 Marioly Müller1,2
Marioly Müller1,2 Ulises Ahumada-Castro1
Ulises Ahumada-Castro1 Mario Sanhueza3
Mario Sanhueza3 Christian Gonzalez-Billault1,4,5
Christian Gonzalez-Billault1,4,5 Felipe A. Court1,3,5
Felipe A. Court1,3,5 César Cárdenas1,4,6,7*
César Cárdenas1,4,6,7*- 1Geroscience Center for Brain Health and Metabolism, Santiago, Chile
- 2Department of Medical Technology, Faculty of Medicine, Universidad de Chile, Santiago, Chile
- 3Center for Integrative Biology, Faculty of Sciences, Universidad Mayor, Santiago, Chile
- 4Department of Biology, Faculty of Sciences, Universidad de Chile, Santiago, Chile
- 5The Buck Institute for Research on Aging, Novato, CA, United States
- 6Anatomy and Developmental Biology Program, Institute of Biomedical Sciences, University of Chile, Santiago, Chile
- 7Department of Chemistry and Biochemistry, University of California, Santa Barbara, Santa Barbara, CA, United States
Age is the main risk factor for the onset of neurodegenerative diseases. A decline of mitochondrial function has been observed in several age-dependent neurodegenerative diseases and may be a major contributing factor in their progression. Recent findings have shown that mitochondrial fitness is tightly regulated by Ca2+ signals, which are altered long before the onset of measurable histopathology hallmarks or cognitive deficits in several neurodegenerative diseases including Alzheimer’s disease (AD), the most frequent cause of dementia. The transfer of Ca2+ from the endoplasmic reticulum (ER) to the mitochondria, facilitated by the presence of mitochondria-associated membranes (MAMs), is essential for several physiological mitochondrial functions such as respiration. Ca2+ transfer to mitochondria must be finely regulated because excess Ca2+ will disturb oxidative phosphorylation (OXPHOS), thereby increasing the generation of reactive oxygen species (ROS) that leads to cellular damage observed in both aging and neurodegenerative diseases. In addition, excess Ca2+ and ROS trigger the opening of the mitochondrial transition pore mPTP, leading to loss of mitochondrial function and cell death. mPTP opening probably increases with age and its activity has been associated with several neurodegenerative diseases. As Ca2+ seems to be the initiator of the mitochondrial failure that contributes to the synaptic deficit observed during aging and neurodegeneration, in this review, we aim to look at current evidence for mitochondrial dysfunction caused by Ca2+ miscommunication in neuronal models of neurodegenerative disorders related to aging, with special emphasis on AD.
Introduction
In the last century, the population aged over 60 years old has rapidly increased around the world (Beard et al., 2016). Aging is the major risk factor for many chronic diseases such as cancer, diabetes, hypertension, and neurodegenerative disorders (Kennedy et al., 2014). In particular, aging has been correlated with the occurrence of several types of dementia, affecting 5–10% of people over 65, and about 50% of people over 85 years old according to the Alzheimer’s Disease (AD) International (Prince et al., 2015). AD, the most common and still incurable form of dementia, shares several similar cellular alterations with brain aging including mitochondrial dysfunction, oxidative stress, Ca2+ dysregulation, and impaired proteostasis (Leuner et al., 2007; Kern and Behl, 2009; Rodrigue et al., 2009; Martinez et al., 2017). Most cases of AD are sporadic (SAD) and characterized by a late onset of symptoms, such as a decline of intellectual and cognitive functions and irreversible memory loss as major features. Several genes have been found to increase the risk of SAD, with the gene for apolipoprotein E (APOE) being the most studied, specifically, the polymorphism that produces the 𝜀4 allele of the APOE, APOE4 variant of the protein (Allen et al., 2012). In addition, nearly 1% of the cases of AD that are dominantly inherited present an early development known as familial AD (FAD) characterized by mutations in presenilin-1 (PS1) and -2 (PS2) or in the amyloid precursor protein (APP; Sherrington et al., 1995). Both SAD and FAD are characterized by neuronal cell death and assumed to be similar to some extent (Hardy and Selkoe, 2002), but the key events prior to cell death are still unclear.
Mitochondria are central organelles in neuronal physiology integrating several crucial functions such as cell respiration, energy metabolism, Ca2+ homeostasis, and reactive oxygen species (ROS) generation, all of which have been found to be dysregulated in aging, AD, and other neurodegenerative disorders such as Parkinson’s disease (PD) and amyotrophic lateral sclerosis (ALS)/frontotemporal dementia (FTD) disease (Winklhofer and Haass, 2010; Schon and Przedborski, 2011; Itoh et al., 2013; Manfredi and Kawamata, 2016). Here we present an overview of selected findings regarding mitochondrial dysfunction in neurodegenerative disease and discuss their potential as therapeutic targets.
Er-Mitochondria Communication and Ca2+ Regulation in Age-Associated Neurodegenerative Diseases
Communication between organelles allows cells to function and adapt in a changing cellular environment. The endoplasmic reticulum (ER) and mitochondria couple at specific sites termed mitochondria-associated membranes (MAMs), which integrate and coordinate several cellular functions, including synthesis and exchange of phospholipid, apoptosis, mitochondrial dynamics, and Ca2+ signaling (Liu and Zhu, 2017; Figure 1A). Remarkably, all these processes are affected early during aging, AD pathogenesis, and other neurodegenerative conditions, suggesting a role for MAMs in the pathogenesis of these diseases (De Vos et al., 2012; Hedskog et al., 2013; Gautier et al., 2016; Area-Gomez et al., 2018). For example, the overexpression of both wild-type and familial ALS/FTD mutant TDP-43 in HEK293, CV-1, and NSC34 cell lines reduces ER–mitochondria associations and Ca2+ exchange between these two organelles (Stoica et al., 2014; Figure 1C). Likewise, loss of Sigma 1 receptor (which is responsible for some familial forms of ALS/FTD) has been shown to interfere with ER–mitochondria associations (Bernard-Marissal et al., 2015; Figure 1C). Conversely, an increase in the lipidic enzymatic function of MAMs and their inter-organelle extension has been described in fibroblasts from patients with SAD, in human SAD brains, and in AD mouse models (Area-Gomez et al., 2012; Hedskog et al., 2013). Remarkably, one of the most common and validated risk factors for SAD, the presence of APOE4 (Holtzman et al., 2012), has recently been associated with an increase in the ER–mitochondrial communication and MAM enzymatic activity (Tambini et al., 2016; Figure 1B). Furthermore, MAMs are highly enriched in PS1 and PS2 proteins (Area-Gomez et al., 2009) which when mutated, as in fibroblasts from patients with FAD, also increase the lipidic enzymatic function of the MAMs and ER–mitochondria communication (Area-Gomez et al., 2012), through a mechanism that involves an interaction between the mutated form of PS2 and mitofusin-2 (Mfn2; Figure 1B), a key protein in the formation of MAMs (Filadi et al., 2016).
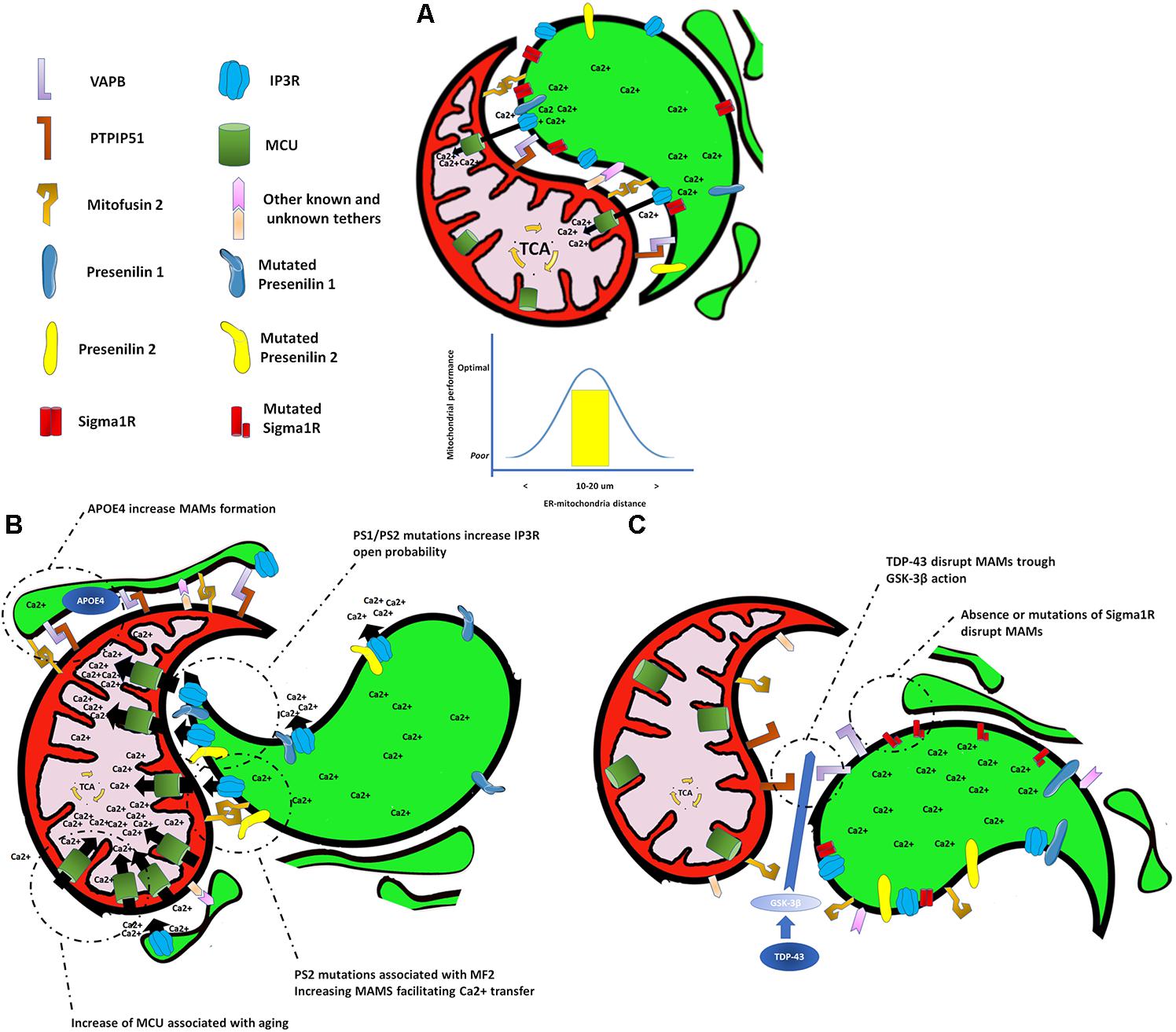
FIGURE 1. Endoplasmic reticulum and mitochondria interactions under different scenarios. (A) Endoplasmic reticulum–mitochondrial interaction at a proper distance allows a correct transfer of calcium to the mitochondrial matrix to sustain bioenergetic function. (B) Shortening of the distance at the endoplasmic reticulum–mitochondrial interaction, or an increase in either the mitochondrial calcium uptake or the calcium release from the endoplasmic reticulum, causes an exaggerated transfer of calcium to mitochondria affecting its normal bioenergetic function that leads to cell death. (C) Separation of the endoplasmic reticulum and mitochondria causes a decrease in the calcium transfer to the mitochondrial matrix generating a drop in the bioenergetic and metabolic activity of the mitochondria.
Conditions that increase or decrease the extension of MAMs will affect the transfer of Ca2+ from the ER to mitochondria, resulting in either a mitochondrial Ca2+ overload, or a lack of Ca2+. If the transfer of Ca2+ is excessive, cell death occurs (Schinder et al., 1996). If Ca2+ transfer to mitochondria is too low, a bioenergetics crisis occurs, also resulting in cell death (Cardenas et al., 2010). Importantly, Ca2+ is dysregulated in the aged brain and in AD (Landfield and Pitler, 1984; Gibson and Peterson, 1987; Khachaturian, 1987). Upregulation of Ca2+ levels can both initiate and accelerate several AD features, from amyloid deposition to synapse loss (Stutzmann et al., 2007). Several mechanisms have been proposed to explain the upregulation of cytoplasmic Ca2+ levels in AD including overexpression of the ryanodine receptor (RyR; Chakroborty et al., 2012), or β-amyloid (Aβ)-triggering release of Ca2+ from both extracellular and intracellular sources (Demuro and Parker, 2013). Another mechanism involves an increase of Ca2+ leak from the ER through sensitization of the inositol 1,4,5-trisphosphate receptor (InsP3R) Ca2+ channel by directed interaction with FAD-linked PS mutants (Cheung et al., 2008, 2010) or indirectly by interaction of FAD-linked PS mutants with the SERCA pump (Green et al., 2008). In agreement with the latter, it has been demonstrated that overexpression of the FAD-linked PS2 mutant leads to an increase in the generation of cytosolic Ca2+ hot spots, ER–mitochondria tethering, and mitochondrial Ca2+ uptake (Zampese et al., 2011). This in turn may result in mitochondrial Ca2+ overload and could explain the metabolic dysfunction and cell death observed in AD (Figure 1B). On the other hand, decreasing intracellular Ca2+ overload, specifically through a reduction of the InsP3R protein expression by 50%, normalizes FAD PS-associated Ca2+ signaling and rescues the biochemical, electrophysiological, and behavioral phenotypes observed in two different PS1-FAD animal models (Shilling et al., 2014; Figure 1B). Altogether, the above findings highlight the importance of MAMs and the transfer of Ca2+ from the ER to mitochondria in AD pathogenesis and their potential as a therapeutic target.
The role of MAMs in aging has just begun to be unveiled. Similar to what has been observed in AD models, aging increases the ER to mitochondria Ca2+ transfer in long-term culture of hippocampal neurons, with correlates with an increase of the mitochondrial Ca2+ uniporter (MCU; Calvo-Rodriguez et al., 2016; Figure 1B). Interestingly, a decrease in Ca2+ transfer to mitochondria and a dissociation of the MAMs have been described in cardiomyocytes from old mice suggesting that the type of modification that MAMs undergo with aging might be cell specific (Fernandez-Sanz et al., 2014).
The Mitochondrial Permeability Transition Pore (Mptp) Formation in Age-Associated Neurodegenerative Diseases
Under conditions of Ca2+ and/or ROS overload, formation of the mitochondrial permeability transition pore (mPTP) takes place, which corresponds to a non-selective channel formed by a protein complex spanning the outer and inner mitochondrial membranes (Bernardi et al., 2006). In physiological conditions, transient opening of the mPTP can regulate Ca2+ levels in the mitochondrial matrix (Ichas et al., 1997). However, dysregulated mPTP opening triggers the release of most matrix metabolites such as ROS, Ca2+, and NAD +, leading to loss of the mitochondrial membrane potential, inhibition of oxidative phosphorylation (OXPHOS), and mitochondrial swelling (Elrod and Molkentin, 2013; Rottenberg and Hoek, 2017). Even though several proteins are known to participate in mPTP formation [anion channel VDAC, adenine nucleotide translocator (ANT), mitochondrial ATP synthase (F0F1), phosphate carrier (PiC), and cyclophilin D (CypD; Bernardi et al., 2006; Rao et al., 2014), its detailed structural configuration is not yet entirely known.
The mPTP has been linked to neurodegeneration in vitro and in vivo. In neural progenitor cells, Aβ-amyloid exposure leads to mPTP opening and a decrease in mitochondrial membrane potential, release of cytochrome C, and cell death (Hou et al., 2014). In human AD brains, Aβ-amyloid binds CypD in mitochondria (Du and Yan, 2010), and CypD deficiency improves mitochondrial function, memory, and learning in an AD mouse model (Du et al., 2008). Aβ-induced neurotoxicity in vitro was also attenuated pharmacologically by inhibition of the mPTP using cyclosporine A (CsA) on neural stem cells (Chen et al., 2016). Interestingly, it has been shown that CypD knock-out mice exhibit delayed axonal degeneration, a common feature of diverse neurodegenerative diseases (Barrientos et al., 2011; Catenaccio et al., 2017; Salvadores et al., 2017). Indeed, genetic deletion of CypD delays disease progression in other mouse models of neurodegenerative disorders, including ALS (Martin et al., 2009), PD (Thomas et al., 2012), and multiple sclerosis (Forte et al., 2007). Therefore, novel compounds that inhibit mPTP opening are currently been developed, including sanglifehrin A, N-Me-Ala-6-cyclosporin A, and antmanide (Rao et al., 2014).
Aging also modifies the opening probability of the mPTP (Rottenberg and Hoek, 2017). During aging, the probability of the mPTP opening increases due to higher expression levels of CypD and the CypD-activator p53 (Priami et al., 2015). Furthermore, the expression of HSP90, a chaperone that binds CypD to trigger its degradation, is decreased in aged cells (Lam et al., 2015), which could also increase the mPTP opening probability. This evidence is further supported by a faster Ca2+-induced mitochondrial swelling in purified liver mitochondria obtained from aged mice (Goodell and Cortopassi, 1998). Interestingly, CypD is inactivated by the deacetylase SIRT3 (Hafner et al., 2010), a known modulator of longevity in diverse species (Jasper, 2013). The reported decline in SIRT3 activity during aging (Brown et al., 2013) may lead to a greater activation of the mPTP, underscoring the role of mitochondria in longevity and onset of age-dependent neurodegenerative diseases. Interestingly, several modulators of longevity, including metformin, mitochondrial UPR, and caloric restriction inhibit the activation of the mPTP (Bhamra et al., 2008; Altieri, 2013; Amigo et al., 2017), and may contribute to lifespan extension (Rottenberg and Hoek, 2017). A key role for mitochondria in age-related disorders has been associated to broad damaging events including increased ROS production and defects in the regulation of intracellular Ca2+ levels, which are directly associated to mPTP activation with profound negative consequences for cell survival. Therefore, mPTP emerges as a potential target for neuroprotection in age-related neurodegenerative conditions.
Mitochondria, Ros, Aging, and Neurodegeneration
Reactive oxygen species are chemical species that are produced by most cell types. The group of molecules that fulfill the criteria for ROS includes hydrogen peroxide, and the highly reactive species superoxide anion and hydroxyl radical (Wilson et al., 2017). The production of ROS in cells is controlled by enzymatic or non-enzymatic mechanisms. The main source for ROS production in terms of quantitative production is the mitochondria (Holmstrom and Finkel, 2014). Mitochondria produces superoxide anion, a by-product of the inefficient transfer of electrons by the electron transport chain (ETC) during OXPHOS, that is quickly converted into hydrogen peroxide by the action of the superoxide dismutases 1–3 (SOD1–3; Quinlan et al., 2013). Of note, despite mitochondria being the main source of ROS in cells, hydrogen peroxide can be produced by more than 30 different enzymes (Go et al., 2015).
While a huge amount of work in the past focused on the deleterious roles for ROS species in cells and organisms, including the “free radical” or “oxidative stress” theory of aging (Harman, 1956) supported by many studies (Harman, 1992; Cadenas and Davies, 2000; Golden et al., 2002), there is growing evidence in the last decades that ROS may serve physiological functions (Zuo et al., 2015; Sies et al., 2017; Wilson et al., 2017). Related to aging, other studies show that unbalanced ROS production does not modify lifespan in mice under tightly controlled conditions (Van Remmen et al., 2003; Ran et al., 2007). Moreover, it was demonstrated that there is no increased oxidative damage with age (Barja and Herrero, 2000; Kauppila et al., 2017). Currently, it has been proposed that adaptive or hormetic production of ROS is required to maintain several cellular mechanisms including stem cell proliferation and fate determination in the brain (Sena and Chandel, 2012; Chaudhari et al., 2014).
In terms of neurodegeneration associated to aging, it has been reported in AD that the Aβ peptide interacts with the mitochondrial protein termed amyloid binding alcohol dehydrogenase (ABAD) in AD mouse models and in post-mortem samples derived from AD human patients. The functional consequence of such an interaction is an increase in ROS production due to abnormal mitochondrial membrane permeability (Lustbader et al., 2004). Altered OXPHOS increases the generation of ROS (Koopman et al., 2013) and is indeed a hallmark for early AD abnormalities in humans. In fact, samples from human subjects show that mitochondrial-encoded OXPHOS genes are altered in aging, mild cognitive impairment, and AD (Mastroeni et al., 2017). Similarly, AD mouse models have shown that both Aβ and tau protein can induce alterations in mitochondrial proteins involved in OXPHOS (Caspersen et al., 2005; Rhein et al., 2009; Eckert et al., 2010), causing an aberrant ROS generation leading to cellular damage. In addition, ROS are known to cause mitochondrial fragmentation (Wang et al., 2014), which reduces mitochondrial performance (Westermann, 2012) favoring the generation of more ROS and cellular damage associated to it.
Mitochondrial Dysfunction and Synaptic Deficits
Synapses are neuronal structures in which mitochondria are fundamental (Li et al., 2004) by providing large amounts of ATP required to fuel synaptic vesicle physiology and by acting as a Ca2+ buffer modulating cytoplasmic Ca2+ signal and hence, neurotransmission (Ghosh and Greenberg, 1995; Verstreken et al., 2005; Gunter and Sheu, 2009; Wan et al., 2012). Synaptic mitochondria are more vulnerable to cumulative damage showing impaired Ca2+ uptake capacity and increased propensity to undergo mPTP compared to non-synaptic mitochondria (Scheff et al., 2006). Likewise, in an APP/PS1 AD mouse, synaptic mitochondrial function was significantly more affected than non-synaptic mitochondria (Dragicevic et al., 2010). Synaptic deficit is an early event in the pathogenesis of several neurodegenerative disorders including AD and worsens with disease progression and age (Wilcox et al., 2011). The extent of cognitive decline in AD patients is tightly associated with the extent of synapse loss in specific brain regions including cortex and hippocampus (Scheff et al., 2006; Mattson, 2010). Post-mortem hippocampus from AD patients shows a considerable decrease in dendritic spine density (Ferrer et al., 1990) and transgenic mouse models of AD show age-dependent reduction in spine density before plaque deposition (Lanz et al., 2003). Along these lines, in aged synapses and from AD models, a decline in mitochondrial respiration and signs of mitochondrial damage such as reduced antioxidant contents and increased oxidative stress markers has been described (Du et al., 2010; Quiroz-Baez et al., 2013). Proteomic analysis of aged synaptic mitochondria reveals changes in ETC proteins, antioxidants, and proteins related to mitochondrial dynamics (Stauch et al., 2014). Just recently, through the use of cytoplasmic hybrid (“cybrid”) technology, Yu et al. (2017) were able to recapitulate mitochondrial structural and functional changes observed in AD-affected brains. In this model, their findings demonstrate that AD-affected mitochondria elicited detrimental effects on synaptic development (Yu et al., 2017). How ER vesicles found in the synaptic region and the transfer of Ca2+ contribute to the impairment of mitochondria and synaptic formation remains to be explored, but given the dysregulation of Ca2+ observed during aging, AD, and other neurodegeneration, an important role is expected. Elucidating the factors that underlie early synaptic dysfunction will be key to prevent the widespread neurodegeneration associated with aging.
Conclusion
Aging continues to be the most relevant risk factor for AD, the most common form of dementia in the elderly, and other neurodegenerative diseases. Both aging and neurodegeneration are accompanied by a loss in the ability of the cells to adjust and rewire their metabolic networks to keep a tight balance between energy production and expenditure in an ever-changing environment. Mitochondria work as an adaptable metabolic control, a “rheostat,” that integrates inputs from the intra and extracellular environment to generate functional outputs that adjust cell behavior and energy production and consumption. Several lines of evidence suggest that mitochondrial function deteriorates with increasing age and the progression of several neurodegenerative diseases. This supports the notion that aging and the neurodegenerative diseases such as AD may share a common root, the failure of the rheostat program. Since Ca2+ is also altered in both conditions and can either energize or overload the rheostat depending on the concentration, understanding how MAM formation is regulated is important. Identifying the players that participate in the regulation to assure a proper Ca2+ transfer to mitochondria is critical in order to determine the real potential of this intracellular signaling platform as an intervention candidate to improve aging and hinder the onset of neurodegenerative disease such as AD.
Author Contributions
MM, UA-C, and CC designed and outlined the structure and contents of the review. MM, UA-C, MS, FC, CG-B, and CC contributed to the literature review, discussion, and writing of the manuscript. All authors contributed equally to the draft revisions and final approval of the version to be published.
Funding
This work was supported by Geroscience Center for Brain Health and Metabolism (FONDAP-15150012; FC, CG-B, and CC) and Fondo Nacional de Desarrollo Científico y Tecnológico (FONDECYT) #1160332 (CC), 1150766 (FC) and #1180419 (CG-B).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors want to thank Dr. Alenka Lovy for her feedback and useful comments.
References
Allen, M., Zou, F., Chai, H. S., Younkin, C. S., Crook, J., Pankratz, V. S., et al. (2012). Novel late-onset Alzheimer disease loci variants associate with brain gene expression. Neurology 79, 221–228. doi: 10.1212/WNL.0b013e3182605801
Altieri, D. C. (2013). Hsp90 regulation of mitochondrial protein folding: from organelle integrity to cellular homeostasis. Cell. Mol. Life Sci. 70, 2463–2472. doi: 10.1007/s00018-012-1177-0
Amigo, I., Menezes-Filho, S. L., Luevano-Martinez, L. A., Chausse, B., and Kowaltowski, A. J. (2017). Caloric restriction increases brain mitochondrial calcium retention capacity and protects against excitotoxicity. Aging Cell 16, 73–81. doi: 10.1111/acel.12527
Area-Gomez, E., De Groof, A., Bonilla, E., Montesinos, J., Tanji, K., Boldogh, I., et al. (2018). A key role for MAM in mediating mitochondrial dysfunction in Alzheimer disease. Cell Death Dis. 9:335. doi: 10.1038/s41419-017-0215-0
Area-Gomez, E., De Groof, A. J., Boldogh, I., Bird, T. D., Gibson, G. E., Koehler, C. M., et al. (2009). Presenilins are enriched in endoplasmic reticulum membranes associated with mitochondria. Am. J. Pathol. 175, 1810–1816. doi: 10.2353/ajpath.2009.090219
Area-Gomez, E., Del Carmen Lara Castillo, M., Tambini, M. D., Guardia-Laguarta, C., De Groof, A. J., Madra, M., et al. (2012). Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. EMBO J. 31, 4106–4123. doi: 10.1038/emboj.2012.202
Barja, G., and Herrero, A. (2000). Oxidative damage to mitochondrial DNA is inversely related to maximum life span in the heart and brain of mammals. FASEB J. 14, 312–318. doi: 10.1096/fasebj.14.2.312
Barrientos, S. A., Martinez, N. W., Yoo, S., Jara, J. S., Zamorano, S., Hetz, C., et al. (2011). Axonal degeneration is mediated by the mitochondrial permeability transition pore. J. Neurosci. 31, 966–978. doi: 10.1523/JNEUROSCI.4065-10.2011
Beard, J. R., Officer, A., De Carvalho, I. A., Sadana, R., Pot, A. M., Michel, J. P., et al. (2016). The world report on ageing and health: a policy framework for healthy ageing. Lancet 387, 2145–2154. doi: 10.1016/S0140-6736(15)00516-4
Bernard-Marissal, N., Medard, J. J., Azzedine, H., and Chrast, R. (2015). Dysfunction in endoplasmic reticulum-mitochondria crosstalk underlies SIGMAR1 loss of function mediated motor neuron degeneration. Brain 138, 875–890. doi: 10.1093/brain/awv008
Bernardi, P., Krauskopf, A., Basso, E., Petronilli, V., Blachly-Dyson, E., Di Lisa, F., et al. (2006). The mitochondrial permeability transition from in vitro artifact to disease target. FEBS J. 273, 2077–2099. doi: 10.1111/j.1742-4658.2006.05213.x
Bhamra, G. S., Hausenloy, D. J., Davidson, S. M., Carr, R. D., Paiva, M., Wynne, A. M., et al. (2008). Metformin protects the ischemic heart by the Akt-mediated inhibition of mitochondrial permeability transition pore opening. Basic Res. Cardiol. 103, 274–284. doi: 10.1007/s00395-007-0691-y
Brown, K., Xie, S., Qiu, X., Mohrin, M., Shin, J., Liu, Y., et al. (2013). SIRT3 reverses aging-associated degeneration. Cell Rep. 3, 319–327. doi: 10.1016/j.celrep.2013.01.005
Cadenas, E., and Davies, K. J. (2000). Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 29, 222–230. doi: 10.1016/S0891-5849(00)00317-8
Calvo-Rodriguez, M., Garcia-Durillo, M., Villalobos, C., and Nunez, L. (2016). In vitro aging promotes endoplasmic reticulum (ER)-mitochondria Ca2+ cross talk and loss of store-operated Ca2+ entry (SOCE) in rat hippocampal neurons. Biochim. Biophys. Acta 1863, 2637–2649. doi: 10.1016/j.bbamcr.2016.08.001
Cardenas, C., Miller, R. A., Smith, I., Bui, T., Molgo, J., Muller, M., et al. (2010). Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 142, 270–283. doi: 10.1016/j.cell.2010.06.007
Caspersen, C., Wang, N., Yao, J., Sosunov, A., Chen, X., Lustbader, J. W., et al. (2005). Mitochondrial abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J. 19, 2040–2041. doi: 10.1096/fj.05-3735fje
Catenaccio, A., Llavero Hurtado, M., Diaz, P., Lamont, D. J., Wishart, T. M., and Court, F. A. (2017). Molecular analysis of axonal-intrinsic and glial-associated co-regulation of axon degeneration. Cell Death Dis. 8:e3166. doi: 10.1038/cddis.2017.489
Chakroborty, S., Kim, J., Schneider, C., Jacobson, C., Molgo, J., and Stutzmann, G. E. (2012). Early presynaptic and postsynaptic calcium signaling abnormalities mask underlying synaptic depression in presymptomatic Alzheimer’s disease mice. J. Neurosci. 32, 8341–8353. doi: 10.1523/JNEUROSCI.0936-12.2012
Chaudhari, P., Ye, Z., and Jang, Y. Y. (2014). Roles of reactive oxygen species in the fate of stem cells. Antioxid. Redox Signal. 20, 1881–1890. doi: 10.1089/ars.2012.4963
Chen, J., Yang, F., Yu, X., Yu, Y., and Gong, Y. (2016). Cyclosporine a promotes cell proliferation, collagen and alpha-smooth muscle actin expressions in rat gingival fibroblasts by Smad3 activation and miR-29b suppression. J. Periodontal Res. 51, 735–747. doi: 10.1111/jre.12350
Cheung, K. H., Shineman, D., Mueller, M., Cardenas, C., Mei, L., Yang, J., et al. (2008). Mechanism of Ca2+ disruption in Alzheimer’s disease by presenilin regulation of InsP(3) receptor channel gating. Neuron 58, 871–883. doi: 10.1016/j.neuron.2008.04.015
Cheung, K. H., Mei, L., Mak, D. O., Hayashi, I., Iwatsubo, T., Kang, D. E., et al. (2010). Gain-of-function enhancement of IP3 receptor modal gating by familial Alzheimer’s disease-linked presenilin mutants in human cells and mouse neurons. Sci. Signal. 3:ra22. doi: 10.1126/scisignal.2000818
De Vos, K. J., Morotz, G. M., Stoica, R., Tudor, E. L., Lau, K. F., Ackerley, S., et al. (2012). VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum. Mol. Genet. 21, 1299–1311. doi: 10.1093/hmg/ddr559
Demuro, A., and Parker, I. (2013). Cytotoxicity of intracellular abeta42 amyloid oligomers involves Ca2+ release from the endoplasmic reticulum by stimulated production of inositol trisphosphate. J. Neurosci. 33, 3824–3833. doi: 10.1523/JNEUROSCI.4367-12.2013
Dragicevic, N., Mamcarz, M., Zhu, Y., Buzzeo, R., Tan, J., Arendash, G. W.,et al. (2010). Mitochondrial amyloid-beta levels are associated with the extent of mitochondrial dysfunction in different brain regions and the degree of cognitive impairment in Alzheimer’s transgenic mice. J. Alzheimers Dis. 20(Suppl. 2), S535–S550. doi: 10.3233/JAD-2010-100342
Du, H., Guo, L., Fang, F., Chen, D., Sosunov, A. A., Mckhann, G. M., et al. (2008). Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat. Med. 14, 1097–1105. doi: 10.1038/nm.1868
Du, H., Guo, L., Yan, S., Sosunov, A. A., Mckhann, G. M., and Yan, S. S. (2010). Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc. Natl. Acad. Sci. U.S.A. 107, 18670–18675. doi: 10.1073/pnas.1006586107
Du, H., and Yan, S. S. (2010). Mitochondrial permeability transition pore in Alzheimer’s disease: cyclophilin D and amyloid beta. Biochim. Biophys. Acta 1802, 198–204. doi: 10.1016/j.bbadis.2009.07.005
Eckert, A., Schulz, K. L., Rhein, V., and Gotz, J. (2010). Convergence of amyloid-beta and tau pathologies on mitochondria in vivo. Mol. Neurobiol. 41, 107–114. doi: 10.1007/s12035-010-8109-5
Elrod, J. W., and Molkentin, J. D. (2013). Physiologic functions of cyclophilin D and the mitochondrial permeability transition pore. Circ. J. 77, 1111–1122. doi: 10.1253/circj.CJ-13-0321
Fernandez-Sanz, C., Ruiz-Meana, M., Miro-Casas, E., Nunez, E., Castellano, J., Loureiro, M., et al. (2014). Defective sarcoplasmic reticulum-mitochondria calcium exchange in aged mouse myocardium. Cell Death Dis. 5:e1573. doi: 10.1038/cddis.2014.526
Ferrer, I., Guionnet, N., Cruz-Sanchez, F., and Tunon, T. (1990). Neuronal alterations in patients with dementia: a Golgi study on biopsy samples. Neurosci. Lett. 114, 11–16. doi: 10.1016/0304-3940(90)90420-E
Filadi, R., Greotti, E., Turacchio, G., Luini, A., Pozzan, T., and Pizzo, P. (2016). Presenilin 2 modulates endoplasmic reticulum-mitochondria coupling by tuning the antagonistic effect of mitofusin 2. Cell Rep. 15, 2226–2238. doi: 10.1016/j.celrep.2016.05.013
Forte, M., Gold, B. G., Marracci, G., Chaudhary, P., Basso, E., Johnsen, D., et al. (2007). Cyclophilin D inactivation protects axons in experimental autoimmune encephalomyelitis, an animal model of multiple sclerosis. Proc. Natl. Acad. Sci. U.S.A. 104, 7558–7563. doi: 10.1073/pnas.0702228104
Gautier, C. A., Erpapazoglou, Z., Mouton-Liger, F., Muriel, M. P., Cormier, F., Bigou, S., et al. (2016). The endoplasmic reticulum-mitochondria interface is perturbed in PARK2 knockout mice and patients with PARK2 mutations. Hum. Mol. Genet. 25, 2972–2984. doi: 10.1093/hmg/ddw148
Ghosh, A., and Greenberg, M. E. (1995). Calcium signaling in neurons: molecular mechanisms and cellular consequences. Science 268, 239–247. doi: 10.1126/science.7716515
Gibson, G. E., and Peterson, C. (1987). Calcium and the aging nervous system. Neurobiol. Aging 8, 329–343. doi: 10.1016/0197-4580(87)90072-8
Go, Y. M., Chandler, J. D., and Jones, D. P. (2015). The cysteine proteome. Free Radic. Biol. Med. 84, 227–245. doi: 10.1016/j.freeradbiomed.2015.03.022
Golden, T. R., Hinerfeld, D. A., and Melov, S. (2002). Oxidative stress and aging: beyond correlation. Aging Cell 1, 117–123. doi: 10.1046/j.1474-9728.2002.00015.x
Goodell, S., and Cortopassi, G. (1998). Analysis of oxygen consumption and mitochondrial permeability with age in mice. Mech. Ageing Dev. 101, 245–256. doi: 10.1016/S0047-6374(97)00182-6
Green, K. N., Demuro, A., Akbari, Y., Hitt, B. D., Smith, I. F., Parker, I., et al. (2008). SERCA pump activity is physiologically regulated by presenilin and regulates amyloid beta production. J. Cell Physiol. 181, 1107–1116.
Gunter, T. E., and Sheu, S. S. (2009). Characteristics and possible functions of mitochondrial Ca2+ transport mechanisms. Biochim. Biophys. Acta 1787, 1291–1308. doi: 10.1016/j.bbabio.2008.12.011
Hafner, A. V., Dai, J., Gomes, A. P., Xiao, C. Y., Palmeira, C. M., Rosenzweig, A., et al. (2010). Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging 2, 914–923. doi: 10.18632/aging.100252
Hardy, J., and Selkoe, D. J. (2002). The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356. doi: 10.1126/science.1072994
Harman, D. (1956). Aging: a theory based on free radical and radiation chemistry. J. Gerontol. 11, 298–300. doi: 10.1093/geronj/11.3.298
Harman, D. (1992). Role of free radicals in aging and disease. Ann. N.Y. Acad. Sci. 673, 126–141. doi: 10.1111/j.1749-6632.1992.tb27444.x
Hedskog, L., Pinho, C. M., Filadi, R., Ronnback, A., Hertwig, L., Wiehager, B., et al. (2013). Modulation of the endoplasmic reticulum-mitochondria interface in Alzheimer’s disease and related models. Proc. Natl. Acad. Sci. U.S.A. 110, 7916–7921. doi: 10.1073/pnas.1300677110
Holmstrom, K. M., and Finkel, T. (2014). Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 15, 411–421. doi: 10.1038/nrm3801
Holtzman, D. M., Herz, J., and Bu, G. (2012). Apolipoprotein E and apolipoprotein E receptors: normal biology and roles in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2:a006312. doi: 10.1101/cshperspect.a006312
Hou, Y., Ghosh, P., Wan, R., Ouyang, X., Cheng, H., Mattson, M. P., et al. (2014). Permeability transition pore-mediated mitochondrial superoxide flashes mediate an early inhibitory effect of amyloid beta1-42 on neural progenitor cell proliferation. Neurobiol. Aging 35, 975–989. doi: 10.1016/j.neurobiolaging.2013.11.002
Ichas, F., Jouaville, L. S., and Mazat, J. P. (1997). Mitochondria are excitable organelles capable of generating and conveying electrical and calcium signals. Cell 89, 1145–1153. doi: 10.1016/S0092-8674(00)80301-3
Itoh, K., Nakamura, K., Iijima, M., and Sesaki, H. (2013). Mitochondrial dynamics in neurodegeneration. Trends Cell Biol. 23, 64–71. doi: 10.1016/j.tcb.2012.10.006
Jasper, H. (2013). Sirtuins: longevity focuses on NAD+. Nat. Chem. Biol. 9, 666–667. doi: 10.1038/nchembio.1369
Kauppila, T. E. S., Kauppila, J. H. K., and Larsson, N. G. (2017). Mammalian mitochondria and aging: an update. Cell Metab. 25, 57–71. doi: 10.1016/j.cmet.2016.09.017
Kennedy, B. K., Berger, S. L., Brunet, A., Campisi, J., Cuervo, A. M., Epel, E. S., et al. (2014). Geroscience: linking aging to chronic disease. Cell 159, 709–713. doi: 10.1016/j.cell.2014.10.039
Kern, A., and Behl, C. (2009). The unsolved relationship of brain aging and late-onset Alzheimer disease. Biochim. Biophys. Acta 1790, 1124–1132. doi: 10.1016/j.bbagen.2009.07.016
Khachaturian, Z. S. (1987). Hypothesis on the regulation of cytosol calcium concentration and the aging brain. Neurobiol. Aging 8, 345–346. doi: 10.1016/0197-4580(87)90073-X
Koopman, W. J., Distelmaier, F., Smeitink, J. A., and Willems, P. H. (2013). OXPHOS mutations and neurodegeneration. EMBO J. 32, 9–29. doi: 10.1038/emboj.2012.300
Lam, C. K., Zhao, W., Liu, G. S., Cai, W. F., Gardner, G., Adly, G., et al. (2015). HAX-1 regulates cyclophilin-D levels and mitochondria permeability transition pore in the heart. Proc. Natl. Acad. Sci. U.S.A. 112, E6466–E6475. doi: 10.1073/pnas.1508760112
Landfield, P. W., and Pitler, T. A. (1984). Prolonged Ca2+-dependent afterhyperpolarizations in hippocampal neurons of aged rats. Science 226, 1089–1092. doi: 10.1126/science.6494926
Lanz, T. A., Carter, D. B., and Merchant, K. M. (2003). Dendritic spine loss in the hippocampus of young PDAPP and Tg2576 mice and its prevention by the ApoE2 genotype. Neurobiol. Dis. 13, 246–253. doi: 10.1016/S0969-9961(03)00079-2
Leuner, K., Hauptmann, S., Abdel-Kader, R., Scherping, I., Keil, U., Strosznajder, J. B., Eckert, A., and Muller, W. E. (2007). Mitochondrial dysfunction: the first domino in brain aging and Alzheimer’s disease? Antioxid. Redox Signal. 9, 1659–1675. doi: 10.1089/ars.2007.1763
Li, Z., Okamoto, K., Hayashi, Y., and Sheng, M. (2004). The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 119, 873–887. doi: 10.1016/j.cell.2004.11.003
Liu, Y., and Zhu, X. (2017). Endoplasmic reticulum-mitochondria tethering in neurodegenerative diseases. Transl. Neurodegener. 6:21. doi: 10.1186/s40035-017-0092-6
Lustbader, J. W., Cirilli, M., Lin, C., Xu, H. W., Takuma, K., Wang, N., et al. (2004). ABAD directly links abeta to mitochondrial toxicity in Alzheimer’s disease. Science 304, 448–452. doi: 10.1126/science.1091230
Manfredi, G., and Kawamata, H. (2016). Mitochondria and endoplasmic reticulum crosstalk in amyotrophic lateral sclerosis. Neurobiol. Dis. 90, 35–42. doi: 10.1016/j.nbd.2015.08.004
Martin, L. J., Gertz, B., Pan, Y., Price, A. C., Molkentin, J. D., and Chang, Q. (2009). The mitochondrial permeability transition pore in motor neurons: involvement in the pathobiology of ALS mice. Exp. Neurol. 218, 333–346. doi: 10.1016/j.expneurol.2009.02.015
Martinez, G., Duran-Aniotz, C., Cabral-Miranda, F., Vivar, J. P., and Hetz, C. (2017). Endoplasmic reticulum proteostasis impairment in aging. Aging Cell 16, 615–623. doi: 10.1111/acel.12599
Mastroeni, D., Khdour, O. M., Delvaux, E., Nolz, J., Olsen, G., Berchtold, N., et al. (2017). Nuclear but not mitochondrial-encoded oxidative phosphorylation genes are altered in aging, mild cognitive impairment, and Alzheimer’s disease. Alzheimers Dement. 13, 510–519. doi: 10.1016/j.jalz.2016.09.003
Mattson, M. P. (2010). ER calcium and Alzheimer’s disease: in a state of flux. Sci. Signal. 3:pe10. doi: 10.1126/scisignal.3114pe10
Priami, C., De Michele, G., Cotelli, F., Cellerino, A., Giorgio, M., Pelicci, P. G., et al. (2015). Modelling the p53/p66Shc aging pathway in the shortest living vertebrate Nothobranchius furzeri. Aging Dis. 6, 95–108. doi: 10.14336/AD.2014.0228
Prince, M., Wimo, A., Guerchet, M., Ali, G., Wu, Y., and Prina, M. (2015). World Alzheimer Report 2015. The Global Impact of Dementia. an Analysis of Prevalence, Incidence, Cost & Trends. London: Alzheimer’s Disease International (ADI).
Quinlan, C. L., Perevoshchikova, I. V., Hey-Mogensen, M., Orr, A. L., and Brand, M. D. (2013). Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 1, 304–312. doi: 10.1016/j.redox.2013.04.005
Quiroz-Baez, R., Flores-Dominguez, D., and Arias, C. (2013). Synaptic aging is associated with mitochondrial dysfunction, reduced antioxidant contents and increased vulnerability to amyloid-beta toxicity. Curr. Alzheimer Res. 10, 324–331. doi: 10.2174/1567205011310030012
Ran, Q., Liang, H., Ikeno, Y., Qi, W., Prolla, T. A., Roberts, L. J., et al. (2007). Reduction in glutathione peroxidase 4 increases life span through increased sensitivity to apoptosis. J. Gerontol. A Biol. Sci. Med. Sci. 62, 932–942. doi: 10.1093/gerona/62.9.932
Rao, V. K., Carlson, E. A., and Yan, S. S. (2014). Mitochondrial permeability transition pore is a potential drug target for neurodegeneration. Biochim. Biophys. Acta 1842, 1267–1272. doi: 10.1016/j.bbadis.2013.09.003
Rhein, V., Song, X., Wiesner, A., Ittner, L. M., Baysang, G., Meier, F., et al. (2009). Amyloid-beta and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer’s disease mice. Proc. Natl. Acad. Sci. U.S.A. 106, 20057–20062. doi: 10.1073/pnas.0905529106
Rodrigue, K. M., Kennedy, K. M., and Park, D. C. (2009). Beta-amyloid deposition and the aging brain. Neuropsychol. Rev. 19, 436–450. doi: 10.1007/s11065-009-9118-x
Rottenberg, H., and Hoek, J. B. (2017). The path from mitochondrial ROS to aging runs through the mitochondrial permeability transition pore. Aging Cell 16, 943–955. doi: 10.1111/acel.12650
Salvadores, N., Sanhueza, M., Manque, P., and Court, F. A. (2017). Axonal degeneration during aging and its functional role in neurodegenerative disorders. Front. Neurosci. 11:451. doi: 10.3389/fnins.2017.00451
Scheff, S. W., Price, D. A., Schmitt, F. A., and Mufson, E. J. (2006). Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol. Aging 27, 1372–1384. doi: 10.1016/j.neurobiolaging.2005.09.012
Schinder, A. F., Olson, E. C., Spitzer, N. C., and Montal, M. (1996). Mitochondrial dysfunction is a primary event in glutamate neurotoxicity. J. Neurosci. 16, 6125–6133. doi: 10.1523/JNEUROSCI.16-19-06125.1996
Schon, E. A., and Przedborski, S. (2011). Mitochondria: the next (neurode)generation. Neuron 70, 1033–1053. doi: 10.1016/j.neuron.2011.06.003
Sena, L. A., and Chandel, N. S. (2012). Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 48, 158–167. doi: 10.1016/j.molcel.2012.09.025
Sherrington, R., Rogaev, E. I., Liang, Y., Rogaeva, E. A., Levesque, G., Ikeda, M., et al. (1995). Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 375, 754–760. doi: 10.1038/375754a0
Shilling, D., Muller, M., Takano, H., Mak, D. O., Abel, T., Coulter, D. A., et al. (2014). Suppression of InsP3 receptor-mediated Ca2+ signaling alleviates mutant presenilin-linked familial Alzheimer’s disease pathogenesis. J. Neurosci. 34, 6910–6923. doi: 10.1523/JNEUROSCI.5441-13.2014
Sies, H., Berndt, C., and Jones, D. P. (2017). Oxidative stress. Annu. Rev. Biochem. 86, 715–748. doi: 10.1146/annurev-biochem-061516-045037
Stauch, K. L., Purnell, P. R., and Fox, H. S. (2014). Aging synaptic mitochondria exhibit dynamic proteomic changes while maintaining bioenergetic function. Aging 6, 320–334. doi: 10.18632/aging.100657
Stoica, R., De Vos, K. J., Paillusson, S., Mueller, S., Sancho, R. M., Lau, K. F., et al. (2014). ER-mitochondria associations are regulated by the VAPB-PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat. Commun. 5:3996. doi: 10.1038/ncomms4996
Stutzmann, G. E., Smith, I., Caccamo, A., Oddo, S., Parker, I., and Laferla, F. (2007). Enhanced ryanodine-mediated calcium release in mutant PS1-expressing Alzheimer’s mouse models. Ann. N. Y. Acad. Sci. 1097, 265–277. doi: 10.1196/annals.1379.025
Tambini, M. D., Pera, M., Kanter, E., Yang, H., Guardia-Laguarta, C., Holtzman, D., et al. (2016). ApoE4 upregulates the activity of mitochondria-associated ER membranes. EMBO Rep. 17, 27–36. doi: 10.15252/embr.201540614
Thomas, R. R., Keeney, P. M., and Bennett, J. P. (2012). Impaired complex-I mitochondrial biogenesis in Parkinson disease frontal cortex. J. Parkinsons Dis. 2, 67–76.
Van Remmen, H., Ikeno, Y., Hamilton, M., Pahlavani, M., Wolf, N., Thorpe, S. R., et al. (2003). Life-long reduction in MnSOD activity results in increased DNA damage and higher incidence of cancer but does not accelerate aging. Physiol. Genomics 16, 29–37. doi: 10.1152/physiolgenomics.00122.2003
Verstreken, P., Ly, C. V., Venken, K. J., Koh, T. W., Zhou, Y., and Bellen, H. J. (2005). Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron 47, 365–378. doi: 10.1016/j.neuron.2005.06.018
Wan, Q. F., Nixon, E., and Heidelberger, R. (2012). Regulation of presynaptic calcium in a mammalian synaptic terminal. J. Neurophysiol. 108, 3059–3067. doi: 10.1152/jn.00213.2012
Wang, X., Wang, W., Li, L., Perry, G., Lee, H. G., and Zhu, X. (2014). Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta 1842, 1240–1247. doi: 10.1016/j.bbadis.2013.10.015
Westermann, B. (2012). Bioenergetic role of mitochondrial fusion and fission. Biochim. Biophys. Acta 1817, 1833–1838. doi: 10.1016/j.bbabio.2012.02.033
Wilcox, J. T., Lai, J. K., Semple, E., Brisson, B. A., Gartley, C., Armstrong, J. N., et al. (2011). Synaptically-competent neurons derived from canine embryonic stem cells by lineage selection with EGF and noggin. PLoS One 6:e19768. doi: 10.1371/journal.pone.0019768
Wilson, C., Munoz-Palma, E., and Gonzalez-Billault, C. (2017). From birth to death: a role for reactive oxygen species in neuronal development. Semin. Cell Dev. Biol. 80, 43–49. doi: 10.1016/j.semcdb.2017.09.012
Winklhofer, K. F., and Haass, C. (2010). Mitochondrial dysfunction in Parkinson’s disease. Biochim. Biophys. Acta 1802, 29–44. doi: 10.1016/j.bbadis.2009.08.013
Yu, Q., Du, F., Douglas, J. T., Yu, H., Yan, S. S., and Yan, S. F. (2017). Mitochondrial dysfunction triggers synaptic deficits via activation of p38 map kinase signaling in differentiated Alzheimer’s disease trans-mitochondrial cybrid cells. J. Alzheimers Dis. 59, 223–239. doi: 10.3233/JAD-170283
Zampese, E., Fasolato, C., Pozzan, T., and Pizzo, P. (2011). Presenilin-2 modulation of ER-mitochondria interactions: FAD mutations, mechanisms and pathological consequences. Commun. Integr. Biol. 4, 357–360. doi: 10.4161/cib.4.3.15160
Keywords: mitochondria, MAMS, calcium, neurodegeneration, ROS, MPTP, aging, endoplasmic reticulum
Citation: Müller M, Ahumada-Castro U, Sanhueza M, Gonzalez-Billault C, Court FA and Cárdenas C (2018) Mitochondria and Calcium Regulation as Basis of Neurodegeneration Associated With Aging. Front. Neurosci. 12:470. doi: 10.3389/fnins.2018.00470
Received: 26 March 2018; Accepted: 20 June 2018;
Published: 13 July 2018.
Edited by:
Victor Tapias, Cornell University, United StatesReviewed by:
Rebecca Jane Rylett, University of Western Ontario, CanadaMartin Lothar Duennwald, University of Western Ontario, Canada
Jeremy Michael Van Raamsdonk, Van Andel Institute, United States
Copyright © 2018 Müller, Ahumada-Castro, Sanhueza, Gonzalez-Billault, Court and Cárdenas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: César Cárdenas, amNlc2FyQHUudWNoaWxlLmNs