 Jean-Martin Beaulieu1*
Jean-Martin Beaulieu1* Thomas Del’Guidice1 Tatyana D. Sotnikova2
Thomas Del’Guidice1 Tatyana D. Sotnikova2 Morgane Lemasson1
Morgane Lemasson1 Raul R. Gainetdinov2*
Raul R. Gainetdinov2*- 1 Department of Psychiatry and Neuroscience, Université Laval-CRULRG, Québec, QC, Canada
- 2 Department of Neuroscience and Brain Technologies, Istituto Italiano di Tecnologia, Genova, Italy
Brain dopamine receptors have been preferred targets for numerous pharmacological compounds developed for the treatment of various neuropsychiatric disorders. Recent discovery that D2 dopamine receptors, in addition to cAMP pathways, can engage also in Akt/GSK3 signaling cascade provided a new framework to understand intracellular signaling mechanisms involved in dopamine-related behaviors and pathologies. Here we review a recent progress in understanding the role of Akt, GSK3, and related signaling molecules in dopamine receptor signaling and functions. Particularly, we focus on the molecular mechanisms involved, interacting partners, role of these signaling events in the action of antipsychotics, psychostimulants, and antidepressants as well as involvement in pathophysiology of schizophrenia, bipolar disorder, and Parkinson’s disease. Further understanding of the role of Akt/GSK3 signaling in dopamine receptor functions could provide novel targets for pharmacological interventions in dopamine-related disorders.
Introduction
Dopamine is a catecholamine neurotransmitter involved in the regulation of multiple functions in the CNS and periphery, including locomotion, cognition, emotional behaviors, and endocrine regulation (Carlsson, 1993; Beaulieu and Gainetdinov, 2011). Consequently, abnormal dopamine signaling could play a role in many neuropsychiatric disorders such as schizophrenia, bipolar disorder, depression, Parkinson’s disease, attention deficit hyperactivity disorder (ADHD), Tourette syndrome, and drug abuse. Dopamine exerts its biological functions by activation and signaling through two different groups of G protein-coupled receptors (GPCRs). The first category, the D1 family, comprises the D1- and D5-receptors (D1R and D5R). The second family, named D2-class receptors, is formed by the D3R, D4R along with the short and long splice variants of the D2R (Missale et al., 1998; Beaulieu and Gainetdinov, 2011).
Historically, it was believed that dopamine receptors signal exclusively through G protein-dependent cellular processes. D1R is mostly coupled to Gαs/olf proteins and stimulate the activity of adenylate cyclase and the production of the second messenger cAMP. In contrast, D2R is associated to Gαi/o protein to inhibit the production of cAMP (Spano et al., 1978; Kebabian and Calne, 1979). However, more recent investigations have revealed that dopamine receptors can exert some of their biological effects through alternative signaling pathways which may or may not involve cAMP (Beaulieu et al., 2004, 2005; Hasbi et al., 2009). For instance, there are indications that both D1R and D2R can transactivate the brain derived neurotrophic factor (BDNF) receptor in neurons (Swift et al., 2011). These two dopamine receptors have also been shown to regulate the internalization of calcium channels through direct protein:protein interaction in vivo (Kisilevsky and Zamponi, 2008; Kisilevsky et al., 2008). Finally, there is strong evidence that dopamine receptors can signal in vivo by activating cAMP-independent mechanisms involving the multifunctional adaptor protein beta-arrestin 2 (βArr2; Beaulieu et al., 2004, 2005). This protein is generally recruited to activated/phosphorylated GPCRs and plays a central role in receptor desensitization through receptor-G protein uncoupling and clathrin-dependent internalization (Lohse et al., 1990; Ferguson et al., 1996). In addition to this well established role, βArr2 can act as a scaffold for kinases and phosphatases by forming a signaling complex that leads to the activation of various G protein-independent intracellular signaling cascades (Figure 1) including the Akt/GSK3 pathway (Luttrell et al., 1999; Beaulieu et al., 2004, 2005; Luttrell and Gesty-Palmer, 2010).
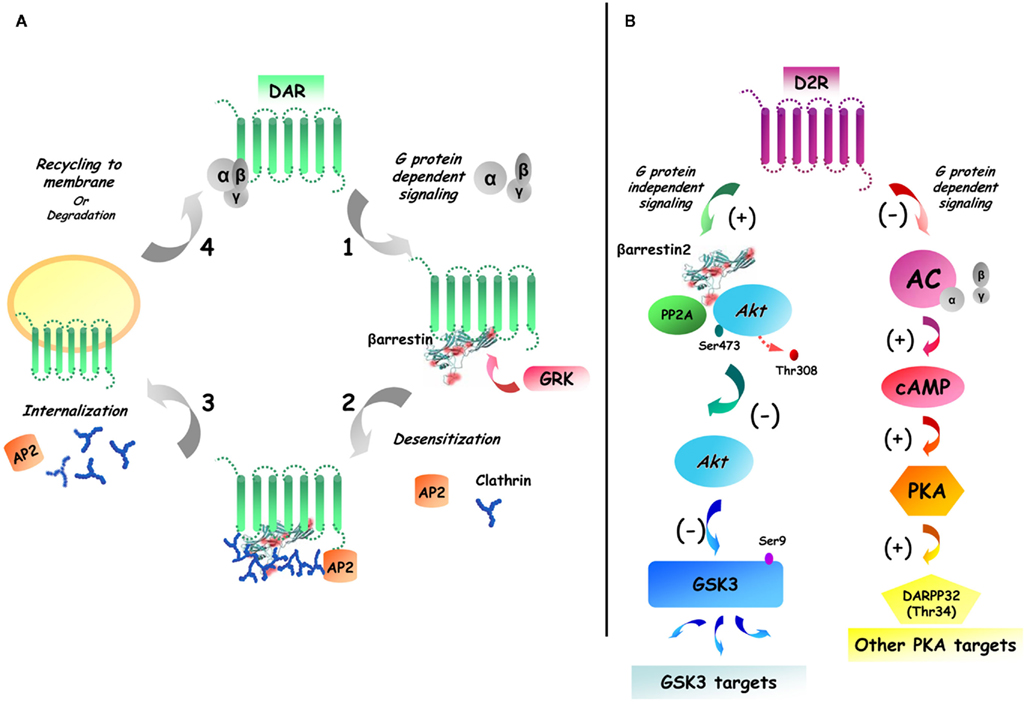
Figure 1. Dual role of beta-arrestin 2 in D2R desensitization and Akt/GSK3 signaling. (A) GPCR activation/desensitization cycle. Following the activation of dopamine receptors (DAR), the receptor is phosphorylated by G protein receptor kinases (GRK), which leads to the recruitment of beta-arrestins. The recruitment of beta-arrestins to the receptors results in clatrin-mediated endocytosis that is followed either by receptor degradation of cell surface recycling. (B) G protein-dependent and G protein-independent beta-arrestin-mediated signaling of D2R. Receptor activation leads to both classical G protein mediated signaling and to the formation of complexes of signaling molecules that are associated together by beta-arrestins. In the specific case of striatal D2R beta-arrestin 2 has been shown to enhance the interaction between Akt and protein phosphatase 2A (PP2A) therefore resulting in Akt inactivation and increased activation of GSK3.
The serine/threonine kinase Akt is known to be regulated by phosphoinositide 3-kinase (PI3K) signaling via the activatory phosphorylation of Akt at its threonine (Thr 308) and its serine (Ser 473) residues by two phosphatidyl-dependant kinases, PDK1 and PDK2/rictor-mTOR, respectively (Scheid and Woodgett, 2001; Jacinto et al., 2006). Once activated, Akt in turn phosphorylates GSK3 isoforms at the single regulatory serine residues serine 21 (GSK3α) and serine 9 (GSK3β) that are located in the N-terminal domains of both GSK3α and GSK3β (Stambolic and Woodgett, 1994; Frame and Cohen, 2001) and thereby causing their inactivation. Over the last 7 years, several independent lines of evidence have demonstrated that dopamine receptors can exert at least some of their biological functions by regulating the activity of Akt and GSK3 isoforms.
Here we provide an overview of the molecular mechanisms by which dopamine receptors can regulate the activity of brain Akt and GSK3. In addition, we evaluate the role played by these kinases in the regulation of various behaviors by dopamine, the role of Akt/GSK3 signaling cascade in the action of psychoactive drugs and potential relevance of the abnormalities found in this pathway for the development of dopamine-related neuropsychiatric diseases.
The Regulation of Akt and GSK3 by D2 Dopamine Receptors
Several lines of evidence have suggested that dopamine may play a role in the regulation of Akt and GSK3 signaling in vivo. For instance, as early as in 1971, behavioral observations in rodents have shown that lithium, which is now known to inhibit GSK3, can antagonize locomotor activity induced by drugs elevating dopaminergic tone or by the direct injection of dopamine in the nucleus accumbens (Cox et al., 1971; Barnes et al., 1986; Aylmer et al., 1987). However, these studies were all conducted before the discovery of the effect of lithium on GSK3 (Klein and Melton, 1996; Stambolic et al., 1996), and the actions of lithium in these experimental systems were generally attributed to other mechanisms including changes in dopamine release or dopamine receptor blockade. More recently, independent lines of research have shown that several antipsychotic drugs, known to share a D2R antagonist action, can activate Akt and increase the inhibitory phosphorylation of GSK3 in the rodent brain (Beaulieu et al., 2004; Emamian et al., 2004; Li et al., 2007). However, since these drugs also have other molecular targets and alternative mechanisms such as an action on serotonin receptors (Beaulieu, 2007; Li et al., 2007; Beaulieu et al., 2008b), these additional mechanisms could also contribute to the effect of these drugs on Akt/GSK3 signaling.
Characterization of cell signaling abnormalities in mice lacking the dopamine transporter (DAT–KO) have provided the first direct in vivo evidence for a role of dopamine in the regulation of Akt/GSK3 signaling in the mouse striatum (Beaulieu et al., 2004). The absence of dopamine reuptake by presynaptic dopamine neurons in these mice lead to about fivefold increase in extracellular dopamine concentration in the striatum (Gainetdinov et al., 1999). This persistent hyperdopaminergia results in reduced phosphorylation of Akt on its regulatory threonine 308 residue leading to a reduction of Akt activity and a concomitant activation of both GSK3α and GSK3β due to a reduction in the phosphorylation of their N-terminal domain by Akt (Beaulieu et al., 2004). A similar effect on Akt and GSK3 has been observed following the administration of indirect dopamine agonist amphetamine, that exerts its effects by elevating extracellular dopamine concentrations (Beaulieu et al., 2004; Polter et al., 2010). Furthermore, the non-selective D1R/D2R agonist apomorphine has also been reported to inhibit Akt phosphorylation in the mouse striatum (Beaulieu et al., 2005, 2007b).
A direct contribution of dopamine to the regulation of Akt and GSK3 was established by inhibiting dopamine synthesis in DAT–KO mice using the irreversible tyrosine hydroxylase inhibitor α-methyl-para-tyrosine (Beaulieu et al., 2004). It is important to note, that since these mice cannot reuptake and recycle dopamine to presynaptic terminal, inhibition of dopamine synthesis in DAT–KO mice leads to a virtual absence of striatal dopamine within minutes following drug administration (Beaulieu et al., 2004; Sotnikova et al., 2005). Two hours after this treatment, DAT–KO mice showed a restoration of Akt activity and enhanced inhibitory N-terminal phosphorylation of GSK3α and GSK3β.
The Role of D2-Class Receptors
Pharmacological characterization of the receptor involved in the inhibition of Akt and activation of GSK3 by dopamine in DAT–KO mice showed that Akt and GSK3 phosphorylation can be restored in DAT–KO mice by the administration of raclopride, a D2-class receptor antagonist (Beaulieu et al., 2004). A contribution of this family of receptors has later been confirmed by an investigation of dopamine-dependent regulation of Akt and GSK3 phosphorylation in mice lacking different subtypes of dopamine receptors. This study showed that D2R is essential for the inhibition of striatal Akt by amphetamine and apomorphine, while the effect of these drugs remained intact in D1R–KO mice. Interestingly, mice lacking the dopamine D3R showed a reduced sensitivity of Akt-mediated signaling to dopaminergic drugs but retained the action of these drugs on Akt at high dose regimens, suggesting that D3R also participates in the regulation of Akt/GSK3 signaling, potentially by enhancing D2R responses (Beaulieu et al., 2007b).
Since these initial characterizations were performed (Beaulieu et al., 2004), several independent studies using different pharmacological approaches have shown that a regulation of Akt and GSK3 by D2R end D3R is not restricted only to the mouse striatum. Indeed, prolonged elevation of dopaminergic tone (Li et al., 2009) or subchronic treatment with the D2R agonist quinpirole have been reported to inactivate Akt and activate GSK3 in the rat frontal cortex (Sutton and Rushlow, 2011). In contrast, administration of the D2R antagonist raclopride or D3R antagonist nafadotride has the opposite effect on Akt activity in different regions of the rat brain (Sutton and Rushlow, 2011). Finally, a recent report has convincingly shown the inactivation of Akt in response to D2R stimulation in the developing zebrafish brain (Souza et al., 2011), thus demonstrating that regulation of Akt and GSK3 is a shared functional property of D2R across several species of vertebrates.
cAMP-Independent Dopamine Receptor Signaling
Dopamine D2-class receptors are coupled to Gαi/o G protein and therefore inhibit adenylate cyclase and the production of cAMP and the activity of protein kinase A (PKA). Importantly, modulation of cAMP levels does not affect Akt and GSK3 activity in the mouse striatum, therefore indicating that D2R regulate this signaling cascade via a different type of mechanism (Beaulieu et al., 2004). Characterization of cell signaling responses in mice lacking βArr2 has shown that this molecule is essential for the inhibition of Akt and concomitant activation of GSK3 by D2R (Beaulieu et al., 2005; Figure 1). Furthermore, the inactivation of Akt by dopamine can also be prevented by inhibiting the serine/threonine protein phosphatase 2A (PP2A), which is known to play a role in mediating Akt inactivation in different biological systems (Ugi et al., 2004; Beaulieu et al., 2005). Further characterizations of the mechanisms through which D2R triggers the inactivation of Akt have shown that dopamine promotes the formation of a signaling complex (Figure 2) composed at least of Akt1, βArr2, and PP2A, thereby facilitating the inactivation of Akt by the phosphatase (Beaulieu et al., 2005, 2008a).
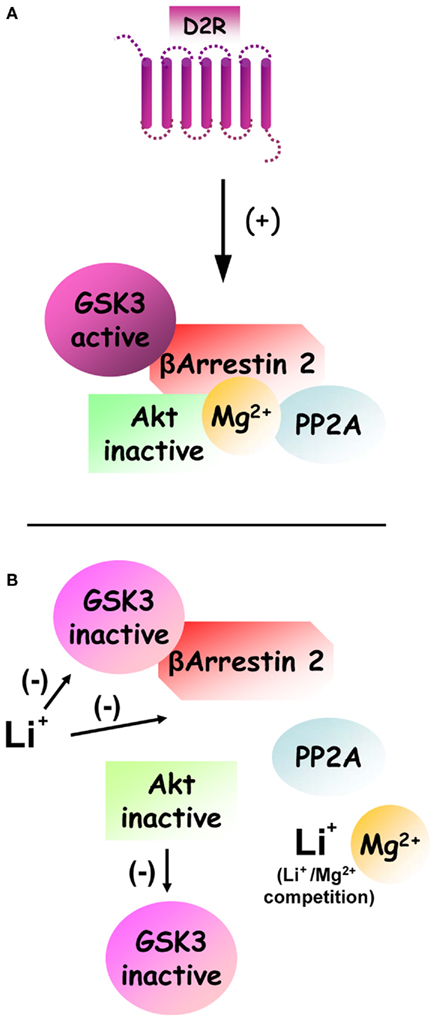
Figure 2. Regulation of dopamine and D2R-dependent Akt:βArr2:PP2A signaling complex by lithium and GSK3. (A) Under basal condition, the activation of D2R stimulates the formation of a signaling complex composed by beta-arrestin 2, PP2A, and Akt. The formation of this complex results in increased inactivation of Akt by PP2A. Furthermore enhanced activation of GSK3 that results from Akt inhibition would act as a positive feedback loop that further stabilizes the signaling complex. Finally, the formation of this complex also appears to be dependent on Mg2+ to allow the interaction of Akt and beta-arrestin 2. (B) Following treatment with lithium, the formation of this signaling complex can be destabilized by two overlapping mechanisms. First a competition between Li+ and Mg2+ ions can disrupt the complex directly by preventing the interaction of Akt and beta-arrestin 2. Second direct inhibition of GSK3 by lithium prevents the stabilization of the complex by activated GSK3.
It is noteworthy, that the formation of the Akt:βArr2:PP2A signaling complex in response to D2R activation represents a mechanism through which an intercellular messenger molecule (i.e., dopamine) can trigger the inactivation of PI3K/Akt signaling in a regulated fashion. Importantly, the Akt:βArr2:PP2A signaling complex can be dissociated in response to lithium (Figure 2), thus providing a likely explanation for early behavioral observations of an antagonistic effect of lithium on dopamine-mediated behaviors as well as a plausible mechanism for the activation of Akt by lithium (Beaulieu et al., 2008a; O’Brien et al., 2011; Pan et al., 2011). The details of the mechanism(s) by which lithium triggers this dissociation are not yet fully understood. Present evidence suggests that lithium may affect the stability of this complex by acting on several of its components, possibly in a synergistic fashion. First, lithium can interfere with the interaction of Akt1 and βArr2 (Beaulieu et al., 2008a). Indeed in vitro experiments conducted on recombinant proteins have shown that this interaction is dependent upon the presence of magnesium ions and that addition of an excess of magnesium can prevent the dissociation of Akt and βArr2 by lithium at therapeutic concentrations (1 mM). Second, GSK3β has also been shown to interact with βArr2. Recent evidence obtained from transgenic mice over-expressing frog GSK3β in neurons indicate that activated GSK3 can act as a feed forward mechanism for its own activation by stabilizing the Akt:βArr2:PP2A signaling complex (O’Brien et al., 2011). According to this model, a direct inhibition of GSK3 by lithium would thus constitute an additional mechanism that can promote the disassembly of the Akt:βArr2:PP2A in response to lithium.
Other Regulation Modes of Akt and GSK3 by Dopamine
While there are several reports that Akt is inhibited following the activation of D2-class receptors, there are also some in vitro and indirect in vivo observations of Akt activation and subsequent inhibition of GSK3 following dopamine receptor stimulation (Brami-Cherrier et al., 2002; Svenningsson et al., 2003; Mannoury La Cour et al., 2011; Rau et al., 2011). Indeed, activation of both D1R and D2R in primary cultures of embryonic striatal neurons has been shown to increase the phosphorylation/activation of Akt in the nucleus (Brami-Cherrier et al., 2002). Similarly, a recent report has shown that transfection of the long splice variants of human D2R or D3R in Chinese hamster ovary (CHO) cells can activate Akt and inhibit GSK3 by increasing their phosphorylation (Mannoury La Cour et al., 2011). In vivo, activation of Akt by cocaine (Brami-Cherrier et al., 2002) and inhibition of GSK3 by amphetamine (Svenningsson et al., 2003) in the mouse striatum, as well as activation of Akt following methamphetamine in a rat model stroke (Rau et al., 2011) have also been reported.
One major difference between these observations and research showing an inactivation of Akt by D2-class receptors resides in the intrinsic temporal dynamics of these signaling mechanisms. In general, increased Akt and/or GSK3 phosphorylation have been reported to occur at short intervals (1–15 min) after receptor activation (Brami-Cherrier et al., 2002; Svenningsson et al., 2003; Mannoury La Cour et al., 2011). In contrast, βArr2-mediated signaling resulting in reduced phosphorylation of Akt and GSK3 occurs at later time periods (30 min or more) following receptor activation (Beaulieu et al., 2004, 2005; Emamian et al., 2004; Li et al., 2009). These variations in response time are important since G protein- and arrestin-mediated signaling (Figure 3) are characterized by different temporal dynamics (Ahn et al., 2004; Shenoy et al., 2006; Beaulieu et al., 2007a). Mechanisms that are mediated by G proteins have a fast onset and a short duration as a result of GPCR desensitization in both cultured cells and in vivo. In contrast, arrestin-mediated signaling is characterized by much slower and progressive onset and does not have a known desensitization mechanism therefore allowing for prolonged signaling responses lasting from several minutes to hours.
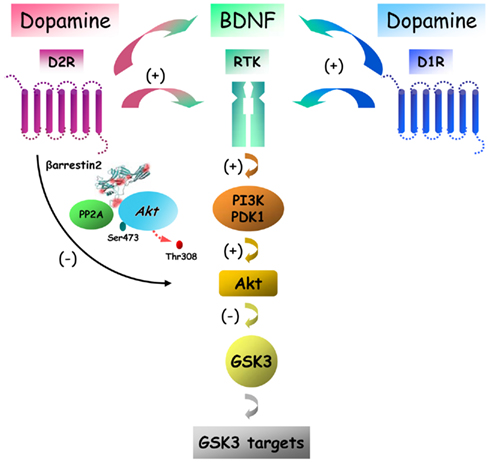
Figure 3. Putative signaling pathways downstream of dopamine receptors with different potential outcomes on Akt/GSK3 signaling. Dopamine receptors can regulate Akt and GSK3 signaling by acting through at least two signaling mechanisms that have an opposite effect on the activity of these two kinases. Activation of D2R leads to a regulated deactivation of Akt by PP2A that is mediated by beta-arrestin 2. In contrast activation of either D1R or D2R can also activate Akt via receptor tyrosine kinase (RTK) transactivation leading to enhancement of Pi3K signaling.
It is thus possible that in addition of the βArr2-mediated signaling that results in the inhibition of Akt and the activation of GSK3, dopamine receptors may also have an opposite effect by activating Akt and inactivating GSK3 via G protein-dependent mechanisms. However, the exact nature of such mechanisms remains mostly unexplained as changes in cAMP production, the hallmark of dopamine receptor G protein-mediated signaling, does not affect the phosphorylation of Akt and GSK3 in the mouse striatum, at least at later time points (Beaulieu et al., 2004). One possibility is that activation of Akt by dopamine under certain conditions may result from alternative G protein signaling mechanisms. Among such potential mechanisms, Gβγ G protein subunits can activate PI3K and therefore result in increased Akt activation, as has been shown following the activation of several different GPCRs (Brock et al., 2003). In addition to this possibility, recent reports also indicate that dopamine receptors may activate Akt by transactivating receptor tyrosine kinase (RTK). For instance, the activation of Akt by D2R and D3R in CHO cells appears to involved the transactivation of the insulin like growth factor 1 receptor (Mannoury La Cour et al., 2011). In a similar way, the development of a new image analysis method to quantify protein dimerization in single cells – spatial intensity distribution analysis (SpIDA) – has shown that stimulation of either transfected D1R or D2R in CHO cells provoques a dose-dependent dimerization and activation of the epidermal growth factor receptor as early as 5 min following dopamine receptor stimulation with apomorphine (Swift et al., 2011). Closer to a naturally occurring situation, SpIDA also revealed that endogenous D1R and D2R can activate endogenous BDNF TrkB receptor within a similar time frame in response to apomorphine in primary neuron cultures obtained from new born mice striatum (Swift et al., 2011). Intriguingly also, a decreased mRNA levels for BDNF and its high affinity receptor TrkB was found in the frontal cortex of hyperdopaminergic DAT-KO mice (Fumagalli et al., 2003). Taken together, these observations indicate that D1R or D2R have the ability to transactivate or interact with at least 3 different types of RTKs and that such a signaling mechanism is compatible with the normal expression of these different receptors in neurons (Figure 3). Despite the interesting possibilities raised by this mechanism, further studies will be needed to evaluate the relative contribution of RTK transactivation by GPCRs in the biological functions of dopamine receptors in in vivo settings.
Involvement of Akt and GSK3 in the Regulation of Behavior by Dopamine
Importance of Akt/GSK3 signaling for dopamine-related behaviors was evident from the pioneering observations uncovering the role of this pathway in dopamine receptor signaling. Pharmacological and genetic manipulations that increased or reduced dopaminergic activity and caused profound effects on locomotor activity in mice, induced also significant alterations in the phosphorylation of Akt and GSK3 (Beaulieu et al., 2004). At the same time, pharmacological or genetic suppression of GSK3 activity inhibited locomotor hyperactivity related to excessive dopaminergic tone in DAT–KO or amphetamine-treated mice (Beaulieu et al., 2004; Gould et al., 2007). Similarly, several GSK3 inhibitors blocked amphetamine-induced hyperactivity in normal animals (Gould et al., 2007; Kalinichev and Dawson, 2011). In contrast, mice over-expressing GSK3β showed pronounced locomotor hyperactivity (Prickaerts et al., 2006) and transgenic mice that express a GSK3β mutant (lacking an inhibitory phosphorylation site and thus have constitutively active GSK3β), demonstrate increased novelty-driven and amphetamine-induced hyperactivity (Polter et al., 2010).
Mice lacking Akt1 demonstrate enhanced sensitivity to amphetamine as regard to disruption of sensorimotor gating in pre-pulse inhibition (PPI) test, which is classically used to model psychosis in rodents (Emamian et al., 2004). As described above, Akt1 is inhibited following the stimulation of D2R, thus the increased behavioral effect of amphetamine in Akt1–KO mice likely results from the involvement of Akt in dopamine-related behavioral responses.
There are several lines of evidence highlighting the role of βArr2-dependent signaling complex in the actions of dopamine. It has been observed, that βArr2–KO mice display spontaneous locomotor hypoactivity, reduced apomorphine-induced climbing and amphetamine-induced hyperlocomotion (Gainetdinov et al., 2004; Beaulieu et al., 2005). These mutants also have a reduced responsiveness to the dopamine-dependent locomotor effects of morphine (Bohn et al., 2003). In addition, novelty-driven locomotor hyperactivity phenotype typical of hyperdopaminergic DAT–KO mice is less pronounced in mice lacking both βArr2 and DAT (Beaulieu et al., 2005). Finally, the antimanic drug lithium that disrupts βArr2/Akt/PP2A signaling complex and can directly inhibit GSK3, exerts multiple actions on behaviors including suppression of spontaneous locomotor activity in DAT–KO and normal mice, but not in βArr2–KO mice (Beaulieu et al., 2004, 2005).
Interestingly, a recent publication (Miller et al., 2010) also points toward a possible involvement of GSK3 in regulation locomotor responses do D1R stimulation. In this article, the authors report that pharmacological inhibition of GSK3 using SB 216763 leads to reduced behavioral responses to the selective D1R agonist SKF-82958. However, this has to be interpreted with caution since SB216763 is also an inhibitor of cyclin dependent kinases (Meijer et al., 2004), and kinases from this family, namely cyclin dependent kinase 5 (CDK5), are known to contribute to D1R signaling responses (Bibb et al., 1999; Cyr et al., 2003). Furthermore, the expression of a full locomotor response to dopamine agonists is known to depend upon a synergism of D1R and D2R stimulation. Under normal conditions the locomotor effects of a D1R agonist results from an exacerbated stimulation of D1R in a context where both D1R and D2R are stimulated by dopamine already present in the brain. Since GSK3 is regulated by D2R, it is therefore possible that the results reported by Miller et al., may be explained by an effect of GSK3 inhibitors on locomotion downstream of D2R. In line with this, it is of interest that the full effects of D1R stimulation on locomotion are significantly curtailed in D2R knockout mice where D1R/D2R synergism is abolished (Kelly et al., 1998).
Given the involvement of dopamine in multiple behavioral manifestations beyond locomotor activity, it is imperative to understand the role of AKT/GSK3 signaling cascade in these behaviors. For example, recent publications have suggested that GSK3 is important for psychostimulant-induced sensitization (Chen et al., 2007b; Miller et al., 2009), a process generally related to intermittent dopaminergic stimulation. Certainly, more studies in various behavioral paradigms could be expected in future on this issue.
Molecular Targets of Dopamine, Regulated by βArr2, Akt, and GSK3
With growing appreciation of a role for Akt and GSK3 in dopamine receptor signaling, understanding of the nature of the molecular targets of these kinases has become a priority. Akt and GSK3 are known to have multiple substrates, many of them being involved in critical cellular processes such as cytoskeleton organization, trafficking, cell survival, apoptosis, and DNA transcription (Frame and Cohen, 2001; Woodgett, 2001). Here, we overview the major targets of GSK3 that have shown a direct role in dopamine receptor signaling and that could be involved in the pathogenesis of neuropsychiatric disorders. These targets include, but are not limited to, the direct GSK3 targets β-catenin, ionotropic glutamate receptors and clock genes involved in the regulation of circadian rhythms.
β-Catenin
β-catenin is a common component of the Wnt and Akt/GSK3 signaling pathway. This protein that has multiple roles in the regulation of various aspects of cell cycle (Freyberg et al., 2010). Among other functions, β-catenin can act as a transcription factor and as a scaffolding protein that anchors the actin cytoskeleton to organize adherent junctions. Generally, in the absence of Wnt stimulation, β-catenin predominantly engages in Akt/GSK3 pathway by forming a complex with GSK3 and several other proteins. In this complex, β-catenin is phosphorylated by GSK3, which results in ubiquitination and proteasomal degradation of this protein (Doble and Woodgett, 2003). At the same time, activation of the Wnt receptor Frizzled causes disruption of this complex via involvement of Disheveled (Dvl) protein. Freed β-catenin then translocates to the nucleus and affects gene expression (Fukumoto et al., 2001). Several reports indicate that the action of dopamine and psychotropic drugs on the gene expression and neuronal morphology can involve the Wnt pathway and the Akt/GSK3 signaling cascade-mediated alterations in β-catenin activity. For example, the antipsychotic drugs haloperidol and clozapine may cause an enhancement of Dvl-3 isoform activity in the striatum which in turn may interact with D2R (Sutton et al., 2007) to modulate Akt, GSK3, and β-catenin activities (Sutton and Rushlow, 2011). Furthermore, antipsychotics haloperidol, risperidone, and clozapine can alter GSK3 and β-catenin expression not only in the striatum but also in the prefrontal cortex (Alimohamad et al., 2005b) and ventral midbrain (Alimohamad et al., 2005c). In contrast, direct and indirect dopamine receptor agonists quinpirole and amphetamine, respectively, induce the opposite response (Alimohamad et al., 2005c; Sutton and Rushlow, 2011). Importantly, chronic treatment of mice with the mood stabilizer lithium results in increased β-catenin expression in the amygdala, hypothalamus, and hippocampus (O’Brien et al., 2004; Beaulieu et al., 2008a). Furthermore, mice over-expressing β-catenin recapitulate the behavioral effects of the GSK3 inhibition by lithium in dopamine-dependent tests such as locomotor hyperactivity and in tests used to evaluate antidepressant and anxiolytic actions (Gould et al., 2008). It is likely, that these effects are mediated by β-catenin in the striatum, since the β-catenin forebrain-specific conditional mutant mice demonstrate only minimal behavioral abnormalities (Gould et al., 2008). Certainly, future studies are necessary to fully understand the role of β-catenin in actions of dopamine and psychotropic drugs.
Ionotropic Glutamate Receptors
The regulation of Akt/GSK3 signaling cascade by dopamine receptors has important modulatory influences on some specific functions of NMDA ionotropic glutamate receptors and related synaptic plasticity (Chen et al., 2007a). For example, alterations in GSK3 activity modulate the development of both long-term potentiation (LTP) and long-term depression (LTD) in rat hippocampal slices, the synaptic processes known to be regulated by ionotropic glutamate receptors (Peineau et al., 2007, 2008; Zhu et al., 2007). Intriguingly, D2R-mediated Akt/GSK3 signaling seems to be primarily responsible for the regulation of NMDA receptor functions in response to excessive levels of dopamine in the rat frontal cortex (Li et al., 2009). It has been reported that high concentrations of dopamine cause a reduction of NMDA currents and internalization of the NMDA receptor subunit NR2B. These effects of excessive dopaminergic stimulations on NMDA receptor functions are dependent of D2R, GSK3, and PP2A and can not be affected by inhibition of G proteins, thus indirectly suggesting an involvement of G protein-independent βArr2-mediated D2R signaling (Li et al., 2009). However, the postulated contribution of βArr2 to the regulation of NMDA receptor function by dopamine has not been validated yet in direct investigations involving βArr2–KO mice. Furthermore, it would be important to determine if similar type of regulation occurs in the striatum and in other brain areas or if it is restricted only to cortical neurons.
Regulation of Clock Gene Signaling
Several psychiatric disorders such as bipolar disorder and particularly seasonal affective disorder could be related, at least in part, to the dysregulation of circadian rhythms (Benedetti et al., 2004; Mansour et al., 2006). Original observations associating GSK3 activity to the regulation of circadian rhythms performed in Drosophila melanogaster (Yuan et al., 2005) revealed that the fly GSK3 ortholog, Shaggy, potently modulates the circadian cycle in response to serotonin. Consequently, several other studies in mammals have demonstrated the regulation of circadian rhythms and clock genes by GSK3 (Iitaka et al., 2005; Lamont et al., 2007). For example, it has been found that lithium affects the transcription of the clock gene Bmal1 presumably via GSK3 inhibition (Lamont et al., 2007). Furthermore, regulation of mammalian circadian protein functions by GSK3β was shown in vitro (Iitaka et al., 2005). Intriguingly, it has been reported that D2R can influence circadian rhythm-regulated gene expression and related behaviors (Doi et al., 2006; Yujnovsky et al., 2006). Also, it is well known that D2R is expressed at high levels in the retina and thus can play an important role in adaptation to light (Doi et al., 2006). Finally, activation of D2R causes stimulation CLOCK:BMAL1 functions potentially via regulation of circadian gene expression through βArr2/Akt/GSK3 signaling cascade (Sahar et al., 2010). Taken together, these observations indicate a putative mechanism connecting circadian rhythm to D2R functions.
Akt and GSK3 in the Action of Antipsychotics
All clinically effective antipsychotics share the ability to antagonize D2R (Snyder, 1976). Several antipsychotics have been shown to be able to activate Akt and inhibit GSK3 further supporting the role of the βArr2/Akt/GSK3 pathway in D2R signaling (Emamian et al., 2004; Alimohamad et al., 2005b; Li et al., 2007; Beaulieu et al., 2009). For example, the typical antipsychotic haloperidol increases phosphorylation of Akt and GSK3 thereby antagonizing action of dopamine (Beaulieu et al., 2004; Emamian et al., 2004). Furthermore, it has been shown that atypical antipsychotics clozapine, olanzapine, quetiapine, risperidone, and ziprasidone can either activate Akt or mimic its activity by increasing phosphorylation of GSK3α and GSK3β (Alimohamad et al., 2005b; Li et al., 2007). While the role of βArr2 in the actions of antipsychotics has not been validated yet in in vivo studies involving mutant mice, in vitro studies in cells have indicated that both typical and atypical antipsychotics are highly effective in antagonizing the recruitment of βArr2 to the D2R long isoform in comparison to their action on cAMP signaling (Masri et al., 2008). Particularly, three different patterns of activity of a panel of antipsychotics on cAMP mediated signaling and βArr2 recruitment were observed. Certain antipsychotics, including haloperidol, demonstrated potent antagonism of both the regulation of cAMP and the recruitment of βArr2 to the D2LR with approximately similar efficacy. Other antipsychotics, such as clozapine showed a stronger antagonist activity on βArr2 recruitment than on the cAMP regulation. Finally, a representative of a third group of antipsychotics, aripiprazole, acted as a partial agonist as regard to cAMP signaling while potently inhibiting βArr2 recruitment to D2R.
In summary, these results indicate that antipsychotics may vary in their ability to antagonize cAMP production but share a common antagonistic action as regard to D2R/βArr2-mediated signaling at least in vitro. Future in vivo studies are thus necessary to compare the physiological action of each of these drugs with regard to their action on D2R/βArr2/Akt/GSK3 signaling cascade. However, it is noteworthy that several atypical antipsychotics such as clozapine are not only D2R antagonists but are also able to affect brain 5-HT neurotransmission via action on 5-HT2A receptors (Kapur and Remington, 2001). Since drugs acting on 5-HT neurotransmission or decreasing 5-HT synthesis are known to affect GSK3 activity in vivo (Li et al., 2007; Beaulieu et al., 2008b), it is likely that atypical antipsychotics can affect GSK3 activity through both dopaminergic and serotonergic mechanisms. Further studies are therefore necessary to delineate the relative contribution of dopamine- and 5-HT-related processes in the modulation of Akt/GSK3 signaling cascade by antipsychotic drugs and thus to define the roles that these mechanisms may play in the treatment of psychotic disorders.
Genetic Associations between Regulators of Akt/GSK3 Signaling and Psychiatric Disorders
A role for D2R/βArr2/Akt/GSK3 signaling in the action of mood stabilizers and antipsychotics is also supported by clinical studies of genetic risk factors for schizophrenia and bipolar disorders. Psychiatric disorders are multifactorial conditions that involve various combinations of genetic, epigenetic, and environmental factors (Karayiorgou and Gogos, 1997). Several recently published meta-analysis studies have examined the results of ≥1000 genetic association studies attempting to identify candidate genes for schizophrenia (Allen et al., 2008; Jia et al., 2010). Interestingly, in some cases the same allelic polymorphisms have been associated with different neuropsychiatric pathologies such as schizophrenia, bipolar disorder, unipolar depression, and autism. To better understand the mechanisms of such possible common etiology, several genetically altered animal models have been created with the focus on genes known to be altered in these mental illnesses (Gottesman and Gould, 2003). Numerous risk genes for these diseases affect or are being affected by Akt and GSK3. Here, we review genetic associations between psychiatric disorders and the products of three of such genes (DISC1, NRG1, and AKT1) that are known to be involved directly or indirectly in the Akt/GSK3 signaling pathways.
Akt1
Accumulating evidence points to an important role for Akt and related signaling molecules in schizophrenia pathogenesis. In the pioneering study, a reduced levels of Akt1, one of the three Akt isoforms, in the hippocampus and frontal cortex of schizophrenia patients was found (Emamian et al., 2004). A major association of Akt1 haplotype with schizophrenia has been reported following transmission-disequilibrium tests in several other cohorts of patients (Ikeda et al., 2004). Supportive of these associations of AKT1 with schizophrenia, another group has reported a coding variation in AKT1 that affects protein expression in lymphoblasts and which can modulate prefrontal cortical structure and function was associated with risk for schizophrenia (Tan et al., 2008). Importantly, the behavioral outcomes of reduced Akt1 expression both in mice and humans can be affected by alterations in D2R functions. In Akt1–KO mice, amphetamine-induced disruption of sensory–motor gating in the PPI test is enhanced but can be reversed by the D2R antagonist haloperidol (Emamian et al., 2004). In humans, interaction between polymorphisms in the D2R (DRD2 rs1076560) and AKT1 (AKT1 rs1130233) genes was associated with deficient attentional processing in normal individuals and response to olanzapine treatment in schizophrenia patients (Blasi et al., 2011). Furthermore, it has been reported that Akt1 levels and Akt-dependent phosphorylation of GSK3α and GSK3β isoforms is reduced in the frontal cortex of schizophrenia patients (Emamian et al., 2004). Because Akt activity is affected by D2R and this kinase seems to be important for certain functions of prefrontal cortex such as cognitive performance (working memory; Lai et al., 2006), it is reasonable to consider this kinase as an important mediator in dopamine-related processes involved in etiology and treatment of schizophrenia. Importantly, Akt activity and functions can be also affected by the products of two other genes reported to be associated with schizophrenia: neuregulin 1 (NRG1) and disrupted in schizophrenia 1 (DISC1; Kanakry et al., 2007).
Neuregulins
A member of growth factors family, NRG, by acting on ErbB (EGF RTK) signaling pathway influences numerous processes that could be involved in the neuropsychiatric disorders, such as cell migration, myelination, NMDA receptor- dependent neuronal plasticity and even expression of monoamine receptors and transporters (Mei and Xiong, 2008; Pitcher et al., 2011). It has been reported, that in patients with schizophrenia, NRG signaling is increased in the hippocampus causing an up-regulation of Erb receptors and concomitant PI3K activation that results in increased phosphorylation of Akt (Law et al., 2007; Jaaro-Peled et al., 2009). Mice heterozygous for NRG1 or ErbB4 receptor deletion demonstrate some “schizophrenia-related” behavioral abnormalities such as hyperactivity, deficient social behaviors, anxiety, memory deficits, and impaired responses in PPI (Chen et al., 2008; Desbonnet et al., 2009). While the mechanisms involved in these pathologies are still poorly understood, several lines of evidence suggest involvement of NRG1-dependent modulation of the striatal dopaminergic system via Akt (Tosato et al., 2005). In fact, NRG1 mutant mice show a similar phenotype to hyperdopaminergic DAT mutant mice. In addition, Akt is involved in NRG1-associated processes such as myelination (Flores et al., 2000; Li et al., 2001) and Akt1 phosphorylation is stimulated by NRG1 (Keri et al., 2009). Further studies are necessary to explore this hypothesis.
DISC1
DISC1 is a gene locus originally identified as disrupted in Scottish family members suffering from schizophrenia, but also from bipolar disorder and major depression (Chubb et al., 2008). Alterations of DISC1 in cell culture and mice induce abnormal processes that may occur in schizophrenia such as impaired neurite outgrowth, abnormal cortical development and incorrect neuronal migration (Kamiya et al., 2005). Recently, DISC1 has been found to be a direct GSK3β regulator thereby providing further support for a role for this kinase in neuropsychiatric disorders (Mao et al., 2009). Genetic suppression of DISC1 in the dentate gyrus of adult mice increased GSK3 activity and caused abnormal behaviors that might have relevance for depression and schizophrenia (Mao et al., 2009). DISC1 mutant mice have also demonstrated deficits in working memory and these behavioral abnormalities could be corrected by an administration of a direct inhibitor of GSK3 (SB216763; Kvajo et al., 2008). In another study, dominant-negative mutant DISC1 influenced the neurobehavioral and molecular effects of methamphetamine in mice likely via interaction with Akt/GSK3 signaling cascade (Pogorelov et al., 2011). Furthermore, DISC1 can be also involved in modulation of Akt and related signaling in newly generated neurons through its interaction with the potential DISC1-interacting partner KIAA1212, known as a direct activator of Akt (Camargo et al., 2007; Kim et al., 2009). In this model, binding of DISC1 to KIAA1212 prevents its action on Akt function. Importantly, both Akt overexpression and DISC1 suppression result in similar developmental abnormalities of newborn neurons. Taken together, these observations suggest a role of DISC1 interaction with Akt/GSK3 signaling in neurogenesis processes that could be important for both neurotransmission- and neurodevelopment-associated neuropsychiatric abnormalities.
In summary, while behavioral abnormalities observed in mice deficient in NRG1 or DISC1 expression suggest a role of these molecules in modulation of dopaminergic transmission, little evidence still exist supporting this association in clinical genetic studies. Nevertheless, it seems likely that monoamine-related processes involved in the etiology of mental disorders or therapeutic actions of drugs may engage in functional and selective crosstalks with others cellular signaling mechanisms such as those involved in apoptosis, neurogenesis, and plasticity and that are also could be affected by genetic and/or environmental factors in patients with these disorders.
Striatal Akt and GSK3 in Parkinson Disease
Striatal Akt/GSK3 signaling and related processes may also contribute to mechanisms leading to neurodegeneration or neuroprotection. In fact, recent studies have indicated association of Akt signaling with neurodegenerative disorders such as Parkinson disease. Two neurotoxins, 6-hydroxydopamine (6-OHDA) and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), induce a massive loss of nigral tyrosine hydroxylase-expressing cells and striatal dopamine depletion. Consequently, these two drugs are routinely used to generate pharmacological animal models of Parkinson’s disease (Lane and Dunnett, 2008). Interestingly, both MPTP and 6-OHDA induced neurodegeneration is associated with alterations in striatal Akt/GSK3 signaling cascade (Quesada et al., 2008; Aleyasin et al., 2010). It has been reported also, that chronic treatment with L-DOPA but not DAR agonist pergolide strongly increases the activity of Akt and inhibits GSK3 in the dopamine-depleted striatum of 6-OHDA-lesioned rats suggesting that Akt pathway may be involved in cronic L-DOPA-induced dyskinesia (Bychkov et al., 2007). Indeed, in monkeys, which show dopamine depletion after MPTP administration, chronic L-DOPA treatment with or without two types of antidyskinetic drugs induced a prolonged changes in Akt and GSK3 phosphorylation levels (Morissette et al., 2010). While it is unclear at present if these effects caused by G protein-mediated or βArr2-mediated dopaminergic signaling mechanisms, other neurotransmitter systems like glutamate may also contribute to L-DOPA-induced dyskinesia (Ouattara et al., 2010) by directly regulating Akt/GSK3 or D1 dopamine receptor mediated signaling may be also involved (Santini et al., 2009).
In summary, these results suggest that Akt/GSK3 signaling may contribute to the development and manifestations of neurodegenerative disorders such as Parkinson’s disease. Thus, it would seem relevant to investigate alterations in activity and molecular targets of these kinases as possible biomarkers of underlying pathology or treatment complications of this disorder. Understanding these mechanisms may provide better understanding of pathological processes and potentially new therapeutic strategy for the treatment of neurodegenerative disorders.
Conclusion and Perspectives
Since the first reports published about 7 years ago on the role Akt/GSK3 signaling cascade in the dopamine receptor functions and behavior, this field showed quite remarkable developments. One reason for such noticeable progress is the growing appreciation of the involvement of Akt, GSK3, and related signaling processes in the pathophysiology of various psychiatric and neurological disorders. The role of this pathway in dopamine signaling has also received considerable attention due to multiple lines of evidence for its physiological significance in vivo. Numerous studies have convincingly shown significant alterations in dopamine-related functions and behaviors caused by manipulations affecting Akt/GSK3 pathway. Conversely, several genetic or pharmacological manipulation of dopamine receptors or their signaling intermediates have been shown to affect Akt/GSK3 signaling (Table 1). Certainly, many outstanding questions still remain unresolved and require further detailed investigations both in vitro and particularly at the level of whole organism. Most noticeable among them: understanding mechanisms of formation of arrestin-dependent signaling complexes in response to dopamine, identification of critical downstream targets of Akt and GSK3, clarification of cross-talk mechanisms between this and other signaling pathways, further understanding of the role of this signaling mode in pathological manifestations and effects of therapeutic treatments. It might be predicted that this line of research will continue to develop in very exciting ways and will eventually bring new molecules and pathways under attention as potential novel targets for treatment of neuropsychiatric disorders.
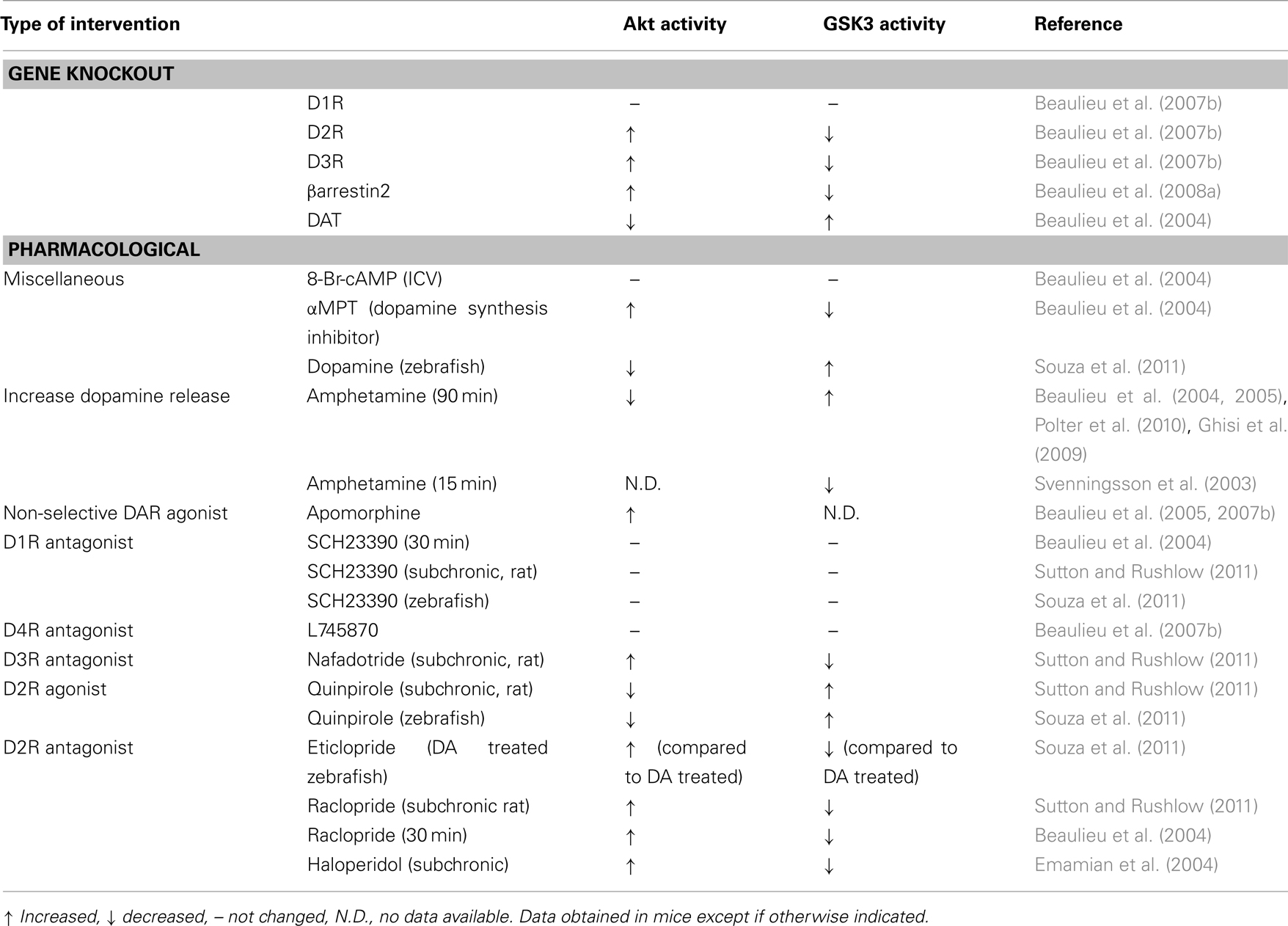
Table 1. Effect of pharmacological and genetic manipulation of dopamine receptor activation on Akt and GSK3 activity in vivo.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This review was supported by an Operating Grant (NSA 93798) from Canadian Institutes of Health Research (CIHR) to Jean-Martin Beaulieu. Jean-Martin Beaulieu is also supported by a National Alliance for Research on Schizophrenia and Depression (NARSAD) Young Investigator Award and hold a Canada research Chair in Molecular Psychiatry. Raul R. Gainetdinov is supported in part by research awards from F. Hoffmann – La Roche Ltd. (Basel, Switzerland) and Fondazione Compagnia di San Paolo (Torino, Italy).
References
Ahn, S., Shenoy, S. K., Wei, H., and Lefkowitz, R. J. (2004). Differential kinetic and spatial patterns of beta-arrestin and G protein-mediated ERK activation by the angiotensin II receptor. J. Biol. Chem. 279, 35518–35525.
Aleyasin, H., Rousseaux, M. W., Marcogliese, P. C., Hewitt, S. J., Irrcher, I., Joselin, A. P., Parsanejad, M., Kim, R. H., Rizzu, P., Callaghan, S. M., Slack, R. S., Mak, T. W., and Park, D. S. (2010). DJ-1 protects the nigrostriatal axis from the neurotoxin MPTP by modulation of the AKT pathway. Proc. Natl. Acad. Sci. U.S.A. 107, 3186–3191.
Alimohamad, H., Rajakumar, N., Seah, Y. H., and Rushlow, W. (2005b). Antipsychotics alter the protein expression levels of beta-catenin and GSK-3 in the rat medial prefrontal cortex and striatum. Biol. Psychiatry 57, 533–542.
Alimohamad, H., Sutton, L., Mouyal, J., Rajakumar, N., and Rushlow, W. J. (2005c). The effects of antipsychotics on beta-catenin, glycogen synthase kinase-3 and dishevelled in the ventral midbrain of rats. J. Neurochem. 95, 513–525.
Allen, N. C., Bagade, S., Mcqueen, M. B., Ioannidis, J. P., Kavvoura, F. K., Khoury, M. J., Tanzi, R. E., and Bertram, L. (2008). Systematic meta-analyses and field synopsis of genetic association studies in schizophrenia: the SzGene database. Nat. Genet. 40, 827–834.
Aylmer, C. G., Steinberg, H., and Webster, R. A. (1987). Hyperactivity induced by dexamphetamine/chlordiazepoxide mixtures in rats and its attenuation by lithium pretreatment: a role for dopamine? Psychopharmacology (Berl.) 91, 198–206.
Barnes, J. C., Costall, B., Domeney, A. M., and Naylor, R. J. (1986). Lithium and bupropion antagonise the phasic changes in locomotor activity caused by dopamine infused into the rat nucleus accumbens. Psychopharmacology (Berl.) 89, 311–316.
Beaulieu, J. M. (2007). Not only lithium: regulation of glycogen synthase kinase-3 by antipsychotics and serotonergic drugs. Int. J. Neuropsychopharmacol. 10, 3–6.
Beaulieu, J. M., and Gainetdinov, R. R. (2011). The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol. Rev. 63, 182–217.
Beaulieu, J. M., Gainetdinov, R. R., and Caron, M. G. (2007a). The Akt-GSK-3 signaling cascade in the actions of dopamine. Trends Pharmacol. Sci. 28, 166–172.
Beaulieu, J. M., Tirotta, E., Sotnikova, T. D., Masri, B., Salahpour, A., Gainetdinov, R. R., Borrelli, E., and Caron, M. G. (2007b). Regulation of Akt signaling by D2 and D3 dopamine receptors in vivo. J. Neurosci. 27, 881–885.
Beaulieu, J. M., Gainetdinov, R. R., and Caron, M. G. (2009). Akt/GSK3 signaling in the action of psychotropic drugs. Annu. Rev. Pharmacol. Toxicol. 49, 327–347.
Beaulieu, J. M., Marion, S., Rodriguiz, R. M., Medvedev, I. O., Sotnikova, T. D., Ghisi, V., Wetsel, W. C., Lefkowitz, R. J., Gainetdinov, R. R., and Caron, M. G. (2008a). A beta-arrestin 2 signaling complex mediates lithium action on behavior. Cell 132, 125–136.
Beaulieu, J. M., Zhang, X., Rodriguiz, R. M., Sotnikova, T. D., Cools, M. J., Wetsel, W. C., Gainetdinov, R. R., and Caron, M. G. (2008b). Role of GSK3beta in behavioral abnormalities induced by serotonin deficiency. Proc. Natl. Acad. Sci. U.S.A. 105, 1333–1338.
Beaulieu, J. M., Sotnikova, T. D., Marion, S., Lefkowitz, R. J., Gainetdinov, R. R., and Caron, M. G. (2005). An Akt/beta-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell 122, 261–273.
Beaulieu, J. M., Sotnikova, T. D., Yao, W. D., Kockeritz, L., Woodgett, J. R., Gainetdinov, R. R., and Caron, M. G. (2004). Lithium antagonizes dopamine-dependent behaviors mediated by an AKT/glycogen synthase kinase 3 signaling cascade. Proc. Natl. Acad. Sci. U.S.A. 101, 5099–5104.
Benedetti, F., Serretti, A., Colombo, C., Lorenzi, C., Tubazio, V., and Smeraldi, E. (2004). A glycogen synthase kinase 3-beta promoter gene single nucleotide polymorphism is associated with age at onset and response to total sleep deprivation in bipolar depression. Neurosci. Lett. 368, 123–126.
Bibb, J. A., Snyder, G. L., Nishi, A., Yan, Z., Meijer, L., Fienberg, A. A., Tsai, L. H., Kwon, Y. T., Girault, J. A., Czernik, A. J., Huganir, R. L., Hemmings, H. C., Nairn, A. C., and Greengard, P. (1999). Phosphorylation of DARPP-32 by Cdk5 modulates dopamine signaling in neurons. Nature 402, 669–671.
Blasi, G., Napolitano, F., Ursini, G., Taurisano, P., Romano, R., Caforio, G., Fazio, L., Gelao, B., Di Giorgio, A., Iacovelli, L., Sinibaldi, L., Popolizio, T., Usiello, A., and Bertolino, A. (2011). DRD2/AKT1 interaction on D2 c-AMP independent signaling, attentional processing, and response to olanzapine treatment in schizophrenia. Proc. Natl. Acad. Sci. U.S.A. 108, 1158–1163.
Bohn, L. M., Gainetdinov, R. R., Sotnikova, T. D., Medvedev, I. O., Lefkowitz, R. J., Dykstra, L. A., and Caron, M. G. (2003). Enhanced rewarding properties of morphine, but not cocaine, in beta(arrestin)-2 knock-out mice. J. Neurosci. 23, 10265–10273.
Brami-Cherrier, K., Valjent, E., Garcia, M., Pages, C., Hipskind, R. A., and Caboche, J. (2002). Dopamine induces a PI3-kinase-independent activation of Akt in striatal neurons: a new route to cAMP response element-binding protein phosphorylation. J. Neurosci. 22, 8911–8921.
Brock, C., Schaefer, M., Reusch, H. P., Czupalla, C., Michalke, M., Spicher, K., Schultz, G., and Nurnberg, B. (2003). Roles of G beta gamma in membrane recruitment and activation of p110 gamma/p101 phosphoinositide 3-kinase gamma. J. Cell Biol. 160, 89–99.
Bychkov, E., Ahmed, M. R., Dalby, K. N., and Gurevich, E. V. (2007). Dopamine depletion and subsequent treatment with L-DOPA, but not the long-lived dopamine agonist pergolide, enhances activity of the Akt pathway in the rat striatum. J. Neurochem. 102, 699–711.
Camargo, L. M., Collura, V., Rain, J. C., Mizuguchi, K., Hermjakob, H., Kerrien, S., Bonnert, T. P., Whiting, P. J., and Brandon, N. J. (2007). Disrupted in schizophrenia 1 interactome: evidence for the close connectivity of risk genes and a potential synaptic basis for schizophrenia. Mol. Psychiatry 12, 74–86.
Chen, P., Gu, Z., Liu, W., and Yan, Z. (2007a). Glycogen synthase kinase 3 regulates N-methyl-D-aspartate receptor channel trafficking and function in cortical neurons. Mol. Pharmacol. 72, 40–51.
Chen, P. C., Lao, C. L., and Chen, J. C. (2007b). Dual alteration of limbic dopamine D1 receptor-mediated signalling and the Akt/GSK3 pathway in dopamine D3 receptor mutants during the development of methamphetamine sensitization. J. Neurochem. 100, 225–241.
Chen, Y. J., Johnson, M. A., Lieberman, M. D., Goodchild, R. E., Schobel, S., Lewandowski, N., Rosoklija, G., Liu, R. C., Gingrich, J. A., Small, S., Moore, H., Dwork, A. J., Talmage, D. A., and Role, L. W. (2008). Type III neuregulin-1 is required for normal sensorimotor gating, memory-related behaviors, and corticostriatal circuit components. J. Neurosci. 28, 6872–6883.
Chubb, J. E., Bradshaw, N. J., Soares, D. C., Porteous, D. J., and Millar, J. K. (2008). The DISC locus in psychiatric illness. Mol. Psychiatry 13, 36–64.
Cox, C., Harrison-Read, P. E., Steinberg, H., and Tomkiewicz, M. (1971). Lithium attenuates drug-induced hyperactivity in rats. Nature 232, 336–338.
Cyr, M., Beaulieu, J. M., Laakso, A., Sotnikova, T. D., Yao, W. D., Bohn, L. M., Gainetdinov, R. R., and Caron, M. G. (2003). Sustained elevation of extracellular dopamine causes motor dysfunction and selective degeneration of striatal GABAergic neurons. Proc. Natl. Acad. Sci. U.S.A. 100, 11035–11040.
Desbonnet, L., Waddington, J. L., and O’Tuathaigh, C. M. (2009). Mutant models for genes associated with schizophrenia. Biochem. Soc. Trans. 37, 308–312.
Doble, B. W., and Woodgett, J. R. (2003). GSK-3: tricks of the trade for a multi-tasking kinase. J. Cell. Sci. 116, 1175–1186.
Doi, M., Yujnovsky, I., Hirayama, J., Malerba, M., Tirotta, E., Sassone-Corsi, P., and Borrelli, E. (2006). Impaired light masking in dopamine D2 receptor-null mice. Nat. Neurosci. 9, 732–734.
Emamian, E. S., Hall, D., Birnbaum, M. J., Karayiorgou, M., and Gogos, J. A. (2004). Convergent evidence for impaired AKT1-GSK3beta signaling in schizophrenia. Nat. Genet. 36, 131–137.
Ferguson, S. S., Downey, W. E., Colapietro, A. M. 3rd, Barak, L. S., Menard, L., and Caron, M. G. (1996). Role of beta-arrestin in mediating agonist-promoted G protein-coupled receptor internalization. Science 271, 363–366.
Flores, A. I., Mallon, B. S., Matsui, T., Ogawa, W., Rosenzweig, A., Okamoto, T., and Macklin, W. B. (2000). Akt-mediated survival of oligodendrocytes induced by neuregulins. J. Neurosci. 20, 7622–7630.
Frame, S., and Cohen, P. (2001). GSK3 takes centre stage more than 20 years after its discovery. Biochem. J. 359, 1–16.
Freyberg, Z., Ferrando, S. J., and Javitch, J. A. (2010). Roles of the Akt/GSK-3 and Wnt signaling pathways in schizophrenia and antipsychotic drug action. Am. J. Psychiatry 167, 388–396.
Fumagalli, F., Racagni, G., Colombo, E., and Riva, M. A. (2003). BDNF gene expression is reduced in the frontal cortex of dopamine transporter knockout mice. Mol. Psychiatry 8, 898–899.
Fukumoto, S., Hsieh, C. M., Maemura, K., Layne, M. D., Yet, S. F., Lee, K. H., Matsui, T., Rosenzweig, A., Taylor, W. G., Rubin, J. S., Perrella, M. A., and Lee, M. E. (2001). Akt participation in the Wnt signaling pathway through dishevelled. J. Biol. Chem. 276, 17479–17483.
Gainetdinov, R. R., Premont, R. T., Bohn, L. M., Lefkowitz, R. J., and Caron, M. G. (2004). Desensitization of G protein-coupled receptors and neuronal functions. Annu. Rev. Neurosci. 27, 107–144.
Gainetdinov, R. R., Wetsel, W. C., Jones, S. R., Levin, E. D., Jaber, M., and Caron, M. G. (1999). Role of serotonin in the paradoxical calming effect of psychostimulants on hyperactivity. Science 283, 397–401.
Ghisi, V., Ramsey, A. J., Masri, B., Gainetdinov, R. R., Caron, M. G., and Salahpour, A. (2009). Reduced D2-mediated activity and trans-synaptic upregulation of D1 and D2 dopamine receptors in mice overexpressing the dopamine transporter. Cell. Signal. 21, 87–94.
Gottesman, I. I., and Gould, T. D. (2003). The endophenotype concept in psychiatry: etymology and strategic intentions. Am. J. Psychiatry 160, 636–645.
Gould, T. D., O’Donnell, K. C., Picchini, A. M., Dow, E. R., Chen, G., and Manji, H. K. (2008). Generation and behavioral characterization of beta-catenin forebrain-specific conditional knock-out mice. Behav. Brain Res. 189, 117–125.
Gould, T. D., O’Donnell, K. C., Picchini, A. M., and Manji, H. K. (2007). Strain differences in lithium attenuation of d-amphetamine-induced hyperlocomotion: a mouse model for the genetics of clinical response to lithium. Neuropsychopharmacology 32, 1321–1333.
Hasbi, A., Fan, T., Alijaniaram, M., Nguyen, T., Perreault, M. L., O’Dowd, B. F., and George, S. R. (2009). Calcium signaling cascade links dopamine D1-D2 receptor heteromer to striatal BDNF production and neuronal growth. Proc. Natl. Acad. Sci. U.S.A. 106, 21377–21382.
Iitaka, C., Miyazaki, K., Akaike, T., and Ishida, N. (2005). A role for glycogen synthase kinase-3beta in the mammalian circadian clock. J. Biol. Chem. 280, 29397–29402.
Ikeda, M., Iwata, N., Suzuki, T., Kitajima, T., Yamanouchi, Y., Kinoshita, Y., Inada, T., and Ozaki, N. (2004). Association of AKT1 with schizophrenia confirmed in a Japanese population. Biol. Psychiatry 56, 698–700.
Jaaro-Peled, H., Hayashi-Takagi, A., Seshadri, S., Kamiya, A., Brandon, N. J., and Sawa, A. (2009). Neurodevelopmental mechanisms of schizophrenia: understanding disturbed postnatal brain maturation through neuregulin-1-ErbB4 and DISC1. Trends Neurosci. 32, 485–495.
Jacinto, E., Facchinetti, V., Liu, D., Soto, N., Wei, S., Jung, S. Y., Huang, Q., Qin, J., and Su, B. (2006). SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 127, 125–137.
Jia, P., Sun, J., Guo, A. Y., and Zhao, Z. (2010). SZGR: a comprehensive schizophrenia gene resource. Mol. Psychiatry 15, 453–462.
Kalinichev, M., and Dawson, L. A. (2011). Evidence for antimanic efficacy of glycogen synthase kinase-3 (GSK3) inhibitors in a strain-specific model of acute mania. Int. J. Neuropsychopharmacol. 14, 1051–1067.
Kamiya, A., Kubo, K., Tomoda, T., Takaki, M., Youn, R., Ozeki, Y., Sawamura, N., Park, U., Kudo, C., Okawa, M., Ross, C. A., Hatten, M. E., Nakajima, K., and Sawa, A. (2005). A schizophrenia-associated mutation of DISC1 perturbs cerebral cortex development. Nat. Cell Biol. 7, 1167–1178.
Kanakry, C. G., Li, Z., Nakai, Y., Sei, Y., and Weinberger, D. R. (2007). Neuregulin-1 regulates cell adhesion via an ErbB2/phosphoinositide-3 kinase/Akt-dependent pathway: potential implications for schizophrenia and cancer. PLoS ONE 2, e1369. doi:10.1371/journal.pone.0001369
Kapur, S., and Remington, G. (2001). Atypical antipsychotics: new directions and new challenges in the treatment of schizophrenia. Annu. Rev. Med. 52, 503–517.
Karayiorgou, M., and Gogos, J. A. (1997). Dissecting the genetic complexity of schizophrenia. Mol. Psychiatry 2, 211–223.
Kelly, M. A., Rubinstein, M., Philips, T. J., Lessov, C. N., Burkhart-Kasch, S., Zhang, G., Bunzow, J. R., Fang, Y., Gerhardt, G. A., Grandy, D. K., and Low, M. J. (1998). Locomotor activity in D2 dopamine receptor-deficient mice is determine by gene dosahe, genetic background, and developmental adaptations. J. Neurosci. 18, 3470–3479.
Keri, S., Seres, I., Kelemen, O., and Benedek, G. (2009). Neuregulin 1-stimulated phosphorylation of AKT in psychotic disorders and its relationship with neurocognitive functions. Neurochem. Int. 55, 606–609.
Kim, J. Y., Duan, X., Liu, C. Y., Jang, M. H., Guo, J. U., Pow-Anpongkul, N., Kang, E., Song, H., and Ming, G. L. (2009). DISC1 regulates new neuron development in the adult brain via modulation of AKT-mTOR signaling through KIAA1212. Neuron 63, 761–773.
Kisilevsky, A. E., Mulligan, S. J., Altier, C., Iftinca, M. C., Varela, D., Tai, C., Chen, L., Hameed, S., Hamid, J., Macvicar, B. A., and Zamponi, G. W. (2008). D1 receptors physically interact with N-type calcium channels to regulate channel distribution and dendritic calcium entry. Neuron 58, 557–570.
Kisilevsky, A. E., and Zamponi, G. W. (2008). D2 dopamine receptors interact directly with N-type calcium channels and regulate channel surface expression levels. Channels (Austin) 2, 269–277.
Klein, P. S., and Melton, D. A. (1996). A molecular mechanism for the effect of lithium on development. Proc. Natl. Acad. Sci. U.S.A. 93, 8455–8459.
Kvajo, M., Mckellar, H., Arguello, P. A., Drew, L. J., Moore, H., Macdermott, A. B., Karayiorgou, M., and Gogos, J. A. (2008). A mutation in mouse Disc1 that models a schizophrenia risk allele leads to specific alterations in neuronal architecture and cognition. Proc. Natl. Acad. Sci. U.S.A. 105, 7076–7081.
Lai, W. S., Xu, B., Westphal, K. G., Paterlini, M., Olivier, B., Pavlidis, P., Karayiorgou, M., and Gogos, J. A. (2006). Akt1 deficiency affects neuronal morphology and predisposes to abnormalities in prefrontal cortex functioning. Proc. Natl. Acad. Sci. U.S.A. 103, 16906–16911.
Lamont, E. W., Legault-Coutu, D., Cermakian, N., and Boivin, D. B. (2007). The role of circadian clock genes in mental disorders. Dialogues Clin. Neurosci. 9, 333–342.
Lane, E., and Dunnett, S. (2008). Animal models of Parkinson’s disease and L-dopa induced dyskinesia: how close are we to the clinic? Psychopharmacology (Berl.) 199, 303–312.
Law, A. J., Kleinman, J. E., Weinberger, D. R., and Weickert, C. S. (2007). Disease-associated intronic variants in the ErbB4 gene are related to altered ErbB4 splice-variant expression in the brain in schizophrenia. Hum. Mol. Genet. 16, 129–141.
Li, X., Rosborough, K. M., Friedman, A. B., Zhu, W., and Roth, K. A. (2007). Regulation of mouse brain glycogen synthase kinase-3 by atypical antipsychotics. Int. J. Neuropsychopharmacol. 10, 7–19.
Li, Y., Tennekoon, G. I., Birnbaum, M., Marchionni, M. A., and Rutkowski, J. L. (2001). Neuregulin signaling through a PI3K/Akt/Bad pathway in Schwann cell survival. Mol. Cell. Neurosci. 17, 761–767.
Li, Y. C., Xi, D., Roman, J., Huang, Y. Q., and Gao, W. J. (2009). Activation of glycogen synthase kinase-3 beta is required for hyperdopamine and D2 receptor-mediated inhibition of synaptic NMDA receptor function in the rat prefrontal cortex. J. Neurosci. 29, 15551–15563.
Lohse, M. J., Benovic, J. L., Codina, J., Caron, M. G., and Lefkowitz, R. J. (1990). Beta-arrestin: a protein that regulates beta-adrenergic receptor function. Science 248, 1547–1550.
Luttrell, L. M., Ferguson, S. S., Daaka, Y., Miller, W. E., Maudsley, S., Della Rocca, G. J., Lin, F., Kawakatsu, H., Owada, K., Luttrell, D. K., Caron, M. G., and Lefkowitz, R. J. (1999). Beta-arrestin-dependent formation of beta2 adrenergic receptor-Src protein kinase complexes. Science 283, 655–661.
Luttrell, L. M., and Gesty-Palmer, D. (2010). Beyond desensitization: physiological relevance of arrestin-dependent signaling. Pharmacol. Rev. 62, 305–330.
Mannoury La Cour, C., Salles, M. J., Pasteau, V., and Millan, M. J. (2011). Signaling pathways leading to phosphorylation of Akt and GSK-3beta by activation of cloned human and rat cerebral Dand D receptors. Mol. Pharmacol. 79, 91–105.
Mansour, H. A., Wood, J., Logue, T., Chowdari, K. V., Dayal, M., Kupfer, D. J., Monk, T. H., Devlin, B., and Nimgaonkar, V. L. (2006). Association study of eight circadian genes with bipolar I disorder, schizoaffective disorder and schizophrenia. Genes Brain Behav. 5, 150–157.
Mao, Y., Ge, X., Frank, C. L., Madison, J. M., Koehler, A. N., Doud, M. K., Tassa, C., Berry, E. M., Soda, T., Singh, K. K., Biechele, T., Petryshen, T. L., Moon, R. T., Haggarty, S. J., and Tsai, L. H. (2009). Disrupted in schizophrenia 1 regulates neuronal progenitor proliferation via modulation of GSK3beta/beta-catenin signaling. Cell 136, 1017–1031.
Masri, B., Salahpour, A., Didriksen, M., Ghisi, V., Beaulieu, J. M., Gainetdinov, R. R., and Caron, M. G. (2008). Antagonism of dopamine D2 receptor/beta-arrestin 2 interaction is a common property of clinically effective antipsychotics. Proc. Natl. Acad. Sci. U.S.A. 105, 13656–13661.
Mei, L., and Xiong, W. C. (2008). Neuregulin 1 in neural development, synaptic plasticity and schizophrenia. Nat. Rev. Neurosci. 9, 437–452.
Meijer, L., Flajolet, M., and Greengard, P. (2004). Pharmacological inhibitors of glycogen synthase kinase 3. Trends Pharmacol. Sci. 25, 471–480.
Miller, J. S., Tallarida, R. J., and Unterwald, E. M. (2009). Cocaine-induced hyperactivity and sensitization are dependent on GSK3. Neuropharmacology 56, 1116–1123.
Miller, J. S., Tallarida, R. J., and Unterwald, E. M. (2010). Inhibition of GSK3 attenuates dopamine D1 receptor agonist-induced hyperactivity in mice. Brain Res. Bull. 82, 184–187.
Missale, C., Nash, S. R., Robinson, S. W., Jaber, M., and Caron, M. G. (1998). Dopamine receptors: from structure to function. Physiol. Rev. 78, 189–225.
Morissette, M., Samadi, P., Hadj Tahar, A., Belanger, N., and Di Paolo, T. (2010). Striatal Akt/GSK3 signaling pathway in the development of L-Dopa-induced dyskinesias in MPTP monkeys. Prog. Neuropsychopharmacol. Biol. Psychiatry 34, 446–454.
O’Brien, W. T., Harper, A. D., Jove, F., Woodgett, J. R., Maretto, S., Piccolo, S., and Klein, P. S. (2004). Glycogen synthase kinase-3beta haploinsufficiency mimics the behavioral and molecular effects of lithium. J. Neurosci. 24, 6791–6798.
O’Brien, W. T., Huang, J., Buccafusca, R., Garskof, J., Valvezan, A. J., Berry, G. T., and Klein, P. S. (2011). Glycogen synthase kinase-3 is essential for beta-arrestin-2 complex formation and lithium-sensitive behaviors in mice. J. Clin. Invest. 121, 3756–3762.
Ouattara, B., Gasparini, F., Morissette, M., Gregoire, L., Samadi, P., Gomez-Mancilla, B., and Di Paolo, T. (2010). Effect of L-Dopa on metabotropic glutamate receptor 5 in the brain of parkinsonian monkeys. J. Neurochem. 113, 715–724.
Pan, J. Q., Lewis, M. C., Ketterman, J. K., Clore, E. L., Riley, M., Richards, K. R., Berry-Scott, E., Liu, X., Wagner, F. F., Holson, E. B., Neve, R. L., Biechele, T. L., Moon, R. T., Scolnick, E. M., Petryshen, T. L., and Haggarty, S. J. (2011). AKT kinase activity is required for lithium to modulate mood-related behaviors in mice. Neuropsychopharmacology 36, 1397–1411.
Peineau, S., Bradley, C., Taghibiglou, C., Doherty, A., Bortolotto, Z. A., Wang, Y. T., and Collingridge, G. L. (2008). The role of GSK-3 in synaptic plasticity. Br. J. Pharmacol. 153(Suppl. 1), S428–S437.
Peineau, S., Taghibiglou, C., Bradley, C., Wong, T. P., Liu, L., Lu, J., Lo, E., Wu, D., Saule, E., Bouschet, T., Matthews, P., Isaac, J. T., Bortolotto, Z. A., Wang, Y. T., and Collingridge, G. L. (2007). LTP inhibits LTD in the hippocampus via regulation of GSK3beta. Neuron 53, 703–717.
Pitcher, G. M., Kalia, L. V., Ng, D., Goodfellow, N. M., Yee, K. T., Lambe, E. K., and Salter, M. W. (2011). Schizophrenia susceptibility pathway neuregulin 1-ErbB4 suppresses Src upregulation of NMDA receptors. Nat. Med. 17, 470–478.
Pogorelov, V. M., Nomura, J., Kim, J., Kannan, G., Ayhan, Y., Yang, C., Taniguchi, Y., Abazyan, B., Valentine, H., Krasnova, I. N., Kamiya, A., Cadet, J. L., Wong, D. F., and Pletnikov, M. V. (2011). Mutant DISC1 affects methamphetamine-induced sensitization and conditioned place preference: a comorbidity model. Neuropharmacology. doi:10.1016/j.neuropharm.2011.02.003. [Epub ahead of print].
Polter, A., Beurel, E., Yang, S., Garner, R., Song, L., Miller, C. A., Sweatt, J. D., Mcmahon, L., Bartolucci, A. A., Li, X., and Jope, R. S. (2010). Deficiency in the inhibitory serine-phosphorylation of glycogen synthase kinase-3 increases sensitivity to mood disturbances. Neuropsychopharmacology 35, 1761–1774.
Prickaerts, J., Moechars, D., Cryns, K., Lenaerts, I., Van Craenendonck, H., Goris, I., Daneels, G., Bouwknecht, J. A., and Steckler, T. (2006). Transgenic mice overexpressing glycogen synthase kinase 3beta: a putative model of hyperactivity and mania. J. Neurosci. 26, 9022–9029.
Quesada, A., Lee, B. Y., and Micevych, P. E. (2008). PI3 kinase/Akt activation mediates estrogen and IGF-1 nigral DA neuronal neuroprotection against a unilateral rat model of Parkinson’s disease. Dev. Neurobiol. 68, 632–644.
Rau, T. F., Kothiwal, A., Zhang, L., Ulatowski, S., Jacobson, S., Brooks, D. M., Cardozo-Pelaez, F., Chopp, M., and Poulsen, D. J. (2011). Low dose methamphetamine mediates neuroprotection through a PI3K-AKT pathway. Neuropharmacology 61, 677–686.
Sahar, S., Zocchi, L., Kinoshita, C., Borrelli, E., and Sassone-Corsi, P. (2010). Regulation of BMAL1 protein stability and circadian function by GSK3beta-mediated phosphorylation. PLoS ONE 5, e8561. doi:10.1371/journal.pone.0008561
Santini, E., Alcacer, C., Cacciatore, S., Heiman, M., Herve, D., Greengard, P., Girault, J. A., Valjent, E., and Fisone, G. (2009). L-DOPA activates ERK signaling and phosphorylates histone H3 in the striatonigral medium spiny neurons of hemiparkinsonian mice. J. Neurochem. 108, 621–633.
Scheid, M. P., and Woodgett, J. R. (2001). PKB/AKT: functional insights from genetic models. Nat. Rev. Mol. Cell Biol. 2, 760–768.
Shenoy, S. K., Drake, M. T., Nelson, C. D., Houtz, D. A., Xiao, K., Madabushi, S., Reiter, E., Premont, R. T., Lichtarge, O., and Lefkowitz, R. J. (2006). beta-arrestin-dependent, G protein-independent ERK1/2 activation by the beta2 adrenergic receptor. J. Biol. Chem. 281, 1261–1273.
Snyder, S. H. (1976). The dopamine hypothesis of schizophrenia: focus on the dopamine receptor. Am. J. Psychiatry 133, 197–202.
Sotnikova, T. D., Beaulieu, J. M., Barak, L. S., Wetsel, W. C., Caron, M. G., and Gainetdinov, R. R. (2005). Dopamine-independent locomotor actions of amphetamines in a novel acute mouse model of Parkinson disease. PLoS Biol. 3, e271. doi:10.1371/journal.pbio.0030271
Souza, B. R., Romano-Silva, M. A., and Tropepe, V. (2011). Dopamine D2 receptor activity modulates Akt signaling and alters GABAergic neuron development and motor behavior in zebrafish larvae. J. Neurosci. 31, 5512–5525.
Spano, P. F., Govoni, S., and Trabucchi, M. (1978). Studies on the pharmacological properties of dopamine receptors in various areas of the central nervous system. Adv. Biochem. Psychopharmacol. 19, 155–165.
Stambolic, V., Ruel, L., and Woodgett, J. R. (1996). Lithium inhibits glycogen synthase kinase-3 activity and mimics wingless signalling in intact cells. Curr. Biol. 6, 1664–1668.
Stambolic, V., and Woodgett, J. R. (1994). Mitogen inactivation of glycogen synthase kinase-3 beta in intact cells via serine 9 phosphorylation. Biochem. J. 303(Pt 3), 701–704.
Sutton, L. P., Honardoust, D., Mouyal, J., Rajakumar, N., and Rushlow, W. J. (2007). Activation of the canonical Wnt pathway by the antipsychotics haloperidol and clozapine involves dishevelled-3. J. Neurochem. 102, 153–169.
Sutton, L. P., and Rushlow, W. J. (2011). The dopamine D2 receptor regulates Akt and GSK-3 via Dvl-3. Int. J. Neuropsychopharmacol. 22, 1–15.
Svenningsson, P., Tzavara, E. T., Carruthers, R., Rachleff, I., Wattler, S., Nehls, M., Mckinzie, D. L., Fienberg, A. A., Nomikos, G. G., and Greengard, P. (2003). Diverse psychotomimetics act through a common signaling pathway. Science 302, 1412–1415.
Swift, J. L., Godin, A. G., Dore, K., Freland, L., Bouchard, N., Nimmo, C., Sergeev, M., De Koninck, Y., Wiseman, P. W., and Beaulieu, J. M. (2011). Quantification of receptor tyrosine kinase transactivation through direct dimerization and surface density measurements in single cells. Proc. Natl. Acad. Sci. U.S.A. 108, 7016–7021.
Tan, H. Y., Nicodemus, K. K., Chen, Q., Li, Z., Brooke, J. K., Honea, R., Kolachana, B. S., Straub, R. E., Meyer-Lindenberg, A., Sei, Y., Mattay, V. S., Callicott, J. H., and Weinberger, D. R. (2008). Genetic variation in AKT1 is linked to dopamine-associated prefrontal cortical structure and function in humans. J. Clin. Invest. 118, 2200–2208.
Tosato, S., Dazzan, P., and Collier, D. (2005). Association between the neuregulin 1 gene and schizophrenia: a systematic review. Schizophr. Bull. 31, 613–617.
Ugi, S., Imamura, T., Maegawa, H., Egawa, K., Yoshizaki, T., Shi, K., Obata, T., Ebina, Y., Kashiwagi, A., and Olefsky, J. M. (2004). Protein phosphatase 2A negatively regulates insulin’s metabolic signaling pathway by inhibiting Akt (protein kinase B) activity in 3T3-L1 adipocytes. Mol. Cell. Biol. 24, 8778–8789.
Yuan, Q., Lin, F., Zheng, X., and Sehgal, A. (2005). Serotonin modulates circadian entrainment in Drosophila. Neuron 47, 115–127.
Yujnovsky, I., Hirayama, J., Doi, M., Borrelli, E., and Sassone-Corsi, P. (2006). Signaling mediated by the dopamine D2 receptor potentiates circadian regulation by CLOCK:BMAL1. Proc. Natl. Acad. Sci. U.S.A. 103, 6386–6391.
Keywords: glycogen synthase kinase 3, Akt, protein kinase B, dopamine, catecholamines, arrestin, G protein coupled receptor, receptor tyrosine kinase
Citation: Beaulieu J-M, Del’Guidice T, Sotnikova TD, Lemasson M and Gainetdinov RR (2011) Beyond cAMP: the regulation of Akt and GSK3 by dopamine receptors. Front. Mol. Neurosci. 4:38. doi: 10.3389/fnmol.2011.00038
Received: 26 August 2011;
Paper pending published: 08 September 2011;
Accepted: 13 October 2011;
Published online: 01 November 2011.
Edited by:
Oksana Kaidanovich-Beilin, Samuel Lunenfeld Research Institute, CanadaReviewed by:
Cristina Missale, University of Brescia, ItalyFabio Fumagalli, University of Milan, Italy
Denis Joseph David, Université Paris-Sud, France
Copyright: © 2011 Beaulieu, Del’Guidice, Sotnikova, Lemasson and Gainetdinov. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Jean-Martin Beaulieu, Department of Psychiatry and Neuroscience, Université Laval-CRULRG, 2601 Chemin de la Canardière, Suite F-6500, Beauport, Québec, QC, Canada G1J 2G3. e-mail:bWFydGluLmJlYXVsaWV1QGNydWxyZy51bGF2YWwuY2E=; Raul R. Gainetdinov, Department of Neuroscience and Brain Technologies, Italian Institute of Technology, Via Morego 30, Genova 16167, Italy. e-mail:cmF1bC5nYWluZXRkaW5vdkBpaXQuaXQ=