 Jayashree Karar1,2 and Amit Maity1,2*
Jayashree Karar1,2 and Amit Maity1,2*- 1 Department of Radiation Oncology, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA, USA
- 2 Abramson Cancer Center, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA, USA
The phosphatidylinositol 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) pathway is activated in the majority of human cancers. This pathway is known to play a key role in numerous cellular functions including proliferation, adhesion, migration, invasion, metabolism, and survival, but in the current review we focus on its role in angiogenesis. PI3K activation may occur via RAS mutation, loss of phosphatase and tensin homolog (PTEN), or by increased expression of growth factor receptors such as epidermal growth factor receptor. There is a connection between the PI3K pathway and angiogenesis. Hypoxia leads to HIF-1α stabilization and is a major stimulus for increased vascular endothelial growth factor (VEGF) production by tumor cells. However, activation of the PI3K/AKT pathway in tumor cells can also increase VEGF secretion, both by hypoxia-inducible factor 1 (HIF-1) dependent and independent mechanisms. The PI3K/AKT pathway also modulates the expression of other angiogenic factors such as nitric oxide and angiopoietins. Numerous inhibitors targeting the PI3K/AKT/mTOR pathway have been developed, and these agents have been shown to decrease VEGF secretion and angiogenesis. The effect of these inhibitors on tumor vasculature can be difficult to predict. The vasculature of tumors is aberrant, leading to sluggish bloodflow and elevated interstitial blood pressure, which can be perpetuated by the high levels of VEGF. Hence, decreasing VEGF expression can paradoxically lead to vascular normalization and improved bloodflow in some tumors. In addition to its importance in cancer, the PI3K pathway also plays an essential role in the formation of normal blood vessels during development. Embryos with kinase-dead p110α catalytic subunit of PI3K develop vascular defects. Stimulation of endothelial cells by VEGF leads to activation of the PI3K pathway within these cells, which is important for cell migration. Sustained endothelial activation of AKT1 has been shown to induce the formation of structurally abnormal blood vessels that recapitulate the aberrations of tumor vessels. Hence, the PI3K pathway plays an important role in regulating angiogenesis both in normal tissues and in cancers.
Introduction
The PI3K/AKT pathway is important not only in the development of cancers but also for signaling in normal cells. This pathway is known to play a key role in numerous cellular functions including proliferation, adhesion, migration, invasion, metabolism, and survival (Bader et al., 2005), but in the current review we focus on its role in angiogenesis. Phosphatidylinositol 3-kinases (PI3K) are a family of enzymes that phosphorylate the 3′-OH of the inositol ring of phosphatidylinositol. There are three classes of PI3K: I, II, and III. Class I PI3Ks are heterodimers composed of a catalytic and a regulatory subunit and are further subdivided into two subclasses: IA and IB. Class II PI3Ks consists of three members, P13KC2α, Pl3KC2β, and P13KC2γ, while Class III contains only one member encoded by the gene VpS34 (Rodriguez-Viciana et al., 1994; Toker and Cantley, 1997; Fruman et al., 1998). Phosphatidylinositol 3,4,5-triphosphate (PIP3) is an important lipid second messenger generated by PI3K, which plays a vital role in several signal transduction pathways (Whitman et al., 1988; Toker and Cantley, 1997). PIP3 activates the serine/threonine kinases PDKl and AKT. The phosphatase and tensin homolog (PTEN) gene encodes a phosphatase that opposes the action of PI3K, thereby reducing the level of activated AKT. AKT controls protein synthesis and cell growth by leading to the phosphorylation of mammalian target of rapamycin (mTOR).
Vascular endothelial growth factor (VEGF) is a potent angiogenic factor whose effects on endothelial cells are mediated in part by the PI3K pathway. The mammalian VEGF family is comprised of five members: VEGFA, VEGFB, VEGFC, VEGFD, and placenta growth factor (PGF), the best characterized of which is VEGFA, commonly referred to as simply VEGF. A number of proteins structurally related to VEGF have also been discovered encoded by parapoxviruses (VEGFE) and in snake venom (VEGFF). The VEGF ligands can bind to three type III receptor tyrosine kinases: VEGFRl/FLT1, VEGFR2/KDR, and VEGFR3/FLT4, of which VEGFR2 has the most important role in VEGF-induced angiogenesis (Ellis and Hicklin, 2008). VEGFA, B and PGF can bind to VEGFR1. VEGFA and E bind to VEGFR2, while VEGFC and D bind to VEGFR3 (Olsson et al., 2006).
Tumor Vasculature and Regulation of VEGF by Hypoxia
The vasculature of tumors is aberrant, resulting in heterogeneity in the tumor microenvironment with variation in oxygen concentration, nutritional factors, and metabolic waste products (Vaupel et al., 1989). Tumor angiogenesis results from an imbalance between pro-angiogenic factors such as VEGF and endogenous anti-angiogenic factors such as angiostatin and endostatin (Folkman, 2002; Nieder et al., 2006). Tumor cells vary in their angiogenic potential, and angiogenic tumor cells generally secrete more bFGF and VEGF than non-angiogenic ones (Naumov et al., 2006). Elevated levels of VEGF can increase vascular permeability, leading to leaky vessels, sluggish bloodflow, and elevated interstitial pressure.
One of the most potent stimuli for increased VEGF production by tumor cells is hypoxia, which commonly occurs in tumors. Hypoxia leads to the induction of hypoxia-inducible factor 1 (HIF-1), a bHLH–PAS transcription factor that consists of both an α and a β subunit (Ratcliffe et al., 2000; Semenza, 2003). Decreased pO2 increases HIF-1α stability through post-translational modification of the protein. Once stabilized, the α subunit binds to the β subunit, which is constitutively present. This heterodimer then binds to numerous promoters containing hypoxia response elements (HREs), leading to the transcription of dozens of target genes including VEGF (Forsythe et al., 1996). HIF-1α is upregulated across a broad range of cancers, and HIF regulates key features of tumor biology such as angiogenesis, invasion, and glucose metabolism (Ratcliffe et al., 2000; Semenza, 2003).
Studies have shown that binding of both STAT3 and HIF-1α to the VEGF promoter is essential for maximum transcription of VEGF mRNA under hypoxia (Gray et al., 2005). STAT3 signaling is required for VEGF and PI3K/AKT mediated HIF-1α expression. Blocking STAT3 by the small molecule inhibitor, CPA-7 or STAT3 siRNA abolished both HIF-1 and VEGF expression (Xu et al., 2005). EGF-induced STAT3 binding to the VEGF promoter can be blocked by the PI3K inhibitor, LY294002, or STAT3 siRNA in colorectal cancer cells (Cascio et al., 2009).
EGFR/PI3K/mTOR Pathway, HIF, and VEGF
While hypoxia is the primary stimulus for HIF-1α upregulation, activation of the epidermal growth factor receptor (EGFR), and the PI3K pathway can also contribute to increased HIF-1α (Figure 1). EGFR is a transmembrane receptor tyrosine kinase that belongs to the HER family of receptors. It is overexpressed and activated in a variety of cancers and provides an attractive target for anti-cancer therapy (Dutta and Maity, 2007). Zhong et al. (2000) were among the first to show that activation of the EGFR/PI3K/AKT/mTOR pathway could increase VEGF expression by upregulating HIF-1α. PI3K/mTOR pathway activation increases HIF-1α protein levels without altering HIF-1α mRNA levels (Jiang et al., 2001), presumably by increasing HIF-1α translation (Laughner et al., 2001).
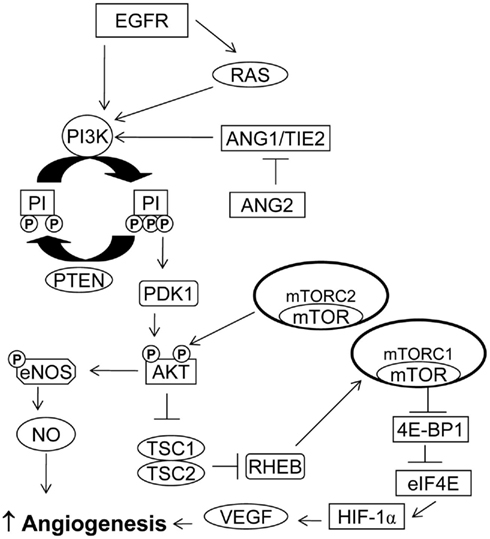
Figure 1. PI3K/AKT/mTOR pathway in angiogenesis. PI3K activation may occur via RAS mutation, by increased expression of growth factor receptors such as EGFR or by loss of PTEN. Activation of the PI3K/AKT/mTOR pathway can increase VEGF secretion. The PI3K/AKT pathway also modulates the expression of other angiogenic factors such as nitric oxide and angiopoietins.
Work from our own lab has shown that in U87MG human glioblastoma cells, EGFR activation can also increase VEGF expression by transcriptional activation of the VEGF promoter via a PI3K dependent pathway that is independent of HIF-1 (Maity et al., 2000). We have also shown that EGFR amplification and PTEN mutation had an additive effect in increasing the VEGF promoter activity in human glioblastoma cells (Pore et al., 2003).
Pharmacological inhibition of EGFR can decrease VEGF expression and reduce angiogenesis in many tumor types (Perrotte et al., 1999; Ciardiello et al., 2001; Huang et al., 2002; Pore et al., 2006b). One of the initial reports examining EGFR inhibitors found that the anti-EGFR antibody C225 could decrease not only levels of VEGF mRNA and protein but that of other angiogenic factors such as interleukin 8 and bFGF (Petit et al., 1997; Perrotte et al., 1999). One report showed that cells could acquire resistance to this antibody by selection of tumor cell subpopulations with increased angiogenic potential (Viloria-Petit et al., 2001). The consequences of decreasing VEGF by blocking EGFR signaling may be complicated. We have also shown that targeting tumor cells with the EGFR inhibitor erlotinib may improve vascular flow by causing vascular normalization, hence leading to improved response to chemotherapy and radiation (Cerniglia et al., 2009).
Epidermal growth factor receptor activation may have effects on VEGF expression independent of PI3K. A recent study exploring the role of MAPK and PI3K signaling in angiogenesis in squamous cell carcinoma of the head and neck found that EGFR activation induces VEGFA expression that requires both PI3K and MAPK signaling whereas VEGFC expression is dependent on MAPK, and not the PI3K–mTOR pathway (Luangdilok et al., 2011).
Another common mechanism of PI3K activation within tumors is through loss of PTEN which in glioblastomas results in increased VEGF expression mediated by HIF-1α stabilization (Zundel et al., 2000). PTEN loss is also seen in advanced prostate cancers. PTEN siRNA transfected pancreatic cancer cells have higher levels of secreted VEGF and are highly proliferative (Ma et al., 2009). Overexpression of wildtype PTEN in PC3 prostate cancer cells, which have PTEN mutations, inhibits angiogenesis, and tumor growth (Fang et al., 2007). A recent study exploring the role of PTEN in hepatocellular carcinoma also found similar inhibition of angiogenesis. Interestingly, this study also suggested that PTEN regulates angiogenesis through both phosphatase-dependent and independent mechanisms (Tian et al., 2010).
RAS Signaling and VEGF
The RAS family of GTPases (H-RAS, N-RAS, and K-RAS) plays an important role in various cellular functions, including proliferation, differentiation, and apoptosis. Mutations in H-RAS, K-RAS, and N-RAS convert them into oncogenes, which commonly occur in various cancers (Bos, 1989). Expression of mutant K-RAS or H-RAS has been associated with increased VEGF expression (Grugel et al., 1995; Rak et al., 1995). Mazure et al. (1997) showed that induction of VEGF by hypoxia is modulated by a PI3K/AKT pathway in H-RAS transformed cells through a HIF-1 transcriptional element. Introduction of oncogenic H-RAS into immortalized endothelial cells increases their angiogenic potential and results in increased VEGF expression. This angiogenic potential could be only partially inhibited by PI3K inhibition, indicating that RAS mediated angiogenesis through a PI3K-independent mechanism as well (Arbiser et al., 1997). H-RAS was found to play an important role in angiogenesis in skin carcinogenesis. In this model, H-RAS was responsible not only for VEGF induction, but also for inducing the expression of other angiogenic factors such as PGF and angiopoietin-2 (Larcher et al., 1996, 2003). Work from our lab has shown that H-RAS upregulates Glut1 promoter activity, at least in part, by increasing HIF-1α protein and that this involves the PI3K pathway (Chen et al., 2001).
Role of PI3K in Normal Development and Endothelial Cells
The PI3K pathway also plays an essential role in the formation of normal blood vessels during development. Embryos with kinase-dead p110α catalytic subunit of PI3K develop gross vascular defects, indicating that this subunit is essential for endothelial cell migration and angiogenesis (Graupera et al., 2008). The same study showed that p110α controls endothelial cell migration by positively regulating RhoA in endothelial cells. The p110α subunit also regulates barrier function and junctional morphology of endothelial cells. Only the p110α subunit decreased TNF-induced endothelial cell permeability, Tyr phosphorylation of the adherens junction protein vascular endothelial cadherin, and leukocyte transendothelial migration (Cain et al., 2010).
When VEGF binds to its receptor on normal endothelial cells, the RAS and PI3K pathways are activated. One study tried to identify vascular signaling in response to H-RAS and its immediate effectors by activating either the ERK/MAPK or PI3K pathways in primary endothelial tissues (Serban et al., 2008). Constitutive activation of the ERK/MAPK pathway could induce angiogenesis but not vascular permeability, while activation of PI3K was required for both angiogenesis and vascular permeability. Pharmacological inhibition of PI3K (α/β) suppressed both RAS or VEGF mediated vascular response in vivo and survival of primary endothelial cells in vitro. However, inhibition of PI3K (γ/δ) suppressed RAS or VEGF mediated vascular permeability in vivo, with no effect on the survival of primary endothelial cells.
Phosphatase and tensin homolog, a phosphatase that negatively regulates PI3K, has been found to be indispensable for normal cardiovascular homeostasis. PTEN-deficient endothelial cells display increased angiogenesis and tumorigenesis (Hamada et al., 2005). The PDKl kinase is an essential player in the PI3K pathway, phosphorylating AKT on the T308 residue. PDKl is vital to endothelial cell migration in response to VEGF stimulation in a PI3K-dependent manner (Primo et al., 2007). The study also identified that phosphorylation at T308 is more important than at S473.
A recent study found that attenuating the expression of RACKl, a ubiquitously expressed scaffolding protein, with siRNA suppressed the VEGF/PI3K/AKT driven migration of endothelial cells (Wang et al., 2011). Survivin, an anti-apoptotic protein, is also regulated by the PI3K pathway (Martinelli et al., 2006; Zhao et al., 2010) and is upregulated in endothelial cells in response to VEGF (Tran et al., 1999). Lack of survivin in endothelial cells has been shown to result in embryonic defects in angiogenesis (Zwerts et al., 2007). VEGF and survivin can act as chemoprotectants in endothelial cells, protecting them from drug-induced dell death. This chemoprotective role requires an active PI3K/AKT pathway (Tran et al., 2002).
Sustained endothelia1 activation of AKTl has been shown to induce the formation of structurally and functionally abnormal blood vessels that recapitulate the aberrations of tumor vessels (Phung et al., 2006). This study also showed that inhibition of AKT signaling by rapamycin could inhibit the pathological vascularization. In a recent study, sustained RAS activation in endothelial cells bypassed senescence and resulted in abnormal vascular morphogenesis, an effect that was regulated by PI3K signaling (Bajaj et al., 2010).
Ephrins and their receptor tyrosine kinases play significant roles in various processes such as vascular development, cell migration, and angiogenesis. There are two classes of Eph receptors, A and B. Class A receptors usually bind to GPI anchored ephrin A ligands, and class B receptors bind to transmembrane-linked ephrin B ligands. Binding of ephrin to the Eph receptor results in bidirectional signaling between the receptor-carrying cell and the ligand-producing cell (Pitulescu and Adams, 2010). Ephrin B2 controls the internalization of VEGFR2 and VEGFR3, a process that is crucial for the growth of blood vessels (Sawamiphak et al., 2010; Wang et al., 2010). In the absence of ligands, EphA2 receptor can be phosphorylated by AKT, promoting cell migration while in the presence of Ephrin A1, both AKT and the EphA2 receptor are dephosphorylated, preventing cell migration (Miao et al., 2009). Slit and Robo proteins are regulators of vascular remodeling. Slit 2 protein can promote angiogenesis via mTORC2-dependent activation of AKT and RAC. However, this process is inhibited in the presence of Ephrin A1, suggesting that the signaling between Ephrin A1 and Slit can regulate angiogenesis and vascular homeostasis (Dunaway et al., 2011).
PI3K/AKT and Nitric Oxide
The PI3K/AKT pathway has also been implicated in angiogenesis through its regulation of nitric oxide (NO) signaling in endothelial cells. Nitric oxide synthesis is regulated by the enzyme NO synthase (NOS). There are three isoforms of NOS, of which nNOS/NOS1 and eNOS/NOS3 are constitutively expressed, while expression of the third isoform, iNOS/NOS2, is inducible. Tumor cells mostly express iNOS, but some tumor types also express eNOS and nNOS. Tumor vascular endothelial cells mostly express eNOS, while the tumor associated stromal fibroblasts and immune cells express iNOS (Fukumura et al., 2006).
Studies using eNOS−/− and iNOS−/− mice have shown that eNOS plays a key role in VEGF-induced angiogenesis and vascular permeability (Fukumura et al., 2001). VEGF stimulation of endothelial cells is required for the migration of these cells and the formation of capillary-like structures in a PI3K–AKT dependent manner (Morales-Ruiz et al., 2000). Earlier studies have shown that VEGF can induce NO production, which is attenuated by PI3K inhibitors (Papapetropoulos et al., 1997). This regulation may occur via phosphorylation of eNOS at the serine 1177 residue by AKT (Dimmeler et al., 1999; Fulton et al., 1999). This phosphorylation is required for VEGF-induced endothelial cell migration (Dimmeler et al., 2000). Hypoxia can also increase eNOS phosphorylation via HSP9O binding to eNOS and activation of the PI3K/AKT pathway (Chen and Meyrick, 2004). Another link between the NO pathway and angiogenesis is suggested by the fact that NO donors can increase the expression and transcriptional activity of HIF-1, thus resulting in the induction of VEGF mRNA (Kasuno et al., 2004). The same study reported that targeting the PI3K pathway using LY294002 or rapamycin inhibited the expression of HIF-1 dependent reporter gene induced by the NO donor.
Angiopoietins
In addition to VEGF, another class of growth factors having angiogenic activity includes the angiopoietins and their receptors, especially the ANGl/TIE2 system. Angiopoietin 1 and 2 (ANG1 and 2) bind to TIE2 receptor tyrosine kinase, which is expressed primarily on endothelial cells. While ANGl is required for endothelial development, ANG2 is an antagonist of the ANGl/TIE2 system and can disrupt blood vessel formation (Davis et al., 1996; Maisonpierre et al., 1997). However, some reports also suggest that ANG2 can be pro-angiogenic in some circumstances. ANG2 can promote proliferation and migration of endothelial cells, sprouting, and neovascularization in the presence of VEGF (Asahara et al., 1998; Lobov et al., 2002). A study using ANGl or ANG2 inhibitors reported that while ANG1 inhibition had little effect on the tumor vasculature, ANG2 inhibition resulted in decreased tumor vascularity and allowed for unrestricted ANGl activity, leading to vessel normalization. The ANGl–ANG2 inhibitor combination did not affect the ANG2-mediated inhibition of vasculature but prevented tumor vessel normalization (Falcon et al., 2009).
ANG1 can promote endothelial cell survival through activation of the PI3K pathway (Kim et al., 2000a), and similar results could be achieved with higher concentrations of ANG2 (Kim et al., 2000b). A recent study by Yuan et al. (2009) showed that ANG2 can act as an agonist of the TIE2 receptor in the absence of ANGl and can activate the PI3K–AKT pathway, although with weaker potency than ANGl. ANGl has been shown to stimulate TIE2, AKT, and eNOS phosphorylation in HUVECs in a PI3K dependent manner (Babaei et al., 2003). Another study reported that HSP90 binding to eNOS and activation of the PI3K pathway contribute to ANGl induced eNOS phosphorylation, NO production, and angiogenesis in coronary artery endothelial cells (Chen et al., 2004). Mice lacking the p110α catalytic subunit of PI3K display vascular defects along with reduced TIE2 protein levels (Lelievre et al., 2005). It has been shown that association between p85 subunit of PI3K and TIE2 is essential for PI3K activity (Kontos et al., 1998). ANGl can induce localization of TIE2 to endothelial cell–cell contacts, however in isolated cells TIE2 localizes to cell–substratum contacts (Fukuhara et al., 2008; Saharinen et al., 2008). TIE2 activation at cell–cell contacts leads to the activation of AKT, which results in phosphorylation of eNOS and inhibition of FOXO1 (Fukuhara et al., 2008). FOXO1 regulates the expression of genes involved in apoptosis such as survivin and also regulates the expression of ANG2 (ANG1 antagonist) in endothelial cells, suggesting the importance of ANGl in maintaining blood vessel stability (Daly et al., 2004). The ANG1–TIE2 association also regulates the expression of D114, a Notch ligand involved in vascular stability in a β-catenin dependent manner. PI3K–AKT inhibits GSK3β leading to β-catenin stability and this potentiates Notch signal-mediated D114 expression (Zhang et al., 2011).
PI3K/AKT/mTOR Inhibitors
Inhibitors targeting the PI3K/AKT pathway have been developed and, as predicted, these agents can decrease VEGF secretion and angiogenesis. The traditional PI3K/AKT inhibitors LY294002 and wortmannin exhibit anti-angiogenic activity. However, LY294002 and wortmannin are unsuitable for human use due to toxicity. Newer PI3K inhibitors including SF1126 (Garlich et al., 2008), ZSTK474 (Kong et al., 2009), and PI103 (Raynaud et al., 2007) have displayed anti-angiogenic activity in xenograft models. The Novartis drug NVP-BEZ235, which is a dual PI3K and mTOR inhibitor (Maira et al., 2008), blocked VEGF-induced neovascularization in mice (Schnell et al., 2008), and inhibited the growth and proliferation of cancer cells with wild type and p110α mutations (Serra et al., 2008). We have previously shown that nelfinavir, a HIV protease inhibitor can also inhibit AKT signaling and decrease VEGF expression. Nelfinavir decreased the VEGF secretion under both normoxic and hypoxic conditions (Pore et al., 2006a).
Inhibitors are also available against mTOR. mTOR kinase can participate in two different complexes: mTORC1 (mTOR/Raptor) and mTORC2 (mTOR/Rictor). Rapamycin is an mTORC1 inhibitor, and treatment with this agent or its analogs induces insulin receptor substrate-1 expression, terminating the feedback inhibition of the pathway, and resulting in AKT activation (O’Reilly et al., 2006). However, with prolonged incubation, rapamycin may inhibit mTORC2 in certain cell types (Sarbassov et al., 2006). Rapamycin inhibits VEGF production and has antiproliferative and antitumor activity (Zhong et al., 2000; Guba et al., 2002). The drug has also been shown to inhibit angiogenesis in vivo (Guba et al., 2002). The TSC2–TSC1 protein complex negatively regulates mTOR. TSC2-null cells have high levels of HIF-1α and VEGF. Rapamycin treatment reduces HIF-1α levels but fails to reduce VEGF levels completely in these cells, indicating that TSC2 regulates VEGF through both mTOR-dependent and -independent pathways (Brugarolas et al., 2003). Treatment of myrAKT1 mice (with sustained AKT activation) with rapamycin has been shown to block blood vessel formation (Phung et al., 2006). The regulatory associated protein of mTOR (Raptor) has been shown to interact with HIF-1α via an mTOR signaling motif located in the N terminus of HIF-1α. HIF-1α lacking this motif had impaired activity under hypoxia and was unable to bind to the co-activator CBP/p300 (Land and Tee, 2007). The dual mTORC1/mTORC2 inhibitors, OSI-027, and OXA-01 have been shown to considerably reduce angiogenesis and regrowth compared to rapamycin (mTORC1 inhibitor) alone. Combining these dual inhibitors with VEGFR antagonists was even more efficient in reducing tumor growth (Falcon et al., 2011).
Summary
Activation of the PI3K/AKT/mTOR pathway in tumor cells can increase VEGF secretion by both HIF-1 dependent and independent mechanisms. This pathway can also regulate angiogenesis by modulating expression of nitric oxide and angiopoietins. Many agents have been developed that can inhibit PI3K and/or mTOR signaling in tumor cells, and these drugs have effects on angiogenesis as well as on tumor cell proliferation and survival. Not only is the PI3K/AKT/mTOR pathway commonly activated in tumor cells, but VEGF binding to receptors on endothelial cells stimulates this pathway which is essential for endothelial cell migration. For this reason the PI3K/AKT pathway is essential for normal blood vessel development during embryogenesis.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Arbiser, J. L., Moses, M. A., Fernandez, C. A., Ghiso, N., Cao, Y., Klauber, N., Frank, D., Brownlee, M., Flynn, E., Parangi, S., Byers, H. R., and Folkman, J. (1997). Oncogenic H-ras stimulates tumor angiogenesis by two distinct pathways. Proc. Natl. Acad. Sci. U.S.A. 94, 861–866.
Asahara, T., Chen, D., Takahashi, T., Fujikawa, K., Kearney, M., Magner, M., Yancopoulos, G. D., and Isner, J. M. (1998). Tie2 receptor ligands, angiopoietin-1 and angiopoietin-2, modulate VEGF-induced postnatal neovascularization. Circ. Res. 83, 233–240.
Babaei, S., Teichert-Kuliszewska, K., Zhang, Q., Jones, N., Dumont, D. J., and Stewart, D. J. (2003). Angiogenic actions of angiopoietin-1 require endothelium-derived nitric oxide. Am. J. Pathol. 162, 1927–1936.
Bader, A. G., Kang, S., Zhao, L., and Vogt, P. K. (2005). Oncogenic PI3K deregulates transcription and translation. Nat. Rev. Cancer 5, 921–929.
Bajaj, A., Zheng, Q., Adam, A., Vincent, P., and Pumiglia, K. (2010). Activation of endothelial ras signaling bypasses senescence and causes abnormal vascular morphogenesis. Cancer Res. 70, 3803–3812.
Brugarolas, J. B., Vazquez, F., Reddy, A., Sellers, W. R., and Kaelin, W. G. Jr. (2003). TSC2 regulates VEGF through mTOR-dependent and -independent pathways. Cancer Cell 4, 147–158.
Cain, R. J., Vanhaesebroeck, B., and Ridley, A. J. (2010). The PI3K p110alpha isoform regulates endothelial adherens junctions via Pyk2 and Rac1. J. Cell Biol. 188, 863–876.
Cascio, S., Ferla, R., D’Andrea, A., Gerbino, A., Bazan, V., Surmacz, E., and Russo, A. (2009). Expression of angiogenic regulators, VEGF and leptin, is regulated by the EGF/PI3K/STAT3 pathway in colorectal cancer cells. J. Cell. Physiol. 221, 189–194.
Cerniglia, G. J., Pore, N., Tsai, J. H., Schultz, S., Mick, R., Choe, R., Xing, X., Durduran, T., Yodh, A. G., Evans, S. M., Koch, C. J., Hahn, S. M., Quon, H., Sehgal, C. M., Lee, W. M., and Maity, A. (2009). Epidermal growth factor receptor inhibition modulates the microenvironment by vascular normalization to improve chemotherapy and radiotherapy efficacy. PLoS ONE 4, e6539. doi:10.1371/journal.pone.0006539
Chen, C., Pore, N., Behrooz, A., Ismail-Beigi, F., and Maity, A. (2001). Regulation of glut1 mRNA by hypoxia-inducible factor-1. Interaction between H-ras and hypoxia. J. Biol. Chem. 276, 9519–9525.
Chen, J. X., Lawrence, M. L., Cunningham, G., Christman, B. W., and Meyrick, B. (2004). HSP90 and Akt modulate Ang-1-induced angiogenesis via NO in coronary artery endothelium. J. Appl. Physiol. 96, 612–620.
Chen, J. X., and Meyrick, B. (2004). Hypoxia increases Hsp90 binding to eNOS via PI3K-Akt in porcine coronary artery endothelium. Lab. Invest. 84, 182–190.
Ciardiello, F., Caputo, R., Bianco, R., Damiano, V., Fontanini, G., Cuccato, S., De, P. S., Bianco, A. R., and Tortora, G. (2001). Inhibition of growth factor production and angiogenesis in human cancer cells by ZD1839 (Iressa), a selective epidermal growth factor receptor tyrosine kinase inhibitor. Clin. Cancer Res. 7, 1459–1465.
Daly, C., Wong, V., Burova, E., Wei, Y., Zabski, S., Griffiths, J., Lai, K. M., Lin, H. C., Ioffe, E., Yancopoulos, G. D., and Rudge, J. S. (2004). Angiopoietin-1 modulates endothelial cell function and gene expression via the transcription factor FKHR (FOXO1). Genes Dev. 18, 1060–1071.
Davis, S., Aldrich, T. H., Jones, P. F., Acheson, A., Compton, D. L., Jain, V., Ryan, T. E., Bruno, J., Radziejewski, C., Maisonpierre, P. C., and Yancopoulos, G. D. (1996). Isolation of angiopoietin-1, a ligand for the TIE2 receptor, by secretion-trap expression cloning. Cell 87, 1161–1169.
Dimmeler, S., Dernbach, E., and Zeiher, A. M. (2000). Phosphorylation of the endothelial nitric oxide synthase at ser-1177 is required for VEGF-induced endothelial cell migration. FEBS Lett. 477, 258–262.
Dimmeler, S., Fleming, I., Fisslthaler, B., Hermann, C., Busse, R., and Zeiher, A. M. (1999). Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 399, 601–605.
Dunaway, C. M., Hwang, Y., Lindsley, C. W., Cook, R. S., Wu, J. Y., Boothby, M., Chen, J., and Brantley-Sieders, D. M. (2011). Cooperative signaling between Slit2 and Ephrin-A1 regulates a balance between angiogenesis and angiostasis. Mol. Cell. Biol. 31, 404–416.
Dutta, P. R., and Maity, A. (2007). Cellular responses to EGFR inhibitors and their relevance to cancer therapy. Cancer Lett. 254, 165–177.
Ellis, L. M., and Hicklin, D. J. (2008). VEGF-targeted therapy: mechanisms of anti-tumour activity. Nat. Rev. Cancer 8, 579–591.
Falcon, B. L., Barr, S., Gokhale, P. C., Chou, J., Fogarty, J., Depeille, P., Miglarese, M., Epstein, D. M., and McDonald, D. M. (2011). Reduced VEGF production, angiogenesis, and vascular regrowth contribute to the antitumor properties of dual mTORC1/mTORC2 inhibitors. Cancer Res. 71, 1573–1583.
Falcon, B. L., Hashizume, H., Koumoutsakos, P., Chou, J., Bready, J. V., Coxon, A., Oliner, J. D., and McDonald, D. M. (2009). Contrasting actions of selective inhibitors of angiopoietin-1 and angiopoietin-2 on the normalization of tumor blood vessels. Am. J. Pathol. 175, 2159–2170.
Fang, J., Ding, M., Yang, L., Liu, L. Z., and Jiang, B. H. (2007). PI3K/PTEN/AKT signaling regulates prostate tumor angiogenesis. Cell. Signal. 19, 2487–2497.
Forsythe, J. A., Jiang, B. H., Iyer, N. V., Agani, F., Leung, S. W., Koos, R. D., and Semenza, G. L. (1996). Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 16, 4604–4613.
Fruman, D. A., Meyers, R. E., and Cantley, L. C. (1998). Phosphoinositide kinases. Annu. Rev. Biochem. 67, 481–507.
Fukuhara, S., Sako, K., Minami, T., Noda, K., Kim, H. Z., Kodama, T., Shibuya, M., Takakura, N., Koh, G. Y., and Mochizuki, N. (2008). Differential function of Tie2 at cell-cell contacts and cell-substratum contacts regulated by angiopoietin-1. Nat. Cell Biol. 10, 513–526.
Fukumura, D., Gohongi, T., Kadambi, A., Izumi, Y., Ang, J., Yun, C. O., Buerk, D. G., Huang, P. L., and Jain, R. K. (2001). Predominant role of endothelial nitric oxide synthase in vascular endothelial growth factor-induced angiogenesis and vascular permeability. Proc. Natl. Acad. Sci. U.S.A. 98, 2604–2609.
Fukumura, D., Kashiwagi, S., and Jain, R. K. (2006). The role of nitric oxide in tumour progression. Nat. Rev. Cancer 6, 521–534.
Fulton, D., Gratton, J. P., McCabe, T. J., Fontana, J., Fujio, Y., Walsh, K., Franke, T. F., Papapetropoulos, A., and Sessa, W. C. (1999). Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature 399, 597–601.
Garlich, J. R., De, P., Dey, N., Su, J. D., Peng, X., Miller, A., Murali, R., Lu, Y., Mills, G. B., Kundra, V., Shu, H. K., Peng, Q., and Durden, D. L. (2008). A vascular targeted pan phosphoinositide 3-kinase inhibitor prodrug, SF1126, with antitumor and antiangiogenic activity. Cancer Res. 68, 206–215.
Graupera, M., Guillermet-Guibert, J., Foukas, L. C., Phng, L. K., Cain, R. J., Salpekar, A., Pearce, W., Meek, S., Millan, J., Cutillas, P. R., Smith, A. J., Ridley, A. J., Ruhrberg, C., Gerhardt, H., and Vanhaesebroeck, B. (2008). Angiogenesis selectively requires the p110alpha isoform of PI3K to control endothelial cell migration. Nature 453, 662–666.
Gray, M. J., Zhang, J., Ellis, L. M., Semenza, G. L., Evans, D. B., Watowich, S. S., and Gallick, G. E. (2005). HIF-1alpha, STAT3, CBP/p300 and Ref-1/APE are components of a transcriptional complex that regulates Src-dependent hypoxia-induced expression of VEGF in pancreatic and prostate carcinomas. Oncogene 24, 3110–3120.
Grugel, S., Finkenzeller, G., Weindel, K., Barleon, B., and Marme, D. (1995). Both v-Ha-Ras and v-Raf stimulate expression of the vascular endothelial growth factor in NIH 3T3 cells. J. Biol. Chem. 270, 25915–25919.
Guba, M., von Breitenbuch, P., Steinbauer, M., Koehl, G., Flegel, S., Hornung, M., Bruns, C. J., Zuelke, C., Farkas, S., Anthuber, M., Jauch, K. W., and Geissler, E. K. (2002). Rapamycin inhibits primary and metastatic tumor growth by antiangiogenesis: involvement of vascular endothelial growth factor. Nat. Med. 8, 128–135.
Hamada, K., Sasaki, T., Koni, P. A., Natsui, M., Kishimoto, H., Sasaki, J., Yajima, N., Horie, Y., Hasegawa, G., Naito, M., Miyazaki, J., Suda, T., Itoh, H., Nakao, K., Mak, T. W., Nakano, T., and Suzuki, A. (2005). The PTEN/PI3K pathway governs normal vascular development and tumor angiogenesis. Genes Dev. 19, 2054–2065.
Huang, S. M., Li, J., Armstrong, E. A., and Harari, P. M. (2002). Modulation of radiation response and tumor-induced angiogenesis after epidermal growth factor receptor inhibition by ZD1839 (Iressa). Cancer Res. 62, 4300–4306.
Jiang, B. H., Jiang, G., Zheng, J. Z., Lu, Z., Hunter, T., and Vogt, P. K. (2001). Phosphatidylinositol 3-kinase signaling controls levels of hypoxia-inducible factor 1. Cell Growth Differ. 12, 363–369.
Kasuno, K., Takabuchi, S., Fukuda, K., Kizaka-Kondoh, S., Yodoi, J., Adachi, T., Semenza, G. L., and Hirota, K. (2004). Nitric oxide induces hypoxia-inducible factor 1 activation that is dependent on MAPK and phosphatidylinositol 3-kinase signaling. J. Biol. Chem. 279, 2550–2558.
Kim, I., Kim, H. G., So, J. N., Kim, J. H., Kwak, H. J., and Koh, G. Y. (2000a). Angiopoietin-1 regulates endothelial cell survival through the phosphatidylinositol 3′-Kinase/Akt signal transduction pathway. Circ. Res. 86, 24–29.
Kim, I., Kim, J. H., Moon, S. O., Kwak, H. J., Kim, N. G., and Koh, G. Y. (2000b). Angiopoietin-2 at high concentration can enhance endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway. Oncogene 19, 4549–4552.
Kong, D., Okamura, M., Yoshimi, H., and Yamori, T. (2009). Antiangiogenic effect of ZSTK474, a novel phosphatidylinositol 3-kinase inhibitor. Eur. J. Cancer 45, 857–865.
Kontos, C. D., Stauffer, T. P., Yang, W. P., York, J. D., Huang, L., Blanar, M. A., Meyer, T., and Peters, K. G. (1998). Tyrosine 1101 of Tie2 is the major site of association of p85 and is required for activation of phosphatidylinositol 3-kinase and Akt. Mol. Cell. Biol. 18, 4131–4140.
Land, S. C., and Tee, A. R. (2007). Hypoxia-inducible factor 1alpha is regulated by the mammalian target of rapamycin (mTOR) via an mTOR signaling motif. J. Biol. Chem. 282, 20534–20543.
Larcher, F., Franco, M., Bolontrade, M., Rodriguez-Puebla, M., Casanova, L., Navarro, M., Yancopoulos, G., Jorcano, J. L., and Conti, C. J. (2003). Modulation of the angiogenesis response through Ha-ras control, placenta growth factor, and angiopoietin expression in mouse skin carcinogenesis. Mol. Carcinog. 37, 83–90.
Larcher, F., Robles, A. I., Duran, H., Murillas, R., Quintanilla, M., Cano, A., Conti, C. J., and Jorcano, J. L. (1996). Up-regulation of vascular endothelial growth factor/vascular permeability factor in mouse skin carcinogenesis correlates with malignant progression state and activated H-ras expression levels. Cancer Res. 56, 5391–5396.
Laughner, E., Taghavi, P., Chiles, K., Mahon, P. C., and Semenza, G. L. (2001). HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1alpha (HIF-1alpha) synthesis: novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol. Cell. Biol. 21, 3995–4004.
Lelievre, E., Bourbon, P. M., Duan, L. J., Nussbaum, R. L., and Fong, G. H. (2005). Deficiency in the p110alpha subunit of PI3K results in diminished Tie2 expression and Tie2(-/-)-like vascular defects in mice. Blood 105, 3935–3938.
Lobov, I. B., Brooks, P. C., and Lang, R. A. (2002). Angiopoietin-2 displays VEGF-dependent modulation of capillary structure and endothelial cell survival in vivo. Proc. Natl. Acad. Sci. U.S.A. 99, 11205–11210.
Luangdilok, S., Box, C., Harrington, K., Rhys-Evans, P., and Eccles, S. (2011). MAPK and PI3K signalling differentially regulate angiogenic and lymphangiogenic cytokine secretion in squamous cell carcinoma of the head and neck. Eur. J. Cancer 47, 520–529.
Ma, J., Sawai, H., Ochi, N., Matsuo, Y., Xu, D., Yasuda, A., Takahashi, H., Wakasugi, T., and Takeyama, H. (2009). PTEN regulates angiogenesis through PI3K/Akt/VEGF signaling pathway in human pancreatic cancer cells. Mol. Cell. Biochem. 331, 161–171.
Maira, S. M., Stauffer, F., Brueggen, J., Furet, P., Schnell, C., Fritsch, C., Brachmann, S., Chène, P., De Pover, A., Schoemaker, K., Fabbro, D., Gabriel, D., Simonen, M., Murphy, L., Finan, P., Sellers, W., and García-Echeverría, C. (2008). Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol. Cancer Ther. 7, 1851–1863.
Maisonpierre, P. C., Suri, C., Jones, P. F., Bartunkova, S., Wiegand, S. J., Radziejewski, C., Compton, D., McClain, J., Aldrich, T. H., Papadopoulos, N., Daly, T. J., Davis, S., Sato, T. N., and Yancopoulos, G. D. (1997). Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science 277, 55–60.
Maity, A., Pore, N., Lee, J., Solomon, D., and O’Rourke, D. M. (2000). Epidermal growth factor receptor transcriptionally up-regulates vascular endothelial growth factor expression in human glioblastoma cells via a pathway involving phosphatidylinositol 3′-kinase and distinct from that induced by hypoxia. Cancer Res. 60, 5879–5886.
Martinelli, S., Kostylina, G., Niggli, V., Baumann, C., Fey, M. F., Wendel, H. G., Lowe, S. W., Yousefi, S., and Simon, H. U. (2006). Targeting survivin via PI3K but not c-akt/PKB by anticancer drugs in immature neutrophils. Oncogene 25, 6915–6923.
Mazure, N. M., Chen, E. Y., Laderoute, K. R., and Giaccia, A. J. (1997). Induction of vascular endothelial growth factor by hypoxia is modulated by a phosphatidylinositol 3-kinase/Akt signaling pathway in Ha-ras-transformed cells through a hypoxia inducible factor-1 transcriptional element. Blood 90, 3322–3331.
Miao, H., Li, D. Q., Mukherjee, A., Guo, H., Petty, A., Cutter, J., Basilion, J. P., Sedor, J., Wu, J., Danielpour, D., Sloan, A. E., Cohen, M. L., and Wang, B. (2009). EphA2 mediates ligand-dependent inhibition and ligand-independent promotion of cell migration and invasion via a reciprocal regulatory loop with Akt. Cancer Cell 16, 9–20.
Morales-Ruiz, M., Fulton, D., Sowa, G., Languino, L. R., Fujio, Y., Walsh, K., and Sessa, W. C. (2000). Vascular endothelial growth factor-stimulated actin reorganization and migration of endothelial cells is regulated via the serine/threonine kinase Akt. Circ. Res. 86, 892–896.
Naumov, G. N., Bender, E., Zurakowski, D., Kang, S. Y., Sampson, D., Flynn, E., Watnick, R. S., Straume, O., Akslen, L. A., Folkman, J., and Almog, N. (2006). A model of human tumor dormancy: an angiogenic switch from the nonangiogenic phenotype. J. Natl. Cancer Inst. 98, 316–325.
Nieder, C., Wiedenmann, N., Andratschke, N., and Molls, M. (2006). Current status of angiogenesis inhibitors combined with radiation therapy. Cancer Treat. Rev. 32, 348–364.
Olsson, A. K., Dimberg, A., Kreuger, J., and Claesson-Welsh, L. (2006). VEGF receptor signalling - in control of vascular function. Nat. Rev. Mol. Cell. Biol. 7, 359–371.
O’Reilly, K. E., Rojo, F., She, Q. B., Solit, D., Mills, G. B., Smith, D., Lane, H., Hofmann, F., Hicklin, D. J., Ludwig, D. L., Baselga, J., and Rosen, N. (2006). mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 66, 1500–1508.
Papapetropoulos, A., Garcia-Cardena, G., Madri, J. A., and Sessa, W. C. (1997). Nitric oxide production contributes to the angiogenic properties of vascular endothelial growth factor in human endothelial cells. J. Clin. Invest. 100, 3131–3139.
Perrotte, P., Matsumoto, T., Inoue, K., Kuniyasu, H., Eve, B. Y., Hicklin, D. J., Radinsky, R., and Dinney, C. P. (1999). Anti-epidermal growth factor receptor antibody C225 inhibits angiogenesis in human transitional cell carcinoma growing orthotopically in nude mice. Clin. Cancer Res. 5, 257–265.
Petit, A. M., Rak, J., Hung, M. C., Rockwell, P., Goldstein, N., Fendly, B., and Kerbel, R. S. (1997). Neutralizing antibodies against epidermal growth factor and ErbB-2/neu receptor tyrosine kinases down-regulate vascular endothelial growth factor production by tumor cells in vitro and in vivo: angiogenic implications for signal transduction therapy of solid tumors. Am. J. Pathol. 151, 1523–1530.
Phung, T. L., Ziv, K., Dabydeen, D., Eyiah-Mensah, G., Riveros, M., Perruzzi, C., Sun, J., Monahan-Earley, R. A., Shiojima, I., Nagy, J. A., Lin, M. I., Walsh, K., Dvorak, A. M., Briscoe, D. M., Neeman, M., Sessa, W. C., Dvorak, H. F., and Benjamin, L. E. (2006). Pathological angiogenesis is induced by sustained Akt signaling and inhibited by rapamycin. Cancer Cell 10, 159–170.
Pitulescu, M. E., and Adams, R. H. (2010). Eph/ephrin molecules–a hub for signaling and endocytosis. Genes Dev. 24, 2480–2492.
Pore, N., Gupta, A. K., Cerniglia, G. J., and Maity, A. (2006a). HIV protease inhibitors decrease VEGF/HIF-1alpha expression and angiogenesis in glioblastoma cells. Neoplasia 8, 889–895.
Pore, N., Jiang, Z., Gupta, A., Cerniglia, G., Kao, G. D., and Maity, A. (2006b). EGFR tyrosine kinase inhibitors decrease VEGF expression by both hypoxia-inducible factor (HIF)-1-independent and HIF-1-dependent mechanisms. Cancer Res. 66, 3197–3204.
Pore, N., Liu, S., Haas-Kogan, D. A., O’Rourke, D. M., and Maity, A. (2003). PTEN mutation and epidermal growth factor receptor activation regulate vascular endothelial growth factor (VEGF) mRNA expression in human glioblastoma cells by transactivating the proximal VEGF promoter. Cancer Res. 63, 236–241.
Primo, L., di, B. L., Roca, C., Droetto, S., Piva, R., Schaffhausen, B., and Bussolino, F. (2007). Essential role of PDK1 in regulating endothelial cell migration. J. Cell Biol. 176, 1035–1047.
Rak, J., Mitsuhashi, Y., Bayko, L., Filmus, J., Shirasawa, S., Sasazuki, T., and Kerbel, R. S. (1995). Mutant ras oncogenes upregulate VEGF/VPF expression: implications for induction and inhibition of tumor angiogenesis. Cancer Res. 55, 4575–4580.
Ratcliffe, P. J., Pugh, C. W., and Maxwell, P. H. (2000). Targeting tumors through the HIF system. Nat. Med. 6, 1315–1316.
Raynaud, F. I., Eccles, S., Clarke, P. A., Hayes, A., Nutley, B., Alix, S., Henley, A., Di-Stefano, F., Ahmad, Z., Guillard, S., Bjerke, L. M., Kelland, L., Valenti, M., Patterson, L., Gowan, S., de Haven Brandon, A., Hayakawa, M., Kaizawa, H., Koizumi, T., Ohishi, T., Patel, S., Saghir, N., Parker, P., Waterfield, M., and Workman, P. (2007). Pharmacologic characterization of a potent inhibitor of class I phosphatidylinositide 3-kinases. Cancer Res. 67, 5840–5850.
Rodriguez-Viciana, P., Warne, P. H., Dhand, R., Vanhaesebroeck, B., Gout, I., Fry, M. J., Waterfield, M. D., and Downward, J. (1994). Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature 370, 527–532.
Saharinen, P., Eklund, L., Miettinen, J., Wirkkala, R., Anisimov, A., Winderlich, M., Nottebaum, A., Vestweber, D., Deutsch, U., Koh, G. Y., Olsen, B. R., and Alitalo, K. (2008). Angiopoietins assemble distinct Tie2 signalling complexes in endothelial cell-cell and cell-matrix contacts. Nat. Cell Biol. 10, 527–537.
Sarbassov, D. D., Ali, S. M., Sengupta, S., Sheen, J. H., Hsu, P. P., Bagley, A. F., Markhard, A. L., and Sabatini, D. M. (2006). Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 22, 159–168.
Sawamiphak, S., Seidel, S., Essmann, C. L., Wilkinson, G. A., Pitulescu, M. E., Acker, T., and Acker-Palmer, A. (2010). Ephrin-B2 regulates VEGFR2 function in developmental and tumour angiogenesis. Nature 465, 487–491.
Schnell, C. R., Stauffer, F., Allegrini, P. R., O’Reilly, T., McSheehy, P. M., Dartois, C., Stumm, M., Cozens, R., Littlewood-Evans, A., García-Echeverría, C., and Maira, S. M. (2008). Effects of the dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235 on the tumor vasculature: implications for clinical imaging. Cancer Res. 68, 6598–6607.
Serban, D., Leng, J., and Cheresh, D. (2008). H-ras regulates angiogenesis and vascular permeability by activation of distinct downstream effectors. Circ. Res. 102, 1350–1358.
Serra, V., Markman, B., Scaltriti, M., Eichhorn, P. J., Valero, V., Guzman, M., Botero, M. L., Llonch, E., Atzori, F., Di Cosimo, S., Maira, M., Garcia-Echeverria, C., Parra, J. L., Arribas, J., and Baselga, J. (2008). NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer Res. 68, 8022–8030.
Tian, T., Nan, K. J., Wang, S. H., Liang, X., Lu, C. X., Guo, H., Wang, W. J., and Ruan, Z. P. (2010). PTEN regulates angiogenesis and VEGF expression through phosphatase-dependent and -independent mechanisms in HepG2 cells. Carcinogenesis 31, 1211–1219.
Toker, A., and Cantley, L. C. (1997). Signalling through the lipid products of phosphoinositide-3-OH kinase. Nature 387, 673–676.
Tran, J., Master, Z., Yu, J. L., Rak, J., Dumont, D. J., and Kerbel, R. S. (2002). A role for survivin in chemoresistance of endothelial cells mediated by VEGF. Proc. Natl. Acad. Sci. U.S.A. 99, 4349–4354.
Tran, J., Rak, J., Sheehan, C., Saibil, S. D., LaCasse, E., Korneluk, R. G., and Kerbel, R. S. (1999). Marked induction of the IAP family antiapoptotic proteins survivin and XIAP by VEGF in vascular endothelial cells. Biochem. Biophys. Res. Commun. 264, 781–788.
Vaupel, P., Kallinowski, F., and Okunieff, P. (1989). Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: a review. Cancer Res. 49, 6449–6465.
Viloria-Petit, A., Crombet, T., Jothy, S., Hicklin, D., Bohlen, P., Schlaeppi, J. M., Rak, J., and Kerbel, R. S. (2001). Acquired resistance to the antitumor effect of epidermal growth factor receptor-blocking antibodies in vivo: a role for altered tumor angiogenesis. Cancer Res. 61, 5090–5101.
Wang, F., Yamauchi, M., Muramatsu, M., Osawa, T., Tsuchida, R., and Shibuya, M. (2011). RACK1 regulates VEGF/Flt1-mediated cell migration via activation of a PI3K/Akt pathway. J. Biol. Chem. 286, 9097–9106.
Wang, Y., Nakayama, M., Pitulescu, M. E., Schmidt, T. S., Bochenek, M. L., Sakakibara, A., Adams, S., Davy, A., Deutsch, U., Lüthi, U., Barberis, A., Benjamin, L. E., Mäkinen, T., Nobes, C. D., and Adams, R. H. (2010). Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature 465, 483–486.
Whitman, M., Downes, C. P., Keeler, M., Keller, T., and Cantley, L. (1988). Type I phosphatidylinositol kinase makes a novel inositol phospholipid, phosphatidylinositol-3-phosphate. Nature 332, 644–646.
Xu, Q., Briggs, J., Park, S., Niu, G., Kortylewski, M., Zhang, S., Gritsko, T., Turkson, J., Kay, H., Semenza, G. L., Cheng, J. Q., Jove, R., and Yu, H. (2005). Targeting Stat3 blocks both HIF-1 and VEGF expression induced by multiple oncogenic growth signaling pathways. Oncogene 24, 5552–5560.
Yuan, H. T., Khankin, E. V., Karumanchi, S. A., and Parikh, S. M. (2009). Angiopoietin 2 is a partial agonist/antagonist of Tie2 signaling in the endothelium. Mol. Cell. Biol. 29, 2011–2022.
Zhang, J., Fukuhara, S., Sako, K., Takenouchi, T., Kitani, H., Kume, T., Koh, G. Y., and Mochizuki, N. (2011). Angiopoietin-1/Tie2 signal augments basal Notch signal controlling vascular quiescence by inducing delta-like 4 expression through AKT-mediated activation of beta-catenin. J. Biol. Chem. 286, 8055–8066.
Zhao, P., Meng, Q., Liu, L. Z., You, Y. P., Liu, N., and Jiang, B. H. (2010). Regulation of survivin by PI3K/Akt/p70S6K1 pathway. Biochem. Biophys. Res. Commun. 395, 219–224.
Zhong, H., Chiles, K., Feldser, D., Laughner, E., Hanrahan, C., Georgescu, M. M., Simons, J. W., and Semenza, G. L. (2000). Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer Res. 60, 1541–1545.
Zundel, W., Schindler, C., Haas-Kogan, D., Koong, A., Kaper, F., Chen, E., Gottschalk, A. R., Ryan, H. E., Johnson, R. S., Jefferson, A. B., Stokoe, D., and Giaccia, A. J. (2000). Loss of PTEN facilitates HIF-1-mediated gene expression. Genes Dev. 14, 391–396.
Zwerts, F., Lupu, F., De Vriese, A., Pollefeyt, S., Moons, L., Altura, R. A., Jiang, Y., Maxwell, P. H., Hill, P., Oh, H., Rieker, C., Collen, D., Conway, S. J., and Conway, E. M. (2007). Lack of endothelial cell survivin causes embryonic defects in angiogenesis, cardiogenesis, and neural tube closure. Blood 109, 4742–4752.
Keywords: angiogenesis, PI3K/AKT/mTOR, VEGF, nitric oxide, angiopoietins
Citation: Karar J and Maity A (2011) PI3K/AKT/mTOR pathway in angiogenesis. Front. Mol. Neurosci. 4:51. doi: 10.3389/fnmol.2011.00051
Received: 01 August 2011;
Accepted: 15 November 2011;
Published online: 02 December 2011.
Edited by:
Michael Fähling, Charité – Universitätsmedizin Berlin, GermanyReviewed by:
Carlo Sala, National Research Council Institute of Neuroscience, ItalyChandan Guha, Albert Einstein College of Medicine, USA
Copyright: © 2011 Karar and Maity. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Amit Maity, Department of Radiation Oncology, Perelman School of Medicine at the University of Pennsylvania, John Morgan Building, Room 195, 3620 Hamilton Walk, Philadelphia, PA 19104, USA. e-mail:bWFpdHlAdXBocy51cGVubi5lZHU=