 Woo-Yang Kim1* and William D. Snider2
Woo-Yang Kim1* and William D. Snider2
- 1 Department of Developmental Neuroscience, Munroe-Meyer Institute, University of Nebraska Medical Center, Omaha, NE, USA
- 2 Neuroscience Center, University of North Carolina, Chapel Hill, NC, USA
Glycogen synthase kinase-3 (GSK-3) is central to multiple intracellular pathways including those activated by Wnt/β-catenin, Sonic Hedgehog, Notch, growth factor/RTK, and G protein-coupled receptor signals. All of these signals importantly contribute to neural development. Early attention on GSK-3 signaling in neural development centered on the regulation of neuronal polarity using in vitro paradigms. However, recent creation of appropriate genetic models has demonstrated the importance of GSK-3 to multiple aspects of neural development including neural progenitor self-renewal, neurogenesis, neuronal migration, neural differentiation, and synaptic development.
Introduction
Glycogen synthase kinase-3 (GSK-3), a serine–threonine kinase, was originally identified as an important component of the glycogen metabolism pathway. Later, however, it was found to be an important regulator in multiple intracellular contexts. Recent studies using pharmacological and genetic approaches have suggested important functions for GSK-3 in an array of neural developmental activities including neural progenitor homeostasis, neuronal migration, neurite growth/specification, and synapse development. Further, recent data indicate that the Disrupted in Schizophrenia protein, DISC1, acts in part via GSK-3 suggesting that dysregulated GSK-3 activity during development may contribute to the pathogenesis of schizophrenia. Multiple proteins that play important roles during neural development have been identified as either GSK-3 substrates or binding partners (Table 1).
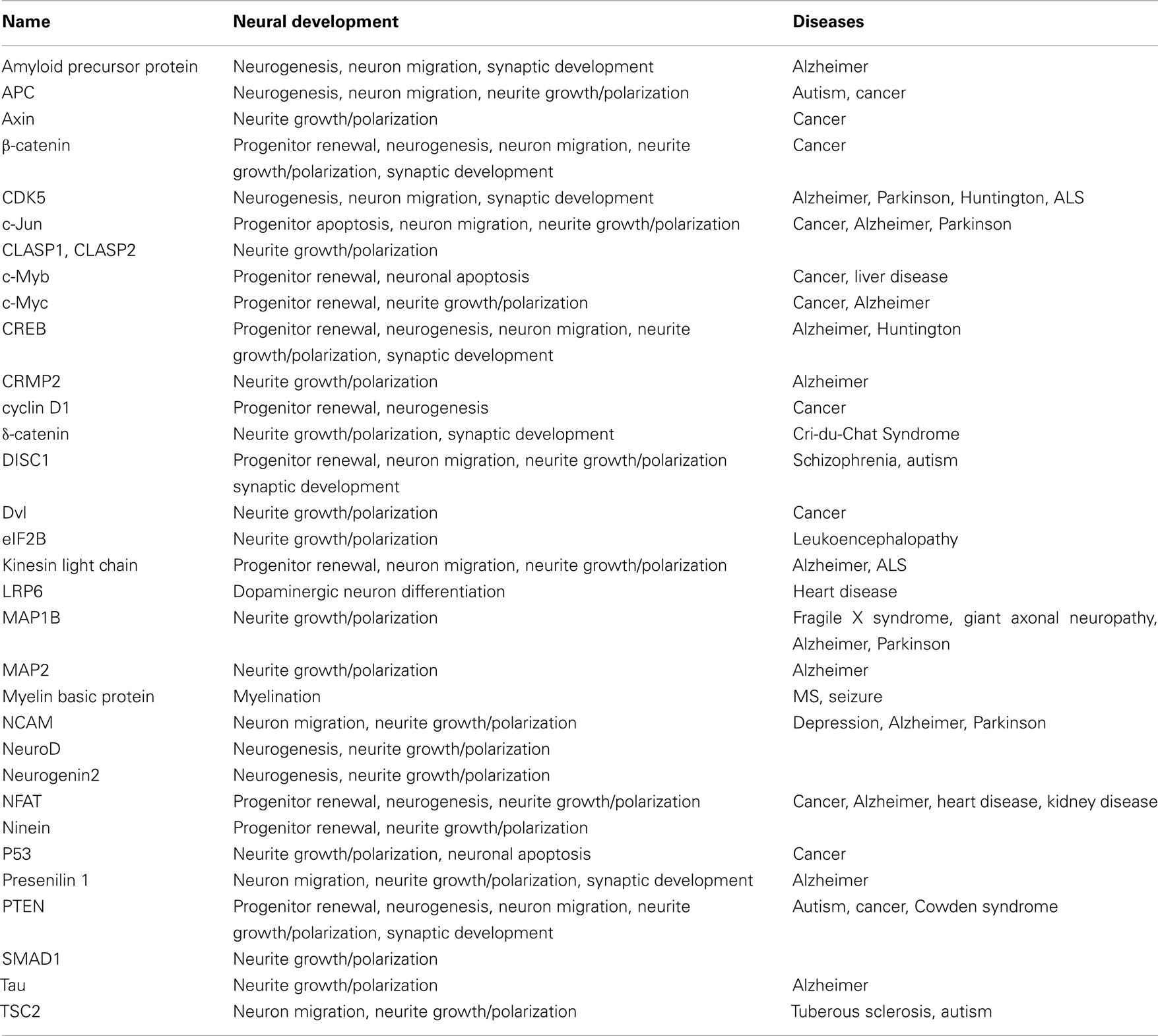
Table 1. Glycogen synthase kinase-3-interacting proteins associated with neural development and diseases.
Isoforms of GSK-3 in the Nervous System
In mammals, the GSK-3 family consists of two members, GSK-3α and GSK-3β that show 98% sequence identity within their kinase domains and overall share 85% amino acid sequence identity (reviewed in Doble and Woodgett, 2003). Both GSK-3 isoforms are highly expressed in the nervous system including brain and spinal cord. There are two splice variants of the GSK-3β and one variant GSK-3β2 is highly expressed in the developing nervous system (Mukai et al., 2002). It is interesting that GSK-3β2 is only expressed in differentiated neurons, not in glial cells while GSK-3β1 is expressed in glial cells (Wood-Kaczmar et al., 2009). Further, GSK-3β2 is upregulated in PC12 cells when induced by nerve growth factor to differentiate into a neuronal phenotype (Goold and Gordon-Weeks, 2001). These findings suggest that differential regulation of GSK-3β isoform levels may be important in the initiation of neuronal differentiation and the maintenance of neurons during nervous system development.
Most functional studies of GSK-3 functions in mammalian neural development have relied on pharmacological inhibitors in dissociated preparations. However, since pharmacological GSK-3 inhibitors target ATP-binding pockets of both isoforms, distinctive functional analysis of each isoform using pharmacological inhibitors is not possible. Recently, studies in gene targeted mice have begun to yield decisive information regarding the roles of GSK-3 isoforms in neural development. Gene targeting of GSK-3β leads to late embryonic lethality via liver apoptosis or heart defects (Hoeflich et al., 2000; Kerkela et al., 2008). Surprisingly, the developing nervous system of GSK-3β null mice including the brain and spinal cord exhibits grossly normal morphology (Kim et al., 2006). GSK-3β heterozygous (+/−) mice, however, show multiple neurobehavioral abnormalities including aggressive behaviors, increased anxiety, reduced movement, and poor memory processing (O’Brien et al., 2004; Beaulieu et al., 2008; Kimura et al., 2008). Further, overexpression of GSK-3β in the brain results in hyperactivity and mania (Prickaerts et al., 2006). Finally GSK-3β is important in signaling downstream of dopamine receptors (Beaulieu et al., 2004). These results demonstrate that the GSK-3β isoform has important functions in regulation of behavior, probably via regulation of signaling downstream of neurotransmitter receptors.
GSK-3α null mice are viable and fertile (MacAulay et al., 2007) and show no abnormalities of gross anatomy in the developing brain (Kim et al., 2009; Yokota et al., 2010). However, young adult GSK-3α null mice show decreases in the size and number of cerebellar Purkinje cells and changes in social interaction and locomotion (Kaidanovich-Beilin et al., 2009). GSK-3α deletion also partially rescues the dendritic abnormality in cortical neurons of DISC1-L100P mutant mice that show reduced spine density compared to wild type mice (Lee et al., 2011). Further, GSK-3α null mice are hypersensitive to insulin (MacAulay et al., 2007), suggesting the potential roles of GSK-3α in hypothalamic regulation. Using brain lysates of GSK-3α null, β null, and double knockout mice, a recent study has demonstrated isoform-specific activities on substrate phosphorylation in the brain. Each isoform produces distinct phosphorylation patterns of CRMP2, CRMP4, and Tau (Soutar et al., 2010). Together, these results indicate that GSK-3α isoform has important specific functions in postnatal neuroanatomy and behavioral regulation.
The lack of gross brain developmental abnormalities in GSK-3α null and GSK-3β null mice suggests that either isoform might compensate for the other in the regulation of differentiation, survival, and morphogenesis of neurons and glia. Indeed, axon and dendritic morphogenesis is normal in cultured hippocampal neurons from GSK-3β null embryonic brains (Kim et al., 2006). However, genetic elimination of GSK-3 using an shRNA that targeted both GSK-3 isoforms lead to strong changes in axon and dendritic morphogenesis in cultured hippocampal neurons (Kim et al., 2006). Further, the possibility of compensation has now been confirmed in vivo. Conditional elimination of both GSK-3α and β at an early stage (∼E10) of mouse nervous system development results in dramatic effects on murine brain development (Kim et al., 2009).
Regulation of Neural Progenitors
There are several types of neural progenitors in rodent developing brains (Figure 1). Radial neural progenitors are a major population of neural progenitors and occupy the ventricular zone in the developing brain. They display long radial processes to form a scaffold covering from apical ventricular membrane to pial surface. This scaffold supports neuronal migration. Intermediate progenitors in the subventricular zone execute only the final neurogenic division. In addition to radial and intermediate progenitors, there are other types of neural progenitors more prominent in higher species. Importantly, recent studies show existence of a greatly expanded oRG (outer subventricular zone progenitors) in human (Hansen et al., 2010) and mice (Wang et al., 2011). oRG progenitors are located in the outer region of the subventricular zone in rodent. Unlike intermediate progenitors, this type of progenitor can undergo self-renewal as well as symmetric neurogenesis.
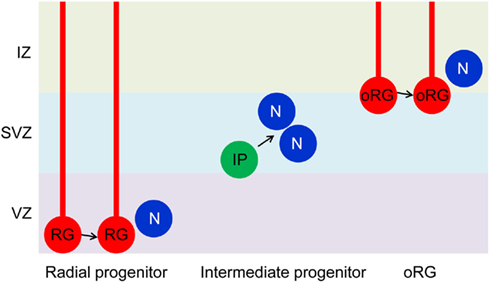
Figure 1. Neural progenitors in the developing brain. Radial glial progenitors (RG) arise early in development from neuroepithelial cells. They are located in the ventricular zone (VZ) and have long radial processes that extend form VZ to the pial surface. A RG divides symmetrically to self-renew and asymmetrically to generate a daughter RG and a post-mitotic neuron or an intermediate progenitor (IP). IPs localize in the subventricular zone (SVZ) and divide symmetrically to produce neurons. There is another type of progenitors (oRG) in the superficial layer of SVZ or in the intermediate zone (IZ). Like RGs, oRGs have glial processes and can self-renew as well as generate neurons. For more information on oRG, see a recent review by Kriegstein and colleagues (Lui et al., 2011). The role of GSK-3 in regulating radial progenitors is now established. Functions related to IPs and oRGs remain to be investigated.
Effects of GSK-3α and β Deletion
Recent pharmacological evidence has strongly implicated GSK-3 signaling in the regulation of embryonic stem cell self-renewal (Sato et al., 2004; Umehara et al., 2007; Ying et al., 2008; Bone et al., 2009). Further, DISC1 implicated in some familial cases of schizophrenia and depression, regulates neural progenitor proliferation and may act in part via regulation of GSK-3 activity (Mao et al., 2009). These observations underscore the importance of determining GSK-3 functions in relation to neural progenitor self-renewal and neurogenesis during brain development in vivo.
The functions of GSK-3 in mammalian brain development remained unclear because GSK-3 single isoform mutants had not been associated with major brain developmental abnormalities (see above). The results, however, did not exclude possible compensatory effects of one GSK-3 isoform for the other in single null mutant animals (Doble et al., 2007). To explore GSK-3 functions definitively, a recent study pursued a conditional knockout strategy in which a GSK-3β floxed allele was bred onto GSK-3α null background to allow tissue specific elimination of GSK-3 signaling (Kim et al., 2009). This study used a mouse strain expressing Cre recombinase under the control of the neural specifying elements of the nestin promoter (Tronche et al., 1999) to induce recombination around E10 in neuroepithelial cells as they convert to radial progenitors and initiate neurogenesis (Yoon et al., 2008; Yokota et al., 2009). The study conclusively demonstrated that elimination of both GSK-3 isoforms in the developing brain lead to a massive increase in neural progenitor proliferation. This increase was indicated by infolding of the developing cortical wall, major increases in numbers of cells that expressed mitotic markers, and a dramatic expansion of the population of neural progenitors expressing Sox2. GSK-3 double knockout brains showed increased progenitor division, premature re-entry and shortening of the cell cycle.
Glycogen synthase kinase-3 deficient cells appeared to be locked into the radial progenitor phase as generation of both intermediate neural progenitors and post-mitotic neurons was markedly suppressed. In contrast to the expansion of Sox2-positive radial progenitors, the population of intermediate neural progenitors marked with Tbr2 was dramatically down-regulated in GSK-3 double knockout brains (Kim et al., 2009). Consistent with this finding, only around one-third of the normal number of neurons was generated in GSK-3 double knockout brains. Although deletion of GSK-3 prevented progression of most cells beyond the radial progenitor phase, a few progenitors were still capable of differentiation as manifested by expression of Tbr2 and markers of post-mitotic neurons. Whether some progenitors can truly progressed independently of GSK-3 signaling or whether the differentiated cells represented a population of radial progenitors that had some persistent GSK-3 proteins due to either late or incomplete deletion of GSK-3 remained unclear.
Mechanisms Associated with GSK-3 Regulation of Neural Progenitors
Multiple extracellular signals and intracellular signaling mechanisms that regulate neural progenitor proliferation and differentiation have been identified (reviewed in Corbin et al., 2008). The mechanisms that have received the most attention in the dorsal telencephalon are β-catenin/LEF1 and Notch signaling (Figure 2A). Whether these mechanisms operate independently or are coordinately regulated is an interesting question. GSK-3 has the potential to influence both mechanisms. It is important to emphasize that GSK-3s are also key components in pathways downstream of two extracellular signals, FGF2 and Shh, which also strongly regulate progenitor proliferation in the developing telencephalon (Figure 2A; Vaccarino et al., 1999; Raballo et al., 2000; Komada et al., 2008; Shimizu et al., 2008).
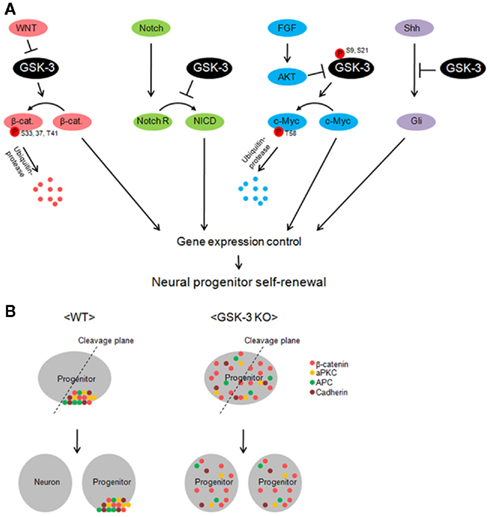
Figure 2. Glycogen synthase kinase-3 is a key regulator of multiple signaling pathways in neural progenitors. (A) GSK-3 controls gene induction by regulating the levels of transcription factors in neural progenitors. β-catenin is degraded via GSK-3 phosphorylation-dependent ubiquitin-protease machinery in the resting state. However, inhibition of GSK-3 by Wnt allows β-catenin accumulation. GSK-3 is also critical in the control of c-Myc levels. c-Myc is phosphorylated at T58 and recruited by ubiquitin-protease for degradation. FGF inhibits GSK-3 via AKT-mediated phosphorylation at S9 (beta) or S21 (alpha) and leads to increase in c-Myc levels. Inhibition of GSK-3 increases Notch intracellular domain (NICD) and Gli proteins in neural progenitors. Thus, GSK-3 activity is critical in the control of transcription factors in multiple signal pathways, which eventually regulates neural progenitors during development. (B) A schematic model of the GSK-3 function in apical–basal polarity and neurogenesis. GSK-3 is required for polarity establishment and possibly for neurogenic division of progenitors. Polarity-associated molecules including β-catenin, atypical PKC, APC, and cadherin are localized in the apical membrane. When progenitors divide, daughter cells that inherit the polarity molecules are thought to become progenitors. Without the polarity protein, cells become neurons. However, in GSK-3-deleted neural progenitors, polarity molecules are distributed throughout the cell. Thus, wherever the cleavage plane is positioned, daughter cells have almost same composition of the molecules. This may be an important factor that leads constant self-renewal of progenitors in GSK-3-deleted brains.
β-Catenin signaling and GSK-3
It is well known that regulation of β-catenin is critical to control of progenitor proliferation in multiple regions of the developing nervous system (Chenn and Walsh, 2002; Machon et al., 2003, 2007; Zechner et al., 2003; Woodhead et al., 2006; Gulacsi and Anderson, 2008). As expected, β-catenin was increased substantially in GSK-3 double knockout brains, consistent with the critical importance of GSK-3 in phosphorylating β-catenin in the destruction complex that mediates canonical Wnt signaling (Kim et al., 2009). However, stabilization of β-catenin alone may not have fully accounted for the effects of progenitor hyperproliferation in GSK-3 double knockout brains. Although it is difficult to compare studies directly, it appears that the enlarged brain observed upon overexpression of a stabilized β-catenin is associated with a more normal pattern of neuronal differentiation than GSK-3 double knockout brains (Chenn and Walsh, 2002, 2003; Kim et al., 2009). Further, elimination of β-catenin in neural progenitors reduces but does not eliminate progenitor proliferation in vivo (Machon et al., 2003; Zechner et al., 2003; Gulacsi and Anderson, 2008). Finally, overexpression of dominant-negative TCF only partially inhibited proliferation in GSK-3 knockout neural progenitors in vitro (Kim et al., 2009). Thus, GSK-3 may regulate self-renewal of neural progenitors by mechanisms in addition to β-catenin signaling.
Notch signaling and GSK-3
Notch signaling is critical to the maintenance and the identity of radial glial cells during development (Gaiano et al., 2000; reviewed in Ohtsuka et al., 2001; Hatakeyama et al., 2004; Yoon and Gaiano, 2005; Mizutani et al., 2007; Shimojo et al., 2008). There are several mechanisms by which GSK-3 may regulate Notch signaling both directly and indirectly (Foltz et al., 2002; Espinosa et al., 2003; Uemura et al., 2007; Hayward et al., 2008; Shimizu et al., 2008; Jin et al., 2009a). For example, GSK-3β is able to bind and phosphorylate Notch2 in vitro and in vivo (Espinosa et al., 2003). The GSK-3α isoform likely has the same function, because the phosphorylation was inhibited by the treatment of Wnt (Espinosa et al., 2003). Thereby, GSK-3 inhibits the transcriptional activation of Notch target genes. Also, β-catenin reportedly enhances the level and transcription activity of Notch1 (Shimizu et al., 2008; Jin et al., 2009b). Meanwhile, there are conflicting studies with regard to the role of GSK-3 in Notch signaling, in which inhibition of GSK-3 using pharmacological drugs or genetic tools decreased NICD levels in neuroblastoma cells (Foltz et al., 2002) and vascular smooth muscle cells (Guha et al., 2011).
Notch signaling was shown to be increased in GSK-3 double knockout brains (Kim et al., 2009). Levels of Notch intracellular domain were markedly enhanced as were levels of the Notch downstream signaling mediators Hes1 and Hes5. Further, immunohistochemical data suggested that most or all of the extra progenitors in GSK-3 double knockout mice express high levels of Hes1 protein, indicative of active Notch signaling in these cells. Levels of Jagged-1, a Notch agonist, were increased in GSK-3-deleted brain lysates, suggesting that the activation of Notch pathway upon GSK-3-deletion requires a Notch agonist. This is different from the activation of β-catenin signal, which is induced in GSK-3-deleted cells in the absence of Wnt ligands (Doble et al., 2007). Overexpression of a dominant-negative Hes1 and pharmacological inhibition of Notch signaling partially inhibited proliferation in GSK-3-deleted progenitors (Kim et al., 2009). Interestingly, Notch signaling levels distinguish radial progenitors from intermediate neural progenitors that have attenuated canonical Notch signaling (Mizutani et al., 2007). Therefore, high levels of Notch signaling in GSK-3 double knockout cells may have suppressed neurogenesis at the stage of radial progenitors such that few cells progress to the stage of Tbr2-positive intermediate neural progenitors. However, whether GSK-3 regulated Notch signaling directly or indirectly via regulation of β-catenin or some other mechanism was not established.
GSK-3 regulation of Myc
GSK-3 is a downstream mediator of FGF/PI3K signaling in neural progenitors (Shimizu et al., 2008). Several recent studies have demonstrated that GSK-3 regulates the stability of Myc family proteins via the PI3K pathway (reviewed in Knoepfler and Kenney, 2006; Bechard and Dalton, 2009; reviewed in Doble and Woodgett, 2009; Otto et al., 2009). It is important to emphasize that GSK-3 can directly regulate the stability of Myc family proteins, independently of β-catenin effects on Myc transcription (Bechard and Dalton, 2009; reviewed in Doble and Woodgett, 2009; Otto et al., 2009). Interestingly, these effects are thought to be downstream of RTK/PI3K rather than Wnt/Frizzled signaling (reviewed in Knoepfler and Kenney, 2006). Myc family proteins are key elements in the maintenance of neural progenitors by inhibiting differentiation (Knoepfler et al., 2002; Wey et al., 2010). Phosphorylation of c-Myc at T58 thought to be a key to its stability is regulated by GSK-3. The GSK-3-mediated phosphorylation at T58 plays an important role in inducing cell differentiation, as inhibition of the phosphorylation maintains embryonic stem cell pluripotency (Cartwright et al., 2005). This phosphorylation was shown to be eliminated in GSK-3 double knockout brains and the level of c-Myc was increased (Kim et al., 2009). Given the short half-life cycle of c-Myc (∼20 min; reviewed in Eilers and Eisenman, 2008), control of c-Myc stability via T58 phosphorylation may be a critical step in GSK-3 regulation of neural progenitor self-renewal.
Myc binds to promoters of many genes including cyclins and regulates their transcription. This traditional role as a transcription factor has been primarily considered in maintaining self-renewal and in inhibiting differentiation of stem cells. But several recent reports suggest that Myc contributes to stem cell maintenance in other ways by globally maintaining the histone acetylation and methylation euchromatin state (Lin et al., 2009; reviewed in Varlakhanova and Knoepfler, 2009; Araki et al., 2011). Whether Myc elicits changes in histone methylation patterns of promoters in neural progenitors and whether GSK-3 is associated with this process remain to be elucidated. Further characterization of functional genomic analysis will likely elucidate Myc functions as epigenetic transcriptional regulator in neural progenitors.
Apical–basal polarity and GSK-3
Cell polarity is a fundamental property of all cells including neural progenitors. GSK-3 is thought to be a key regulator of cell polarity and microtubule-cytoskeleton regulation based on in vitro studies (Etienne-Manneville and Hall, 2003; reviewed in Buttrick and Wakefield, 2008). GSK-3 directly binds to polarity-associated proteins including APC (Adenomatous polyposis coli), EB1 (End binding 1), and CLASPs (Cytoplasmic linker-associated proteins) and regulates their activities by phosphorylation. For example, APC and CLASPs promote microtubule stability and their phosphorylation by GSK-3 inhibits microtubule binding, thus destabilizing microtubules (Akhmanova et al., 2001; Zumbrunn et al., 2001). Asymmetric cell division results from polarized deposition of polarity proteins and it is required for neurogenic division of radial neural progenitors (Gotz and Huttner, 2005). Apical–basal polarity indicated by the distribution patterns of cell polarity/adhesion proteins including APC, EB1, cadherin, and aPKC was disrupted in GSK-3-deleted developing brains (Kim et al., 2009). Thus, the expansion of the radial progenitor pool and inhibition of neurogenesis in GSK-3-deficient brains may be attributed in part to the dysregulation of polarity-associated proteins (Figure 2B). Localized distribution patterns of apical molecules may be coordinated by GSK-3 regulation of the microtubule apparatus or motor proteins. In this regard, it is notable that elimination of polarity-associated molecules, Lgl1 and αE-catenin results in a major hyperproliferation of neural progenitors (Klezovitch et al., 2004; Lien et al., 2006).
It has also been demonstrated that GSK-3 regulates centrosome reorientation and microtubule spindle formation during cell division (Wakefield et al., 2003; Cheng et al., 2008; Izumi et al., 2008). Dysregulation of centrosome positioning and cleavage planes may favor divisions that result in additional progenitors rather than post-mitotic neurons (reviewed in Higginbotham and Gleeson, 2007). An important implication of these findings is that apical–basal polarity and centrosome positioning may be among of the mechanisms responsible for GSK-3-mediated neural progenitor homeostasis.
DISC1 and GSK-3 signaling
A recent study has shown that GSK-3 binds to DISC1 and that pharmacological inhibition of GSK-3 can mitigate effects of DISC1 shRNA knockdown on neural progenitor proliferation in vitro (Mao et al., 2009). DISC1 protein does not have enzymatic activity, rather it functions as a physiological scaffold by interacting with multiple proteins to modulate their functional activities. For example, DISC1 binds to NUDEL (Duan et al., 2007) and girdin (Enomoto et al., 2009), and modulates the activities of these proteins in adult neural progenitors and their progeny. Likewise, DISC1 may serve as an important scaffold to bridge GSK-3 and its regulators in neural progenitors during development.
Homeostatic Controls in Neural Progenitors
During nervous system development, an over-abundance of cells is produced presumably to accommodate the significant loss of cells that fail to achieve functionality. This overproduction allows for remarkable plasticity in nervous system development but requires exquisite inductive coordination. Temporal and spatial regulation of GSK-3 activity during nervous system development must be achieved in order to terminate neural progenitor proliferation and allow differentiation at just the right moment to yield the correct number of neurons. It is remarkable that regulation of the activity of a single kinase provides a powerful and simple mechanism to control neural progenitor homeostasis.
The functions of GSK-3 in radial neural progenitors have been discussed above. However, whether GSK-3 has significant roles in intermediate progenitors and oRGs remains to be elucidated. It will be important to see if GSK-3-deletion also results in self-renewal of these progenitors.
GSK-3 Signaling in Neuronal Migration
After progenitor proliferation and neurogenesis, post-mitotic neurons migrate to final locations which in the cortex results in proper lamination. There is increasing evidence that GSK-3 may be important in regulating neuronal migration.
Wnt Signal and Neuron Migration in C. elegans
Recent studies have shown that Wnt family molecules play important roles in controlling anterior/posterior (AP)-directed neuronal cell migration and polarity in C. elegans (reviewed in Silhankova and Korswagen, 2007). Multiple Wnt proteins function in parallel as instructive cues or permissive signals to control neuronal migration along the AP axis. EGL-20/Wnt is one of the five Wnt proteins of C. elegans and specifies posterior direction of Q neuroblast migration (Harris et al., 1996; Maloof et al., 1999). This protein activates a canonical Wnt pathway (β-catenin-dependent) in the Q neuroblast (Salser and Kenyon, 1992; reviewed in Korswagen, 2002). Accordingly, overexpression of SGG-1, the C. elegans GSK-3β ortholog induced defects in Q cell migration far toward anterior region (Korswagen et al., 2002). Together, these suggest that GSK-3 signaling plays an important role in C. elegans neuron migration by negatively regulating Wnt signaling.
Wnt Signaling Molecules and Neuronal Migration in Mice
Considerable evidence both direct and indirect now implicates GSK-3 in regulation of neuronal migration in mice. Some evidence has come from functional studies of the GSK-3 target and destruction complex component, APC. In addition to its role in the destruction complex, APC is a microtubule-associated protein and participates in microtubule-based cytoskeleton dynamics (Barth et al., 2008). GSK-3 directly binds to APC and phosphorylates it in order to regulate its activity. APC phosphorylation by GSK-3 dissociates APC from microtubules. A recent study has demonstrated that APC serves an essential function in the maintenance of the polarized radial glial scaffold during mouse brain development (Yokota et al., 2009). Elimination of APC in radial progenitors by conditional gene targeting lead to striking defects in cortical lamination, suggesting an important role for APC in neuronal migration. However, it is not clear whether migration defects were entirely due to disruption of the radial glia scaffold or whether APC regulation of microtubules in neurons also contributed. It will be important to settles this issue with conditional elimination of APC specifically in early post-mitotic cortical neurons.
GSK-3 may also affect neuronal migration via regulation of β-catenin. Several studies have shown that genetic manipulation of β-catenin by either deletion or overexpression of a stabilized form in mice leads to the disruption of brain and spinal cord formation (Chenn and Walsh, 2002, 2003; Zechner et al., 2003). The malformation is mostly attributed to abnormal neural progenitor development. However, there were also abnormally localized neurons found in the mutant nervous system. Further, a separate study has demonstrated that β-catenin levels in progenitors influenced cortical neuron positioning (Mutch et al., 2009). A caveat of this interpretation is that the abnormal neuronal positioning may be due to a delay in progenitor fate determination. However, the β-catenin mutation studies again strongly suggest potential roles for GSK-3 in neuronal migration.
DISC1 regulates neuronal migration during brain development (Kamiya et al., 2005) and is strongly associated with GSK-3. DISC1 binds to GSK-3 and the interaction interferes with GSK-3 inhibition of the β-catenin signal in neural progenitors, resulting in progenitor proliferation (Mao et al., 2009). Recently, the interaction between DISC1 and GSK-3 has been implicated in control of cortical neuronal migration (Ishizuka et al., 2011). DISC1 bound to GSK-3 tightly during mid-embryonic stage (E14) in which neural progenitor proliferation is active (Figure 3A). But, DISC1 was dissociated with GSK-3 during later embryonic stages (E18) when neuronal migration and neuronal differentiation become dominant. These findings suggest that the DISC1/GSK-3 interaction determines transition of neural progenitor self-renewal to neuronal migration in the developing brain.
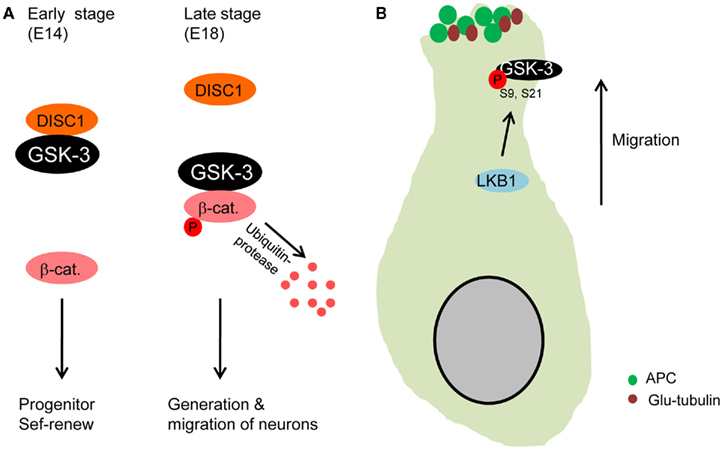
Figure 3. Glycogen synthase kinase-3 in neuronal migration. (A) GSK-3 is required for neuronal migration. During early stages of development, DISC1 that is an important regulator of neuron migration binds to GSK-3, resulting in dissociation of GSK-3 from β-catenin. The increased β-catenin level directs progenitors to self-renew. However, the binding activity of DISC1 decreases at later stages of embryogenesis, which induces reduction in β-catenin level via GSK-3 phosphorylation. Progenitors then produce more neurons that subsequently migrate from the ventricular zone (Ishizuka et al., 2011). (B) Microtubules are stabilized at the leading edge of migrating neurons by GSK-3 inactivation. LKB1 inactivates GSK-3 by the phosphorylation at S9. APC is dissociated from GSK-3 and binds to microtubules at the leading edge of migrating neurons (Asada and Sanada, 2010), which stabilizes microtubules (glu-tubulin).
Additional relevant evidence about the role of GSK-3 in neuron migration has been recently reported. Radial migration and placement of cortical neurons were aberrant in GSK-3/nestin-cre mutant mice (Yokota et al., 2010). This study showed that, compared to control brains, fewer deeper layer neurons (Tbr1-positive) were found in the cortical plate of GSK-3 double knockout brains. Substantial numbers of Tbr1 neurons were trapped within the intermediate zone. Yet, GSK-3/nestin-cre mutant brains also show disrupted radial glial development (Yokota et al., 2010). The disruption of radial glial platform may indirectly create radial neuron migration defects in GSK-3/nestin-cre brains. Therefore, elimination of GSK-3 only in neuronal population needs to be done to establish whether or not GSK-3 has a direct role in neuronal migration in developing mammalian brains.
A separate study has recently shown that in utero electroporation with active GSK-3β plasmid into cortical ventricular zone elicits neuronal migration defects (Asada and Sanada, 2010). LKB1 that is an important regulator of neuronal migration in the developing cortex (Asada et al., 2007) mediated GSK-3β phosphorylation at Serine 9 residue. The phosphorylation induced high levels of APC and Glu-tubulin (stabilized microtubule) at the leading process of neurons, thereby directing centrosomal movement. However, inhibition of the Serine 9 phosphorylation by overexpressing active GSK-3β (S9A) resulted in very low levels of APC and Glu-tubulin at the leading process of migrating neurons. These results suggest that GSK-3 inactivation via Serine 9 phosphorylation is required for directional migration of cortical neurons by rearranging APC and stabilizing microtubules at the leading process (Figure 3B). An important point of this study is that only sub-population of cortical ventricular cells are manipulated by the gene transfection, leaving surrounding environment including radial glial platform mostly intact.
Although this initial work outlined above has provided valuable information on GSK-3 functions in neuronal migration, it is important now to define the molecular and cellular mechanisms of GSK-3 regulation. For example, GSK-3 may regulate neuronal migration during development by directing gene transcription and/or by rearranging the intracellular cytoskeleton. Results from β-catenin and active GSK-3 overexpression studies strongly suggest that both mechanisms may be significant to GSK-3 regulation in neuronal migration. Defining GSK-3 functions and associated mechanisms has important clinical implications since defects in neuron migration are implicated in a range of neurodevelopmental disorders. Mutations in DISC1 are causative in some cases of schizophrenia.
Neuronal Morphogenesis
After migration, GSK-3 is critically involved in neuronal morphological development including neurite outgrowth and axon/dendritic specification. The roles of GSK-3 on axon/dendrite development are described well in the article in the same issue from Zhou and Snider.
Glial Development
Although not yet tested, it can be anticipated that GSK-3 will also have important functions related to glial development. IGF1 and FGF2 pathways coordinately control the cell cycle progression of oligodendrocyte progenitors via cyclin D1 expression and localization (Frederick and Wood, 2004). The turnover of this protein is regulated by GSK-3-dependent proteasome degradation (Diehl et al., 1998). GSK-3β is phosphorylated in the presence of IGF1 downstream of PI3K/AKT signaling in oligodendrocyte progenitors (Frederick et al., 2007). This suggests that GSK-3 activity is inhibited by IGF1 in oligodendrocyte progenitors, which then enhances the stability and nuclear localization of cyclin D1 protein. Furthermore, inhibition of GSK-3 in postnatal mice by injecting pharmacological inhibitors increased oligodendrocyte differentiation and myelination (Azim and Butt, 2011). This effect was via canonical Wnt signaling stimulating β-catenin. Thus, these findings suggest that GSK-3 may be a crucial to oligodendrocyte differentiation.
Activation of PI3K and inhibition of GSK-3 using lithium promote Schwann cell differentiation (Ogata et al., 2004). This finding suggests that GSK-3 as a downstream of PI3K–AKT pathway is crucial for initiation of myelination during development. It will be important in future work to define aspects of GSK-3 signaling in gliogenesis during neural development that might have additional implications in the context of glial tumors and demyelinating diseases.
Synaptic Development
Synapses connect neurons to transmit signals and synapse formation is an important step in later stages of development. Recent studies have suggested important functions for GSK-3 in synaptic development. Shaggy, the fly homolog of GSK-3, is present at synapses and negatively influences AP-1-dependent synaptic growth, by modulating the Jun-N-terminal kinase pathway, and regulates presynaptic neurotransmitter release at the larval neuromuscular junction (Franciscovich et al., 2008). In fractionation studies, GSK-3β is readily detected within the synaptosomal fraction (Hooper et al., 2007). Activation of GSK-3 by transfecting GSK-3 genes in cultured neurons reduces expression and clustering of a synaptic protein Synapsin I (Zhu et al., 2007). In contrast, pharmacological inhibition of GSK-3 activity induces Synapsin I clustering in developing neurons (Hall et al., 2002). Further, GSK-3 activation reduces presynaptic glutamate release by inhibiting the synaptic vesicle exocytosis in response to membrane depolarization (Zhu et al., 2010). GSK-3β negatively regulates synaptic vesicle fusion events via interfering with Ca2+-dependent SNARE complex formation. Finally, in rat, there is strong GSK-3 expression in dendritic spines of cultured hippocampal neurons (Peineau et al., 2007).
Glycogen synthase kinase-3 activity is also implicated in regulation of adult synapses. GSK-3 plays a role in amyloid-induced synaptic alternation and dendritic spine shortening (reviewed in Knobloch and Mansuy, 2008). All these demonstrate that GSK-3 signaling is likely critical to synaptic development, but the precise functions regulated by GSK-3 will require further study.
A recent study has reported functional roles of GSK-3 in the regulation of N-Methyl-D-aspartic acid (NMDA) receptor-dependent synaptic plasticity (Peineau et al., 2008). Activation of GSK-3 mediates NMDA-dependent long term depression (LTD) induction. In contrast, inhibition of GSK-3 activity prevents LTD induction and participates in long term potentiation (LTP) induction (Hooper et al., 2007). For further information on the roles of GSK-3 in synaptic plasticity and LTP/LTD formation, please refer the review from Collingridge group in this issue.
GSK-3 in Tumorigenesis
Dual Functions of GSK-3 in Tumorigenesis
Glycogen synthase kinase-3 is associated with neoplastic transformation and tumor development (reviewed in Ougolkov and Billadeau, 2006; reviewed in Luo, 2009). Since GSK-3 negatively regulates oncoproteins such as Myc and β-catenin, it may act as a suppressor of tumorigenesis. Indeed, downregulation of GSK-3β and upregulation of phospho-GSK-3β (inactive form) have been found in skin (Leis et al., 2002; Ding et al., 2007; Ma et al., 2007) and mammary tumors (Farago et al., 2005) in mice and humans. Furthermore, overexpression of wild type or constitutively active GSK-3β inhibits the progression of skin tumors in mice (Ma et al., 2007), while mice overexpressing dominant-negative GSK-3 develop mammary tumors (Farago et al., 2005). The promotion of mammary tumorigenesis by GSK-3 inactivation is accompanied by the accumulation of β-catenin and cyclin D1, suggesting the dysregulation of the Wnt pathway. These findings demonstrate the suppressive role of GSK-3 in tumorigenesis. However, some studies have shown the contrary role of GSK-3, that it may positively regulate tumorigenesis in other types of cancers. Increased GSK-3 levels have been found in human ovarian, colon, liver, thyroid, and pancreatic carcinomas (reviewed in Ougolkov and Billadeau, 2006; reviewed in Luo, 2009). Inhibition of GSK-3 activity by pharmacological inhibitors or RNA interference suppresses proliferation of the human ovarian cancer (Cao et al., 2006), colon cancer (Shakoori et al., 2007), pancreatic cancer (Ougolkov et al., 2005), and prostate cancer cells (Zhu et al., 2011). Conversely, overexpressing constitutively active GSK-3β induces cell cycle progression of ovarian cancer cells (Cao et al., 2006). Therefore, GSK-3 also functions as a tumor promoter in certain types of tumors. In this case, inhibition of GSK-3 could be an attractive therapeutic strategy for the treatment of ovarian, colon, and pancreatic cancers. Taken together, GSK-3 plays opposing roles as a tumor suppressor or a promoter, depending on cell-types and cellular contexts.
Tumorigenesis in the Nervous System
Recent studies suggest important roles of GSK-3 in the formation and maintenance of tumors in the nervous system. Pharmacological inhibition of GSK-3 using the indirubin family reduces growth and invasion of human glioma cells both in vitro and in mice (Nowicki et al., 2008; Williams et al., 2011). β-catenin signaling plays an important role in mediating these effects. Another study using a different GSK-3 inhibitor, TDZD-8, has also shown that inhibition of GSK-3 activity suppresses proliferation of mouse glioma cells and induces apoptosis in vitro (Aguilar-Morante et al., 2010). The inhibitory effects of TDZD-8 in culture were confirmed in mice, which showed delay in tumor growth after glioma cell injection, and an increase in survival (Aguilar-Morante et al., 2010). Additionally, GSK-3 is involved in the differentiation of human glioma cells (Korur et al., 2009). Interference of GSK-3 activity with siRNA or pharmacological inhibitors induces differentiation of glioma cells. The capacity of GSK-3 in inducing glioma cell differentiation could be considered for the differentiation therapy that has emerged as a promising treatment tool for brain tumors. However, progression of this idea needs careful assessments, as contrary effects of GSK-3 on glioma differentiation are also reported (Li et al., 2010). GSK-3 also plays a promoting role in the majority of medulloblastoma, the most common pediatric malignant tumor of the central nervous system. Lithium, a clinically available GSK-3 inhibitor, suppresses proliferation of medulloblastoma cells and induces non-apoptotic cell death (Ronchi et al., 2010). Interestingly, the inhibitory effect of GSK-3 is independent of changes in intracellular levels of β-catenin. Together, GSK-3 signaling plays important roles in tumorigenesis and differentiation in the nervous system, and may be a promising target in the establishment of therapeutic strategies. However, before considering clinical application, it is necessary to determine the suppressive and promoting functions of GSK-3 in each tumor type, as both activities have been evident in non-nervous system tumors. This dual role of GSK-3 in tumorigenesis may not be surprising since GSK-3 is involved in multiple pathways and regulates many substrates. Different environments and/or epigenetic programs around different tissues/cells may regulate GSK-3 in different ways, which subsequently influences distinct downstream targets. In this regard, it is important to define and compare GSK-3 signaling mechanisms in each cell-type. This will delineate how the suppressive and promoting activities of GSK-3 are counterbalanced and help to develop therapeutic potentials.
Implications in Neurodevelopmental Disorders
Recent studies suggest that changes in GSK-3 activity may be an important aspect of pathophysiologies of neurodevelopmental disorders, including autism spectrum disorders (ASDs), schizophrenia, mental retardation, and bipolar disorders. There are two types of key observations in animal models of neurodevelopmental disorders: (1) The levels and activity of GSK-3 are altered in the brain samples. (2) Pharmacological or genetic inhibition of GSK-3 activity reduces the pathological symptoms. In humans, lithium, a GSK-3 inhibitor is used for controlling abnormal mood changes in patients with bipolar disorders.
ASDs are a spectrum of psychological conditions characterized by abnormalities in social interactions and communication as well as stereotypic behaviors and motor dysfunctions. Neurodevelopmental abnormalities are found in ASD brains. Studies using magnetic resonance imaging and postmortem brains show abnormalities in cortical minicolumn organization and neuron migration in patients with ASDs (Piven et al., 1990; Casanova et al., 2003, 2006). Etiology of autism has yet to be determined. However, recent studies suggest that genetic factors may be the most significant cause for ASDs. Gene linkage studies and array analyses have found multiple ASD-associated genes that encode FMR1 (fragile X mental retardation 1), PTEN (phosphatase and tensin homolog), APC, DISC1, SHANK3 (SH3 and multiple ankyrin repeat domains 3), TSC (tuberous sclerosis), MeCP2 (methyl CpG binding protein 2), and Reelin (reviewed in Abrahams and Geschwind, 2008). All these genes play important roles in functional and anatomical development of the nervous system.
Importantly, GSK-3, a critical regulator in broad aspects of neural development, is associated with many ASD-related genes mentioned above. GSK-3 activity measured by the levels of phospho-GSK-3 is increased in the brains of FMR1-deleted mice (Min et al., 2009; Yuskaitis et al., 2010). Inhibition of GSK-3 activity by treatment with lithium reduces abnormal social behaviors and seizures in the FMR1 mice. Additionally, GSK-3 knock-in mice, in which inhibitory serine 9 (β) and 21 (α) residues are substituted by alanine (McManus et al., 2005), show abnormal social behaviors similar to those of FMR1-deleted mice (Mines et al., 2010). Mice with conditional PTEN deletion in the cortex and hippocampus show macrocephaly and abnormal social interaction (Kwon et al., 2006). The levels of inactive GSK-3β (phospho-GSK-3β) are elevated in the brains. Furthermore, recent studies suggest that GSK-3 is a critical downstream mediator of the DISC1 signal. DISC1 regulates proliferation of neural progenitors by modulating GSK-3 activity (Mao et al., 2009). DISC1 directly binds to GSK-3 and participates in the regulation of β-catenin stability. Inhibition of GSK-3 with lithium or overexpression of a stable β-catenin rescues DISC1-deletion effects in neural progenitors. However, whether DISC1 directly controls GSK-3/β-catenin or serves as a scaffold remains unknown. Together, GSK-3 is associated with genes that are mutated in neurodevelopmental disorders and may play a critical role in the pathogenesis of the diseases. The association of GSK-3 with these genes suggests a therapeutic intervention point in the development of tools for neurodevelopmental disorders.
GSK-3 in Epigenetic Control
Epigenetic control has become an important cell-intrinsic program in the regulation of various processes during neural development. DNA methylation and histone modification (i.e., phosphorylation, acetylation, and methylation) change chromatin structures for transcription factors to gain access to gene promoters (Fischle et al., 2003). Non-coding RNAs also functionally participate in neural development. For example, microRNAs regulate neural progenitor proliferation and differentiation (Aranha et al., 2011; Chen et al., 2011), neural migration (Delaloy et al., 2010), oligodendrocyte differentiation and myelination (Dugas et al., 2010), hippocampal axon growth (Sanuki et al., 2011), and dendritic spine morphogenesis (Lippi et al., 2011).
The role of GSK-3 in epigenetic control has recently been evident. GSK-3 modulates the activity of epigenetic factors. GSK-3 directly phosphorylates histone deacetylase 3 that promotes the death of cortical neurons (Bardai and D’Mello, 2011), suggesting that the neuronal death-promoting activity of GSK-3 could be mediated via an epigenetic mechanism. Pharmacological inhibition of GSK-3 increases methylation at several sites on histone H3 and histone H4 bound to the promoters of the NFκB target genes Bcl-2 and XIAP (Ougolkov et al., 2007). This leads to the suppression of NFkB binding to its target promoters and thereby epigenetic silencing of XIAP and Bcl-2. Additionally, a recent study demonstrates that GSK-3 also participates in epigenetic control by regulating the levels of a DNA methyltransferase. Genetic deletion of both GSK-3α and GSK-3β in mouse embryonic stem cells reduces expression of DNA methyltransferase 3a2, which results in hypomethylation and misexpression of the imprinted genes Igf2, H19, and Igf2r (Popkie et al., 2010). Our understanding of this subject is at an initial stage, but the information thus far strongly suggests important roles of GSK-3 in epigenetic control. This field will be expanded by identifying more epigenetic molecules that are regulated by GSK-3.
Conclusion
All of the studies discussed here implicate GSK-3 as a multifunctional regulator of neural development. At early stages, GSK-3 serves as a master switch regulating neural progenitor homeostasis and appears to act via regulation of β-catenin as well as other key signaling pathways. It now seems quite clear that after progenitors differentiate into neurons, GSK-3 signaling is also key to neuronal migration and lamination. Although some evidence implicates β-catenin in these processes, GSK-3 regulation of the cytoskeleton via APC and other microtubule-associated proteins may be particularly important. Finally, functions of GSK-3 in neuronal morphogenesis, synaptogenesis, and behavior are only now being defined. Future studies defining cell-type specific effects in vivo with a variety of Cre drivers will rapidly enhance our understanding of GSK-3 functions.
A key regulator of GSK-3 function in neural development is an important protein in genetic causes of schizophrenia. Details of GSK-3 regulation by DISC1 have already provided key insights into regulation of key developmental events with important implications to the pathogenesis of some cases of schizophrenia.
Finally, our current understanding of GSK-3 in neural development largely reflects phenotypic characterization of the loss of the protein. It will be important to learn more cellular and molecular mechanisms of GSK-3 signaling related to these striking phenotypes. Further works should distinguish GSK-3 functions between gene induction activity and cytoskeleton regulation and how these processes are linked in various neurodevelopmental events. Also, key to defining GSK-3 functions will be to understand how GSK-3 activity is regulated. Understanding regulation of GSK-3 activity will be a particularly important goal, as we can expect that even relative small changes might have a large impact on important developmental outcomes such as determination of numbers of neurons in the brain.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abrahams, B. S., and Geschwind, D. H. (2008). Advances in autism genetics: on the threshold of a new neurobiology. Nat. Rev. Genet. 9, 341–355.
Aguilar-Morante, D., Morales-Garcia, J. A., Sanz-SanCristobal, M., Garcia-Cabezas, M. A., Santos, A., and Perez-Castillo, A. (2010). Inhibition of glioblastoma growth by the thiadiazolidinone compound TDZD-8. PLoS ONE 5, e13879. doi:10.1371/journal.pone.0013879
Akhmanova, A., Hoogenraad, C. C., Drabek, K., Stepanova, T., Dortland, B., Verkerk, T., Vermeulen, W., Burgering, B. M., De Zeeuw, C. I., Grosveld, F., and Galjart, N. (2001). Clasps are CLIP-115 and -170 associating proteins involved in the regional regulation of microtubule dynamics in motile fibroblasts. Cell 104, 923–935.
Araki, R., Hoki, Y., Uda, M., Nakamura, M., Jincho, Y., Tamura, C., Sunayama, M., Ando, S., Sugiura, M., Yoshida, M. A., Kasama, Y., and Abe, M. (2011). Crucial role of C-myc in the generation of induced pluripotent stem cells. Stem Cells 29, 1362–1370.
Aranha, M. M., Santos, D. M., Sola, S., Steer, C. J., and Rodrigues, C. M. (2011). miR-34a regulates mouse neural stem cell differentiation. PLoS ONE 6, e21396. doi:10.1371/journal.pone.0021396
Asada, N., and Sanada, K. (2010). LKB1-mediated spatial control of GSK3beta and adenomatous polyposis coli contributes to centrosomal forward movement and neuronal migration in the developing neocortex. J. Neurosci. 30, 8852–8865.
Asada, N., Sanada, K., and Fukada, Y. (2007). LKB1 regulates neuronal migration and neuronal differentiation in the developing neocortex through centrosomal positioning. J. Neurosci. 27, 11769–11775.
Azim, K., and Butt, A. M. (2011). GSK3beta negatively regulates oligodendrocyte differentiation and myelination in vivo. Glia 59, 540–553.
Bardai, F. H., and D’Mello, S. R. (2011). Selective toxicity by HDAC3 in neurons: regulation by Akt and GSK3beta. J. Neurosci. 31, 1746–1751.
Barth, A. I., Caro-Gonzalez, H. Y., and Nelson, W. J. (2008). Role of adenomatous polyposis coli (APC) and microtubules in directional cell migration and neuronal polarization. Semin. Cell Dev. Biol. 19, 245–251.
Beaulieu, J. M., Sotnikova, T. D., Yao, W. D., Kockeritz, L., Woodgett, J. R., Gainetdinov, R. R., and Caron, M. G. (2004). Lithium antagonizes dopamine-dependent behaviors mediated by an AKT/glycogen synthase kinase 3 signaling cascade. Proc. Natl. Acad. Sci. U.S.A. 101, 5099–5104.
Beaulieu, J. M., Zhang, X., Rodriguiz, R. M., Sotnikova, T. D., Cools, M. J., Wetsel, W. C., Gainetdinov, R. R., and Caron, M. G. (2008). Role of GSK3 beta in behavioral abnormalities induced by serotonin deficiency. Proc. Natl. Acad. Sci. U.S.A. 105, 1333–1338.
Bechard, M., and Dalton, S. (2009). Subcellular localization of glycogen synthase kinase 3beta controls embryonic stem cell self-renewal. Mol. Cell. Biol. 29, 2092–2104.
Bone, H. K., Damiano, T., Bartlett, S., Perry, A., Letchford, J., Ripoll, Y. S., Nelson, A. S., and Welham, M. J. (2009). Involvement of GSK-3 in regulation of murine embryonic stem cell self-renewal revealed by a series of bisindolylmaleimides. Chem. Biol. 16, 15–27.
Buttrick, G. J., and Wakefield, J. G. (2008). PI3-K and GSK-3: Akt-ing together with microtubules. Cell Cycle 7, 2621–2625.
Cao, Q., Lu, X., and Feng, Y. J. (2006). Glycogen synthase kinase-3beta positively regulates the proliferation of human ovarian cancer cells. Cell Res. 16, 671–677.
Cartwright, P., McLean, C., Sheppard, A., Rivett, D., Jones, K., and Dalton, S. (2005). LIF/STAT3 controls ES cell self-renewal and pluripotency by a Myc-dependent mechanism. Development 132, 885–896.
Casanova, M. F., Buxhoeveden, D., and Gomez, J. (2003). Disruption in the inhibitory architecture of the cell minicolumn: implications for autism. Neuroscientist 9, 496–507.
Casanova, M. F., van Kooten, I. A., Switala, A. E., van Engeland, H., Heinsen, H., Steinbusch, H. W., Hof, P. R., Trippe, J., Stone, J., and Schmitz, C. (2006). Minicolumnar abnormalities in autism. Acta Neuropathol. 112, 287–303.
Chen, J. A., Huang, Y. P., Mazzoni, E. O., Tan, G. C., Zavadil, J., and Wichterle, H. (2011). Mir-17-3p controls spinal neural progenitor patterning by regulating Olig2/Irx3 cross-repressive loop. Neuron 69, 721–735.
Cheng, T. S., Hsiao, Y. L., Lin, C. C., Yu, C. T., Hsu, C. M., Chang, M. S., Lee, C. I., Huang, C. Y., Howng, S. L., and Hong, Y. R. (2008). Glycogen synthase kinase 3beta interacts with and phosphorylates the spindle-associated protein astrin. J. Biol. Chem. 283, 2454–2464.
Chenn, A., and Walsh, C. A. (2002). Regulation of cerebral cortical size by control of cell cycle exit in neural precursors. Science 297, 365–369.
Chenn, A., and Walsh, C. A. (2003). Increased neuronal production, enlarged forebrains and cytoarchitectural distortions in beta-catenin overexpressing transgenic mice. Cereb. Cortex 13, 599–606.
Corbin, J. G., Gaiano, N., Juliano, S. L., Poluch, S., Stancik, E., and Haydar, T. F. (2008). Regulation of neural progenitor cell development in the nervous system. J. Neurochem. 106, 2272–2287.
Delaloy, C., Liu, L., Lee, J. A., Su, H., Shen, F., Yang, G. Y., Young, W. L., Ivey, K. N., and Gao, F. B. (2010). MicroRNA-9 coordinates proliferation and migration of human embryonic stem cell-derived neural progenitors. Cell Stem Cell 6, 323–335.
Diehl, J. A., Cheng, M., Roussel, M. F., and Sherr, C. J. (1998). Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 12, 3499–3511.
Ding, Q., He, X., Xia, W., Hsu, J. M., Chen, C. T., Li, L. Y., Lee, D. F., Yang, J. Y., Xie, X., Liu, J. C., and Hung, M. C. (2007). Myeloid cell leukemia-1 inversely correlates with glycogen synthase kinase-3beta activity and associates with poor prognosis in human breast cancer. Cancer Res. 67, 4564–4571.
Doble, B. W., Patel, S., Wood, G. A., Kockeritz, L. K., and Woodgett, J. R. (2007). Functional redundancy of GSK-3alpha and GSK-3beta in Wnt/beta-catenin signaling shown by using an allelic series of embryonic stem cell lines. Dev. Cell 12, 957–971.
Doble, B. W., and Woodgett, J. R. (2003). GSK-3: tricks of the trade for a multi-tasking kinase. J. Cell. Sci. 116, 1175–1186.
Doble, B. W., and Woodgett, J. R. (2009). Exploring pluripotency with chemical genetics. Cell Stem Cell 4, 98–100.
Duan, X., Chang, J. H., Ge, S., Faulkner, R. L., Kim, J. Y., Kitabatake, Y., Liu, X. B., Yang, C. H., Jordan, J. D., Ma, D. K., Liu, C. Y., Ganesan, S., Cheng, H. J., Ming, G. L., Lu, B., and Song, H. (2007). Disrupted-in-schizophrenia 1 regulates integration of newly generated neurons in the adult brain. Cell 130, 1146–1158.
Dugas, J. C., Cuellar, T. L., Scholze, A., Ason, B., Ibrahim, A., Emery, B., Zamanian, J. L., Foo, L. C., McManus, M. T., and Barres, B. A. (2010). Dicer1 and miR-219 Are required for normal oligodendrocyte differentiation and myelination. Neuron 65, 597–611.
Enomoto, A., Asai, N., Namba, T., Wang, Y., Kato, T., Tanaka, M., Tatsumi, H., Taya, S., Tsuboi, D., Kuroda, K., Kaneko, N., Sawamoto, K., Miyamoto, R., Jijiwa, M., Murakumo, Y., Sokabe, M., Seki, T., Kaibuchi, K., and Takahashi, M. (2009). Roles of disrupted-in-schizophrenia 1-interacting protein girdin in postnatal development of the dentate gyrus. Neuron 63, 774–787.
Espinosa, L., Ingles-Esteve, J., Aguilera, C., and Bigas, A. (2003). Phosphorylation by glycogen synthase kinase-3 beta down-regulates Notch activity, a link for Notch and Wnt pathways. J. Biol. Chem. 278, 32227–32235.
Etienne-Manneville, S., and Hall, A. (2003). Cdc42 regulates GSK-3beta and adenomatous polyposis coli to control cell polarity. Nature 421, 753–756.
Farago, M., Dominguez, I., Landesman-Bollag, E., Xu, X., Rosner, A., Cardiff, R. D., and Seldin, D. C. (2005). Kinase-inactive glycogen synthase kinase 3beta promotes Wnt signaling and mammary tumorigenesis. Cancer Res. 65, 5792–5801.
Fischle, W., Wang, Y., and Allis, C. D. (2003). Histone and chromatin cross-talk. Curr. Opin. Cell Biol. 15, 172–183.
Foltz, D. R., Santiago, M. C., Berechid, B. E., and Nye, J. S. (2002). Glycogen synthase kinase-3beta modulates Notch signaling and stability. Curr. Biol. 12, 1006–1011.
Franciscovich, A. L., Mortimer, A. D., Freeman, A. A., Gu, J., and Sanyal, S. (2008). Overexpression screen in Drosophila identifies neuronal roles of GSK-3 beta/shaggy as a regulator of AP-1-dependent developmental plasticity. Genetics 180, 2057–2071.
Frederick, T. J., Min, J., Altieri, S. C., Mitchell, N. E., and Wood, T. L. (2007). Synergistic induction of cyclin D1 in oligodendrocyte progenitor cells by IGF-I and FGF-2 requires differential stimulation of multiple signaling pathways. Glia 55, 1011–1022.
Frederick, T. J., and Wood, T. L. (2004). IGF-I and FGF-2 coordinately enhance cyclin D1 and cyclin E-cdk2 association and activity to promote G1 progression in oligodendrocyte progenitor cells. Mol. Cell. Neurosci. 25, 480–492.
Gaiano, N., Nye, J. S., and Fishell, G. (2000). Radial glial identity is promoted by Notch1 signaling in the murine forebrain. Neuron 26, 395–404.
Goold, R. G., and Gordon-Weeks, P. R. (2001). Microtubule-associated protein 1B phosphorylation by glycogen synthase kinase 3beta is induced during PC12 cell differentiation. J. Cell. Sci. 114, 4273–4284.
Gotz, M., and Huttner, W. B. (2005). The cell biology of neurogenesis. Nat. Rev. Mol. Cell Biol. 6, 777–788.
Guha, S., Cullen, J. P., Morrow, D., Colombo, A., Lally, C., Walls, D., Redmond, E. M., and Cahill, P. A. (2011). Glycogen synthase kinase 3 beta positively regulates Notch signaling in vascular smooth muscle cells: role in cell proliferation and survival. Basic Res. Cardiol. 106, 773–785.
Gulacsi, A. A., and Anderson, S. A. (2008). Beta-catenin-mediated Wnt signaling regulates neurogenesis in the ventral telencephalon. Nat. Neurosci. 11, 1383–1391.
Hall, A. C., Brennan, A., Goold, R. G., Cleverley, K., Lucas, F. R., Gordon-Weeks, P. R., and Salinas, P. C. (2002). Valproate regulates GSK-3-mediated axonal remodeling and synapsin I clustering in developing neurons. Mol. Cell. Neurosci. 20, 257–270.
Hansen, D. V., Lui, J. H., Parker, P. R., and Kriegstein, A. R. (2010). Neurogenic radial glia in the outer subventricular zone of human neocortex. Nature 464, 554–561.
Harris, J., Honigberg, L., Robinson, N., and Kenyon, C. (1996). Neuronal cell migration in C. elegans: regulation of Hox gene expression and cell position. Development 122, 3117–3131.
Hatakeyama, J., Bessho, Y., Katoh, K., Ookawara, S., Fujioka, M., Guillemot, F., and Kageyama, R. (2004). Hes genes regulate size, shape and histogenesis of the nervous system by control of the timing of neural stem cell differentiation. Development 131, 5539–5550.
Hayward, P., Kalmar, T., and Arias, A. M. (2008). Wnt/Notch signalling and information processing during development. Development 135, 411–424.
Higginbotham, H. R., and Gleeson, J. G. (2007). The centrosome in neuronal development. Trends Neurosci. 30, 276–283.
Hoeflich, K. P., Luo, J., Rubie, E. A., Tsao, M. S., Jin, O., and Woodgett, J. R. (2000). Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature 406, 86–90.
Hooper, C., Markevich, V., Plattner, F., Killick, R., Schofield, E., Engel, T., Hernandez, F., Anderton, B., Rosenblum, K., Bliss, T., Cooke, S. F., Avila, J., Lucas, J. J., Giese, K. P., Stephenson, J., and Lovestone, S. (2007). Glycogen synthase kinase-3 inhibition is integral to long-term potentiation. Eur. J. Neurosci. 25, 81–86.
Ishizuka, K., Kamiya, A., Oh, E. C., Kanki, H., Seshadri, S., Robinson, J. F., Murdoch, H., Dunlop, A. J., Kubo, K., Furukori, K., Huang, B., Zeledon, M., Hayashi-Takagi, A., Okano, H., Nakajima, K., Houslay, M. D., Katsanis, N., and Sawa, A. (2011). DISC1-dependent switch from progenitor proliferation to migration in the developing cortex. Nature 473, 92–96.
Izumi, N., Fumoto, K., Izumi, S., and Kikuchi, A. (2008). GSK-3beta regulates proper mitotic spindle formation in cooperation with a component of the gamma-tubulin ring complex, GCP5. J. Biol. Chem. 283, 12981–12991.
Jin, Y. H., Kim, H., Oh, M., Ki, H., and Kim, K. (2009a). Regulation of Notch1/NICD and Hes1 expressions by GSK-3alpha/beta. Mol. Cells 27, 15–19.
Jin, Y. H., Kim, H., Ki, H., Yang, I., Yang, N., Lee, K. Y., Kim, N., Park, H. S., and Kim, K. (2009b). Beta-catenin modulates the level and transcriptional activity of Notch1/NICD through its direct interaction. Biochim. Biophys. Acta 1793, 290–299.
Kaidanovich-Beilin, O., Lipina, T. V., Takao, K., van Eede, M., Hattori, S., Laliberte, C., Khan, M., Okamoto, K., Chambers, J. W., Fletcher, P. J., MacAulay, K., Doble, B. W., Henkelman, M., Miyakawa, T., Roder, J., and Woodgett, J. R. (2009). Abnormalities in brain structure and behavior in GSK-3alpha mutant mice. Mol. Brain 2, 35.
Kamiya, A., Kubo, K., Tomoda, T., Takaki, M., Youn, R., Ozeki, Y., Sawamura, N., Park, U., Kudo, C., Okawa, M., Ross, C. A., Hatten, M. E., Nakajima, K., and Sawa, A. (2005). A schizophrenia-associated mutation of DISC1 perturbs cerebral cortex development. Nat. Cell Biol. 7, 1167–1178.
Kerkela, R., Kockeritz, L., Macaulay, K., Zhou, J., Doble, B. W., Beahm, C., Greytak, S., Woulfe, K., Trivedi, C. M., Woodgett, J. R., Epstein, J. A., Force, T., and Huggins, G. S. (2008). Deletion of GSK-3beta in mice leads to hypertrophic cardiomyopathy secondary to cardiomyoblast hyperproliferation. J. Clin. Invest. 118, 3609–3618.
Kim, W. Y., Wang, X., Wu, Y., Doble, B. W., Patel, S., Woodgett, J. R., and Snider, W. D. (2009). GSK-3 is a master regulator of neural progenitor homeostasis. Nat. Neurosci. 12, 1390–1397.
Kim, W. Y., Zhou, F. Q., Zhou, J., Yokota, Y., Wang, Y. M., Yoshimura, T., Kaibuchi, K., Woodgett, J. R., Anton, E. S., and Snider, W. D. (2006). Essential roles for GSK-3s and GSK-3-primed substrates in neurotrophin-induced and hippocampal axon growth. Neuron 52, 981–996.
Kimura, T., Yamashita, S., Nakao, S., Park, J. M., Murayama, M., Mizoroki, T., Yoshiike, Y., Sahara, N., and Takashima, A. (2008). GSK-3beta is required for memory reconsolidation in adult brain. PLoS ONE 3, e3540. doi:10.1371/journal.pone.0003540
Klezovitch, O., Fernandez, T. E., Tapscott, S. J., and Vasioukhin, V. (2004). Loss of cell polarity causes severe brain dysplasia in Lgl1 knockout mice. Genes Dev. 18, 559–571.
Knobloch, M., and Mansuy, I. M. (2008). Dendritic spine loss and synaptic alterations in Alzheimer’s disease. Mol. Neurobiol. 37, 73–82.
Knoepfler, P. S., Cheng, P. F., and Eisenman, R. N. (2002). N-myc is essential during neurogenesis for the rapid expansion of progenitor cell populations and the inhibition of neuronal differentiation. Genes Dev. 16, 2699–2712.
Knoepfler, P. S., and Kenney, A. M. (2006). Neural precursor cycling at sonic speed: N-Myc pedals, GSK-3 brakes. Cell Cycle 5, 47–52.
Komada, M., Saitsu, H., Kinboshi, M., Miura, T., Shiota, K., and Ishibashi, M. (2008). Hedgehog signaling is involved in development of the neocortex. Development 135, 2717–2727.
Korswagen, H. C. (2002). Canonical and non-canonical Wnt signaling pathways in Caenorhabditis elegans: variations on a common signaling theme. Bioessays 24, 801–810.
Korswagen, H. C., Coudreuse, D. Y., Betist, M. C., van de Water, S., Zivkovic, D., and Clevers, H. C. (2002). The Axin-like protein PRY-1 is a negative regulator of a canonical Wnt pathway in C. elegans. Genes Dev. 16, 1291–1302.
Korur, S., Huber, R. M., Sivasankaran, B., Petrich, M., Morin, P. Jr., Hemmings, B. A., Merlo, A., and Lino, M. M. (2009). GSK3beta regulates differentiation and growth arrest in glioblastoma. PLoS ONE 4, e7443. doi:10.1371/journal.pone.0007443
Kwon, C. H., Luikart, B. W., Powell, C. M., Zhou, J., Matheny, S. A., Zhang, W., Li, Y., Baker, S. J., and Parada, L. F. (2006). Pten regulates neuronal arborization and social interaction in mice. Neuron 50, 377–388.
Lee, F. H., Kaidanovich-Beilin, O., Roder, J. C., Woodgett, J. R., and Wong, A. H. (2011). Genetic inactivation of GSK3alpha rescues spine deficits in Disc1-L100P mutant mice. Schizophr. Res. 129, 74–79.
Leis, H., Segrelles, C., Ruiz, S., Santos, M., and Paramio, J. M. (2002). Expression, localization, and activity of glycogen synthase kinase 3beta during mouse skin tumorigenesis. Mol. Carcinog. 35, 180–185.
Li, Y., Lu, H., Huang, Y., Xiao, R., Cai, X., He, S., and Yan, G. (2010). Glycogen synthase kinases-3beta controls differentiation of malignant glioma cells. Int. J. Cancer 127, 1271–1282.
Lien, W. H., Klezovitch, O., Fernandez, T. E., Delrow, J., and Vasioukhin, V. (2006). alphaE-catenin controls cerebral cortical size by regulating the hedgehog signaling pathway. Science 311, 1609–1612.
Lin, C. H., Lin, C., Tanaka, H., Fero, M. L., and Eisenman, R. N. (2009). Gene regulation and epigenetic remodeling in murine embryonic stem cells by c-Myc. PLoS ONE 4, e7839. doi: 10.1371/journal.pone.0007839
Lippi, G., Steinert, J. R., Marczylo, E. L., D’Oro, S., Fiore, R., Forsythe, I. D., Schratt, G., Zoli, M., Nicotera, P., and Young, K. W. (2011). Targeting of the Arpc3 actin nucleation factor by miR-29a/b regulates dendritic spine morphology. J. Cell Biol. 194, 889–904.
Lui, J. H., Hansen, D. V., and Kriegstein, A. R. (2011). Development and evolution of the human neocortex. Cell 146, 18–36.
Luo, J. (2009). Glycogen synthase kinase 3beta (GSK3beta) in tumorigenesis and cancer chemotherapy. Cancer Lett. 273, 194–200.
Ma, C., Wang, J., Gao, Y., Gao, T. W., Chen, G., Bower, K. A., Odetallah, M., Ding, M., Ke, Z., and Luo, J. (2007). The role of glycogen synthase kinase 3beta in the transformation of epidermal cells. Cancer Res. 67, 7756–7764.
MacAulay, K., Doble, B. W., Patel, S., Hansotia, T., Sinclair, E. M., Drucker, D. J., Nagy, A., and Woodgett, J. R. (2007). Glycogen synthase kinase 3alpha-specific regulation of murine hepatic glycogen metabolism. Cell Metab. 6, 329–337.
Machon, O., Backman, M., Machonova, O., Kozmik, Z., Vacik, T., Andersen, L., and Krauss, S. (2007). A dynamic gradient of Wnt signaling controls initiation of neurogenesis in the mammalian cortex and cellular specification in the hippocampus. Dev. Biol. 311, 223–237.
Machon, O., van den Bout, C. J., Backman, M., Kemler, R., and Krauss, S. (2003). Role of beta-catenin in the developing cortical and hippocampal neuroepithelium. Neuroscience 122, 129–143.
Maloof, J. N., Whangbo, J., Harris, J. M., Jongeward, G. D., and Kenyon, C. (1999). A Wnt signaling pathway controls hox gene expression and neuroblast migration in C. elegans. Development 126, 37–49.
Mao, Y., Ge, X., Frank, C. L., Madison, J. M., Koehler, A. N., Doud, M. K., Tassa, C., Berry, E. M., Soda, T., Singh, K. K., Biechele, T., Petryshen, T. L., Moon, R. T., Haggarty, S. J., and Tsai, L. H. (2009). Disrupted in schizophrenia 1 regulates neuronal progenitor proliferation via modulation of GSK3beta/beta-catenin signaling. Cell 136, 1017–1031.
McManus, E. J., Sakamoto, K., Armit, L. J., Ronaldson, L., Shpiro, N., Marquez, R., and Alessi, D. R. (2005). Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by knockin analysis. EMBO J. 24, 1571–1583.
Min, W. W., Yuskaitis, C. J., Yan, Q., Sikorski, C., Chen, S., Jope, R. S., and Bauchwitz, R. P. (2009). Elevated glycogen synthase kinase-3 activity in Fragile X mice: key metabolic regulator with evidence for treatment potential. Neuropharmacology 56, 463–472.
Mines, M. A., Yuskaitis, C. J., King, M. K., Beurel, E., and Jope, R. S. (2010). GSK3 influences social preference and anxiety-related behaviors during social interaction in a mouse model of fragile X syndrome and autism. PLoS ONE 5, e9706. doi:10.1371/journal.pone.0009706
Mizutani, K., Yoon, K., Dang, L., Tokunaga, A., and Gaiano, N. (2007). Differential Notch signalling distinguishes neural stem cells from intermediate progenitors. Nature 449, 351–355.
Mukai, F., Ishiguro, K., Sano, Y., and Fujita, S. C. (2002). Alternative splicing isoform of tau protein kinase I/glycogen synthase kinase 3beta. J. Neurochem. 81, 1073–1083.
Mutch, C. A., Funatsu, N., Monuki, E. S., and Chenn, A. (2009). Beta-catenin signaling levels in progenitors influence the laminar cell fates of projection neurons. J. Neurosci. 29, 13710–13719.
Nowicki, M. O., Dmitrieva, N., Stein, A. M., Cutter, J. L., Godlewski, J., Saeki, Y., Nita, M., Berens, M. E., Sander, L. M., Newton, H. B., Chiocca, E. A., and Lawler, S. (2008). Lithium inhibits invasion of glioma cells; possible involvement of glycogen synthase kinase-3. Neuro-oncology 10, 690–699.
O’Brien, W. T., Harper, A. D., Jove, F., Woodgett, J. R., Maretto, S., Piccolo, S., and Klein, P. S. (2004). Glycogen synthase kinase-3beta haploinsufficiency mimics the behavioral and molecular effects of lithium. J. Neurosci. 24, 6791–6798.
Ogata, T., Iijima, S., Hoshikawa, S., Miura, T., Yamamoto, S., Oda, H., Nakamura, K., and Tanaka, S. (2004). Opposing extracellular signal-regulated kinase and Akt pathways control Schwann cell myelination. J. Neurosci. 24, 6724–6732.
Ohtsuka, T., Sakamoto, M., Guillemot, F., and Kageyama, R. (2001). Roles of the basic helix-loop-helix genes Hes1 and Hes5 in expansion of neural stem cells of the developing brain. J. Biol. Chem. 276, 30467–30474.
Otto, T., Horn, S., Brockmann, M., Eilers, U., Schuttrumpf, L., Popov, N., Kenney, A. M., Schulte, J. H., Beijersbergen, R., Christiansen, H., Berwanger, B., and Eilers, M. (2009). Stabilization of N-Myc is a critical function of Aurora A in human neuroblastoma. Cancer Cell 15, 67–78.
Ougolkov, A. V., and Billadeau, D. D. (2006). Targeting GSK-3: a promising approach for cancer therapy? Future Oncol. 2, 91–100.
Ougolkov, A. V., Bone, N. D., Fernandez-Zapico, M. E., Kay, N. E., and Billadeau, D. D. (2007). Inhibition of glycogen synthase kinase-3 activity leads to epigenetic silencing of nuclear factor kappaB target genes and induction of apoptosis in chronic lymphocytic leukemia B cells. Blood 110, 735–742.
Ougolkov, A. V., Fernandez-Zapico, M. E., Savoy, D. N., Urrutia, R. A., and Billadeau, D. D. (2005). Glycogen synthase kinase-3beta participates in nuclear factor kappaB-mediated gene transcription and cell survival in pancreatic cancer cells. Cancer Res. 65, 2076–2081.
Peineau, S., Bradley, C., Taghibiglou, C., Doherty, A., Bortolotto, Z. A., Wang, Y. T., and Collingridge, G. L. (2008). The role of GSK-3 in synaptic plasticity. Br. J. Pharmacol. 153(Suppl. 1), S428–S437.
Peineau, S., Taghibiglou, C., Bradley, C., Wong, T. P., Liu, L., Lu, J., Lo, E., Wu, D., Saule, E., Bouschet, T., Matthews, P., Isaac, J. T., Bortolotto, Z. A., Wang, Y. T., and Collingridge, G. L. (2007). LTP inhibits LTD in the hippocampus via regulation of GSK3beta. Neuron 53, 703–717.
Piven, J., Berthier, M. L., Starkstein, S. E., Nehme, E., Pearlson, G., and Folstein, S. (1990). Magnetic resonance imaging evidence for a defect of cerebral cortical development in autism. Am. J. Psychiatry 147, 734–739.
Popkie, A. P., Zeidner, L. C., Albrecht, A. M., D’Ippolito, A., Eckardt, S., Newsom, D. E., Groden, J., Doble, B. W., Aronow, B., McLaughlin, K. J., White, P., and Phiel, C. J. (2010). Phosphatidylinositol 3-kinase (PI3K) signaling via glycogen synthase kinase-3 (Gsk-3) regulates DNA methylation of imprinted loci. J. Biol. Chem. 285, 41337–41347.
Prickaerts, J., Moechars, D., Cryns, K., Lenaerts, I., van Craenendonck, H., Goris, I., Daneels, G., Bouwknecht, J. A., and Steckler, T. (2006). Transgenic mice overexpressing glycogen synthase kinase 3beta: a putative model of hyperactivity and mania. J. Neurosci. 26, 9022–9029.
Raballo, R., Rhee, J., Lyn-Cook, R., Leckman, J. F., Schwartz, M. L., and Vaccarino, F. M. (2000). Basic fibroblast growth factor (Fgf2) is necessary for cell proliferation and neurogenesis in the developing cerebral cortex. J. Neurosci. 20, 5012–5023.
Ronchi, A., Salaroli, R., Rivetti, S., Della Bella, E., Di Tomaso, T., Voltattorni, M., Cammelli, S., Ceccarelli, C., Giangaspero, F., Barbieri, E., and Cenacchi, G. (2010). Lithium induces mortality in medulloblastoma cell lines. Int. J. Oncol. 37, 745–752.
Salser, S. J., and Kenyon, C. (1992). Activation of a C. elegans Antennapedia homologue in migrating cells controls their direction of migration. Nature 355, 255–258.
Sanuki, R., Onishi, A., Koike, C., Muramatsu, R., Watanabe, S., Muranishi, Y., Irie, S., Uneo, S., Koyasu, T., Matsui, R., Cherasse, Y., Urade, Y., Watanabe, D., Kondo, M., Yamashita, T., and Furukawa, T. (2011). miR-124a is required for hippocampal axogenesis and retinal cone survival through Lhx2 suppression. Nat. Neurosci. 14, 1125–1134.
Sato, N., Meijer, L., Skaltsounis, L., Greengard, P., and Brivanlou, A. H. (2004). Maintenance of pluripotency in human and mouse embryonic stem cells through activation of Wnt signaling by a pharmacological GSK-3-specific inhibitor. Nat. Med. 10, 55–63.
Shakoori, A., Mai, W., Miyashita, K., Yasumoto, K., Takahashi, Y., Ooi, A., Kawakami, K., and Minamoto, T. (2007). Inhibition of GSK-3 beta activity attenuates proliferation of human colon cancer cells in rodents. Cancer Sci. 98, 1388–1393.
Shimizu, T., Kagawa, T., Inoue, T., Nonaka, A., Takada, S., Aburatani, H., and Taga, T. (2008). Stabilized beta-catenin functions through TCF/LEF proteins and the Notch/RBP-Jkappa complex to promote proliferation and suppress differentiation of neural precursor cells. Mol. Cell. Biol. 28, 7427–7441.
Shimojo, H., Ohtsuka, T., and Kageyama, R. (2008). Oscillations in notch signaling regulate maintenance of neural progenitors. Neuron 58, 52–64.
Silhankova, M., and Korswagen, H. C. (2007). Migration of neuronal cells along the anterior-posterior body axis of C. elegans: Wnts are in control. Curr. Opin. Genet. Dev. 17, 320–325.
Soutar, M. P., Kim, W. Y., Williamson, R., Peggie, M., Hastie, C. J., McLauchlan, H., Snider, W. D., Gordon-Weeks, P. R., and Sutherland, C. (2010). Evidence that glycogen synthase kinase-3 isoforms have distinct substrate preference in the brain. J. Neurochem. 115, 974–983.
Tronche, F., Kellendonk, C., Kretz, O., Gass, P., Anlag, K., Orban, P. C., Bock, R., Klein, R., and Schutz, G. (1999). Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat. Genet. 23, 99–103.
Uemura, K., Kuzuya, A., Shimozono, Y., Aoyagi, N., Ando, K., Shimohama, S., and Kinoshita, A. (2007). GSK3beta activity modifies the localization and function of presenilin 1. J. Biol. Chem. 282, 15823–15832.
Umehara, H., Kimura, T., Ohtsuka, S., Nakamura, T., Kitajima, K., Ikawa, M., Okabe, M., Niwa, H., and Nakano, T. (2007). Efficient derivation of embryonic stem cells by inhibition of glycogen synthase kinase-3. Stem Cells 25, 2705–2711.
Vaccarino, F. M., Schwartz, M. L., Raballo, R., Nilsen, J., Rhee, J., Zhou, M., Doetschman, T., Coffin, J. D., Wyland, J. J., and Hung, Y. T. (1999). Changes in cerebral cortex size are governed by fibroblast growth factor during embryogenesis. Nat. Neurosci. 2, 848.
Varlakhanova, N. V., and Knoepfler, P. S. (2009). Acting locally and globally: Myc’s ever-expanding roles on chromatin. Cancer Res. 69, 7487–7490.
Wakefield, J. G., Stephens, D. J., and Tavare, J. M. (2003). A role for glycogen synthase kinase-3 in mitotic spindle dynamics and chromosome alignment. J. Cell. Sci. 116, 637–646.
Wang, X., Tsai, J. W., Lamonica, B., and Kriegstein, A. R. (2011). A new subtype of progenitor cell in the mouse embryonic neocortex. Nat. Neurosci. 14, 555–561.
Wey, A., Martinez Cerdeno, V., Pleasure, D., and Knoepfler, P. S. (2010). c- and N-myc regulate neural precursor cell fate, cell cycle, and metabolism to direct cerebellar development. Cerebellum 9, 537–547.
Williams, S. P., Nowicki, M. O., Liu, F., Press, R., Godlewski, J., Abdel-Rasoul, M., Kaur, B., Fernandez, S. A., Chiocca, E. A., and Lawler, S. E. (2011). Indirubins decrease glioma invasion by blocking migratory phenotypes in both the tumor and stromal endothelial cell compartments. Cancer Res. 71, 5374–5380.
Woodhead, G. J., Mutch, C. A., Olson, E. C., and Chenn, A. (2006). Cell-autonomous beta-catenin signaling regulates cortical precursor proliferation. J. Neurosci. 26, 12620–12630.
Wood-Kaczmar, A., Kraus, M., Ishiguro, K., Philpott, K. L., and Gordon-Weeks, P. R. (2009). An alternatively spliced form of glycogen synthase kinase-3beta is targeted to growing neurites and growth cones. Mol. Cell. Neurosci. 42, 184–194.
Ying, Q. L., Wray, J., Nichols, J., Batlle-Morera, L., Doble, B., Woodgett, J., Cohen, P., and Smith, A. (2008). The ground state of embryonic stem cell self-renewal. Nature 453, 519–523.
Yokota, Y., Eom, T. Y., Stanco, A., Kim, W. Y., Rao, S., Snider, W. D., and Anton, E. S. (2010). Cdc42 and Gsk3 modulate the dynamics of radial glial growth, inter-radial glial interactions and polarity in the developing cerebral cortex. Development 137, 4101–4110.
Yokota, Y., Kim, W. Y., Chen, Y., Wang, X., Stanco, A., Komuro, Y., Snider, W., and Anton, E. S. (2009). The adenomatous polyposis coli protein is an essential regulator of radial glial polarity and construction of the cerebral cortex. Neuron 61, 42–56.
Yoon, K., and Gaiano, N. (2005). Notch signaling in the mammalian central nervous system: insights from mouse mutants. Nat. Neurosci. 8, 709–715.
Yoon, K. J., Koo, B. K., Im, S. K., Jeong, H. W., Ghim, J., Kwon, M. C., Moon, J. S., Miyata, T., and Kong, Y. Y. (2008). Mind bomb 1-expressing intermediate progenitors generate notch signaling to maintain radial glial cells. Neuron 58, 519–531.
Yuskaitis, C. J., Mines, M. A., King, M. K., Sweatt, J. D., Miller, C. A., and Jope, R. S. (2010). Lithium ameliorates altered glycogen synthase kinase-3 and behavior in a mouse model of fragile X syndrome. Biochem. Pharmacol. 79, 632–646.
Zechner, D., Fujita, Y., Hulsken, J., Muller, T., Walther, I., Taketo, M. M., Crenshaw, E. B. III, Birchmeier, W., and Birchmeier, C. (2003). beta-Catenin signals regulate cell growth and the balance between progenitor cell expansion and differentiation in the nervous system. Dev. Biol. 258, 406–418.
Zhu, L. Q., Liu, D., Hu, J., Cheng, J., Wang, S. H., Wang, Q., Wang, F., Chen, J. G., and Wang, J. Z. (2010). GSK-3 beta inhibits presynaptic vesicle exocytosis by phosphorylating P/Q-type calcium channel and interrupting SNARE complex formation. J. Neurosci. 30, 3624–3633.
Zhu, L. Q., Wang, S. H., Liu, D., Yin, Y. Y., Tian, Q., Wang, X. C., Wang, Q., Chen, J. G., and Wang, J. Z. (2007). Activation of glycogen synthase kinase-3 inhibits long-term potentiation with synapse-associated impairments. J. Neurosci. 27, 12211–12220.
Zhu, Q., Yang, J., Han, S., Liu, J., Holzbeierlein, J., Thrasher, J. B., and Li, B. (2011). Suppression of glycogen synthase kinase 3 activity reduces tumor growth of prostate cancer in vivo. Prostate 71, 835–845.
Keywords: GSK-3, neural progenitor, neuronal migration
Citation: Kim W-Y and Snider WD (2011) Functions of GSK-3 signaling in development of the nervous system. Front. Mol. Neurosci. 4:44. doi: 10.3389/fnmol.2011.00044
Received: 30 August 2011;
Accepted: 31 October 2011;
Published online: 17 November 2011.
Edited by:
Jim Robert Woodgett, Mount Sinai Hospital, CanadaReviewed by:
Jim Robert Woodgett, Mount Sinai Hospital, CanadaTatiana Lipina, Samuel Lunenfeld Research Institute, Canada
Copyright: © 2011 Kim and Snider. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Woo-Yang Kim, Department of Developmental Neuroscience, University of Nebraska Medical Center, 985960 Nebraska Medical Center, Omaha, NE 68198-5960, USA. e-mail:d29veWFuZy5raW1AdW5tYy5lZHU=