 Silvia Murillo-Cuesta1,2,3
Silvia Murillo-Cuesta1,2,3 Lourdes Rodríguez-de la Rosa1,3
Lourdes Rodríguez-de la Rosa1,3 Rafael Cediel4,5
Rafael Cediel4,5 Luis Lassaletta6,7
Luis Lassaletta6,7 Isabel Varela-Nieto3,5,7*
Isabel Varela-Nieto3,5,7*- 1 Servicio de Evaluación Neurofuncional no Invasiva, Instituto de Investigaciones Biomédicas Alberto Sols, Consejo Superior de Investigaciones Científicas-Universidad Autónoma de Madrid, Madrid, Spain
- 2 Servicio de Fenotipado de Animal de Laboratorio en Red, Centro de Investigación Biomédica en Red de Enfermedades Raras, Valencia, Spain
- 3 U761, Centro de Investigación Biomédica en Red de Enfermedades Raras, Valencia, Spain
- 4 Facultad de Veterinaria, Universidad Complutense, Madrid, Spain
- 5 Grupo de Neurobiología de la Audición, Instituto de Investigaciones Biomédicas Alberto Sols, Consejo Superior de Investigaciones Científicas-Universidad Autónoma de Madrid, Madrid, Spain
- 6 Servicio de Otorrinolaringología, Hospital La Paz, Madrid, Spain
- 7 Instituto de Investigación Sanitaria Hospital Universitario La Paz, Madrid, Spain
Insulin-like growth factor-I (IGF-I) belongs to the family of polypeptides of insulin, which play a central role in embryonic development and adult nervous system homeostasis by endocrine, autocrine, and paracrine mechanisms. IGF-I is fundamental for the regulation of cochlear development, growth, and differentiation, and its mutations are associated with hearing loss in mice and men. Low levels of IGF-I have been shown to correlate with different human syndromes showing hearing loss and with presbyacusis. Animal models are fundamental to understand the genetic, epigenetic, and environmental factors that contribute to human hearing loss. In the mouse, IGF-I serum levels decrease with aging and there is a concomitant hearing loss and retinal degeneration. In the Igf1−/− null mouse, hearing loss is due to neuronal loss, poor innervation of the sensory hair cells, and age-related stria vascularis alterations. In the inner ear, IGF-I actions are mediated by intracellular signaling networks, RAF, AKT, and p38 MAPK protein kinases modulate the expression and activity of transcription factors, as AP1, MEF2, FoxM1, and FoxP3, leading to the regulation of cell cycle and metabolism. Therapy with rhIGF-I has been approved in humans for the treatment of poor linear growth and certain neurodegenerative diseases. This review will discuss these findings and their implications in new IGF-I-based treatments for the protection or repair of hearing loss.
IGF System and Signaling Pathways
Early studies have provided evidences that the insulin-like growth factor (IGF) system exerts influence on almost every organ system in the body, playing an important function in growth, development, and metabolism. The mammalian IGF system is a complex family of proteins that includes three factors (insulin, IGF-I, and IGF-II) and their cell surface receptors, which are the insulin receptor (IR), the IGF receptors 1 and 2 (IGF1R, IGF2R), and hybrid IGF1R/IR receptors. Six high-affinity binding proteins (IGFBP1–6) and their IGFBP proteases modulate the interactions between factors and receptors (Figure 1).
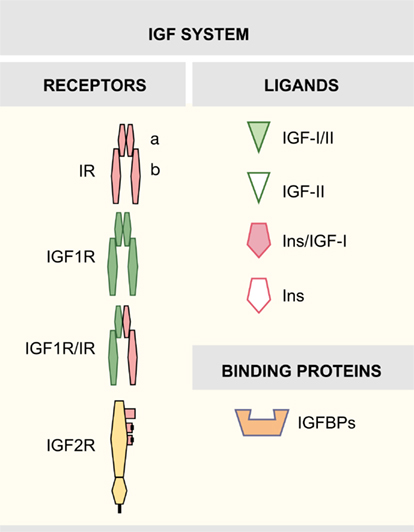
Figure 1. Insulin-like growth factor system.
Insulin-like growth factor-I is a single-chain 70 amino acid peptide, mainly secreted by the liver (Foulstone et al., 2005). At the surface of target cells, the biological actions of the IGFs are mediated by receptors responsible for the transmission to a highly regulated intracellular signaling network. IGF1R, a transmembrane tyrosine kinase receptor, shares functional and structural homology with the IR, and can bind IGF-I and insulin, the latter only with a 100-fold lower affinity than IGF-I (Annunziata et al., 2010). The IGF1R is a tetramer with an intracellular protein tyrosine kinase domain (Adams et al., 2000; De Meyts and Whittaker, 2002). The close homology between IGF1R and IR allows the formation of functional hybrid receptors (Pandini et al., 2002; Torres-Aleman, 2010). The biological role of hybrid receptors has not been extensively tested and needs further investigation (Belfiore et al., 2009; Beauchamp et al., 2010). Finally, IGF2R, the cation-independent mannose-6-phosphate receptor, is a transmembrane glycoprotein without tyrosine kinase activity (Morgan et al., 1987). The IGF2R binds IGF-II but is unable to bind IGF-I (Brown et al., 2009). IGF system actions are regulated by a family of six high-affinity secreted binding proteins (IGFBPs1–6) that act modulating the half-life and distribution of the IGFs and competing with receptors for free ligands (Federici et al., 1997; Firth and Baxter, 2002). The IGFBP family also includes a group of low-affinity IGFBP-related proteins (Jiang et al., 2008).
Insulin-like growth factor signaling occurs via a complex intracellular network of molecules, where the cascade(s) triggered depend on the cellular spatiotemporal context and is determined by the ligands and receptors involved (Figure 2). It was already known that signaling mediated by these tyrosine kinase receptors regulates pancreatic β cell function, but it has been recently concluded that IGF1R and IR signaling modulate insulin secretion and cell proliferation differently through their actions on IRS1 or IRS2, respectively (Xuan et al., 2010). IGF-I binding to IGF1R results in its autophosphorylation and activation of docking sites for receptor substrates (Shc/IRS; Laviola et al., 2008). Activation of IGF1R triggers two main downstream pathways: the PI3K–Akt and the RAF/ERK cascade. Both have been shown to play an important role in the transmission of signals from cell membrane receptors to the nucleus through interactions with other proteins (Mccubrey et al., 2011). On the first pathway, one of the critical targets is the serine/threonine kinase Akt that regulates protein synthesis and antiapoptotic effects of IGF1R (Laviola et al., 2008). In turn, activation of the RAF/ERK cascade participates in cell growth, proliferation, and differentiation (Downward, 2003; Mebratu and Tesfaigzi, 2009). Other well-documented kinases modulated in response to IGF-I are SAPK (stress-activated protein kinase) and the p38 MAP kinase activated by different stress stimuli (Cuenda and Nebreda, 2009; Vardatsikos et al., 2009).
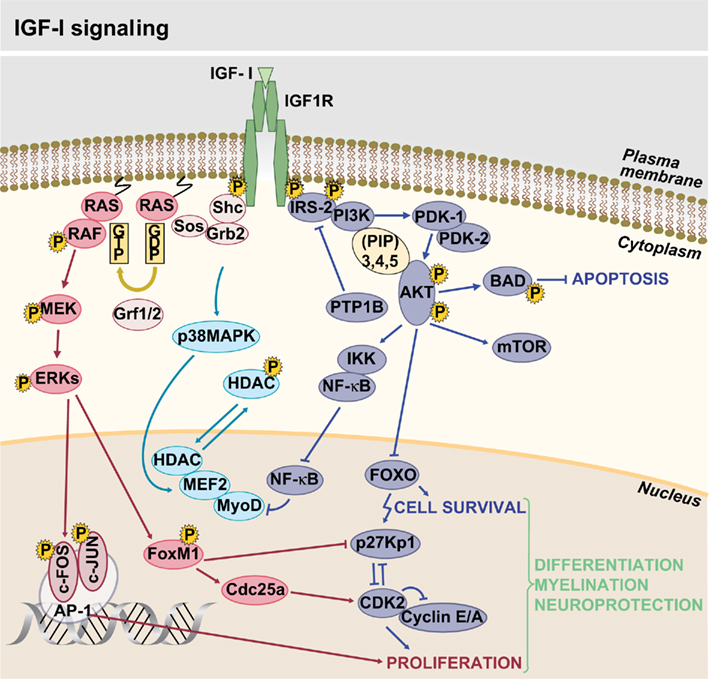
Figure 2. Insulin-like growth factor-I signaling.
Mutations of the Genes of the IGF System and Human Deafness
Main molecular defects in the IGF system include mutations of the IGF1 gene and mutations of the IGF-I high-affinity receptor gene (IGF1R).
Mutations of the IGF1 Gene
Homozygote mutations of the IGF1 gene are extremely rare and only four cases have been reported (Woods et al., 1996; Bonapace et al., 2003; Walenkamp et al., 2005; Netchine et al., 2009). These patients have in common the features of severe intrauterine growth retardation, and some degree of microcephaly. However, sensorineural deafness is not constant and the intensity of delayed psychomotor development is variable. Severe or profound hearing loss has been described in three of the four cases. Table 1 shows the main features of homozygous mutations of the IGF1 gene. In the fourth case reported, an extensive hearing test performed at 9 years of age was normal. This child also showed the mildest phenotype since the mutation allows IGF-I synthesis although with reduced affinity for its receptor (Netchine et al., 2009).
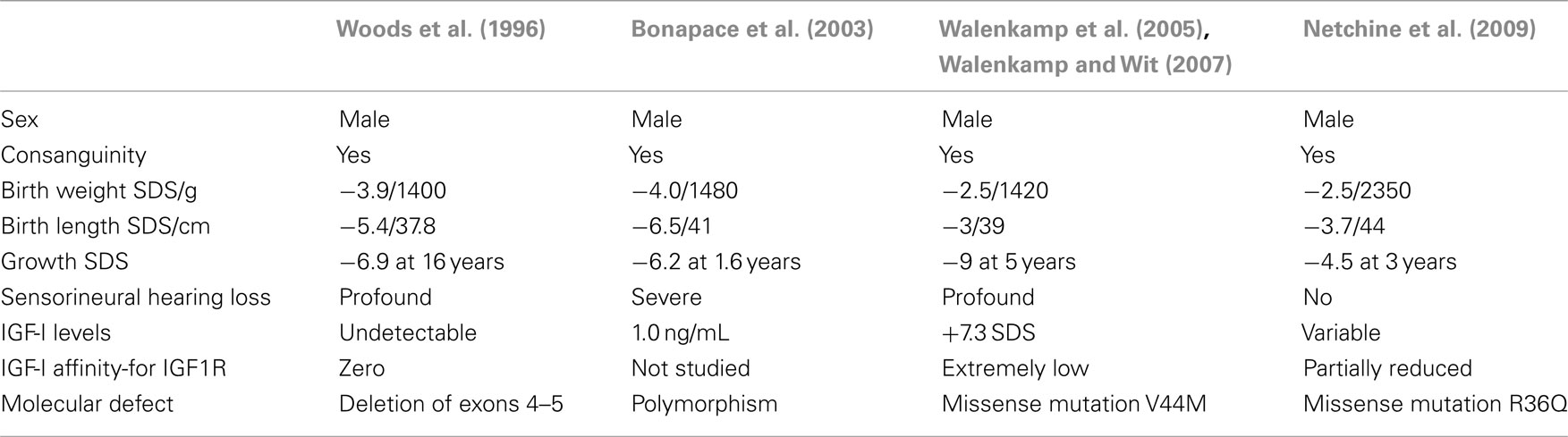
Table 1. Reported cases with homozygous mutations of the IGF1 gene.
Accordingly, heterozygosis for IGF1 mutations is associated with a modest decrease of height, but hearing loss has not been reported in these patients. A case of partial IGF-I deficiency has recently been described and was associated with pre and postnatal growth retardation and microcephaly, but the developmental delay was mild and hearing tests were normal (Van Duyvenvoorde et al., 2010). Sensorineural hearing loss is associated with poor growth rates in infancy and adolescence (Welch and Dawes, 2007), adult short stature (Barrenas et al., 2005), and Turner’s syndrome (Barrenas et al., 2000).
Mutations of the IGF-I Receptor Gene (IGF1R)
IGF1R mutations are characterized by IGF-I resistance causing impaired fetal and postnatal growth. Ester et al. (2009) reported hearing problems in two cases of IGF1R mutations. No data about audiograms or electrophysiological tests were provided. The fact that craniofacial anomalies were present, and tympanostomy tubes were inserted in both cases suggests that hearing loss could be conductive, and the link to IGF-I resistance remains unproved. Other authors reporting human IGF1R mutations did not find marked hearing loss (Klammt et al., 2011). The fact that no profound sensorineural deafness has been reported in patients with heterozygous IGF1 or IGF1R defects suggest that partial IGF-I signaling is sufficient for normal development of the inner ear.
Nervous System Alterations Associated with IGF1 Mutations in Mice
Studies of mutant mice with altered IGF-I expression show that this factor exerts a variety of actions that take place in the nervous system during neural cell development as well as in adulthood, and highlights the fact that IGF-I contributes to neurogenesis by promoting proliferation, differentiation, and survival of neural cells (D’ercole et al., 2002; Liu et al., 2009).
The Igf1−/− null mice present reduced brain size and altered brain regions, where the hippocampus, cerebellum, and cerebral cortex are the most affected, along with alteration of myelination processes (Ye et al., 2002a,b). Postnatal in vivo imaging by RMN has shown that the olfactory bulb is reduced and disorganized, in addition to reduced brain size and other brain and cranium malformations (Riquelme et al., 2010). The number of myelinated fibbers is normal, but whole fibber size is reduced (Gao et al., 1999). In mice with absent or reduced IGF-I levels, motor, and sensory conduction velocities are significantly decreased (Sullivan et al., 2008). These mice also present a decrease in dendrite length and complexity in the cortex, together with a smaller pyramidal neuron soma size (Cheng et al., 2003). Previous reports showed that the IGF-I knockout has a lower number of neurons and oligodendrocytes in the olfactory bulb, dentate gyrus, and striatum (Liu et al., 1993; Beck et al., 1995; Cheng et al., 1998) along with a decrease in the cochlear ganglion neurons (Camarero et al., 2001). This appears to be the result of various processes such as decreased cell proliferation, delayed development, and increased cell death (Beck et al., 1995; Camarero et al., 2001; Ye et al., 2002b; Ye and D’ercole, 2006; Sanchez-Calderon et al., 2010). Studies with Igf1−/− mice during embryonic and adult neurogenesis suggest possible new functions of IGF-I as the regulation of neuronal migration and disposition in the olfactory bulb, the neuroblast output from the subventricular zone and incorporation into the rostral migratory stream, as well as the entry of new neurons from subventricular zone to the olfactory bulb (Hurtado-Chong et al., 2009). A conditional mouse mutant (ΔIgf1r), with targeted manipulation of Igf1r gene by CaMKIIα-Cre recombination in the central nervous system, in which the oligodendrocyte progenitors do not accumulate, proliferate, or survive, has been the preferred model to study the role of IGF-I signaling in remyelinating lesions (Mason et al., 2003).
Otherwise the transgenic mouse with IGF-I overexpression in the brain presents increased brain size and cell numbers in all neural lineages during embryonic and early postnatal development, whereas hippocampus and cerebellum sizes are increased postnatally. Previous studies have shown a proportional increase in the number of neurons, astrocytes, and oligodendrocytes in the dentate gyra of mice expressing an IGF-I transgene specifically in astrocytes, indicating that the development of these cell lineages was similarly affected by the IGF-I transgene (Ye and D’ercole, 2006). The potential role of IGF-I during embryonic and early postnatal brain development was investigated in a transgenic mice (IGF-I nestin Tg mice) with IGF-I overexpression driven from early in embryogenesis by regulatory sequences from the nestin gene. In this case, the neuron progenitor proliferation in the cerebral cortical ventricular and subventricular zones is performed by shortening the length of the cell cycle G1 phase and an accumulation of progenitors in the mitotic cell cycle. IGF-I promotes growth of the central nervous system in these stages of development by mechanisms that involve both neural progenitor proliferation and reduced apoptosis during the postnatal phase (Hodge et al., 2004; Popken et al., 2004).
On the other hand, single and double mutant mice for IGF-I and leukemia inhibitory factor (LIF) have been used to analyze the neural embryonic phenotype, and the results suggest that both factors decrease motoneuron number in specific brain stem nuclei (Vicario-Abejon et al., 2004). Finally, the comparison of the brain phenotype of Igf1 mutants carrying a deletion either in the liver gene or in the forebrain neurons have lead to the conclusion that circulating IGF-I plays an important role in brain function (reviewed in Torres-Aleman, 2010).
In conclusion, transgenic mouse models are valuable tools for better understanding of IGF-I mechanisms in the nervous system, albeit further investigation is needed to understand the organ-specific actions and to explore possible therapies.
Sensory systems are also affected by IGF-I deficit, particular attention has been devoted to the study of hearing because of the human phenotype (Figure 3).
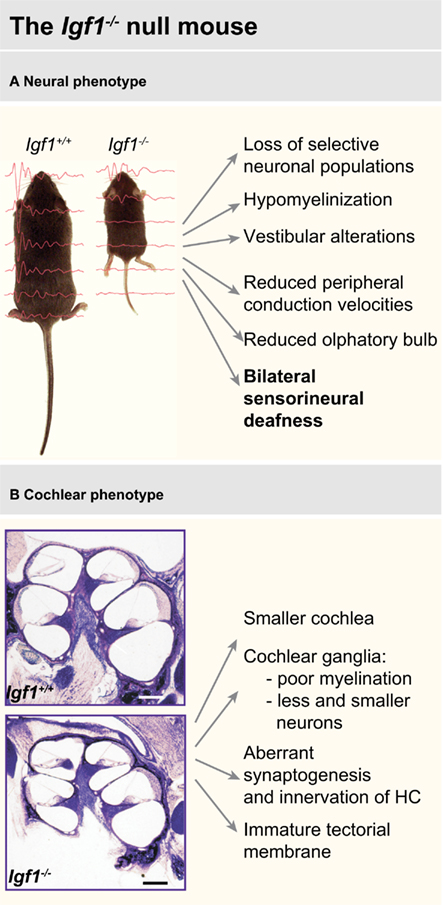
Figure 3. Main features of the neural (A) and cochlear (B) phenotype of the Igf1−/− null mouse. Data have been compiled from Liu et al. (1993), Beck et al. (1995), Cheng et al. (1998), Gao et al. (1999), Camarero et al. (2001, 2002), Ye et al. (2002a,b), Cediel et al. (2006), Sullivan et al. (2008), Riquelme et al. (2010).
Animal Models for the Study of IGF-I Actions on the Inner Ear
The mammalian inner ear develops from an ectodermal patch, the otic placode, which invaginates to form the otic cup that subsequently closes and forms the otic vesicle. This autonomous structure contains the information required to generate the adult inner ear (Bissonnette and Fekete, 1996; Sanchez-Calderon et al., 2007). Organogenesis involves a dynamic balance of cell proliferation, differentiation, survival, and death, biological processes that are tightly regulated by a network of extrinsic and intrinsic factors (Sanchez-Calderon et al., 2007; Magarinos et al., 2010). Among them, the IGF-I plays a key role in promoting proliferation and survival of otic progenitor cells and in supporting neurogenesis and late differentiation (Frago et al., 2003; Varela-Nieto et al., 2007). During cochlear development, this factor and its specific receptor IGF1R are expressed in abundance in several species (Groigno et al., 1999; Varela-Nieto et al., 2003; Sanchez-Calderon et al., 2007, 2010; Gross et al., 2008); however the transcription of both ligand and receptor declines significantly after birth (Baker et al., 1993; Stratikopoulos et al., 2008; Riquelme et al., 2010; Sanchez-Calderon et al., 2010). In a variety of neural cell types, IGF-I is able to induce growth and DNA synthesis, i.e., in the chicken otic precursors (Leon et al., 1995; Rodriguez-De La Rosa et al., submitted), to short the length of the cell cycle, to increase the number of progenitors and to prevent apoptosis (Varela-Nieto et al., 2003; Ye and D’ercole, 2006). Thus IGF-I is essential for the proliferation, survival, and early differentiation of inner ear neuronal precursors (Varela-Nieto et al., 2004, 2007).
The lack of IGF-I causes significant defects in the inner ear in different species, including zebra fish (Schlueter et al., 2007), chicken (Camarero et al., 2003), rat (Zheng et al., 1997), mice (Camarero et al., 2001, 2002; Cediel et al., 2006), and men (Walenkamp and Wit, 2007). Currently, there are several animal models available with mutations in different IGF-I signaling pathway components, which could be used to study the effects of the deficit in inner ear development. Regarding to mice, there are genetically modified strains with mutations in the factors (IGF-I and II), the receptor (IGF1R, IGF2R, and insulin receptor), and the IGF binding proteins (IGFBP1–5 and IGFBP7). Detailed information about the category, allelic composition and phenotype of the mutant lines can be obtained at the Mouse Genome Database at the Mouse Genome Informatics website (http://www.informatics.jax.org). Most of the IGF system mutant mice present a phenotype that includes some of these features: increased mortality, premature aging, growth deficits small size, infertility, endocrine changes, muscle–skeletal abnormalities, and neural alterations. Hearing impairment has been described only in the mutant mouse with a targeted (knockout) mutation in the Igf1 gene (Igf1tm1Arge/Igf1tm1Arge; Liu et al., 1993). It was generated by Argiris Efstratiadis’ group by replacement of sequences encoding part of exon 4 with a neomycin resistance gene. The homozygous mutant mouse maintained in a hybrid background (129S/SvEv*MF1) presents delayed inner ear development, abnormal cochlear, and otic capsule morphology, degeneration of the cochlear ganglion, aberrant innervation patterns, increased neural apoptosis, and deficits in myelination (Camarero et al., 2001, 2002; Sanchez-Calderon et al., 2010). As a consequence, Igf1−/− null mice develop a profound syndromic sensorineural hearing loss with increased hearing threshold assessed by auditory brainstem response recording. Furthermore, as discussed above for the nervous system, the cochlea and the auditory central pathway present poor myelinization and this is reflected in the delayed latencies of the auditory brainstem responses (Cediel et al., 2006; Riquelme et al., 2010). IGF-I deficiency induces caspase-dependent neuronal apoptosis in the brain and also in the cochlea (Sanchez-Calderon et al., 2007). However, the impact of IGF-I deficit on the neuronal populations of the central nuclei of the auditory pathway have not been yet studied.
In addition to deafness, Igf1−/− null mice also show other sensorial deficits including olfactory and visual function (Vicario-Abejon et al., 2003; Otaegi et al., 2006; Scolnick et al., 2008; Hurtado-Chong et al., 2009; Riquelme et al., 2010; Rodriguez-De La Rosa et al., submitted). Accordingly, the transcriptome of the Igf1−/− cochlea when compared to that of the wild type mouse showed expression changes in multiple genes associated with hearing and seeing (Table A1 in Appendix). It is remarkable that genes as harmonin, Rp1h, and tubby, whose mutations have been associated to retinitis pigmentosa and to the human Usher syndrome are IGF-I targets in the mouse cochlea (Sanchez-Calderon et al., 2010). Unfortunately, there are no data available on the hearing phenotype of other IGF system mouse mutants.
Perspectives of IGF-I Therapy in the Treatment of Deafness
In recent years, advances in the fields of human genetics and the sequencing of the human and other species genomes had prompted a step forward in the knowledge of hearing physiopathology (Dror and Avraham, 2009). In parallel, the use of animal models of deafness (Lewis and Steel, 2010) and the developments in the fields of nanotechnology (Danti et al., 2009) and stem cells (Rivolta, 2010) have opened new possibilities of therapeutic intervention. Finally, advances in microsurgery and cochlear implants technology have facilitated research on local drug delivery to the cochlea for hearing loss treatment (Rivera et al., submitted). Local recombinant human IGF-I application has been reported to be useful for the treatment of noise induced hearing loss in animals (Iwai et al., 2006; Lee et al., 2007). Recently, Nakagawa et al. (2010) have demonstrated that topical IGF-I application using gelatine hydrogels is well tolerated and may be efficacious for hearing recovery in patients with sudden sensorineural hearing loss that is resistant to systemic glucocorticoids. These recent data open new perspectives for IGF-I in the treatment of deafness. Still, further studies are required to confirm the therapeutic potential of IGF-I in hearing loss and the prognostic value of plasmatic IGF-I levels.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Professor M. O. Savage for sharing unpublished data on human IGF-I defects. We warmly appreciate the help of Javier Pérez with the illustrations. This work was supported by the “Ministerio de Ciencia e Innovacion” (SAF2008-00470) and the “Fundacion Mutua Madrileña” to Isabel Varela-Nieto.
References
Adams, T. E., Epa, V. C., Garrett, T. P., and Ward, C. W. (2000). Structure and function of the type 1 insulin-like growth factor receptor. Cell. Mol. Life Sci. 57, 1050–1093.
Baker, J., Liu, J. P., Robertson, E. J., and Efstratiadis, A. (1993). Role of insulin-like growth factors in embryonic and postnatal growth. Cell 75, 73–82.
Barrenas, M., Landin-Wilhelmsen, K., and Hanson, C. (2000). Ear and hearing in relation to genotype and growth in Turner syndrome. Hear. Res. 144, 21–28.
Barrenas, M. L., Bratthall, A., and Dahlgren, J. (2005). The association between short stature and sensorineural hearing loss. Hear. Res. 205, 123–130.
Beauchamp, M. C., Yasmeen, A., Knafo, A., and Gotlieb, W. H. (2010). Targeting insulin and insulin-like growth factor pathways in epithelial ovarian cancer. J. Oncol. 2010, 257058.
Beck, K. D., Powell-Braxton, L., Widmer, H. R., Valverde, J., and Hefti, F. (1995). Igf1 gene disruption results in reduced brain size, CNS hypomyelination, and loss of hippocampal granule and striatal parvalbumin-containing neurons. Neuron 14, 717–730.
Belfiore, A., Frasca, F., Pandini, G., Sciacca, L., and Vigneri, R. (2009). Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr. Rev. 30, 586–623.
Bissonnette, J. P., and Fekete, D. M. (1996). Standard atlas of the gross anatomy of the developing inner ear of the chicken. J. Comp. Neurol. 368, 620–630.
Bonapace, G., Concolino, D., Formicola, S., and Strisciuglio, P. (2003). A novel mutation in a patient with insulin-like growth factor 1 (IGF1) deficiency. J. Med. Genet. 40, 913–917.
Brown, J., Jones, E. Y., and Forbes, B. E. (2009). Keeping IGF-II under control: lessons from the IGF-II-IGF2R crystal structure. Trends Biochem. Sci. 34, 612–619.
Camarero, G., Avendano, C., Fernandez-Moreno, C., Villar, A., Contreras, J., De Pablo, F., Pichel, J. G., and Varela-Nieto, I. (2001). Delayed inner ear maturation and neuronal loss in postnatal Igf-1-deficient mice. J. Neurosci. 21, 7630–7641.
Camarero, G., Leon, Y., Gorospe, I., De Pablo, F., Alsina, B., Giraldez, F., and Varela-Nieto, I. (2003). Insulin-like growth factor 1 is required for survival of transit-amplifying neuroblasts and differentiation of otic neurons. Dev. Biol. 262, 242–253.
Camarero, G., Villar, M. A., Contreras, J., Fernandez-Moreno, C., Pichel, J. G., Avendano, C., and Varela-Nieto, I. (2002). Cochlear abnormalities in insulin-like growth factor-1 mouse mutants. Hear. Res. 170, 2–11.
Cediel, R., Riquelme, R., Contreras, J., Diaz, A., and Varela-Nieto, I. (2006). Sensorineural hearing loss in insulin-like growth factor I-null mice: a new model of human deafness. Eur. J. Neurosci. 23, 587–590.
Cheng, C. M., Joncas, G., Reinhardt, R. R., Farrer, R., Quarles, R., Janssen, J., Mcdonald, M. P., Crawley, J. N., Powell-Braxton, L., and Bondy, C. A. (1998). Biochemical and morphometric analyses show that myelination in the insulin-like growth factor 1 null brain is proportionate to its neuronal composition. J. Neurosci. 18, 5673–5681.
Cheng, C. M., Mervis, R. F., Niu, S. L., Salem, N. Jr., Witters, L. A., Tseng, V., Reinhardt, R., and Bondy, C. A. (2003). Insulin-like growth factor 1 is essential for normal dendritic growth. J. Neurosci. Res. 73, 1–9.
Cuenda, A., and Nebreda, A. R. (2009). p38delta and PKD1: kinase switches for insulin secretion. Cell 136, 209–210.
Danti, S., D’alessandro, D., Pietrabissa, A., Petrini, M., and Berrettini, S. (2009). Development of tissue-engineered substitutes of the ear ossicles: PORP-shaped poly(propylene fumarate)-based scaffolds cultured with human mesenchymal stromal cells. J. Biomed. Mater. Res. A 92, 1343–1356.
De Meyts, P., and Whittaker, J. (2002). Structural biology of insulin and IGF1 receptors: implications for drug design. Nat. Rev. Drug Discov. 1, 769–783.
D’ercole, A. J., Ye, P., and O’kusky, J. R. (2002). Mutant mouse models of insulin-like growth factor actions in the central nervous system. Neuropeptides 36, 209–220.
Downward, J. (2003). Role of receptor tyrosine kinases in G-protein-coupled receptor regulation of Ras: transactivation or parallel pathways? Biochem. J. 376, e9–e10.
Dror, A. A., and Avraham, K. B. (2009). Hearing loss: mechanisms revealed by genetics and cell biology. Annu. Rev. Genet. 43, 411–437.
Ester, W. A., Van Duyvenvoorde, H. A., De Wit, C. C., Broekman, A. J., Ruivenkamp, C. A., Govaerts, L. C., Wit, J. M., Hokken-Koelega, A. C., and Losekoot, M. (2009). Two short children born small for gestational age with insulin-like growth factor 1 receptor haploinsufficiency illustrate the heterogeneity of its phenotype. J. Clin. Endocrinol. Metab. 94, 4717–4727.
Federici, M., Porzio, O., Zucaro, L., Fusco, A., Borboni, P., Lauro, D., and Sesti, G. (1997). Distribution of insulin/insulin-like growth factor-I hybrid receptors in human tissues. Mol. Cell. Endocrinol. 129, 121–126.
Firth, S. M., and Baxter, R. C. (2002). Cellular actions of the insulin-like growth factor binding proteins. Endocr. Rev. 23, 824–854.
Foulstone, E., Prince, S., Zaccheo, O., Burns, J. L., Harper, J., Jacobs, C., Church, D., and Hassan, A. B. (2005). Insulin-like growth factor ligands, receptors, and binding proteins in cancer. J. Pathol. 205, 145–153.
Frago, L. M., Canon, S., De La Rosa, E. J., Leon, Y., and Varela-Nieto, I. (2003). Programmed cell death in the developing inner ear is balanced by nerve growth factor and insulin-like growth factor I. J. Cell. Sci. 116, 475–486.
Gao, W. Q., Shinsky, N., Ingle, G., Beck, K., Elias, K. A., and Powell-Braxton, L. (1999). IGF-I deficient mice show reduced peripheral nerve conduction velocities and decreased axonal diameters and respond to exogenous IGF-I treatment. J. Neurobiol. 39, 142–152.
Groigno, L., Richard-Parpaillon, L., and Boujard, D. (1999). Expression pattern of insulin receptor mRNA during Xenopus laevis embryogenesis. Mech. Dev. 86, 151–154.
Gross, J., Machulik, A., Moller, R., Fuchs, J., Amarjargal, N., Ungethum, U., Kuban, R. J., Szczepek, A. J., Haupt, H., and Mazurek, B. (2008). MRNA expression of members of the IGF system in the organ of Corti, the modiolus and the stria vascularis of newborn rats. Growth Factors 26, 180–191.
Hodge, R. D., D’ercole, A. J., and O’kusky, J. R. (2004). Insulin-like growth factor-I accelerates the cell cycle by decreasing G1 phase length and increases cell cycle reentry in the embryonic cerebral cortex. J. Neurosci. 24, 10201–10210.
Hurtado-Chong, A., Yusta-Boyo, M. J., Vergano-Vera, E., Bulfone, A., De Pablo, F., and Vicario-Abejon, C. (2009). IGF-I promotes neuronal migration and positioning in the olfactory bulb and the exit of neuroblasts from the subventricular zone. Eur. J. Neurosci. 30, 742–755.
Iwai, K., Nakagawa, T., Endo, T., Matsuoka, Y., Kita, T., Kim, T. S., Tabata, Y., and Ito, J. (2006). Cochlear protection by local insulin-like growth factor-1 application using biodegradable hydrogel. Laryngoscope 116, 529–533.
Jiang, W., Xiang, C., Cazacu, S., Brodie, C., and Mikkelsen, T. (2008). Insulin-like growth factor binding protein 7 mediates glioma cell growth and migration. Neoplasia 10, 1335–1342.
Klammt, J., Kiess, W., and Pfaffle, R. (2011). IGF1R mutations as cause of SGA. Best Pract. Res. Clin. Endocrinol. Metab. 25, 191–206.
Laviola, L., Natalicchio, A., Perrini, S., and Giorgino, F. (2008). Abnormalities of IGF-I signaling in the pathogenesis of diseases of the bone, brain, and fetoplacental unit in humans. Am. J. Physiol. Endocrinol. Metab. 295, E991–E999.
Lee, K. Y., Nakagawa, T., Okano, T., Hori, R., Ono, K., Tabata, Y., Lee, S. H., and Ito, J. (2007). Novel therapy for hearing loss: delivery of insulin-like growth factor 1 to the cochlea using gelatin hydrogel. Otol. Neurotol. 28, 976–981.
Leon, Y., Vazquez, E., Sanz, C., Vega, J. A., Mato, J. M., Giraldez, F., Represa, J., and Varela-Nieto, I. (1995). Insulin-like growth factor-I regulates cell proliferation in the developing inner ear, activating glycosyl-phosphatidylinositol hydrolysis and Fos expression. Endocrinology 136, 3494–3503.
Lewis, M. A., and Steel, K. P. (2010). MicroRNAs in mouse development and disease. Semin. Cell Dev. Biol. 21, 774–780.
Liu, J. P., Baker, J., Perkins, A. S., Robertson, E. J., and Efstratiadis, A. (1993). Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r). Cell 75, 59–72.
Liu, W., Ye, P., O’kusky, J. R., and D’ercole, A. J. (2009). Type 1 insulin-like growth factor receptor signaling is essential for the development of the hippocampal formation and dentate gyrus. J. Neurosci. Res. 87, 2821–2832.
Magarinos, M., Aburto, M. R., Sanchez-Calderon, H., Munoz-Agudo, C., Rapp, U. R., and Varela-Nieto, I. (2010). RAF kinase activity regulates neuroepithelial cell proliferation and neuronal progenitor cell differentiation during early inner ear development. PLoS ONE 5, e14435.
Mason, J. L., Xuan, S., Dragatsis, I., Efstratiadis, A., and Goldman, J. E. (2003). Insulin-like growth factor (IGF) signaling through type 1 IGF receptor plays an important role in remyelination. J. Neurosci. 23, 7710–7718.
Mccubrey, J. A., Steelman, L. S., Kempf, C. R., Chappell, W., Abrams, S. L., Stivala, F., Malaponte, G., Nicoletti, F., Libra, M., Basecke, J., Maksimovic-Ivanic, D., Mijatovic, S., Montalto, G., Cervello, M., Cocco, L., and Martelli, A. M. (2011). Therapeutic resistance resulting from mutations in Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR signaling pathways. J. Cell. Physiol.[Epub ahead of print].
Mebratu, Y., and Tesfaigzi, Y. (2009). How ERK1/2 activation controls cell proliferation and cell death: is subcellular localization the answer? Cell Cycle 8, 1168–1175.
Morgan, D. O., Edman, J. C., Standring, D. N., Fried, V. A., Smith, M. C., Roth, R. A., and Rutter, W. J. (1987). Insulin-like growth factor II receptor as a multifunctional binding protein. Nature 329, 301–307.
Nakagawa, T., Sakamoto, T., Hiraumi, H., Kikkawa, Y. S., Yamamoto, N., Hamaguchi, K., Ono, K., Yamamoto, M., Tabata, Y., Teramukai, S., Tanaka, S., Tada, H., Onodera, R., Yonezawa, A., Inui, K., and Ito, J. (2010). Topical insulin-like growth factor 1 treatment using gelatin hydrogels for glucocorticoid-resistant sudden sensorineural hearing loss: a prospective clinical trial. BMC Med. 8, 76.
Netchine, I., Azzi, S., Houang, M., Seurin, D., Perin, L., Ricort, J. M., Daubas, C., Legay, C., Mester, J., Herich, R., Godeau, F., and Le Bouc, Y. (2009). Partial primary deficiency of insulin-like growth factor (IGF)-I activity associated with IGF1 mutation demonstrates its critical role in growth and brain development. J. Clin. Endocrinol. Metab. 94, 3913–3921.
Otaegi, G., Yusta-Boyo, M. J., Vergano-Vera, E., Mendez-Gomez, H. R., Carrera, A. C., Abad, J. L., Gonzalez, M., De La Rosa, E. J., Vicario-Abejon, C., and De Pablo, F. (2006). Modulation of the PI 3-kinase-Akt signalling pathway by IGF-I and PTEN regulates the differentiation of neural stem/precursor cells. J. Cell. Sci. 119, 2739–2748.
Pandini, G., Frasca, F., Mineo, R., Sciacca, L., Vigneri, R., and Belfiore, A. (2002). Insulin/insulin-like growth factor I hybrid receptors have different biological characteristics depending on the insulin receptor isoform involved. J. Biol. Chem. 277, 39684–39695.
Popken, G. J., Hodge, R. D., Ye, P., Zhang, J., Ng, W., O’kusky, J. R., and D’ercole, A. J. (2004). In vivo effects of insulin-like growth factor-I (IGF-I) on prenatal and early postnatal development of the central nervous system. Eur. J. Neurosci. 19, 2056–2068.
Riquelme, R., Cediel, R., Contreras, J., La Rosa Lourdes, R. D., Murillo-Cuesta, S., Hernandez-Sanchez, C., Zubeldia, J. M., Cerdan, S., and Varela-Nieto, I. (2010). A comparative study of age-related hearing loss in wild type and insulin-like growth factor I deficient mice. Front. Neuroanat. 4:27.
Rivolta, M. N. (2010). Stem cells and cell lines from the human auditory organ: applications, hurdles and bottlenecks in the development of regenerative therapies for deafness. Drug Discov. Today 15, 283–286.
Rodriguez-De La Rosa, L., Murillo-Cuesta, S., Camarero, G., and Varela-Nieto, I. (2011). “Regulation of cochlear development and function: a scenario for growth factor actions,” in Usher Syndrome: Pathogenesis, Diagnosis and Therapy, 1st Edn, ed. S. Ahuja (Hauppauge, NY: Nova Science Publishers), 131–154.
Sanchez-Calderon, H., Milo, M., Leon, Y., and Varela-Nieto, I. (2007). A network of growth and transcription factors controls neuronal differentiation and survival in the developing ear. Int. J. Dev. Biol. 51, 557–570.
Sanchez-Calderon, H., Rodriguez-De La Rosa, L., Milo, M., Pichel, J. G., Holley, M., and Varela-Nieto, I. (2010). RNA microarray analysis in prenatal mouse cochlea reveals novel IGF-I target genes: implication of MEF2 and FOXM1 transcription factors. PLoS ONE 5, e8699.
Schlueter, P. J., Peng, G., Westerfield, M., and Duan, C. (2007). Insulin-like growth factor signaling regulates zebrafish embryonic growth and development by promoting cell survival and cell cycle progression. Cell Death Differ. 14, 1095–1105.
Scolnick, J. A., Cui, K., Duggan, C. D., Xuan, S., Yuan, X. B., Efstratiadis, A., and Ngai, J. (2008). Role of IGF signaling in olfactory sensory map formation and axon guidance. Neuron 57, 847–857.
Stratikopoulos, E., Szabolcs, M., Dragatsis, I., Klinakis, A., and Efstratiadis, A. (2008). The hormonal action of IGF1 in postnatal mouse growth. Proc. Natl. Acad. Sci. U.S.A. 105, 19378–19383.
Sullivan, K. A., Kim, B., and Feldman, E. L. (2008). Insulin-like growth factors in the peripheral nervous system. Endocrinology 149, 5963–5971.
Torres-Aleman, I. (2010). Toward a comprehensive neurobiology of IGF-I. Dev. Neurobiol. 70, 384–396.
Van Duyvenvoorde, H. A., Van Setten, P. A., Walenkamp, M. J., Van Doorn, J., Koenig, J., Gauguin, L., Oostdijk, W., Ruivenkamp, C. A., Losekoot, M., Wade, J. D., De Meyts, P., Karperien, M., Noordam, C., and Wit, J. M. (2010). Short stature associated with a novel heterozygous mutation in the insulin-like growth factor 1 gene. J. Clin. Endocrinol. Metab. 95, E363–E367.
Vardatsikos, G., Sahu, A., and Srivastava, A. K. (2009). The insulin-like growth factor family: molecular mechanisms, redox regulation, and clinical implications. Antioxid. Redox Signal. 11, 1165–1190.
Varela-Nieto, I., De La Rosa, E. J., Valenciano, A. I., and Leon, Y. (2003). Cell death in the nervous system: lessons from insulin and insulin-like growth factors. Mol. Neurobiol. 28, 23–50.
Varela-Nieto, I., Hartl, M., Gorospe, I., and Leon, Y. (2007). Anti-apoptotic actions of insulin-like growth factors: lessons from development and implications in neoplastic cell transformation. Curr. Pharm. Des. 13, 687–703.
Varela-Nieto, I., Morales-Garcia, J. A., Vigil, P., Diaz-Casares, A., Gorospe, I., Sanchez-Galiano, S., Canon, S., Camarero, G., Contreras, J., Cediel, R., and Leon, Y. (2004). Trophic effects of insulin-like growth factor-I (IGF-I) in the inner ear. Hear. Res. 196, 19–25.
Vicario-Abejon, C., Fernandez-Moreno, C., Pichel, J. G., and De Pablo, F. (2004). Mice lacking IGF-I and LIF have motoneuron deficits in brain stem nuclei. Neuroreport 15, 2769–2772.
Vicario-Abejon, C., Yusta-Boyo, M. J., Fernandez-Moreno, C., and De Pablo, F. (2003). Locally born olfactory bulb stem cells proliferate in response to insulin-related factors and require endogenous insulin-like growth factor-I for differentiation into neurons and glia. J. Neurosci. 23, 895–906.
Walenkamp, M. J., Karperien, M., Pereira, A. M., Hilhorst-Hofstee, Y., Van Doorn, J., Chen, J. W., Mohan, S., Denley, A., Forbes, B., Van Duyvenvoorde, H. A., Van Thiel, S. W., Sluimers, C. A., Bax, J. J., De Laat, J. A., Breuning, M. B., Romijn, J. A., and Wit, J. M. (2005). Homozygous and heterozygous expression of a novel insulin-like growth factor-I mutation. J. Clin. Endocrinol. Metab. 90, 2855–2864.
Walenkamp, M. J., and Wit, J. M. (2007). Genetic disorders in the GH IGF-I axis in mouse and man. Eur. J. Endocrinol. 157(Suppl. 1), S15–S26.
Welch, D., and Dawes, P. J. (2007). Childhood hearing is associated with growth rates in infancy and adolescence. Pediatr. Res. 62, 495–498.
Woods, K. A., Camacho-Hubner, C., Savage, M. O., and Clark, A. J. (1996). Intrauterine growth retardation and postnatal growth failure associated with deletion of the insulin-like growth factor I gene. N. Engl. J. Med. 335, 1363–1367.
Xuan, S., Szabolcs, M., Cinti, F., Perincheri, S., Accili, D., and Efstratiadis, A. (2010). Genetic analysis of type-1 insulin-like growth factor receptor signaling through insulin receptor substrate-1 and -2 in pancreatic beta cells. J. Biol. Chem. 285, 41044–41050.
Ye, P., and D’ercole, A. J. (2006). Insulin-like growth factor actions during development of neural stem cells and progenitors in the central nervous system. J. Neurosci. Res. 83, 1–6.
Ye, P., Li, L., Lund, P. K., and D’ercole, A. J. (2002a). Deficient expression of insulin receptor substrate-1 (IRS-1) fails to block insulin-like growth factor-I (IGF-I) stimulation of brain growth and myelination. Brain Res. Dev. Brain Res. 136, 111–121.
Ye, P., Li, L., Richards, R. G., Diaugustine, R. P., and D’ercole, A. J. (2002b). Myelination is altered in insulin-like growth factor-I null mutant mice. J. Neurosci. 22, 6041–6051.
Keywords: animal models, deafness, human genetics, insulin-like factors, IGF1R signaling, organ of Corti
Citation: Murillo-Cuesta S, Rodríguez-de la Rosa L, Cediel R, Lassaletta L and Varela-Nieto I (2011) The role of insulin-like growth factor-I in the physiopathology of hearing. Front. Mol. Neurosci. 4:11. doi: 10.3389/fnmol.2011.00011
Received: 17 June 2011;
Paper pending published: 06 July 2011;
Accepted: 11 July 2011;
Published online: 25 July 2011.
Edited by:
Julie A. Chowen, University of Washington, USAReviewed by:
Ye He, University of California San Francisco, USASmita Jha, Baylor College of Medicine, USA
Copyright: © 2011 Murillo-Cuesta, Rosa, Cediel, Lassaletta and Varela-Nieto. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Isabel Varela-Nieto, Instituto de Investigaciones Biomédicas Alberto Sols, Consejo Superior de Investigaciones Científicas-Universidad Autónoma de Madrid, Arturo Duperier, 4, 28029 Madrid, Spain. e-mail:aXZhcmVsYUBpaWIudWFtLmVz