 Holger Scholz* and Karin M. Kirschner
Holger Scholz* and Karin M. Kirschner
- Institut für Vegetative Physiologie, Charité – Universitätsmedizin Berlin, Berlin, Germany
Adequate tissue oxygenation is a prerequisite for normal development of the embryo. Most fetal organs are exquisitely susceptible to hypoxia which occurs when the delivery of oxygen is exceeded by the actual demand. Developmental abnormalities due to insufficient supply with oxygen can result from the impaired expression of genes with essential functions during embryogenesis. As such, the Wilms’ tumor gene, WT1, is among the fetal genes that are regulated by the local oxygen tension. WT1 was originally discovered as a tumor suppressor gene owing to loss-of-function mutations in a subset of pediatric renal neoplasias, known as nephroblastomas or Wilms’ tumors. Wilms’ tumors can arise when pluripotent progenitor cells in the embryonic kidney continue to proliferate rather than differentiating to glomeruli and tubules. WT1 encodes a zinc finger protein, of which multiple isoforms exist due to alternative mRNA splicing in addition to translational and post-translational modifications. While some WT1 isoforms function as transcription factors, other WT1 proteins are presumably involved in post-transcriptional mRNA processing. However, the role of WT1 reaches far beyond that of a tumor suppressor as homozygous disruption of Wt1 in mice caused embryonic lethality with a failure of normal development of the kidneys, gonads, heart, and other tissues. WT1 mutations in humans are associated with malformation of the genitourinary system. A common paradigm of WT1 expressing cells is their capacity to switch between a mesenchymal and epithelial state. Thus, WT1 likely acts as a master switch that enables cells to undergo reciprocal epithelial-to-mesenchymal transition. Impairment of renal precursor cells to differentiate along the epithelial lineage due to WT1 mutations may favor malignant tumor growth. This article shall provide a concise review of the function of WT1 in development and disease with special consideration of its regulation by molecular oxygen.
The Fetus and Hypoxia
During no other period of life our organism bears such a high risk of insufficient tissue oxygenation as it does prenatally. Fetal hypoxia in mammals can lead to intrauterine growth restriction and reduced birth weight, or may even impair normal organ formation (Webster and Abela, 2007; Hutter et al., 2010). Fetal programming refers to the situation that an early stimulus or insult operating at a sensitive period of prenatal development results in long-term structural and functional changes of an individual (Barker, 2004). Placental pathologies that can lead to fetal programming are frequently associated with hypoxia and oxidative stress. For example, placental villous explants from patients with preeclampsia, a severe form of pregnancy-induced arterial hypertension, secreted fourfold higher amounts of the soluble receptor-1 for vascular endothelial growth factor (VEGF) than tissue from healthy pregnant women (Ahmad and Ahmed, 2004). These findings suggested that villous explants in vitro retain a hypoxia memory, which may reflect long-term fetal programming (Ahmad and Ahmed, 2004). This and other investigations also provided evidence that elevated levels of the soluble VEGF receptor-1 can inhibit angiogenesis in preeclampsia thereby increasing the morbidity and mortality for mother and fetus due to endothelial dysfunction (reviewed in Mutter and Karumanchi, 2008). As the consequences of intrauterine oxygen restriction may extend into adulthood, adaptive mechanisms have evolved in the embryo to overcome the devastating effects of inadequate supply with oxygen. These include the synthesis of a fetal form of hemoglobin with improved oxygen binding affinity, a high cardiac output in relation to the effective oxygen needs, and a relatively low specific metabolic rate of most embryonic tissues (Martin, 2008; Thein et al., 2009).
Besides imposing the risk of hypoxia on the growing embryo tissue oxygenation may also serve as a physiological signal that directs the temporal and spatial gene expression during intrauterine maturation. The importance of oxygen as a signaling molecule in development becomes evident from the lethality of mouse embryos with targeted disruption of the genes encoding the hypoxia-inducible factors (HIFs). HIFs are heterodimeric basic helix–loop–helix transcription factors which bind to the RCGTG consensus sequence and function as master regulators of gene expression in hypoxia (Semenza et al., 1991; Webb et al., 2009). HIFs are composed of an oxygen-sensitive α-subunit and the constitutive aryl hydrocarbon receptor nuclear translocator (Arnt, also called HIF-β). Three HIF-α genes exist in mammalian cells, which are expressed either ubiquitously (HIF-1α) or in a more restricted manner (HIF-2α, HIF-3α; Chen et al., 2009). The HIF-α subunits are highly unstable under normoxic conditions: hydroxylation of HIF-α at two conserved proline residues by the oxygen-dependent enzyme HIF prolyl-hydroxylase allows for the interaction of HIF with the von Hippel–Lindau E3 ubiquitin ligase complex and subsequent proteasomal degradation (Jaakkola et al., 2001; reviewed in Semenza, 2004). Additionally, the factor inhibiting HIF-1 (FIH-1) hydroxylates an asparagine residue in the C-terminal transactivation domain (TAD) of HIF-α in normoxia thereby preventing its interaction with the transcriptional co-activator p300 (Koivunen et al., 2004). These molecular regulators of HIFs function as oxygen sensors in various tissues (Webb et al., 2009).
Strikingly, complete disruption of HIF-1α in mice resulted in a developmental arrest and embryonic lethality by E11 (Iyer et al., 1998; Ryan et al., 1998). Among other abnormalities Hif-1α-deficient murine embryos (Hif-1α−/−) exhibited impaired vascularization of various tissues in addition to neural tube defects and marked cell death within the cephalic mesenchyme (Iyer et al., 1998; Ryan et al., 1998). Likewise, inactivation of the genes encoding HIF-2α and HIF-1β caused embryonic lethality due to vascular defects, impaired hematopoiesis, and a failure of normal placental differentiation (Kozak et al., 1997; Peng et al., 2000; Scortegagna et al., 2003). The preterm death and the phenotypic abnormalities of murine embryos with targeted disruption of the genes encoding HIF-1α, HIF-2α, or HIF-1β indicate that oxygen sensing is indeed necessary for normal intrauterine development.
The Wilms’ Tumor Gene, WT1
The Wilms’ tumor gene, WT1, is among the molecular downstream targets of HIF. Loss-of-function mutations in a childhood cancer of the kidney known as nephroblastoma or Wilms’ tumor testified WT1 as a tumor suppressor gene (Huff et al., 1991; Pelletier et al., 1991a,b; Park et al., 1993). The disease affects approximately 1:10000 children worldwide, usually around age five, but individuals with genetic predisposition can develop bilateral tumors even earlier in life. Since the WT1 gene is inactivated in only ∼15% of sporadic Wilms’ tumors, the genetic abnormalities underlying the majority of nephroblastomas are currently unknown. Recent findings indicate that monoallelic mutations of the X-chromosome linked gene WTX (APC membrane recruitment 1 also called AMER1) may account for approximately one-third of Wilms’ tumors (Rivera et al., 2007).
The WT1 gene, which is in the focus of this article, spans more than 50 kb on the short arm of human chromosome 11 (11p13) and contains 10 exons (Gessler et al., 1992). It encodes a ∼55 kDa protein with four C-terminal Cys2-His2 zinc fingers allowing for sequence-specific nucleic acid binding (Call et al., 1990; Gessler et al., 1990; reviewed in Scharnhorst et al., 2001). Other functional domains include a proline/glutamine-rich N-terminal sequence implicated in transcriptional activation, a self-association domain, and a putative RNA recognition motif (Kennedy et al., 1996). Overall, more than two dozens WT1 isoforms are generated by alternative mRNA splicing, translation initiation at variable start points, RNA editing, and post-translational modifications (reviewed in Scharnhorst et al., 2001). Of particular importance are two alternative splicing events that give rise to four different WT1 splice isoforms (Haber et al., 1991; Gessler et al., 1992). Alternative splicing of exon 5, which occurs only in mammals but in no other vertebrates analyzed so far (Kent et al., 1995), interposes 17 amino acids between the proline/glutamine-rich N-terminus and the zinc finger domain of the WT1 protein. Unexpectedly, selective ablation of the exon 5 containing Wt1 isoforms caused no obvious phenotype in mice suggesting functional redundancy among the different Wt1 molecules during embryonic development (Natoli et al., 2002). The usage of an alternative splice donor site at the end of exon 9 results in the insertion of three extra amino acids, lysine, threonine, serine (KTS), between the third and fourth zinc fingers (Haber et al., 1991). WT1 proteins with the KTS tripeptide have a reduced DNA binding affinity compared to the −KTS forms (Laity et al., 2000). Evidence obtained from morphological and biochemical studies suggests that the prevalent WT1(+KTS) proteins are involved in post-transcriptional processes, whereas the WT1(−KTS) molecules act predominantly as transcription factors (Larsson et al., 1995; Caricasole et al., 1996; Davies et al., 1998; Niksic et al., 2004; Bor et al., 2006). Several dozens downstream candidate targets of WT1 have been identified including the genes for growth factors and their receptors, extracellular matrix and cell adhesion molecules, transcription factors, and signal transducers (for review see Scharnhorst et al., 2001; Roberts, 2005). However, the majority of these genes were assessed by co-transfecting promoter–reporter constructs with WT1 expression plasmids, and only few candidates can be considered as bona fide targets that are regulated by WT1 in vivo.
Besides its function as a tumor suppressor WT1 is also required for normal embryogenesis. Mice with homozygous disruption of Wt1 (Wt1−/−) are embryonic lethal and lack kidneys and gonads (Kreidberg et al., 1993). Other defects in Wt1 null mutant embryos pertain to the mesothelium, i.e., the epicardium and coelomic epithelium (Moore et al., 1999), spleen (Herzer et al., 1999), adrenal glands (Moore et al., 1999), retina (Wagner et al., 2002b), and the olfactory epithelium (Wagner et al., 2005a). A frequent observation in Wt1 deficiency is an increased number of apoptotic cells in tissues that would normally express Wt1 in wild-type animals (Kreidberg et al., 1993; Wagner et al., 2002b, 2005b). It has therefore been proposed that WT1 proteins may rescue cells from apoptosis and permit other factors to promote cell differentiation. Hemizygosity for WT1 in humans is associated with genitourinary malformations suggesting that the WT1 gene dosage is relevant for normal development (Pelletier et al., 1991a).
Despite the importance of WT1 in embryogenesis and tumor formation little is known about the physiological mechanisms that control its expression. Several transcription factors have been reported to regulate the proximal WT1 promoter in transfection experiments including activation by Sp1 (Cohen et al., 1997) and Pax2 (Dehbi et al., 1996), and negative feedback inhibition by WT1 itself (Malik et al., 1994; Rupprecht et al., 1994). Transgenic expression from a yeast artificial chromosome (YAC) carrying ∼280 kb of flanking sequence of the human WT1 gene was performed in an attempt to rescue the phenotype of Wt1-null embryos (Moore et al., 1999). While this strategy proved successful in restoring the morphology of the epicardium and diaphragm of Wt1-deficient mice, the kidney and gonad defects could not be rescued (Moore et al., 1999). Consistently, in vitro studies confirmed that cis-regulatory elements at far distance from the transcription start site are important for normal WT1 levels (Scholz et al., 1997). Interestingly, recent findings indicate that WT1 expression is regulated in vitro and in vivo by the local oxygen tension. This notion is based on the observation that exposure of adult rats at low ambient oxygen (8% O2) significantly increased Wt1 mRNA and protein levels in kidneys and hearts (Wagner et al., 2002a). Furthermore, HIF-1 protein in nuclear extracts from hypoxic cells bound to a hypoxia responsive cis-element in the proximal WT1 promoter (Wagner et al., 2003). Mutation of the HIF-1 binding site abrogated stimulation of the WT1 promoter by hypoxia in transient reporter gene assays (Wagner et al., 2003). While these findings identified tissue hypoxia as a stimulatory signal for WT1, the functional significance of an oxygen-dependent control of WT1 expression is just beginning to emerge. The following paragraphs highlight possible implications of this regulatory switch in embryonic development and tumor formation.
Oxygen-Dependent WT1 Expression in the Heart
The epicardium constitutes an epithelial sheath that is spread on the outer surface of the heart. It belongs to the few tissues which express WT1 continuously from early embryogenesis on throughout adulthood. Remarkably, the hearts of Wt1-null mutant mouse embryos (Wt1−/−) exhibit severe muscular wall thinning with frequent bleeding into the pericardial cavity (Kreidberg et al., 1993; Moore et al., 1999). Myocardial hypoplasia as in Wt1−/− embryos was also observed upon targeted disruption of other epicardial genes including those for vascular cell adhesion molecule-1 (VCAM-1; Kwee et al., 1995), α4integrin (Yang et al., 1995; Sengbusch et al., 2002), RARα (Kastner et al., 1997), RXRα (Sucov et al., 1994), and FOG2 (Tevosian et al., 2000). Thus, normal myocardial growth during embryogenesis appears to depend on mitogenic signals emanating from the WT1 expressing epicardium (Figure 1). This raises the interesting question of whether compensatory heart growth in the adult, e.g., in response to arterial hypertension, is related to changes in WT1 expression. A study addressing this issue yielded intriguing results: While no correlation between epicardial Wt1 RNA and left ventricular heart weights was found in several disease models of compensatory cardiac hypertrophy, Wt1 was significantly up-regulated in heart tissue after myocardial infarction in rats (Wagner et al., 2002a). In contrast to the situation in sham-operated animals, which expressed Wt1 exclusively in the epicardium but in no other cell types in the heart, de novo expression of Wt1 was detected in myocardial blood vessels in the infarct border zone (Wagner et al., 2002a). Further on, Wt1 in vascular cells was associated with a local increase of pro-angiogenic molecules, e.g., VEGF. Wt1 expression in the vasculature of the ischemic hearts could be mimicked by exposure of rats to either 8% ambient oxygen or 0.1% carbon monoxide (Wagner et al., 2002a). Although these and other results (Wagner et al., 2003) disclosed WT1 as an oxygen-regulated gene, at least in the heart, the functional importance of this regulatory mechanism remained elusive. A short recapitulation of the developmental events underlying formation of the epicardium and the presumed role of WT1 herein may bring some light into the dark.
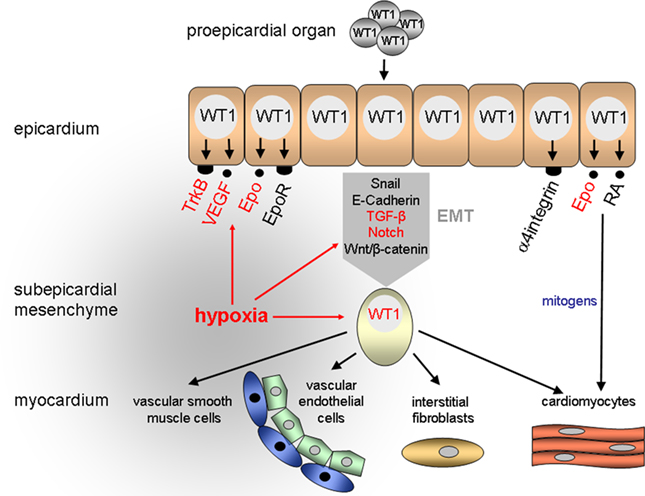
Figure 1. Proposed role of hypoxia and WT1 in the developing heart. A major function of WT1 is the control of epithelial-to-mesenchymal transition (EMT) of epicardial cells. Epicardium-derived mesenchymal cells give rise to vascular endothelial and smooth muscle cells as well as interstitial fibroblasts and cardiomyocytes in the heart. RA, retinoic acid.
WT1 in the Developing Epicardium
The epicardium originates from the proepicardial organ (PEO) which arises from the mesothelium of the septum transversum in mammalian embryos and the sinus venosus near the embryonic liver in birds (Männer et al., 2001; Ratajska et al., 2008). Expansion of the spreading epicardium on the outer heart surface occurs by cell proliferation. In the following stages a subset of epicardial cells loosen their contacts, detach from the basement membrane, and migrate in the space directly beneath the epicardium (reviewed in Männer et al., 2001; Gittenberger-de Groot et al., 2010). The delamination of epicardial cells is closely linked to a change in the differentiation state of these cells, which lose their apico-basal polarity and acquire a mesenchymal phenotype (Figure 1). This phenomenon, which has been referred to as epithelial-to-mesenchymal transition (EMT) is associated with a profound cytoskeletal remodeling that results in a spindle-shaped cell morphology and improved cell migration capacity (reviewed in Thiery and Sleeman, 2006; Guarino, 2007). Epicardium-derived mesenchymal cells populate the myocardial tissue and differentiate there to interstitial fibroblasts and coronary vascular smooth muscle and endothelial cells (Mikawa and Gourdie, 1996; Dettman et al., 1998; Pérez-Pomares et al., 2002a; Wilting et al., 2007). Since epicardium-derived mesenchymal cells can also differentiate to cardiac myocytes (Cai et al., 2008; Zhou et al., 2008) they have been considered as cardiovascular progenitor cells in the developing heart (Figure 1).
Where in this sequence of events does WT1 come into play and what is its function during epicardial development relating to the local oxygen concentration? WT1 is initially expressed in the proepicardium and the coelomic epithelium which gives rise to the parietal pericardium and the germinal epithelium (Carmona et al., 2001; Pérez-Pomares et al., 2002b). Alpha4integrin, whose product associates with β1- and β7-integrin subunits to form heterodimeric cell adhesion molecules (reviewed in Liu et al., 2000), was identified as a first candidate target gene of WT1 in (pro)epicardial cells (Kirschner et al., 2006). Remarkably, embryos which lack α4integrin have similar epicardial defects as Wt1−/− mice suggesting that impaired α4integrin expression accounts at least in part for the abnormalities in Wt1-deficient hearts (Sengbusch et al., 2002). Thus, a physiological function of WT1 during early heart development may consist in facilitating the attachment of epicardial progenitor cells to the outer surface of the heart through enhanced expression of cell adhesion molecules (Figure 1).
The developing epicardium is highly susceptible to hypoxia, and epicardial detachment was the first sign of hypoxic damage in mouse fetuses with reduced tissue oxygenation (Ream et al., 2008). Further on lowering the inspiratory oxygen concentration of pregnant mice from 21 to 8% O2 caused intrauterine growth restriction and severe myocardial thinning by E12.5 (Ream et al., 2008). These phenotypic abnormalities of hypoxic fetal hearts are reminiscent of the myocardial hypoplasia in Wt1-deficient murine embryos (Kreidberg et al., 1993; Moore et al., 1999). It is thus likely that myocardial growth promoting signals are generated in the developing epicardium, which requires a balanced oxygen environment and intact WT1 expression. Accordingly, mechanical removal of the epicardium from explants of chicken embryonic hearts caused a proliferation arrest of cardiac myocytes (Stuckmann et al., 2003). Likewise, blockade of retinoic acid or erythropoietin (Epo) signaling from the epicardium inhibited cardiac myocyte proliferation and survival, and this effect could be reversed by administration of either exogenous Epo or retinoic acid, respectively (Stuckmann et al., 2003). Consistently, murine embryos which lack the genes encoding Epo and its receptor (Epo-R) exhibit thin walled ventricles, partial detachment of the epicardium, and abnormal coronary vascular plexus (Wu et al., 1999). In wild-type murine embryos Epo-R is expressed in the epicardium and the endocardium of the heart but not in cardiomyocytes (Wu et al., 1999). These combined findings suggest that cardiac myocyte proliferation is stimulated by epicardium-derived mitogens which are under the reciprocal control of Epo and retinoic acid (Figure 1). Interestingly, transcription of the genes encoding Epo, which is also oxygen-regulated, and its cognate receptor is activated by WT1 (Dame et al., 2006; Kirschner et al., 2008). Moreover, WT1 has been reported to repress the human but not the murine RARα1 promoter through binding to a GC-rich consensus motif (Goodyer et al., 1995). A highly conserved retinoic acid binding element was recently discovered in the wt1a gene from zebrafish (Bollig et al., 2009). This retinoic acid sensitive element is located in an enhancer region which directs promoter gene expression to the intermediate mesoderm (Bollig et al., 2009). Thus, oxygen-regulated WT1 may function as a major component within the complex network of molecules that control the signal transfer between epicardial cells and cardiomyocytes in the developing heart.
WT1 and Epithelial-to-Mesenchymal Transition
Epithelial-to-mesenchymal transition of epicardial cells is crucial for the generation of cardiovascular progenitor cells in the heart (Wessels and Perez-Pomares, 2004). In an elegant study it was shown recently that WT1 is necessary for repression of the epithelial phenotype in epicardial cells and during embryonic stem cell differentiation. Transcriptional activation by WT1 of the genes encoding Snail (Snai1) and E-cadherin (Cdh1) was proven as a critical step during EMT (Martinez-Estrada et al., 2010). Lack of EMT in Wt1−/− mutant embryonic stem cells caused a failure in the differentiation of some mesothelial lineages, which could be rescued by forced expression of Snail (Martinez-Estrada et al., 2010). Notably, Wt1 is thought to regulate the reverse process, i.e., mesenchymal-to-epithelial transition, in the developing kidney (Davies et al., 2004; Hohenstein and Hastie, 2006). Induction of the permanent kidney occurs through reciprocal interactions between the metanephric mesenchyme and the invading mesonephric (Wolffian) duct, which gives rise to the ureter (Quaggin and Kreidberg, 2008; Constantini and Kopan, 2010). Another important event during nephron formation is the condensation and subsequent epithelialization of the metanephric mesenchyme around the branching ureteric bud tips (Quaggin and Kreidberg, 2008; Constantini and Kopan, 2010). Epithelial differentiation is disrupted in WT1-deficient embryonic kidneys, which fail to mature beyond the early inductive stages (Kreidberg et al., 1993). The highest levels of WT1 are usually found in podocytes of the kidneys, Sertoli cells of the testis, and mesothelial cells (reviewed in Scharnhorst et al., 2001). These cell types share the capability of switching between an epithelial and mesenchymal phenotype, a process which is likely controlled by WT1. Interestingly, proteomic analysis of renal podocytes from a Denys–Drash syndrome patient with a WT1 gene mutation revealed that of the 75 misexpressed proteins 43% were related to cytoskeletal components (Viney et al., 2007). This finding supports the notion that WT1 directs the expression of components of the cytoskeleton and regulators of its assembly during reciprocal transition between a mesenchymal and epithelial differentiation state.
Evidence has been provided that EMT is associated with a low oxygen microenvironment. For example, exposure of breast cancer cells to 1% O2 induced changes, e.g., down-regulation of epithelial marker proteins, enhanced vimentin expression, and cell migration, that are consistent with EMT (Lester et al., 2007). Hypoxia and HIFs can modulate EMT-triggering pathways through several mechanisms (Figure 2). These include direct regulation of EMT inducers and their receptors, changes in the expression of signaling molecules acting on EMT, and enhancement of functional interaction with EMT-associated nuclear factors (reviewed in Haase, 2009). EMT inducers belong to the TGF-β growth factor family (Zavadil et al., 2004) and the Notch and Wnt/β-catenin signaling pathways (Gustafsson et al., 2005; Neth et al., 2007; Sahlgren et al., 2008). Upon binding to its cognate receptors TGF-β stimulates EMT via SMAD-dependent expression of genes involved in cell proliferation, differentiation, and migration (reviewed in Zavadil and Bottinger, 2005). Non-SMAD-dependent pathways of TGF-β in EMT act along the MAPK-signaling cascade, which controls small Rho family GTPases thereby influencing cell motility (reviewed in Zavadil and Bottinger, 2005). Activation of Notch promotes EMT through induction of Snai1 and Snai2 (Timmerman et al., 2004), and through interfering with TGF-β/SMAD-mediated EMT via the hairy/enhancer-of-split-related transcriptional repressor Hey1 (Zavadil et al., 2004). The role of the Wnt/β-catenin signaling pathway as an inducer of EMT in development and cancer is also well established (Huber et al., 2005; Wu and Zhou, 2008).
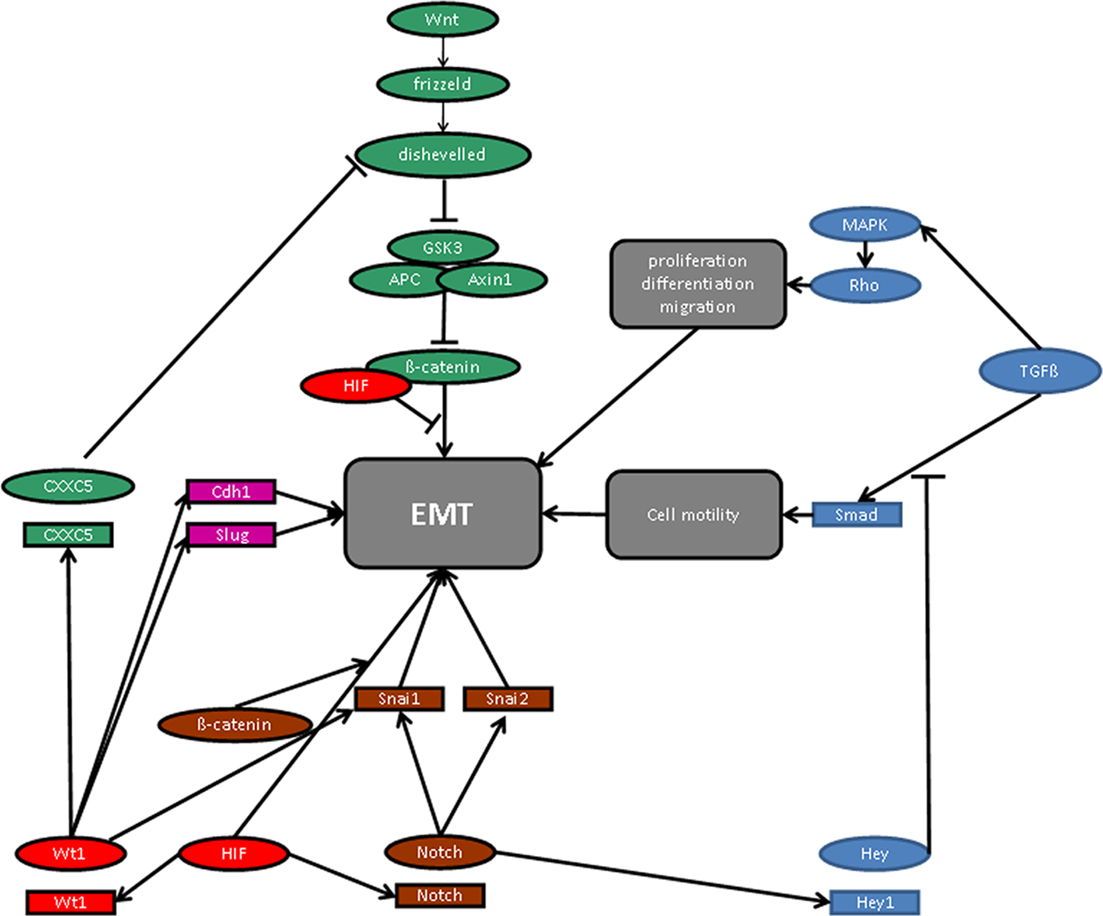
Figure 2. Molecular signaling pathways in epithelial-to-mesenchymal transition (EMT). For details see manuscript text.
Remarkably, the molecular signaling pathways of hypoxia and WT1, whose expression is increased at low oxygen tension via HIF-1 activation (Wagner et al., 2003), converge on the level of EMT regulatory molecules (Figure 2). Thus, β-catenin enhances HIF-1 target gene transcription, whereas physical interaction with HIF inhibits the transcriptional activity of β-catenin (Kaidi et al., 2007). Wnt/β-catenin downstream signaling is also negatively regulated by the product of the recently identified WT1 target gene CXXC5 (syn. WID), which interacts with the immediate downstream effector of the frizzled receptor, Disheveled (Kim et al., 2010). The local oxygen environment controls the Notch pathway and the expression of TGF-β (Scheid et al., 2002; Schäffer et al., 2003; Chen et al., 2007; Sahlgren et al., 2008; Zheng et al., 2008), which is also regulated by WT1 (Dey et al., 1994; Jin et al., 1999). TGF-β has been reported to stimulate EMT in proepicardial tissue explants (Olivey et al., 2006), and mice with targeted inactivation either of the TGF-β type III receptor (Compton et al., 2007) or the TGF-β type I receptor (Alk5; (Sridurongrit et al., 2008) exhibit cardiac defects resembling those in Wt1 deficiency (Moore et al., 1999; Wagner et al., 2005b). Thus, TGF-β signaling is a possible pathway along which WT1 may regulate EMT in epicardial cells in relation to the local oxygen concentration.
WT1 and Coronary Blood Vessel Development
As outlined above, cells destined to assemble the myocardial vasculature originate from the proepicardium. The epicardium and the subepicardial mesenchyme constitute a source for various cell lineages in the embryonic heart including coronary endothelial and smooth muscle cells, cardiac fibroblasts, and also cardiomyocytes (Mikawa and Gourdie, 1996; Dettman et al., 1998; Pérez-Pomares et al., 2002a; Wilting et al., 2007; Cai et al., 2008; Zhou et al., 2008). Considering the crucial role of WT1 in the formation of the epicardium (Kreidberg et al., 1993; Moore et al., 1999) it is no longer surprising that normal vascularization of the heart is impaired in the absence of WT1. Indeed, blood vessel density was significantly reduced in the hearts of Wt1-null murine embryos at 15.5 d.p.c. compared to their wild-type littermates (Wagner et al., 2005b). Apoptosis of a significant number of cells in the Wt1-deficient epicardium and subepicardial tissue suggests that Wt1 acts as a survival factor for mesenchymal cells in the heart (Wagner et al., 2005b). Detection of Wt1 protein in the vessel walls of wild-type embryonic hearts indicates that Wt1 may also fulfill a role in stabilizing the newly formed coronary vasculature (Wagner et al., 2005b). A likely downstream target of Wt1 in this function is the Ntrk2 gene, which encodes the high affinity neurotrophin receptor, TrkB (Klein et al., 1992; Wagner et al., 2005b). The promoter of the Ntrk2 gene was trans-activated by Wt1 in vitro and in the embryonic epicardium in vivo (Wagner et al., 2005b). Inactivation of the genes encoding either TrkB or its ligand, brain-derived neurotrophic factor (BDNF), caused myocardial vessel destabilization, and subsequent bleeding in the subepicardial mesenchyme (Donovan et al., 2000; Wagner et al., 2005b). BDNF–TrkB signaling is assumed to promote cell contact formation in the nascent vasculature through maintaining the clustering of the adherens junction molecule β-catenin (Dejana et al., 1999). It is noteworthy that TrkB expression, at least in neurons, is enhanced in hypoxia through activation by HIF-1 (Martens et al., 2007).
Remarkably, intravenous injection of the hypoxia marker EF5 [2-(2-nitro-1H-imidazol-1-yl)-N-(2,2,3,3,3-pentafluoropropyl) acetamide] revealed severely hypoxic tissue regions close to the origin of the major coronary arteries in avian embryos (Sugishita et al., 2004; Wikenheiser et al., 2006). Incubation of chicken embryos at Hamburger and Hamilton (HH) stage 25 at low (15% O2) or high (40–75%) ambient oxygen caused malformations of coronary arteries at their proximal connections to the aorta (Wikenheiser et al., 2009). Local over-expression of HIF-1α increased the number of hemangioblasts and blood vessels at the base of the outflow tract (Wikenheiser et al., 2009). The vascular abnormalities in oxygen-depleted chicken embryos are presumably due to the recruitment of epicardium-derived vascular progenitor cells to ectopic sites in the developing heart. Thus, a picture emerges according to which the formation of the coronary vascular system involves differential oxygen levels within the developing heart leading to regional activation of oxygen-dependent transcription factors such as HIF-1, WT1, and others. In the absence of a normal coronary vessel system myocardial growth restriction may occur when the diffusion capacity for oxygen and nutrients from the ventricular blood is exceeded by the actual needs of the embryonic heart. Hence, disturbed myocardial vascularization likely contributes to the ventricular wall thinning in Wt1-deficient murine embryos (Kreidberg et al., 1993; Moore et al., 1999).
WT1 and Vascularization of Non-Cardiac Tissues
It is still unclear whether the pro-vasculogenic role of WT1 is confined to the heart, or whether WT1 plays a role in blood vessel formation in other tissues as well. Remarkable structural and functional analogies exist in the development of the heart and the gut. Similar to the heart, the gut is covered by a mesothelial envelope which is thought to provide the source of vasculogenic cells (reviewed in Ratajska et al., 2008; Olivey and Svensson, 2010). By the combinatorial use of molecular biology techniques and vital cell fate tracing it has been demonstrated that Wt1 expressing serosal mesothelial cells are spread on the outer surface of the developing mouse gut, which is initially devoid of blood vessels (Wilm et al., 2005). Subsequently, a subpopulation of cells undergoes EMT, migrates throughout the gut, and differentiates to smooth muscle cells of all major blood vessels in the intestine (Wilm et al., 2005). Considering these similarities of developmental events and the presumed role of hypoxia in EMT (Lester et al., 2007) it is tempting to speculate whether Wt1 fulfills related functions during blood vessel formation in the embryonic gut and heart. However, definite proof for a critical role of WT1 in vasculogenesis of the intestine still needs to be provided. In this regard, transgenic animal models which enable a developmental stage and tissue-specific inactivation of Wt1 will provide a major technical advancement.
Wilms’ tumor gene protein was recently also detected in the vasculature of diverse tumors arising in the skin (Timár et al., 2005), breast, endometrium, lung, and other organs (Wagner et al., 2008; Dohi et al., 2010). In most cases, WT1 was not detected in blood vessels of adjacent normal tissue samples, but was restricted to endothelial cells of the tumor vasculature (Wagner et al., 2008). At these sites WT1 was frequently associated with proliferating cell nuclear antigen (PCNA), and WT1 silencing by RNAi significantly lowered vascular endothelial cell proliferation and migration in vitro (Wagner et al., 2008). Vascular endothelial cells undergo profound structural and functional changes during branching angiogenesis and it will be a challenging task to elucidate a possible role of WT1 in endothelial cell plasticity.
WT1 in Tumors
Wilms’ tumor gene has been proven to function as a suppressor of Wilms’ tumor growth by a multitude of genetic and experimental studies (reviewed in Scharnhorst et al., 2001; Rivera and Haber, 2005). However, during the past few years it became increasingly clear that WT1 is expressed at a rather high level in various malignancies including carcinomas of the lung (Oji et al., 2002), colon (Kösters et al., 2004), thyroid gland (Oji et al., 2003), breast (Loeb et al., 2001), pancreas (Oji et al., 2004a), brain (Oji et al., 2004b), and the majority of Wilms’ tumors (Rivera and Haber, 2005). The molecular mechanisms that account for the robust expression of WT1 in these diseases are not well understood. However, considering the low oxygen tensions in rapidly growing tumors (reviewed in Hockel and Vaupel, 2001), one can assume that local tissue hypoxia contributes to the increased WT1 levels in various carcinomas.
Most WT1 expressing tumors originate from tissues, which do normally not contain substantial amounts of WT1. The absence of WT1 gene mutations from these neoplasms has therefore been considered as circumstantial evidence for an oncogenic potential of WT1 (reviewed in Yang et al., 2007). Highly elevated WT1 levels were also found in the bone marrow and peripheral blood of many leukemias compared to normal bone marrow and hematopoietic progenitor cells (Miwa et al., 1992; Miyagi et al., 1993; Menssen et al., 1995). Increased WT1 expression correlated with a relatively low responsiveness to standard chemotherapy and a poor prognosis of most leukemias suggesting that wild-type WT1 contributes to the disease phenotype (Tamaki et al., 1999; Barragan et al., 2004; Chiusa et al., 2006). Consistently, in most but not all studies down-regulation of WT1 was associated with a reduced growth and rapid apoptosis of acute myeloic leukemia cells (Yamagami et al., 1996; Ito et al., 2006). The molecular signaling pathways underlying the dual role (oncogene vs. tumor suppressor) of WT1 in tumor formation are not well understood but may involve variable target gene selection in different tissues and/or changes in protein function due to physical interaction with partner molecules in a cell type-specific manner (Johnstone et al., 1996, reviewed in Roberts, 2006). For example, interaction with prostate apoptosis response-4 (par-4) through the zinc finger DNA binding domain shifted the effect of WT1 from transcriptional activation to repression (Johnstone et al., 1996). Co-transfection of wild-type par-4, but not of a mutant form defective of WT1 interaction, abrogated growth suppression caused by WT1 (Johnstone et al., 1996).
Interestingly, the mitochondrial serine protease HtrA2 was recently identified as a novel WT1 binding partner (Hartkamp et al., 2010). HtrA2 is thought to function as a tumor suppressor by limiting the activity of prosurvival oncogenes through proteolytic degradation. All four major WT1 protein isoforms were cleaved by HtrA2 at multiple sites in response to cytotoxic stress, and blockade of HtrA2 activity prevented the proteolysis of WT1 under proapoptotic conditions (Hartkamp et al., 2010). The latter observations are remarkable as a recent proteome-wide screen of more than 1000 proteins detected only 15 substrates of HtrA2 (Vande Walle et al., 2007), with very few apoptosis-related mitochondrial proteins among those (Johnson and Kaplitt, 2009). The release of HtrA2 from mitochondria and subsequent induction of apoptosis was stimulated by reactive oxygen species in vascular endothelial cells (Liu et al., 2010). Moreover, oxidative stress resulting from tissue reperfusion after ischemic injury activated the HtrA2 pathway in the brain (Saito et al., 2004). It is thus conceivable that tissue oxygenation does not only regulate the expression of WT1, but may also modulate its function through interfering with HtrA2 signaling (Figure 3). A potential downstream mechanism of WT1 in drug-induced cell death involves the proapoptotic genes c-Myc and JunB, which are normally suppressed by WT1 (Hartkamp et al., 2010). Proteolytic cleavage of WT1 by HtrA2 in response to cytotoxic stress resulted in high levels of c-Myc and JunB and subsequent apoptosis of cancer cells (Hartkamp et al., 2010). Accordingly, HtrA2-mediated cleavage may represent a critical switch that alleviates the oncogenic potential of WT1 in the presence of cytotoxic stress (Figure 3). It will be a thrilling task for future studies to determine whether cleavage-resistant WT1 variants exist, and to explore their potential functions in normal development and cancer. Further on, the effect of other sorts of cellular stress on WT1-HtrA2 interaction needs to be investigated.
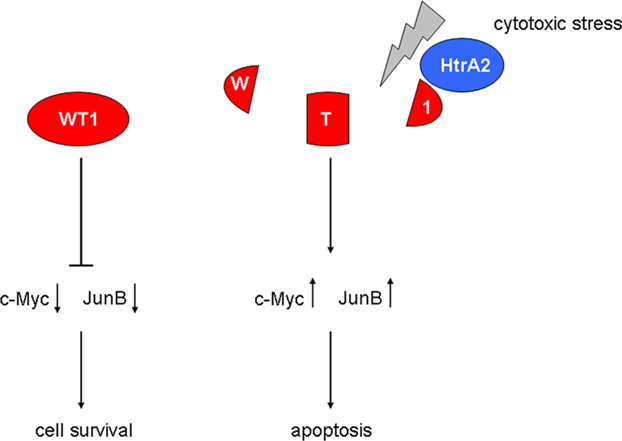
Figure 3. Role of the mitochondrial serine protease HtrA2 in the regulation of WT1 function. Activation of HtrA2 by cytotoxic stress causes degradation of WT1 protein, thereby increasing the proapoptotic transcription factors c-Myc and JunB.
WT1 has recently been reported to act downstream of the KRAS oncogene in the control of cell proliferation and senescence (Vicent et al., 2010). The KRAS gene, which belongs to the RAS gene family, encodes a membrane-bound 21 kDa guanosine triphosphate (GTP)-binding protein. Inactivation of Wt1 caused senescence of KRAS expressing mouse embryonic fibroblasts and inhibited tumor progression in an animal model of KRAS-driven lung cancer (Vicent et al., 2010; Figure 4). In contrast, deletion or suppression of Wt1 had no effect on wild-type cells in the absence of oncogenic KRAS (Vicent et al., 2010; Figure 4). Moreover, decreased WT1 target gene expression correlated with improved prognosis only in lung cancer patients whose genetic signature was indicative of oncogenic KRAS activation (Vicent et al., 2010). In another study, deletion of HIF-2α, but not of HIF-1α, was found to promote tumor formation in human KRAS-driven lung cancer cells (Mazumdar et al., 2010). This effect was attributed, at least in part, to a reduced expression of the candidate tumor suppressor gene Scgb3a1 in the absence of HIF-2α (Mazumdar et al., 2010; Figure 4). Enhanced levels of the activated, GTP-bound K-ras proto-oncogene in response to hypoxia were found in colon cancer cells with a wild-type KRAS gene (Zeng et al., 2010). These combined findings testify that the oncogenic properties of WT1 are closely related to the KRAS signaling pathway which is also under control of the local oxygen environment. Hence, WT1 and hypoxia may functionally synergize on the level of KRAS-dependent cell cycle control.
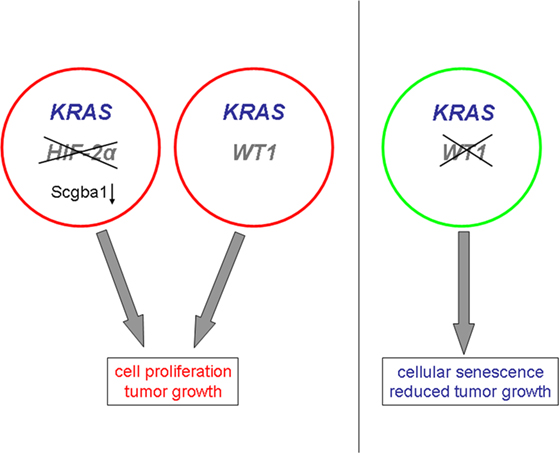
Figure 4. Proposed roles of WT1 and hypoxia-inducible factor HIF-2α in KRAS-driven tumor formation. The prooncogenic effects of WT1 are closely linked to KRAS signaling, and inactivation of WT1 inhibits KRAS-dependent tumor formation. Deletion of HIF-2α promotes KRAS-driven tumorigenesis through down-regulation of the tumor suppressor Scgba1.
Concluding Remarks
The product of the Wilms’ tumor gene, WT1, acts as a critical gatekeeper at the crossroad of normal development and cancer. The back and forth transition between a mesenchymal and epithelial cellular state appears to be a major target event for WT1 action. To gain a complete picture of the multiple functions of WT1, it is mandatory to identify bona fide downstream effectors of the various protein isoforms in conjunction with their interacting partner molecules.
Moreover, it will necessary to decipher the physiological mechanisms that regulate WT1 in normal and tumor cells. Previous findings indicated that tissue oxygenation determines WT1 expression, and that HIFs can stimulate transcription of the WT1 gene at low ambient oxygen. Future studies will need to address the question of whether oxygen-dependent regulation of WT1 also pertains to the post-transcriptional level, e.g., through changes in mRNA stability and/or translation. Notably, the transcripts of many “classical” HIF target genes including those for Epo (McGary et al., 1997), collagen prolyl-4-hydroxylase alpha(I) chain (Fähling et al., 2006a,b), and VEGF (Levy et al., 1998), are stabilized in hypoxia. Moreover, modulation of the mRNA translation rate emerges as a novel paradigm in oxygen-dependent gene regulation (reviewed in Fähling, 2009).
The multiple functions of WT1 in development and disease rely at least in part on the various protein isoforms that are generated by alternative mRNA splicing and other post-transcriptional events. Accumulating evidence indicates that the transcripts of many oxygen-regulated genes, e.g., those for VEGFs (Houck et al., 1991; Tischer et al., 1991), HIFs (Gothié et al., 2000; Maynard et al., 2003), and carbonic anhydrase IX (Malentacchi et al., 2009), are alternatively spliced. Hypoxia can regulate alternative mRNA splicing in several ways which include changes in splicing factor levels as well as activities (Webby et al., 2009). It is therefore tempting to speculate whether oxygen-dependent regulation of alternative WT1 mRNA splicing contributes to the functional complexity of WT1 proteins. A thorough investigation of these issues may provide novel insights into important regulatory mechanisms during embryonic development and tissue repair, and may eventually pave the way for innovative therapeutic strategies in a broad range of diseases.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors acknowledge the continued funding of their work by grants from the Charité – Universitätsmedizin Berlin and the Deutsche Forschungsgemeinschaft.
References
Ahmad, S., and Ahmed, A. (2004). Elevated placental soluble vascular endothelial growth factor receptor-1 inhibits angiogenesis in preeclampsia. Circ. Res. 95, 884–891.
Barker, D. J. (2004). The developmental origins of adult disease. J. Am. Coll. Nutr. 23(Suppl.), 588S–595S.
Barragan, E., Cervera, J., Bolufer, P., Ballester, S., Martin, G., Fernandez, P., Collado, R., Sayas, M. J., and Sanz, M. A. (2004). Prognostic implications of Wilms’ tumor gene (WT1) expression in patients with de novo acute myeloid leukemia. Haematologica 89, 926–933.
Bollig, F., Perner, B., Besenbeck, B., Köthe, S., Ebert, C., Taudien, S., and Englert, C. (2009). A highly conserved retinoic acid responsive element controls wt1a expression in the zebrafish pronephros. Development 136, 2883–2892.
Bor, Y. C., Swartz, J., Morrison, A., Rekosh, D., Ladomery, M., and Hammarskjöld, M. L. (2006). The Wilms’ tumor 1 (WT1) gene (+KTS isoform) functions with a CTE to enhance translation from an unspliced RNA with a retained intron. Genes Dev. 20, 1597–1608.
Cai, C. L., Martin, J. C., Sun, Y., Cui, L., Wang, L., Ouyang, K., Yang, L., Bu, L., Liang, X., Zhang, X., Stallcup, W. B., Denton, C. P., McCulloch, A., Chen, J., and Evans, S. M. (2008). A myocardial lineage derives from Tbx18 epicardial cells. Nature 454, 104–108.
Call, K. M., Glaser, T., Ito, C. Y., Buckler, A. J., Pelletier, J., Haber, D. A., Rose, E. A., Kral, A., Yeger, H., Lewis, W. H., Jones, C., and Housman, D. E. (1990). Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms’ tumor locus. Cell 60, 509–520.
Caricasole, A., Duarte, A., Larsson, S. H., Hastie, N. D., Little, M., Holmes, G., Todorov, I., and Ward, A. (1996). RNA binding by the Wilms tumor suppressor zinc finger protein. Proc. Natl. Acad. Sci. U.S.A. 93, 7562–7566.
Carmona, R., González-Iriarte, M., Pérez-Pomares, J. M., and Muñoz-Chápuli, R. (2001). Localization of the Wilms’ tumour protein WT1 in avian embryos. Cell Tissue Res. 303, 173–186.
Chen, L., Endler, A., and Shibasaki, F. (2009). Hypoxia and angiogenesis: regulation of hypoxia-inducible factors via novel binding factors. Exp. Mol. Med. 41, 849–857.
Chen, Y., De Marco, M. A., Graziani, I., Gazdar, A. F., Strack, P. R., Miele, L., and Bocchetta, M. (2007). Oxygen concentration determines the biological effects of NOTCH-1 signaling in adenocarcinoma of the lung. Cancer Res. 67, 7954–7959.
Chiusa, L., Francia di Celle, P., Campisi, P., Ceretto, C., Marmont, F., and Pich, A. (2006). Prognostic value of quantitative analysis of WT1 gene transcripts in adult acute lymphoblastic leukemia. Haematologica 91, 270–271.
Cohen, H. T., Bossone, S. A., Zhu, G., McDonald, G. A., and Sukhatme, V. P. (1997). Sp1 is a critical regulator of the Wilms’ tumor-1 gene. J. Biol. Chem. 272, 2901–2913.
Compton, L. A., Potash, D. A., Brown, C. B., and Barnett, J. V. (2007). Coronary vessel development is dependent on the type III transforming growth factor beta receptor. Circ. Res. 101, 784–791.
Constantini, F., and Kopan, R. (2010). Patterning a complex organ: branching morphogenesis and nephron segmentation in kidney development. Dev. Cell 18, 698–712.
Dame, C., Kirschner, K. M., Bartz, K. V., Wallach, T., Hussels, C. S., and Scholz, H. (2006). Wilms’ tumor suppressor, Wt1, is a transcriptional activator of the erythropoietin gene. Blood 107, 4282–4290.
Davies, J. A., Ladomery, M., Hohenstein, P., Michael, L., Shafe, A., Spraggon, L., and Hastie, N. (2004). Development of an siRNA-based method for repressing specific genes in renal organ culture and its use to show that the Wt1 tumour suppressor is required for nephron differentiation. Hum. Mol. Genet. 13, 235–246.
Davies, R. C., Calvio, C., Bratt, E., Larsson, S. H., Lamond, A. I., and Hastie, N. D. (1998). WT1 interacts with the splicing factor U2AF65 in an isoform-dependent manner and can be incorporated into spliceosomes. Genes Dev. 12, 3217–3225.
Dehbi, M., Ghahremani, M., Lechner, M., Dressler, G., and Pelletier, J. (1996). The paired-box transcription factor, PAX2, positively modulates expression of the Wilms’ tumor suppressor gene (WT1). Oncogene 13, 447–453.
Dejana, E., Bazzoni, J., and Lampugnani, M. G. (1999). The role of endothelial cell-to-cell junctions in vascular morphogenesis. Thromb. Haemost. 82, 755–761.
Dettman, R. W., Denetclaw, W., Ordahl, C. P., and Bristow, J. (1998). Common epicardial origin of coronary vascular smooth muscle, perivascular fibroblasts, and intermyocardial fibroblasts in the avian heart. Dev. Biol. 193, 169–181.
Dey, B. R., Sukhatme, V. P., Roberts, A. B., Sporn, M. B., Rauscher, F. J. III, and Kim, S. J. (1994). Repression of the transforming growth factor-beta 1 gene by the Wilms’ tumor suppressor WT1 gene product. Mol. Endocrinol. 8, 595–602.
Dohi, S., Ohno, S., Ohno, Y., Kyo, S., Soma, G., Sugiyama, H., and Inoue, M. (2010). WT1 expression correlates with angiogenesis in endometrial cancer tissue. Anticancer Res. 30, 3187–3192.
Donovan, M. J., Lin, M. I., Wiegn, P., Ringstedt, T., Kraemer, R., Hahn, R., Wang, S., Ibañez, C. F., Rafii, S., and Hempstead, B. L. (2000). Brain derived neurotrophic factor is an endothelial cell survival factor required for intramyocardial vessel stabilization. Development 127, 4531–4540.
Fähling, M. (2009). Surviving hypoxia by modulation of mRNA translation rate. J. Cell. Mol. Med. 13, 2770–2779.
Fähling, M., Mrowka, R., Steege, A., Nebrich, G., Perlewitz, A., Persson, P. B., and Thiele, B. J. (2006a). Translational control of collagen prolyl 4-hydroxylase-alpha(I) gene expression under hypoxia. J. Biol. Chem. 281, 26089–126101.
Fähling, M., Mrowka, R., Steege, A., Martinka, P., Persson, P. B., and Thiele, B. J. (2006b). Heterogeneous nuclear ribonucleoprotein-A2/B1 modulate collagen prolyl 4-hydroxylase, alpha (I) mRNA stability. J. Biol. Chem. 281, 9279–9286.
Gessler, M., König, A., and Bruns, G. A. (1992). The genomic organization and expression of the WT1 gene. Genomics 12, 807–813.
Gessler, M., Poustka, A., Cavenee, W., Neve, R. L., Orkin, S. H., and Bruns, G. A. P. (1990). Homozygous deletion in Wilms tumours of a zinc-finger gene identified by chromosome jumping. Nature 343, 774–778.
Gittenberger-de Groot, A. C., Winter, E. M., and Poelmann, R. E. (2010). Epicardium-derived cells (EPDCs) in development, cardiac disease and repair of ischemia. J. Cell. Mol. Med. 14, 1056–1060.
Goodyer, P., Dehbi, M., Torban, E., Bruening, W., and Pelletier, J. (1995). Repression of the retinoic acid receptor-alpha gene by the Wilms’ tumor suppressor gene product, Wt1. Oncogene 10, 1125–1129.
Gothié, E., Richard, D. E., Berra, E., Pagès, G., and Pouysségur, J. (2000). Identification of alternative spliced variants of human hypoxia-inducible factor-1alpha. J. Biol. Chem. 275, 6922–6927.
Guarino, M. (2007). Epithelial-mesenchymal transition and tumour invasion. Int. J. Biochem. Cell Biol. 39, 2153–2160.
Gustafsson, M. V., Zheng, X., Pereira, T., Gradin, K., Jin, S., Lundkvist, J., Ruas, J. L., Poellinger, L., Lendahl, U., and Bondesson, M. (2005). Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev. Cell 9, 617–628.
Haase, V. H. (2009). Oxygen regulates epithelial-to-mesenchymal transition: insights into molecular mechanisms and relevance to disease. Kidney Int. 76, 492–499.
Haber, D. A., Sohn, R. L., Buckler, A. J., Pelletier, J., Call, K. M., and Housman, D. E. (1991). Alternative splicing and genomic structure of the Wilms tumor gene WT1. Proc. Natl. Acad. Sci. U.S.A. 88, 9618–9622.
Hartkamp, J., Carpenter, B., and Roberts, S. G. E. (2010). The Wilms’ tumor suppressor protein WT1 is processed by the serine protease HtrA2/Omi. Mol. Cell 37, 159–171.
Herzer, U., Crocoll, A., Barton, D., Howells, N., and Englert, C. (1999). The Wilms tumor suppressor gene Wt1 is required for development of the spleen. Curr. Biol. 9, 837–840.
Hockel, M., and Vaupel, P. (2001). Tumor hypoxia: definitions and current clinical, biologic, and molecular aspects. J. Natl. Cancer Inst. 93, 266–276.
Hohenstein, P., and Hastie, N. D. (2006). The many facets of the Wilms’ tumour gene, WT1. Hum. Mol. Genet. 15, R196–R201.
Houck, K. A., Ferrara, N., Winer, J., Cachianes, G., Li, B., and Leung, D. W. (1991). The vascular endothelial growth factor family: identification of a fourth molecular species and characterization of alternative splicing of RNA. Mol. Endocrinol. 5, 1806–1814.
Huber, M. A., Kraut, N., and Beug, H. (2005). Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr. Opin. Cell Biol. 17, 548–558.
Huff, V., Miwa, H., Haber, D. A., Call, K. M., Housman, D., Strong, L. C., and Saunders, G. F. (1991). Evidence for WT1 as a Wilms tumor (WT) gene: intragenic germinal deletion in bilateral WT. Am. J. Hum. Genet. 48, 997–1003.
Hutter, D., Kingdom, J., and Jaeggi, E. (2010). Causes and mechanisms of intrauterine hypoxia and its impact on the fetal cardiovascular system: a review. Int. J. Paediatr. 2010, 401323.
Ito, K., Oji, Y., Tatsumi, N., Shimizu, S., Kanai, Y., Nakazawa, T., Asada, M., Jomgeow, T., Aoyagi, S., Nakano, Y. Tamaki, H., Sakaguchi, N., Shirakata, T., Nishida, S., Kawakami, M., Tsuboi, A., Oka, Y., Tsujimoto, Y., and Sugiyama, H. (2006). Antiapoptotic function of 17AA( + )WT1 (Wilms’ tumor gene) isoforms on the intrinsic apoptosis pathway. Oncogene 25, 4217–4229.
Iyer, N. V., Kotch, L. E., Agani, F., Leung, S. W., Laughner, E., Wenger, R. H., Gassmann, M., Gearhart, J. D., Lawler, A. M., Yu, A. Y., and Semenza, G. L. (1998). Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 12, 149–162.
Jaakkola, P., Mole, D. R., Tian, Y. M., Wilson, M. I., Gielbert, J., Gaskell, S. J., Kriegsheim, A. V., Hebestreit, H. F., Mukherji, M., Schofield, C. J., Maxwell, P. H., Pugh, C. W., and Ratcliffe, P. J. (2001). Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 292, 468–472.
Jin, D. K., Kang, S. J., Kim, S. J., Bang, E. H., Hwang, H. Z., Tadokoro, K., Yamada, M., and Kohsaka, T. (1999). Transcriptional regulation of PDGF-A and TGF-beta by +KTS WT1 deletion mutants and a mutant mimicking Denys-Drash syndrome. Ren. Fail. 21, 685–694.
Johnson, F., and Kaplitt, M. G. (2009). Novel mitochondrial substrates of omi indicate a new regulatory role in neurodegenerative disorders. PLoS ONE 4, e7100. doi: 10.1371/journal.pone.0007100
Johnstone, R. W., See, R. H., Sells, S. F., Wang, J., Muthukkumar, S., Englert, C., Haber, D. A., Licht, J. D., Sugrue, S. P., Roberts, T., Rangnekar, V. M., and Shi, Y. (1996). A novel repressor, par-4, modulates transcription and growth suppression functions of the Wilms’ tumor suppressor WT1. Mol. Cell. Biol. 16, 6945–6956.
Kaidi, A., Williams, A. C., and Paraskeva, C. (2007). Interaction between beta-catenin and HIF-1 promotes cellular adaption to hypoxia. Nat. Cell Biol. 9, 210–217.
Kastner, P., Messaddeq, N., Mark, M., Wendling, O., Grondona, J. M., Ward, S., Ghyselinck, N., and Chambon, P. (1997). Vitamin A deficiency and mutations of RXRalpha, RXRbeta and RARalpha lead to early differentiation of embryonic ventricular cardiomyocytes. Development 124, 4749–4758.
Kennedy, D., Ramsdale, T., Mattick, J., and Little, M. (1996). An RNA recognition motif in Wilms’ tumour protein (WT1) revealed by structural modelling. Nat. Genet. 12, 329–331.
Kent, J., Coriat, A. M., Sharpe, P. T., Hastie, N. D., and van Heyningen, V. (1995). The evolution of WT1 sequence and expression pattern in the vertebrates. Oncogene 11, 1781–1792.
Kim, M. S., Yoon, S. K., Bollig, F., Kitagaki, J., Hur, W., Whye, N. J., Wu, Y. P., Rivera, M. N., Park, J. Y., Kim, H. S., Malik, K., Bell, D. W., Englert, C., Perantoni, A. O., and Lee, S. B. (2010). A novel Wilms tumor 1 (WT1) target gene negatively regulates the WNT signaling pathway. J. Biol. Chem. 285, 14585–14593.
Kirschner, K. M., Hagen, P., Hussels, C. S., Ballmaier, M., Scholz, H., and Dame, C. (2008). The Wilms’ tumor suppressor Wt1 activates transcription of the erythropoietin receptor in hematopoietic progenitor cells. FASEB J. 22, 2690–2701.
Kirschner, K. M., Wagner, N., Wagner, K. D., Wellmann, S., and Scholz, H. (2006). The Wilms’ tumor suppressor Wt1 promotes cell adhesion through transcriptional activation of the alpha4integrin gene. J. Biol. Chem. 281, 31930–31939.
Klein, R., Lamballe, F., Bryant, S., and Barbacid, M. (1992). The TrkB tyrosine protein kinase is a receptor for neurotrophin-4. Neuron 8, 947–956.
Koivunen, P., Hirsilä, M., Günzler, V., Kivirikko, K. I., and Myllyharju, J. (2004). Catalytic properties of the asparaginyl hydroxylase (FIH) in the oxygen sensing pathway are distinct from those of its prolyl 4-hydroxylases. J. Biol. Chem. 279, 9899–9904.
Kösters, R., Linnebacher, M., Coy, J. F., Germann, A., Schwitalle, Y., Findeisen, P., and von Knebel Doeberitz, M. (2004). WT1 is a tumor-associated antigen in colon cancer that can be recognized by in vitro stimulated cytotoxic T cells. Int. J. Cancer 109, 385–392.
Kozak, K. R., Abbott, B., and Hankinson, O. (1997). ARNT-deficient mice and placental differentiation. Dev. Biol. 191, 297–305.
Kreidberg, J. A., Sariola, H., Loring, J. M., Maeda, M., Pelletier, J., Housman, D., and Jaenisch, R. (1993). WT-1 is required for early kidney development. Cell 74, 679–691.
Kwee, L., Baldwin, H. S., Shen, H. M., Stewart, C. L., Buck, C., Buck, C. A., and Labow, M. A. (1995). Defective development of the embryonic and extraembryonic circulatory systems in vascular cell adhesion molecule (VCAM-1) – deficient mice. Development 121, 489–503.
Laity, J. H., Dyson, H. J., and Wright, P. E. (2000). Molecular basis for modulation of biological function by alternate splicing of the Wilms’ tumor suppressor protein. Proc. Natl. Acad. Sci. U.S.A. 97, 11932–11935.
Larsson, S. H., Charlieu, J. P., Miyagawa, K., Engelkamp, D., Rassoulzadegan, M., Ross, A., Cuzin, F., van Heyningen, V., and Hastie, N. D. (1995). Subnuclear localization of WT1 in splicing or transcription factor domains is regulated by alternative splicing. Cell 81, 391–401.
Lester, R. D., Jo, M., Montel, V., Takimoto, S., and Gonias, S. L. (2007). uPAR induces epithelial-mesenchymal transition in hypoxic breast cancer cells. J. Cell. Biol. 178, 425–436.
Levy, N. S., Chung, S., Furneaux, H., and Levy, A. P. (1998). Hypoxic stabilization of vascular endothelial growth factor mRNA by the RNA-binding protein HuR. J. Biol. Chem. 273, 6417–6423.
Liu, Q. B., Liu, L. L., Lu, Y. M., Tao, R. R., Huang, J. Y., Han, F., and Lou, Y. J. (2010). The induction of reactive oxygen species and loss of mitochondrial Omi/HtrA2 is associated with S-nitrosoglutathione-induced apoptosis in human endothelial cells. Toxicol. Appl. Pharmacol. 244, 374–384.
Liu, S., Rose, D. M., Han, J., and Ginsberg, M. H. (2000). Alpha4 integrins in cardiovascular development and diseases. Trends Cardiovasc. Med. 10, 253–257.
Loeb, D. M., Evron, E., Patel, C. B., Sharma, P. M., Niranjan, B., Buluwela, L., Weitzman, S. A., Korz, D., and Sukumar, S. (2001). Wilms’ tumor suppressor gene (WT1) is expressed in primary breast tumors despite tumor-specific promoter methylation. Cancer Res. 61, 921–925.
Malentacchi, F., Simi, L., Nannelli, C., Andreani, M., Janni, A., Pastorekova, S., and Orlando, C. (2009). Alternative splicing variants of carbonic anhydrase IX in human non-small cell lung cancer. Lung Cancer 64, 271–276.
Malik, K. T., Poirier, V., Ivins, S. M., and Brown, K. W. (1994). Autoregulation of the human WT1 gene promoter. FEBS Lett. 349, 75–78.
Männer, J., Pérez-Pomares, J. M., Macias, S. D., and Muñoz-Chápuli, R. (2001). The origin, formation and developmental significance of the epicardium: a review. Cells Tissues Organs 169, 89–103.
Martens, L. K., Kirschner, K. M., Warnecke, C., and Scholz, H. (2007). Hypoxia-inducible factor-1 (HIF-1) is a transcriptional activator of the TrkB neurotrophin receptor gene. J. Biol. Chem. 282, 14379–14388.
Martin, C. B. Jr. (2008). Normal fetal physiology and behaviour, and adaptive responses with hypoxemia. Semin. Perinatol. 32, 239–242.
Martinez-Estrada, O. M., Lettice, L. A., Essafi, A., Guadix, J. A., Slight, J., Velecela, V., Hall, E., Reichmann, J., Devenney, P. S., Hohenstein, P., Hosen, N., Hill, R. E., Muñoz-Chápuli, R., and Hastie, N. D. (2010). Wt1 is required for cardiovascular progenitor cell formation through transcriptional control of Snail and E-cadherin. Nat. Genet. 42, 89–93.
Maynard, M. A., Qi, H., Chung, J., Lee, E. H., Kondo, Y., Hara, S., Conaway, R. C., Conaway, J. W., and Ohh, M. (2003). Multiple splice variants of the human HIF-3 alpha locus are targets of the von Hippel-Lindau E3 ubiquitin ligase complex. J. Biol. Chem. 278, 11032–11040.
Mazumdar, J., Hickey, M. M., Pant, D. K., Durham, A. C., Sweet-Cordero, A., Vachani, A., Jacks, T., Chodosh, L. A., Kissil, J. L., Simon, M. C., and Keith, B. (2010). HIF-2alpha deletion promotes Kras-driven lung tumor development. Proc. Natl. Acad. Sci. U.S.A. 107, 14182–14187.
McGary, E. C., Rondon, I. J., and Beckman, B. S. (1997). Post-transcriptional regulation of erythropoietin mRNA stability by erythropoietin mRNA-binding protein. J. Biol. Chem. 272, 8628–8634.
Menssen, H. D., Renkl, H. J., Rodeck, U., Maurer, J., Notter, M., Schwartz, S., Reinhardt, R., and Thiel, E. (1995). Presence of Wilms’ tumor gene (wt1) transcripts and the WT1 nuclear protein in the majority of human acute leukemias. Leukemia 9, 1060–1067.
Mikawa, T., and Gourdie, R. G. (1996). Pericardial mesoderm generates a population of coronary smooth muscle cells migrating into the heart along with ingrowth of the epicardial organ. Dev. Biol. 174, 221–232.
Miwa, H., Beran, M., and Saunders, G. F. (1992). Expression of the Wilms’ tumor gene (WT1) in human leukemias. Leukemia 6, 405–409.
Miyagi, T., Ahuja, H., Kubota, T., Kubonishi, I., Koeffler, H. P., and Miyoshi, I. (1993). Expression of the candidate Wilms’ tumor gene, WT1, in human leukemia cells. Leukemia 7, 970–977.
Moore, A. W., McInnes, L., Kreidberg, J., Hastie, N. D., and Schedl, A. (1999). YAC complementation shows a requirement for Wt1 in the development of epicardium, adrenal gland and throughout nephrogenesis. Development 126, 1845–1857.
Mutter, W. P., and Karumanchi, S. A. (2008). Molecular mechanisms of preeclampsia, Microvasc. Res. 75, 1–8.
Natoli, T. A., McDonald, A., Alberta, J. A., Taglienti, M. E., Housman, D. E., and Kreidberg, J. A. (2002). A mammal-specific exon of WT1 is not required for development of fertility. Mol. Cell. Biol. 22, 4433–4438.
Neth, P., Ries, C., Karow, M., Egea, V., Ilmer, M., and Jochum, M. (2007). The Wnt signal transduction pathway in stem cells and cancer cells: influence on cellular invasion. Stem Cell Rev. 3, 18–29.
Niksic, M., Slight, J., Sanford, J. R., Caceres, J. F., and Hastie, N. D. (2004). The Wilms’ tumour protein (WT1) shuttles between nucleus and cytoplasm and is present in functional polysomes. Hum. Mol. Genet. 13, 463–471.
Oji, Y., Miyoshi, S., Maeda, H., Hayashi, S., Tamaki, H., Nakatsuka, S., Yao, M., Takahashi, E., Nakano, Y., Hirabayashi, H., Shintani, Y., Oka, Y., Tsuboi, A., Hosen, N., Asada, M., Fujioka, T., Murakami, M., Kanato, K., Motomura, M., Kim, E. H., Kawakami, M., Ikegame, K., Ogawa, H., Aozasa, K., Kawase, I., and Sugiyama, H. (2002). Overexpression of the Wilms’ tumor gene WT1 in de novo lung cancers. Int. J. Cancer 100, 297–303.
Oji, Y., Miyoshi, Y., Koga, S., Nakano, Y., Ando, A., Nakatsuka, S., Ikeba, A., Takahashi, E., Sakaguchi, N., Yokota, A., Hosen, N., Ikegame, K., Kawakami, M., Tsuboi, A., Oka, Y., Ogawa, H., Aozasa, K., Noguchi, S., and Sugiyama, H. (2003). Overexpression of the Wilms’ tumor gene WT1 in primary thyroid cancer. Cancer Sci. 94, 606–611.
Oji, Y., Nakamori, S., Fujikawa, M., Nakatsuka, S., Yokota, A., Tatsumi, N., Abeno, S., Ikeba, A., Takashima, S., Tsujie, M., Yamamoto, H., Sakon, M., Nezu, R., Kawano, K., Nishida, S., Ikegame, K., Kawakami, M., Tsuboi, A., Oka, Y., Yoshikawa K., Aozasa, K., Monden, M., and Sugiyama, H. (2004a). Overexpression of the Wilms’ tumor gene WT1 in pancreatic ductal adenocarcinoma. Cancer Sci. 95, 583–587.
Oji, Y., Suzuki, T., Nakano, Y., Maruno, M., Nakatsuka, S., Jomgeow, T., Abeno, S., Tatsumi, N., Yokota, A., Aoyagi, S., Nakazawa, T., Ito, K., Kanato, K., Shirakata, T., Nishida, S., Hosen, N., Kawakami, M., Tsuboi, A., Oka, Y., Aozasa, K., Yoshimine, T., and Sugiyama, H. (2004b). Overexpression of the Wilms’ tumor gene W T1 in primary astrocytic tumors. Cancer Sci. 95, 822–827.
Olivey, H. E., and Svensson, E. C. (2010). Epicardial-myocardial signaling directing coronary vasculogenesis. Circ. Res. 106, 818–832.
Olivey, H. E., Mundell, N. A., Austin, A. F., and Barnett, J. V. (2006). Transforming growth factor-beta stimulates epithelial-mesenchymal transformation in the proepicardium. Dev. Dyn. 235, 50–509.
Park, S., Bernard, A., Bove, K. E., Sens, D. A., Hazen-Martin, D. J., Garvin, A. J., and Haber, D. A. (1993). Inactivation of WT1 in nephrogenic rests, genetic precursors to Wilms’ tumour. Nat. Genet. 5, 363–367.
Pelletier, J., Bruening, W., Kashtan, C. E., Mauer, S. M., Manivel, J. C., Striegel, J. E., Houghton, D. C., Junien, C., Habib, R., Fouser, L., Fine, R. N., Silverman, B. L., Haber, D. A., and Housman, D. E. (1991a). Germline mutations in the Wilms’ tumor suppressor gene are associated with abnormal urogenital development in Denys-Drash syndrome. Cell 67, 437–447.
Pelletier, J., Bruening, W., Li, F. P., Haber, D. A., Glaser, T., and Housman, D. E. (1991b). WT1 mutations contriubte to abnormal genital system development and hereditary Wilms’ tumour. Nature 353, 431–434.
Peng, J., Zhang, L., Drysdale, L., and Fong, G. H. (2000). The transcription factor EPAS-1/hypoxia-inducible factor 2alpha plays an important role in vascular remodelling. Proc. Natl. Acad. Sci. U.S.A. 97, 8386–8391.
Pérez-Pomares, J. M., Carmona, R., Gonzalez-Iriarte, M., Atencia, G., Wessels, A., and Muñoz-Chápuli, R. (2002a). Origin of coronary endothelial cells from epicardial mesothelium in avian embryos. Int. J. Dev. Biol. 46, 1005–1013.
Pérez-Pomares, J. M., Phelps, A., Sedmerova, M., Carmona, R., Gonzalez-Iriarte, M., Muñoz-Chápuli, R., and Wessels, A. (2002b). Experimental studies on the spatiotemporal expression of WT1 and RALDH2 in the embryonic avian heart: a model for the regulation of myocardial and valvuloseptal development by epicardially derived cells (EPDCs). Dev. Biol. 247, 307–323.
Quaggin, S. E., and Kreidberg, J. A. (2008). Development of the renal glomerulus: good neighbors and good fences. Development 135, 609–620.
Ratajska, A., Czarnowska, E., and Ciszek, B. (2008). Embryonic development of the proepicardium and coronary vessels. Int. J. Dev. Biol. 52, 229–236.
Ream, M., Ray, A. M., Chandra, R., and Chikaraishi, D. M. (2008). Early fetal hypoxia leads to growth restriction and myocardial thinning. Am. J. Physiol. Regul. Integr. Comp. Physiol. 295, R583–R595.
Rivera, M. N., and Haber, D. A. (2005). Wilms’ tumour: connecting tumorigenesis and organ development in the kidney. Nat. Rev. Cancer 5, 699–712.
Rivera, M. N., Kim, W. J., Wells, J., Driscoll, D. R., Brannigan, B. W., Han, M., Kim, J. C., Feinberg, A. P., Gerald, W. L., Vargas, S. O., Chin, L., Iafrate, A. J., Bell, D. W., and Haber, D. A. (2007). An X chromosome gene, WTX, is commonly inactivated in Wilms tumor. Science 315, 642–645.
Roberts, S. G. (2005). Transcriptional regulation by WT1 in development. Curr. Opin. Genet. Dev. 15, 542–547.
Roberts, S. G. (2006). The modulation of WT1 transcription function by cofactors. Biochem. Soc. Symp. 73, 191–201.
Rupprecht, H. D., Drummond, I. A., Madden, S. L., Rauscher, F. J. III, and Sukhatme, V. P. (1994). The Wilms’ tumor suppressor gene WT1 is negatively autoregulated. J. Biol. Chem. 269, 6198–6206.
Ryan, H. E., Lo, J., and Johnson, R. S. (1998). HIF-1 alpha is required for solid tumor formation and embryonic vascularization. EMBO J. 17, 3005–3015.
Sahlgren, C., Gustafsson, M. V., Jin, S., Poellinger, L., and Lendahl, U. (2008). Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc. Natl. Acad. Sci. U.S.A. 105, 6392–6397.
Saito, A., Hayashi, T., Okuno, S., Nishi, T., and Chan, P. H. (2004). Modulation of the Omi/HtrA2 signaling pathway after transient focal cerebral ischemia in mouse brains that overexpress SOD1. Brain Res. Mol. Brain Res. 127, 89–95.
Schäffer, L., Scheid, A., Spielmann, P., Breymann, C., Zimmermann, R., Meuli, M., Gassmann, M., Marti, H. H., and Wenger, R. H. (2003). Oxygen-regulated expression of TGF-beta 3, a growth factor involved in trophoblast differentiation. Placenta 24, 941–950.
Scharnhorst, V., van der Eb, A. J., and Jochemsen, A. G. (2001). WT1 proteins: functions in growth and differentiation. Gene 273, 141–161.
Scheid, A., Wenger, R. H., Schäffer, L., Camenisch, I., Distler, O., Ferenc, A., Cristina, H., Ryan, H. E., Johnson, R. S., Wagner, K. F., Stauffer, U. G., Bauer, C., Gassmann, M., and Meuli, M. (2002). Physiologically low oxygen concentrations in fetal skin regulate hypoxia-inducible factor 1 and transforming growth factor-beta3. FASEB J. 16, 411–413.
Scholz, H., Bossone, S. A., Cohen, H. T., Akella, U., Strauss, W. M., and Sukhatme, V. P. (1997). A far upstream element is required for Wilms tumor-1 (WT1) gene expression in renal cell culture. J. Biol. Chem. 272, 32836–32846.
Scortegagna, M., Morris, M. A., Oktay, Y., Bennett, M., and Garcia, J. A. (2003). The HIF family member EPAS1/HIF-2alpha is required for normal hematopoiesis in mice. Blood 102, 1634–1640.
Semenza, G. L. (2004). Hydroxylation of HIF-1: oxygen sensing at the molecular level. Physiology (Bethesda) 19, 176–182.
Semenza, G. L., Nejfelt, M. K., Chi, S. M., and Antonarakis, S. E. (1991). Hypoxia-inducible nuclear factors bind to an enhancer element located 30 to the human erythropoietin gene. Proc. Natl. Acad. Sci. U.S.A. 88, 5680–5684.
Sengbusch, J. K., He, W., Pinco, K. A., and Yang, J. T. (2002). Dual functions of a4β1 integrin in epicardial development: initial migration and long-term attachment. J. Cell Biol. 157, 873–882.
Sridurongrit, S., Larsson, J., Schwartz, R., Ruiz-Lozano, P., and Kaartinen, V. (2008). Signaling via the Tgf-beta type I receptor Alk5 in heart development. Dev. Biol. 322, 208–218.
Stuckmann, I., Evans, S., and Lassar, A. B. (2003). Erythropoietin and retinoic acid, secreted from the epicardium, are required for cardiac myocyte proliferation. Dev. Biol. 255, 334–349.
Sucov, H. M., Dyson, E., Gumeringer, C. L., Price, J., Chien, K. R., and Evans, R. M. (1994). RXR alpha mutant mice establish a genetic basis for vitamin A signaling in heart morphogenesis. Genes Dev. 8, 1007–1018.
Sugishita, Y., Watanabe, M., and Fisher, S. A. (2004). Role of myocardial hypoxia in the remodeling of the embryonic avian cardiac outflow tract. Dev. Biol. 267, 294–308.
Tamaki, H., Ogawa, H., Ohyashiki, K., Ohyashiki, J. H., Iwama, H., Inoue, K., Soma, T., Oka, Y., Tatekawa, T., Oji, Y., Tsuboi, A., Kim, E. H., Kawakami, M., Fuchigami, K., Tomonaga, M., Toyama, K., Aozasa, K., Kishimoto, T., and Sugiyama, H. (1999). The Wilms’ tumor gene WT1 is a good marker for diagnosis of disease progression of myelodysplastic syndromes. Leukemia 13, 393–399.
Tevosian, S. G., Deconinck, A. E., Tanaka, M., Schinke, M., Litovsky, S. H., Izumo, S., Fujiwara, Y., and Orkin, S. H. (2000). FOG-2, a cofactor for GATA transcription factors, is essential for heart morphogenesis and development of coronary vessels from epicardium. Cell 101, 729–739.
Thein, S. L., Menzel, S., Lathrop, M., and Garner, C. (2009). Control of fetal haemoglobin: new insights emerging from genomics and clinical implications. Hum. Mol. Genet. 18, R216–R223.
Thiery, J. P., and Sleeman, J. P. (2006). Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 7, 131–142.
Timár, J., Mészáros, L., Orosz, Z., Albini, A., and Rásó, E. (2005). WT1 expression in angiogenic tumours of the skin. Histopathology 47, 67–73.
Timmerman, L. A., Grego-Bessa, J., Raya, A., Bertrán, E., Pérez-Pomares, J. M., Díez, J., Aranda, S., Palomo, S., McCormick, F., Izpisúa-Belmonte, J. C., and de la Pompa, J. L. (2004). Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes Dev. 18, 99–115.
Tischer, E., Mitchell, R., Hartman, T., Silva, M., Gospodarowicz, D., Fiddes, J. C., and Abraham, J. A. (1991). The human gene for vascular endothelial growth factor. Multiple protein forms are encoded through alternative exon splicing. J. Biol. Chem. 1266, 11947–11954.
Vande Walle, L., Van Damme, P., Lamkanfi, M., Saelens, X., Vandekerckhove, J., Gevaert, K., and Vandenabeele, P. (2007). Proteome-wide identification of HtrA2/Omi substrates. J. Proteome Res. 6, 1006–1015.
Vicent, S., Chen, R., Sayles, L. C., Lin, C., Walker, R. G., Gillespie, A. K., Subramanian, A., Hinkle, G., Yang, X., Saif, S., Root, D. E., Huff, V., Hahn, W. C., and Sweet-Cordero, E. A. (2010). Wilms tumor 1 (WT1) regulates KRAS-driven oncogenesis and senescence in mouse and human models. J. Clin. Invest. 120, 3940–3952.
Viney, R. L., Morrison, A. A., van den Heuvel, L. P., Ni, L., Mathieson, P. W., Saleem, M. A., and Ladomery, M. R. (2007). A proteomic investigation of glomerular podocytes from a Denys-Drash syndrome patient with a mutation in the Wilms tumour suppressor gene WT1. Proteomics 7, 804–815.
Wagner, K. D., Wagner, N., Bondke, A., Nafz, B., Flemming, B., Theres, H., and Scholz, H. (2002a). The Wilms’ tumor suppressor Wt1 is expressed in the coronary vasculature after myocardial infarction. FASEB J. 16, 1117–1119.
Wagner, K. D, Wagner, N., Vidal, V. P., Schley, G., Wilhelm, D., Schedl, A., Englert, C., and Scholz, H. (2002b). The Wilms’ tumor gene Wt1 is required for normal development of the retina. EMBO J. 21, 1398–1405.
Wagner, K. D., Wagner, N., Wellmann, S., Schley, G., Bondke, A., Theres, H., and Scholz, H. (2003). Oxygen-regulated expression of the Wilms’ tumor suppressor Wt1 involves hypoxia-inducible factor-1 (HIF-1). FASEB J. 17, 1364–1366.
Wagner, N., Michiels, J. F., Schedl, A., and Wagner, K. D. (2008). The Wilms’ tumour suppressor WT1 is involved in endothelial cell proliferation and migration: expression in tumour vessels in vivo. Oncogene 27, 3662–3672.
Wagner, N., Wagner, K. D., Hammes, A., Kirschner, K. M., Vidal, V. P., Schedl, A., and Scholz, H. (2005a). A splice variant of the Wilms’ tumour suppressor Wt1 is required for normal development of the olfactory system. Development 132, 1327–1336.
Wagner, N., Wagner, K. D., Theres, H., Englert, C., Schedl, A., and Scholz, H. (2005b). Coronary vessel development requires activation of the TrkB neurotrophin receptor by the Wilms’ tumor transcription factor Wt1. Genes Dev. 19, 2631–2642.
Webb, J. D., Coleman, M. L., and Pugh, C. W. (2009). Hypoxia, hypoxia-inducible factors (HIF), HIF hydroxylases and oxygen sensing. Cell. Mol. Life Sci. 66, 3539–3554.
Webby, C. J., Wolf, A., Gromak, N., Dreger, M., Kramer, H., Kessler, B., Nielsen, M. L., Schmitz, C., Butler, D. S., Yates, J. R. III, Delahunty, C. M., Hahn, P., Lengeling, A., Mann, M., Proudfoot, N. J., Schofield, C. J., and Böttger, A. (2009). Jmjd6 catalyses lysyl-hydroxylation of U2AF65, a protein associated with RNA splicing. Science 325, 90–93.
Webster, W. S., and Abela, D. A. (2007). The effect of hypoxia in development. Birth Defects Res. C Embryo Today 81, 215–228.
Wessels, A., and Perez-Pomares, J. M. (2004). The epicardium and epicardially derived cells (EPDCs) as cardiac stem cells. Anat. Rec. A Discov. Mol. Cell. Evol. Biol. 276, 43–57.
Wikenheiser, J., Doughman, Y. Q., Fisher, S. A., and Watanabe, M. (2006). Differential levels of tissue hypoxia in the developing chicken heart. Dev. Dyn. 235, 115–123.
Wikenheiser, J., Wolfram, J. A., Gargesha, M., Yang, K., Karunamuni, G., Wilson, D. L., Semenza, G. L., Agani, F., Fisher, S. A., Ward, N., and Watanabe, M. (2009). Altered hypoxia-inducible factor-1 alpha expression levels correlate with coronary vessel anomalies. Dev. Dyn. 238, 2688–2700.
Wilm, B., Ipenberg, A., Hastie, N. D., Burch, J. B., and Bader, D. M. (2005). The serosal mesothelium is a major source of smooth muscle cells of the gut vasculature. Development 132, 5317–5328.
Wilting, J., Buttler, K., Schulte, I., Papoutsi, M., Schweigerer, L., and Männer, J. (2007). The proepicardium delivers hemangioblasts but not lymphangioblasts to the developing heart. Dev. Biol. 305, 451–459.
Wu, H., Lee, S. H., Gao, J., Liu, X., and Iruela-Arispe, M. L. (1999). Inactivation of erythropoietin leads to defects in cardiac morphogenesis. Development 126, 3597–3605.
Wu, Y., and Zhou, B. P. (2008). New insights of epithelial-mesenchymal transition in cancer metastasis. Acta Biochim. Biophys. Sin. 40, 643–650.
Yamagami, T., Sugiyama, H., Inoue, K., Ogawa, H., Tatekawa, T., Hirata, M., Kudoh, T., Akiyama, T., Murakami, A., and Maekawa, T. (1996). Growth inhibition of human leukemic cells by WT1 (Wilms tumor gene) antisense oligodeoxynucleotides: implications for the involvement of WT1 in leukemogenesis. Blood 87, 2878–2884.
Yang, J. T., Rayburn, H., and Hynes, R. O. (1995). Cell adhesion events mediated by alpha 4 integrins are essential in placental and cardiac development. Development 121, 549–560.
Yang, L., Han, Y., Suarez Saiz, F., and Minden, M. D. (2007). A tumor suppressor and oncogene: the WT1 story. Leukemia 21, 868–876.
Zavadil, J., and Bottinger, E. P. (2005). TGF-beta and epithelial-to-mesenchymal transitions. Oncogene 24, 5764–5774.
Zavadil, J., Cermak, L., Soto-Nieves, N., and Bottinger, E. P. (2004). Integration of TGF-beta/Smad and Jagged1/Notch signalling in epithelial-to-mesenchymal transition. EMBO J. 23, 1155–1165.
Zeng, M., Kikuchi, H., Pino, M. S., and Chung, D. C. (2010). Hypoxia activates the K-ras proto-oncogene to stimulate angiogenesis and inhibit apoptosis in colon cancer cells. PLoS ONE 5, e10966. doi: 10.1371/journal.pone.0010966
Zheng, X., Linke, S., Dias, J. M., Zheng, X., Gradin, K., Wallis, T. P., Hamilton, B. R., Gustafsson, M., Ruas, J. L., Wilkins, S., Bilton, R. L., Brismar, K., Whitelaw, M. L., Pereira, T., Gorman, J. J., Ericson, J., Peet, D. J., Lendahl, U., and Poellinger, L. (2008). Interaction with factor inhibiting HIF-1 defines an additional mode of cross-coupling between the Notch and hypoxia signaling pathways. Proc. Natl. Acad. Sci. U.S.A. 105, 3368–3373.
Keywords: hypoxia, Wilms’ tumor gene, epithelial-to-mesenchymal transition, angiogenesis, epicardium, transcription factor, oncogene, HIF
Citation: Scholz H and Kirschner KM (2011) Oxygen-dependent gene expression in development and cancer: lessons learned from the Wilms’ tumor gene, WT1. Front. Mol. Neurosci. 4:4. doi: 10.3389/fnmol.2011.00004
Received: 03 February 2011;
Paper pending published: 04 February 2011;
Accepted: 11 February 2011;
Published online: 24 February 2011
Edited by:
Michael Fähling, Charite – University Medicine Berlin, GermanyReviewed by:
Michael Fähling, Charite – University Medicine Berlin, GermanyMichael Ladomery, University of the West of England Bristol, UK
Copyright: © 2011 Scholz and Kirschner. This is an open-access article subject to an exclusive license agreement between the authors and Frontiers Media SA, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Holger Scholz, Institut für Vegetative Physiologie, Charité – Universitätsmedizin Berlin, Hessische Straße 3-4, 10115 Berlin, Germany. e-mail:aG9sZ2VyLnNjaG9sekBjaGFyaXRlLmRl