 Qing Zhu1†
Qing Zhu1† Han Huang
Han Huang Zhimin Liao
Zhimin Liao- 1Department of Anesthesiology, Key Laboratory of Birth Defects and Related Diseases of Women and Children, West China Second University Hospital, Sichuan University, Chengdu, China
- 2Department of Medical Genetics/Prenatal Diagnostic Center Nursing and Key Laboratory of Birth Defects and Related Diseases of Women and Children, West China Second University Hospital of Sichuan University, Chengdu, China
Sepsis is a leading cause of death resulting from an uncontrolled inflammatory response to an infectious agent. Multiple organ injuries, including brain injuries, are common in sepsis. The underlying mechanism of sepsis-associated encephalopathy (SAE), which is associated with neuroinflammation, is not yet fully understood. Recent studies suggest that the release of interleukin-1β (IL-1β) following activation of microglial cells plays a crucial role in the development of long-lasting neuroinflammation after the initial sepsis episode. This review provides a comprehensive analysis of the recent literature on the molecular signaling pathways involved in microglial cell activation and interleukin-1β release. It also explores the physiological and pathophysiological role of IL-1β in cognitive function, with a particular focus on its contribution to long-lasting neuroinflammation after sepsis. The findings from this review may assist healthcare providers in developing novel interventions against SAE.
1 Introduction
Sepsis is one of the leading causes of death worldwide and is characterized by multiple organ injuries resulting from dysregulated host response to infections (Liu et al., 2014; Singer et al., 2016). In addition to its high mortality rate, sepsis can also cause to long-lasting, even permanent, organ damages in septic survivors. It has been estimated that up to 40% of sepsis patients experience neurological and/or cognitive complications even after being discharged (Rudd et al., 2020; Fleischmann-Struzek et al., 2021; Sonneville et al., 2023). Some researchers prefer these prolonged and disabling forms of cognitive impairment in sepsis survivors as sepsis-associated encephalopathy (SAE) (Catarina et al., 2021). Although the exact underlying mechanism is not yet fully understood, it is widely accepted that neuroinflammation plays a significant role (Annane and Sharshar, 2015; Rengel et al., 2019; Pan et al., 2022; Sekino et al., 2022). During the initial systemic inflammation of sepsis, a large amount of cytokines are released, which could enter the central nervous system (CNS) via circumventricular organ and choroid plexus devoid of a blood–brain barrier (Shimada and Hasegawa-Ishii, 2017). Additional, systemic inflammation can also disrupt the blood–brain barriers (Gao and Hernandes, 2021; Sekino et al., 2022; Zou et al., 2022). Once inside the CNS, these cytokines activate microglia, the innate immune cells in the brain, to produce more pro-inflammatory cytokines such as IL-1β, tumor necrosis factor-α (TNF-α), and interferon-γ (INF-γ) (Hoogland et al., 2015; Zrzavy et al., 2019; Hu et al., 2023), ultimately resulting in long-lasting neuroinflammation that leads to neuron loss and cognitive impairment (Sekino et al., 2022; Hu et al., 2023). Among these pro-inflammatory cytokines, IL-1β is particularly important within the CNS after sepsis (Wang et al., 2012; Pozzi et al., 2018). Previous studies have shown that IL-1β, but not TNF-α or IL-6, is the exclusive cytokine within the CNS that has been verified to remain elevated for up to 70 days in rats survived neonatal sepsis but subsequently developed cognitive impairment (Lan et al., 2015; Liao et al., 2023). Furthermore, neurodevelopment of neonatal septic rats could be improved by directly antagonizing IL-1β (Zhang et al., 2022). This review summarizes recent advances in microglia activation-induced excessive IL-1β release and the molecular signaling pathways involved in IL-1β-induced cognitive dysfunction among sepsis survivors.
2 Activation of microglia during sepsis
As the primary immune cell within the CNS, microglia are activated by the recognition of danger signals such as invading pathogens and cytokines during systemic inflammation. For instance, inflammatory cytokines like TNF-α, IL-1β and IL-6 activate microglia through their specific receptors (van Gool et al., 2010).
The nuclear factor kappa B (NF-κB) signaling pathway is a key signal pathway that modulates inflammatory responses. Studies have shown that the NF-κB signaling pathway is involved in the activation of microglia during sepsis (Mulero et al., 2019; Zhao et al., 2019; Nguyen et al., 2021; Wang H. et al., 2022). The NF-κB family consists of subunits p105/p50, p100/p52, p65/RelA, RelB, and c-Rel. These subunits reside in cytoplasm and bind with inhibitory IκB proteins to form an inactive complex (Mitchell et al., 2016; Mulero et al., 2019). The NF-κB signal can be activated through the canonical pathway or the noncanonical pathway. In the canonical pathway, inflammatory stimulation activates a complex containing NF-κB essential modulator (NEMO) and IκB kinases (IKK1/2). This complex phosphorylates IκB proteins that are bound to NF-κB in the cytoplasm, leading to the proteasomal degradation of IκBs. With the rapid degradation of IκBs, the p65:p50 heterodimer is released and translocates into nucleus, where it binds to κB site-containing DNA to enhance the transcription of target genes (Mitchell et al., 2016; Mulero et al., 2019). In the noncanonical pathway, signals such as the activation of TNF receptors by TNF-α or CD40 ligand, activate the NF-κB-inducing kinase (NIK) complex, which is identified as a mitogen-activated protein kinase (MAPK) kinase (Coope et al., 2002; Razani et al., 2011). NIK activates IKK1 through phosphorylation at Ser and Thr residues (Ling et al., 1998; Razani et al., 2011). The activated NIK/IKK1 complex phosphorylates inhibitor IκB and leads to the processing of precursor forms of NF-κB family members, particularly p100. p100 triggers IκB degradation and the release of the active p52 subunit, which forms active RelB:p52 heterodimers. These heterodimers then translocate into the nucleus and regulate specific gene transcription for immune responses (Ling et al., 1998; Coope et al., 2002; Mulero et al., 2019) (Figure 1).
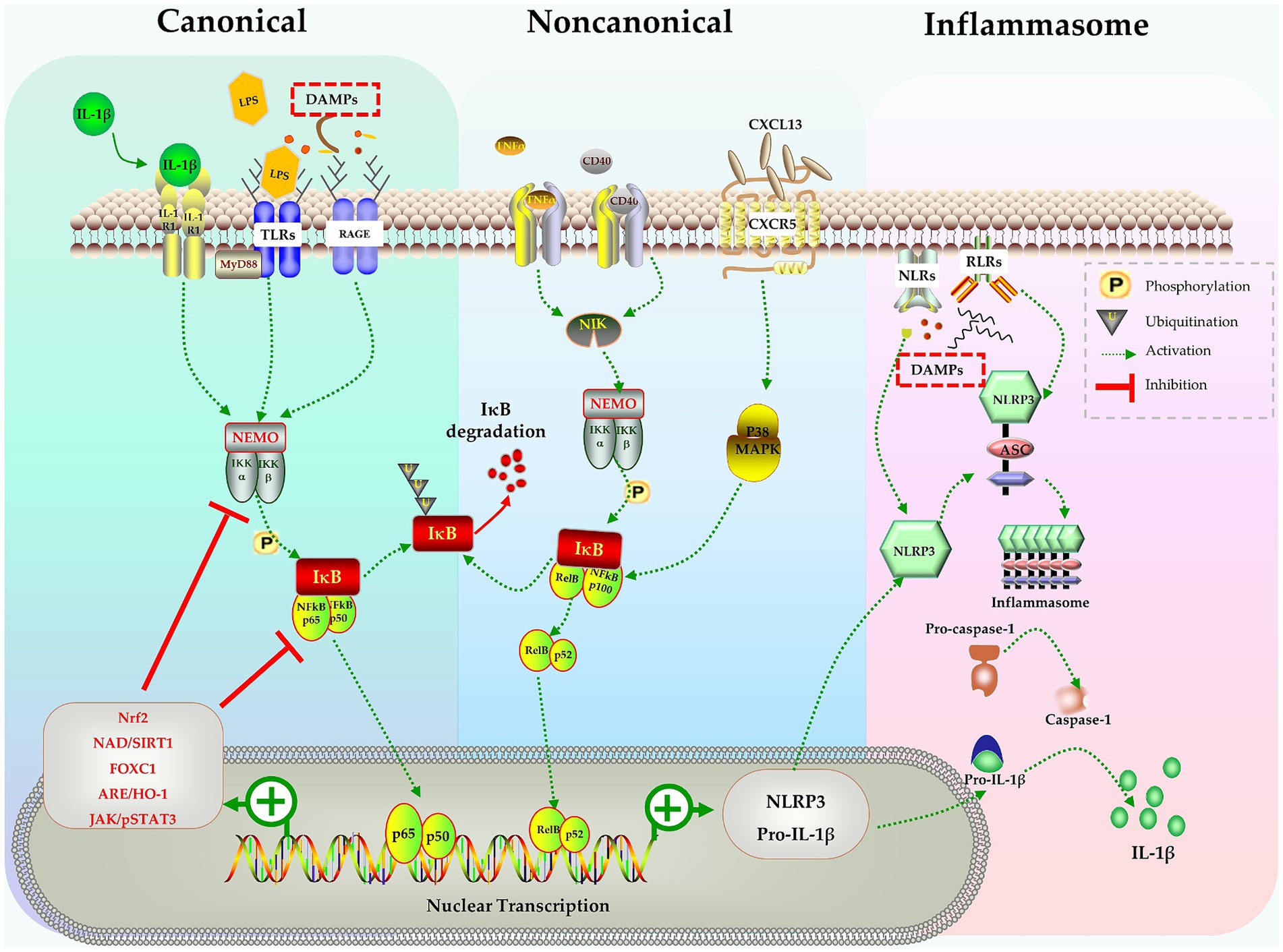
Figure 1. The NF-κB pathway in activation of microglia, inflammasomes formation and production of IL-1β. Stimulates like IL-1β, LPS, DAMPs trigger the canonical pathway, TNF-α and CD40 activate the noncanonical pathway, both pathways activate the NF-κB complex and causes nuclear transcription of NLRP3 and pro-IL-1β. NLRP3 assembles into inflammasome and activates caspase-1, finally leads to release of mature IL-1β. Many molecules in various signaling pathways can negatively regulate the NF-B pathway and inhibit the activation of microglia.
Considering the crucial role of the NF-κB signaling pathway in microglia activation, numerous studies have indeed explored the potential of attenuating microglia activation by targeting the NF-κB signaling pathway. Zhao et al. demonstrated in mice that lipopolysaccharide (LPS) induced cognitive impairment and neuroinflammation occurred due to microglia activation via activation of the NF-kB signaling pathway. By pharmacological suppressing TLR-4/MyD88, they were able to abolish LPS-induced NF-κB activation and the release of pro-inflammatory cytokines, which in turn mitigated cognitive impairment (Zhao et al., 2019).
The nuclear erythroid 2-related factor 2 (Nrf2) is a transcription factor, which, under normal homeostatic conditions, is expressed with Kelch-like ECH-associated protein 1 (Keap1) in the cytoplasm. When stimulated by hazardous signals like oxidative or inflammatory stress, Nrf2 is released from Keap1 and translocates into the nucleus, where it binds to antioxidant response elements (AREs). This leads to the transcription of ARE genes involved in cellular defense and antioxidant responses, ultimately promoting cellular adaptation and survival under stress conditions (Saha et al., 2020). NF-κB is one of the downstream targets of Nrf2 that has been extensively studied. Nrf2 restricts the degradation of IκBα, thereby indirectly inhibiting the nuclear translocation of NF-κB and blocking the activation of the NF-κB signaling pathway (Yerra et al., 2013; Chen et al., 2018; Saha et al., 2020; Yu et al., 2020). Previous studies have highlighted the neuroprotective role of Nrf2 in various conditions, such as traumatic and ischemic brain injury (Dong A. et al., 2018; Dong W. et al., 2018; Yang et al., 2018). Chen et al. further demonstrated thathydrogen gas improved neuronal injury or cognitive dysfunction in mice with SAE through the activation of the Nrf2 pathway (Chen et al., 2021).
Forkhead box C1 (Foxc1) is another transcription factor implicated in the defense processes against oxidative damage, inflammation, and apoptosis (Xia et al., 2019; Zhao et al., 2020). Wang et al. showed with a cecal ligation perforation (CLP) sepsis model that overexpression of Foxc1 suppressed microglia activation related to the NF-κB pathway by enhancing IκBα expression, stabilizing the IκBα/p65 complex and reducing transcription driven by p65. This ultimately resulted in improved cognitive function in mice with SAE (Wang H. et al., 2022).
The Janus kinase/signal transducer and activator of transcription (JAK/STAT) signaling system is an essential cascade involved in cell proliferation, differentiation, and immune modulation (Xin et al., 2020). Therefore, it is not surprising that JAK/STAT signaling pathway interacts with the NF-κB pathway, and these interactions regulate immune responses during sepsis (Cai et al., 2015). Persistently activated JAK/STAT signaling is associated with prolonged inflammation and organ damage observed in sepsis (Cai et al., 2015; Wu et al., 2020; Xin et al., 2020). The complex interactions with other signaling pathways make the biological effects of JAK/STAT signaling more intricate. Septic cytokine-mediated activation of JAK/STAT3 signaling enhances STAT3 binding to the telomerase reverse transcriptase (TERT) promoter and leads to increased TERT protein expression (Cai et al., 2015; Wu et al., 2020). TERT, the telomerase protein, provides neuroprotection against LPS-induced brain injury by converting microglial polarization from M1 to M2 phenotype (Xin et al., 2020; Zhou et al., 2021). Furthermore, JAK2/STAT3 also modulates inflammatory responses through the α7 nicotinic receptor (α7nAChR), suppressing NF-κB and cytokine production and improving survival in sepsis (Peña et al., 2010; Zhang et al., 2017).
The chemokine C-X-C motif ligand 13 (CXCL13) and its receptor CXCR5 also play important roles in various immune and inflammatory processes (Kazanietz et al., 2019). Activation of the CXCL13/CXCR5 axis in microglia results in p38MAPK/NF-κB signal activation and promotes microglial polarization into the M1 phenotype. Conversely, down-regulation of CXCR5 suppresses neuroinflammation caused by microglial activation and alleviates cognitive dysfunction in a mouse model of sepsis (Li et al., 2017; Shen et al., 2021).
Silent information regulator 1 (SIRT1), a deacetylase-dependent nicotinamide adenine dinucleotide (NAD), is a key regulator in energy metabolism and tissue survival (Houtkooper et al., 2012; Jiao and Gong, 2020). The SIRT1 protein, which mediates oxidative respiration and anti-inflammatory responses, directly inhibits NF-κB signaling and mitigates SAE (Kauppinen et al., 2013). Specifically, SIRT1 deacetylates the RelA/p65 complex at Lys310, inhibits the nuclear translocation of NF-κB, and leads to IκBα-dependent nuclear export of NF-κB (Yeung et al., 2004; Han et al., 2019; Tian et al., 2019; Yang et al., 2020).
These studies have revealed the critical role of the NF-κB signaling pathway in microglial activation, neuroinflammation and cognitive impairment following sepsis. Moreover, they suggest that targeting the NF-κB signaling may be a potential intervention for improving cognitive dysfunction induced by sepsis (Figure 1).
In summary, the activation of NF-κB plays a pivotal role in initiating the transcription of pro-inflammatory cytokines, such as IL-1β. This NF-κB pathway is a component of a larger inflammatory response encompassing the activation of other signaling pathways and the release of inflammatory mediators. It is imperative to comprehend the molecular events that connect NF-κB activation and to the release of IL-1β in order to understand the mechanisms that drive inflammation and to devise therapeutic strategies to manipulating these processes in diverse diseases.
3 M1 microglia as the primary source of brain IL-1β during sepsis
Microglia, the main type of resident macrophages in the CNS, play a crucial role in immunological surveillance. Equipped with an array of pattern recognition receptors (PRRs), they detect changes in cytokine and chemokine levels as well as identify infections and damages (Ransohoff and Perry, 2009; Cherry et al., 2014; Roh and Sohn, 2018; Wesselingh et al., 2019). Therefore, it is not surprising that microglia can be activated by septic cytokine storms reaching the brain, utilizing the aforementioned pathways and receptors (Yan et al., 2022). Activation of microglia has been observed as early as 6 h following sepsis induced by CLP (Ye et al., 2019). Prolonged activation of microglia following sepsis has been associated with SAE. Wang et al. and Lan KM et al. discovered that following exposure to LPS in newborn rats resulted in sustained activation of microglia until postnatal day 71, leading to severe cognitive dysfunction (Wang et al., 2013; Lan et al., 2015). Numerous studies have indicated that inflammation caused by activated microglia is the main contributor to cognitive impairment following sepsis (Goshen et al., 2007; Ye et al., 2019; Zrzavy et al., 2019; Yan et al., 2022; Hu et al., 2023; Mordelt and de Witte, 2023). Neuroprotective benefits and reduced cognitive impairment induced by sepsis or surgery have been observed when microglia activation is prevented using the antibiotic minocycline (Michels et al., 2015; Tan et al., 2023). Unfortunately, minocycline failed to prevent postoperative cognitive dysfunction in a recent clinical trial, suggesting that the mechanisms underlying neuroinflammation-related brain dysfunction extend well beyond microglial activation (Takazawa et al., 2023).
Even in the context of microglial activation, there are two distinct phenotypes that can occur. Microglia can be activated and polarized into the M1 phenotype through the pathogen associated molecular patterns (PAMPs) and damage associated molecular patterns (DAMPs), which occurs via PRRs such as membrane Toll-like receptors (TLRs), Scavenger receptors (RAGE) and cytoplasmic Nod-like receptors (NLRs) and RIG-like receptors (RLRs) (Boche et al., 2013; Roh and Sohn, 2018; Hu et al., 2023). The M1 phenotype of microglia releases large amount of pro-inflammatory cytokines, including IL-1β, which induces inflammation to combat invading pathogens (Cherry et al., 2014; Wang et al., 2023). Morphologically, M1 phenotype microglia is characterized by larger amoeboid-like somata, with expression of Iba-1, CD68 and CD11b as the surface markers (Hoogland et al., 2015; Orihuela et al., 2016). Another activated phenotype of microglia is the M2 phenotype, which releases anti-inflammatory mediators and contributes to the resolution of inflammatory responses and tissue repair. The M2 phenotype microglia have a rod-like shape and express CD206 as a surface marker (Orihuela et al., 2016; Zhuang et al., 2020). Microglia play a crucial role in maintaining brain homeostasis through the balanced regulation of M1 and M2 phenotypes, as well as regulating immune response within the CNS. An imbalance in the M1/M2 ratio can have significant implications for neuroinflammation following sepsis (Orihuela et al., 2016; Hu et al., 2023; Wang et al., 2023). As demonstrated in previous studies, excessive production of IL-1β following M1 activation may lead to sustained neuroinflammation, contributing to the progression of cognitive dysfunction in neonatal sepsis. Conversely, an overactive M2 response may suppress necessary immune responses, impairing the ability to eliminate pathogens or clear cellular debris.
4 Activation of inflammasome and release of IL-1β
Inflammasomes are multiprotein complex modulating the inflammatory response and host defense against pathogens (Broz and Dixit, 2016). The inflammasome comprises several subunits, including nucleotide-binding oligomerization domain leucine-rich-repeat-containing pyrin 3 (NLRP3), the adaptor apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC), and the effector molecule pro-caspase-1 (Paerewijck and Lamkanfi, 2022). Inflammasomes are assembled when microglia are stimulated by a variety of stimuli, such as PAMPs (viral, bacterial, or fungal pathogens) and DAMPs (extracellular ATP, mtDNA, ROS, etc.) (Broz and Dixit, 2016; Paerewijck and Lamkanfi, 2022). Upon autoproteolytic activation inside the complex, pro-caspase-1 is hydrolyzed into active caspase-1, which then processes pro-IL-1β and pro-IL-18 into their mature, active forms (Broz and Dixit, 2016; Paerewijck and Lamkanfi, 2022) (Figure 1). Active caspase-1 also cleaves gasdermin D (GSDMD) and triggers pyroptosis, resulting in release of mature IL-1β (Shi et al., 2017). It has been found that activated microglia, not astrocytes, are the sole source of NLRP3 inflammasome in CNS of rodents (Gustin et al., 2015; Moraes et al., 2023). Numerous studies have demonstrated that NLRP3-induced IL-1β release is the main cause of the brain dysfunction following sepsis (Sui et al., 2016; Xie et al., 2020; Yan et al., 2022). Pharmacological inhibition of NLRP3 not only reduces the expression of IL-1β, but also alleviates cognition impairment after sepsis (Sui et al., 2016; Xie et al., 2020; Li et al., 2022; Sekino et al., 2022; Moraes et al., 2023).
5 Effect of IL-1β on learning and memory
As a pro-inflammatory cytokine, IL-1β is involved in inflammation and the host’s defensive response. Once it binds with its receptor (IL-1R), IL-1β can increase the expression of adhesion factors and facilitate the chemotaxis of leukocytes to infection sites, intensifying the cascade of immune reaction (Weber et al., 2010). Aside from regulating immune responses, IL-1β in the CNS is also associated with learning and memory. The hippocampus is a critical structure located in the medial temporal lobe of the brain, playing a central role in learning and memory formation. It is particularly involved in the consolidation of short-term memory into long-term memory, as well as spatial memory and navigation. Both IL-1β and IL-1R are expressed in the hippocampus under normal physiological conditions (Huang and Sheng, 2010). Previous studies have demonstrated that a physiological level of IL-1β is necessary for memory formation, especially for hippocampus-dependent memory (Goshen et al., 2007; Yirmiya and Goshen, 2010; Wang et al., 2012). Disruption of IL-1 signaling through either the use of IL-1R antagonist (IL-1ra) or genetic deletion of IL-1R (IL-1rKO) impairs hippocampus-dependent memory (Avital et al., 2003; Goshen et al., 2007). Additionally, a physiological level of IL-1β is also essential for high-frequency stimulation-induced long-term potentiation (LTP) and normal hippocampal development (Goshen et al., 2007). Treatment with IL-1rKO or IL-1ra can impair memory formation and the maintenance of LTP (Schneider et al., 1998; Weber et al., 2010). On the contrary, excessive IL-1β produced detrimental effect on hippocampus-dependent memory formation and LTP maintenance. However, this effect can be reversed by blocking IL-1β signaling with IL-1ra, which improves cognitive function (Goshen et al., 2007; Weber et al., 2010). In summary, current data suggests that the effect of IL-1β on memory follows a U-shaped pattern: physiological level of IL-1β is necessary for memory formation and a slight increase in IL-1β can improve memory function. However, deviations from physiological range of IL-1β levels, whether through inhibition of IL-1β signaling or excessive IL-1β expression, lead to impaired memory formation and consolidation (Goshen et al., 2007; Weber et al., 2010).
6 IL-1β impairs cognitive function after sepsis
In addition to serving as pivotal pro-inflammatory cytokine in the innate immune response, IL-1β also plays a critical role in the pathogenesis of a variety of CNS diseases associated with cognitive impairment, such as epilepsy, stroke, schizophrenia, and autism (Iori et al., 2016; Krakowiak et al., 2017; Tsai, 2017). Research has provided evidence demonstrating that elevated expression of IL-1β can impair synaptic formation and function, thereby interfering normal neuronal communication and disrupting cognitive function.
6.1 Synapse formation
Synapse formation and maintenance encompass a series of sequential events that commence with the differentiation of neural precursor cells, followed by axonal migration and guidance, axonal and dendritic branch formation, and the maturation of synaptic circuits (Taverna et al., 2014; Harris and Littleton, 2015; Kay, 2016; Ulloa et al., 2018). At each stage, an elevated level of IL-1β has the potential to disrupt the establishment of functional synapses.
6.1.1 Differentiation of neural precursor cell
Neurogenesis plays a crucial role in memory function. Neural precursor cells (NPCs) can differentiate into neurons, astrocytes, and oligodendrocytes (Lazutkin et al., 2019). Sustained expression of IL-1β inhibits neurogenesis and triggers apoptosis in NPCs (Wang et al., 2007; Lin Q. et al., 2019). IL-1β activates stress-activated protein kinase/c-Jun N-terminal kinase (SAPK/JNK), which phosphorylates and cleaves the Notch receptor. This results in the release and translocation of the Notch intracellular domain (NICD) into nucleus (Lin Q. et al., 2019). Once NICD binds to DNA, it promotes the expression of Hes1 and Hes5 genes, which induce NPCs to differentiate into glial cells. At the same time, IL-1β suppresses the expression of Mash1 and Ngn1 genes, thereby inhibiting NPCs from differentiating into neurons (Kageyama et al., 2008; Teng et al., 2009; Lin Q. et al., 2019). By inhibiting the IL-1β or SAPK/JNK signaling pathway, neurogenesis in NPCs can be restored and excessive gliogenesis can be prevented (Wang et al., 2007; Lin Q. et al., 2019).
6.1.2 Synapse structure
To establish a sophisticated network, neurons extend their axonal and dendritic processes to connect with other neurons. Once inter-neuron contact is established, neurons undergo structural change to form new synapse. Synaptophysin (SYN) and post-synaptic density-95 (PSD-95), located in the pre-and post-synaptic membranes respectively, are commonly used molecular markers for evaluating synapse morphology (Dore and Malinow, 2021; Zhang and Augustine, 2021). Studies have demonstrated that microglia regulated formation and development of synapses. Stimulation with LPS increases the release of IL-1β and reduces production of IL-10 and TGF-α in activated microglia, leading to a decrease in the number of synapses between neurons as measured by SYN/PSD-95 colocalized puncta (Mishra et al., 2012; Lim et al., 2013; Hu et al., 2023). Additionally, excessive IL-1β suppresses the function of interleukin-1 receptor accessory protein like 1 (IL1RAPL1), which normally activates JNK and phosphorylates PSD-95. Suppressed IL1RAPL1 results in mis-location of PSD-95 in post-synaptic membranes and impair synaptic function (Pavlowsky et al., 2010; Pozzi et al., 2018).
Oligodendrocytes wrap around the axon to form myelinated axons and Ranvier nodes, increasing axonal conduction velocity. The differentiation and maturation of oligodendrocyte progenitor cells (OPCs) are essential for axonal remyelination. IL-1β also inhibits the normal differentiation of OPCs in postnatal sepsis rats (Ohtomo et al., 2018). Myelin basic protein (MBP), 2′, 3′-cyclic-nucleotide 3-phosphodiesterase (CNPase), and proteolipid (PLP) are well-recognized protein markers for mature myelination, and their expression can be significantly suppressed by IL-1β, resulting in axonal hypomyelination (Xie et al., 2016; Zhou et al., 2021). Furthermore, neurofilament proteins neurofilament-68(NFL), neurofilament-160(NFM), and neurofilament-200(NFH) play a vital role in providing structural support to neurons, as well as determining axonal diameter and conduction velocity. IL-1β can inhibit these neurofilament proteins by activating the p38MAPK pathway and suppressing FYN/MEK/ERK phosphorylation (Xie et al., 2016; Zhou et al., 2021). This leads to hypomyelination, destruction of myelin sheaths, and damage to Ranvier nodes, resulting in small axon diameter and abnormal axon morphology (Wang et al., 2013; Xie et al., 2016; Han et al., 2017; Zhou et al., 2021).
In addition, IL-1β inhibits the formation of dendrites and dendritic spines. Sustained elevations of IL-1β in the hippocampus lead to a decrease in the complexity of dendritic trees, including reduced branch length, dendritic spine density, and dendritic branch density (Liu et al., 2018; Lin L. et al., 2019). Methyl CpG binding protein 2 (MeCP2) is a transcriptional regulator involved in controlling spine morphogenesis and plasticity. IL-1β-induced up-regulation of MeCP2, through the activation of the PI3K/AKT/mTOR signaling pathway, decreases the expression of PSD-95 and spine density (Chao et al., 2007; Cheng and Qiu, 2014; Tomasoni et al., 2017; Pozzi et al., 2018). Brain-derived neurotrophic factor (BDNF), a member of the neurotrophin family, is known to play a vital role in plasticity, neuronal survival, synaptic development, dendritic branching, and long-term potentiation (LTP) (Wang C. S. et al., 2022). When BDNF binds to its high-affinity receptor TrkB, it activates cAMP Response Element-Binding Protein (CREB) via the PI3K/Akt signaling pathway and promotes the transcription of Arc and Homer1. Arc and Homer1 facilitate the organization of filamentous actin (F-actin) and remodeling of spines, which are crucial processes for structural alterations of spines and stabilization of the potentiation effect (Tong et al., 2012; Giacobbo et al., 2019; Eriksen and Bramham, 2022). IL-1β disrupts cytoskeletal alterations and reduces the density of mature spines through the aforementioned p38MAPK signaling pathway (Tong et al., 2012; Herrera et al., 2019) (Figure 2A).
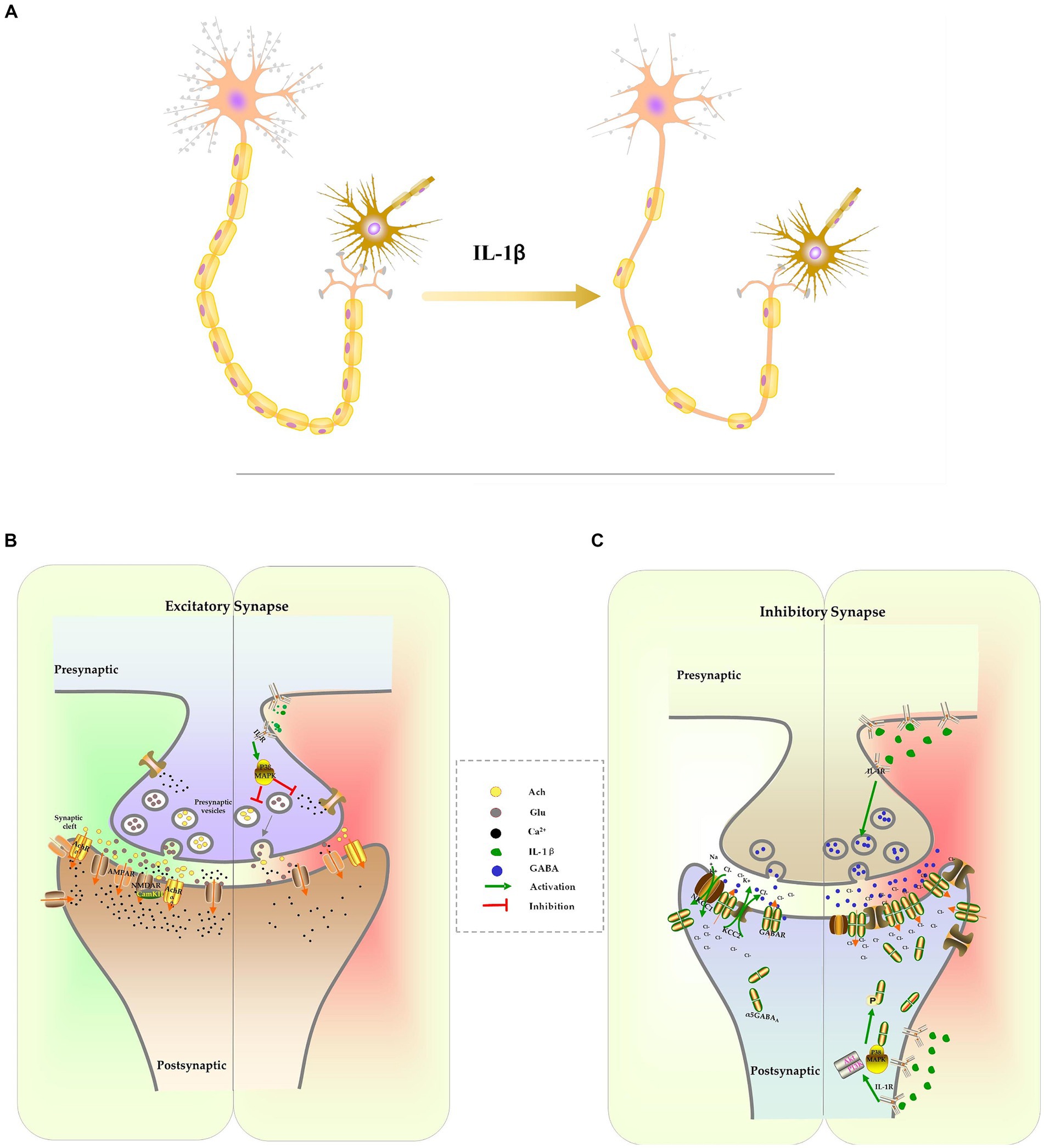
Figure 2. The effect of IL-1β on neuron development and synapse formation. Excessive IL-1β suppresses the formation of dendrites and dendritic spines, which results in a reduction of the complexity of dendrites and the density of spines. It also inhibits the expression of myelination protein, resulting in axonal hypomyelination (A). IL-1β suppresses expression of receptors and release of excitatory neurotransmitters, culminating in inhibition of the glutamatergic system and cholinergic system (B). IL-1β increases the expression of the GABA receptor and the release of GABA. IL-1β interferes with chloride homeostasis by changing NKCC1/KCC2, which delays the E/I switch (C).
6.2 Neurotransmitter and receptor
6.2.1 Glutamatergic system
Glutamate is one of the excitatory neurotransmitters in CNS. It is primarily synthesized in glial cells as glutamine and transported into neurons, where it is converted into glutamate by glutaminase, and stored in secretory vesicles. Following stimulation, the vesicles merge with the cellular membrane, allowing glutamate to be released into the synaptic space (Marmiroli and Cavaletti, 2012). Once it binds to its receptors, primarily the N-methyl-D-aspartate receptor (NMDAR) or the alpha-amino-3-hydroxy-5-methylisoxazole-4-propinonic acid receptor (AMPAR), glutamate induces calcium influx, activates intracellular kinases and phosphatases, and facilitate synaptic transmission. These processes are involved in various neuronal activities, including neuronal maturation, synaptogenesis, learning, and memory (Marmiroli and Cavaletti, 2012). The suppression of the glutamatergic system has been associated with cognitive dysfunction. Pro-inflammatory cytokine IL-1β inhibits glutamate release in presynaptic terminals and impairs memory consolidation by activating p38MAPK, inhibiting ERK phosphorylation and calcium influx (Kelly et al., 2003; Gonzalez et al., 2013; Machado et al., 2015; Chakraborty et al., 2023). It has been observed that excessive IL-1β also suppresses the function and expression of glutamatergic receptors in sepsis models. The reconsolidation of memories following contextual fear memory reactivation was impaired as IL-1β reduces surface expression of AMPA receptor subunit GluA1 and decreased its phosphorylation at Serine 831/845 (Machado et al., 2018). Additionally, IL-1β inhibits the expression of the NR2A and NR2B NMDAR subunits. This effect can be reversed by inhibiting IL-1β signaling with IL-1ra or by activating NMDAR with an agonist (Di Filippo et al., 2013; Taoro-González et al., 2019) (Figure 2B).
Other studies have shown that IL-1β can produce opposite effects on the glutamatergic system in different CNS diseases and chronic pain (Dong et al., 2017; Qiu et al., 2020; Choi et al., 2021). Activated microglia and excessive IL-1β can enhance glutamate release, increase NMDAR expression, and result in significant calcium inflow, leading to excitotoxicity and oxidative stress damage (Mishra et al., 2012; Dong et al., 2017; Qiu et al., 2020). To accurately determine the impact of IL-1β on the glutamatergic system in various neuroinflammatory diseases, more detailed and disease-specific research is required.
6.2.2 Cholinergic system
Another crucial system involved in cognitive functions is the cholinergic signaling system. In neurons, the choline acetyltransferase enzyme converts acetyl-CoA and choline into acetylcholine (ACh). ACh is stored in vesicles and released into the synaptic cleft in response to specific stimuli. ACh activates heterotrimeric G proteins, resulting in the gating of cation channels and the activation of downstream signaling pathway through binding to muscarinic (mAChRs) or nicotinic ACh receptors (nAChRs) (Maurer and Williams, 2017). ACh is degraded by the enzyme acetylcholinesterase (AChE) into choline and acetic acid and then re-uptaken by neurons. Studies have demonstrated that cholinergic transmission through M1-type mAChRs and 7nAChRs activates calcium influx, leading to increased stability of F-actin and dendritic spines, thereby modifying synaptic plasticity and stabilizing LTP (Maurer and Williams, 2017; Solari and Hangya, 2018). Elevated levels of IL-1β in the CNS impair cognition by upregulating AChE expression and activity, as well as suppressing ACh synthesis and release (Main et al., 1993; Li et al., 2000; Zhang et al., 2014). Inhibiting AChE can raise ACh levels in the CNS, thereby improving cognitive function in mice exposed to LPS (Liu et al., 2018). The cholinergic anti-inflammatory pathway, which is activated by increased ACh binding to 7nAChR, activates both STAT3/Jak2 and Nrf2 signaling pathways to prevent NF-κB-induced release of proinflammatory cytokines. This pathway is another mechanism responsible for the observed protective effect of AChE inhibitors on cognitive function (Peña et al., 2010; Zhang et al., 2017; Liu et al., 2018; Benfante et al., 2021) (Figure 2B).
6.2.3 GABAergic system
Gamma-aminobutyric acid (GABA) is the principal inhibitory neurotransmitter in the brain. Glutamate is converted to GABA by the enzyme glutamic acid decarboxylase. GABA is then converted to succinic semialdehyde by GABA-transaminase, which is ultimately converted to succinate by the enzyme succinic semialdehyde dehydrogenase (Sears and Hewett, 2021). GABA is released into the synaptic cleft from presynaptic vesicles and binds to postsynaptic receptors allowing chloride to enter the neuron and causing postsynaptic hyperpolarization. The GABAergic system plays a significant role in regulating synaptic plasticity, learning, and memory (Sakimoto et al., 2021; Sears and Hewett, 2021). Dysfunction of the GABAergic system, particularly with reduced GABA system function, leads to cognitive impairment (Prévot and Sibille, 2021). In the hippocampal CA1 subfield, activation of the alpha5 subtype of GABAA receptors (alpha5GABAA) primarily produces tonic inhibitory conductance (Sakimoto et al., 2021). Increased alpha5GABAA activity impairs memory performance (Martin et al., 2009, 2010).
IL-1β increases GABA levels in the brain by inhibiting reuptake and enhancing GABA release from activated glial cells (Wu et al., 2007; Lee et al., 2010). Additionally, IL-1β upregulates the expression of alpha5GABAA receptors on the surface of proximal dendrites. Activation of IL-1β leads to phosphorylation of the 2/3 subunits of GABAA receptors at Ser-408-409 through the p38MAPK and PI3K-Akt signaling pathways, resulting in the translocation of intracellular receptors to the neuronal membrane, and an increase in tonic inhibitory conductance (Serantes et al., 2006; Luscher et al., 2011; Wang et al., 2012; Prévot and Sibille, 2021).
IL-1β also disrupts the excitatory-to-inhibitory GABA switch during early CNS development in the context of neonatal sepsis. The balance between the chloride importer Na+-K+-2Cl-cotransporter (NKCC1) and the chloride exporter K+-Cl− cotransporter (KCC1) determines the direction of chloride ion flow across the cellular membrane, which in turn determines the depolarization or hyperpolarization of the neuron following GABA stimulation. In the early postnatal stage, NKCC1 is the primary chloride transporter, leading to the accumulation of high chloride concentration inside the neuron and mediating the depolarizing effects of GABA (Peerboom et al., 2023). As the CNS matures, KCC2 becomes the dominant chloride transporter, resulting in a decrease in intracellular chloride ion concentration. This causes chloride influx following GABA activation, which further hyperpolarizes the neuron (Peerboom et al., 2023). The excitatory-to-inhibitory (E/I) switch in GABA function during CNS development is essential for cell proliferation, differentiation, early network wiring, synapse development, and neural plasticity (Deidda et al., 2015a; Virtanen et al., 2021; Peerboom et al., 2023). Our recent research has shown that increased IL-1β in the hippocampus, caused by severe inflammation, upregulates KCC2 expression during early CNS development, accelerating the GABA E/I switch and causing long-lasting cognitive dysfunction. Specific knockdowns of IL-1β or KCC2 expression can reverse this severe inflammation-induced cognitive impairment (Zhang et al., 2022). Interestingly, others have reported the opposite effect of IL-1β on the GABA E/I switch, where overexpression of IL-1β following maternal immune activation (MIA) leads to increased expression of the NKCC1 transporter and delayed GABA E/I switch in offspring. The defected GABA E/I switch has been associated with CNS diseases such as epilepsy (Corradini et al., 2018). Furthermore, IL-1β-mediated E/I switch defects was also involved in other neurodevelopmental disorders, including Down syndrome and autism spectrum disorders (ASD) (Tyzio et al., 2014; Deidda et al., 2015b; Inui et al., 2015). Inhibiting IL-1β signaling can be improve cognitive impairment in certain disorders (Figure 2C).
6.3 LTP
Long-term potentiation (LTP) is a process where synaptic efficacy is enhanced following high-frequency stimulation. It is considered as the cellular foundation for memory and learning (Dringenberg, 2020). Excitatory neurotransmitters activate postsynaptic NMDAR, resulting in the influx of calcium into the postsynaptic compartment. This calcium influx triggers a series of events, including the translocation of actin and its regulators to the dendritic spine. The translocation of actin to the spine is followed by the translocation of AMPA receptors (AMPARs), actin-binding proteins, and kinases, ultimately leading to the expansion of the dendritic spine (Bosch et al., 2014). AMPARs are ionotropic transmembrane receptors for glutamate that facilitate rapid synaptic transmission in the central nervous system (Armstrong et al., 2006). The translocation of these components to the synapse is essential for synaptic plasticity (Hanley, 2018). Furthermore, an increase in postsynaptic Ca2+ concentration promotes Ca2+/calmodulin-dependent protein kinase II (CaMKII) to phosphorylate various substrates, including the transcription factors CREB, GluA, and GluN2A/2B, as well as the synaptic scaffolding protein Homer and PSD-95. These substrates can be phosphorylated to activate multiple downstream signaling pathways and expedite the synaptic consolidation process (Hayashi, 2021).
In the CNS, dendritic spines, which resemble tiny mushrooms-like protrusions on dendrites, generally serve as the sites for the formation of excitatory synapses (Bosch and Hayashi, 2012; Bosch et al., 2014). In summary, the formation and maintenance of LTP, a key mechanism in learning and memory, depend on the normal structure and function of synapses, particularly at the sites of dendritic spines in the CNS (Choi and Kaang, 2023). Therefore, it is unsurprising to find that excessive expression of IL-1β in CNS affects the development of the synapse’s axon, dendrite, and dendritic spine as well as its cholinergic, glutamatergic, and GABAergic neurotransmitter systems, thereby inhibiting long-term potentiation (LTP) and impairing cognition (Bosch et al., 2014; Hayashi, 2021). The specific mechanisms by which IL-1β affects LTP are not fully understood and should be investigated within a disease-specific context.
7 Feed-forward loop in neuroinflammation
The sustained elevation of IL-1β levels in the CNS after a single dose of LPS treatment raises another important and interesting question: what is the underlying mechanism for prolonged microglia activation and IL-1β release after transient systemic inflammation? In the context of neuroinflammation, researchers have identified a feed-forward loop between neurons and microglia (Terrando et al., 2010; Lan et al., 2015). They have discovered that the IL-1 receptor antagonist (IL-1ra) not only decreases IL-1β-induced long-lasting cognitive impairments but also suppresses microglia activation and IL-1β production (Terrando et al., 2010; Wang et al., 2013; Lan et al., 2015). In the absence of pathogen or PAMPs, microglia are activated by DAMPs, which are only produced by dying cells (Lehnardt, 2010; Ransohoff and Brown, 2012). Together, they have concluded that IL-1β-mediated neuroinflammation caused neuronal death, which then released DAMPs to activate microglia and release more IL-1β, creating a feed-forward loop that leads to chronic neuroinflammation and long-term cognitive dysfunction (Terrando et al., 2010; Wang et al., 2013; Lan et al., 2015).
Endogenous DAMPs are molecules that promote inflammation and passively leak out of dying cells when membranes rupture. Numerous cell death processes, including necrosis/necroptosis, pyroptosis, and ferroptosis, are connected to the release of DAMPs (Murao et al., 2021). All these different types of cell death have been reported in the pathological development of sepsis and cognitive dysfunction associated with sepsis (Fu et al., 2019; Zhou et al., 2019; Chu et al., 2022; Liao et al., 2022; Wang J. et al., 2022; Liao et al., 2023). Our recent research showed that necrostatin-1 and xenon were protective against sepsis-induced brain injury by preventing necroptosis. Necroptosis was activated via receptor interacting protein 1/3 (RIP1/3), which contributed to impaired neurodevelopment in neonatal sepsis survivors (Liao et al., 2022, 2023). The NLRP3/caspase-1 pathway-induced pyroptosis has also been linked to cognitive impairment following sepsis. Inhibiting pyroptosis by inhibiting NLRP3 and caspase-1 expression has shown protective effects on cognitive impairment following sepsis in newborn rats (Fu et al., 2019; Zhou et al., 2019). According to Wang (Wang J. et al., 2022) and Chu (Chu et al., 2022), ferroptosis contributed to cognitive dysfunction following SAE via the Nrf2/ferroptosis-related protein (GPX4) signaling pathway. In conclusion, all these types of cell death that release DAMPs may create a feed-forward loop that eventually results in chronic neuroinflammation and cognitive impairment after sepsis. To break the feed-forward cycle, future research should focus on the molecular mechanisms linking cell death and persistent neuroinflammation. It should also investigate possible therapeutic interventions for sepsis-related cognitive impairment (Figure 3).
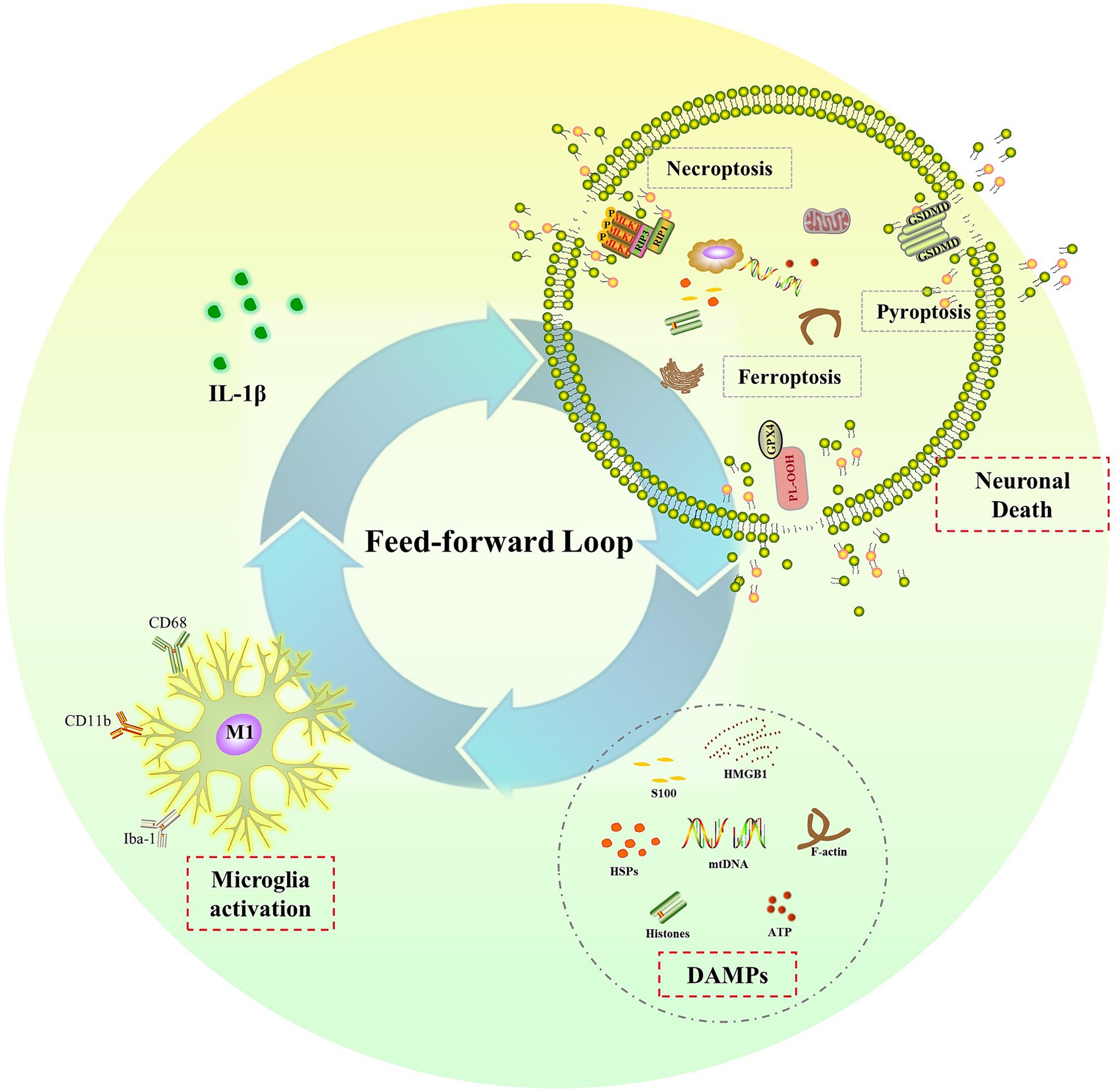
Figure 3. The feed-forward loop in chronic neuroinflammation. Elevated IL-1β induces neuronal death, such as necroptosis, pyroptosis and ferroptosis, leading to the release of DAMPs, which in turn activate microglia and result in more IL-1β release and chronic neuroinflammation.
8 Conclusion
Significant advancements have been made in the past two decades regarding our understanding of the correlation between neuroinflammation and post-septic cognitive impairment. This review encompasses an examination of the diverse effects of IL-1β generated following microglial activation on cognitive impairment, as well as delves into the underlying molecular mechanisms behind microglia activation, the influence of IL-1β on synapse development, and the impact of impairment on synaptic function. Furthermore, we explore the feed-forward loop mechanism, which relies on different types of cell death and releases endogenous DAMPs to activate the innate immune system and create persistent neuroinflammation. Researchers have conducted experimented utilizing several approaches based on these mechanisms, yielding encouraging results. Given the intricate interplay among these processes, a multifaceted approach should be employed in future preclinical and clinical trials to target various pathways in order to prevent or treat cognitive impairment following sepsis.
Author contributions
QZ: Writing – original draft, Writing – review & editing. LW: Writing – original draft, Writing – review & editing. HH: Writing – original draft, Writing – review & editing. ZL: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Annane, D., and Sharshar, T. (2015). Cognitive decline after sepsis. Lancet Respir. Med. 3, 61–69. doi: 10.1016/S2213-2600(14)70246-2
Armstrong, N., Jasti, J., Beich-Frandsen, M., and Gouaux, E. (2006). Measurement of conformational changes accompanying desensitization in an ionotropic glutamate receptor. Cell 127, 85–97. doi: 10.1016/j.cell.2006.08.037
Avital, A., Goshen, I., Kamsler, A., Segal, M., Iverfeldt, K., Richter-Levin, G., et al. (2003). Impaired interleukin-1 signaling is associated with deficits in hippocampal memory processes and neural plasticity. Hippocampus 13, 826–834. doi: 10.1002/hipo.10135
Benfante, R., Di Lascio, S., Cardani, S., and Fornasari, D. (2021). Acetylcholinesterase inhibitors targeting the cholinergic anti-inflammatory pathway: a new therapeutic perspective in aging-related disorders. Aging Clin. Exp. Res. 33, 823–834. doi: 10.1007/s40520-019-01359-4
Boche, D., Perry, V. H., and Nicoll, J. A. (2013). Review: activation patterns of microglia and their identification in the human brain. Neuropathol. Appl. Neurobiol. 39, 3–18. doi: 10.1111/nan.12011
Bosch, M., Castro, J., Saneyoshi, T., Matsuno, H., Sur, M., and Hayashi, Y. (2014). Structural and molecular remodeling of dendritic spine substructures during long-term potentiation. Neuron 82, 444–459. doi: 10.1016/j.neuron.2014.03.021
Bosch, M., and Hayashi, Y. (2012). Structural plasticity of dendritic spines. Curr. Opin. Neurobiol. 22, 383–388. doi: 10.1016/j.conb.2011.09.002
Broz, P., and Dixit, V. M. (2016). Inflammasomes: mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 16, 407–420. doi: 10.1038/nri.2016.58
Cai, B., Cai, J. P., Luo, Y. L., Chen, C., and Zhang, S. (2015). The specific roles of JAK/STAT signaling pathway in Sepsis. Inflammation 38, 1599–1608. doi: 10.1007/s10753-015-0135-z
Catarina, A. V., Branchini, G., Bettoni, L., De Oliveira, J. R., and Nunes, F. B. (2021). Sepsis-associated encephalopathy: from pathophysiology to Progress in experimental studies. Mol. Neurobiol. 58, 2770–2779. doi: 10.1007/s12035-021-02303-2
Chakraborty, P., Dey, A., Gopalakrishnan, A. V., Swati, K., Ojha, S., Prakash, A., et al. (2023). Glutamatergic neurotransmission: a potential pharmacotherapeutic target for the treatment of cognitive disorders. Ageing Res. Rev. 85:101838. doi: 10.1016/j.arr.2022.101838
Chao, H. T., Zoghbi, H. Y., and Rosenmund, C. (2007). MeCP2 controls excitatory synaptic strength by regulating glutamatergic synapse number. Neuron 56, 58–65. doi: 10.1016/j.neuron.2007.08.018
Chen, H., Dong, B., Shi, Y., Yu, Y., and Xie, K. (2021). Hydrogen alleviates neuronal injury and Neuroinflammation induced by microglial activation via the nuclear factor erythroid 2-related factor 2 pathway in Sepsis-associated encephalopathy. Neuroscience 466, 87–100. doi: 10.1016/j.neuroscience.2021.05.003
Chen, L. G., Zhang, Y. Q., Wu, Z. Z., Hsieh, C. W., Chu, C. S., and Wung, B. S. (2018). Peanut arachidin-1 enhances Nrf 2-mediated protective mechanisms against TNF-α-induced ICAM-1 expression and NF-κB activation in endothelial cells. Int. J. Mol. Med. 41, 541–547. doi: 10.3892/ijmm.2017.3238
Cheng, T. L., and Qiu, Z. (2014). MeCP2: multifaceted roles in gene regulation and neural development. Neurosci. Bull. 30, 601–609. doi: 10.1007/s12264-014-1452-6
Cherry, J. D., Olschowka, J. A., and O’Banion, M. K. (2014). Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. J. Neuroinflammation 11:98. doi: 10.1186/1742-2094-11-98
Choi, S. R., Han, H. J., Beitz, A. J., and Lee, J. H. (2021). Intrathecal interleukin-1β decreases sigma-1 receptor expression in spinal astrocytes in a murine model of neuropathic pain. Biomed. Pharmacother. 144:112272. doi: 10.1016/j.biopha.2021.112272
Choi, J. E., and Kaang, B. K. (2023). Plasticity of dendritic spines underlies fear memory. Neuroscientist :10738584231185530. doi: 10.1177/10738584231185530 [E-pub ahead of print].
Chu, J., Jiang, Y., Zhou, W., Zhang, J., Li, H., Yu, Y., et al. (2022). Acetaminophen alleviates ferroptosis in mice with sepsis-associated encephalopathy via the GPX4 pathway. Hum. Exp. Toxicol. 41:9603271221133547. doi: 10.1177/09603271221133547
Coope, H. J., Atkinson, P. G., Huhse, B., Belich, M., Janzen, J., Holman, M. J., et al. (2002). CD40 regulates the processing of NF-κB2 p 100 to p52. EMBO J. 21, 5375–5385. doi: 10.1093/emboj/cdf542
Corradini, I., Focchi, E., Rasile, M., Morini, R., Desiato, G., Tomasoni, R., et al. (2018). Maternal immune activation delays excitatory-to-inhibitory gamma-aminobutyric acid switch in offspring. Biol. Psychiatry 83, 680–691. doi: 10.1016/j.biopsych.2017.09.030
Deidda, G., Allegra, M., Cerri, C., Naskar, S., Bony, G., Zunino, G., et al. (2015a). Early depolarizing GABA controls critical-period plasticity in the rat visual cortex. Nat. Neurosci. 18, 87–96. doi: 10.1038/nn.3890
Deidda, G., Parrini, M., Naskar, S., Bozarth, I. F., Contestabile, A., and Cancedda, L. (2015b). Reversing excitatory GABAAR signaling restores synaptic plasticity and memory in a mouse model of down syndrome. Nat. Med. 21, 318–326. doi: 10.1038/nm.3827
Di Filippo, M., Chiasserini, D., Gardoni, F., Viviani, B., Tozzi, A., Giampà, C., et al. (2013). Effects of central and peripheral inflammation on hippocampal synaptic plasticity. Neurobiol. Dis. 52, 229–236. doi: 10.1016/j.nbd.2012.12.009
Dong, Y., Kalueff, A. V., and Song, C. (2017). N-methyl-d-aspartate receptor-mediated calcium overload and endoplasmic reticulum stress are involved in interleukin-1beta-induced neuronal apoptosis in rat hippocampus. J. Neuroimmunol. 307, 7–13. doi: 10.1016/j.jneuroim.2017.03.005
Dong, W., Yang, B., Wang, L., Li, B., Guo, X., Zhang, M., et al. (2018). Curcumin plays neuroprotective roles against traumatic brain injury partly via Nrf2 signaling. Toxicol. Appl. Pharmacol. 346, 28–36. doi: 10.1016/j.taap.2018.03.020
Dong, A., Yu, Y., Wang, Y., Li, C., Chen, H., Bian, Y., et al. (2018). Protective effects of hydrogen gas against sepsis induced acute lung injury via regulation of mitochondrial function and dynamics. Int. Immunopharmacol. 65, 366–372. doi: 10.1016/j.intimp.2018.10.012
Dore, K., and Malinow, R. (2021). Elevated PSD-95 blocks ion-flux independent LTD: a potential new role for PSD-95 in synaptic plasticity. Neuroscience 456, 43–49. doi: 10.1016/j.neuroscience.2020.02.020
Dringenberg, H. C. (2020). The history of long-term potentiation as a memory mechanism: controversies, confirmation, and some lessons to remember. Hippocampus 30, 987–1012. doi: 10.1002/hipo.23213
Eriksen, M. S., and Bramham, C. R. (2022). Molecular physiology of arc/Arg3.1: the oligomeric state hypothesis of synaptic plasticity. Acta Physiol (Oxf.) 236:e13886. doi: 10.1111/apha.13886
Fleischmann-Struzek, C., Rose, N., Freytag, A., Spoden, M., Prescott, H. C., Schettler, A., et al. (2021). Epidemiology and costs of Postsepsis morbidity, nursing care dependency, and mortality in Germany, 2013 to 2017. JAMA Netw. Open 4:e2134290. doi: 10.1001/jamanetworkopen.2021.34290
Fu, Q., Wu, J., Zhou, X. Y., Ji, M. H., Mao, Q. H., Li, Q., et al. (2019). NLRP3/Caspase-1 pathway-induced Pyroptosis mediated cognitive deficits in a mouse model of Sepsis-associated encephalopathy. Inflammation 42, 306–318. doi: 10.1007/s10753-018-0894-4
Gao, Q., and Hernandes, M. S. (2021). Sepsis-associated encephalopathy and blood-brain barrier dysfunction. Inflammation 44, 2143–2150. doi: 10.1007/s10753-021-01501-3
Giacobbo, B. L., Doorduin, J., Klein, H. C., Dierckx, R. A. J. O., Bromberg, E., and de Vries, E. F. J. (2019). Brain-derived neurotrophic factor in brain disorders: focus on neuroinflammation. Mol. Neurobiol. 56, 3295–3312. doi: 10.1007/s12035-018-1283-6
Gonzalez, P., Machado, I., Vilcaes, A., Caruso, C., Roth, G. A., Schiöth, H., et al. (2013). Molecular mechanisms involved in interleukin 1-beta (IL-1β)-induced memory impairment. Modulation by alpha-melanocyte-stimulating hormone (α-MSH). Brain Behav. Immun. 34, 141–150. doi: 10.1016/j.bbi.2013.08.007
Goshen, I., Kreisel, T., Ounallah-Saad, H., Renbaum, P., Zalzstein, Y., Ben-Hur, T., et al. (2007). A dual role for interleukin-1 in hippocampal-dependent memory processes. Psychoneuroendocrinology 32, 1106–1115. doi: 10.1016/j.psyneuen.2007.09.004
Gustin, A., Kirchmeyer, M., Koncina, E., Felten, P., Losciuto, S., Heurtaux, T., et al. (2015). NLRP3 inflammasome is expressed and functional in mouse brain microglia but not in astrocytes. PLoS One 10:e0130624. doi: 10.1371/journal.pone.0130624
Han, S., Li, Z., Han, F., Jia, Y., Qi, L., Wu, G., et al. (2019). ROR alpha protects against LPS-induced inflammation by down-regulating SIRT1/NF-kappa B pathway. Arch. Biochem. Biophys. 668, 1–8. doi: 10.1016/j.abb.2019.05.003
Han, Q., Lin, Q., Huang, P., Chen, M., Hu, X., Fu, H., et al. (2017). Microglia-derived IL-1β contributes to axon development disorders and synaptic deficit through p38-MAPK signal pathway in septic neonatal rats. J. Neuroinflammation 14:52. doi: 10.1186/s12974-017-0805-x
Hanley, J. G. (2018). The regulation of AMPA receptor endocytosis by dynamic protein-protein interactions. Front. Cell. Neurosci. 12:362. doi: 10.3389/fncel.2018.00362
Harris, K. P., and Littleton, J. T. (2015). Transmission, development, and plasticity of synapses. Genetics 201, 345–375. doi: 10.1534/genetics.115.176529
Hayashi, Y. (2021). Molecular mechanism of hippocampal long-term potentiation - towards multiscale understanding of learning and memory. Neurosci. Res. 175, 3–15. doi: 10.1016/j.neures.2021.08.001
Herrera, G., Calfa, G., Schiöth, H. B., Lasaga, M., and Scimonelli, T. (2019). Memory consolidation impairment induced by interleukin-1β is associated with changes in hippocampal structural plasticity. Behav. Brain Res. 370:111969. doi: 10.1016/j.bbr.2019.111969
Hoogland, I. C., Houbolt, C., van Westerloo, D. J., van Gool, W. A., and van de Beek, D. (2015). Systemic inflammation and microglial activation: systematic review of animal experiments. J. Neuroinflamm. 12:114. doi: 10.1186/s12974-015-0332-6
Houtkooper, R. H., Pirinen, E., and Auwerx, J. (2012). Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell Biol. 13, 225–238. doi: 10.1038/nrm3293
Hu, J., Xie, S., Zhang, H., Wang, X., Meng, B., and Zhang, L. (2023). Microglial activation: key players in Sepsis-associated encephalopathy. Brain Sci. 13:1453. doi: 10.3390/brainsci13101453
Huang, Z. B., and Sheng, G. Q. (2010). Interleukin-1β with learning and memory. Neurosci. Bull. 26, 455–468. doi: 10.1007/s12264-010-6023-5
Inui, T., Kumagaya, S., and Myowa-Yamakoshi, M. (2015). Neurodevelopmental hypothesis about the etiology of autism Spectrum disorders. Front. Hum. Neurosci. 11:354. doi: 10.3389/fnhum.2017.00354
Iori, V., Frigerio, F., and Vezzani, A. (2016). Modulation of neuronal excitability by immune mediators in epilepsy. Curr. Opin. Pharmacol. 26, 118–123. doi: 10.1016/j.coph.2015.11.002
Jiao, F., and Gong, Z. (2020). The beneficial roles of SIRT1 in Neuroinflammation-related diseases. Oxidative Med. Cell. Longev. 2020, 1–19. doi: 10.1155/2020/6782872
Kageyama, R., Ohtsuka, T., and Kobayashi, T. (2008). Roles of Hes genes in neural development. Develop. Growth Differ. 50, S97–S103. doi: 10.1111/j.1440-169X.2008.00993.x
Kauppinen, A., Suuronen, T., Ojala, J., Kaarniranta, K., and Salminen, A. (2013). Antagonistic crosstalk between NF-κB and SIRT1 in the regulation of inflammation and metabolic disorders. Cell. Signal. 25, 1939–1948. doi: 10.1016/j.cellsig.2013.06.007
Kay, J. N. (2016). Radial migration: retinal neurons hold on for the ride. J. Cell Biol. 215, 147–149. doi: 10.1083/jcb.201609135
Kazanietz, M. G., Durando, M., and Cooke, M. (2019). CXCL13 and its receptor CXCR5 in Cancer: inflammation, immune response, and beyond. Front. Endocrinol. 10:471. doi: 10.3389/fendo.2019.00471
Kelly, A., Vereker, E., Nolan, Y., Brady, M., Barry, C., Loscher, C. E., et al. (2003). Activation of p38 plays a pivotal role in the inhibitory effect of lipopolysaccharide and interleukin-1 beta on long term potentiation in rat dentate gyrus. J. Biol. Chem. 278, 19453–19462. doi: 10.1074/jbc.M301938200
Krakowiak, P., Goines, P. E., Tancredi, D. J., Ashwood, P., Hansen, R. L., Hertz-Picciotto, I., et al. (2017). Neonatal cytokine profiles associated with autism spectrum disorder. Biol. Psychiatry 81, 442–451. doi: 10.1016/j.biopsych.2015.08.007
Lan, K. M., Tien, L. T., Pang, Y., Bhatt, A. J., and Fan, L. W. (2015). IL-1 receptor antagonist attenuates neonatal lipopolysaccharide-induced long-lasting learning impairment and hippocampal injury in adult rats. Toxicol. Lett. 234, 30–39. doi: 10.1016/j.toxlet.2015.02.002
Lazutkin, A., Podgorny, O., and Enikolopov, G. (2019). Modes of division and differentiation of neural stem cells. Behav. Brain Res. 374:112118. doi: 10.1016/j.bbr.2019.112118
Lee, S., Yoon, B. E., Berglund, K., Oh, S. J., Park, H., Shin, H. S., et al. (2010). Channel-mediated tonic GABA release from glia. Science 330, 790–796. doi: 10.1126/science.1184334
Lehnardt, S. (2010). Innate immunity and neuroinflammation in the CNS: the role of microglia in toll-like receptor-mediated neuronal injury. Glia 58, 253–263. doi: 10.1002/glia.20928
Li, Y., Ji, M., and Yang, J. (2022). Current understanding of long-term cognitive impairment after Sepsis. Front. Immunol. 13:855006. doi: 10.3389/fimmu.2022.855006
Li, Y., Liu, L., Kang, J., Sheng, J. G., Barger, S. W., Mrak, R. E., et al. (2000). Neuronal-glial interactions mediated by interleukin-1 enhance neuronal acetylcholinesterase activity and mRNA expression. J. Neurosci. 20, 149–155. doi: 10.1523/JNEUROSCI.20-01-00149.2000
Li, R., Ma, L., Huang, H., Ou, S., Yuan, J., Xu, T., et al. (2017). Altered expression of CXCL13 and CXCR5 in intractable temporal lobe epilepsy patients and pilocarpine-induced epileptic rats. Neurochem. Res. 42, 526–540. doi: 10.1007/s11064-016-2102-y
Liao, Z., Ou, X., Zhou, C., Ma, D., Zhao, H., and Huang, H. (2022). Xenon attenuated neonatal lipopolysaccharide exposure induced neuronal necroptosis and subsequently improved cognition in juvenile rats. Front. Pharmacol. 13:1002920. doi: 10.3389/fphar.2022.1002920
Liao, Z., Zhu, Q., and Huang, H. (2023). Involvement of IL-1β-mediated necroptosis in neurodevelopment impairment after neonatal Sepsis in rats. Int. J. Mol. Sci. 24:14693. doi: 10.3390/ijms241914693
Lim, S. H., Park, E., You, B., Jung, Y., Park, A. R., Park, S. G., et al. (2013). Neuronal synapse formation induced by microglia and interleukin 10. PLoS One 8:e81218. doi: 10.1371/journal.pone.0081218
Lin, L., Chen, X., Zhou, Q., Huang, P., Jiang, S., Wang, H., et al. (2019). Synaptic structure and alterations in the hippocampus in neonatal rats exposed to lipopolysaccharide. Neurosci. Lett. 709:134364. doi: 10.1016/j.neulet.2019.134364
Lin, Q., Shen, F., Zhou, Q., Huang, P., Lin, L., Chen, M., et al. (2019). Interleukin-1β disturbs the proliferation and differentiation of neural precursor cells in the hippocampus via activation of notch signaling in postnatal rats exposed to lipopolysaccharide. ACS Chem. Neurosci. 10, 2560–2575. doi: 10.1021/acschemneuro.9b00051
Ling, L., Cao, Z., and Goeddel, D. V. (1998). NF-κB-inducing kinase activates IKK-α by phosphorylation of Ser-176. Proc. Natl. Acad. Sci. USA 95, 3792–3797. doi: 10.1073/pnas.95.7.3792
Liu, V., Escobar, G. J., Greene, J. D., Soule, J., Whippy, A., Angus, D. C., et al. (2014). Hospital deaths in patients with sepsis from 2 independent cohorts. JAMA 312, 90–92. doi: 10.1001/jama.2014.5804
Liu, Y., Zhang, Y., Zheng, X., Fang, T., Yang, X., Luo, X., et al. (2018). Galantamine improves cognition, hippocampal inflammation, and synaptic plasticity impairments induced by lipopolysaccharide in mice. J. Neuroinflammation 15:112. doi: 10.1186/s12974-018-1141-5
Luscher, B., Fuchs, T., and Kilpatrick, C. L. (2011). GABAA receptor trafficking-mediated plasticity of inhibitory synapses. Neuron 70, 385–409. doi: 10.1016/j.neuron.2011.03.024
Machado, I., Gonzalez, P. V., Vilcaes, A., Carniglia, L., Schiöth, H. B., Lasaga, M., et al. (2015). Interleukin-1β-induced memory reconsolidation impairment is mediated by a reduction in glutamate release and zif 268 expression and α-melanocyte-stimulating hormone prevented these effects. Brain Behav. Immun. 46, 137–146. doi: 10.1016/j.bbi.2015.01.012
Machado, I., Schiöth, H. B., Lasaga, M., and Scimonelli, T. (2018). IL-1β reduces GluA1 phosphorylation and its surface expression during memory reconsolidation and α-melanocyte-stimulating hormone can modulate these effects. Neuropharmacology 128, 314–323. doi: 10.1016/j.neuropharm.2017.09.041
Main, C., Blennerhassett, P., and Collins, S. M. (1993). Human recombinant interleukin 1 beta suppresses acetylcholine release from rat myenteric plexus. Gastroenterology 104, 1648–1654. doi: 10.1016/0016-5085(93)90641-o
Marmiroli, P., and Cavaletti, G. (2012). The glutamatergic neurotransmission in the central nervous system. Curr. Med. Chem. 19, 1269–1276. doi: 10.2174/092986712799462711
Martin, L. J., Oh, G. H., and Orser, B. A. (2009). Etomidate targets alpha 5 gamma-aminobutyric acid subtype a receptors to regulate synaptic plasticity and memory blockade. Anesthesiology 111, 1025–1035. doi: 10.1097/ALN.0b013e3181bbc961
Martin, L. J., Zurek, A. A., MacDonald, J. F., Roder, J. C., Jackson, M. F., and Orser, B. A. (2010). Alpha5GABAA receptor activity sets the threshold for long-term potentiation and constrains hippocampus-dependent memory. J. Neurosci. 30, 5269–5282. doi: 10.1523/JNEUROSCI.4209-09.2010
Maurer, S. V., and Williams, C. L. (2017). The cholinergic system modulates memory and hippocampal plasticity via its interactions with non-neuronal cells. Front. Immunol. 8:1489. doi: 10.3389/fimmu.2017.01489
Michels, M., Vieira, A. S., Vuolo, F., Zapelini, H. G., Mendonça, B., Mina, F., et al. (2015). The role of microglia activation in the development of sepsis-induced long-term cognitive impairment. Brain Behav. Immun. 43, 54–59. doi: 10.1016/j.bbi.2014.07.002
Mishra, A., Kim, H. J., Shin, A. H., and Thayer, S. A. (2012). Synapse loss induced by interleukin-1β requires pre-and post-synaptic mechanisms. J. Neuroimmune Pharmacol. 7, 571–578. doi: 10.1007/s11481-012-9342-7
Mitchell, S., Vargas, J., and Hoffmann, A. (2016). Signaling via the NFκB system. Wiley Interdiscip. Rev. Syst. Biol. Med. 8, 227–241. doi: 10.1002/wsbm.1331
Moraes, C. A., Hottz, E. D., Dos Santos Ornellas, D., Adesse, D., de Azevedo, C. T., d’Avila, J. C., et al. (2023). Microglial NLRP3 inflammasome induces excitatory synaptic loss through IL-1β-enriched microvesicle release: implications for sepsis-associated encephalopathy. Mol. Neurobiol. 60, 481–494. doi: 10.1007/s12035-022-03067-z
Mordelt, A., and de Witte, L. D. (2023). Microglia-mediated synaptic pruning as a key deficit in neurodevelopmental disorders: hype or hope? Curr. Opin. Neurobiol. 79:102674. doi: 10.1016/j.conb.2022.102674
Mulero, M. C., Huxford, T., and Ghosh, G. (2019). NF-κB, IκB, and IKK: integral components of immune system signaling. Adv. Exp. Med. Biol. 1172, 207–226. doi: 10.1007/978-981-13-9367-9_10
Murao, A., Aziz, M., Wang, H., Brenner, M., and Wang, P. (2021). Release mechanisms of major DAMPs. Apoptosis 26, 152–162. doi: 10.1007/s10495-021-01663-3
Nguyen, P. L., Bui, B. P., Lee, H., and Cho, J. (2021). A novel 1,8-naphthyridine-2-carboxamide derivative attenuates inflammatory responses and cell migration in LPS-treated BV2 cells via the suppression of ROS generation and TLR4/Myd88/NF-κB signaling pathway. Int. J. Mol. Sci. 22:2527. doi: 10.3390/ijms22052527
Ohtomo, R., Iwata, A., and Arai, K. (2018). Molecular mechanisms of oligodendrocyte regeneration in white matter-related diseases. Int. J. Mol. Sci. 19:1743. doi: 10.3390/ijms19061743
Orihuela, R., McPherson, C. A., and Harry, G. J. (2016). Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 173, 649–665. doi: 10.1111/bph.13139
Paerewijck, O., and Lamkanfi, M. (2022). The human inflammasomes. Mol. Asp. Med. 88:101100. doi: 10.1016/j.mam.2022.101100
Pan, S., Lv, Z., Wang, R., Shu, H., Yuan, S., Yu, Y., et al. (2022). Sepsis-induced brain dysfunction: pathogenesis, diagnosis, and treatment. Oxidative Med. Cell. Longev. 2022, 1328729–1328713. doi: 10.1155/2022/1328729
Pavlowsky, A., Gianfelice, A., Pallotto, M., Zanchi, A., Vara, H., Khelfaoui, M., et al. (2010). A postsynaptic signaling pathway that may account for the cognitive defect due to IL1RAPL1 mutation. Curr. Biol. 20, 103–115. doi: 10.1016/j.cub.2009.12.030
Peerboom, C., de Kater, S., Jonker, N., Rieter, M. P. J. M., Wijne, T., and Wierenga, C. J. (2023). Delaying the GABA shift indirectly affects membrane properties in the developing Hippocampus. J. Neurosci. 43, 5483–5500. doi: 10.1523/JNEUROSCI.0251-23.2023
Peña, G., Cai, B., Liu, J., van der Zanden, E. P., Deitch, E. A., de Jonge, W. J., et al. (2010). Unphosphorylated STAT3 modulates alpha 7 nicotinic receptor signaling and cytokine production in sepsis. Eur. J. Immunol. 40, 2580–2589. doi: 10.1002/eji.201040540
Pozzi, D., Menna, E., Canzi, A., Desiato, G., Mantovani, C., and Matteoli, M. (2018). The communication between the immune and nervous systems: the role of IL-1β in synaptopathies. Front. Mol. Neurosci. 11:111. doi: 10.3389/fnmol.2018.00111
Prévot, T., and Sibille, E. (2021). Altered GABA-mediated information processing and cognitive dysfunctions in depression and other brain disorders. Mol. Psychiatry 26, 151–167. doi: 10.1038/s41380-020-0727-3
Qiu, L. L., Pan, W., Luo, D., Zhang, G. F., Zhou, Z. Q., Sun, X. Y., et al. (2020). Dysregulation of BDNF/TrkB signaling mediated by NMDAR/Ca2+/calpain might contribute to postoperative cognitive dysfunction in aging mice. J. Neuroinflammation 17:23. doi: 10.1186/s12974-019-1695-x
Ransohoff, R. M., and Brown, M. A. (2012). Innate immunity in the central nervous system. J. Clin. Invest. 122, 1164–1171. doi: 10.1172/JCI58644
Ransohoff, R. M., and Perry, V. H. (2009). Microglial physiology: unique stimuli, specialized responses. Annu. Rev. Immunol. 27, 119–145. doi: 10.1146/annurev.immunol.021908.132528
Razani, B., Reichardt, A. D., and Cheng, G. (2011). Non-canonical NF-κB signaling activation and regulation: principles and perspectives. Immunol. Rev. 244, 44–54. doi: 10.1111/j.1600-065X.2011.01059.x
Rengel, K. F., Hayhurst, C. J., Pandharipande, P. P., and Hughes, C. G. (2019). Long-term cognitive and functional impairments after critical illness. Anesth. Analg. 128, 772–780. doi: 10.1213/ANE.0000000000004066
Roh, J. S., and Sohn, D. H. (2018). Damage-associated molecular patterns in inflammatory diseases. Immune Netw. 18:e27. doi: 10.4110/in.2018.18.e27
Rudd, K. E., Johnson, S. C., Agesa, K. M., Shackelford, K. A., Tsoi, D., Kievlan, D. R., et al. (2020). Global, regional, and national sepsis incidence and mortality, 1990-2017: analysis for the global burden of disease study. Lancet 395, 200–211. doi: 10.1016/S0140-6736(19)32989-7
Saha, S., Buttari, B., Panieri, E., Profumo, E., and Saso, L. (2020). An overview of Nrf2 signaling pathway and its role in inflammation. Molecules 25:5474. doi: 10.3390/molecules25225474
Sakimoto, Y., Oo, P. M., Goshima, M., Kanehisa, I., Tsukada, Y., and Mitsushima, D. (2021). Significance of GABAA receptor for cognitive function and hippocampal pathology. Int. J. Mol. Sci. 22:12456. doi: 10.3390/ijms222212456
Schneider, H., Pitossi, F., Balschun, D., Wagner, A., del Rey, A., and Besedovsky, H. O. (1998). A neuromodulatory role of interleukin-1beta in the hippocampus. Proc. Natl. Acad. Sci. USA 95, 7778–7783. doi: 10.1073/pnas.95.13.7778
Sears, S. M., and Hewett, S. J. (2021). Influence of glutamate and GABA transport on brain excitatory/inhibitory balance. Exp. Biol. Med. (Maywood) 246, 1069–1083. doi: 10.1177/1535370221989263
Sekino, N., Selim, M., and Shehadah, A. (2022). Sepsis-associated brain injury: underlying mechanisms and potential therapeutic strategies for acute and long-term cognitive impairments. J. Neuroinflammation 19:101. doi: 10.1186/s12974-022-02464-4
Serantes, R., Arnalich, F., Figueroa, M., Salinas, M., Andrés-Mateos, E., Codoceo, R., et al. (2006). Interleukin-1beta enhances GABAA receptor cell-surface expression by a phosphatidylinositol 3-kinase/Akt pathway: relevance to sepsis-associated encephalopathy. J. Biol. Chem. 281, 14632–14643. doi: 10.1074/jbc.M512489200
Shen, Y., Zhang, Y., Du, J., Jiang, B., Shan, T., Li, H., et al. (2021). CXCR5 down-regulation alleviates cognitive dysfunction in a mouse model of sepsis-associated encephalopathy: potential role of microglial autophagy and the p38MAPK/NF-κB/STAT3 signaling pathway. J. Neuroinflammation 18:246. doi: 10.1186/s12974-021-02300-1
Shi, J., Gao, W., and Shao, F. (2017). Pyroptosis: Gasdermin-mediated programmed necrotic cell death. Trends Biochem. Sci. 42, 245–254. doi: 10.1016/j.tibs.2016.10.004
Shimada, A., and Hasegawa-Ishii, S. (2017). Histological architecture underlying brain-immune cell-cell interactions and the cerebral response to systemic inflammation. Front. Immunol. 8:17. doi: 10.3389/fimmu.2017.00017
Singer, M., Deutschman, C. S., Seymour, C. W., Shankar-Hari, M., Annane, D., Bauer, M., et al. (2016). The third international consensus definitions for Sepsis and septic shock (Sepsis-3). JAMA 315, 801–810. doi: 10.1001/jama.2016.0287
Solari, N., and Hangya, B. (2018). Cholinergic modulation of spatial learning, memory and navigation. Eur. J. Neurosci. 48, 2199–2230. doi: 10.1111/ejn.14089
Sonneville, R., Benghanem, S., Jeantin, L., de Montmollin, E., Doman, M., Gaudemer, A., et al. (2023). The spectrum of sepsis-associated encephalopathy: a clinical perspective. Crit. Care 27:386. doi: 10.1186/s13054-023-04655-8
Sui, D. M., Xie, Q., Yi, W. J., Gupta, S., Yu, X. Y., Li, J. B., et al. (2016). Resveratrol protects against sepsis-associated encephalopathy and inhibits the NLRP3/IL-1β axis in microglia. Mediat. Inflamm. 2016:1045657. doi: 10.1155/2016/1045657
Takazawa, T., Horiuchi, T., Orihara, M., Nagumo, K., Tomioka, A., Ideno, Y., et al. (2023). Prevention of postoperative cognitive dysfunction by minocycline in elderly patients after Total knee arthroplasty: a randomized, double-blind, placebo-controlled clinical trial. Anesthesiology 138, 172–183. doi: 10.1097/ALN.0000000000004439
Tan, X., Wang, J., Yao, J., Yuan, J., Dai, Y., Sun, M., et al. (2023). Microglia participate in postoperative cognitive dysfunction by mediating the loss of inhibitory synapse through the complement pathway. Neurosci. Lett. 796:137049. doi: 10.1016/j.neulet.2023.137049
Taoro-González, L., Cabrera-Pastor, A., Sancho-Alonso, M., Arenas, Y. M., Meseguer-Estornell, F., Balzano, T., et al. (2019). Differential role of interleukin-1β in neuroinflammation-induced impairment of spatial and nonspatial memory in hyperammonemic rats. FASEB J. 33, 9913–9928. doi: 10.1096/fj.201900230RR
Taverna, E., Götz, M., and Huttner, W. B. (2014). The cell biology of neurogenesis: toward an understanding of the development and evolution of the neocortex. Annu. Rev. Cell Dev. Biol. 30, 465–502. doi: 10.1146/annurev-cellbio-101011-155801
Teng, F. Y., Hor, C. H., and Tang, B. L. (2009). Emerging cues mediating astroglia lineage restriction of progenitor cells in the injured/diseased adult CNS. Differentiation 77, 121–127. doi: 10.1016/j.diff.2008.09.013
Terrando, N., Rei Fidalgo, A., Vizcaychipi, M., Cibelli, M., Ma, D., Monaco, C., et al. (2010). The impact of IL-1 modulation on the development of lipopolysaccharide-induced cognitive dysfunction. Crit. Care 14:R88. doi: 10.1186/cc9019
Tian, M., Qingzhen, L., Zhiyang, Y., Chunlong, C., Jiao, D., Zhang, L., et al. (2019). Attractylone attenuates sepsis-associated encephalopathy and cognitive dysfunction by inhibiting microglial activation and neuroinflammation. J. Cell. Biochem. 120, 7101–7108. doi: 10.1002/jcb.27983
Tomasoni, R., Morini, R., Lopez-Atalaya, J. P., Corradini, I., Canzi, A., Rasile, M., et al. (2017). Lack of IL-1R8 in neurons causes hyperactivation of IL-1 receptor pathway and induces MECP2-dependent synaptic defects. eLife 6:e21735. doi: 10.7554/eLife.21735
Tong, L., Prieto, G. A., Kramár, E. A., Smith, E. D., Cribbs, D. H., Lynch, G., et al. (2012). Brain-derived neurotrophic factor-dependent synaptic plasticity is suppressed by interleukin-1β via p38 mitogen-activated protein kinase. J. Neurosci. 32, 17714–17724. doi: 10.1523/JNEUROSCI.1253-12.201
Tsai, S. J. (2017). Effects of interleukin-1beta polymorphisms on brain function and behavior in healthy and psychiatric disease conditions. Cytokine Growth Factor Rev. 37, 89–97. doi: 10.1016/j.cytogfr.2017.06.001
Tyzio, R., Nardou, R., Ferrari, D. C., Tsintsadze, T., Shahrokhi, A., Eftekhari, S., et al. (2014). Oxytocin-mediated GABA inhibition during delivery attenuates autism pathogenesis in rodent offspring. Science 343, 675–679. doi: 10.1126/science.1247190
Ulloa, F., Cotrufo, T., Ricolo, D., Soriano, E., and Araújo, S. J. (2018). SNARE complex in axonal guidance and neuroregeneration. Neural Regen. Res. 13, 386–392. doi: 10.4103/1673-5374.228710
van Gool, W. A., van de Beek, D., and Eikelenboom, P. (2010). Systemic infection and delirium: when cytokines and acetylcholine collide. Lancet 375, 773–775. doi: 10.1016/S0140-6736(09)61158-2
Virtanen, M. A., Uvarov, P., Mavrovic, M., Poncer, J. C., and Kaila, K. (2021). The multifaceted roles of KCC2 in cortical development. Trends Neurosci. 44, 378–392. doi: 10.1016/j.tins.2021.01.004
Wang, K. C., Fan, L. W., Kaizaki, A., Pang, Y., Cai, Z., and Tien, L. T. (2013). Neonatal lipopolysaccharide exposure induces long-lasting learning impairment, less anxiety-like response and hippocampal injury in adult rats. Neuroscience 234, 146–157. doi: 10.1016/j.neuroscience.2012.12.049
Wang, X., Fu, S., Wang, Y., Yu, P., Hu, J., Gu, W., et al. (2007). Interleukin-1beta mediates proliferation and differentiation of multipotent neural precursor cells through the activation of SAPK/JNK pathway. Mol. Cell. Neurosci. 36, 343–354. doi: 10.1016/j.mcn.2007.07.005
Wang, J., He, W., and Zhang, J. (2023). A richer and more diverse future for microglia phenotypes. Heliyon 9:e14713. doi: 10.1016/j.heliyon.2023.e14713
Wang, C. S., Kavalali, E. T., and Monteggia, L. M. (2022). BDNF signaling in context: from synaptic regulation to psychiatric disorders. Cell 185, 62–76. doi: 10.1016/j.cell.2021.12.003
Wang, H., Wang, H., Song, Y., Liu, C., Qian, X., Zhang, D., et al. (2022). Overexpression of Foxc1 ameliorates sepsis-associated encephalopathy by inhibiting microglial migration and neuroinflammation through the IκBα/NF-κB pathway. Mol. Med. Rep. 25:107. doi: 10.3892/mmr.2022.12623
Wang, J., Zhu, Q., Wang, Y., Peng, J., Shao, L., and Li, X. (2022). Irisin protects against sepsis-associated encephalopathy by suppressing ferroptosis via activation of the Nrf2/GPX4 signal axis. Free Radic. Biol. Med. 187, 171–184. doi: 10.1016/j.freeradbiomed.2022.05.023
Wang, D. S., Zurek, A. A., Lecker, I., Yu, J., Abramian, A. M., Avramescu, S., et al. (2012). Memory deficits induced by inflammation are regulated by α5-subunit-containing GABAA receptors. Cell Rep. 2, 488–496. doi: 10.1016/j.celrep.2012.08.022
Weber, A., Wasiliew, P., and Kracht, M. (2010). Interleukin-1beta (IL-1beta) processing pathway. Sci. Signal. 3:cm 2. doi: 10.1126/scisignal.3105cm2
Wesselingh, R., Butzkueven, H., Buzzard, K., Tarlinton, D., O’Brien, T. J., and Monif, M. (2019). Innate immunity in the central nervous system: a missing piece of the autoimmune encephalitis puzzle? Front. Immunol. 10:2066. doi: 10.3389/fimmu.2019.02066
Wu, L., Fidan, K., Um, J. Y., and Ahn, K. S. (2020). Telomerase: key regulator of inflammation and cancer. Pharmacol. Res. 155:104726. doi: 10.1016/j.phrs.2020.104726
Wu, Y., Wang, W., Díez-Sampedro, A., and Richerson, G. B. (2007). Nonvesicular inhibitory neurotransmission via reversal of the GABA transporter GAT-1. Neuron 56, 851–865. doi: 10.1016/j.neuron.2007.10.021
Xia, S., Qu, J., Jia, H., He, W., Li, J., Zhao, L., et al. (2019). Overexpression of Forkhead box C1 attenuates oxidative stress, inflammation and apoptosis in chronic obstructive pulmonary disease. Life Sci. 216, 75–84. doi: 10.1016/j.lfs.2018.11.023
Xie, D., Shen, F., He, S., Chen, M., Han, Q., Fang, M., et al. (2016). IL-1β induces hypomyelination in the periventricular white matter through inhibition of oligodendrocyte progenitor cell maturation via FYN/MEK/ERK signaling pathway in septic neonatal rats. Glia 64, 583–602. doi: 10.1002/glia.22950
Xie, K., Zhang, Y., Wang, Y., Meng, X., Wang, Y., Yu, Y., et al. (2020). Hydrogen attenuates sepsis-associated encephalopathy by NRF2 mediated NLRP3 pathway inactivation. Inflamm. Res. 69, 697–710. doi: 10.1007/s00011-020-01347-9
Xin, P., Xu, X., Deng, C., Liu, S., Wang, Y., Zhou, X., et al. (2020). The role of JAK/STAT signaling pathway and its inhibitors in diseases. Int. Immunopharmacol. 80:106210. doi: 10.1016/j.intimp.2020.106210
Yan, X., Yang, K., Xiao, Q., Hou, R., Pan, X., and Zhu, X. (2022). Central role of microglia in sepsis-associated encephalopathy: from mechanism to therapy. Front. Immunol. 13:929316. doi: 10.3389/fimmu.2022.929316
Yang, Y., Liu, Y., He, X., Yang, F., Han, S., Qin, A., et al. (2020). ING4 alleviated lipopolysaccharide-induced inflammation by regulating the NF-κB pathway via a direct interaction with SIRT1. Immunol. Cell Biol. 98, 127–137. doi: 10.1111/imcb.12308
Yang, T., Sun, Y., Mao, L., Zhang, M., Li, Q., Zhang, L., et al. (2018). Brain ischemic preconditioning protects against ischemic injury and preserves the blood-brain barrier via oxidative signaling and Nrf2 activation. Redox Biol. 17, 323–337. doi: 10.1016/j.redox.2018.05.001
Ye, B., Tao, T., Zhao, A., Wen, L., He, X., Liu, Y., et al. (2019). Blockade of IL-17A/IL-17R pathway protected mice from Sepsis-associated encephalopathy by inhibition of microglia activation. Mediat. Inflamm. 2019, 1–11. doi: 10.1155/2019/8461725
Yerra, V. G., Negi, G., Sharma, S. S., and Kumar, A. (2013). Potential therapeutic effects of the simultaneous targeting of the Nrf2 and NF-κB pathways in diabetic neuropathy. Redox Biol. 1, 394–397. doi: 10.1016/j.redox.2013.07.005
Yeung, F., Hoberg, J. E., Ramsey, C. S., Keller, M. D., Jones, D. R., Frye, R. A., et al. (2004). Modulation of NF-kappa B-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 23, 2369–2380. doi: 10.1038/sj.emboj.7600244
Yirmiya, R., and Goshen, I. (2010). Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav. Immun. 25, 181–213. doi: 10.1016/j.bbi.2010.10.015
Yu, Y., Feng, J., Lian, N., Yang, M., Xie, K., Wang, G., et al. (2020). Hydrogen gas alleviates blood-brain barrier impairment and cognitive dysfunction of septic mice in an Nrf2-dependent pathway. Int. Immunopharmacol. 85:106585. doi: 10.1016/j.intimp.2020.106585
Zhang, M., and Augustine, G. J. (2021). Synapsins and the synaptic vesicle reserve Pool: floats or anchors? Cells 10:658. doi: 10.3390/cells10030658
Zhang, Q., Lu, Y., Bian, H., Guo, L., and Zhu, H. (2017). Activation of the α7 nicotinic receptor promotes lipopolysaccharide-induced conversion of M1 microglia to M2. Am. J. Transl. Res. 9, 971–985.
Zhang, Q. H., Sheng, Z. Y., and Yao, Y. M. (2014). Septic encephalopathy: when cytokines interact with acetylcholine in the brain. Mil. Med. Res. 1:20. doi: 10.1186/2054-9369-1-20
Zhang, D., Yang, Y., Yang, Y., Liu, J., Zhu, T., Huang, H., et al. (2022). Severe inflammation in new-borns induces long-term cognitive impairment by activation of IL-1β/KCC2 signaling during early development. BMC Med. 20:235. doi: 10.1186/s12916-022-02434-w
Zhao, J., Bi, W., Xiao, S., Lan, X., Cheng, X., Zhang, J., et al. (2019). Neuroinflammation induced by lipopolysaccharide causes cognitive impairment in mice. Sci. Rep. 9:5790. doi: 10.1038/s41598-019-42286-8
Zhao, L., Zhang, R., Su, F., Dai, L., Wang, J., Cui, J., et al. (2020). FoxC1-induced vascular niche improves survival and myocardial repair of mesenchymal stem cells in infarcted hearts. Oxidative Med. Cell. Longev. 2020, 7865395–7865317. doi: 10.1155/2020/7865395
Zhou, Q., Lin, L., Li, H., Wang, H., Jiang, S., Huang, P., et al. (2021). Melatonin reduces Neuroinflammation and improves axonal Hypomyelination by modulating M1/M2 microglia polarization via JAK2-STAT3-telomerase pathway in postnatal rats exposed to lipopolysaccharide. Mol. Neurobiol. 58, 6552–6576. doi: 10.1007/s12035-021-02568-7
Zhou, R., Yang, X., Li, X., Qu, Y., Huang, Q., Sun, X., et al. (2019). Recombinant CC16 inhibits NLRP3/caspase-1-induced pyroptosis through p38 MAPK and ERK signaling pathways in the brain of a neonatal rat model with sepsis. J. Neuroinflammation 16:239. doi: 10.1186/s12974-019-1651-9
Zhuang, X., Yu, Y., Jiang, Y., Zhao, S., Wang, Y., Su, L., et al. (2020). Molecular hydrogen attenuates sepsis-induced neuroinflammation through regulation of microglia polarization through an mTOR-autophagy-dependent pathway. Int. Immunopharmacol. 81:106287. doi: 10.1016/j.intimp.2020.106287
Zou, L., He, J., Gu, L., Shahror, R. A., Li, Y., Cao, T., et al. (2022). Brain innate immune response via miRNA-TLR7 sensing in polymicrobial sepsis. Brain Behav. Immun. 100, 10–24. doi: 10.1016/j.bbi.2021.11.007
Keywords: sepsis, IL-1β, sepsis-associated encephalopathy, cognitive impairment, microglia activate
Citation: Zhu Q, Wan L, Huang H and Liao Z (2024) IL-1β, the first piece to the puzzle of sepsis-related cognitive impairment? Front. Neurosci. 18:1370406. doi: 10.3389/fnins.2024.1370406
Edited by:
Roberta Rizzo, University of Ferrara, ItalyReviewed by:
Hsiuying Wang, National Yang Ming Chiao Tung University, TaiwanSongyun Deng, Central South University, China
Copyright © 2024 Zhu, Wan, Huang and Liao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhimin Liao, zhimin-liao@qq.com
†These authors have contributed equally to this work