli M. Keshavan M. S. Minshew N. J. (2006). An MRI study of increased cortical thickness in autism. Am. J. Psychiatry163, 1290–1292.10.1176/appi.ajp.163.7.1290
122
Hashimoto T. Tayama M. Murakawa K. Yoshimoto T. Miyazaki M. Harada M. Kuroda Y. (1995). Development of the brainstem and cerebellum in autistic patients. J. Autism Dev. Disord.25, 1–18.10.1007/BF02178163
123
Haxby J. V. Horwitz B. Ungerleider L. G. Maisog J. M. Pietrini P. Grady C. L. (1994). The functional organization of human extrastriate cortex: a PET-rCBF study of selective attention to faces and locations. J. Neurosci.14, 6336–6353.
124
Hayashi M. L. Rao B. S. Seo J. S. Choi H. S. Dolan B. M. Choi S. Y. Chattarji S. Tonegawa S. (2007). Inhibition of p21-activated kinase rescues symptoms of fragile X syndrome in mice. Proc. Natl. Acad. Sci. U.S.A.104, 11489–11494.10.1073/pnas.0705003104
125
Herbert M. R. Ziegler D. A. Deutsch C. K. O'Brien L. M. Lange N. Bakardjiev A. Hodgson J. Adrien K. T. Steele S. Makris N. Kennedy D. Harris G. J. Caviness V. S. Jr. (2003). Dissociations of cerebral cortex, subcortical and cerebral white matter volumes in autistic boys. Brain126, 1182–1192.10.1093/brain/awg110
126
Herbert M. R. Ziegler D. A. Makris N. Filipek P. A. Kemper T. L. Normandin J. J. Sanders H. A. Kennedy D. N. Caviness V. S. Jr. (2004). Localization of white matter volume increase in autism and developmental language disorder. Ann. Neurol.55, 530–540.10.1002/ana.20032
127
Hirstein W. Iversen P. Ramachandran V. S. (2001). Autonomic responses of autistic children to people and objects. Proc. Biol. Sci.268, 1883–1888.10.1098/rspb.2001.1724
128
Horwitz B. Rumsey J. M. Grady C. L. Rapoport S. I. (1988). The cerebral metabolic landscape in autism. Intercorrelations of regional glucose utilization. Arch. Neurol.45, 749–755.
129
Hubel D. H. Wiesel T. N. (1962). Receptive fields, binocular interaction and functional architecture in the cat's visual cortex. J. Physiol.160, 106–154.
130
Huber K. M. Gallagher S. M. Warren S. T. Bear M. F. (2002). Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc. Natl. Acad. Sci. U.S.A.99, 7746–7750.10.1073/pnas.122205699
131
Hubl D. Bolte S. Feineis-Matthews S. Lanfermann H. Federspiel A. Strik W. Poustka F. Dierks T. (2003). Functional imbalance of visual pathways indicates alternative face processing strategies in autism. Neurology61, 1232–1237.
132
Huppke P. Laccone F. Kramer N. Engel W. Hanefeld F. (2000). Rett syndrome: analysis of MECP2 and clinical characterization of 31 patients. Hum. Mol. Genet.9, 1369–1375.10.1093/hmg/9.9.1369
133
Hussman J. P. (2001). Suppressed GABAergic inhibition as a common factor in suspected etiologies of autism. J. Autism Dev. Disord.31, 247–248.10.1023/A:1010715619091
134
Hutt C. Hutt S. J. (1965). Effects of environmental complexity on stereotyped behaviours of children. Anim. Behav.13, 1–4.10.1016/0003-3472(65)90064-3
135
Hutt C. Hutt S. J. Lee D. Ounsted C. (1964). Arousal and childhood autism. Nature204, 908–909.10.1038/204908a0
136
Hutt S. J. Hutt C. Lee D. Ounsted C. (1965). A behavioural and electroencephalographic study of autistic children. J. Psychiatr. Res.3, 181–197.10.1016/0022-3956(65)90028-2
137
Ingram J. L. Peckham S. M. Tisdale B. Rodier P. M. (2000). Prenatal exposure of rats to valproic acid reproduces the cerebellar anomalies associated with autism. Neurotoxicol. Teratol.22, 319–324.10.1016/S0892-0362(99)00083-5
138
Irwin S. A. Idupulapati M. Gilbert M. E. Harris J. B. Chakravarti A. B. Rogers E. J. Crisostomo R. A. Larsen B. P. Mehta A. Alcantara C. J. Patel B. Swain R. A. Weiler I. J. Oostra B. A. Greenough W. T. (2002). Dendritic spine and dendritic field characteristics of layer V pyramidal neurons in the visual cortex of fragile-X knockout mice. Am. J. Med. Genet.111, 140–146.10.1002/ajmg.10500
139
Jamain S. Betancur C. Quach H. Philippe A. Fellous M. Giros B. Gillberg C. Leboyer M. Bourgeron T. (2002). Linkage and association of the glutamate receptor 6 gene with autism. Mol. Psychiatry7, 302–310.10.1038/sj.mp.4000979
140
Jamain S. Quach H. Betancur C. Rastam M. Colineaux C. Gillberg I. C. Soderstrom H. Giros B. Leboyer M. Gillberg C. Bourgeron T. (2003). Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet.34, 27–29.10.1038/ng1136
141
James A. L. Barry R. J. (1984). Cardiovascular and electrodermal responses to simple stimuli in autistic, retarded and normal children. Int. J. Psychophysiol.1, 179–193.10.1016/0167-8760(84)90037-0
142
Jarrold C. Gilchrist I. D. Bender A. (2005). Embedded figures detection in autism and typical development: preliminary evidence of a double dissociation in relationships with visual search. Dev. Sci.8, 344–351.10.1111/j.1467-7687.2005.00422.x
143
Johnson K. A. Robertson I. H. Kelly S. P. Silk T. J. Barry E. Daibhis A. Watchorn A. Keavey M. Fitzgerald M. Gallagher L. Gill M. Bellgrove M. A. (2007). Dissociation in performance of children with ADHD and high-functioning autism on a task of sustained attention. Neuropsychologia45, 2234–2245.10.1016/j.neuropsychologia.2007.02.019
144
Jolliffe T. Baron-Cohen S. (1997). Are people with autism and Asperger syndrome faster than normal on the embedded figures test?J. Child Psychol. Psychiatry38, 527–534.10.1111/j.1469-7610.1997.tb01539.x
145
Just M. A. Cherkassky V. L. Keller T. A. Kana R. K. Minshew N. J. (2007). Functional and anatomical cortical underconnectivity in autism: evidence from an FMRI study of an executive function task and corpus callosum morphometry. Cereb. Cortex17, 951–961.10.1093/cercor/bhl006
146
Just M. A. Cherkassky V. L. Keller T. A. Minshew N. J. (2004). Cortical activation and synchronization during sentence comprehension in high-functioning autism: evidence of underconnectivity. Brain127, 1811–1821.10.1093/brain/awh199
147
Kana R. K. Keller T. A. Cherkassky V. L. Minshew N. J. Just M. A. (2006). Sentence comprehension in autism: thinking in pictures with decreased functional connectivity. Brain129, 2484–2493.10.1093/brain/awl164
148
Kana R. K. Keller T. A. Minshew N. J. Just M. A. (2007). Inhibitory control in high-functioning autism: decreased activation and underconnectivity in inhibition networks. Biol. Psychiatry62, 198–206.10.1016/j.biopsych.2006.08.004
149
Kanner L. (1943). Autistic disturbances of affective contact. Nerv. Child2, 217–250.
150
Kanwisher N. (2000). Domain specificity in face perception. Nat. Neurosci.3, 759–763.10.1038/77664
151
Kemper T. L. Bauman M. (1998). Neuropathology of infantile autism. J. Neuropathol. Exp. Neurol.57, 645–652.10.1097/00005072-199807000-00001
152
Kenworthy L. E. Black D. O. Wallace G. L. Ahluvalia T. Wagner A. E. Sirian L. M. (2005). Disorganization: the forgotten executive dysfunction in high-functioning autism (HFA) spectrum disorders. Dev. Neuropsychol.28, 809–827.10.1207/s15326942dn2803_4
153
King B. H. Wright D. M. Handen B. L. Sikich L. Zimmerman A. W. McMahon W. Cantwell E. Davanzo P. A. Dourish C. T. Dykens E. M. Hooper S. R. Jaselskis C. A. Leventhal B. L. Levitt J. Lord C. Lubetsky M. J. Myers S. M. Ozonoff S. Shah B. G. Snape M. Shernoff E. W. Williamson K. Cook E. H. Jr. (2001a). Double-blind, placebo-controlled study of amantadine hydrochloride in the treatment of children with autistic disorder. J. Am. Acad. Child Adolesc. Psychiatry40, 658–665.10.1097/00004583-200106000-00010
154
King B. H. Wright D. M. Snape M. Dourish C. T. (2001b). Case series: amantadine open-label treatment of impulsive and aggressive behavior in hospitalized children with developmental disabilities. J. Am. Acad. Child Adolesc. Psychiatry40, 654–657.10.1097/00004583-200106000-00009
155
Kleinhans N. M. Johnson L. C. Richards T. Mahurin R. Greenson J. Dawson G. Aylward E. (2009). Reduced neural habituation in the amygdala and social impairments in autism spectrum disorders. Am. J. Psychiatry166, 467–475.10.1176/appi.ajp.2008.07101681
156
Knaus T. A. Silver A. M. Lindgren K. A. Hadjikhani N. Tager-Flusberg H. (2008). fMRI activation during a language task in adolescents with ASD. J. Int. Neuropsychol. Soc.14, 967–979.10.1017/S1355617708081216
157
Koekkoek S. K. Yamaguchi K. Milojkovic B. A. Dortland B. R. Ruigrok T. J. Maex R. De Graaf W. Smit A. E. VanderWerf F. Bakker C. E. Willemsen R. Ikeda T. Kakizawa S. Onodera K. Nelson D. L. Mientjes E. Joosten M. De Schutter E. Oostra B. A. Ito M. De Zeeuw C. I. (2005). Deletion of FMR1 in Purkinje cells enhances parallel fiber LTD, enlarges spines, and attenuates cerebellar eyelid conditioning in Fragile X syndrome. Neuron47, 339–352.10.1016/j.neuron.2005.07.005
158
Koshino H. Carpenter P. A. Minshew N. J. Cherkassky V. L. Keller T. A. Just M. A. (2005). Functional connectivity in an fMRI working memory task in high-functioning autism. Neuroimage24, 810–821.10.1016/j.neuroimage.2004.09.028
159
Koshino H. Kana R. K. Keller T. A. Cherkassky V. L. Minshew N. J. Just M. A. (2008). fMRI investigation of working memory for faces in autism: visual coding and underconnectivity with frontal areas. Cereb. Cortex18, 289–300.10.1093/cercor/bhm054
160
Krupa D. J. Matell M. S. Brisben A. J. Oliveira L. M. Nicolelis M. A. (2001). Behavioral properties of the trigeminal somatosensory system in rats performing whisker-dependent tactile discriminations. J. Neurosci.21, 5752–5763.
161
Kurup R. K. Kurup P. A. (2003). A hypothalamic digoxin-mediated model for autism. Int. J. Neurosci.113, 1537–1559.10.1080/00207450390231482
162
Lake C. R. Ziegler M. G. Murphy D. L. (1977). Increased norepinephrine levels and decreased dopamine-beta-hydroxylase activity in primary autism. Arch. Gen. Psychiatry34, 553–556.
163
Lam C. W. Yeung W. L. Ko C. H. Poon P. M. Tong S. F. Chan K. Y. Lo I. F. Chan L. Y. Hui J. Wong V. Pang C. P. Lo Y. M. Fok T. F. (2000). Spectrum of mutations in the MECP2 gene in patients with infantile autism and Rett syndrome. J. Med. Genet.37, E41.10.1136/jmg.37.12.e41
164
Lam K. S. Aman M. G. Arnold L. E. (2006). Neurochemical correlates of autistic disorder: a review of the literature. Res. Dev. Disabil. 27, 254–289.10.1016/j.ridd.2005.03.003
165
Landry R. Bryson S. E. (2004). Impaired disengagement of attention in young children with autism. J. Child Psychol. Psychiatry45, 1115–1122.10.1111/j.1469-7610.2004.00304.x
166
Larson J. Jessen R. E. Kim D. Fine A. K. du Hoffmann J. (2005). Age-dependent and selective impairment of long-term potentiation in the anterior piriform cortex of mice lacking the fragile X mental retardation protein. J. Neurosci.25, 9460–9469.10.1523/JNEUROSCI.2638-05.2005
167
Laumonnier F. Bonnet-Brilhault F. Gomot M. Blanc R. David A. Moizard M. P. Raynaud M. Ronce N. Lemonnier E. Calvas P. Laudier B. Chelly J. Fryns J. P. Ropers H. H. Hamel B. C. Andres C. Barthelemy C. Moraine C. Briault S. (2004). X-linked mental retardation and autism are associated with a mutation in the NLGN4 gene, a member of the neuroligin family. Am. J. Hum. Genet.74, 552–557.10.1086/382137
168
Le Be J. V. Markram H. (2006). Spontaneous and evoked synaptic rewiring in the neonatal neocortex. Proc. Natl. Acad. Sci. U.S.A.103, 13214–13219.10.1073/pnas.0604691103
169
LeDoux J. (2003). The emotional brain, fear, and the amygdala. Cell. Mol. Neurobiol.23, 727–738.10.1023/A:1025048802629
170
Lepagnol-Bestel A. M. Maussion G. Boda B. Cardona A. Iwayama Y. Delezoide A. L. Moalic J. M. Muller D. Dean B. Yoshikawa T. Gorwood P. Buxbaum J. D. Ramoz N. Simonneau M. (2008). SLC25A12 expression is associated with neurite outgrowth and is upregulated in the prefrontal cortex of autistic subjects. Mol. Psychiatry13, 385–397.10.1038/sj.mp.4002120
171
Levinson J. N. Chery N. Huang K. Wong T. P. Gerrow K. Kang R. Prange O. Wang Y. T. El-Husseini A. (2005). Neuroligins mediate excitatory and inhibitory synapse formation: involvement of PSD-95 and neurexin-1beta in neuroligin-induced synaptic specificity. J. Biol. Chem.280, 17312–17319.10.1074/jbc.M413812200
172
Levy L. M. Lehre K. P. Walaas S. I. Storm-Mathisen J. Danbolt N. C. (1995). Down-regulation of glial glutamate transporters after glutamatergic denervation in the rat brain. Eur. J. Neurosci.7, 2036–2041.10.1111/j.1460-9568.1995.tb00626.x
173
Li J. Pelletier M. R. Perez Velazquez J. L. Carlen P. L. (2002). Reduced cortical synaptic plasticity and GluR1 expression associated with fragile X mental retardation protein deficiency. Mol. Cell. Neurosci.19, 138–151.10.1006/mcne.2001.1085
174
Liao D. Hessler N. A. Malinow R. (1995). Activation of postsynaptically silent synapses during pairing-induced LTP in CA1 region of hippocampal slice. Nature375, 400–404.10.1038/375400a0
175
Lisman J. Schulman H. Cline H. (2002). The molecular basis of CaMKII function in synaptic and behavioural memory. Nat. Rev. Neurosci.3, 175–190.10.1038/nrn753
176
Liss M. Saulnier C. Fein D. Kinsbourne M. (2006). Sensory and attention abnormalities in autistic spectrum disorders. Autism10, 155–172.10.1177/1362361306062021
177
Lord C. Spence S. (2006). “Autism spectrum disorder: phenotype and diagnosis,” in Understanding Autism: From Basic Neuroscience to Treatment, eds MoldinS. O.RubensteinJ. R. L. (Boca Raton: CRC Press, Taylor and Francis Group), 1–23.10.1201/9781420004205.ch1
178
Luna B. Doll S. K. Hegedus S. J. Minshew N. J. Sweeney J. A. (2007). Maturation of executive function in autism. Biol. Psychiatry61, 474–481.10.1016/j.biopsych.2006.02.030
179
Luna B. Minshew N. J. Garver K. E. Lazar N. A. Thulborn K. R. Eddy W. F. Sweeney J. A. (2002). Neocortical system abnormalities in autism: an fMRI study of spatial working memory. Neurology59, 834–840.
180
Markram H. Tsodyks M. (1996). Redistribution of synaptic efficacy between neocortical pyramidal neurons. Nature382, 807–810.10.1038/382807a0
181
Markram K. Kosten J. Tate A. Gervasoni D. Nicolelis M. (2007a). “Tactile discrimination learning in the valproic acid rat model of autism,” in Society for Neuroscience 37th Annual Meeting, San Diego, CA.
182
Markram K. Rinaldi T. Markram H. (2007b). The intense world syndrome – an alternative hypothesis for autism. Front. Neurosci.1: 1.10.3389/neuro.01.1.1.006.2007
183
Markram K. LaMendola D. Rinaldi T. Sandi C. Markram H. (2005). Enhanced Fear-Conditioned Memory and Reduced Fear-Conditioned Extinction in an Animal Model of Autism. Paper Presented at the Society of Neuroscience Abstract, Washington, DC.
184
Markram K. Rinaldi T. LaMendola D. Sandi C. Markram H. (2008). Abnormal fear conditioning and amygdala processing in an animal model of autism. Neuropsychopharmacology33, 901–912.10.1038/sj.npp.1301453
185
McAlonan G. M. Daly E. Kumari V. Critchley H. D. van Amelsvoort T. Suckling J. Simmons A. Sigmundsson T. Greenwood K. Russell A. Schmitz N. Happé F. Howlin P. Murphy D. G. (2002). Brain anatomy and sensorimotor gating in Asperger's syndrome. Brain125, 1594–1606.10.1093/brain/awf150
186
McGaugh J. L. (2004). The amygdala modulates the consolidation of memories of emotionally arousing experiences. Annu. Rev. Neurosci.27, 1–28.10.1146/annurev.neuro.27.070203.144157
187
McKinney B. C. Grossman A. W. Elisseou N. M. Greenough W. T. (2005). Dendritic spine abnormalities in the occipital cortex of C57BL/6 Fmr1 knockout mice. Am. J. Med. Genet. B Neuropsychiatr. Genet.136B, 98–102.10.1002/ajmg.b.30183
188
Medrihan L. Tantalaki E. Aramuni G. Sargsyan V. Dudanova I. Missler M. Zhang W. (2008). Early defects of GABAergic synapses in the brain stem of a MeCP2 mouse model of Rett syndrome. J. Neurophysiol.99, 112–121.10.1152/jn.00826.2007
189
Meyer G. Varoqueaux F. Neeb A. Oschlies M. Brose N. (2004). The complexity of PDZ domain-mediated interactions at glutamatergic synapses: a case study on neuroligin. Neuropharmacology47, 724–733.10.1016/j.neuropharm.2004.06.023
190
Meyer-Lindenberg A. Hariri A. R. Munoz K. E. Mervis C. B. Mattay V. S. Morris C. A. Berman K. F. (2005). Neural correlates of genetically abnormal social cognition in Williams syndrome. Nat. Neurosci.8, 991–993.10.1038/nn1494
191
Micali N. Chakrabarti S. Fombonne E. (2004). The broad autism phenotype: findings from an epidemiological survey. Autism8, 21–37.10.1177/1362361304040636
192
Ming X. Julu P. O. Brimacombe M. Connor S. Daniels M. L. (2005). Reduced cardiac parasympathetic activity in children with autism. Brain Dev.27, 509–516.10.1016/j.braindev.2005.01.003
193
Minshew N. J. Goldstein G. (1998). Autism as a disorder of complex information processing. Ment. Retard. Dev. Disabil. Res. Rev.4, 129–136.10.1002/(SICI)1098-2779(1998)4:2<129::AID-MRDD10>3.0.CO;2-X
194
Minshew N. J. Goldstein G. (2001). The pattern of intact and impaired memory functions in autism. J. Child Psychol. Psychiatry42, 1095–1101.10.1111/1469-7610.00808
195
Minshew N. J. Goldstein G. Muenz L. R. Payton J. B. (1992). Neuropsychological functioning in non-mentally retarded autistic individuals. J. Clin. Exp. Neuropsychol.14, 749–761.10.1080/01688639208402860
196
Minshew N. J. Goldstein G. Siegel D. J. (1997). Neuropsychologic functioning in autism: profile of a complex information processing disorder. J. Int. Neuropsychol. Soc.3, 303–316.
197
Minshew N. J. Williams D. L. (2007). The new neurobiology of autism: cortex, connectivity, and neuronal organization. Arch. Neurol.64, 945–950.10.1001/archneur.64.7.945
198
Miyazaki K. Narita N. Narita M. (2005). Maternal administration of thalidomide or valproic acid causes abnormal serotonergic neurons in the offspring: implication for pathogenesis of autism. Int. J. Dev. Neurosci.23, 287–297.10.1016/j.ijdevneu.2004.05.004
199
Monk C. S. Weng S. J. Wiggins J. L. Kurapati N. Louro H. M. Carrasco M. Maslowsky J. Risi S. Lord C. (2010). Neural circuitry of emotional face processing in autism spectrum disorders. J. Psychiatry Neurosci.35, 105–114.10.1503/jpn.090085
200
Moore S. J. Turnpenny P. Quinn A. Glover S. Lloyd D. J. Montgomery T. Dean J. C. (2000). A clinical study of 57 children with fetal anticonvulsant syndromes. J. Med. Genet.37, 489–497.10.1136/jmg.37.7.489
201
Moreno-Fuenmayor H. Borjas L. Arrieta A. Valera V. Socorro-Candanoza L. (1996). Plasma excitatory amino acids in autism. Invest. Clin.37, 113–128.
202
Moretti P. Levenson J. M. Battaglia F. Atkinson R. Teague R. Antalffy B. Armstrong D. Arancio O. Sweatt J. D. Zoghbi H. Y. (2006). Learning and memory and synaptic plasticity are impaired in a mouse model of Rett syndrome. J. Neurosci.26, 319–327.10.1523/JNEUROSCI.2623-05.2006
203
Mosconi M. W. Cody-Hazlett H. Poe M. D. Gerig G. Gimpel-Smith R. Piven J. (2009). Longitudinal study of amygdala volume and joint attention in 2- to 4-year-old children with autism. Arch. Gen. Psychiatry66, 509–516.10.1001/archgenpsychiatry.2009.19
204
Mottron L. Belleville S. Menard E. (1999). Local bias in autistic subjects as evidenced by graphic tasks: perceptual hierarchization or working memory deficit?J. Child Psychol. Psychiatry40, 743–755.10.1111/1469-7610.00490
205
Mottron L. Burack J. (2001). “Enhanced perceptual functioning in the development of autism,” in The Development of Autism: Perspectives from Theory and Research, eds BurackJ. A.CharmanT.YirmiyaN.ZelazoP. R. (Mahwah, NY: Erlbaum), 131–148.
206
Mottron L. Burack J. A. Iarocci G. Belleville S. Enns J. T. (2003). Locally oriented perception with intact global processing among adolescents with high-functioning autism: evidence from multiple paradigms. J. Child Psychol. Psychiatry44, 904–913.10.1111/1469-7610.00174
207
Mottron L. Dawson M. Soulieres I. (2010). A different memory: are distinctions drawn from the study of nonautistic memory appropriate to describe memory in autism? In Memory in Autism – Theory and Evidence, eds BoucherJ.BowlerD. (Cambridge: Cambridge University Press), 311–329.
208
Mottron L. Dawson M. Soulieres I. Hubert B. Burack J. (2006). Enhanced perceptual functioning in autism: an update, and eight principles of autistic perception. J. Autism Dev. Disord.36, 27–43.10.1007/s10803-005-0040-7
209
Mottron L. Peretz I. Menard E. (2000). Local and global processing of music in high-functioning persons with autism: beyond central coherence?J. Child Psychol. Psychiatry41, 1057–1065.10.1111/1469-7610.00693
210
Muller D. Nikonenko I. Jourdain P. Alberi S. (2002). LTP, memory and structural plasticity. Curr. Mol. Med.2, 605–611.10.2174/1566524023362041
211
Muller R. A. Behen M. E. Rothermel R. D. Chugani D. C. Muzik O. Mangner T. J. Chugani H. T. (1999). Brain mapping of language and auditory perception in high-functioning autistic adults: a PET study. J. Autism Dev. Disord.29, 19–31.10.1023/A:1025914515203
212
Muller R. A. Chugani D. C. Behen M. E. Rothermel R. D. Muzik O. Chakraborty P. K. Chugani H. T. (1998). Impairment of dentato-thalamo-cortical pathway in autistic men: language activation data from positron emission tomography. Neurosci. Lett.245, 1–4.10.1016/S0304-3940(98)00151-7
213
Mullins M. Rincover A. (1985). Comparing autistic and normal children along the dimensions of reinforcement maximization, stimulus sampling, and responsiveness to extinction. J. Exp. Child Psychol.40, 350–374.10.1016/0022-0965(85)90095-5
214
Muris P. Steerneman P. Merckelbach H. Holdrinet I. Meesters C. (1998). Comorbid anxiety symptoms in children with pervasive developmental disorders. J. Anxiety Disord.12, 387–393.10.1016/S0887-6185(98)00022-X
215
Naisbitt S. Kim E. Tu J. C. Xiao B. Sala C. Valtschanoff J. Weinberg R. J. Worley P. F. Sheng M. (1999). Shank, a novel family of postsynaptic density proteins that binds to the NMDA receptor/PSD-95/GKAP complex and cortactin. Neuron23, 569–582.10.1016/S0896-6273(00)80809-0
216
Nakamura K. (2002). Aniracetam: its novel therapeutic potential in cerebral dysfunctional disorders based on recent pharmacological discoveries. CNS Drug Rev.8, 70–89.10.1111/j.1527-3458.2002.tb00216.x
217
Nam C. I. Chen L. (2005). Postsynaptic assembly induced by neurexin-neuroligin interaction and neurotransmitter. Proc. Natl. Acad. Sci. U.S.A.102, 6137–6142.10.1073/pnas.0502038102
218
Narita N. Kato M. Tazoe M. Miyazaki K. Narita M. Okado N. (2002). Increased monoamine concentration in the brain and blood of fetal thalidomide- and valproic acid-exposed rat: putative animal models for autism. Pediatr. Res. 52, 576–579.
219
Negri S. Oberson A. Steinmann M. Sauser C. Nicod P. Waeber G. Schorderet D.F. Bonny C. (2000). cDNA cloning and mapping of a novel islet-brain/JNK-interacting protein. Genomics64, 324–330.10.1006/geno.2000.6129
220
Nicoll R. A. Malenka R. C. (1999). Expression mechanisms underlying NMDA receptor-dependent long-term potentiation. Ann. N. Y. Acad. Sci.868, 515–525.10.1111/j.1749-6632.1999.tb11320.x
221
Noterdaeme M. Amorosa H. Mildenberger K. Sitter S. Minow F. (2001). Evaluation of attention problems in children with autism and children with a specific language disorder. Eur. Child Adolesc. Psychiatry10, 58–66.10.1007/s007870170048
222
Oberman L. Ifert-Miller F. Najib U. Bashir S. Woollacott I. Gonzalez-Heydrich J. Picker J. Rotenberg A. Pascual-Leone A. (2010). Transcranial magnetic stimulation provides means to assess cortical plasticity and excitability in humans with fragile X syndrome and autism spectrum disorder. Front. Syn. Neurosci.2: 26.10.3389/fnsyn.2010.00026.
223
Oberman L. M. Gautam S. Eldaief M. Fecteau S. Tormos J. M. Pascual-Leone A. (2009). “Mechanisms of cortical plasticity are pathologically enhanced in autism and suppressed in schizophrenia,” in Neuroscience Meeting Planner (Chicago, IL: Society for Neuroscience), Online.
224
O'Riordan M. Plaisted K. (2001). Enhanced discrimination in autism. Q. J. Exp. Psychol.54, 961–979.10.1080/02724980042000543
225
O'Riordan M. A. Plaisted K. C. Driver J. Baron-Cohen S. (2001). Superior visual search in autism. J. Exp. Psychol. Hum. Percept. Perform.27, 719–730.10.1037/0096-1523.27.3.719
226
Ozonoff S. (1995). Reliability and validity of the Wisconsin card sorting test in studies of autism. Neuropsychology9, 491–500.10.1037/0894-4105.9.4.491
227
Ozonoff S. Cook I. Coon H. Dawson G. Joseph R. M. Klin A. McMahon W. M. Minshew N. Munson J. A. Pennington B. F. Rogers S. J. Spence M. A. Tager-Flusberg H. Volkmar F. R. Wrathall D. (2004). Performance on Cambridge Neuropsychological Test Automated Battery subtests sensitive to frontal lobe function in people with autistic disorder: evidence from the collaborative programs of excellence in autism network. J. Autism Dev. Disord.34, 139–150.10.1023/B:JADD.0000022605.81989.cc
228
Ozonoff S. Pennington B. F. Rogers S. J. (1991). Executive function deficits in high-functioning autistic individuals: relationship to theory of mind. J. Child Psychol. Psychiatry32, 1081–1105.10.1111/j.1469-7610.1991.tb00351.x
229
Ozonoff S. Strayer D. L. (1997). Inhibitory function in non-retarded children with autism. J. Autism Dev. Disord.27, 59–77.10.1023/A:1025821222046
230
Ozonoff S. Strayer D. L. McMahon W. M. Filloux F. (1994). Executive function abilities in autism and Tourette syndrome: an information processing approach. J. Child Psychol. Psychiatry35, 1015–1032.10.1111/j.1469-7610.1994.tb01807.x
231
Page L. A. Daly E. Schmitz N. Simmons A. Toal F. Deeley Q. Ambery F. McAlonan G. M. Murphy K. C. Murphy D. G. (2006). In vivo 1H-magnetic resonance spectroscopy study of amygdala-hippocampal and parietal regions in autism. Am. J. Psychiatry163, 2189–2192.10.1176/appi.ajp.163.12.2189
232
Palkovitz R. J. Wiesenfeld A. R. (1980). Differential autonomic responses of autistic and normal children. J. Autism Dev. Disord.10, 347–360.10.1007/BF02408294
233
Palmen S. J. van Engeland H. Hof P. R. Schmitz C. (2004). Neuropathological findings in autism. Brain127, 2572–2583.10.1093/brain/awh287
234
Paradee W. Melikian H. E. Rasmussen D. L. Kenneson A. Conn P. J. Warren S. T. (1999). Fragile X mouse: strain effects of knockout phenotype and evidence suggesting deficient amygdala function. Neuroscience94, 185–192.10.1016/S0306-4522(99)00285-7
235
Pascualvaca D. M. Fantie B. D. Papageorgiou M. Mirsky A. F. (1998). Attentional capacities in children with autism: is there a general deficit in shifting focus?J. Autism Dev. Disord.28, 467–478.10.1023/A:1026091809650
236
Perry W. Minassian A. Lopez B. Maron L. Lincoln A. (2007). Sensorimotor gating deficits in adults with autism. Biol. Psychiatry61, 482–486.10.1016/j.biopsych.2005.09.025
237
Persico A. M. Bourgeron T. (2006). Searching for ways out of the autism maze: genetic, epigenetic and environmental clues. Trends Neurosci.29, 349–358.10.1016/j.tins.2006.05.010
238
Petersen C. C. (2007). The functional organization of the barrel cortex. Neuron56, 339–355.10.1016/j.neuron.2007.09.017
239
Phiel C. J. Zhang F. Huang E. Y. Guenther M. G. Lazar M. A. Klein P. S. (2001). Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J. Biol. Chem.276, 36734–36741.10.1074/jbc.M101287200
240
Pierce K. Courchesne E. (2000). Exploring the neurofunctional organization of face processing in autism. Arch. Gen. Psychiatry57, 344–346.10.1001/archpsyc.57.4.344
241
Pierce K. Haist F. Sedaghat F. Courchesne E. (2004). The brain response to personally familiar faces in autism: findings of fusiform activity and beyond. Brain127, 2703–2716.10.1093/brain/awh289
242
Pierce K. Muller R. A. Ambrose J. Allen G. Courchesne E. (2001). Face processing occurs outside the fusiform “face area” in autism: evidence from functional MRI. Brain124, 2059–2073.10.1093/brain/124.10.2059
243
Piggot J. Kwon H. Mobbs D. Blasey C. Lotspeich L. Menon V. Bookheimer S. Reiss A. L. (2004). Emotional attribution in high-functioning individuals with autistic spectrum disorder: a functional imaging study. J. Am. Acad. Child Adolesc. Psychiatry43, 473–480.10.1097/00004583-200404000-00014
244
Plaisted K. O'Riordan M. Baron-Cohen S. (1998). Enhanced visual search for a conjunctive target in autism: a research note. J. Child Psychol. Psychiatry39, 777–783.10.1017/S0021963098002613
245
Plaisted K. Saksida L. Alcantara J. Weisblatt E. (2003). Towards an understanding of the mechanisms of weak central coherence effects: experiments in visual configural learning and auditory perception. Philos. Trans. R. Soc. Lond. B Biol. Sci.358, 375–386.10.1098/rstb.2002.1211
246
Plaisted K. Swettenham J. Rees L. (1999). Children with autism show local precedence in a divided attention task and global precedence in a selective attention task. J. Child Psychol. Psychiatry40, 733–742.10.1111/1469-7610.00489
247
Posey D. J. Kem D. L. Swiezy N. B. Sweeten T. L. Wiegand R. E. McDougle C. J. (2004). A pilot study of D-cycloserine in subjects with autistic disorder. Am. J. Psychiatry161, 2115–2117.10.1176/appi.ajp.161.11.2115
248
Posner M. I. Petersen S. E. (1990). The attention system of the human brain. Annu. Rev. Neurosci.13, 25–42.10.1146/annurev.ne.13.030190.000325
249
Prather M. D. Lavenex P. Mauldin-Jourdain M. L. Mason W. A. Capitanio J. P. Mendoza S. P. Amaral D. G. (2001). Increased social fear and decreased fear of objects in monkeys with neonatal amygdala lesions. Neuroscience106, 653–658.10.1016/S0306-4522(01)00445-6
250
Pring L. (2005). Savant talent. Dev. Med. Child Neurol.47, 500–503.10.1017/S0012162205000976
251
Prior M. Hoffmann W. (1990). Brief report: neuropsychological testing of autistic children through an exploration with frontal lobe tests. J. Autism Dev. Disord.20, 581–590.10.1007/BF02216063
252
Puce A. Allison T. Gore J. C. McCarthy G. (1995). Face-sensitive regions in human extrastriate cortex studied by functional MRI. J. Neurophysiol.74, 1192–1199.
253
Purcell A. E. Jeon O. H. Zimmerman A. W. Blue M. E. Pevsner J. (2001). Postmortem brain abnormalities of the glutamate neurotransmitter system in autism. Neurology57, 1618–1628.
254
Rabionet R. McCauley J. L. Jaworski J. M. Ashley-Koch A. E. Martin E. R. Sutcliffe J. S. Haines J. L. DeLong G. R. Abramson R. K. Wright H. H. Cuccaro M. L. Gilbert J. R. Pericak-Vance M. A. (2006). Lack of association between autism and SLC25A12. Am. J. Psychiatry163, 929–931.10.1176/appi.ajp.163.5.929
255
Ramoz N. Reichert J. G. Smith C. J. Silverman J. M. Bespalova I. N. Davis K. L. Buxbaum J. D. (2004). Linkage and association of the mitochondrial aspartate/glutamate carrier SLC25A12 gene with autism. Am. J. Psychiatry161, 662–669.10.1176/appi.ajp.161.4.662
256
Rasalam A. D. Hailey H. Williams J. H. Moore S. J. Turnpenny P. D. Lloyd D. J. Dean J. C. (2005). Characteristics of fetal anticonvulsant syndrome associated autistic disorder. Dev. Med. Child Neurol.47, 551–555.10.1017/S0012162205001076
257
Raven J. Raven J. C. Court J. H. (1998). Raven Manual: Section 3. Standard Progressive MatricesOxford: Oxford Psychologists Press.
258
Renner P. Grofer Klinger L. Klinger M. R. (2006). Exogenous and endogenous attention orienting in autism spectrum disorders. Child Neuropsychol.12, 361–382.10.1080/09297040600770753
259
Renner P. Klinger L. G. Klinger M. R. (2000). Implicit and explicit memory in autism: is autism an amnesic disorder?J. Autism Dev. Disord.30, 3–14.10.1023/A:1005487009889
260
Rinaldi T. Kulangara K. Antoniello K. Markram H. (2007). Elevated NMDA receptor levels and enhanced postsynaptic long-term potentiation induced by prenatal exposure to valproic acid. Proc. Natl. Acad. Sci. U.S.A.104, 13501–13506.10.1073/pnas.0704391104
261
Rinaldi T. Perrodin C. Markram H. (2008a). Hyper-connectivity and hyper-plasticity in the medial prefrontal cortex in the valproic acid animal model of autism. Front. Neural Circuits2: 4.10.3389/neuro.04.004.2008
262
Rinaldi T. Silberberg G. Markram H. (2008b). Hyperconnectivity of local neocortical microcircuitry induced by prenatal exposure to valproic acid. Cereb. Cortex18, 163–170.
263
Rinehart N. J. Bradshaw J. L. Moss S. A. Brereton A. V. Tonge B. J. (2001). A deficit in shifting attention present in high-functioning autism but not Asperger's disorder. Autism5, 67–80.10.1177/1362361301005001007
264
Ring H. A. Baron-Cohen S. Wheelwright S. Williams S. C. Brammer M. Andrew C. Bullmore E. T. (1999). Cerebral correlates of preserved cognitive skills in autism: a functional MRI study of embedded figures task performance. Brain122 (Pt 7), 1305–1315.10.1093/brain/122.7.1305
265
Ritvo E. R. Freeman B. J. Scheibel A. B. Duong T. Robinson H. Guthrie D. Ritvo A. (1986). Lower Purkinje cell counts in the cerebella of four autistic subjects: initial findings of the UCLA-NSAC autopsy research report. Am. J. Psychiatry143, 862–866.
266
Rodier P. M. Ingram J. L. Tisdale B. Croog V. J. (1997). Linking etiologies in humans and animal models: studies of autism. Reprod. Toxicol.11, 417–422.10.1016/S0890-6238(97)80001-U
267
Rodier P. M. Ingram J. L. Tisdale B. Nelson S. Romano J. (1996). Embryological origin for autism: developmental anomalies of the cranial nerve motor nuclei. J. Comp. Neurol.370, 247–261.10.1002/(SICI)1096-9861(19960624)370:2<247::AID-CNE8>3.0.CO;2-2
268
Rogers S. J. Wehner D. E. Hagerman R. (2001). The behavioral phenotype in fragile X: symptoms of autism in very young children with fragile X syndrome, idiopathic autism, and other developmental disorders. J. Dev. Behav. Pediatr.22, 409–417.
269
Rojas D. C. Smith J. A. Benkers T. L. Camou S. L. Reite M. L. Rogers S. J. (2004). Hippocampus and amygdala volumes in parents of children with autistic disorder. Am. J. Psychiatry161, 2038–2044.10.1176/appi.ajp.161.11.2038
270
Rolf L. H. Haarmann F. Y. Grotemeyer K. H. Kehrer H. (1993). Serotonin and amino acid content in platelets of autistic children. Acta Psychiatr. Scand.87, 312–316.10.1111/j.1600-0447.1993.tb03378.x
271
Rubenstein J. L. Merzenich M. M. (2003). Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav.2, 255–267.10.1034/j.1601-183X.2003.00037.x
272
Rumsey J. M. Hamburger S. D. (1988). Neuropsychological findings in high-functioning men with infantile autism, residual state. J. Clin. Exp. Neuropsychol.10, 201–221.10.1080/01688638808408236
273
Sanders J. Johnson K. A. Garavan H. Gill M. Gallagher L. (2008). A review of neuropsychological and neuroimaging research in autistic spectrum disorders: attention, inhibition and cognitive flexibility. Res. Autism Spectr. Disord.2, 1–16.10.1016/j.rasd.2007.03.005
274
Scheiffele P. Fan J. Choih J. Fetter R. Serafini T. (2000). Neuroligin expressed in non-neuronal cells triggers presynaptic development in contacting axons. Cell101, 657–669.
275
Schneider T. Labuz D. Przewlocki R. (2001). Nociceptive changes in rats after prenatal exposure to valproic acid. Pol. J. Pharmacol.53, 531–534.
276
Schneider T. Przewlocki R. (2005). Behavioral alterations in rats prenatally exposed to valproic acid: animal model of autism. Neuropsychopharmacology30, 80–89.10.1038/sj.npp.1300518
277
Schneider T. Ziolkowska B. Gieryk A. Tyminska A. Przewlocki R. (2007). Prenatal exposure to valproic acid disturbs the enkephalinergic system functioning, basal hedonic tone, and emotional responses in an animal model of autism. Psychopharmacology193, 547–555.10.1007/s00213-007-0795-y
278
Schulkin J. (2007). Autism and the amygdala: an endocrine hypothesis. Brain Cogn. 65, 87–99.10.1016/j.bandc.2006.02.009
279
Schultz R. T. (2005). Developmental deficits in social perception in autism: the role of the amygdala and fusiform face area. Int. J. Dev. Neurosci.23, 125–141.10.1016/j.ijdevneu.2004.12.012
280
Schultz R. T. Gauthier I. Klin A. Fulbright R. K. Anderson A. W. Volkmar F. Skudlarski P. Lacadie C. Cohen D. J. Gore J. C. (2000). Abnormal ventral temporal cortical activity during face discrimination among individuals with autism and Asperger syndrome. Arch. Gen. Psychiatry57, 331–340.10.1001/archpsyc.57.4.331
281
Schumann C. M. Amaral D. G. (2006). Stereological analysis of amygdala neuron number in autism. J. Neurosci.26, 7674–7679.10.1523/JNEUROSCI.1285-06.2006
282
Schumann C. M. Hamstra J. Goodlin-Jones B. L. Lotspeich L. J. Kwon H. Buonocore M. H. Lammers C. R. Reiss A. L. Amaral D. G. (2004). The amygdala is enlarged in children but not adolescents with autism; the hippocampus is enlarged at all ages. J. Neurosci.24, 6392–6401.10.1523/JNEUROSCI.1297-04.2004
283
Sears L. L. Finn P. R. Steinmetz J. E. (1994). Abnormal classical eye-blink conditioning in autism. J. Autism Dev. Disord.24, 737–751.10.1007/BF02172283
284
Segurado R. Conroy J. Meally E. Fitzgerald M. Gill M. Gallagher L. (2005). Confirmation of association between autism and the mitochondrial aspartate/glutamate carrier SLC25A12 gene on chromosome 2q31. Am. J. Psychiatry162, 2182–2184.10.1176/appi.ajp.162.11.2182
285
Serajee F. J. Zhong H. Nabi R. Huq A. H. (2003). The metabotropic glutamate receptor 8 gene at 7q31: partial duplication and possible association with autism. J. Med. Genet.40, e42.10.1136/jmg.40.4.e42
286
Shah A. Frith U. (1983). An islet of ability in autistic children: a research note. J. Child Psychol. Psychiatry24, 613–620.10.1111/j.1469-7610.1983.tb00137.x
287
Shah A. Frith U. (1993). Why do autistic individuals show superior performance on the block design task?J. Child Psychol. Psychiatry34, 1351–1364.10.1111/j.1469-7610.1993.tb02095.x
288
Shayegan D. K. Stahl S. M. (2005). Emotion processing, the amygdala, and outcome in schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry29, 840–845.10.1016/j.pnpbp.2005.03.002
289
Shinohe A. Hashimoto K. Nakamura K. Tsujii M. Iwata Y. Tsuchiya K. J. Sekine Y. Suda S. Suzuki K. Sugihara G. Matsuzaki H. Minabe Y. Sugiyama T. Kawai M. Iyo M. Takei N. Mori N. (2006). Increased serum levels of glutamate in adult patients with autism. Prog. Neuropsychopharmacol. Biol. Psychiatry30, 1472–1477.10.1016/j.pnpbp.2006.06.013
290
Shu B. C. Lung F. W. Tien A. Y. Chen B. C. (2001). Executive function deficits in non-retarded autistic children. Autism5, 165–174.10.1177/1362361301005002006
291
Shuang M. Liu J. Jia M. X. Yang J. Z. Wu S. P. Gong X. H. Ling Y. S. Ruan Y. Yang X. L. Zhang D. (2004). Family-based association study between autism and glutamate receptor 6 gene in Chinese han trios. Am. J. Med. Genet. B Neuropsychiatr. Genet.131, 48–50.10.1002/ajmg.b.30025
292
Siegel B. V. Jr. Nuechterlein K. H. Abel L. Wu J. C. Buchsbaum M. S. (1995). Glucose metabolic correlates of continuous performance test performance in adults with a history of infantile autism, schizophrenics, and controls. Schizophr. Res.17, 85–94.10.1016/0920-9964(95)00033-I
293
Silberberg G. Gupta A. Markram H. (2002). Stereotypy in neocortical microcircuits. Trends Neurosci.25, 227–230.10.1016/S0166-2236(02)02151-3
294
Silva A. J. Wang Y. Paylor R. Wehner J. M. Stevens C. F. Tonegawa S. (1992). Alpha calcium/calmodulin kinase II mutant mice: deficient long-term potentiation and impaired spatial learning. Cold Spring Harb. Symp. Quant. Biol.57, 527–539.
295
Silva G. T. LeBe J.-V. Riachi I. Rinaldi T. Markram K. Markram H. (2009). Enhanced long term microcircuit plasticity in the valproic acid animal model of autism. Front. Syn. Neurosci.1: 1.10.3389/neuro.3319.3001.2009.
296
Silverman J. M. Buxbaum J. D. Ramoz N. Schmeidler J. Reichenberg A. Hollander E. Angelo G. Smith C. J. Kryzak L. A. (2008). Autism-related routines and rituals associated with a mitochondrial aspartate/glutamate carrier SLC25A12 polymorphism. Am. J. Med. Genet. B Neuropsychiatr. Genet.147, 408–410.10.1002/ajmg.b.30614
297
Sparks B. F. Friedman S. D. Shaw D. W. Aylward E. H. Echelard D. Artru A. A. Maravilla K. R. Giedd J. N. Munson J. Dawson G. Dager S. R. (2002). Brain structural abnormalities in young children with autism spectrum disorder. Neurology59, 184–192.
298
Stanton M. E. Peloso E. Brown K. L. Rodier P. (2007). Discrimination learning and reversal of the conditioned eyeblink reflex in a rodent model of autism. Behav. Brain Res.176, 133–140.10.1016/j.bbr.2006.10.022
299
Stein M. B. Goldin P. R. Sareen J. Zorrilla L. T. Brown G. G. (2002). Increased amygdala activation to angry and contemptuous faces in generalized social phobia. Arch. Gen. Psychiatry59, 1027–1034.10.1001/archpsyc.59.11.1027
300
Stromland K. Nordin V. Miller M. Akerstrom B. Gillberg C. (1994). Autism in thalidomide embryopathy: a population study. Dev. Med. Child Neurol.36, 351–356.10.1111/j.1469-8749.1994.tb11856.x
301
Struss D. Knight R. (2002). Principles of Frontal Lobe Function. Oxford: Oxford University Press.10.1093/acprof:oso/9780195134971.001.0001
302
Sweeten T. L. Posey D. J. Shekhar A. McDougle C. J. (2002). The amygdala and related structures in the pathophysiology of autism. Pharmacol. Biochem. Behav.71, 449–455.10.1016/S0091-3057(01)00697-9
303
Szatmari P. Bartolucci G. Bremner R. Bond S. Rich S. (1989). A follow-up study of high-functioning autistic children. J. Autism Dev. Disord.19, 213–225.10.1007/BF02211842
304
Toichi M. Kamio Y. (2002). Long-term memory and levels-of-processing in autism. Neuropsychologia40, 964–969.10.1016/S0028-3932(01)00163-4
305
Tootell R. B. Silverman M. S. De Valois R. L. (1981). Spatial frequency columns in primary visual cortex. Science214, 813–815.10.1126/science.7292014
306
Tordjman S. Anderson G. M. McBride P. A. Hertzig M. E. Snow M. E. Hall L. M. Thompson S. M. Ferrari P. Cohen D. J. (1997). Plasma beta-endorphin, adrenocorticotropin hormone, and cortisol in autism. J. Child Psychol. Psychiatry38, 705–715.10.1111/j.1469-7610.1997.tb01697.x
307
Townsend J. Courchesne E. Covington J. Westerfield M. Harris N. S. Lyden P. Lowry T. P. Press G. A. (1999). Spatial attention deficits in patients with acquired or developmental cerebellar abnormality. J. Neurosci.19, 5632–5643.
308
Townsend J. Harris N. S. Courchesne E. (1996). Visual attention abnormalities in autism: delayed orienting to location. J. Int. Neuropsychol. Soc.2, 541–550.10.1017/S1355617700001715
309
Treffert D. A. (1999). The savant syndrome and autistic disorder. CNS Spectr.4, 57–60.
310
Tsujino N. Nakatani Y. Seki Y. Nakasato A. Nakamura M. Sugawara M. Arita H. (2007). Abnormality of circadian rhythm accompanied by an increase in frontal cortex serotonin in animal model of autism. Neurosci. Res.57, 289–295.10.1016/j.neures.2006.10.018
311
van Engeland H. (1984). The electrodermal orienting response to auditive stimuli in autistic children, normal children, mentally retarded children, and child psychiatric patients. J. Autism Dev. Disord.14, 261–279.10.1007/BF02409578
312
Verte S. Geurts H. M. Roeyers H. Oosterlaan J. Sergeant J. A. (2005). Executive functioning in children with autism and Tourette syndrome. Dev. Psychopathol.17, 415–445.10.1017/S0954579405050200
313
Voelbel G. T. Bates M. E. Buckman J. F. Pandina G. Hendren R. L. (2006). Caudate nucleus volume and cognitive performance: Are they related in childhood psychopathology?Biol. Psychiatry60, 942–950.10.1016/j.biopsych.2006.03.071
314
Vorhees C. V. (1987a). Behavioral teratogenicity of valproic acid: selective effects on behavior after prenatal exposure to rats. Psychopharmacology (Berl.)92, 173–179.10.1007/BF00177911
315
Vorhees C. V. (1987b). Teratogenicity and developmental toxicity of valproic acid in rats. Teratology35, 195–202.10.1002/tera.1420350205
316
Vourc'h P. Bienvenu T. Beldjord C. Chelly J. Barthelemy C. Muh J. P. Andres C. (2001). No mutations in the coding region of the Rett syndrome gene MECP2 in 59 autistic patients. Eur. J. Hum. Genet.9, 556–558.10.1038/sj.ejhg.5200660
317
Wagner G. C. Reuhl K. R. Cheh M. McRae P. Halladay A. K. (2006). A new neurobehavioral model of autism in mice: pre- and postnatal exposure to sodium valproate. J. Autism Dev. Disord.36, 779–793.10.1007/s10803-006-0117-y
318
Wainwright J. A. Bryson S. E. (1996). Visual-spatial orienting in autism. J. Autism Dev. Disord.26, 423–438.10.1007/BF02172827
319
Wainwright-Sharp J. A. Bryson S. E. (1993). Visual orienting deficits in high-functioning people with autism. J. Autism Dev. Disord.23, 1–13.10.1007/BF01066415
320
Wang A. T. Dapretto M. Hariri A. R. Sigman M. Bookheimer S. Y. (2004). Neural correlates of facial affect processing in children and adolescents with autism spectrum disorder. J. Am. Acad. Child Adolesc. Psychiatry43, 481–490.10.1097/00004583-200404000-00015
321
Welchew D. E. Ashwin C. Berkouk K. Salvador R. Suckling J. Baron-Cohen S. Bullmore E. (2005). Functional disconnectivity of the medial temporal lobe in Asperger's syndrome. Biol. Psychiatry57, 991–998.10.1016/j.biopsych.2005.01.028
322
Williams D. L. Goldstein G. Carpenter P. A. Minshew N. J. (2005). Verbal and spatial working memory in autism. J. Autism Dev. Disord.35, 747–756.10.1007/s10803-005-0021-x
323
Williams D. L. Goldstein G. Minshew N. J. (2006). The profile of memory function in children with autism. Neuropsychology20, 21–29.10.1037/0894-4105.20.1.21
324
Williams G. King J. Cunningham M. Stephan M. Kerr B. Hersh J. H. (2001). Fetal valproate syndrome and autism: additional evidence of an association. Dev. Med. Child Neurol.43, 202–206.
325
Williams P. G. Hersh J. H. (1997). A male with fetal valproate syndrome and autism. Dev. Med. Child Neurol.39, 632–634.10.1111/j.1469-8749.1997.tb07500.x
326
Woolsey T. A. Van der Loos H. (1970). The structural organization of layer IV in the somatosensory region (SI) of mouse cerebral cortex. The description of a cortical field composed of discrete cytoarchitectonic units. Brain Res.17, 205–242.10.1016/0006-8993(70)90079-X
327
Yoo J. H. Valdovinos M. G. Williams D. C. (2007). Relevance of donepezil in enhancing learning and memory in special populations: a review of the literature. J. Autism Dev. Disord.37, 1883–1901.10.1007/s10803-006-0322-8
328
Zald D. H. (2003). The human amygdala and the emotional evaluation of sensory stimuli. Brain Res. Brain Res. Rev.41, 88–123.10.1016/S0165-0173(02)00248-5
329
Zalfa F. Giorgi M. Primerano B. Moro A. Di Penta A. Reis S. Oostra B. Bagni C. (2003). The fragile X syndrome protein FMRP associates with BC1 RNA and regulates the translation of specific mRNAs at synapses. Cell112, 317–327.
Summary
Keywords
autism, neocortex, amygdala, neural circuitry, connectivity, plasticity, NMDA, glutamate, perception, attention, memory, emotion, valproic acid, animal model
Citation
Markram K and Markram H (2010) The Intense World Theory – A Unifying Theory of the Neurobiology of Autism. Front. Hum. Neurosci. 4:224. doi: 10.3389/fnhum.2010.00224
Received
12 July 2010
Accepted
19 November 2010
Published
21 December 2010
Volume
4 - 2010
Edited by
Silvia A. Bunge, University of California Berkeley, USA
Reviewed by
Matthew K. Belmonte, National Brain Research Centre Manesar, India; Egidio D'Angelo, University of Pavia, Italy
Copyright
© 2010 Markram and Markram.
This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Kamila Markram, Laboratory of Neural Microcircuits, Brain Mind Institute, School of Life Sciences, Ecole Polytechnique Fédérale de Lausanne, Building AAB - Office 201 - Station 15, 1015 Lausanne, Switzerland. e-mail: kamila.markram@epfl.ch
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.