
- Department of Psychiatry and Behavioral Neurobiology, University of Alabama at Birmingham, Birmingham, AL, USA
The mood disorders major depressive disorder and bipolar disorder are prevalent, are inadequately treated, and little is known about their etiologies. A better understanding of the causes of mood disorders would benefit from improved animal models of mood disorders, which now rely on behavioral measurements. This review considers the limitations in relating measures of rodent behaviors to mood disorders, and the evidence from behavioral assessments indicating that glycogen synthase kinase-3 (GSK3) dysregulation promotes mood disorders and is a potential target for treating mood disorders. The classical mood stabilizer lithium was identified by studying animal behaviors and later was discovered to be an inhibitor of GSK3. Several mood-relevant behavioral effects of lithium in rodents have been identified, and most have now been shown to be due to its inhibition of GSK3. An extensive variety of pharmacological and molecular approaches for manipulating GSK3 are discussed, the results of which strongly support the proposal that inhibition of GSK3 reduces both depression-like and manic-like behaviors. Studies in human postmortem brain and peripheral cells also have identified correlations between alterations in GSK3 and mood disorders. Evidence is reviewed that depression may be associated with impaired inhibitory control of GSK3, and mania by hyper-stimulation of GSK3. Taken together, these studies provide substantial support for the hypothesis that inhibition of GSK3 activity is therapeutic for mood disorders. Future research should identify the causes of dysregulated GSK3 in mood disorders and the actions of GSK3 that contribute to these diseases.
Introduction
Major depressive disorder and bipolar disorder, in which patients experience manic episodes typically interspersed with depressive episodes, are commonly referred to as mood disorders. These are debilitating and prevalent illnesses, with a lifetime incidence of approximately 20% in the United States, and they are life-threatening due to suicide as well as other causes (Wong and Licinio, 2001; Nestler et al., 2002; Berns and Nemeroff, 2003; Belmaker, 2004). The pathophysiological underpinnings of mood disorders are unknown. Research into the causative mechanisms has been greatly hampered by the lack of adequate animal models of these diseases. However, studies of behaviors in rodents, including investigations of the mechanisms of action of therapeutic agents, have provided substantial evidence of a number of connections between glycogen synthase kinase-3 (GSK3) and mood disorders. Altogether, these findings suggest that dysregulation of GSK3 that causes its increased activity in specific cellular locations, pathways, and circuits, promotes susceptibility to mood disorders, and that inhibition of GSK3 is an important component of the therapeutic actions of interventions used to treat mood disorders.
Assessing Characteristics of Mood Disorders in Rodents
Progress in understanding and treating mood disorders faces a difficult paradox. On one hand, there is no doubt that progress would be enhanced by mood disorder models in animals, preferably rodents. On the other hand, it is unlikely that rodents are capable of experiencing mood states that are equivalent to mania or major depression. Thus, unlike most other diseases, it may not be possible to precisely model the defining characteristics of mood disorders in animals. This has led to measurements of behaviors, instead of mood, in rodents, although it remains unclear to what extent assessments of rodent behaviors can be useful for clarifying the causes of mood disorders and for developing new therapeutics (Chen et al., 2010; Nestler and Hyman, 2010). Nonetheless, much effort has been exerted in the pursuit of measuring behavioral characteristics in rodents because of the high value of such models in developing better treatments for mood disorders (Matthews et al., 2005; Gould and Einat, 2007). In this regard, it is important to remember that the classical mood stabilizer lithium was initially conceived as a possible therapeutic for psychiatric diseases based on its behavioral effects in guinea pigs (Cade, 1949).
The rationale for using rodent behaviors to estimate mood is strengthened when it is possible to obtain predictable actions of drugs, such as by administration of antidepressants or mood stabilizers. However, care must be taken that administration of these drugs reasonably models therapeutic usage in human patients. For example, antidepressants have differential effects when administered acutely or chronically, and the latter have traditionally been favored as modeling actions in patients because of their delayed therapeutic effects in major depression. Regarding lithium, many studies originally used injections that produce large fluctuations in serum levels of lithium. More recently, relatively stable therapeutic levels have been achieved by administering lithium in the food of rodents in a manner that produces serum lithium concentrations that are within the therapeutic range in humans. Generally, the range of 0.4–1.2 mM lithium is achieved in rodent serum using food pellets that contain 0.2–0.4% lithium, which can be maintained for months as long as the rodents are provided with a source of extra sodium to prevent hyponatremia (Thomsen and Olesen, 1974).
Quite a few models have been developed for studying antidepressant actions and depression-like behavior in rodents. Hereafter this will be referred to as depression for simplicity with the understanding that there are immense limitations in this categorization since behavior, rather than mood, is measured. Extensive reviews of these developments and their limitations have been published previously (Nestler et al., 2002; Cryan and Mombereau, 2004; O’Donnell and Gould, 2007; Chen et al., 2010). Therefore, only limited examples will be discussed here with a focus on those that have been employed in studies of the role of GSK3 in depression. One widely used model is the learned helplessness model of depressive-like behavior (Chourbaji et al., 2005). In this test, exposure to uncontrollable and inescapable shock will afterward cause depressed rodents to exhibit a deficit in escape when it is available, and this failure to escape can be ameliorated by chronic administration of antidepressants. The forced swim test (FST) and tail suspension test (TST) also have been used to identify in rodents drugs with antidepressant effects (Porsolt et al., 1977). These are only a few examples of a growing number of tests that have been applied to measure depressive-like behavior in rodents and antidepressant actions (Nestler et al., 2002; Cryan and Mombereau, 2004; O’Donnell and Gould, 2007).
In contrast to depression, progress has been more limited in developing rodent models of bipolar disorder. Furthermore, these studies have primarily focused on the manic-like component of behavior. This will be referred to as mania for simplicity, again with the understanding that there is currently not a true rodent model of the condition that humans experience as mania. Mania is a complex group of symptoms and no single measure can identify a manic rodent. Therefore, investigators have attempted to model characteristics of mania in rodents while understanding the limitations of these models. These models are most useful when disturbances in several rodent behaviors are observed that fall within the definition of mania, although not unique to mania, and that are ameliorated by a mood stabilizer (Machado-Vieira et al., 2004; Gould and Gottesman, 2006; Einat, 2007; Kovacsics et al., 2009). The two most common assessments of mania in rodents are basal locomotor activity, either in the home cage or in a novel environment, and amphetamine-induced hyperactivity. The latter has been used in part because amphetamine can worsen symptoms or induce mania relapse in patients, and mood stabilizers can alleviate these responses (reviewed in Einat et al., 2003; O’Donnell and Gould, 2007). Further behavioral measurements have been employed to attempt to study additional components of manic-like behavior in rodents. Recent evidence was reported that increased preference for sweet solutions provides a model for increased reward seeking, a central component of manic behavior that can be reduced by administration of mood stabilizers (Flaisher-Grinberg et al., 2009). Enhanced sucrose preference was displayed by CLOCK mutant mice that were characterized as exhibiting mania-like behavior (Roybal et al., 2007) and by heterozygote bcl-2 deficient mice (Lien et al., 2008). Furthermore, CLOCK mutant mice exhibiting characteristics of manic-like behavior were resistant to learned helplessness-induced depression-like behavior (Roybal et al., 2007). Increased acoustic startle response has also been observed in mice characterized as manic-like or bipolar-like, including mice expressing neuron-specific mutant mitochondrial DNA polymerase (Kasahara et al., 2006) and mice postnatally overexpressing constitutively active S9A–GSK3β in neurons (Prickaerts et al., 2006).
Thus, several approaches have been developed for measuring behaviors in rodents that have provided a wealth of novel information and that may be relevant for studying depression and mania. However, still needed are more behavioral approaches and methods to validate the applicability of these behavioral measurements to mood disorders. Particularly lacking are animal models that display the episodic and progressive natures of mood disorders.
Actions of Lithium on Rodent Behaviors
Since lithium is an effective mood stabilizer for bipolar disorder, its effects on locomotor activity of rodents has been the focus of much research. The activity of normal rodents in a familiar environment has generally been found to be unaffected by lithium, although this depends on the administration protocol (Smith, 1980; Gould et al., 2007b; O’Donnell and Gould, 2007). Additionally, strain-selective actions of lithium on basal and drug-induced behaviors is an important variable (Can et al., 2011; Pan et al., 2011). The absence of lithium effects in wild-type rodent locomotor activity heightens interest in the reduced locomotor activity induced by lithium in transgenic mice that exhibit increased locomotor activity and in mice given stimulant drugs that increase locomotor activity. For example, rodents that spontaneously exhibit increased locomotor activity that is reduced by lithium treatment include sleep-deprived rats (Gessa et al., 1995), rats administered ouabain (Jornada et al., 2010; Gao et al., 2011), mouse knockouts of the kainate receptor subunit GluR6 (Shaltiel et al., 2008), mouse knockouts of the fmr1 gene that model Fragile X Syndrome (Yuskaitis et al., 2010), diacylglycerol kinase β knockout mice (Kakefuda et al., 2010), muscarinic M1 receptor knockout mice (Creson et al., 2011), and mouse knockouts of the AMPA receptor GluA1 subunit (Fitzgerald et al., 2010). This differential effect of lithium on normal compared with abnormal states of locomotor activity provides some support for the rationale that studies of rodent locomotor behaviors may be useful for examining therapeutic interventions for bipolar disorder. These studies also exemplify the well-known fact that multiple single alterations (e.g., various genetic manipulations) can cause equivalent behavioral outcomes in mice (locomotor hyperactivity), which is undoubtedly also the case for mood disorders. Altogether, it is remarkable that lithium is capable of normalizing behavioral locomotor hyperactivity in such a variety of mutant mice.
Studies of drug-induced increased locomotor activity that is reduced by lithium have predominantly used amphetamine, but other drugs also have been examined. Long ago, lithium was found to reduce hyperactivity and/or stereotypic behavior induced by amphetamine (Cox et al., 1971; Berggren et al., 1978; Borison et al., 1978) or by cocaine (Flemenbaum, 1977; Antelman et al., 1998). There has recently been a resurgence in studies examining lithium’s control of amphetamine-induced hyperactivity (reviewed in Einat et al., 2003; O’Donnell and Gould, 2007). For example, lithium antagonized locomotor behaviors induced by amphetamine and dopamine D2 receptor stimulation (Beaulieu et al., 2004, 2005, 2008a). Lithium administration also reversed heightened amphetamine-induced hyperactivity or sensitization to amphetamine displayed by ERK1 knockout mice that exhibit increased behavioral excitement (Engel et al., 2009), heterozygote bcl-2 deficient mice (Lien et al., 2008), and omega-3 fatty acid deficient mice (McNamara et al., 2008). In addition to locomotor hyperactivity, lithium treatment also has been reported to reduce heightened sweet solution preference in CLOCK mutant mice that were characterized as exhibiting mania-like behavior (Roybal et al., 2007) and in heterozygote bcl-2 deficient mice (Lien et al., 2008).
Strategies to Study the Actions of GSK3 as a Therapeutic Target of Lithium
The discovery that lithium directly inhibits GSK3 raised the possibility that this action contributes to the mood stabilizing action of lithium in bipolar disorder (Klein and Melton, 1996; Stambolic et al., 1996). In addition to directly inhibiting the activity of GSK3, in vivo treatment with a therapeutically relevant level of lithium also increases the inhibitory serine-phosphorylation of GSK3, which was suggested to amplify the direct inhibitory action of lithium on GSK3 (De Sarno et al., 2002). This amplification mechanism has received support from a variety of studies (Eom and Jope, 2009; Polter et al., 2010; Pan et al., 2011). There is increasing evidence that many of the behavioral actions of lithium in rodents results from inhibiting GSK3 (Jope, 1999; Manji et al., 2000; Phiel and Klein, 2001; Harwood and Agam, 2003; Jope and Johnson, 2004), and that the diverse effects of lithium may largely be due to the numerous substrates of GSK3 and its consequential influences on many cellular functions. Evidence has been reported that GSK3 phosphorylates more than 100 substrates, and projections suggest that there may be many more proteins that are phosphorylated by GSK3 (Pilot-Storck et al., 2010; Taelman et al., 2010). Thus, it is inevitable that an inhibitor of GSK3, such as lithium, would have many effects on cellular functions. However, GSK3 is clearly not the only target of lithium (Chiu and Chuang, 2010), which also directly inhibits phosphoglucomutase (Ray et al., 1978), bisphosphate 3′-nucleotidase 1 (Spiegelberg et al., 1999), inositol monophosphatase (Hallcher and Sherman, 1980), and other inositol polyphosphatases (Inhorn and Majerus, 1987). Another proposed target of lithium, destabilization of the Akt-β-arrestin–protein phosphatase 2A protein complex (Beaulieu et al., 2004), was recently attributed to inhibition of GSK3 (O’Brien et al., 2011). Thus, multiple targets of lithium must be considered as potential contributors to its behavioral effects, although many of the major advances in recent years have identified the outcomes of GSK3 inhibition.
The identification of behavioral, or mood-altering, effects of lithium raised the possibility that these may be mediated by inhibition of GSK3, a potential causal relationship that requires verification. The two major strategies for doing so utilize pharmacological and molecular approaches. The pharmacological approach has been strengthened in recent years because many academic and pharmaceutical laboratories have engaged in intensive efforts to develop new small molecule selective inhibitors of GSK3 (Meijer et al., 2004; Martinez et al., 2006). This was particularly driven by the discovery that lithium inhibits GSK3 and the abundant evidence that GSK3 contributes to prevalent diseases, such as diabetes and Alzheimer’s disease, as well as mood disorders. This has resulted in the availability of several selective agents for testing if other GSK3 inhibitors cause effects similar, or not, to lithium, and several of these new inhibitors have been widely used. Frequently used inhibitors include indirubin derivatives (Leclerc et al., 2001), L803-mts (Plotkin et al., 2003), SB216763, with care taken concerning its solubility as originally described (Coghlan et al., 2000), TDZD derivatives (Martinez et al., 2002), paullone derivatives (Leost et al., 2000), and AR-A014418 (Bhat et al., 2003), although the reports of behavioral effects of AR-A014418 are mitigated by other studies indicating that it does not significantly enter the CNS (Vasdev et al., 2005; Selenica et al., 2007; Hicks et al., 2010). Particularly valuable are studies of the kinase specificities of several GSK3 inhibitors (Davies et al., 2000; Murray et al., 2004; Bain et al., 2007), which enable investigators to utilize a panel of GSK3 inhibitors with differing off-target actions to provide reasonable confidence in ascribing overlapping effects of the inhibitors to their common target GSK3. The kinase specificity studies (Davies et al., 2000; Murray et al., 2004; Bain et al., 2007) identified CT99021 (Wagman et al., 2004) as the most specific GSK3 inhibitor of those tested. This information is also valuable for finding if GSK3 may be an off-target effect of inhibitors of other kinases. Also valuable is an important report of the in vivo CNS penetration and actions of a panel of GSK3 inhibitors (Selenica et al., 2007). Thus, many tools are available for pharmacologically identifying actions of GSK3.
Molecular approaches also have begun to be used to test if increased GSK3 activity has effects opposite to lithium treatment, and if reducing GSK3 molecularly has outcomes similar to lithium treatment. Increased GSK3 activity has been studied by overexpressing GSK3 and by using GSK3 knockin mice. However, although overexpression of GSK3 has provided a wealth of information about the actions of GSK3 in cells, in vivo CNS studies are hampered because GSK3 overexpression can cause neurodegeneration (Lucas et al., 2001; Spittaels et al., 2002). However, targeted overexpression of GSK3β in the nucleus accumbens induced a depression-like phenotype in multiple behavioral measurements (Wilkinson et al., 2011). Instead of overexpression of GSK3, another approach was taken by Alessi and colleagues to increase GSK3 activity by developing GSK3 knockin mice (McManus et al., 2005). The two GSK3 isoforms, GSK3α and GSK3β, are mainly regulated by inhibitory phosphorylation on Ser21–GSK3α and Ser9–GSK3β (Figure 1). This is normally maintained by signaling pathways, such as serotonergic activity (Li et al., 2004), that may be deficient in mood disorders, resulting in inadequately inhibited GSK3. The importance of inhibitory control of GSK3 can be studied using GSK3α21A/21A/β9A/9A knockin mice, with the regulatory serines of one or both GSK3 isoforms mutated to alanines (McManus et al., 2005). These mutations maintain GSK3 maximally active, but importantly within the physiological range since both GSK3 isoforms are expressed at normal levels. GSK3 knockin mice develop and reproduce normally with no overt phenotype. For studying the outcomes of molecular deficiencies in GSK3, the Woodgett laboratory has provided invaluable leadership by first producing all of the transgenic mice available for these studies (MacAulay and Woodgett, 2008; Force and Woodgett, 2009). They found that GSK3β knockout mice cannot be used because they are embryonically lethal (Hoeflich et al., 2000), but GSK3β± heterozygote knockout mice (O’Brien et al., 2004) and GSK3α knockout mice (Kaidanovich-Beilin et al., 2009) have been used to study the effects of reduced GSK3 expression. Thus, there are now multiple pharmacological and molecular tools available to modify GSK3 activity in vivo in order to examine effects on behavior, which are discussed below.
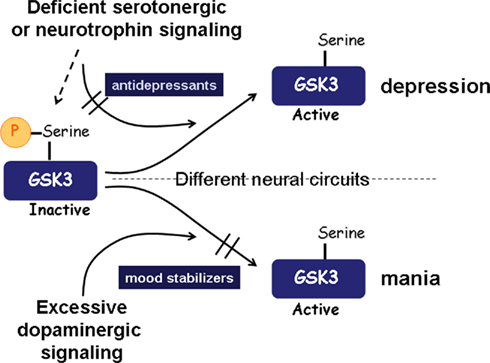
Figure 1. Summary of the involvement of GSK3 in mood disorders. A simplified scheme is shown to represent examples of how GSK3 may be dysregulated in mood disorders. In depression, deficiencies in signals that normally maintain inhibition of GSK3, such as signaling induced by serotonin or neurotrophins, can cause up-regulation of GSK3 activity, which is capable of promoting susceptibility to depression. Mania may involve excessive dopaminergic signaling, which induces activation of GSK3. Part of the therapeutic actions of antidepressants and mood stabilizers may be derived from their direct or indirect effects that cause inhibition of GSK3.
Depression and GSK3 in Rodents
Many of the neuromodulators that are widely thought to be deficient in depression, such as serotonin, BDNF (brain-derived growth factor), and VEGF (vascular endothelial growth factor), normally stimulate signaling pathways that maintain inhibitory control of GSK3. Thus, we hypothesize that deficiencies in these signals in depression leave GSK3 inadequately inhibited, and that restoration of the inhibitory control of GSK3 by therapeutic drugs is an important part of their therapeutic mechanism of action (Figure 1).
The concept that dysregulated GSK3 promotes depression-like behavior in rodents has been extensively supported by multiple pharmacological approaches. Early studies indicated that lithium, when administered properly, attenuated several measures of depression in rodents and enhanced the antidepressant effects of serotonin reuptake inhibitors (Faria and Teixeira, 1993; Nixon et al., 1994; Redrobe and Bourin, 1999). Following the 1996 discovery that lithium inhibits GSK3 (Klein and Melton, 1996; Stambolic et al., 1996), many findings supported the conclusion these responses to lithium were likely due to inhibition of GSK3. These include findings of clear antidepressant effects in rodents of a variety of new small molecule inhibitors of GSK3 in addition to lithium (Gould et al., 2004; Kaidanovich-Beilin et al., 2004; O’Brien et al., 2004; Shapira et al., 2007; Beaulieu et al., 2008a; Silva et al., 2008), including on depressive behavior exhibited by tryptophan hydroxylase 2 mutant mice with deficient serotonin (Beaulieu et al., 2008b). Other studies have shown that classical antidepressants, as well as atypical antipsychotics, also inhibit GSK3 in mouse brain after in vivo administration of clinically relevant doses (Li et al., 2004, 2007a; Alimohamad et al., 2005; Roh et al., 2007; Beaulieu et al., 2008b; Okamoto et al., 2010). Furthermore, blocked inhibitory serine-phosphorylation of GSK3 in GSK3 knockin mice abrogated the neurogenesis-stimulating effect of fluoxetine and lithium administration, suggesting that at least some of the responses to fluoxetine and lithium depend on their induction of inhibitory serine-phosphorylation of GSK3 (Eom and Jope, 2009). Additionally, inhibition of GSK3 is required for the rapid antidepressant effect of ketamine in the learned helplessness model of depression in mice (Beurel et al., 2011b). In addition to inhibiting GSK3 via serine-phosphorylation, antidepressants also increase signaling by Wnt2, which inhibits GSK3 in the Wnt signaling pathway (Okamoto et al., 2010). Expressing or depleting disheveled, a protein capable of inhibiting GSK3 in the Wnt signaling pathway, induced multiple behavioral outcomes consistent with the concept that inhibiting GSK3 counteracts disrupted mood-relevant behaviors with the important emphasis that these effects resulted from alterations in the nucleus accumbens (Wilkinson et al., 2011). Conversely, GSK3 was found to be activated via decreased inhibitory serine-phosphorylation in the brains of mice exhibiting the learned helplessness model of depression (Polter et al., 2010). This activation of GSK3 during depression-like behavior was further shown to occur in the nucleus accumbens of mice exhibiting social defeat stress (Wilkinson et al., 2011). Taken together, these multiple approaches support the concept that impaired inhibition of GSK3 promotes depression-like behavior, and inhibition of GSK3 promotes resistance to depression.
In conjunction with these pharmacological studies, strategies using molecular modifications of GSK3 have firmly established that GSK3 promotes depression in rodents. The reduced GSK3β level in heterozygote GSK3β± knockout mice was demonstrated to be sufficient to reduce depression-like immobility in the FST (O’Brien et al., 2004) and the TST (Beaulieu et al., 2008a), and reduced immobility in the TST in tryptophan hydroxylase 2 mutant mice (Beaulieu et al., 2008b). Decreasing GSK3β levels by bilateral intra-hippocampal injections of lentivirus-expressing short-hairpin RNA targeting GSK3β decreased depression-like immobility times in both the forced swim and TSTs (Omata et al., 2011). Decreased immobility times in the FST and the TST also were displayed by mice lacking a functional GSK3α gene (Kaidanovich-Beilin et al., 2009). These studies demonstrate that lowered expression of either isoform of GSK3 reduces vulnerability to depression-like behaviors in rodents. Additionally, overexpression of Wnt2, which inhibits GSK3 in the Wnt signaling pathway, reduced susceptibility to depression-like behavior in the learned helplessness paradigm (D:Okamoto:2010]. Conversely, increased GSK3 activity in GSK3 knockin mice was associated with increased susceptibility to the learned helplessness model of depression and increased immobility time in the FST and TST (Polter et al., 2010). Definitive evidence that GSK3 is the causal target of lithium in modifying behaviors in mice was shown by the finding that transgenic expression of GSK3β in mouse brain rescued lithium-sensitive behaviors, including immobility in the FST, exploratory behavior, and open arm time in the elevated zero maze (O’Brien et al., 2011).
Studies of mice expressing mutant DISC1 or with deficient DISC1 expression have further supported the concept that impaired inhibitory control of GSK3 promotes susceptibility to depression. DISC1 mutations have been implicated as a risk factor for schizophrenia, bipolar disorder, and recurrent major depression. Mice deficient in functional DISC1 exhibit depression-like behavior, as well as other behavioral abnormalities (Clapcote et al., 2007; Hikida et al., 2007; Li et al., 2007; Pletnikov et al., 2008; Shen et al., 2008). Dysregulated GSK3 may contribute to some of these behaviors because wild-type DISC1 directly binds and inhibits GSK3, actions lost with mutated DISC1 (Mao et al., 2009; Lipina et al., 2011a). Importantly, administration of GSK3 inhibitors normalized depression-like behavior in the FST and improved the impaired neurogenesis in DISC1 deficient mice (Mao et al., 2009; Lipina et al., 2011b), and genetic inactivation of GSK3α rescued spine deficits in DISC1 mutant mice (Lee et al., 2011). These are exciting findings because they provide a specific mechanism whereby a molecular variant associated with susceptibility to mood disorders may cause inadequate inhibitory regulation of GSK3.
Mania and GSK3 in Rodents
As noted above, basal locomotor hyperactivity is a commonly used measure of manic-like activity in rodents, although this is likely only a marginally adequate model. Increased locomotor activity was exhibited by mice postnatally overexpressing constitutively active S9A–GSK3β in neurons (Prickaerts et al., 2006) and GSK3 knockin mice displayed increased locomotor activity in a novel open field (Polter et al., 2010), whereas decreased locomotion was displayed by mice lacking a functional GSK3α gene (Kaidanovich-Beilin et al., 2009). Mice deficient in functional DISC1 that impairs its ability to inhibit GSK3 also exhibit spontaneous hyperactivity in the open field (Hikida et al., 2007; Pletnikov et al., 2008) that was normalized by reducing GSK3 activity (Mao et al., 2009; Lipina et al., 2011a,b). These findings are consistent with the concept that increased GSK3 activity is directly correlated with locomotor hyperactivity.
Locomotor hyperactivity induced by drugs, particularly amphetamine, is also widely used to model manic behavior in rodents. Particularly interesting are the studies by Beaulieu et al. 2004 that identified an important role for GSK3 in the locomotor response to amphetamine. They demonstrated that amphetamine administration activated cortical and striatal GSK3 (Beaulieu et al., 2004, 2005, 2008a), and GSK3 was activated in mouse striatum in dopamine transporter knockout mice (DAT-KO) due to reduced Akt activity (Beaulieu et al., 2004). Locomotor hyperactivity displayed by mice lacking the dopamine transporter was reduced by administration of five different GSK3 inhibitors (Beaulieu et al., 2004). GSK3β± heterozygote knockout mice displayed attenuated locomotor activation after amphetamine administration compared with wild-type mice, while basal locomotor activity of the two cohorts was equivalent (Beaulieu et al., 2004). Others have also shown that inhibitors of GSK3 reduce amphetamine-induced locomotor hyperactivity, further strengthening the conclusion that active GSK3 is a critical mediator of this response (Kozikowski et al., 2007; Kalinichev and Dawson, 2011). Increased amphetamine-induced locomotor hyperactivity was exhibited by mice deficient in functional DISC1, which eliminates its inhibition of GSK3 (Lipina et al., 2010), and by GSK3 knockin mice (Polter et al., 2010). Conversely, overexpression of β-catenin, which partially models reduced GSK3 activity, attenuated amphetamine-induced hyperactivity in mice (Gould et al., 2007a). These findings clearly demonstrated that GSK3 is activated after amphetamine administration and that GSK3 mediates some of the behavioral effects of dopamine, supporting a relationship between GSK3 activity and certain manic-like behaviors.
GSK3 in Subjects with Mood Disorders
It is a challenging task to confirm in humans, hypotheses that are developed from in vitro and animal studies. This is particularly true for mood disorders because the target tissue is inaccessible and there is no clear pathological parameter or biomarker that can be assessed. Nonetheless, significant progress has been made investigating potential alterations of GSK3 in humans with mood disorders. The most direct assessment reported is the elevated GSK3 activity, associated with decreased Akt activity, in postmortem samples from ventral prefrontal cortex from patients with major depression disorder (Karege et al., 2007, 2011). In contrast, lower GSK3β expression was reported in prefrontal cortex of teenage suicide victims (Pandey et al., 2009). There is also evidence of fluctuations in the inhibitory serine-phosphorylation of GSK3 in peripheral blood mononuclear cells, generally with decreases associated with disease and increases following therapy (Li et al., 2007b, 2010). These findings suggest that it may be possible to develop measurements of phosphorylated GSK3 as a biomarker to reflect disease state and/or treatment responses. Thus, limited information is available concerning the functional status of GSK3 in humans with mood disorders. However, these findings generally support the concept developed in animal studies that GSK3 is inadequately inhibited in association with mood disorders and is inhibited in humans treated with lithium. This is an area in great need of further research in order to evaluate whether new GSK3 inhibitors should be tested as therapies in mood disorder patients.
Genetic studies have explored GSK3 and related genes in patients with mood disorders. Associations that have been identified include an increase in copy number variations affecting the GSK3β gene locus in bipolar disorder (Lachman et al., 2007), and GSK3β polymorphisms linked to the age of onset of bipolar disorder and of major depressive disorder and the therapeutic responses to lithium (Benedetti et al., 2004a,b, 2005; Szczepankiewicz et al., 2006a; Saus et al., 2010) or to antidepressants (Tsai et al., 2008), and to gray matter volume in patients with major depressive disorder (Inkster et al., 2009). However, others have not found GSK3β polymorphisms associated with bipolar disorder (Lee et al., 2006; Nishiguchi et al., 2006) or lithium response (Szczepankiewicz et al., 2006b). Since cellular signals regulating GSK3, rather than GSK3 expression, have been implicated in most studies of mood disorder models, examinations of signaling pathways linked to GSK3 may prove most informative. This approach is exemplified by a recent report that polymorphisms in several genes encoding proteins directly related to the function of GSK3 are associated with regional gray matter volume changes in major depressive disorder patients (Inkster et al., 2010).
Actions of GSK3 That May Contribute to Mood Disorders
Discussions of the role of GSK3 in mood disorders inevitably lead to the question: What is the target phosphorylated by hyperactive GSK3 that causes increased susceptibility to mood disorders? Ten years ago this was a relevant question because relatively little was known about the actions of GSK3 and few substrates had been identified. However, this question may now be less pressing considering our greater understanding of the regulatory roles of GSK3 in cellular functions and the numerous substrates of GSK3 that are known, and projections of many more that remain to be verified (Pilot-Storck et al., 2010; Taelman et al., 2010). Thus, it may be more pertinent to identify cellular processes regulated by GSK3 that are also dysregulated in mood disorders, rather than identifying individual substrates abnormally phosphorylated by GSK3 in mood disorders. This is somewhat analogous to studies of how GSK3 promotes intrinsic apoptotic signaling (Beurel and Jope, 2006). Although the mechanisms mediating apoptotic signaling are much better understood than mechanisms regulating mood disorders, a single target of GSK3 that underlies its promotion of apoptosis has not been identified. Instead a variety of actions appear to account for GSK3 lowering the threshold for apoptosis that are, in part, related to the initial insult. In this regard, studies of abnormal cellular functions that may contribute to mood disorders and studies of the actions of GSK3 have converged on several common themes. These include, but are not limited to, cellular stress response mechanisms, neurogenesis, and immune system abnormalities, particularly inflammation. There is evidence that alterations in each of these may promote the onset or severity of mood disorders, and that GSK3 has a strong regulatory role in each.
Substantial evidence indicates that mood disorders are associated with neuronal stress, such as oxidative stress and endoplasmic reticulum (ER) stress (Kato and Kato, 2000; Wang, 2007; Andreazza et al., 2008; Ng et al., 2008; Steckert et al., 2010). Initiating causes for increases in neuronal stress associated with mood disorders remain largely undetermined. However, it is well-established that many cell stressors can increase the activity of GSK3 in specific cellular compartments, such as insults causing ER stress (Song et al., 2002) or causing DNA damage (Watcharasit et al., 2002). Furthermore, a well-established characteristic of hyperactive GSK3 is its promotion of detrimental cellular responses to multiple types of insults, including oxidative stress and ER stress, which can be alleviated by GSK3 inhibitors (Beurel and Jope, 2006). Thus, these insults may contribute to abnormal activation of GSK3, and hyperactive GSK3 may contribute to reduced neuronal resilience in stressful environments. However, it is also important to consider that such stresses also affect glia cells, and there is growing evidence for glia abnormalities in mood disorders (Rajkowska and Miguel-Hidalgo, 2007), which may be associated with the increased inflammatory markers associated with mood disorders, as discussed below.
Neurogenesis, the proliferation, and neuronal differentiation of neural precursor cells, may be impaired in mood disorders (Lie et al., 2004). This is supported by findings that antidepressants (Malberg et al., 2000; Manev et al., 2001; Malberg and Duman, 2003; Santarelli et al., 2003; Warner-Schmidt and Duman, 2007; David et al., 2009) and lithium (Chen et al., 2000; Hashimoto et al., 2003; Silva et al., 2008; Wexler et al., 2008) increase neurogenesis in mice, and chronic stress associated with depression-like behaviors decreases neurogenesis (Malberg and Duman, 2003; Dranovsky and Hen, 2006; McEwen, 2008). Dysregulated GSK3 in mood disorders may contribute to deficient neurogenesis because neurogenesis is impaired by 40% in GSK3 knockin mice (Eom and Jope, 2009), neurogenesis was increased by GSK3 deletion (Kim et al., 2009), and GSK3 overexpression inhibited, and the GSK3 inhibitor SB216763 increased, neural precursor cell proliferation that is impaired in mice with DISC1 mutations (Mao et al., 2009). Furthermore, the stimulatory actions of fluoxetine and lithium on neurogenesis were blocked in GSK3 knockin mice in which the drugs could not increase the inhibitory serine-phosphorylation (Eom and Jope, 2009). These results indicate that hyperactive GSK3 impairs neurogenesis and that fluoxetine and lithium need to inhibit GSK3 by serine-phosphorylation to promote neurogenesis. These findings raise the possibilities that impaired neurogenesis by dysregulated GSK3 contributes to increasing susceptibility to mood disorders, and that the rescue of neurogenesis contributes to responses to therapeutic drugs.
Substantial evidence has accumulated demonstrating that mood disorders are associated with activation of the inflammatory system and other alterations of the immune system (Chourbaji et al., 2008; Dantzer et al., 2008; Miller et al., 2009; Rivest, 2009; Miller, 2010). Additionally, inflammatory cytokines impair glucocorticoid responsiveness, raising the possibility that the chronic inflammation associated with mood disorders contributes to reduced glucocorticoid responses in these disorders (Pace et al., 2007). A crucial role for GSK3 in promoting inflammation was first established by the finding that GSK3 promotes the production of several pro-inflammatory cytokines following stimulation of multiple types of Toll-like receptors in human monocytes (Martin et al., 2005). GSK3 deficiency induced with GSK3 inhibitors or by molecular means greatly reduced the production of several pro-inflammatory cytokines. Remarkably, GSK3 regulates oppositely the anti-inflammatory cytokine IL-10, so GSK3 inhibition increased IL-10 levels (Martin et al., 2005). Lithium and other GSK3 inhibitors also reduced by >90% inflammatory cytokine production by mouse primary astrocytes (Beurel and Jope, 2009a) and microglia (Yuskaitis and Jope, 2009), similarly to peripheral cells (Beurel and Jope, 2009b), and GSK3 counteracts down-regulation of inflammation (Beurel and Jope, 2010). Mechanisms for these actions include the findings that GSK3 is required for activation of NF-κB (Hoeflich et al., 2000; Martin et al., 2005) and STAT3 and STAT5 (Beurel and Jope, 2008), critical transcription factors in inflammation. In vivo, chronic administration of a therapeutically relevant dose of lithium rescued 70% of mice from an otherwise 100% lethal inflammatory response to lipopolysaccharide (Martin et al., 2005). In vivo chronic lithium treatment also markedly suppressed EAE (experimental autoimmune encephalomyelitis), an animal model of multiple sclerosis that involves substantial neuroinflammation, and the production of inflammatory Th17 cells that contribute to EAE pathogenesis (De Sarno et al., 2008; Beurel et al., 2011a). These results demonstrate that a therapeutically relevant dose of lithium effectively ameliorates CNS inflammatory diseases in vivo. These and other immune-regulating actions of GSK3 (Beurel et al., 2010) demonstrate that dysregulated GSK3 may contribute to immune system alterations that are associated with mood disorders and suggest that the immune system actions of lithium may contribute to its therapeutic effects in mood disorders.
Thus, stress response mechanisms, neurogenesis, and immune system abnormalities, are examples of processes strongly regulated by GSK3 that may be disrupted in mood disorders. There are multiple additional cellular functions regulated by GSK3 that may be crucial in mood disorders, such as circadian rhythm alterations, mitochondrial function, neurotransmitter synthesis and receptor-induced signaling, that are equally important potential targets. These three are only presented as representative examples of the concept that focusing on GSK3-regulated processes, rather than individual substrates of GSK3, may be most informative for advancing the understanding of how dysregulated GSK3 promotes susceptibility to mood disorders.
Summary
Figure 1 presents a simplified summary of how GSK3 is proposed to be involved in mood disorders. Clusters of symptoms define these disorders, which are undoubtedly induced by multiple combinations of genetic and environmental influences. An important outcome is impaired inhibitory regulation of GSK3, albeit not globally, but in particular circuits, cells, and signaling pathways. Substantial evidence now supports the concept that depression is associated with reduced signaling that otherwise would maintain GSK3 inhibited, which may involve deficient serotonin or BDNF, for example, and genetic changes, such as diminished functional DISC1 that otherwise contributes to GSK3 inhibition. Increased activation of GSK3 also may occur in mania, although in this case it may not result from deficient inhibitory signaling to GSK3 but from excessive activating signaling, such as can be mediated by increased dopaminergic signaling through dopamine D2 receptors. Because many signals converge on GSK3, which integrates these to modulate cellular responses, pharmacologically bolstering the inhibition of GSK3 can compensate for multiple combinations of genetic and environmental influences to promote the re-establishment of mood stability, thus counteracting conditions that would otherwise induce extremes in mood fluctuations. Therefore, although alterations in GSK3 activity may not constitute a primary insult in mood disorders, the role of GSK3 as an integrator of multiple signals allows therapies directed toward inhibiting GSK3 to compensate for a variety of genetic and environmental conditions that disturb mood homeostasis.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Research in the author’s laboratory was funded by the NIMH (MH038752).
References
Alimohamad, H., Rajakumar, N., Seah, Y. H., and Rushlow, W. (2005). Antipsychotics alter the protein expression levels of β-catenin and GSK-3 in the rat medial prefrontal cortex and striatum. Biol. Psychiatry 57, 533–542.
Andreazza, A. C., Kauer-Sant’Anna, M., Frey, B. N., Bond, D. J., Kapczinski, F., Young, L. T., and Yatham, L. N. (2008). Oxidative stress markers in bipolar disorder: a meta-analysis. J. Affect. Disord. 111, 135–144.
Antelman, S. M., Caggiula, A. R., Kucinski, B. J., Fowler, H., Gershon, S., Edwards, D. J., Austin, M. C., Stiller, R., Kiss, S., and Kocan, D. (1998). The effects of lithium on a potential cycling model of bipolar disorder. Prog. Neuropsychopharmacol. Biol. Psychiatry 22, 495–510.
Bain, J., Plater, L., Elliott, M., Shpiro, N., Hastie, C. J., McLauchlan, H., Klevernic, I., Arthur, J. S., Alessi, D. R., and Cohen, P. (2007). The selectivity of protein kinase inhibitors: a further update. Biochem. J. 408, 297–315.
Beaulieu, J. M., Marion, S., Rodriguiz, R. M., Medvedev, I. O., Sotnikova, T. D., Ghisi, V., Wetsel, W. C., Lefkowitz, R. J., Gainetdinov, R. R., and Caron, M. G. (2008a). A β-arrestin 2 signaling complex mediates lithium action on behavior. Cell 132, 125–136.
Beaulieu, J. M., Zhang, X., Rodriguiz, R. M., Sotnikova, T. D., Cools, M. J., Wetsel, W. C., Gainetdinov, R. R., and Caron, M. G. (2008b). Role of GSK3β in behavioral abnormalities induced by serotonin deficiency. Proc. Natl. Acad. Sci. U.S.A. 105, 1333–1338.
Beaulieu, J. M., Sotnikova, T. D., Marion, S., Lefkowitz, R. J., Gainetdinov, R. R., and Caron, M. G. (2005). An Akt/beta-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell 122, 261–273.
Beaulieu, J. M., Sotnikova, T. D., Yao, W. D., Kockeritz, L., Woodgett, J. R., Gainetdinov, R. R., and Caron, M. G. (2004). Lithium antagonizes dopamine-dependent behaviors mediated by an AKT/glycogen synthase kinase 3 signaling cascade. Proc. Natl. Acad. Sci. U.S.A. 101, 5099–5104.
Benedetti, F., Bernasconi, A., Lorenzi, C., Pontiggia, A., Serretti, A., Colombo, C., and Smeraldi, E. (2004a). A single nucleotide polymorphism in glycogen synthase kinase 3β promoter gene influences onset of illness in patients affected by bipolar disorder. Neurosci. Lett. 355, 37–40.
Benedetti, F., Serretti, A., Colombo, C., Lorenzi, C., Tubazio, V., and Smeraldi, E. (2004b). A glycogen synthase kinase 3β promoter gene single nucleotide polymorphism is associated with age at onset and response to total sleep deprivation in bipolar depression. Neurosci. Lett. 368, 123–126.
Benedetti, F., Serretti, A., Pontiggia, A., Bernasconi, A., Lorenzi, C., Colombo, C., and Smeraldi, E. (2005). Long-term response to lithium salts in bipolar illness is influenced by the glycogen synthase kinase 3β-50 T/C SNP. Neurosci. Lett. 376, 51–55.
Berggren, U., Tallstedt, L., Ahlenius, S., and Engel, J. (1978). The effect of lithium on amphetamine-induced locomotor stimulation. Psychopharmacology (Berl.) 59, 41–45.
Berns, G. S., and Nemeroff, C. B. (2003). The neurobiology of bipolar disorder. Am. J. Med. Genet. C Semin. Med. Genet. 123C, 76–84.
Beurel, E., and Jope, R. S. (2006). The paradoxical pro- and anti-apoptotic actions of GSK3 in the intrinsic and extrinsic apoptosis signaling pathways. Prog. Neurobiol. 79, 173–189.
Beurel, E., and Jope, R. S. (2008). Differential regulation of STAT family members by glycogen synthase kinase-3. J. Biol. Chem. 283, 21934–21944.
Beurel, E., and Jope, R. S. (2009a). Lipopolysaccharide-induced interleukin-6 production is controlled by glycogen synthase kinase-3 and STAT3 in the brain. J. Neuroinflammation 6, 9.
Beurel, E., and Jope, R. S. (2009b). Glycogen synthase kinase-3 promotes the synergistic action of interferon-γ on lipopolysaccharide-induced IL-6 production in RAW264.7 cells. Cell. Signal. 21, 978–985.
Beurel, E., and Jope, R. S. (2010). Glycogen synthase kinase-3 controls inflammatory tolerance in astrocytes. Neuroscience 169, 1063–1070.
Beurel, E., Michalek, S. M., and Jope, R. S. (2010). Innate and adaptive immune responses regulated by glycogen synthase kinase-3. Trends Immunol. 31, 24–31.
Beurel, E., Wen, Y.-I., Michalek, S. M., Harrington, L. E., and Jope, R. S. (2011a). Glycogen synthase kinase-3 is an early determinant in the differentiation of pathogenic Th17 cells. J. Immunol. 186, 1391–1398.
Beurel, E., Song, L., and Jope, R. S. (2011b). Inhibition of glycogen synthase kinase-3 is necessary for the rapid antidepressant effect of ketamine in mice. Mol. Psychiatry (in press).
Bhat, R., Xue, Y., Berg, S., Hellberg, S., Ormö, M., Nilsson, Y., Radesäter, A. C., Jerning, E., Markgren, P. O., Borgegård, T., Nylöf, M., Giménez-Cassina, A., Hernández, F., Lucas, J. J., Díaz-Nido, J., and Avila, J. (2003). Structural insights and biological effects of glycogen synthase kinase 3-specific inhibitor AR-A014418. J. Biol. Chem. 278, 45937–45945.
Borison, R. L., Sabelli, H. C., Maple, P. J., Havdala, H. S., and Diamond, B. I. (1978). Lithium prevention of amphetamine-induced “manic” excitement and of reserpine-induced “depression” in mice: possible role of 2-phenylethylamine. Psychopharmacology (Berl.) 59, 259–262.
Cade, J. F. (1949). Lithium salts in the treatment of psychotic excitement. Med. J. Aust. 2, 349–35.
Can, A., Blackwell, R. A., Piantadosi, S. C., Dao, D. T., O’Donnell, K. C., and Gould, T. D. (2011). Antidepressant-like responses to lithium in genetically diverse mouse strains. Genes Brain Behav. 10, 434–443.
Chen, G., Henter, I. D., and Manji, H. K. (2010). Translational research in bipolar disorder: emerging insights from genetically based models. Mol. Psychiatry 15, 883–895.
Chen, G., Rajkowska, G., Du, F., Seraji-Bozorgzad, N., and Manji, H. K. (2000). Enhancement of hippocampal neurogenesis by lithium. J. Neurochem. 75, 1729–1734.
Chiu, C. T., and Chuang, D. M. (2010). Molecular actions and therapeutic potential of lithium in preclinical and clinical studies of CNS disorders. Pharmacol. Ther. 128, 281–304.
Chourbaji, S., Vogt, M. A., and Gass, P. (2008). Mice that under- or overexpress glucocorticoid receptors as models for depression or posttraumatic stress disorder. Prog. Brain Res. 167, 65–77.
Chourbaji, S., Zacher, C., Sanchis-Segura, C., Dormann, C., Vollmayr, B., and Gass, P. (2005). Learned helplessness: validity and reliability of depressive-like states in mice. Brain Res. Brain Res. Protoc. 16, 70–78.
Clapcote, S. J., Lipina, T. V., Millar, J. K., Mackie, S., Christie, S., Ogawa, F., Lerch, J. P., Trimble, K., Uchiyama, M., Sakuraba, Y., Kaneda, H., Shiroishi, T., Houslay, M. D., Henkelman, R. M., Sled, J. G., Gondo, Y., Porteous, D. J., and Roder, J. C. (2007). Behavioral phenotypes of Disc1 missense mutations in mice. Neuron 54, 387–402.
Coghlan, M. P., Culbert, A. A., Cross, D. A., Corcoran, S. L., Yates, J. W., Pearce, N. J., Rausch, O. L., Murphy, G. J., Carter, P. S., Roxbee Cox, L., Mills, D., Brown, M. J., Haigh, D., Ward, R. W., Smith, D. G., Murray, K. J., Reith, A. D., and Holder, J. C. (2000). Selective small molecule inhibitors of glycogen synthase kinase-3 modulate glycogen metabolism and gene transcription. Chem. Biol. 7, 793–803.
Cox, C., Harrison-Read, P. E., Steinberg, H., and Tomkiewicz, M. (1971). Lithium attenuates drug-induced hyperactivity in rats. Nature 232, 336–338.
Creson, T. K., Austin, D. R., Shaltiel, G., McCammon, J., Wess, J., Manji, H. K., and Chen, G. (2011). Lithium treatment attenuates muscarinic M(1) receptor dysfunction. Bipolar Disord. 13, 238–249.
Cryan, J. F., and Mombereau, C. (2004). In search of a depressed mouse: utility of models for studying depression-related behavior in genetically modified mice. Mol. Psychiatry 9, 326–357.
Dantzer, R., O’Connor, J. C., Freund, G. G., Johnson, R. W., and Kelley, K. W. (2008). From inflammation to sickness and depression: when the immune system subjugates the brain. Nat. Rev. Neurosci. 9, 46–56.
David, D. J., Samuels, B. A., Rainer, Q., Wang, J. W., Marsteller, D., Mendez, I., Drew, M., Craig, D. A., Guiard, B. P., Guilloux, J. P., Artymyshyn, R. P., Gardier, A. M., Gerald, C., Antonijevic, I. A., Leonardo, E. D., and Hen, R. (2009). Neurogenesis-dependent and -independent effects of fluoxetine in an animal model of anxiety/depression. Neuron 62, 479–493.
Davies, S. P., Reddy, H., Caivano, M., and Cohen, P. (2000). Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 351, 95–105.
De Sarno, P., Axtell, R. C., Raman, C., Roth, K. A., Alessi, D. R., and Jope, R. S. (2008). Lithium prevents and ameliorates experimental autoimmune encephalomyelitis (EAE). J. Immunol. 181, 338–345.
De Sarno, P., Li, X., and Jope, R. S. (2002). Regulation of Akt and glycogen synthase kinase-3β phosphorylation by sodium valproate and lithium. Neuropharmacology 43, 1158–1164.
Dranovsky, A., and Hen, R. (2006). Hippocampal neurogenesis: regulation by stress and antidepressants. Biol. Psychiatry 59, 1136–1143.
Einat, H. (2007). Establishment of a battery of simple models for facets of bipolar disorder: a practical approach to achieve increased validity, better screening and possible insights into endophenotypes of disease. Behav. Genet. 37, 244–255.
Einat, H., Manji, H. K., and Belmaker, R. H. (2003). New approaches to modeling bipolar disorder. Psychopharmacol. Bull. 37, 47–63.
Engel, S. R., Creson, T. K., Hao, Y., Shen, Y., Maeng, S., Nekrasova, T., Landreth, G. E., Manji, H. K., and Chen, G. (2009). The extracellular signal-regulated kinase pathway contributes to the control of behavioral excitement. Mol. Psychiatry 14, 448–461.
Eom, T. Y., and Jope, R. S. (2009). Blocked inhibitory serine-phosphorylation of glycogen synthase kinase-3α/β impairs in vivo neural precursor cell proliferation. Biol. Psychiatry 66, 494–502.
Faria, M. S., and Teixeira, N. A. (1993). Reversal of learned helplessness by chronic lithium treatment at a prophylactic level. Braz. J. Med. Biol. Res. 26, 1201–1212.
Fitzgerald, P. J., Barkus, C., Feyder, M., Wiedholz, L. M., Chen, Y. C., Karlsson, R. M., Machado-Vieira, R., Graybeal, C., Sharp, T., Zarate, C., Harvey-White, J., Du, J., Sprengel, R., Gass, P., Bannerman, D., and Holmes, A. (2010). Does gene deletion of AMPA GluA1 phenocopy features of schizoaffective disorder? Neurobiol. Dis. 40, 608–621.
Flaisher-Grinberg, S., Overgaard, S., and Einat, H. (2009). Attenuation of high sweet solution preference by mood stabilizers: a possible mouse model for the increased reward-seeking domain of mania. J. Neurosci. Methods 177, 44–50.
Flemenbaum, A. (1977). Antagonism of behavioral effects of cocaine by lithium. Pharmacol. Biochem. Behav. 7, 83–85.
Force, T., and Woodgett, J. R. (2009). Unique and overlapping functions of GSK-3 isoforms in cell differentiation and proliferation and cardiovascular development. J. Biol. Chem. 284, 9643–9647.
Gao, Y., Payne, R. S., Schurr, A., Hougland, T., Lord, J., Herman, L., Lei, Z., Banerjee, P., and El-Mallakh, R. S. (2011). Memantine reduces mania-like symptoms in animal models. Psychiatry Res. 188, 366–371.
Gessa, G. L., Pani, L., Fadda, P., and Fratta, W. (1995). Sleep deprivation in the rat: an animal model of mania. Eur. Neuropsychopharmacol. 5(Suppl.), 89–93.
Gould, T. D., and Einat, H. (2007). Animal models of bipolar disorder and mood stabilizer efficacy: a critical need for improvement. Neurosci. Biobehav. Rev. 31, 825–831.
Gould, T. D., Einat, H., Bhat, R., and Manji, H. K. (2004). AR-A014418, a selective GSK-3 inhibitor, produces antidepressant-like effects in the forced swim test. Int. J. Neuropsychopharmacol. 7, 387–390.
Gould, T. D., Einat, H., O’Donnell, K. C., Picchini, A. M., Schloesser, R. J., and Manji, H. K. (2007a). Beta-catenin overexpression in the mouse brain phenocopies lithium-sensitive behaviors. Neuropsychopharmacology 32, 2173–2183.
Gould, T. D., O’Donnell, K. C., Picchini, A. M., and Manji, H. K. (2007b). Strain differences in lithium attenuation of d-amphetamine-induced hyperlocomotion: a mouse model for the genetics of clinical response to lithium. Neuropsychopharmacology 32, 1321–1333.
Gould, T. D., and Gottesman, I. I. (2006). Psychiatric endophenotypes and the development of valid animal models. Genes Brain Behav. 5, 113–119.
Hallcher, L. M., and Sherman, W. R. (1980). The effects of lithium ion and other agents on the activity of myo-inositol-1-phosphatase from bovine brain. J. Biol. Chem. 255, 10896–108901.
Harwood, A. J., and Agam, G. (2003). Search for a common mechanism of mood stabilizers. Biochem. Pharmacol. 66, 179–189.
Hashimoto, R., Senatorov, V., Kanai, H., Leeds, P., and Chuang, D. M. (2003). Lithium stimulates progenitor proliferation in cultured brain neurons. Neuroscience 117, 55–61.
Hicks, J. W., VanBrocklin, H. F., Wilson, A. A., Houle, S., and Vasdev, N. (2010). Radiolabeled small molecule protein kinase inhibitors for imaging with PET or SPECT. Molecules 15, 8260–8278.
Hikida, T., Jaaro-Peled, H., Seshadri, S., Oishi, K., Hookway, C., Kong, S., Wu, D., Xue, R., Andradé, M., Tankou, S., Mori, S., Gallagher, M., Ishizuka, K., Pletnikov, M., Kida, S., and Sawa, A. (2007). Dominant-negative DISC1 transgenic mice display schizophrenia-associated phenotypes detected by measures translatable to humans. Proc. Natl. Acad. Sci. U.S.A. 104, 14501–14506.
Hoeflich, K. P., Luo, J., Rubie, E. A., Tsao, M. S., Jin, O., and Woodgett, J. R. (2000). Requirement for glycogen synthase kinase-3β in cell survival and NF-κB activation. Nature 406, 86–90.
Inhorn, R. C., and Majerus, P. W. (1987). Inositol polyphosphate 1-phosphatase from calf brain. Purification and inhibition by Li+, Ca2+, and Mn2+. J. Biol. Chem. 262, 15946–15952.
Inkster, B., Nichols, T. E., Saemann, P. G., Auer, D. P., Holsboer, F., Muglia, P., and Matthews, P. M. (2009). Association of GSK3β polymorphisms with brain structural changes in major depressive disorder. Arch. Gen. Psychiatry 66, 721–728.
Inkster, B., Nichols, T. E., Saemann, P. G., Auer, D. P., Holsboer, F., Muglia, P., and Matthews, P. M. (2010). Pathway-based approaches to imaging genetics association studies: Wnt signaling, GSK3β substrates and major depression. Neuroimage 53, 908–917.
Jope, R. S. (1999). Anti-bipolar therapy: mechanism of action of lithium. Mol. Psychiatry 4, 117–128.
Jope, R. S., and Johnson, G. V. W. (2004). The glamour and gloom of glycogen synthase kinase-3 (GSK3). Trends Biochem. Sci. 29, 95–102.
Jornada, L. K., Moretti, M., Valvassori, S. S., Ferreira, C. L., Padilha, P. T., Arent, C. O., Fries, G. R., Kapczinski, F., and Quevedo, J. (2010). Effects of mood stabilizers on hippocampus and amygdala BDNF levels in an animal model of mania induced by ouabain. J. Psychiatr. Res. 44, 506–510.
Kaidanovich-Beilin, O., Lipina, T. V., Takao, K., van Eede, M., Hattori, S., Laliberté, C., Khan, M., Okamoto, K., Chambers, J. W., Fletcher, P. J., MacAulay, K., Doble, B. W., Henkelman, M., Miyakawa, T., Roder, J., and Woodgett, J. R. (2009). Abnormalities in brain structure and behavior in GSK-3α mutant mice. Mol Brain 19, 35.
Kaidanovich-Beilin, O., Milman, A., Weizman, A., Pick, C. G., and Eldar-Finkelman, H. (2004). Rapid antidepressive-like activity of specific glycogen synthase kinase-3 inhibitor and its effect on β-catenin in mouse hippocampus. Biol. Psychiatry 55, 781–784.
Kakefuda, K., Oyagi, A., Ishisaka, M., Tsuruma, K., Shimazawa, M., Yokota, K., Shirai, Y., Horie, K., Saito, N., Takeda, J., and Hara, H. (2010). Diacylglycerol kinase β knockout mice exhibit lithium-sensitive behavioral abnormalities. PLoS ONE 5, e13447. doi: 10.1371/journal.pone.0013447
Kalinichev, M., and Dawson, L. A. (2011). Evidence for antimanic efficacy of glycogen synthase kinase-3 (GSK3) inhibitors in a strain-specific model of acute mania. Int. J. Neuropsychopharmacol. 6, 1–17.
Karege, F., Perroud, N., Burkhardt, S., Fernandez, R., Ballmann, E., La Harpe, R., and Malafosse, A. (2011). Alterations in phosphatidylinositol 3-kinase activity and PTEN phosphatase in the prefrontal cortex of depressed suicide victims. Neuropsychobiology 63, 224–231.
Karege, F., Perroud, N., Burkhardt, S., Schwald, M., Ballmann, E., La Harpe, R., and Malafosse, A. (2007). Alteration in kinase activity but not in protein levels of protein kinase B and glycogen synthase kinase-3β in ventral prefrontal cortex of depressed suicide victims. Biol. Psychiatry 61, 240–245.
Kasahara, T., Kubota, M., Miyauchi, T., Noda, Y., Mouri, A., Nabeshima, T., and Kato, T. (2006). Mice with neuron-specific accumulation of mitochondrial DNA mutations show mood disorder-like phenotypes. Mol. Psychiatry 11, 577–593.
Kato, T., and Kato, N. (2000). Mitochondrial dysfunction in bipolar disorder. Bipolar Disord. 2, 180–190.
Kim, W. Y., Wang, X., Wu, Y., Doble, B. W., Patel, S., Woodgett, J. R., and Snider, W. D. (2009). GSK-3 is a master regulator of neural progenitor homeostasis. Nat. Neurosci. 12, 1390–1397.
Klein, P. S., and Melton, D. A. (1996). A molecular mechanism for the effect of lithium on development. Proc. Natl. Acad. Sci. U.S.A. 93, 8455–8459.
Kovacsics, C. E., Gottesman, I. I., and Gould, T. D. (2009). Lithium’s antisuicidal efficacy: elucidation of neurobiological targets using endophenotype strategies. Annu. Rev. Pharmacol. Toxicol. 49, 175–198.
Kozikowski, A. P., Gaisina, I. N., Yuan, H., Petukhov, P. A., Blond, S. Y., Fedolak, A., Caldarone, B., and McGonigle, P. (2007). Structure-based design leads to the identification of lithium mimetics that block mania-like effects in rodents. Possible new GSK-3β therapies for bipolar disorders. J. Am. Chem. Soc. 129, 8328–8332.
Lachman, H. M., Pedrosa, E., Petruolo, O. A., Cockerham, M., Papolos, A., Novak, T., Papolos, D. F., and Stopkova, P. (2007). Increase in GSK3β gene copy number variation in bipolar disorder. Am. J. Med. Genet. B Neuropsychiatr. Genet. 144B, 259–265.
Leclerc, S., Garnier, M., Hoessel, R., Marko, D., Bibb, J. A., Snyder, G. L., Greengard, P., Biernat, J., Wu, Y. Z., Mandelkow, E. M., Eisenbrand, G., and Meijer, L. (2001). Indirubins inhibit glycogen synthase kinase-3β and CDK5/p25, two protein kinases involved in abnormal tau phosphorylation in Alzheimer’s disease. A property common to most cyclin-dependent kinase inhibitors? J. Biol. Chem. 276, 251–260.
Lee, F. H., Kaidanovich-Beilin, O., Roder, J. C., Woodgett, J. R., and Wong, A. H. (2011). Genetic inactivation of GSK3α rescues spine deficits in Disc1-L100P mutant mice. Schizophr. Res. 129, 74–79.
Lee, K. Y., Ahn, Y. M., Joo, E. J., Jeong, S. H., Chang, J. S., Kim, S. C., and Kim, Y. S. (2006). No association of two common SNPs at position -1727 A/T, -50 C/T of GSK-3β polymorphisms with schizophrenia and bipolar disorder of Korean population. Neurosci. Lett. 395, 175–178.
Leost, M., Schultz, C., Link, A., Wu, Y. Z., Biernat, J., Mandelkow, E. M., Bibb, J. A., Snyder, G. L., Greengard, P., Zaharevitz, D. W., Gussio, R., Senderowicz, A. M., Sausville, E. A., Kunick, C., and Meijer, L. (2000). Paullones are potent inhibitors of glycogen synthase kinase-3β and cyclin-dependent kinase 5/p25. Eur. J. Biochem. 267, 5983–5994.
Li, W., Zhou, Y., Jentsch, J. D., Brown, R. A., Tian, X., Ehninger, D., Hennah, W., Peltonen, L., Lönnqvist, J., Huttunen, M. O., Kaprio, J., Trachtenberg, J. T., Silva, A. J., and Cannon, T. D. (2007). Specific developmental disruption of disrupted-in-schizophrenia-1 function results in schizophrenia-related phenotypes in mice. Proc. Natl. Acad. Sci. U.S.A. 104, 18280–18285.
Li, X., Liu, M., Cai, Z., Wang, G., and Li, X. (2010). Regulation of glycogen synthase kinase-3 during bipolar mania treatment. Bipolar Disord. 12, 741–752.
Li, X., Rosborough, K. M., Friedman, A. B., Zhu, W., and Roth, K. A. (2007a). Regulation of mouse brain glycogen synthase kinase-3 by atypical antipsychotics. Int. J. Neuropsychopharmacol. 10, 7–19.
Li, X., Friedman, A. B., Zhu, W., Wang, L., Boswell, S., May, R. S., Davis, L. L., and Jope, R. S. (2007b). Lithium regulates glycogen synthase kinase-3β in human peripheral blood mononuclear cells: implications in the treatment of bipolar disorder. Biol. Psychiatry 61, 216–222.
Li, X., Zhu, W., Roh, M. S., Friedman, A. B., Rosborough, K., and Jope, R. S. (2004). In vivo regulation of glycogen synthase kinase-3β (GSK3β) by serotonergic activity in mouse brain. Neuropsychopharmacology 29, 1426–1431.
Lie, D. C., Song, H., Colamarino, S. A., Ming, G. L., and Gage, F. H. (2004). Neurogenesis in the adult brain: new strategies for central nervous system diseases. Annu. Rev. Pharmacol. Toxicol. 44, 399–421.
Lien, R., Flaisher-Grinberg, S., Cleary, C., Hejny, M., and Einat, H. (2008). Behavioral effects of Bcl-2 deficiency: implications for affective disorders. Pharmacol Rep 60, 490–498.
Lipina, T. V., Kaidanovich-Beilin, O., Patel, S., Wang, M., Clapcote, S. J., Liu, F., Woodgett, J. R., and Roder, J. C. (2011a). Genetic and pharmacological evidence for schizophrenia-related Disc1 interaction with GSK-3. Synapse 65, 234–248.
Lipina, T. V., Wang, M., Liu, F., and Roder, J. C. (2011b). Synergistic interactions between PDE4B and GSK-3: DISC1 mutant mice. Neuropharmacology. doi: 10.1016/j.neuropharm.2011.02.020. [Epub ahead of print].
Lipina, T. V., Niwa, M., Jaaro-Peled, H., Fletcher, P. J., Seeman, P., Sawa, A., and Roder, J. C. (2010). Enhanced dopamine function in DISC1-L100P mutant mice: implications for schizophrenia. Genes Brain Behav. 9, 777–789.
Lucas, J. J., Hernandez, F., Gomez-Ramos, P., Moran, M. A., Hen, R., and Avila, J. (2001). Decreased nuclear β-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3β conditional transgenic mice. EMBO J. 20, 27–39.
MacAulay, K., and Woodgett, J. R. (2008). Targeting glycogen synthase kinase-3 (GSK-3) in the treatment of Type 2 diabetes. Expert Opin. Ther. Targets 12, 1265–1274.
Machado-Vieira, R., Kapczinski, F., and Soares, J. C. (2004). Perspectives for the development of animal models of bipolar disorder. Prog. Neuropsychopharmacol. Biol. Psychiatry 28, 209–224.
Malberg, J. E., and Duman, R. S. (2003). Cell proliferation in adult hippocampus is decreased by inescapable stress: reversal by fluoxetine treatment. Neuropsychopharmacology 28, 1562–1571.
Malberg, J. E., Eisch, A. J., Nestler, E. J., and Duman, R. S. (2000). Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J. Neurosci. 20, 9104–9110.
Manev, H., Uz, T., Smalheiser, N. R., and Manev, R. (2001). Antidepressants alter cell proliferation in the adult brain in vivo and in neural cultures in vitro. Eur. J. Pharmacol. 411, 67–70.
Manji, H. K., Moore, G. J., Rajkowska, G., and Chen, G. (2000). Neuroplasticity and cellular resilience in mood disorders. Mol. Psychiatry 5, 578–593.
Mao, Y., Ge, X., Frank, C. L., Madison, J. M., Koehler, A. N., Doud, M. K., Tassa, C., Berry, E. M., Soda, T., Singh, K. K., Biechele, T., Petryshen, T. L., Moon, R. T., Haggarty, S. J., and Tsai, L. H. (2009). Disrupted in schizophrenia 1 regulates neuronal progenitor proliferation via modulation of GSK3β/β-catenin signaling. Cell 136, 1017–1031.
Martin, M., Rehani, K., Jope, R. S., and Michalek, S. M. (2005). Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat. Immunol. 6, 777–784.
Martinez, A., Alonso, M., Castro, A., Pérez, C., and Moreno, F. J. (2002). First non-ATP competitive glycogen synthase kinase 3β (GSK-3β) inhibitors: thiadiazolidinones (TDZD)as potential drugs for the treatment of Alzheimer’s disease. J. Med. Chem. 45, 1292–1299.
Martinez, A., Castro, A., and Medina, M. (eds). (2006). Glycogen Synthase Kinase 3 (GSK-3) and its Inhibitors. Hoboken, NJ: John Wiley and Sons, Inc.
Matthews, K., Christmas, D., Swan, J., and Sorrell, E. (2005). Animal models of depression: navigating through the clinical fog. Neurosci. Biobehav. Rev. 29, 503–513.
McEwen, B. S. (2008). Central effects of stress hormones in health and disease: understanding the protective and damaging effects of stress and stress mediators. Eur. J. Pharmacol. 583, 174–185.
McManus, E. J., Sakamoto, K., Armit, L. J., Ronaldson, L., Shpiro, N., Marquez, R., and Alessi, D. R. (2005). Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by knockin analysis. EMBO J. 24, 1571–1583.
McNamara, R. K., Sullivan, J., and Richtand, N. M. (2008). Omega-3 fatty acid deficiency augments amphetamine-induced behavioral sensitization in adult mice: prevention by chronic lithium treatment. J. Psychiatr. Res. 42, 458–468.
Meijer, L., Flajolet, M., and Greengard, P. (2004). Pharmacological inhibitors of glycogen synthase kinase 3. Trends Pharmacol. Sci. 25, 471–480.
Miller, A. H., Maletic, V., and Raison, C. L. (2009). Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol. Psychiatry 65, 732–741.
Murray, J. T., Campbell, D. G., Morrice, N., Auld, G. C., Shpiro, N., Marquez, R., Peggie, M., Bain, J., Bloomberg, G. B., Grahammer, F., Lang, F., Wulff, P., Kuhl, D., and Cohen, P. (2004). Exploitation of KESTREL to identify NDRG family members as physiological substrates for SGK1 and GSK3. Biochem. J. 384, 477–488.
Nestler, E. J., Barrot, M., DiLeone, R. J., Eisch, A. J., Gold, S. J., and Monteggia, L. M. (2002). Neurobiology of depression. Neuron 34, 13–25.
Nestler, E. J., and Hyman, S. E. (2010). Animal models of neuropsychiatric disorders. Nat. Neurosci. 13, 1161–1169.
Ng, F., Berk, M., Dean, O., and Bush, A. I. (2008). Oxidative stress in psychiatric disorders: evidence base and therapeutic implications. Int. J. Neuropsychopharmacol. 11, 851–876.
Nishiguchi, N., Breen, G., Russ, C., St Clair, D., and Collier, D. (2006). Association analysis of the glycogen synthase kinase-3β gene in bipolar disorder. Neurosci. Lett. 394, 243–245.
Nixon, M. K., Hascoet, M., Bourin, M., and Colombel, M. C. (1994). Additive effects of lithium and antidepressants in the forced swimming test: further evidence for involvement of the serotoninergic system. Psychopharmacology (Berl.) 115, 59–64.
O’Brien, W. T., Harper, A. D., Jove, F., Woodgett, J. R., Maretto, S., Piccolo, S., and Klein, P. S. (2004). Glycogen synthase kinase-3β haploinsufficiency mimics the behavioral and molecular effects of lithium. J. Neurosci. 24, 6791–6798.
O’Brien, W. T., Huang, J., Buccafusca, R., Garskof, J., Valvezan, J. A., Berry, G. T., and Klein, P. S. (2011). Essential role of glycogen synthase kinase-3 in β-arrestin-2 complex formation and lithium-sensitive behaviors. J. Clin. Invest. (in press).
O’Donnell, K. C., and Gould, T. D. (2007). The behavioral actions of lithium in rodent models: leads to develop novel therapeutics. Neurosci. Biobehav. Rev. 31, 932–962.
Okamoto, H., Voleti, B., Banasr, M., Sarhan, M., Duric, V., Girgenti, M. J., Dileone, R. J., Newton, S. S., and Duman, R. S. (2010). Wnt2 expression and signaling is increased by different classes of antidepressant treatments. Biol. Psychiatry 68, 521–527.
Omata, N., Chiu, C. T., Moya, P. R., Leng, Y., Wang, Z., Hunsberger, J. G., Leeds, P., and Chuang, D. M. (2011). Lentivirally mediated GSK-3β silencing in the hippocampal dentate gyrus induces antidepressant-like effects in stressed mice. Int. J. Neuropsychopharmacol. 14, 711–717.
Pace, T. W., Hu, F., and Miller, A. H. (2007). Cytokine-effects on glucocorticoid receptor function: relevance to glucocorticoid resistance and the pathophysiology and treatment of major depression. Brain Behav. Immun. 21, 9–19.
Pan, J. Q., Lewis, M. C., Ketterman, J. K., Clore, E. L., Riley, M., Richards, K. R., Berry-Scott, E., Liu, X., Wagner, F. F., Holson, E. B., Neve, R. L., Biechele, T. L., Moon, R. T., Scolnick, E. M., Petryshen, T. L., and Haggarty, S. J. (2011). AKT kinase activity is required for lithium to modulate mood-related behaviors in mice. Neuropsychopharmacology 36, 1397–1411.
Pandey, G. N., Dwivedi, Y., Rizavi, H. S., Teppen, T., Gaszner, G. L., Roberts, R. C., and Conley, R. R. (2009). GSK-3β gene expression in human postmortem brain: regional distribution, effects of age and suicide. Neurochem. Res. 34, 274–285.
Phiel, C. J., and Klein, P. S. (2001). Molecular targets of lithium action. Annu. Rev. Pharmacol. Toxicol. 41, 789–813.
Pilot-Storck, F., Chopin, E., Rual, J. F., Baudot, A., Dobrokhotov, P., Robinson-Rechavi, M., Brun, C., Cusick, M. E., Hill, D. E., Schaeffer, L., Vidal, M., and Goillot, E. (2010). Interactome mapping of the phosphatidylinositol 3-kinase-mammalian target of rapamycin pathway identifies deformed epidermal autoregulatory factor-1 as a new glycogen synthase kinase-3 interactor. Mol. Cell Proteomics 9, 1578–1593.
Pletnikov, M. V., Ayhan, Y., Nikolskaia, O., Xu, Y., Ovanesov, M. V., Huang, H., Mori, S., Moran, T. H., and Ross, C. A. (2008). Inducible expression of mutant human DISC1 in mice is associated with brain and behavioral abnormalities reminiscent of schizophrenia. Mol. Psychiatry 13, 173–186.
Plotkin, B., Kaidanovich, O., Talior, I., and Eldar-Finkelman, H. (2003). Insulin mimetic action of synthetic phosphorylated peptide inhibitors of glycogen synthase kinase-3. J. Pharmacol. Exp. Ther. 305, 974–980.
Polter, A., Beurel, E., Yang, S., Garner, R., Song, L., Miller, C. A., Sweatt, J. D., McMahon, L., Bartolucci, A. A., Li, X., and Jope, R. S. (2010). Deficiency in the inhibitory serine-phosphorylation of glycogen synthase kinase-3 increases sensitivity to mood disturbances. Neuropsychopharmacology 35, 1761–1774.
Porsolt, R. D., Le Pichon, M., and Jalfre, M. (1977). Depression: a new animal model sensitive to antidepressant treatments. Nature 266, 730–732.
Prickaerts, J., Moechars, D., Cryns, K., Lenaerts, I., van Craenendonck, H., Goris, I., Daneels, G., Bouwknecht, J. A., and Steckler, T. (2006). Transgenic mice overexpressing glycogen synthase kinase 3β: a putative model of hyperactivity and mania. J. Neurosci. 26, 9022–9029.
Rajkowska, G., and Miguel-Hidalgo, J. J. (2007). Gliogenesis and glial pathology in depression. CNS Neurol. Disord. Drug Targets 6, 219–233.
Ray, W. J. Jr., Szymanki, E. S., and Ng, L. (1978). The binding of lithium and of anionic metabolites to phosphoglucomutase. Biochim. Biophys. Acta 522, 434–442.
Redrobe, J. P., and Bourin, M. (1999). The effect of lithium administration in animal models of depression: a short review. Fundam. Clin. Pharmacol. 13, 293–299.
Rivest, S. (2009). Regulation of innate immune responses in the brain. Nat. Rev. Immunol. 9, 429–439.
Roh, M. S., Seo, M. S., Kim, Y., Kim, S. H., Jeon, W. J., Ahn, Y. M., Kang, U. G., Juhnn, Y. S., and Kim, Y. S. (2007). Haloperidol and clozapine differentially regulate signals upstream of glycogen synthase kinase 3 in the rat frontal cortex. Exp. Mol. Med. 39, 353–360.
Roybal, K., Theobold, D., Graham, A., DiNieri, J. A., Russo, S. J., Krishnan, V., Chakravarty, S., Peevey, J., Oehrlein, N., Birnbaum, S., Vitaterna, M. H., Orsulak, P., Takahashi, J. S., Nestler, E. J., Carlezon, W. A. Jr., and McClung, C. A. (2007). Mania-like behavior induced by disruption of CLOCK. Proc. Natl. Acad. Sci. U.S.A. 104, 6406–6411.
Santarelli, L., Saxe, M., Gross, C., Surget, A., Battaglia, F., Dulawa, S., Weisstaub, N., Lee, J., Duman, R., Arancio, O., Belzung, C., and Hen, R. (2003). Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 301, 805–809.
Saus, E., Soria, V., Escaramís, G., Crespo, J. M., Valero, J., Gutiérrez-Zotes, A., Martorell, L., Vilella, E., Menchón, J. M., Estivill, X., Gratacòs, M., and Urretavizcaya, M. (2010). A haplotype of glycogen synthase kinase 3β is associated with early onset of unipolar major depression. Genes Brain Behav. 9, 799–807.
Selenica, M. L., Jensen, H. S., Larsen, A. K., Pedersen, M. L., Helboe, L., Leist, M., and Lotharius, J. (2007). Efficacy of small-molecule glycogen synthase kinase-3 inhibitors in the postnatal rat model of tau hyperphosphorylation. Br. J. Pharmacol. 152, 959–979.
Shaltiel, G., Maeng, S., Malkesman, O., Pearson, B., Schloesser, R. J., Tragon, T., Rogawski, M., Gasior, M., Luckenbaugh, D., Chen, G., and Manji, H. K. (2008). Evidence for the involvement of the kainate receptor subunit GluR6 (GRIK2) in mediating behavioral displays related to behavioral symptoms of mania. Mol. Psychiatry 13, 858–872.
Shapira, M., Licht, A., Milman, A., Pick, C. G., Shohami, E., and Eldar-Finkelman, H. (2007). Role of glycogen synthase kinase-3β in early depressive behavior induced by mild traumatic brain injury. Mol. Cell. Neurosci. 34, 571–577.
Shen, S., Lang, B., Nakamoto, C., Zhang, F., Pu, J., Kuan, S. L., Chatzi, C., He, S., Mackie, I., Brandon, N. J., Marquis, K. L., Day, M., Hurko, O., McCaig, C. D., Riedel, G., and St. Clair, D. (2008). Schizophrenia-related neural and behavioral phenotypes in transgenic mice expressing truncated Disc1. J. Neurosci. 28, 10893–108904.
Silva, R., Mesquita, A. R., Bessa, J., Sousa, J. C., Sotiropoulos, I., Leão, P., Almeida, O. F., and Sousa, N. (2008). Lithium blocks stress-induced changes in depressive-like behavior and hippocampal cell fate: the role of glycogen-synthase-kinase-3β. Neuroscience 152, 656–669.
Smith, D. F. (1980). Lithium and motor activity of animals: effects and possible mechanism of action. Int. Pharmacopsychiatry 13, 197–217.
Song, L., De Sarno, P., and Jope, R. S. (2002). Central role of glycogen synthase kinase-3β in endoplasmic stress-induced caspase-3 activation. J. Biol. Chem. 277, 44701–44708.
Spiegelberg, B. D., Xiong, J. P., Smith, J. J., Gu, R. F., and York, J. D. (1999). Cloning and characterization of a mammalian lithium-sensitive bisphosphate 3′-nucleotidase inhibited by inositol 1,4-bisphosphate. J. Biol. Chem. 274, 13619–13628.
Spittaels, K., Van den Haute, C., Van Dorpe, J., Terwel, D., Vandezande, K., Lasrado, R., Bruynseels, K., Irizarry, M., Verhoye, M., Van Lint, J., Vandenheede, J. R., Ashton, D., Mercken, M., Loos, R., Hyman, B., Van der Linden, A., Geerts, H., and Van Leuven, F. (2002). Neonatal neuronal overexpression of glycogen synthase kinase-3β reduces brain size in transgenic mice. Neuroscience 113, 797–808.
Stambolic, V., Ruel, L., and Woodgett, J. R. (1996). Lithium inhibits glycogen synthase kinase-3 activity and mimics wingless signalling in intact cells. Curr. Biol. 6, 1664–1668.
Steckert, A. V., Valvassori, S. S., Moretti, M., Dal-Pizzol, F., and Quevedo, J. (2010). Role of oxidative stress in the pathophysiology of bipolar disorder. Neurochem. Res. 35, 1295–1301.
Szczepankiewicz, A., Skibinska, M., Hauser, J., Slopien, A., Leszczynska-Rodziewicz, A., Kapelski, P., Dmitrzak-Weglarz, M., Czerski, P. M., and Rybakowski, J. K. (2006a). Association analysis of the GSK-3β T-50C gene polymorphism with schizophrenia and bipolar disorder. Neuropsychobiology 53, 51–56.
Szczepankiewicz, A., Rybakowski, J. K., Suwalska, A., Skibinska, M., Leszczynska-Rodziewicz, A., Dmitrzak-Weglarz, M., Czerski, P. M., and Hauser, J. (2006b). Association study of the glycogen synthase kinase-3β gene polymorphism with prophylactic lithium response in bipolar patients. World J. Biol. Psychiatry 7, 158–161.
Taelman, V. F., Dobrowolski, R., Plouhinec, J. L., Fuentealba, L. C., Vorwald, P. P., Gumper, I., Sabatini, D. D., and De Robertis, E. M. (2010). Wnt signaling requires sequestration of glycogen synthase kinase 3 inside multivesicular endosomes. Cell 143, 1136–1148.
Thomsen, K., and Olesen, O. V. (1974). Long-term lithium administration to rats. Lithium and sodium dosage and administration, avoidance of intoxication, polyuric control rats. Int. Pharmacopsychiatry 9, 118–124.
Tsai, S. J., Liou, Y. J., Hong, C. J., Yu, Y. W., and Chen, T. J. (2008). Glycogen synthase kinase-3β gene is associated with antidepressant treatment response in Chinese major depressive disorder. Pharmacogenomics J. 8, 384–390.
Vasdev, N., Garcia, A., Stableford, W. T., Young, A. B., Meyer, J. H., Houle, S., and Wilson, A. A. (2005). Synthesis and ex vivo evaluation of carbon-11 labelled N-(4-methoxybenzyl)-N′-(5-nitro-1,3-thiazol-2-yl)urea ([11C]AR-A014418): a radiolabelled glycogen synthase kinase-3β specific inhibitor for PET studies. Bioorg. Med. Chem. Lett. 15, 5270–5273.
Wagman, A. S., Johnson, K. W., and Bussiere, D. E. (2004). Discovery and development of GSK3 inhibitors for the treatment of type 2 diabetes. Curr. Pharm. Des. 10, 1105–1137.
Wang, J. F. (2007). Defects of mitochondrial electron transport chain in bipolar disorder: implications for mood-stabilizing treatment. Can. J. Psychiatry 52, 753–762.
Warner-Schmidt, J. L., and Duman, R. S. (2007). VEGF is an essential mediator of the neurogenic and behavioral actions of antidepressants. Proc. Natl. Acad. Sci. U.S.A. 104, 4647–4652.
Watcharasit, P., Bijur, G. N., Zmijewski, J. W., Song, L., Zmijewska, A., Chen, X., Johnson, G. V. W., and Jope, R. S. (2002). Direct, activating interaction between glycogen synthase kinase-3β and p53 after DNA damage. Proc. Natl. Acad. Sci. U.S.A. 99, 7951–7955.
Wexler, E. M., Geschwind, D. H., and Palmer, T. D. (2008). Lithium regulates adult hippocampal progenitor development through canonical Wnt pathway activation. Mol. Psychiatry 13, 285–292.
Wilkinson, M. B., Dias, C., Magida, J., Mazei-Robison, M., Lobo, M., Kennedy, P., Dietz, D., Covington, H. III, Russo, S., Neve, R., Ghose, S., Tamminga, C., and Nestler, E. J. (2011). A novel role of the WNT-dishevelled-GSK3β signaling cascade in the mouse nucleus accumbens in a social defeat model of depression. J. Neurosci. 31, 9084–9092.
Wong, M.-L., and Licinio, J. (2001). Research and treatment approaches to depression. Nat. Rev. Neurosci. 2, 343–351.
Yuskaitis, C. J., and Jope, R. S. (2009). Glycogen synthase kinase-3 regulates microglial migration, inflammation, and inflammation-induced neurotoxicity. Cell. Signal. 21, 264–273.
Keywords: glycogen synthase kinase-3, lithium, mood stabilizers, depression, antidepressants
Citation: Jope RS (2011) Glycogen synthase kinase-3 in the etiology and treatment of mood disorders. Front. Mol. Neurosci. 4:16. doi: 10.3389/fnmol.2011.00016
Received: 19 June 2011; Paper pending published: 08 July 2011;
Accepted: 26 July 2011; Published online: 09 August 2011.
Edited by:
Oksana Kaidanovich-Beilin, Samuel Lunenfeld Research Institute, CanadaReviewed by:
Urs Albrecht, University of Fribourg, SwitzerlandYe He, University of California San Francisco, USA
Copyright: © 2011 Jope. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Richard Scott Jope, Department of Psychiatry and Behavioral Neurobiology, University of Alabama at Birmingham, 1720 Seventh Avenue South, SC1057, Birmingham, AL 35294-0017, USA. e-mail:am9wZUB1YWIuZWR1