 Ronald E. van Kesteren1*
Ronald E. van Kesteren1* Matthew R. J. Mason2
Matthew R. J. Mason2 Harold D. MacGillavry1†
Harold D. MacGillavry1† August B. Smit1
August B. Smit1 Joost Verhaagen1,2
Joost Verhaagen1,2
- 1 Center for Neurogenomics and Cognitive Research, Neuroscience Campus Amsterdam, VU University, Amsterdam, Netherlands
- 2 Laboratory for Neuroregeneration, Netherlands Institute for Neuroscience, Amsterdam, Netherlands
The regenerative capacity of injured neurons in the central nervous system is limited due to the absence of a robust neuron-intrinsic injury-induced gene response that supports axon regeneration. In peripheral neurons axotomy induces a large cohort of regeneration-associated genes (RAGs). The forced expression of some of these RAGs in injured neurons has some beneficial effect on axon regeneration, but the reported effects are rather small. Transcription factors (TFs) provide a promising class of RAGs. TFs are hubs in the regeneration-associated gene network, and potentially control the coordinate expression of many RAGs simultaneously. Here we discuss the use of combined experimental and computational methods to identify novel regeneration-associated TFs with a key role in initiating and maintaining the RAG-response in injured neurons. We propose that a relatively small number of hub TFs with multiple functional connections in the RAG network might provide attractive new targets for gene-based and/or pharmacological approaches to promote axon regeneration in the central nervous system.
Regeneration-Associated Genes: A Historical Perspective
The extent to which neurons can regenerate injured axons varies dramatically in the adult mammalian nervous system. Axotomy of peripheral neurons results in the activation of hundreds of neuron-intrinsic regeneration-associated genes (RAGs), whereas in many central neurons a very limited or no RAG-response is observed, except following very proximal lesions (Jenkins et al., 1993; Fernandes et al., 1999; Mason et al., 2003). In addition to the inhibitory extracellular environment of the CNS, the absence of a coordinated RAG-response is a major reason for regenerative failure in the CNS. Early findings that particular axonal proteins, such as GAP-43 and alpha-tubulin, are induced in injured peripheral neurons led to the hypothesis that peripheral nerve injury triggers an intrinsic cell body response that includes the expression of a set of growth-associated proteins (GAPs) that together promote axon regeneration (Skene and Willard, 1981; Skene, 1989; Tetzlaff et al., 1991).
The idea that successful regeneration of injured axons also requires an appropriate intrinsic RAG-response is further supported by several other observations. Firstly, although CNS neurons do not regenerate spontaneously, if a peripheral nerve graft is implanted into the spinal cord, some spinal neurons (on average 100–200 per animal), actually can regenerate injured axons into the graft (David and Aguayo, 1981; Richardson et al., 1982), indicating that certain central neurons do have the intrinsic capacity to regenerate injured axons when provided with a suitable growth-stimulating environment. CNS neurons that are able to regenerate into nerve grafts also upregulate RAGs following axotomy (Anderson et al., 1998; Mason et al., 2002). It is important to note, however, that most injured neurons in the spinal cord do not show such a response, and that the majority of neurons that do are located very close to the lesion site. Secondly, treatment of rubrospinal neurons with BDNF at their cell bodies results in upregulation of GAP expression and promotes regeneration of axons into peripheral nerve transplants in the spinal cord (Kobayashi et al., 1997), even when neurons are chronically lesioned and severely atrophied (Kwon et al., 2002). Thus, exogenous treatments that boost RAG expression in these neurons also increase the number of regeneration-competent cells. Thirdly, when dorsal root ganglion (DRG) neurons are conditioned by a peripheral nerve transection, a lesion which induces RAG expression, their central axons show an enhanced regenerative response that can result in significant sprouting into the spinal cord, even beyond the lesion site (Richardson and Issa, 1984; Chong et al., 1999; Neumann and Woolf, 1999). Finally, this conditioning lesion effect was shown to be transcription dependent (Smith and Skene, 1997). Together these findings all indicate that a coordinated neuron-intrinsic regeneration-associated transcriptional response is critical for successful axonal regeneration to occur, and are in support of the RAG-hypothesis.
By now, genome-wide gene expression profiling studies have provided a near-complete view of the complex changes in neuronal gene expression in diverse PNS and CNS injury paradigms (Costigan et al., 2002; Xiao et al., 2002; Schmitt et al., 2003; Kury et al., 2004; Di Giovanni et al., 2005; Bosse et al., 2006; Stam et al., 2007; Geeven et al., 2011). Meta-analyses of these transcriptional profiling studies revealed the core set of RAGs that is consistently upregulated following neuronal injury (Szpara et al., 2007; Stam et al., 2008). These studies have further supported the original RAG-hypothesis by showing that many genes are strongly upregulated during successful regeneration, whereas types of injury (e.g., dorsal root injury) that are not followed by successful regeneration, do not robustly induce their expression.
The identification of RAGs led to the idea that axon regeneration could be promoted by manipulating the expression of specific RAGs in injured neurons. Initial studies using transgenic GAP-43 mice were promising and showed an enhancement of long-distance regenerative axon growth in the PNS (Aigner et al., 1995). In the CNS, however, overexpression of GAP-43 only induced nerve sprouting, but failed to induce long-distance regeneration (Aigner et al., 1995; Buffo et al., 1997; Neumann and Woolf, 1999), even into growth-permissive peripheral nerve grafts (Mason et al., 2000). Also, GAP-43 knockout mice have no gross axonal growth deficits, but show profound defects in axonal guidance (Strittmatter et al., 1995; Sretavan and Kruger, 1998; Maier et al., 1999; Zhu and Julien, 1999). Other classical RAGs that have only limited effects on axon regeneration include Sprr1a and Itga7 (Werner et al., 2000; Bonilla et al., 2002; Raivich et al., 2004). These findings demonstrate that manipulating the expression of individual RAGs does not induce robust axon regeneration in the CNS, and therefore research started to focus on transcriptional regulators that can affect the expression of multiple RAGs jointly.
The RAG Regulatory Network: Regeneration-Associated Transcription Factors
All cellular processes are controlled by genes that act together in regulatory networks. Gene regulatory networks are inherently robust against random perturbation (Albert et al., 2000), which may explain why manipulating the expression of individual RAGs has a limited effect on axon regeneration. One way to alter the state of the network is to jointly manipulate the expression of multiple genes in the network. Indeed, overexpression of both Gap-43 and Cap23 in the RAG network resulted in enhanced DRG regeneration into peripheral nerve grafts implanted in the spinal cord (Bomze et al., 2001). There are however practical limitations to the number of RAGs that can be simultaneously overexpressed. As an alternative, one could change the expression of so-called hub genes in the RAG network. Hub genes are highly connected nodes in a gene network and regulate the coordinated expression or activity of many other genes. They are therefore attractive candidate targets for perturbation at the network level (Batada et al., 2006; Figure 1).
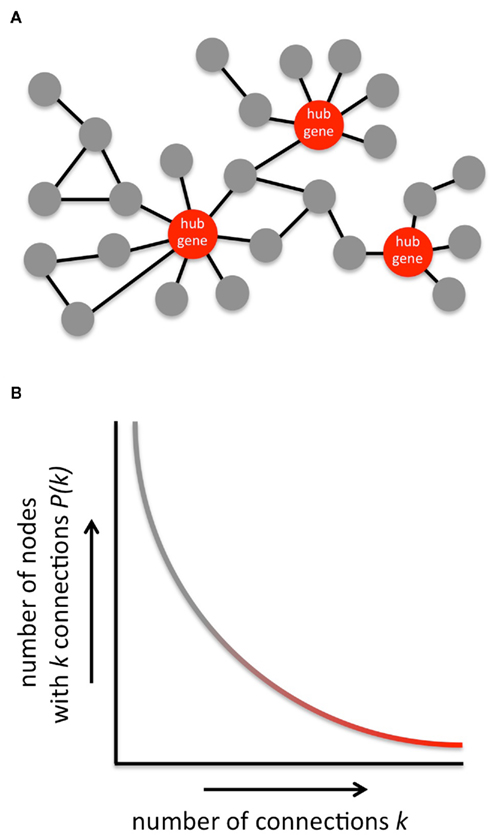
Figure 1. General structure of gene networks. (A) Typical example of a gene network. Highly connected genes (indicated in red) are referred to as hub genes. (B) Gene networks, like most other biological networks and many non-biological networks too, are scale-free. The connectivity distribution P(k) in a scale-free network follows an inverse power relation with number of connections k per node. Consequently, gene networks contain many nodes with low connectivity (gray part of the curve), whereas only a few nodes are highly connected and qualify as hub genes (red part of the curve). Scale-free networks are robust against random perturbation. This protects gene networks from falling apart as a result of random mutations, but it is at the same time the Achilles’ heel of a gene network because targeted attacks at network hubs will quickly result in network fragmentation and deregulation of large groups of genes. Hub genes are therefore interesting candidate targets to efficiently interfere with cellular function.
Hubs in gene regulatory networks are often transcription factors (TFs). C-JUN was the first TF to be identified in the RAG network (Herdegen et al., 1991; Jenkins and Hunt, 1991), and plays an important role in the successful regeneration of facial nerve motor neurons (Raivich et al., 2004). Later, additional regeneration-associated TFs were identified, including ATF3, SOX11, CREB, p53, STAT3, KLF4, and SMAD1 (Schwaiger et al., 2000; Tsujino et al., 2000; Gao et al., 2004; Di Giovanni et al., 2006; Jankowski et al., 2006; Seijffers et al., 2006; Moore et al., 2009; Zou et al., 2009). Some of these TFs are now recognized as important regulators of specific subsets of RAGs. For instance, C-JUN induces expression of Itga7 (with Itgb1 the receptor for laminin), the cell adhesion molecule Cd44, and the neuropeptide Gal (Raivich et al., 2004), and ATF3 induces expression of c-Jun, the heat-shock protein Hsp27 and Sprr1a (a marker specific for regenerative axon growth; Seijffers et al., 2007). The key question now is: which of these TFs are actual network hubs that can be targeted successfully to “reprogram” injured neurons so that they acquire a RAG network that enhances the rate of regeneration of injured central neurons?
We and others recently addressed this question using a combination of various experimental and biocomputational approaches (Macgillavry et al., 2009, 2011; Michaelevski et al., 2010; Geeven et al., 2011). First, we used medium-throughput cellular screening to test the effects of 62 candidate regeneration-associated TFs on axon growth in vitro. Each of these 62 TFs was previously found to be differentially regulated in peripherally injured DRG neurons compared with centrally injured DRG neurons (Stam et al., 2007), however, only 10 affected axon growth after knockdown in a DRG-like cell line (Macgillavry et al., 2009). Importantly, there was a discrepancy between the direction of regulation and the observed effects upon knockdown for several of these TFs. In particular one TF, NFIL3, was strongly upregulated during successful regeneration, but reducing its expression significantly enhanced axon growth (Macgillavry et al., 2009). This rather paradoxical finding is further discussed below. We also developed and used a computational approach to predict TFs that bind to and regulate the expression of RAGs. Using this approach, we identified several novel TFs whose expression levels are not changed upon injury, but that are predicted to affect the coordinated expression of multiple RAGs sharing similar functions (Geeven et al., 2011). One of these TFs, PPAR gamma, was shown to repress the expression of several axon growth-inhibiting genes, and may be involved in fatty acid signaling from the injury site to the nucleus of the injured neurons (Geeven et al., 2011). Together, these two approaches have helped to identify novel candidate TFs in the RAG network. In a related study, Michaelevski et al. (2010) combined gene expression profiling with phosphoproteome analysis of retrograde injury signals derived from axoplasm and in silico promoter-binding analysis, and they were able to identify many novel regeneration-associated TFs in DRG neurons. Further signaling network analysis led them to identify four protein kinases that may act as central regulators of these transcriptional pathways. Manipulation of these kinases by pharmacological inhibition had some effect on neurite outgrowth in vitro, but these effects were largely non-additive, which the authors took to be indicative of the robustness of the RAG network.
Not all RAGs Promote Axon Regeneration: Evolutionary Constraints on the Network
We were intrigued by the fact that NFIL3 is upregulated in DRG sensory neurons by peripheral nerve injury (Stam et al., 2007), but that suppressing NFIL3 expression strongly enhances axon growth of cultured adult DRG neurons (Macgillavry et al., 2009). The relation between the injury-induced expression of a TF and its apparent role in regeneration can indeed be quite paradoxical, and at least three other studies have shown examples of RAGs that repress axon growth. SOCS3, for instance, is induced in DRG neurons by a peripheral nerve lesion, but represses axon growth by inhibiting the growth-promoting TF STAT3 (Miao et al., 2006). The regeneration-promoting effects of cAMP on injured retinal ganglion cells appear to be mediated by a downregulation of SOCS3 (Park et al., 2009). Genetic deletion of SOCS3 promotes optic nerve regeneration (Smith et al., 2009), whereas overexpression of SOCS3 inhibits optic nerve regeneration (Hellstrom et al., 2011). Another study demonstrated that NFAT-3 is a transcriptional repressor of Gap-43 expression (Nguyen et al., 2009). This was unexpected, as it is long known that NFATs are activated in response to growth factors and mediate axon growth and pre-synaptic differentiation (Graef et al., 2003). Finally, KLF4 was identified as a repressor of axon growth (Moore et al., 2009; Blackmore et al., 2010). Although KLF4 expression is not upregulated after injury (Szpara et al., 2007; Stam et al., 2008), in vivo deletion of KLF4 does enhance optic nerve regeneration (Moore et al., 2009). Moreover, the same study showed that several other KLF family members also suppress axon growth, while two others, KLF6 and KLF7, promote axon growth.
In an attempt to understand the role of NFIL3 in the RAG regulatory network, we further studied how NFIL3 regulates RAG expression. We showed that NFIL3 expression follows cAMP induction and CREB activation, and that NFIL3 suppresses CREB target genes such as Arg1 and Gap-43, by competing for the same DNA binding sites (Macgillavry et al., 2009; Figure 2). Such so-called incoherent feed-forward loops occur in many other gene regulatory networks, and allow efficient control of target gene dynamics (Kaplan et al., 2008). CREB and NFIL3 are widely expressed in the brain, and their feed-forward interactions may have evolved to create strict temporal control over the expression of plasticity genes. In a subsequent study, we identified C/EBP TFs as part of this regulatory loop. CREB induces C/EBP expression in neurons, and NFIL3 also suppresses many C/EBP target genes (Macgillavry et al., 2011; Figure 2). Together, these findings for the first time reveal a part of the gene regulatory network architecture underlying neuronal regeneration, and suggest that genetic mechanisms to fine-tune neuronal plasticity form an evolutionary constraint on a neuron’s ability to regenerate injured axons.
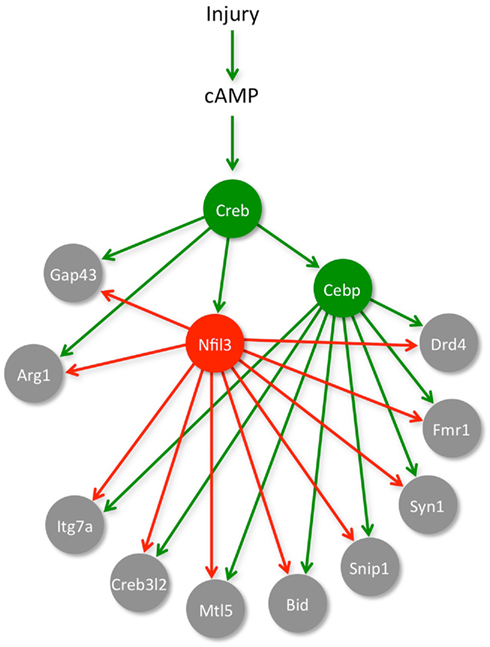
Figure 2. A gene network for neuronal regeneration. Induction of cAMP levels and subsequent activation of CREB play an important role in starting up a RAG-response in injured neurons. We showed that one of the targets of CREB is another TF named NFIL3, and that NFIL3 functions as a feed-forward repressor of several CREB target genes (e.g., Arg1 and Gap-43). In another study, we identified a set of RAGs (i.e., Itg7a, Creb3l2, Mtl5, Bid, Snip1, Syn1, Fmr1, Drd4) that are activated by C/EBP TFs, which are known downstream targets of CREB, and we showed that these RAGs are also repressed by NFIL3. We thus characterized a small part of the gene network that controls neuronal regeneration, and identified CREB, C/EBP, and NFIL3 as hub genes in this network. Downregulation of NFIL3 in neuronal cell lines or in adult primary DRG neurons significantly enhanced their regenerative capacity.
RAG Network Hubs: Therapeutic Perspective
Insights into RAG network architecture, and the identification of hub TFs within the network, may aid the future development of gene therapeutic and pharmacological strategies to enhance the intrinsic regenerative potential of injured central neurons. Although direct translation of current knowledge into clinically relevant therapeutic approaches is not possible yet, we are tempted to make some predictions. NFIL3, for instance, is widely expressed in the CNS, and may serve to suppress or fine-tune CREB-dependent plasticity. If NFIL3 indeed also represses regenerative axon growth in the CNS, then our findings would predict that a transient relief of NFIL3-mediated repression might temporarily enhance the effects of CREB and stimulate axonal regeneration. Gene therapy is an ideal method to deliver or inactivate hub TFs, as it can selectively target the desired neurons and allows simultaneous overexpression of growth-promoting TFs and suppression of growth-inhibiting TFs, at least for pre-clinical studies. We, and others, have explored the use of vectors based on lentivirus and adeno-associated virus (AAV) for gene delivery to the nervous system. AAV vectors are highly effective for gene delivery to DRG neurons and rubrospinal neurons (Hollis et al., 2008; Blits et al., 2010; Mason et al., 2010; Parikh et al., 2011; Figure 3). AAV vectors are also promising as vectors that can be delivered via the CSF intrathecally (Storek et al., 2008; Towne et al., 2009), although such an untargeted delivery method may be unsuitable when the intervention is required only in specific neuronal populations.
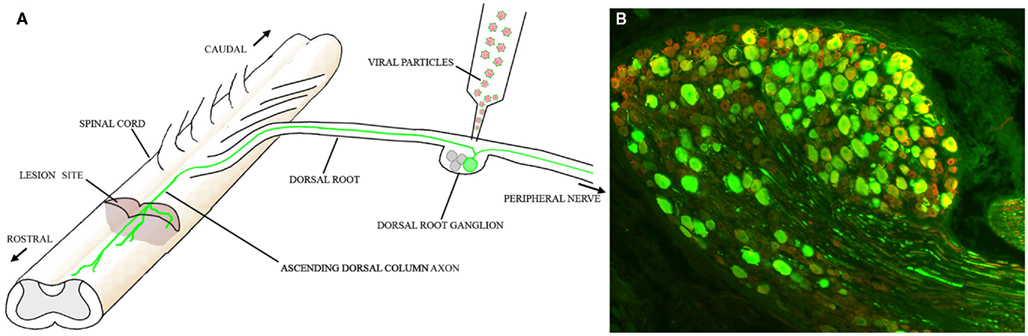
Figure 3. Gene therapy to promote regeneration in the spinal cord. (A) Schematic diagram of how AAV vectors might be used to promote regeneration of ascending dorsal column axons. One or more AAV5 vectors expressing key hub TFs are injected into the DRG. These TFs induce RAG expression in the transduced neurons. (B) Expression of GFP in an L5 DRG 12 weeks after injection of AAV5-CMV-GFP. Almost the entire neuronal population is transduced. Green: GFP; Red: βIII tubulin.
A major challenge is how to regulate gene expression so that it can be turned off when no longer required, whether this is days or months after application of the vector (Mason et al., 2011), as a continued growth response may eventually have undesirable consequences. Other developments will include the methods to deliver multiple transgenes simultaneously to the same neuron, for example using AAV vectors with multiple promoters. AAV vectors in particular, hold great promise as clinically acceptable gene therapy vectors for the promotion of CNS regeneration since they have proven to be well-tolerated and safe in patients.
Concluding Remarks
Hub TFs in the RAG regulatory network provide an attractive class of molecular targets for promoting the regenerative capacity of injured neurons because they jointly regulate the expression of many RAGs simultaneously. Interestingly, recent studies into RAG network architecture showed that some regeneration-associated TFs actually suppress the regenerative capacity of neurons. Such intrinsic inhibitors of neuronal regeneration may provide interesting targets to inactivate using viral vector approaches. Ideally, the combined expression and/or inactivation of a few hub TFs in the RAG network will be sufficient to enhance the regeneration of axotomized central neurons, and we expect that further elucidation of RAG network architecture will aid the selection of therapeutically relevant TF targets in the future.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aigner, L., Arber, S., Kapfhammer, J. P., Laux, T., Schneider, C., Botteri, F., Brenner, H. R., and Caroni, P. (1995). Overexpression of the neural growth-associated protein GAP-43 induces nerve sprouting in the adult nervous system of transgenic mice. Cell 83, 269–278.
Albert, R., Jeong, H., and Barabasi, A. L. (2000). Error and attack tolerance of complex networks. Nature 406, 378–382.
Anderson, P. N., Campbell, G., Zhang, Y., and Lieberman, A. R. (1998). Cellular and molecular correlates of the regeneration of adult mammalian CNS axons into peripheral nerve grafts. Prog. Brain Res. 117, 211–232.
Batada, N. N., Hurst, L. D., and Tyers, M. (2006). Evolutionary and physiological importance of hub proteins. PLoS Comput. Biol. 2, e88. doi: 10.1371/journal.pcbi.0020088
Blackmore, M. G., Moore, D. L., Smith, R. P., Goldberg, J. L., Bixby, J. L., and Lemmon, V. P. (2010). High content screening of cortical neurons identifies novel regulators of axon growth. Mol. Cell. Neurosci. 44, 43–54.
Blits, B., Derks, S., Twisk, J., Ehlert, E., Prins, J., and Verhaagen, J. (2010). Adeno-associated viral vector (AAV)-mediated gene transfer in the red nucleus of the adult rat brain: comparative analysis of the transduction properties of seven AAV serotypes and lentiviral vectors. J. Neurosci. Methods 185, 257–263.
Bomze, H. M., Bulsara, K. R., Iskandar, B. J., Caroni, P., and Skene, J. H. (2001). Spinal axon regeneration evoked by replacing two growth cone proteins in adult neurons. Nat. Neurosci. 4, 38–43.
Bonilla, I. E., Tanabe, K., and Strittmatter, S. M. (2002). Small proline-rich repeat protein 1A is expressed by axotomized neurons and promotes axonal outgrowth. J. Neurosci. 22, 1303–1315.
Bosse, F., Hasenpusch-Theil, K., Kury, P., and Muller, H. W. (2006). Gene expression profiling reveals that peripheral nerve regeneration is a consequence of both novel injury-dependent and reactivated developmental processes. J. Neurochem. 96, 1441–1457.
Buffo, A., Holtmaat, A. J., Savio, T., Verbeek, J. S., Oberdick, J., Oestreicher, A. B., Gispen, W. H., Verhaagen, J., Rossi, F., and Strata, P. (1997). Targeted overexpression of the neurite growth-associated protein B-50/GAP-43 in cerebellar Purkinje cells induces sprouting after axotomy but not axon regeneration into growth-permissive transplants. J. Neurosci. 17, 8778–8791.
Chong, M. S., Woolf, C. J., Haque, N. S., and Anderson, P. N. (1999). Axonal regeneration from injured dorsal roots into the spinal cord of adult rats. J. Comp. Neurol. 410, 42–54.
Costigan, M., Befort, K., Karchewski, L., Griffin, R. S., D’urso, D., Allchorne, A., Sitarski, J., Mannion, J. W., Pratt, R. E., and Woolf, C. J. (2002). Replicate high-density rat genome oligonucleotide microarrays reveal hundreds of regulated genes in the dorsal root ganglion after peripheral nerve injury. BMC Neurosci. 3, 16. doi: 10.1186/1471-2202-3-16
David, S., and Aguayo, A. J. (1981). Axonal elongation into peripheral nervous system “bridges” after central nervous system injury in adult rats. Science 214, 931–933.
Di Giovanni, S., De Biase, A., Yakovlev, A., Finn, T., Beers, J., Hoffman, E. P., and Faden, A. I. (2005). In vivo and in vitro characterization of novel neuronal plasticity factors identified following spinal cord injury. J. Biol. Chem. 280, 2084–2091.
Di Giovanni, S., Knights, C. D., Rao, M., Yakovlev, A., Beers, J., Catania, J., Avantaggiati, M. L., and Faden, A. I. (2006). The tumor suppressor protein p53 is required for neurite outgrowth and axon regeneration. EMBO J. 25, 4084–4096.
Fernandes, K. J., Fan, D. P., Tsui, B. J., Cassar, S. L., and Tetzlaff, W. (1999). Influence of the axotomy to cell body distance in rat rubrospinal and spinal motoneurons: differential regulation of GAP-43, tubulins, and neurofilament-M. J. Comp. Neurol. 414, 495–510.
Gao, Y., Deng, K., Hou, J., Bryson, J. B., Barco, A., Nikulina, E., Spencer, T., Mellado, W., Kandel, E. R., and Filbin, M. T. (2004). Activated CREB is sufficient to overcome inhibitors in myelin and promote spinal axon regeneration in vivo. Neuron 44, 609–621.
Geeven, G., Macgillavry, H. D., Eggers, R., Sassen, M. M., Verhaagen, J., Smit, A. B., De Gunst, M. C., and Van Kesteren, R. E. (2011). LLM3D: a log-linear modeling-based method to predict functional gene regulatory interactions from genome-wide expression data. Nucleic Acids Res. 39, 5313–5327.
Graef, I. A., Wang, F., Charron, F., Chen, L., Neilson, J., Tessier-Lavigne, M., and Crabtree, G. R. (2003). Neurotrophins and netrins require calcineurin/NFAT signaling to stimulate outgrowth of embryonic axons. Cell 113, 657–670.
Hellstrom, M., Muhling, J., Ehlert, E. M., Verhaagen, J., Pollett, M. A., Hu, Y., and Harvey, A. R. (2011). Negative impact of rAAV2 mediated expression of SOCS3 on the regeneration of adult retinal ganglion cell axons. Mol. Cell. Neurosci. 46, 507–515.
Herdegen, T., Kummer, W., Fiallos, C. E., Leah, J., and Bravo, R. (1991). Expression of c-JUN, JUN B and JUN D proteins in rat nervous system following transection of vagus nerve and cervical sympathetic trunk. Neuroscience 45, 413–422.
Hollis, E. R. II, Kadoya, K., Hirsch, M., Samulski, R. J., and Tuszynski, M. H. (2008). Efficient retrograde neuronal transduction utilizing self-complementary AAV1. Mol. Ther. 16, 296–301.
Jankowski, M. P., Cornuet, P. K., Mcilwrath, S., Koerber, H. R., and Albers, K. M. (2006). SRY-box containing gene 11 (Sox11) transcription factor is required for neuron survival and neurite growth. Neuroscience 143, 501–514.
Jenkins, R., and Hunt, S. P. (1991). Long-term increase in the levels of c-jun mRNA and jun protein-like immunoreactivity in motor and sensory neurons following axon damage. Neurosci. Lett. 129, 107–110.
Jenkins, R., Tetzlaff, W., and Hunt, S. P. (1993). Differential expression of immediate early genes in rubrospinal neurons following axotomy in rat. Eur. J. Neurosci. 5, 203–209.
Kaplan, S., Bren, A., Dekel, E., and Alon, U. (2008). The incoherent feed-forward loop can generate non-monotonic input functions for genes. Mol. Syst. Biol. 4, 203.
Kobayashi, N. R., Fan, D. P., Giehl, K. M., Bedard, A. M., Wiegand, S. J., and Tetzlaff, W. (1997). BDNF and NT-4/5 prevent atrophy of rat rubrospinal neurons after cervical axotomy, stimulate GAP-43 and Talpha1-tubulin mRNA expression, and promote axonal regeneration. J. Neurosci. 17, 9583–9595.
Kury, P., Abankwa, D., Kruse, F., Greiner-Petter, R., and Muller, H. W. (2004). Gene expression profiling reveals multiple novel intrinsic and extrinsic factors associated with axonal regeneration failure. Eur. J. Neurosci. 19, 32–42.
Kwon, B. K., Liu, J., Messerer, C., Kobayashi, N. R., Mcgraw, J., Oschipok, L., and Tetzlaff, W. (2002). Survival and regeneration of rubrospinal neurons 1 year after spinal cord injury. Proc. Natl. Acad. Sci. U.S.A. 99, 3246–3251.
Macgillavry, H. D., Cornelis, J., Van Der Kallen, L. R., Sassen, M. M., Verhaagen, J., Smit, A. B., and Van Kesteren, R. E. (2011). Genome-wide gene expression and promoter binding analysis identifies NFIL3 as a repressor of C/EBP target genes in neuronal outgrowth. Mol. Cell. Neurosci. 46, 460–468.
Macgillavry, H. D., Stam, F. J., Sassen, M. M., Kegel, L., Hendriks, W. T., Verhaagen, J., Smit, A. B., and Van Kesteren, R. E. (2009). NFIL3 and cAMP response element-binding protein form a transcriptional feedforward loop that controls neuronal regeneration-associated gene expression. J. Neurosci. 29, 15542–15550.
Maier, D. L., Mani, S., Donovan, S. L., Soppet, D., Tessarollo, L., Mccasland, J. S., and Meiri, K. F. (1999). Disrupted cortical map and absence of cortical barrels in growth-associated protein (GAP)-43 knockout mice. Proc. Natl. Acad. Sci. U.S.A. 96, 9397–9402.
Mason, M. R., Campbell, G., Caroni, P., Anderson, P. N., and Lieberman, A. R. (2000). Overexpression of GAP-43 in thalamic projection neurons of transgenic mice does not enable them to regenerate axons through peripheral nerve grafts. Exp. Neurol. 165, 143–152.
Mason, M. R., Ehlert, E. M., Eggers, R., Pool, C. W., Hermening, S., Huseinovic, A., Timmermans, E., Blits, B., and Verhaagen, J. (2010). Comparison of AAV serotypes for gene delivery to dorsal root ganglion neurons. Mol. Ther. 18, 715–724.
Mason, M. R., Lieberman, A. R., and Anderson, P. N. (2003). Corticospinal neurons up-regulate a range of growth-associated genes following intracortical, but not spinal, axotomy. Eur. J. Neurosci. 18, 789–802.
Mason, M. R., Lieberman, A. R., Grenningloh, G., and Anderson, P. N. (2002). Transcriptional upregulation of SCG10 and CAP-23 is correlated with regeneration of the axons of peripheral and central neurons in vivo. Mol. Cell. Neurosci. 20, 595–615.
Mason, M. R., Tannemaat, M. R., Malessy, M. J., and Verhaagen, J. (2011). Gene therapy for the peripheral nervous system: a strategy to repair the injured nerve? Curr. Gene Ther. 11, 75–89.
Miao, T., Wu, D., Zhang, Y., Bo, X., Subang, M. C., Wang, P., and Richardson, P. M. (2006). Suppressor of cytokine signaling-3 suppresses the ability of activated signal transducer and activator of transcription-3 to stimulate neurite growth in rat primary sensory neurons. J. Neurosci. 26, 9512–9519.
Michaelevski, I., Segal-Ruder, Y., Rozenbaum, M., Medzihradszky, K. F., Shalem, O., Coppola, G., Horn-Saban, S., Ben-Yaakov, K., Dagan, S. Y., Rishal, I., Geschwind, D. H., Pilpel, Y., Burlingame, A. L., and Fainzilber, M. (2010). Signaling to transcription networks in the neuronal retrograde injury response. Sci. Signal. 3, ra53.
Moore, D. L., Blackmore, M. G., Hu, Y., Kaestner, K. H., Bixby, J. L., Lemmon, V. P., and Goldberg, J. L. (2009). KLF family members regulate intrinsic axon regeneration ability. Science 326, 298–301.
Neumann, S., and Woolf, C. J. (1999). Regeneration of dorsal column fibers into and beyond the lesion site following adult spinal cord injury. Neuron 23, 83–91.
Nguyen, T., Lindner, R., Tedeschi, A., Forsberg, K., Green, A., Wuttke, A., Gaub, P., and Di Giovanni, S. (2009). NFAT-3 is a transcriptional repressor of the growth-associated protein 43 during neuronal maturation. J. Biol. Chem. 284, 18816–18823.
Parikh, P., Hao, Y., Hosseinkhani, M., Patil, S. B., Huntley, G. W., Tessier-Lavigne, M., and Zou, H. (2011). Regeneration of axons in injured spinal cord by activation of bone morphogenetic protein/Smad1 signaling pathway in adult neurons. Proc. Natl. Acad. Sci. U.S.A. 108, E99–E107.
Park, K. K., Hu, Y., Muhling, J., Pollett, M. A., Dallimore, E. J., Turnley, A. M., Cui, Q., and Harvey, A. R. (2009). Cytokine-induced SOCS expression is inhibited by cAMP analogue: impact on regeneration in injured retina. Mol. Cell. Neurosci. 41, 313–324.
Raivich, G., Bohatschek, M., Da Costa, C., Iwata, O., Galiano, M., Hristova, M., Nateri, A. S., Makwana, M., Riera-Sans, L., Wolfer, D. P., Lipp, H. P., Aguzzi, A., Wagner, E. F., and Behrens, A. (2004). The AP-1 transcription factor c-Jun is required for efficient axonal regeneration. Neuron 43, 57–67.
Richardson, P. M., and Issa, V. M. (1984). Peripheral injury enhances central regeneration of primary sensory neurones. Nature 309, 791–793.
Richardson, P. M., Mcguinness, U. M., and Aguayo, A. J. (1982). Peripheral nerve autografts to the rat spinal cord: studies with axonal tracing methods. Brain Res. 237, 147–162.
Schmitt, A. B., Breuer, S., Liman, J., Buss, A., Schlangen, C., Pech, K., Hol, E. M., Brook, G. A., Noth, J., and Schwaiger, F. W. (2003). Identification of regeneration-associated genes after central and peripheral nerve injury in the adult rat. BMC Neurosci. 4, 8. doi: 10.1186/1471-2202-4-8
Schwaiger, F. W., Hager, G., Schmitt, A. B., Horvat, A., Streif, R., Spitzer, C., Gamal, S., Breuer, S., Brook, G. A., Nacimiento, W., and Kreutzberg, G. W. (2000). Peripheral but not central axotomy induces changes in Janus kinases (JAK) and signal transducers and activators of transcription (STAT). Eur. J. Neurosci. 12, 1165–1176.
Seijffers, R., Allchorne, A. J., and Woolf, C. J. (2006). The transcription factor ATF-3 promotes neurite outgrowth. Mol. Cell. Neurosci. 32, 143–154.
Seijffers, R., Mills, C. D., and Woolf, C. J. (2007). ATF3 increases the intrinsic growth state of DRG neurons to enhance peripheral nerve regeneration. J. Neurosci. 27, 7911–7920.
Skene, J. H., and Willard, M. (1981). Axonally transported proteins associated with axon growth in rabbit central and peripheral nervous systems. J. Cell Biol. 89, 96–103.
Smith, D. S., and Skene, J. H. (1997). A transcription-dependent switch controls competence of adult neurons for distinct modes of axon growth. J. Neurosci. 17, 646–658.
Smith, P. D., Sun, F., Park, K. K., Cai, B., Wang, C., Kuwako, K., Martinez-Carrasco, I., Connolly, L., and He, Z. (2009). SOCS3 deletion promotes optic nerve regeneration in vivo. Neuron 64, 617–623.
Sretavan, D. W., and Kruger, K. (1998). Randomized retinal ganglion cell axon routing at the optic chiasm of GAP-43-deficient mice: association with midline recrossing and lack of normal ipsilateral axon turning. J. Neurosci. 18, 10502–10513.
Stam, F. J., Macgillavry, H. D., Armstrong, N. J., De Gunst, M. C., Zhang, Y., Van Kesteren, R. E., Smit, A. B., and Verhaagen, J. (2007). Identification of candidate transcriptional modulators involved in successful regeneration after nerve injury. Eur. J. Neurosci. 25, 3629–3637.
Stam, F. J., Mason, M. R., Smit, A. B., and Verhaagen, J. (2008). “A meta-analysis of large-scale gene expression studies of the injured PNS: toward the genetic networks that govern successful regeneration,” in Neural Degeneration and Repair: Gene Expression Profiling, Proteomics, Glycomics, and Systems Biology, ed. H. W. Muller (Hoboken, NJ: Wiley), 35–60.
Storek, B., Reinhardt, M., Wang, C., Janssen, W. G., Harder, N. M., Banck, M. S., Morrison, J. H., and Beutler, A. S. (2008). Sensory neuron targeting by self-complementary AAV8 via lumbar puncture for chronic pain. Proc. Natl. Acad. Sci. U.S.A. 105, 1055–1060.
Strittmatter, S. M., Fankhauser, C., Huang, P. L., Mashimo, H., and Fishman, M. C. (1995). Neuronal pathfinding is abnormal in mice lacking the neuronal growth cone protein GAP-43. Cell 80, 445–452.
Szpara, M. L., Vranizan, K., Tai, Y. C., Goodman, C. S., Speed, T. P., and Ngai, J. (2007). Analysis of gene expression during neurite outgrowth and regeneration. BMC Neurosci. 8, 100. doi: 10.1186/1471-2202-8-100
Tetzlaff, W., Alexander, S. W., Miller, F. D., and Bisby, M. A. (1991). Response of facial and rubrospinal neurons to axotomy: changes in mRNA expression for cytoskeletal proteins and GAP-43. J. Neurosci. 11, 2528–2544.
Towne, C., Pertin, M., Beggah, A. T., Aebischer, P., and Decosterd, I. (2009). Recombinant adeno-associated virus serotype 6 (rAAV2/6)-mediated gene transfer to nociceptive neurons through different routes of delivery. Mol. Pain 5, 52.
Tsujino, H., Kondo, E., Fukuoka, T., Dai, Y., Tokunaga, A., Miki, K., Yonenobu, K., Ochi, T., and Noguchi, K. (2000). Activating transcription factor 3 (ATF3) induction by axotomy in sensory and motoneurons: a novel neuronal marker of nerve injury. Mol. Cell. Neurosci. 15, 170–182.
Werner, A., Willem, M., Jones, L. L., Kreutzberg, G. W., Mayer, U., and Raivich, G. (2000). Impaired axonal regeneration in alpha7 integrin-deficient mice. J. Neurosci. 20, 1822–1830.
Xiao, H. S., Huang, Q. H., Zhang, F. X., Bao, L., Lu, Y. J., Guo, C., Yang, L., Huang, W. J., Fu, G., Xu, S. H., Cheng, X. P., Yan, Q., Zhu, Z. D., Zhang, X., Chen, Z., Han, Z. G., and Zhang, X. (2002). Identification of gene expression profile of dorsal root ganglion in the rat peripheral axotomy model of neuropathic pain. Proc. Natl. Acad. Sci. U.S.A. 99, 8360–8365.
Zhu, Q., and Julien, J. P. (1999). A key role for GAP-43 in the retinotectal topographic organization. Exp. Neurol. 155, 228–242.
Keywords: axon regeneration, sciatic nerve injury, gene regulatory network, gene therapy
Citation: van Kesteren RE, Mason MRJ, MacGillavry HD, Smit AB and Verhaagen J (2011) A gene network perspective on axonal regeneration. Front. Mol. Neurosci. 4:46. doi: 10.3389/fnmol.2011.00046
Received: 04 October 2011; Paper pending published: 16 October 2011;
Accepted: 02 November 2011; Published online: 22 November 2011.
Edited by:
Simone Di Giovanni, University of Tuebingen, GermanyReviewed by:
Darcie Leann Moore, ETH Zurich, SwitzerlandAndrea Tedeschi, Children’s Hospital Boston, USA
Copyright: © 2011 van Kesteren, Mason, MacGillavry, Smit and Verhaagen. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Ronald E. van Kesteren, Center for Neuroscience and Cognitive Research, Neuroscience Campus Amsterdam, VU University, De Boelelaan 1085, 1081HV Amsterdam, Netherlands. e-mail:cm9uYWxkLnZhbi5rZXN0ZXJlbkBjbmNyLnZ1Lm5s
†Present address: Harold D. MacGillavry, Department of Physiology, University of Maryland School of Medicine, 516 Howard Hall, 660 West Redwood Street, Baltimore, MD 21201, USA.