1
Institute of Life Science, School of Medicine, Swansea University, Singleton Park, Swansea, UK
2
Department of Pharmacology, The School of Pharmacy, London, UK
3
Institute of Medical Genetics, School of Medicine, Cardiff University, Cardiff, UK
Human startle disease, also known as hyperekplexia (OMIM 149400), is a paroxysmal neurological disorder caused by defects in glycinergic neurotransmission. Hyperekplexia is characterised by an exaggerated startle reflex in response to tactile or acoustic stimuli which first presents as neonatal hypertonia, followed in some with episodes of life-threatening infantile apnoea. Genetic screening studies have demonstrated that hyperekplexia is genetically heterogeneous with several missense and nonsense mutations in the postsynaptic glycine receptor (GlyR) α1 subunit gene (GLRA1) as the primary cause. More recently, missense, nonsense and frameshift mutations have also been identified in the glycine transporter GlyT2 gene, SLC6A5, demonstrating a presynaptic component to this disease. Further mutations, albeit rare, have been identified in the genes encoding the GlyR β subunit (GLRB), collybistin (ARHGEF9) and gephyrin (GPHN) – all of which are postsynaptic proteins involved in orchestrating glycinergic neurotransmission. In this review, we describe the clinical ascertainment aspects, phenotypic considerations and the downstream molecular genetic tools utilised to analyse both presynaptic and postsynaptic components of this heterogeneous human neurological disorder. Moreover, we will describe how the ancient startle response is the preserve of glycinergic neurotransmission and how animal models and human hyperekplexia patients have provided synergistic evidence that implicates this inhibitory system in the control of startle reflexes.
Glycine receptors (GlyR) are heteropentameric ligand-gated chloride ion channels that facilitate fast inhibitory neurotransmission in the human central nervous system (CNS) (Lynch, 2009
). In humans, GlyRs have four functional subunits, GlyR α1–α3 and β that exist in heteromeric αβ combinations although the exact stoichiometry is a matter of intense debate (Grudzinska et al., 2005
). Dysfunction of inhibitory glycinergic neurotransmission causes startle disease/hyperekplexia in humans (OMIM 149400), characterised by neonatal hypertonia and an exaggerated startle reflex in response to tactile or acoustic stimuli. In some instances, this can result in life-threatening infantile apnoea episodes. The most common disease-causing genes are those encoding the postsynaptic GlyR α1 subunit (GLRA1) on chromosome 5q33.1 (Shiang et al., 1993
, 1995
; Rees et al., 1994
, 2001
) and the presynaptic glycine transporter GlyT2 (SLC6A5) on chromosome 11p15.2 (Eulenburg et al., 2006
; Rees et al., 2006
). Both disease loci have revealed dozens of pathogenic missense, nonsense and frameshift mutations that can be inherited in either a dominant or recessive manner, with several recurrent mutations in GLRA1 (Figure 1
).
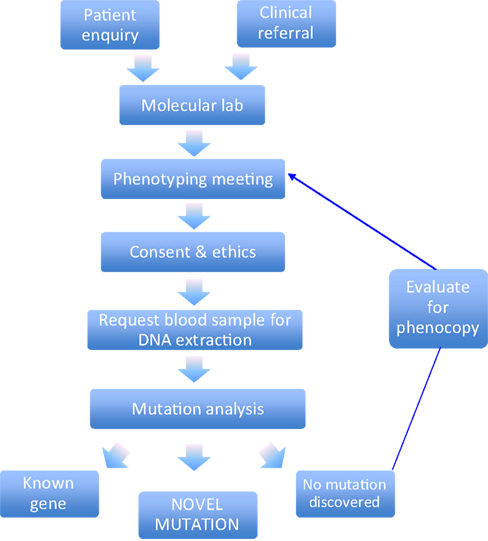
Figure 1. Molecular genetic screening pipeline. This schematic diagram highlights the key stages involved in the clinical ascertainment and molecular genetic diagnosis.
The description of mutations in the GlyR α1 subunit gene by Shiang et al. (1993)
was the first channelopathy associated with ligand-gated chloride channels, whilst the mutations in the GlyT2 gene reported by Rees et al. (2006)
defined the first neurological disorder linked to a defect in presynaptic transporter for a classical fast-acting neurotransmitter. However, both studies owe a great debt to murine models of hyperekplexia which revealed key candidate genes for the human screening programme (Becker et al., 1992
; Buckwalter et al., 1994
; Kingsmore et al., 1994
; Mülhardt et al., 1994
; Ryan et al., 1994
; Gomeza et al., 2003a
). Screening of genes encoding additional postsynaptic proteins involved in GlyR-mediated transmission including the GlyR β subunit (GLRB; Rees et al., 2002
), gephyrin (GPHN; Rees et al., 2003
) and collybistin (ARHGEF9; Harvey et al., 2004
) has revealed only single individuals with potential mutations in these genes. Although mutations in GLRB are a clear cause of exaggerated startle in humans and animals (Kingsmore et al., 1994
; Mülhardt et al., 1994
; Rees et al., 2002
; Hirata et al., 2005
), deletions in GPHN have been associated with molybdenum co-factor deficiency (Reiss et al., 2001
), whilst gene rearrangements in ARHGEF9, encoding the RhoGEF collybistin are more commonly associated with X-linked mental retardation(Harvey et al., 2008b
; Marco et al., 2008
; Kalscheuer et al., 2009
). Despite our successes in disease gene discovery, approximately 35% of the hyperekplexia patients we recruited to our cohort studies are devoid of mutations in genes encoding postsynaptic GlyRs, associated proteins and the presynaptic transporter GlyT2. Although our knowledge of the proteomics of glycinergic synapses is extremely limited, this suggests that other genes encoding presynaptic transporters or receptor/transporter-associated proteins are suitable candidates for mutation screening in hyperekplexia. In this review, we describe the genetic screening/structure-function approaches utilised in our collaborative group to study hyperekplexia, and discuss future genetic screening approaches for the analysis of this genetically heterogenous neurological disorder.
Since 1994, we have received over 260 referrals from clinicians located world-wide to perform genetic testing for hyperekplexia. This is currently performed at the Molecular Neuroscience Laboratory within the Institute of Life Science at Swansea University
1
. Once an anonymous patient clinical summary is received from the referring clinician, data is reviewed by a research committee with a view to assessing whether the patient phenotype fulfils the clinical criteria for mutation screening (Figure 1
).
Alternative diagnoses could include paroxysmal extreme pain disorder, startle epilepsy or acquired(autoimmune) hyperekplexia. If the clinical data is suggestive of inherited hyperekplexia then a study information sheet, consent form and clinical pro-forma are issued to the referring clinician. If the patient and/or their family wish to pursue genetic testing on a research basis, then the referring clinician will obtain consent and collect a blood sample. The completed consent form and clinical pro-forma are then returned to the research team along with the blood sample, which is marked with a unique identifier and transferred for extraction of genomic DNA for mutation screening. Details from the clinical pro-forma are then incorporated into an anonymous dataset to allow the capture of standardised clinical information for each hyperekplexia sample received. The clinical pro-forma, along with evidence of consent, is stored securely on-site at ILS Swansea.
Since new UK-based ethical approval was confirmed in June 2006 (previous ethical approval in Cardiff University, UK, 1994–1999 and Auckland University, New Zealand, 2000–2006), 41 index patient samples have been accepted for hyperekplexia mutation screening and another 11 are currently undergoing the validation/ascertainment. An additional, 13 familial samples have been received for cascade screening – samples which require verification of identified mutations in close relatives. Testing of parental samples of index cases also enables confirmation of the origin of mutations in cases where compound heterozygosity is suspected.
There are potential risks associated with running an international genetic screening service in this way. Since clinical observations are essential to describe the phenotype of the patients referred to our laboratory, we are dependent upon the detail of information provided by the referring clinicians. Occasionally we receive messages from families, index cases and carers where they have received a clinical diagnosis of hyperekplexia. We strongly believe that giving a genetic diagnosis in the absence of genetic counselling is bad practice, so we encourage these individuals to make contact with us via a clinical referral.
Deciding which patient samples to screen can be challenging. If entry criteria are too narrow then the phenotype is never expanded, too broad and the service can be overwhelmed and yet not produce valuable results (see Table 1
). Hyperekplexia is almost certainly a rare condition, but of unknown prevalence and the true phenotype is complicated by cases which report association with co-morbid conditions such as sudden infant death (Giacoia and Ryan, 1994
), epilepsy (Lerman-Sagie et al., 2004
), abdominal herniae (Eppright and Mayhew, 2007
) or developmental delay (Praveen et al., 2001
). The early descriptions of hyperekplexia focussed on the shared clinical features (stiffness, startle and falls), the hereditary nature of the condition and the degree of phenotypic variability (which were qualified as ‘major’ and ‘minor’ variants) (Kirstein and Silfverskiold, 1958
; Kok and Bruyn, 1962
). Although many cases were autosomal dominant, this is not exclusively the case and this misconception represents a bias towards studying large families with many affected members (Rees et al., 1994
; Masri and Hamamy, 2007
). A useful sign particularly in neonatal hyperekplexia (occasionally seen in cerebral palsy) is the nose-tap response: the root of the nose is lightly tapped, provoking a brief and involuntary backwards-retraction of the head.
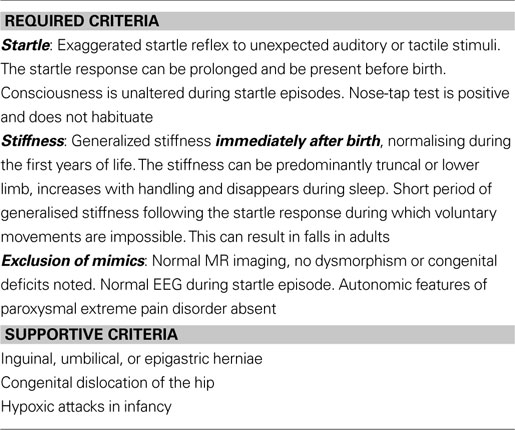
Table 1. Diagnostic criteria for human hyperekplexia.
Neonatal apnoea episodes show an important and potentially life- threatening association with hyperekplexia. These are paroxysmal attacks of abdominal hypertonia with resulting hypoxia. It is important that parents are therefore taught the Vigevano manoeuvre (flexing of the head and limbs toward the trunk) which counteracts these attacks (Vigevano et al., 1989
). Whether these hypoxic spells are related to simple intercostal muscle stiffness, brainstem abnormalities or developmental delay remains unknown.
Hyperekplexia is (not unsurprisingly) most commonly confused with seizure disorders (see Table 2
) such as benign neonatal convulsions. However, in hyperekplexia, consciousness is retained and there is no EEG correlate to either the startle or hypertonic posturing. False positive results are reported, since exaggerated limb jerking can result in a degree of artefact on the EEG trace. However, symptomatic improvement is often seen with benzodiazepines such as clonazepam and less frequently with other anti-convulsant drug therapies.

Table 2. Clinical phenocopies of human hyperekplexia.
Despite the classical features being present from birth or even earlier (some reports suggest that exaggerated startle responses can be felt in utero (Badr El-Din, 1960
; Dalla Bernardina et al., 1988
; Leventer et al., 1995
) the majority of definitive diagnoses are made during infancy. A positive test in an infant can result in older family members receiving an explanation for previously unexplained symptoms. Alternatively, a proportion of early neonatal referrals with primary hypertonia can sometimes develop into more sinister degenerative disorders, which is not typical of inherited hyperekplexia and these cases are invariably negative for GlyR and GlyT2 mutations. However, we are also eager to differentiate between these early onset congenital cases and adult onset cases of hyperekplexia. Since autoimmunity to GlyRs has recently been found in a single case of acquired hyperekplexia (Hutchinson et al., 2008
) we can now process samples from such individuals for testing for anti-GlyR or GlyT2 antibodies, using immortalised human cell lines expressing recombinant GlyRs or GlyTs. Hypertonia (predominantly in the lower limbs) is the key presenting feature of stiff-person syndrome, which is associated with either autoimmunity to gephyrin (Butler et al., 2000
) orglutamic acid decarboxylase 65 (GAD65; Duddy and Baker, 2009
). Furthermore, attempts to definitively describe clinical symptoms and prevalence of hyperekplexia are challenged by potential referral bias. To our knowledge, hyperekplexia has not been diagnosed at the genetic level in people who are ethnically Slavic, South or Central American and very few cases have been reported from Polynesia (see Figure 2
). This may be of importance, since there are certain cultural neuropsychiatric conditions such as the ‘Jumping Frenchmen of Maine’ and ‘Latah Syndrome’, which share some characteristics with hereditary hyperekplexia (Kurczynski, 1983
). Whether these conditions represent the exaggerated startle described in certain anxiety conditions is unknown, since some investigators describe these cultural startle conditions as predominantly psychosomatic in nature (Bartholomew, 1994
). Exaggerated startle may also be secondary to predominately pontine pathology: brainstem infarction, infection, haemorrhage or hypoxia can all produce hyperekplexia-like symptoms. In addition, individuals with paraneoplastic syndromes, multisystem atrophy and multiple sclerosis can all exhibit exaggerated startle responses (Bakker et al., 2006
).
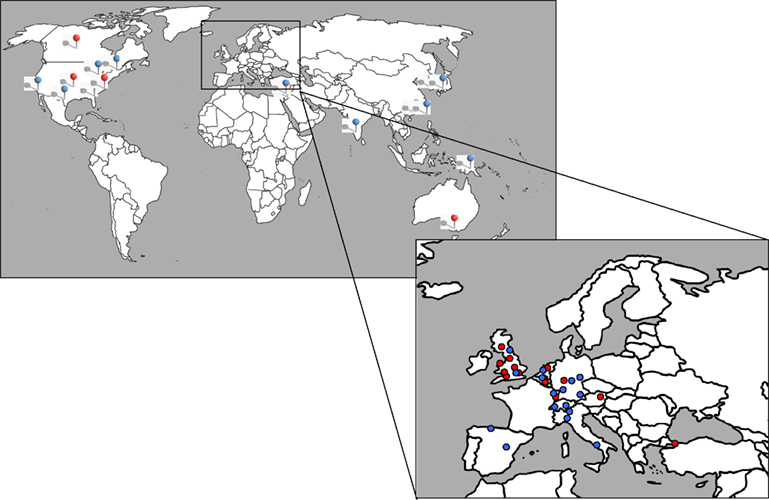
Figure 2. Global origin of hyperekplexia referrals. Upper panel: A geographical representation of the global origin of hyperekplexia patients recruited to our laboratory (red) and other laboratories (blue – Information obtained from the NCBI PubMed search engine). Lower panel: Detail showing the origin of patients recruited from central Europe.
The most common genetic causes of human hyperekplexia are mutations in the GlyR α1 subunit (GLRA1) and GlyT2 (SLC6A5) genes (see Harvey et al., 2008b
for a review and comprehensive mutation data). Therefore, new patient samples that conform to our diagnostic criteria are analysed by PCR amplification of individual exons and dideoxy DNA sequencing on an Applied Biosystems 3100 capillary sequencing platform to detect sequence variants in GLRA1 and SLC6A5 (Rees et al., 2006
). For all candidate genes, analysis includes exons and flanking splice branch points, donor and acceptor sequences, 5′ and 3′ untranslated regions and splice variants are derived insilico, using the Human Genome Browser at the University of California, Santa Cruz
2
. One future aim is also to capture sequence variants in the GLRA1 and SLC6A5 gene promoters, although only the former has been characterised in any detail (Morris et al., 2004
), and the minimal promoter encompasses 5.4 kb of sequence. A robust readout of promoter activity is also required in order to assess any detrimental effect of potential sequence variations. By contrast, GlyT2 transcripts are extensively alternatively spliced at the N-terminus (Ponce et al., 1998
; Ebihara et al., 2004
), and it is possible that multiple tissue-specific or developmentally regulated promoters exist, complicating potential sequence analysis. Candidate genes with rare or as yet unknown mutation frequencies undergo medium-throughput gene variation detection by analysing PCR amplimers with a high-resolution melting (HRM) platform (LightScanner, Idaho Technologies, USA). HRM is a highly-sensitive method of analysing genetic variations in short PCR amplicons generated in the presence of the saturating double-stranded DNA binding dye LCGreen® Plus (Idaho Technologies). The technique allows the PCR amplicons to be distinguished based on their dissociation during rapid melting (Figure 3
). HRM analysis has a published detection rate of 100% for heterozygous mutations (Lonie et al., 2006
; Kennerson et al., 2007
) and has the added advantages of high speed (∼15 min per 96 well plate run), no post-PCR handling and greatly reduced costs (9 p per amplicon) compared to sequencing (∼£5 per read from commercial services)
3
. Thus, HRM allows for rapid and cost-effective mutation discovery in dsDNA with improved sensitivity compared to other screening platforms. However, limitations include the optimisation of amplification conditions in the presence of LCGreen® Plus, limitations on the size of amplicons (<400bp) and non-specific amplicons or primer dimers significantly reduce HRM performance. For a comprehensive review of HRM see White and Potts (2006)
.
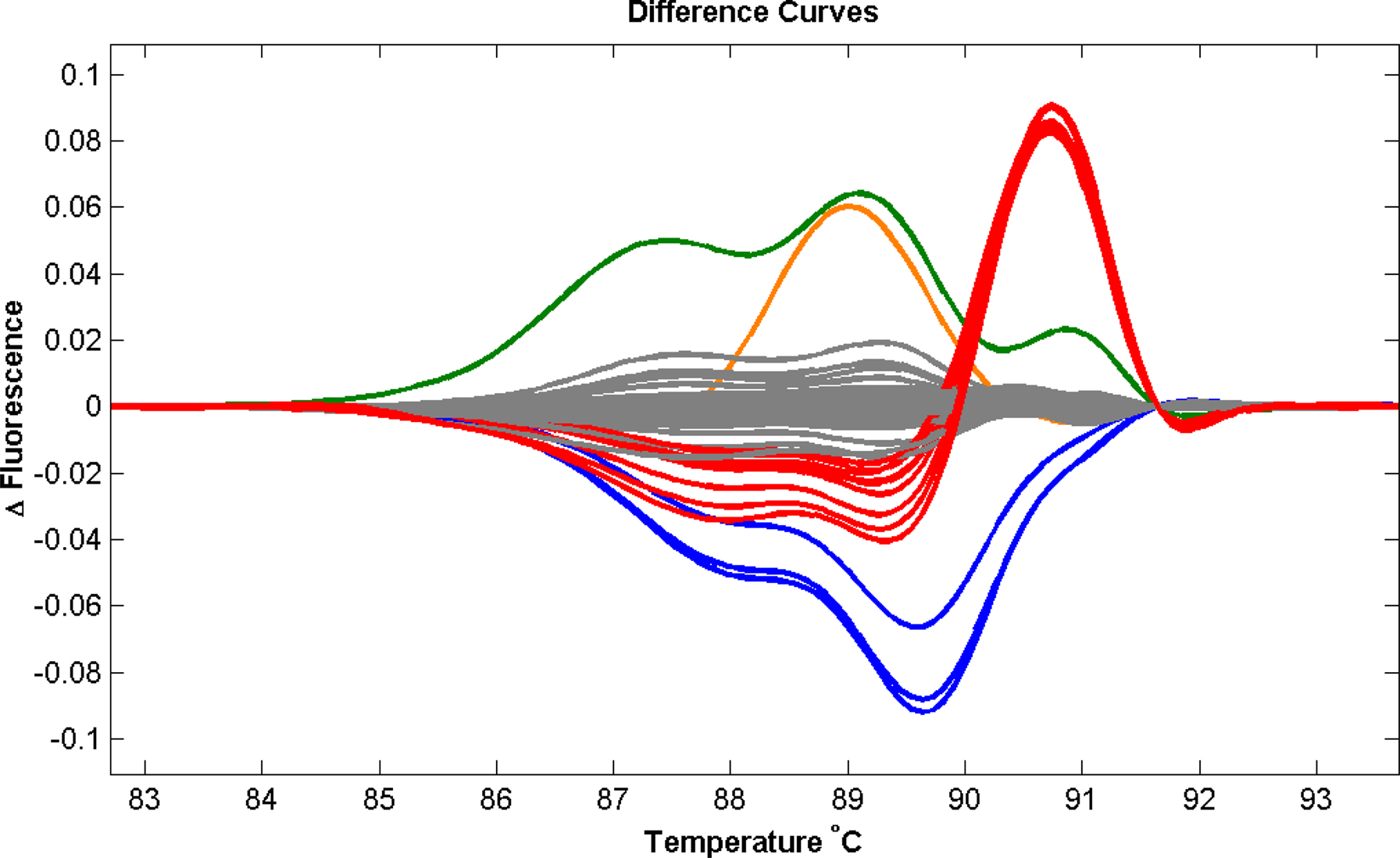
Figure 3. Validation of the Light Scanner technology. To assess the application of this method we performed a series of validation assays utilising known genetic variations in the GLRA1 gene in a population of hyperekplexia patients. This screening method accurately detected all heterozygous (red and orange) and homozygous (blue) missense mutations present. The most common GLRA1 mutation associated with hyperekplexia, R271Q, is shown in red. The assay also detected a false-positive variant (green) generated due to the poor quality of amplified PCR product. Population control samples are shown in grey.
PCR products showing variant melting profiles suggestive of allelic heterogeneity undergo purification and direct sequencing to identify the genetic variation. Population studies of genomic variation are performed using agarose gel restriction fragment length polymorphisms (RFLPs) to confirm the absence of the mutation in the healthy population. This can also be confirmed byassessing HRM profiles of control DNA samples alongside a mutation-positive sample. DNA sequencing chromatograms are then assessed using an automated mutation surveyor programme (MutationSurveyor, SoftGenetics®, USA) which is particularly useful for detecting homozygous changes in the sequence data. After this systematic analysis in our laboratory, 53 single, unrelated, sporadic cases of hyperekplexia have proved gene-negative in screening approaches for GLRA1, GLRB, GPHN, ARHGEF9 and SLC6A5.
Both dominant and recessive mutations in GLRA1 are associated with sporadic and familial startle disease, whilst a proportion of sporadic hyperekplexia is accounted for by the homozygous inheritance of recessive or compound heterozygous GLRA1 mutations (Rees et al., 2001
). By contrast, the vast majority of mutations in SLC6A5 are inherited as compound heterozygotes or show recessive inheritance of a single mutation from consanguineous parents. To date, there is only a single example of a dominant mutation in SLC6A5 (Rees et al., 2006
). Hence, the parental carriers of SLC6A5 mutations are typically asymptomatic indicating that dominant negative effects are not a common mutational mechanism. Other explanations for a single mutation could include an undetected second-hit event, not easily detectable by PCR and sequencing. For example, promoter mutations, large deletions or intronic SNPs causing allelic dropout due to PCR primer mismatches are possible. Even synonymous SNPs that do not cause an amino acid substitution could cause creation of an ectopic splice donor or acceptor site, causing missplicing of gene transcripts. We have begun to assess these issues by using multiplex ligation-dependent probe amplification (MLPA) analysis in index cases, to assay for possible deletions in SLC6A5 and GLRA1. MLPA provides the means for the quantitative analysis of various changes in gene structure and/or gene copy number of several dozens (40–50) of DNA targets in a single reaction containing small amounts (∼20ng) of human chromosomal DNA. Binary probes containing a sequence-specific part and a universal part are hybridized to their DNA targets and ligated. Each ligated probe is then PCR amplified with a universal primer pair and gives rise to an amplicon of unique size. The relative amount of each amplicon reflects the quantity of the corresponding target that is present in the nucleic acid sample. While allelic drop-out can be assessed by designing an additional primer set, changes in the promoter sequences and splicing patterns of GLRA1 and SLC6A5 are more challenging. In particular, we are also limited by the lack of patient-specific RNA resource or the assurance that the genes are expressed in peripheral leukocytes. Affordable next generation re-sequencing (e.g. 454 Roche, Illumina Solexa, AB SOLiD) along with high-throughput splicing assays may resolve these current limitations. These resources would also allow us to assess changes in RNA editing of GlyR transcripts, an important consideration for studies of GlyR α2 and α3 subunit genes in health and disease (Meier et al., 2005
; Eichler et al., 2009
).
Potentially disease-causing mutations can also be subjected to functional and molecular modelling analysis to provide insight into the precise mechanisms underlying pathogenicity. In the case of SLC6A5, mutations were analysed by homology modelling of GlyT2 using the crystal structure of the bacterial leucine transporter (LeuT) (Yamashita et al., 2005
). Simple sequence alignments of LeuT with GlyT1 and GlyT2 allowed us to identify residues potentially involved in coordinating glycine and Na+ binding (Rees et al., 2006
), which were later confirmed by assays on recombinant wild-type and mutant GlyT2 including [3H]glycine uptake assays, sophisticated electrophysiological analyses in Xenopus oocytes and molecular modelling (Rees et al., 2006
; Harvey et al., 2008a
).
Structural modelling is carried out using a homology modelling pipeline built with the Biskit structural bioinformatics platform (Grunberg et al., 2007
). Our pipeline workflow incorporates the NCBI tools platform (Wheeler et al., 2007
), including the BLAST program (Altschul et al., 1990
) for similarity searching of sequence databases. Protein sequences corresponding to the Protein Databank of protein structures were searched for homology with the gene of interest in order to identify putative structural homologues. T-COFFEE (Notredame et al., 2000
) was used for alignment of the test sequence with the template, followed by 10 iterations of the MODELLER homology modelling program (Eswar et al., 2003
) (Figure 4
).
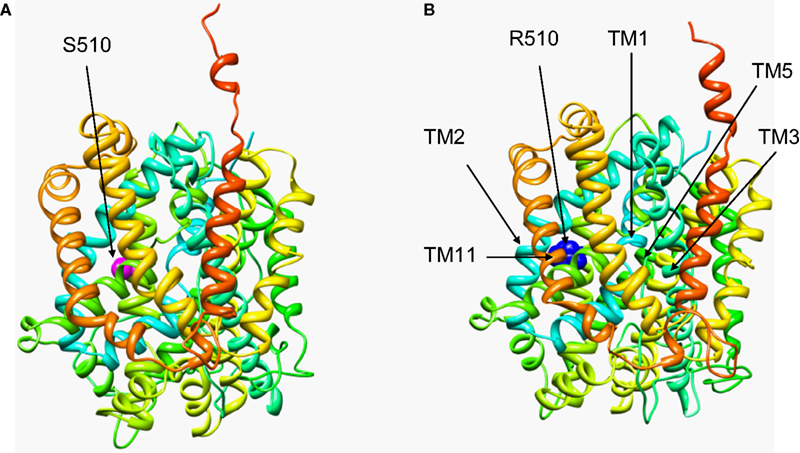
Figure 4. Structural modelling of human GlyT2. The wild-type (A) and the S510R mutant (B) model were generated based on homology with the crystal structure of LeuT, a bacterial Na+/Cl−-dependent neurotransmitter transporter homologue (PDB: 2A65). The models cover residues 191–754 of GlyT2 and show the position of the S510R mutation in TM7 along with the extensive re-arrangement of other transmembrane regions in the S510R that results in defective membrane trafficking of S510R and trapping of wild-type GlyT2 (see Rees et al., 2006
). Models were visualized using the molecular graphics program Chimera (http://www.cgl.ucsf.edu/chimera/
).
Our recent research into glycinergic transmission has clearly demonstrated the importance of both presynaptic (GlyT2) and postsynaptic (GlyR α1β) mechanisms in health and disease. This work has also suggested that our search for other suitable candidate genes for mutation analysis in human hyperekplexia should be broadened (Table 3
). Potential target genes are discussed below in the context of their putative roles in glycinergic synaptic physiology.
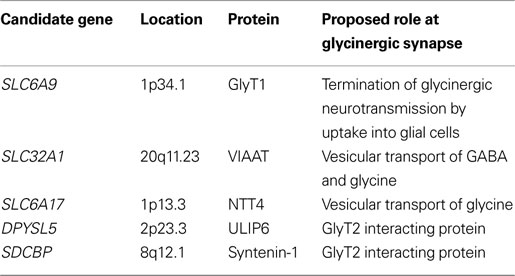
Table 3. Candidate genes for mutation screening in hyperekplexia. Chromosomal locations were obtained from http://ncbi.nlm.nih.gov/
GlyT1
Whilst inhibitory glycinergic neurotransmission is dependent upon agonist-mediated activation of postsynaptic GlyRs, this function is dependent upon having a sufficient pool of releasable glycine within the presynaptic terminal and prompt termination of glycinergic transmission. GlyT1, encoded by SLC6A9 (1p34.1) fulfils the latter function, and is predominately expressed on glial cells. Studies on GlyT1 knockout mice (Gomeza et al., 2003b
; Tsai et al., 2004
) suggest that loss of function of GlyT1 results in a pathological accumulation of synaptic glycine, causing severe motor deficits and premature death as a result of respiratory failure. The phenotype of the GlyT1 knockout mouse resembles a devastating neurological disorder known as glycine encephalopathy (OMIM 605899) although no SLC6A9 mutations have been found to date in this disorder in humans. However, it is possible that more subtle missense mutations could result in a gain of function of GlyT1, causing enhanced clearance of glycine from the synaptic cleft into neighbouring glial cells. This could cause depletion of glycine in neighbouring presynaptic neurones, which may result in hyperekplexia in humans (Harvey et al., 2008a
).
GlyT2 Interactors
Further presynaptic candidates for genetic analysis include proteins interacting with GlyT2, such as syntenin-1 and ULIP6. ULIP6 is encoded by DPYSL5 (2p23.3) and is a brain-specific phosphoprotein of the Ulip/collapsing response mediator protein family. ULIP6 interacts with extended intracellular N-terminus of GlyT2 in a phosphorylation dependent manner (Horiuchi et al., 2005
). Since ULIP6 has been implicated in GlyT2 endocytosis and recycling (Eulenburg et al., 2005
; Horiuchi et al., 2005
), it is plausible that mutations in ULIP6 could cause hyperekplexia by altering levels of presynaptic GlyT2. By contrast, the PDZ containing protein syntenin-1, encoded by the gene SDCBP (8q12.1) is thought to regulate the trafficking or presynaptic localisation of GlyT2 in glycinergic neurones (Ohno et al., 2004
). Mutations that affect syntenin PDZ binding domains could cause mislocalisation of GlyT2 and other interacting partners, since GlyT2 localisation in the presynaptic terminal is dependent on the C-terminal PDZ binding motif (Armsen et al., 2007
). However, due to the wide range of proteins interacting with syntenin-1, we consider it unlikely that defects in syntenin-1 will give rise to classical hyperekplexia (Harvey et al., 2008b
).
VIAAT
Another obvious hyperekplexia candidate gene is the vesicular inhibitory amino acid transporter (VIAAT), encoded by SLC32A1 (20q11.23). VIAAT is expressed in both GABAergic and glycinergic neurones (Chaudhry et al., 1998
), and is responsible for the chloride-dependent loading of presynaptic vesicles with GABA and glycine (Jin et al., 2003
; Juge et al., 2009
). We suggest that certain mutations in VIAAT could lead to the specific loss of glycine (but not GABA) loading into synaptic vesicles, thus resulting in hyperekplexia. VIAAT was first identified as a mammalian homologue of the ‘uncoordinated’ C. elegans mutant unc-47, which was known to be defective in a presynaptic component of GABA release. Despite the fact that C. elegans do not appear to use glycine as a neurotransmitter, in a sophisticated cellular assay Aubrey et al. (2007)
were able to show that UNC-47 is able to readily transport both GABA and glycine into vesicles. Importantly, the UNC-47 mutation G462R was shown to abolish GABA, but not glycine uptake. Since residue G462 is conserved at the equivalent position (G500) in rodent and human VIAAT sequences, mutations in SLC32A1 could theoretically compromise GABA uptake into synaptic vesicles while leaving glycine uptake intact. Since it is clearly possible to separate GABA and glycine transport by a single missense mutation, our hypothesis is that other missense mutations could also result in a GABA-specific VIAAT, potentially resulting in hyperekplexia.
NTT4
The orphan transporter NTT4 (also known as Rxt1), encoded by SLC6A17 (1p13.3) has recently been implicated as a vesicular transporter for glycine, proline, leucine and alanine (Parra et al., 2008
). This novel finding suggests that NTT4, which is highly expressed in several brain regions, including the spinal cord (Liu et al., 1993
), may have an important role in glycinergic transmission and possibly hyperekplexia.
Despite the advances in the molecular genetics of hyperekplexia of the last 20 years, we are no closer to describing certain clinical aspects than the original pioneering study by Andermann et al. (1980)
. Hyperekplexia is rare, easily misdiagnosed and clonazepam is still the current treatment of choice. We have been unable to find a genetic basis to the original delineation – the so called ‘major’ and ‘minor’ variants. Collections of large cohorts, particularly containing sporadic cases may help us move away from the inherent bias caused by studying large families with a single gene mutation. Future challenges include improving testing turnaround times and accessibility, and collecting comprehensive clinical data to improve our understanding of possible differences in clinical phenotypes caused by mutations in GLRA1 versus SLC6A5. This will also enable us to identify potential hyperekplexia-associated co-morbidities and identify potential phenocopy referrals.
On the horizon is the prospect of third generation sequence platforms which will facilitate automated re-sequencing of large genomic segments containing disease-causing genes and new candidate loci and possibly even whole genomes of affected individuals. These include the pyrosequencing-based platform of next generation 454 sequencing (see Rothberg and Leamon, 2008
) which was recently used for rapid genome re-sequencing of an individual genome at a fraction of the cost of previous platforms (Wheeler et al., 2008
). Such coverage will provide not only the sequence of coding exons and flanking spice sites, but information on intragenic DNA, SNP haplotype risk factors and copy number variables. Affordability is at present the main barrier and as a medium-term solution enrichment of individual chromosomal regions containing GLRA1 and SLC6A5 may provide an interim solution. For example, Zheng and colleagues have recently developed a method for high-throughput variant detection, utilising specific genomic regions for target amplification by capture and ligation (TACL), allele enrichment and array resequencing (Zheng et al., 2009
). This platform has identified rare and novel variants, and will undoubtedly lead to improvements in our understanding of complex genetic disorders.
Molecular biology, animal models of glycinergic function and detailed proteomic studies will continue to provide further candidates for genetic screening in hyperekplexia and other potential disorders of glycinergic synapses. The new targets described above are being screened at present in patients lacking mutations in GLRA1 and SLC6A5. Molecular genetic studies of unresolved ENU-induced mutations in zebrafish (Granato et al., 1996
) may also tease out novel determinants of glycinergic function (Hirata et al., 2010
). These leads suggest that new genes of major effect could shortly join GLRA1 and SLC6A5 in our molecular screening programme.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The clinical ascertainment aspects described in this review were reviewed and approved by the Local Research Ethics Committee, the South West Wales REC. Research on hyperekplexia in our laboratories is supported by grants from the Medical Research Council (G0601585 to Robert J. Harvey, Kirsten Harvey and Mark I. Rees) and the Wales Office of Research and Development (Wales Epilepsy Research Network to Mark I. Rees). We would also like to thank Jean-Francois Vanbellinghen (University of Liege, Belgium) for providing information for Figure 2
.
Crisponi, L., Crisponi, G., Meloni, A., Toliat, M. R., Nurnberg, G., Usala, G., Uda, M., Masala, M., Hohne, W., Becker, C., Marongiu, M., Chiappe, F., Kleta, R., Rauch, A., Wollnik, B., Strasser, F., Reese, T., Jakobs, C., Kurlemann, G., Cao, A., Nurnberg, P., and Rutsch, F. (2007). Crisponi syndrome is caused by mutations in the CRLF1 gene and is allelic to cold-induced sweating syndrome type 1. Am. J. Hum. Genet. 80, 971–981.
Fertleman, C. R., Ferrie, C. D., Aicardi, J., Bednarek, N. A., Eeg-Olofsson, O., Elmslie, F. V., Griesemer, D. A., Goutières, F., Kirkpatrick, M., Malmros, I. N., Pollitzer, M., Rossiter, M., Roulet-Perez, E., Schubert, R., Smith, V. V., Testard, H., Wong, V., and Stephenson, J. B. (2007). Paroxysmal extreme pain disorder (previously familial rectal pain syndrome). Neurology 69, 586–595.
Granato, M., van Eeden, F. J., Schach, U., Trowe, T., Brand, M., Furutani-Seiki, M., Haffter, P., Hammerschmidt, M., Heisenberg, C. P., Jiang, Y. J., Kane, D. A., Kelsh, R. N., Mullins, M. C., Odenthal, J., and Nüsslein-Volhard, C. (1996). Genes controlling and mediating locomotion behavior of the zebrafish embryo and larva. Development 123, 399–413.
Harvey, K., Duguid, I. C., Alldred, M. J., Beatty, S. E., Ward, H., Keep, N. H., Lingenfelter, S. E., Pearce, B. R., Lundgren, J., Owen, M. J., Smart, T. G., Lüscher, B., Rees, M. I., and Harvey, R. J. (2004). The GDP-GTP exchange factor collybistin: an essential determinant of neuronal gephyrin clustering. J. Neurosci. 24, 5816–5826.
Horiuchi, M., Horiuchi, M., Loebrich, S., Brandstaetter, J. H., Kneussel, M., and Betz, H. (2005). Cellular localisation and subcellular distribution of Unc-33-like protein 6, a brain-specific protein of the collapsin response mediator protein family that interacts with the neuronal glycine transporter 2. J. Neurochem. 94, 307–315.
Kalscheuer, V. M., Musante, L., Fang, C., Hoffmann, K., Fuchs, C., Carta, E., Deas, E., Venkateswarlu, K., Menzel, C., Ullmann, R., Tommerup, N., Dalprà, L., Tzschach, A., Selicorni, A., Lüscher, B., Ropers, H. H., Harvey, K., and Harvey, R. J. (2009). A balanced chromosomal translocation disrupting ARHGEF9 is associated with epilepsy, anxiety, aggression, and mental retardation. Hum. Mutat. 30, 61–68.
Lonie, L., Porter, D. E., Fraser, M., Cole, T., Wise, C., Yates, L., Wakeling, E., Blair, E., Morava, E., Monaco, A. P., and Ragoussis, J. (2006). Determination of the mutation spectrum of the EXT1/EXT2 genes in British Caucasian patients with multiple osteochondromas, and exclusion of six candidate genes in EXT negative cases. Hum. Mutat. 27, 1160.
Parra, L. A., Baust, T., Mestikawy, S. E., Quiroz, M., Hoffman, B., Haflett, J. M., Yao, J. K., and Torres, G. E. (2008). The orphan transporter RXT1/NTT4 (SLC6A17) functions as a synaptic vesicle amino acid transporter selective for proline, glycine, leucine, and alanine. Mol. Pharmacol. 74, 1521–1532.
Rees, M. I., Harvey, K., Pearce, B. R., Chung, S. K., Duguid, I. C., Thomas, P., Beatty, S., Graham, G. E., Armstrong, L., Shiang, R., Abbott, K. J., Zuberi, S. M., Stephenson, J. B., Owen, M. J., Tijssen, M. A., van den Maagdenberg, A. M., Smart, T. G., Supplisson, S., and Harvey, R. J. (2006). Mutations in the GlyT2 gene define a presynaptic component of human startle disease. Nat. Genet. 38, 801–806.
Rees, M. I., Harvey, K., Ward, H., White, J. H., Evans, L., Duguid, I. C., Hsu, C. C., Coleman, S. L., Miller, J., Baer, K., Waldvogel, H. J., Gibbon, F., Smart, T. G., Owen, M. J., Harvey, R. J., and Snell, R. G. (2003). Isoform heterogeneity of the human gephyrin gene (GPHN), binding domains to the glycine receptor, and mutation analysis in hyperekplexia. J. Biol. Chem. 278, 24688–24696.
Rees, M. I., Lewis, T. M., Vafa, B., Ferrie, C., Corry, P., Muntoni, F., Jungbluth, H., Stephenson, J. B., Kerr, M., Snell, R. G., Schofield, P. R., and Owen, M. J. (2001). Compound heterozygosity and nonsense mutations in the α1 subunit of the inhibitory glycine receptor in hyperekplexia. Hum. Genet. 109, 267–270.
Wheeler, D. A., Srinivasan, M., Egholm, M., Shen, Y., Chen, L., McGuire, A., He, W., Chen, Y. J., Makhijani, V., Roth, G. T., Gomes, X., Tartaro, K., Niazi, F., Turcotte, C. L., Irzyk, G. P., Lupski, J. R., Chinault, C., Song, X. Z., Liu, Y., Yuan, Y., Nazareth, L., Qin, X., Muzny, D. M., Margulies, M., Weinstock, G. M., Gibbs, R. A., and Rothberg, J. M. (2008). The complete genome of an individual by massively parallel DNA sequencing. Nature 454, 872–877.
Wheeler, D. L., Barrett, T., Benson, D. A., Bryant, S. H., Canese, K., Chetvernin, V., Church, D. M., Dicuccio, M., Edgar, R., Federhen, S., Feolo, M., Geer, L. Y., Helmberg, W., Kapustin, Y., Khovayko, O., Landsman, D., Lipman, D. J., Madden, T. L., Maglott, D. R., Miller, V., Ostell, J., Pruitt, K. D., Schuler, G. D., Shumway, M., Sequeira, E., Sherry, S. T., Sirotkin, K., Souvorov, A., Starchenko, G., Tatusov, R. L., Tatusova, T. A., Wagner, L., and Yaschenko, E. (2007). Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 36 (Database issue), 13–21.
White, H., and Potts, G. (2006). Mutation scanning by high resolution melt analysis. Evaluation of rotor-gene 6000 (Corbett Life Science), HR-1 and 384-well lightscanner (Idaho Technology). National Genetics Reference Laboratory (Wessex). Available at: http://www.gene-quantification.de/white-pott-hrm-comp-2006.pdf
.