

- Department of Biology, The University of Iowa, Iowa City, IA, USA
The Pcdh-γ gene cluster encodes 22 protocadherin adhesion molecules that interact as homophilic multimers and critically regulate synaptogenesis and apoptosis of interneurons in the developing spinal cord. Unlike interneurons, the two primary components of the monosynaptic stretch reflex circuit, dorsal root ganglion sensory neurons and ventral motor neurons (MNs), do not undergo excessive apoptosis in Pcdh-γdel/del null mutants, which die shortly after birth. However, as we show here, mutants exhibit severely disorganized Ia proprioceptive afferent terminals in the ventral horn. In contrast to the fine net-like pattern observed in wild-type mice, central Ia terminals in Pcdh-γ mutants appear clumped, and fill the space between individual MNs; quantitative analysis shows a ~2.5-fold increase in the area of terminals. Concomitant with this, there is a 70% loss of the collaterals that Ia afferents extend to ventral interneurons (vINs), many of which undergo apoptosis in the mutants. The Ia afferent phenotype is ameliorated, though not entirely rescued, when apoptosis is blocked in Pcdh-γ null mice by introduction of a Bax null allele. This indicates that loss of vINs, which act as collateral Ia afferent targets, contributes to the disorganization of terminals on motor pools. Restricted mutation of the Pcdh-γ cluster using conditional mutants and multiple Cre transgenic lines (Wnt1-Cre for sensory neurons; Pax2-Cre for vINs; Hb9-Cre for MNs) also revealed a direct requirement for the γ-Pcdhs in Ia neurons and vINs, but not in MNs themselves. Together, these genetic manipulations indicate that the γ-Pcdhs are required for the formation of the Ia afferent circuit in two ways: First, they control the survival of vINs that act as collateral Ia targets; and second, they provide a homophilic molecular cue between Ia afferents and target vINs.
Introduction
The formation of complex neuronal circuits essential for normal behavior depends on a series of sequential events that include neuron subtype differentiation, axon guidance, terminal formation, target selection and synapse formation. Studies have shown that expression of specific transcription factors (reviewed by Dalla Torre di Sanguinetto et al., 2008), trophic factors (reviewed by da Silva and Wang, 2011) and semaphorin/Plexin signaling (Messersmith et al., 1995; Fu et al., 2000; Cheng et al., 2001; Cohen et al., 2005; Yoshida et al., 2006; Pecho-Vrieseling et al., 2009) all play important roles in this process. Cell adhesion molecules, particularly those of the large and diverse immunoglobulin and cadherin superfamilies, have been shown to regulate multiple steps in the process of circuit assembly, including axon fasciculation, axon pathfinding, terminal arborization, and synaptic specificity (reviewed by Takeichi, 2007; Arikkath and Reichardt, 2008; Margeta et al., 2008; Giagtzoglou et al., 2009). A major goal of developmental neurobiology today is to identify the molecular cues that guide the formation of distinct neuronal circuits.
One of the most basic of CNS circuits is the monosynaptic spinal stretch reflex circuit, which is made up of two distinct functional units: a sensory unit and an effector unit. The sensory unit is composed of muscle spindles, which are mechanoreceptors embedded in skeletal muscles, and a specific subpopulation of dorsal root ganglion (DRG) sensory neurons (termed “Ia afferents”) that peripherally innervate these muscle spindles and relay proprioceptive information into the CNS. The central Ia axons interact with components of the effector unit, which is made up of α-motor neurons (MNs) along with particular interneurons located in the ventral horn of the spinal cord. The proprioceptive Ia afferents make precise excitatory monosynaptic connections with the MNs, which project axons to the same target muscle from which they receive sensory feedback. In addition, the Ia afferents send collateral branches to a group of ventral interneurons (vINs), which then inhibit MNs that project to antagonistic muscles; the addition of this collateral projection allows for coordinated muscle movement in response to proprioceptive input (Chen et al., 2003). Proprioceptive afferents thus make very selective monosynaptic connections with MNs supplying the same muscles, and avoid making connections with MNs supplying antagonistic muscles. The formation of their collateral branches onto vINs is also restricted to particular groups, including a population of V1-derived interneurons (Sapir et al., 2004; Alvarez et al., 2005).
In addition to proprioceptive Ia afferents, the DRG neurons extend axons conveying three other sensory modalities into the spinal cord: touch, pain (nociception), and temperature (Brown, 1981). The central projections of all DRG neurons extend dorsally to enter the spinal cord through the dorsal root with their branches terminating in specific target regions of the dorsal or ventral spinal cord. The Ia afferents extend from large DRG neurons that express parvalbumin, vesicular glutamate transporter 1 (VGLUT1), and the TrkC neurotrophin receptor (which selectively binds NT3). Smaller DRG neurons express TrkA (which selectively binds NGF) and project small diameter unmyelinated axons that convey pain, touch, and temperature information into distinct laminae of the dorsal horn (Brown, 1981; Koerber and Mendell, 1992; Mu et al., 1993). While genetic studies to define factors that regulate the differentiation of sensory neurons and the targeting of their axons have revealed roles for several transcription factors such as Ngn-1, Runx, Erg1, Pea3, and others (Dalla Torre di Sanguinetto et al., 2008), there is perhaps less information on cell surface receptors with more direct roles in controlling sensory axonal projection pattern as well as in forming specific synaptic connections with their target neurons. In chick embryos, the immunoglobulin superfamily members F11 and axonin-1 are required for the correct spinal cord pathfinding of, respectively, proprioceptive Ia afferents and nociceptive axons (Perrin et al., 2001). Elegant genetic studies have shown that expression of Sema3e by specific MNs and its high affinity receptor PlexinD1 by Ia afferents is a critical mediator of sensory-motor synaptic connectivity in mice. Genetic manipulation of either Sema3e or PlexinD1 expression results in an aberrant pattern of sensory-motor monosynaptic connections but has no effect on the segregation of MNs into specific pools (Pecho-Vrieseling et al., 2009). PlexinA1 is also specifically expressed in the proprioceptive sensory axons and is a receptor for Sema6c and Sema6d repulsive cues, which are dynamically expressed in the dorsal horn. Loss of plexinA1 signaling resulted in proprioceptive axons following an aberrant pathway through the medial region of the dorsal horn, thereby disrupting the organization of cutaneous sensory afferents (Yoshida et al., 2006). Despite this aberrant pathway followed by the central projection of Ia afferent axons, they still reach their ventral horn target areas and form connections in the spinal cord, however (Yoshida et al., 2006). It has also been found that type II classical cadherins are expressed by distinct subpopulations of DRG sensory neurons and for at least two of them, T-cad and MN-cad, this expression pattern is correlated in subsets of MNs supplying the same muscle (Price et al., 2002).
Over 50 cadherin superfamily genes are present in three clusters, termed Protocadherin- (Pcdh-)α, β, and γ, that together encompass ~900 kb on human chromosome 5q31 and mouse chromosome 18 (Wu and Maniatis, 1999; Wu et al., 2001). We have previously shown that the 22-gene Pcdh-γ cluster is critically required for the development of the CNS. Each γ-Pcdh isoform is encoded by a unique large “variable” exon encoding six extracellular cadherin repeats, a transmembrane domain, and a ~90 amino acid cytoplasmic domain; each variable exon is spliced to three small “constant” exons that encode a further 125 amino acid shared C-terminal domain (Wu and Maniatis, 1999; Tasic et al., 2002; Wang et al., 2002a; a schematic of the locus is shown in Figure 7A). The 22 γ-Pcdh isoforms form cis-homo- or hetero-tetramers that interact in a strictly homophilic manner in trans, indicating that the Pcdh-γ locus could specify at least 104 distinct adhesive interfaces (Schreiner and Weiner, 2010). The γ-Pcdhs are expressed throughout the embryonic and postnatal CNS with individual neurons expressing different subsets of the γ-Pcdh isoforms (Wang et al., 2002b; Kaneko et al., 2006; Zou et al., 2007). The γ-Pcdh proteins are preferentially localized to synaptic and perisynaptic sites, and are expressed by astrocytes as well as by neurons (Wang et al., 2002b; Phillips et al., 2003; Garrett and Weiner, 2009). Mice in which the entire Pcdh-γ gene cluster has been deleted (Pcdh-γdel/del) lack voluntary movements and spinal reflexes and die within a few hours of birth (Wang et al., 2002b). Null mutants exhibit extensive interneuron cell death and reduced synaptic density in the spinal cord during late embryonic development (Wang et al., 2002b; Weiner, 2006; Prasad et al., 2008). We previously showed that loss of the γ-Pcdhs exacerbates an underlying, normal developmental pattern of spinal interneuron apoptosis, the extent of which differs among the many molecularly defined interneuron subsets (Prasad et al., 2008). Intriguingly, neither DRG sensory neurons nor MNs undergo increased apoptosis in Pcdh-γ null mice (Wang et al., 2002b; Prasad et al., 2008; see Figure 2 below), leaving open the question of whether these molecules might regulate the formation of the monosynaptic stretch reflex circuit.
Here, we have uncovered a role for the γ-Pcdhs in the formation of Ia afferent terminals onto their target cells. We conducted a genetic analysis including Pcdh-γdel/del null mutants, Pcdh-γdel/del; Bax−/− double mutants, and a conditional Pcdh-γ mutant allele (Pcdh-γfcon3) along with four Cre transgenic lines to bring about selective loss of the γ-Pcdhs in discrete neuronal populations. We show that γ-Pcdhs are required in a cell autonomous manner in sensory neurons to bring about a normal arborization pattern of Ia afferent terminals onto MNs. Our manipulations further suggest that loss of the γ-Pcdhs from vINs affects the Ia afferent terminal field both directly, by disrupting contacts made by sensory axons, and indirectly, by increasing apoptosis and removing potential target neurons. Interestingly, the loss of γ-Pcdhs in the MNs, which are the primary targets, does not significantly affect the arborization pattern of Ia afferent neurons. Our data suggest that the Pcdh-γ family plays essential roles in specifying the connectivity between Ia afferent collaterals and vINs, which in turn regulates the formation of the primary terminal field on MNs.
Materials and Methods
Mouse Strains
The Pcdh-γdel, Pcdh-γfus (Wang et al., 2002b), and Pcdh-γfcon3 alleles (Prasad et al., 2008) and Bax−/− mutants (Knudson et al., 1995; Deckwerth et al., 1996; White et al., 1998) were described previously. Actin-Cre (Lewandoski et al., 1997), Wnt1-Cre (Danielian et al., 1998), and Hb9-Cre (Arber et al., 1999) mouse lines were obtained from The Jackson Laboratory (Bar Harbor, ME). Pax2-Cre mice (Ohyama and Groves, 2004) were the kind gift of Dr. Andy Groves (House Ear Institute, Los Angeles, CA). All lines utilized were congenic or nearly congenic with C57BL/6; all were backcrossed onto this strain for at least 6–10 generations. All animal procedures were performed in accordance with the University of Iowa’s Institutional Animal Care and Use Committee and NIH guidelines.
Antibodies
The following antibodies were utilized: rabbit anti-cleaved caspase-3 (Cell Signaling Technologies); rabbit anti-GFP (Invitrogen); mouse anti-NeuN (Chemicon); rabbit anti-parvalbumin (Swant); mouse anti-parvalbumin (Sigma); mouse anti-PSD-95 (Affinity BioReagents); guinea pig anti-VGLUT1 (Chemicon). Anti-TrkA antibody was the kind gift of Dr. Louis Reichardt (UCSF). The mouse anti-γ-Pcdh constant domain (N159/5) and mouse anti-γ-Pcdh-B2 (N148/30) were generated by the UC Davis/NIH NeuroMab Facility using antigens produced in our laboratory, and are characterized in detail in Lobas et al. (2011). They are available commercially through Antibodies, Inc.
Immunofluorescence
Embryonic and neonatal mice were killed by decapitation, and vertebral columns were exposed and removed. Spinal columns were prepared for immunofluorescence using one of two methods: (1) Fixation for 2 h in 4% paraformaldehyde in phosphate buffered saline (PBS) at 4°C, followed by washes with cold PBS, cryoprotection in 30% sucrose in PBS at 4°C, and freezing on dry ice in OCT compound (Tissue Tek/Sakura); or (2) Snap freezing in OCT using dry ice/ethanol-cooled isopentane. In either case, transverse cryostat sections were cut at 12 μm. Slides containing fresh-frozen sections were fixed in 100% methanol for 10 min at −20°C. Sections were blocked in 2.5% BSA, 0.1% Triton X100 in PBS for 1 h followed by overnight incubation at 4°C with primary antibodies diluted in the same blocking solution. Sections were washed in PBS and incubated for 1 h at room temperature with the appropriate secondary antibodies conjugated to Alexa 488, 568, or 647 (Molecular Probes/Invitrogen). Sections were washed in PBS containing the nuclear counterstain DAPI (4′,6-diamidino-2-phenylindole), and mounted in Gel/Mount (Biomeda) aqueous mounting media. Some sections were counterstained with NeuroTrace 435 fluorescent Nissl stain (Invitrogen).
In situ Hybridization
In situ hybridization using an antisense riboprobe corresponding to the Pcdh-γ constant exons was performed as described (Wang et al., 2002b). Vertebral columns collected from neonatal mice were snap frozen in OCT using dry ice/ethanol-cooled isopentane. Twenty micron cryostat sections were cut and post-fixed for 5 min at room temperature. Sections were rinsed and then immersed in acetylation solution (295 ml of H2O, 4 ml of triethanolamine, 0.525 ml of concentrated HCl, 0.75 ml of acetic anhydride) for 10 min at room temperature. The sections were then rinsed and incubated with hybridization solution (50% formamide, 5× SSC, 5× Denhardt’s solution, 250 mg/ml yeast tRNA, 500 mg/ml salmon sperm DNA, 50 mg/ml heparin) at room temperature for 1 h followed by overnight hybridization at 65°C with fluorescein–UTP labeled riboprobe diluted in hybridization solution. The next day, the sections were washed for several hours at 65°C in 0.2× SSC, rinsed at room temperature in TBS, and blocked for 1 h with 0.2% Blocking Reagent (Roche) in TBS. Probes were detected by overnight incubation at 4°C with anti-fluorescein antibodies (1:1000) conjugated to peroxidase (Roche), followed by amplification using the TSA Plus system (Perkin-Elmer Life sciences) according to manufacturer’s protocol.
Image Acquisition and Analysis
For interneuron counts, synapse and terminal density quantification 12 μm thick sections were cut from mouse spinal cords. Each quantification was performed on six to eight sections from at least three animals (i.e., at least 18 sections each per genotype per time point). Images were taken of control and mutant spinal cord sections at equivalent thoracolumbar locations and camera exposures using 10× (for cell counts), 20×, or 63× (for synapse and terminal density quantification) PlanApo objectives on a Leica DM5000B digital epifluorescence microscope or on a Leica SP2 AOBS laser scanning confocal microscope. Digital images were captured in Adobe Photoshop and similarly adjusted for brightness and contrast. Cell counts were performed manually. For synaptic puncta and terminal density quantifications images were thresholded in NIH Image/J and the number of puncta and/or total area occupied by terminals was quantified by using the Analyze Particles function. Statistical significance was determined by two-way ANOVA followed by Bonferroni-corrected post hoc tests using Prism software.
RT-PCR
Control and mutant neonatal mice were killed by decapitation, vertebral columns were exposed and removed and the dorsal root ganglia were dissected out and collected in RNAlater (Ambion). Total RNA was extracted by using the RNAqueous-4PCR kit (Ambion) from thoracic (T2–T6) DRGs that were pooled together to get a sufficient amount of RNA. RNA concentration was quantified spectrophotometrically and cDNA was synthesized from 2 μg of RNA using random hexamer primers and Superscript III reverse transcriptase (Invitrogen). An array of PCR reactions was performed to amplify various Pcdh-γ variable exon-constant exon spliced transcripts. The forward and reverse primer sequences utilized were the same as those reported in Prasad et al. (2008). Cycling parameters were: 94°C, 1 min; 55°C, 1 min; 72°C 3 min, for 30 cycles.
Results
Expression of Pcdh-γ Isoforms by DRG Sensory Neurons
Earlier studies have shown that the Pcdh-γ gene cluster is expressed throughout the developing and adult CNS, and that individual neurons express varying subsets of Pcdh-γ isoforms (Wang et al., 2002a,b; Phillips et al., 2003; Frank et al., 2005; Kaneko et al., 2006; Zou et al., 2007). To confirm that sensory neurons in the DRG express the γ-Pcdhs, we examined tissues from Pcdh-γfusg mice, in which the third constant exon is fused to GFP (Wang et al., 2002b), and performed in situ hybridization using a probe against the shared constant exons, which detects expression of all 22 possible Pcdh-γ transcripts (see schematic of the cluster in Figure 7A). Both methods showed that all cells within the DRG expressed the γ-Pcdhs between E12 and P0 (Figures 1A,B, and data not shown). To investigate further if all 22 distinct Pcdh-γ RNA transcripts are expressed by DRG neurons we performed RT-PCR using a reverse primer in the shared constant exons and 22 forward primers specific to each variable exon (Prasad et al., 2008; Garrett and Weiner, 2009). At least 20 of the 22 possible transcripts were readily detectable in RNA from P0 thoracic DRG (Figure 1C). Double immunostaining with antibodies against the γ-Pcdh constant domain (shared by all 22 isoforms) and VGLUT1 indicated that all Ia afferent neurons (as well as all other DRG neurons) express γ-Pcdh proteins (Figures 1D–F). Previous analyses have suggested that individual neurons express a subset of the 22 Pcdh-γ genes (Wang et al., 2002b; Frank et al., 2005; Kaneko et al., 2006). Using a novel γ-Pcdh-B2-specific antibody (Lobas et al., 2011), we found that, indeed, a small percentage of DRG neurons express this γ-Pcdh isoform (Figures 1G–I). Double-labeling for VGLUT1 identified a small number of γ-Pcdh-B2+, and many more γ-Pcdh-B2−, Ia afferent neurons, consistent with the overall occurrence of B2+ cells. Though limited to a single isoform, these data are consistent with the expression of distinct γ-Pcdh isoform subsets in individual sensory neurons.
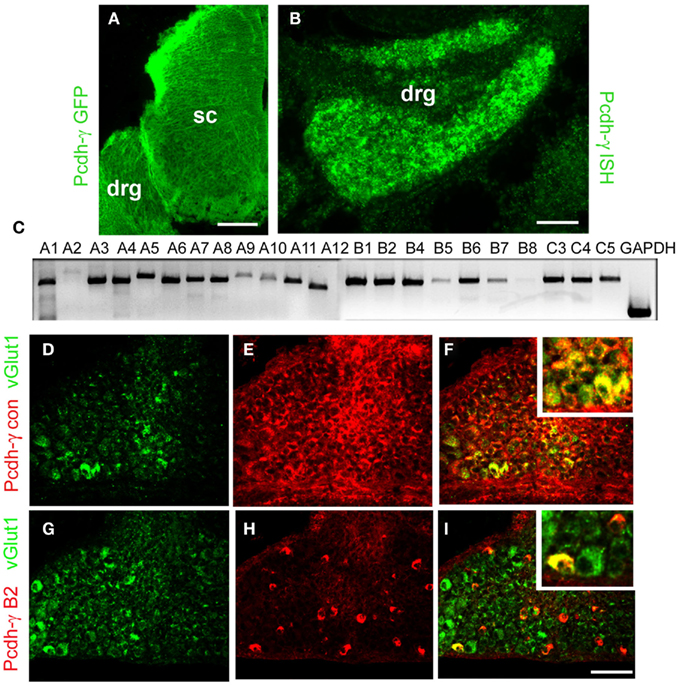
Figure 1. Expression of γ-Pcdhs by DRG sensory neurons. (A) Anti-GFP staining of E12 Pcdh-γfus spinal cord demonstrates uniform expression of γ-Pcdh-GFP fusion proteins throughout the DRG and spinal cord (sc). (B) In situ hybridization using a riboprobe against the Pcdh-γ constant exon also yields uniform labeling in P0 DRG. Sense control riboprobes gave no signal (data not shown). (C) RT-PCR analysis of P0 thoracic (T2–T6) DRG using a constant exon reverse primer and forward primers in each of the indicated variable exons demonstrates that all, or nearly all, of the 22 possible Pcdh-γ transcripts are expressed. (D–F) Immunostaining of P0 control DRG’s with antibodies against the γ-Pcdh constant region (red) and the Ia afferent neuron marker VGLUT1 (green) shows that all Ia neurons express some γ-Pcdh isoforms. (G–I) Immunostaining with antibodies specific for a single γ-Pcdh isoform (B2, red) and VGLUT1 (green) demonstrates that only a subset of sensory neurons, including a small number of Ia afferent neurons, expresses a given γ-Pcdh isoform. Insets, higher magnification view of a region in the main panel. Scale bar: 100 μm in (A), 50 μm in (B) and (D–I).
Normal DRG Neuron Survival in Pcdh-γ Null Mice
Our previous study had shown that in Pcdh-γ null mutant spinal cord there is an accentuation of apoptotic cell death in discrete spinal interneuron populations with a corresponding reduction in excitatory and inhibitory synaptic density (Prasad et al., 2008). However, loss of γ-Pcdhs did not have any effect on the survival of MNs (Wang et al., 2002b; Prasad et al., 2008). To investigate if the γ-Pcdhs are required for survival of sensory neurons, we stained DRG sections taken from postnatal day 0 (P0) control and null mutant mice with antibodies against cleaved caspase-3, a marker for apoptotic cell death, and either NeuN, to label all neurons, or parvalbumin, a marker specific for Ia afferent neurons in the DRG (Honda, 1995; Arber et al., 2000). There was no increase in the number of cleaved caspase-3 stained cells in DRG from Pcdh-γdel/del mice compared to control mice (Figures 2A,B); at this age, which is after the main period of developmental apoptosis in the DRG (Coggeshall et al., 1994; White et al., 1998) few if any cells were labeled at all. In addition, there was no change in the number of parvalbumin+ (Figures 2A–D; control = 13.9 per section, mutant = 14.1 per section, N = 18 sections per genotype taken from three sets of animals) or VGLUT1+ (data not shown) neurons, and a similar overall density of NeuN+ cells (Figures 2C,D). These data confirm our earlier preliminary finding (Wang et al., 2002b) that loss of the γ-Pcdhs does not affect the survival of DRG sensory neurons.
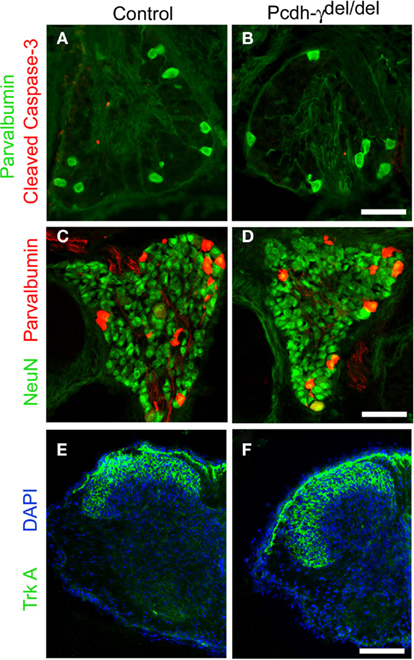
Figure 2. Normal survival and differentiation of DRG sensory neurons in Pcdh-γ null mutants. DRGs from P0 control and Pcdh-γdel/del mice were immunostained with antibodies against cleaved caspase-3 [red; (A,B)] or against the neuronal marker NeuN [green (C,D)], along with those against the Ia neuron marker parvalbumin [green, (A,B); red, (C,D)]. No excessive apoptosis of mutant sensory neurons was observed and the neuronal density, Ia neuron number, and overall size of mutant DRGs are all similar to controls. (E) P0 Control and (F) Pcdh-γ null mutant spinal cords stained with antibodies against TrkA+ cutaneous sensory axons indicate that these terminate normally in the superficial part of the dorsal horn in the absence of the γ-Pcdhs. Scale bar: 50 μm in (A–D); 100 μm in (E,F).
Pcdh-γ Null Mutation Disrupts the Formation of Ia Afferent Terminals in the Ventral Horn
We next asked if the ingrowth and trajectory of sensory axons is affected by loss of the γ-Pcdhs. To assess this, we stained sensory axons with antibodies that detect cutaneous axons projecting to the dorsal horn (TrkA) or Ia afferents projecting to the ventral horn (parvalbumin, VGLUT1) in control and Pcdh-γdel/del spinal cord between embryonic day 14 (E14) and P0, when the mutant mice die. Our earlier preliminary analysis had suggested that loss of the γ-Pcdhs did not have any obvious effect on the dorsal horn projection of substance P+ cutaneous sensory neurons (Wang et al., 2002b). Here, we similarly found that mutant TrkA+ axons projected to the upper dorsal horn as in controls, and appeared normal in their density and spatial restriction at all ages (Figures 2E,F and data not shown). The neonatal lethality of Pcdh-γ null mice (and more restricted mutants as described below), however, prevented us from analyzing any potential role for the γ-Pcdhs in the maturation and formation of specific synapses by cutaneous sensory axons, which takes place during the early postnatal period (Fitzgerald, 1987; Mirnics and Koerber, 1995).
In contrast to cutaneous axons, Ia afferents project to the ventral horn and form synaptic connections with MNs and vINs during the late embryonic period (Mears and Frank, 1997; Chen et al., 2003). By E14 in Pcdh-γ null mutant embryos, the Ia afferent axons had entered the spinal cord through the dorsal root entry zone and were localized medially, just as in controls (Figures 3A,B). The Ia afferent axons enter the ventral horn by E15 in both the Pcdh-γ mutants and controls, and begin to extend collateral branches toward vINs (Figures 3C,D). At no age were any aberrant collateral branches into the dorsal horn observed in mutants, indicating that the γ-Pcdhs are not required for normal Ia afferent axon segregation and guidance into the ventral horn. By E17, a disruption in the pattern of Ia afferent terminals is apparent in the Pcdh-γ mutants: While control terminals have attained a fine net-like pattern of parvalbumin+ puncta around the MN and vIN regions, mutant presynaptic terminals appear clumped together in the MN region (Figures 3E,F). By P0, this phenotype has become more severe, and the lack of laterally positioned collateral branches off of the main MN-contacting axons is clear (Figures 3G,H and 4A–D).
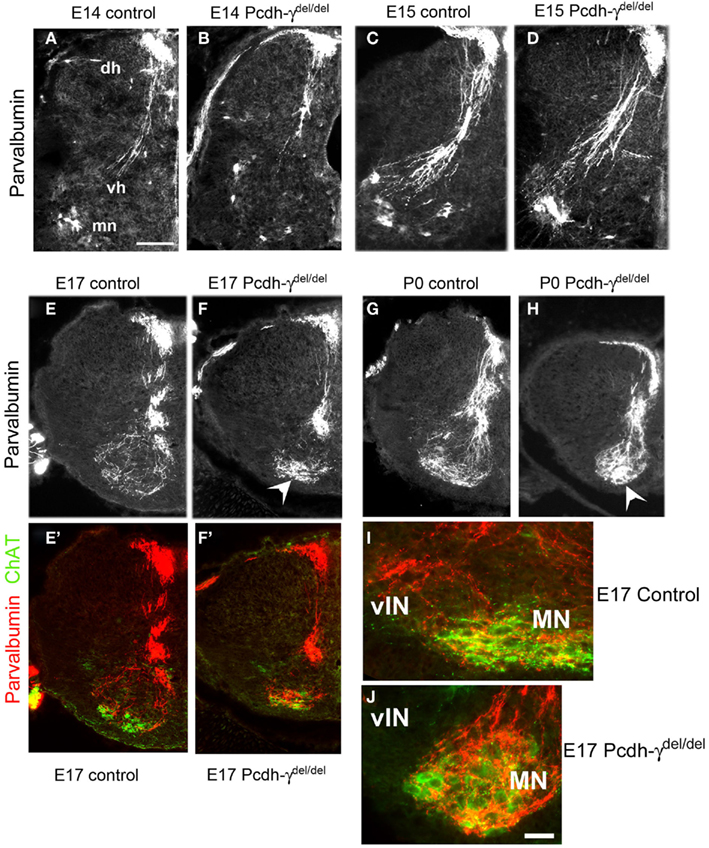
Figure 3. The pattern of Ia afferent terminal arbors in the ventral horn is disrupted in Pcdh-γ null mutant mice. (A–H) Hemi-spinal cords from control (A,C,E,G) and Pcdh-γdel/del(B,D,F,H) mice were immunostained with antibodies against parvalbumin at the indicated time points during embryonic development. Mutant parvalbumin+ Ia axons grow into the spinal cord at the correct time and penetrate as normal into the ventral horn, with no pathfinding errors. By E17, however, as the pattern of Ia afferent terminals emerges as a fine net-like pattern in controls, the mutant terminals appear clumped around motor neurons (arrowheads), with a loss of collateral projections to ventral interneurons. This phenotype has worsened by P0. (E′,F′) show the same fields as (E,F), but with ChAT staining added to indicate where the motor neuron pools are located. Higher magnification views of parvalbumin/ChAT-double stained ventral horn shows that while control Ia axons send collaterals laterally to the ventral interneuron region [vIN, (I)], these collaterals are absent in mutants (J) (dh, dorsal horn; vh, ventral horn; mn, motor neurons). Scale bar: 100 μm in (A–H), 50 in (I,J).
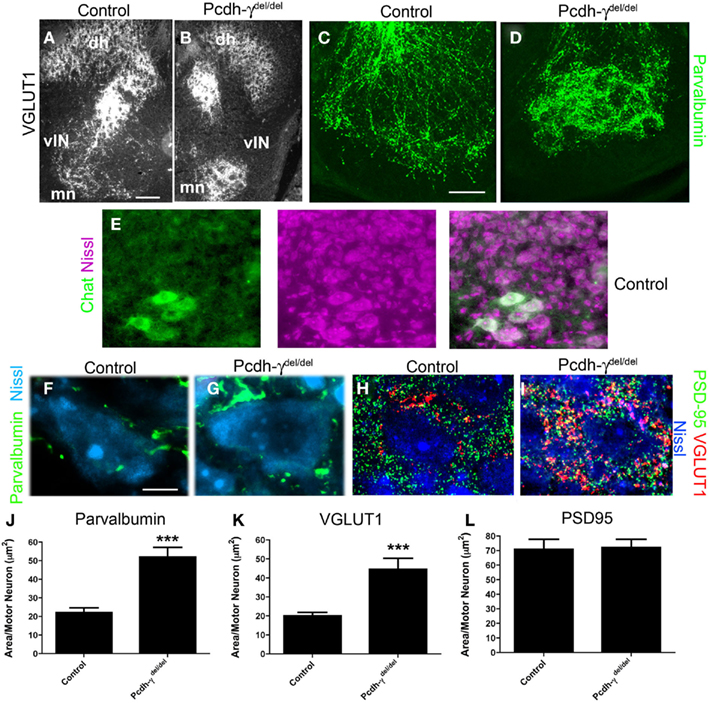
Figure 4. Ia afferent terminals on motor neurons are significantly increased in area in Pcdh-γ null mutants. (A,B) P0 control and Pcdh-γdel/del spinal cord immunostained with antibodies against VGLUT1 shows clumping of terminals around motor neurons (mn) with a corresponding reduction of collateral terminals (lateral to motor neurons) on ventral interneurons (vIN), similar to the parvalbumin staining in Figure 3, and at higher magnification in (C,D). (E) Motor neurons can be reliably identified by their distinctive Nissl staining profile, as confirmed by double staining with antibodies against ChAT. (F–H) High magnification views of P0 control and Pcdh-γdel/del spinal cord in the ventral motor neuron pools, stained with the indicated antibodies (parvalbumin and VGLUT1 for Ia terminals; PSD-95 for glutamatergic postsynaptic sites on motor neurons, which are Nissl stained), show expansion of Ia terminals on individual motor neurons but no change in the density of PSD-95 puncta. (I–K) Quantification of the density of Parvalbumin or VGLUT1-stained Ia terminals per motor neuron indicates a significant increase in Pcdh-γ null mutant when compared to control, while quantification of PSD-95 density is identical. ***p < 0.001. Scale bar: 100 μm in (A,B); 40 μm in (C,D); 10 μm in (E–H).
To quantify the disruption of Ia afferent terminal formation on MN pools, we examined high magnification images of P0 sections stained for parvalbumin, VGLUT1, and PSD-95 (a postsynaptic marker of glutamatergic synapses). We counterstained sections with NeuroTrace 435, a fluorescent Nissl stain that easily distinguishes MNs, with their large soma size and prominent nucleoli, from the much smaller surrounding vINs and glia (confirmed in a set of experiments utilizing the Nissl stain and a specific marker of MNs, ChAT; Figure 4E). We measured the area taken up by parvalbumin and VGLUT1 staining in small microscope fields containing several MNs, and divided this by the number of MNs to obtain a terminal area/MN measure. We found an increase of ~2.5-fold in the area of both parvalbumin- and VGLUT1+ terminals per MN in Pcdh-γ mutants (Figures 4E–J; Table 1). We next asked if there was a corresponding increase in postsynaptic sites by measuring the density of PSD-95+ puncta in the same fields in which VGLUT1 staining was analyzed. Surprisingly, we found that the area of PSD-95 staining per MN was unaffected in Pcdh-γ null mutants (Figures 4G,H,K), suggesting that many of the mutant Ia terminals do not properly contact a postsynaptic site. This would be consistent with the overt phenotype of the neonatal mutants in the hours prior to death, which includes a lack of voluntary movement and of reflexes (Wang et al., 2002b).
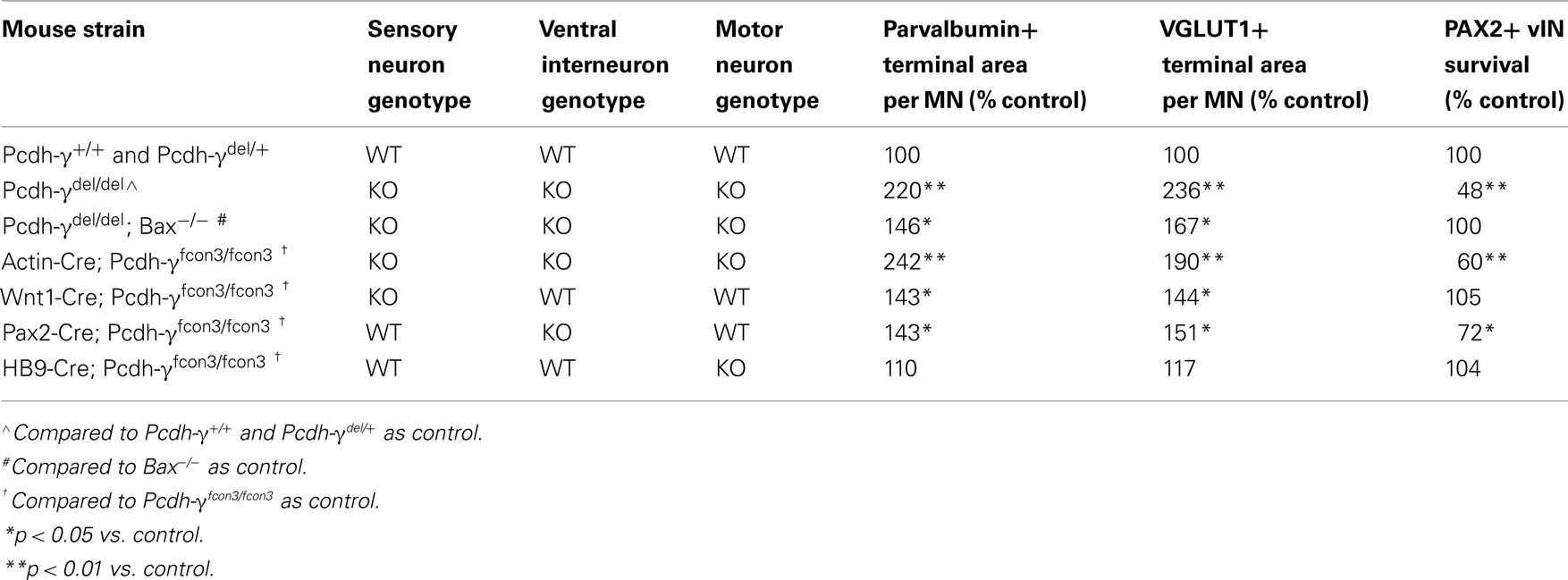
Table 1. Summary of phenotypes in the various Pcdh- γ mouse strains examined.
Loss of Ia Afferent Collaterals to Ventral Interneurons in Pcdh-γ Null Mutants
Ia afferent axons not only make specific connections with MNs; they also send collaterals to discrete vIN populations including V1-derived En1+ “Ia interneurons,” V1-derived calbindin+ Renshaw cells, and several V0-derived interneurons, many of which express Pax2 at some time during development (Burrill et al., 1997; Sapir et al., 2004; Lewis, 2006). These interneurons are a part of the central pattern generator and are, for the most part, inhibitory GABAergic neurons that mediate presynaptic and postsynaptic inhibition, thereby playing a critical role in controlling the amount of excitatory input that passes from sensory neurons onto MNs (reviewed by Goulding, 2009). Very little is known about the molecular mechanisms involved in the formation of specific connections between Ia afferents and vINs, which act as collateral targets. We quantified the apparent loss of Ia collaterals (Figure 3) in Pcdh-γ null mutant spinal cords in two ways. First, because many of the vINs receiving Ia collaterals lie lateral to the MN pools in the thoracolumbar sections we examined, we determined the extent of the spread of parvalbumin+ Ia afferent terminals in the ventral horn, normalized to the total width of the spinal cord (which is smaller in the Pcdh-γ mutants due to excessive vIN apoptosis; Prasad et al., 2008; see schematic, Figure 5A). Second, we quantified the area per neuron of parvalbumin+ Ia terminals in microscope fields containing either MN pools or vINs (see schematic, Figure 5B). Both measures indicated that between E17 and P0, there is a significant reduction in Ia collaterals projecting to vINs in the mutants (Figure 5A) accompanying the corresponding increase in the density of terminals surrounding MNs (Figures 4 and 5B). In our previous work (Prasad et al., 2008), we showed that between E17 and P0, there is massive aberrant apoptosis of vIN populations in Pcdh-γ null mutants. The fact that the reduction in Ia terminals in fields occupied by vINs is observed even when normalized to the density of these cells (Figure 5B), suggests that apoptosis is not the only cause of the lost collateral branches (see below).
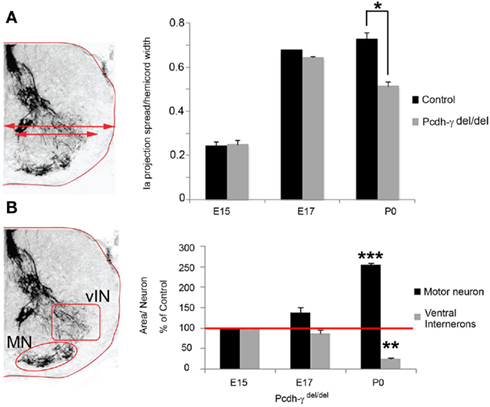
Figure 5. Loss of Ia axon collateral projections to ventral interneurons in Pcdh-γ null mutants. (A) The extent of the spread in parvalbumin+ Ia terminal arbors, normalized to hemi-cord width, was quantified as one measure of collateral formation (schematized at left). A significant reduction in the extent of Ia afferent terminal spread becomes apparent at P0. (B) Quantification of the area covered by parvalbumin+ Ia terminals in the motor neuron (MN) and ventral interneuron (vIN) fields was normalized to neuron number (schematized at left). As mutant terminal density increases around MNs, it decreases around vINs, becoming significant at P0 (data are graphed as percent of E15 control levels). Red line indicates 100%. *p < 0.05; **p < 0.01;***p < 0.001.
Blocking Ventral Interneuron Apoptosis Only Partially Rescues Ia Afferent Terminals
The loss of γ-Pcdhs affects vIN subsets differentially during late embryogenesis: while 80% of V1-derived En1+ neurons are lost, calbindin+ Renshaw cells, which are also V1-derived, are not affected at all. About 52% of all Pax2+ vINs, which derive from both V0 and V1 and include an En1+ population, are lost in Pcdh-γ null embryos (Prasad et al., 2008). To examine if the excessive loss of vINs is solely responsible for the aberrant pattern of Ia afferent terminals in Pcdh-γ null mutants, we crossed these mice with Bax−/− mice. Loss of Bax, a proapoptotic Bcl-2 family member protein, prevents neurons from undergoing apoptotic cell death (Deckwerth et al., 1996) and rescues vIN numbers on a Pcdh-γ null background (Prasad et al., 2008). Quantification of Ia afferent terminals showed that blocking apoptosis does partially rescue the phenotype: in Pcdh-γdel/del; Bax−/− double mutants, parvalbumin- or VGLUT1+ terminal area per MN was increased by only 40% (compared to ~2.5-fold in Pcdh-γ single mutants). This increase was, however, still significant compared to controls (note that the double mutants were compared to Bax−/− mice, to control for increased neuronal numbers when apoptosis is blocked; Figure 6). These results suggest that γ-Pcdhs can affect the formation of Ia terminals indirectly, due to their effect on interneuron survival, but that this indirect effect cannot fully account for the defects observed in Pcdh-γ null mutants.
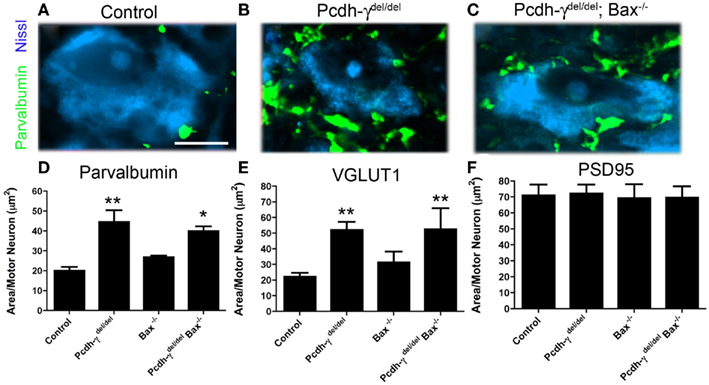
Figure 6. Genetic block of apoptosis ameliorates, but does not completely rescue, the Ia afferent phenotype observed in Pcdh-γ null mutants. (A–C) Spinal cords from P0 control, Pcdh-γdel/del and Pcdh-γdel/del; Bax−/− mice were immunostained with antibodies against parvalbumin and imaged at high magnification to show Ia terminals around motor neurons, which were Nissl stained. (D–F) The aberrant expansion of parvalbumin- or VGLUT1+ terminals in the absence of the γ-Pcdhs is ameliorated when apoptosis is blocked by the additional mutation of Bax. Double mutant terminals are, however, still significantly affected compared to Bax−/− single mutant controls. No effect on the area covered by PSD-95+ puncta is observed. Scale bar: 20 μm. *p < 0.05 and **p < 0.01, compared to appropriate control mice.
Ia Afferent Sensory Neurons Require γ-Pcdhs in a Cell Autonomous Manner for Normal Central Terminal Formation
To ask whether γ-Pcdhs have a direct role in the formation of Ia afferent terminals, we mutated the Pcdh-γ cluster in a cell type-specific fashion, using the conditional Pcdh-γfcon3 allele (Lefebvre et al., 2008; Prasad et al., 2008). In this allele, the third constant exon is fused to GFP and flanked by loxP sites (Figure 7A). Cre-mediated excision of the Pcdh-γfcon3 allele results in a null or near-null: Ubiquitous deletion using Actin-Cre transgenics essentially phenocopies Pcdh-γdel/del spinal cord phenotypes (Table 1; Figure 7D), and no truncated γ-Pcdh proteins can be detected, presumably because deletion of constant exon 3, including the polyadenylation site and 3′UTR for all Pcdh-γ mRNAs, leads to reduced transcript stability (Prasad et al., 2008). We crossed Pcdh-γfcon3 mice to three previously characterized Cre transgenic lines: 1) Wnt1-Cre (Danielian et al., 1998), to target all DRG neurons; 2) Pax2-Cre (Ohyama and Groves, 2004), to target Pax2+ spinal interneurons including Ia-contacted vINs; and 3) Hb9-Cre (Arber et al., 1999), to target MNs (see schematic, Figure 7B). We also crossed all of these Cre lines to Z/EG reporter mice to confirm that the pattern of Cre activity was as expected from the published literature [Prasad et al., 2008, and data not shown; note that in our prior work we utilized a different Hb9-Cre line (Umemori et al., 2004) that is not as cleanly restricted to MNs as the superior line utilized here]. We first assessed the survival of Pax2+ vINs, and found no difference from control in Wnt1-Cre; Pcdh-γfcon3/fcon3 and Hb9-Cre; Pcdh-γfcon3/fcon3 mice (Table 1). As we reported previously (Prasad et al., 2008), survival of V0/V1-derived Pax2+ vINs in Pax2-Cre; Pcdh-γfcon3/fcon3 neonates is intermediate between wild-type and Pcdh-γdel/del null mutant levels (Table 1).
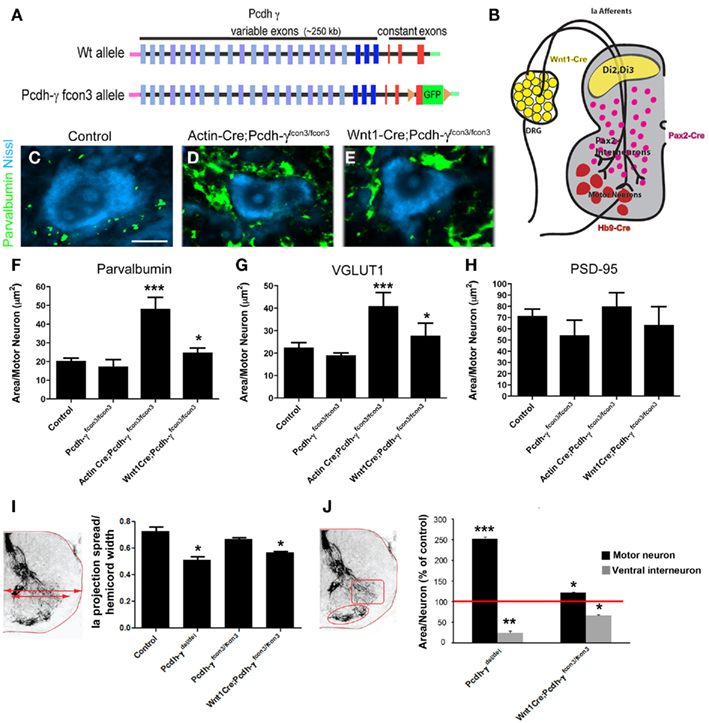
Figure 7. Restricted mutation of the Pcdh-γ gene cluster in DRG sensory neurons reveals a cell autonomous requirement for the γ-Pcdhs in Ia afferent terminal arborization. (A) Schematic diagram showing the wild-type (wt) and conditional mutant Pcdh-γ alleles (Pcdh-γfcon3). The fcon3 alleles harbors loxP sites on either side of the third constant exon to which GFP is fused. (B) Schematic diagram of a P0 spinal hemi-cord showing the neuronal subsets in which the Pcdh-γ gene cluster is disrupted when crossed with the indicated Cre transgenic lines. (C–E) Individual motor neurons from control, Actin-cre; Pcdh-γ fcon3/fcon3 (ubiquitous excision) and Wnt1-Cre; Pcdh-γfcon3/fcon3 (DRG and dorsal horn-specific excision) spinal cords stained for parvalbumin and Nissl counterstain. Ubiquitous excision of the conditional allele phenocopies Pcdh-γdel/del null mutants (compare to Figure 4F), while Wnt1-Cre-restricted mice show an intermediate Ia phenotype. (F–H) Quantification of the area occupied by parvalbumin- or VGLUT1+ Ia terminals and PSD-95+ postsynaptic puncta per motor neuron. (I,J) Measures of Ia terminal spread and area of terminals per neuron in both the MN and vIN fields, quantified as in Figure 5. Scale bar: 20 μm in (C–E). *p < 0.05 and **p < 0.01; ***p < 0.001.
Wnt1-Cre-mediated mutation of Pcdh-γ in DRG neurons resulted in an approximately 40% increase in the density of Ia afferent terminals on MNs (Figures 7C,E–H; Table 1) and a corresponding ~40% reduction in the density of the terminals on vINs (Figures 7I,J). Because in Wnt1-Cre; Pcdh-γfcon3/fcon3 mice Ia afferent neurons lack the γ-Pcdhs while the two target neuronal populations (vINs and MNs) do not, this effect can be ascribed to a cell autonomous requirement for the γ-Pcdhs in the afferents. In Pax2-Cre; Pcdh-γfcon3/fcon3 mice, Ia afferent terminals exhibit a phenotype nearly identical to that seen in the Wnt1-Cre-restricted mutants: a ~40% increase in area per MN (Table 1). This could be due either to the partial loss of V0/V1-derived vINs in Pax2-restricted mutants (Table 1), or to a cell autonomous requirement for the γ-Pcdhs in Pax2+ vINs, which due to the homophilic nature of γ-Pcdh interactions (Schreiner and Weiner, 2010) would be expected given their requirement in Ia neurons themselves. The fact that the ubiquitous loss of the γ-Pcdhs in the absence of apoptosis in Pcdh-γdel/del; Bax−/− double mutants yields a phenotype quantitatively similar to both Wnt1- and Pax2-restricted mutants (Table 1) supports the latter conclusion. Finally, we analyzed Hb9-Cre; Pcdh-γfcon3/fcon3 mice, and, surprisingly, found no significant change in the density of Ia afferent terminals on MNs (Table 1). This suggests that the γ-Pcdhs act primarily to facilitate interactions between Ia afferents and vINs, and that disruptions in these interactions indirectly affect the formation of the primary Ia terminals on MNs.
Discussion
The clustered protocadherins have been suggested to play an important role in providing molecular identity to neuronal subpopulations, which in turn could control the specificity of neuronal connectivity as well as neuronal survival (Morishita and Yagi, 2007; Schreiner and Weiner, 2010; Zipursky and Sanes, 2010). Consistent with this, in mice lacking the α-Pcdhs there is a failure of olfactory axons bearing a single type of odorant receptor to coalesce properly into glomeruli in olfactory bulbs (Hasegawa et al., 2008). Pcdh-α mutant mice also exhibit disruptions in the arborization pattern of serotonergic terminals in the brain (Katori et al., 2009). We earlier showed that deletion of the Pcdh-γ cluster results in widespread interneuron apoptosis in the embryonic spinal cord that reflects an exacerbation of a normal developmental pattern of cell death (Wang et al., 2002b; Prasad et al., 2008). The affected vIN populations include those involved in stereotyped motor circuits (the central pattern generator), which receive inputs from DRG sensory neurons as well as from higher brain centers and play a critical role in controlling motor output (Goulding, 2009). Intriguingly, the survival of DRG sensory neurons and ventral horn MNs is not affected by the loss of the γ-Pcdhs (Wang et al., 2002b; Prasad et al., 2008, the present study). This, along with the observation that Pcdh-γ null mutant neonates lack spinal reflexes, suggested that the γ-Pcdhs might regulate the formation of spinal proprioceptive circuits.
Here, we took advantage of genetic techniques to disrupt γ-Pcdh function in the neurons comprising the proprioceptive stretch reflex circuit in the developing spinal cord. Our data show that loss of γ-Pcdhs results in the expansion and disorganization of Ia afferent terminals on MNs, but does not affect axon pathfinding, as shown by the developmentally appropriate growth of Ia axons into the ventral horn and the lack of aberrant branching into dorsal horn. Because Ia terminal area per MN is increased without any corresponding increase in the area occupied by PSD-95+ puncta, the clumped terminals do not appear to represent functional synaptic connections, consistent with mutant neonates’ outward phenotype. Along with expansion of terminals onto MNs there is a simultaneous loss of terminals formed onto vINs. As the disruption in the pattern of Ia afferent terminals occurs during the same time period when vINs are undergoing aberrant apoptotic cell death in Pcdh-γ null mutants (Prasad et al., 2008), we asked whether the disrupted Ia terminals were due to an extensive loss of vIN targets. Our data from the Pcdh-γdel/del; Bax−/− double mutants show that blocking apoptosis ameliorates, but does not completely rescue, the Ia phenotype. Further analyses of mice in which Pcdh-γ mutation is restricted to sensory neurons, interneurons, or MNs showed that the γ-Pcdhs are required in sensory and interneuron populations, but not in MNs, for the normal pattern of Ia terminal arborization. Taking these data together with our recent demonstration of homophilic γ-Pcdh interactions (Schreiner and Weiner, 2010), we thus suggest that the γ-Pcdhs regulate the formation of spinal proprioceptive circuits both directly and indirectly: Directly via homophilic interactions between incoming Ia axons and vINs, and indirectly by controlling the survival of these vIN target neurons.
Our results raise the possibility that collateral vIN targets somehow regulate the formation of monosynaptic Ia terminals on MNs. As some vINs are located at slightly more dorsal positions than MNs, it may be that collateral branches of the Ia afferents make contact with them first. If so, Ia afferent-vIN contacts mediated by the γ-Pcdhs could provide an instructive signal that regulates the arborization of the main axonal branches onto MNs. Alternatively, it may simply be that Ia axons need to make a particular number of terminals, or to establish a particular area of terminal arborization, and if vIN targets are missing or non-receptive due to γ-Pcdh loss, the MN terminal field is increased in area accordingly to make up for this deficit. Intriguingly, though MNs do express the γ-Pcdhs, this expression is not required for the formation of the Ia circuit, and the number of postsynaptic PSD-95+ puncta does not change even in null mutants. The question of whether the γ-Pcdhs regulate the specificity of proprioceptive synapse formation, as Sema3e-PlexinD1 signaling has been shown to do (Pecho-Vrieseling et al., 2009), remains unclear. The neonatal lethality of Pcdh-γdel/del, Wnt1-Cre; Pcdh-γfcon3/fcon3, and Pax2-Cre; Pcdh-γfcon3/fcon3 mutants unfortunately precludes the kind of electrophysiological studies that can address this issue directly. Although limited to the DRG and the superficial dorsal horn in the spinal sensorimotor system, Wnt1-Cre activity is also found in other brain regions (Rico et al., 2002), including brainstem and basal forebrain nuclei in which loss of the γ-Pcdhs results in increased apoptosis (Wang et al., 2002b), a likely cause of the observed lethality. Future studies utilizing more restricted Cre lines may allow us to circumvent this neonatal lethality and allow for electrophysiological studies of sensorimotor circuit specificity to be performed.
The cell signaling mechanisms by which the γ-Pcdhs might regulate the elaboration of Ia afferent terminals is unknown. One intriguing possibility is that they negatively regulate Wnt signaling. Wnt-3, expressed by MNs, can promote branching and increase the growth cone size of growing NT3-responsive (i.e., Ia afferent), but not NGF-responsive (i.e., cutaneous), DRG neurons in vitro (Krylova et al., 2002). Recently, in studies of kidney tumor cell lines, it was shown that the γ-Pcdhs act to repress Wnt signaling-induced gene transcription by β-catenin/TCF (Dallosso et al., 2009). Although the mechanisms by which the γ-Pcdhs might do so remain unclear, it is an intriguing possibility that Wnts, released by MNs and/or by vINs, promote the elaboration of Ia terminal arbors, while the γ-Pcdhs serve to limit this signal. If so, we might expect just the results that we describe here: loss of the γ-Pcdhs on sensory neurons leads to expansion of terminals and arbors on MNs. The fact that the γ-Pcdhs are not required in MNs themselves raises the possibility that, in addition to their role as homophilic adhesion molecules (Schreiner and Weiner, 2010), this diverse family can cell autonomously regulate signaling pathways during neural development. Future studies aimed at testing this hypothesis may uncover unexpected new roles for the γ-Pcdh family.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Leah Fuller and Claire Vernon for expert assistance with animal breeding and genotyping, and Dr. Louis Reichardt (UCSF) for the anti-TrkA antibody. This work was funded by a grant from the NIH to Joshua A. Weiner (R01 NS055272).
References
Alvarez, F. J., Jonas, P. C., Sapir, T., Hartley, R., Berrocal, M. C., Geiman, E. J., Todd, A. J., and Goulding, M. (2005). Postnatal phenotype and localization of spinal cord V1 derived interneurons. J. Comp. Neurol. 493, 177–192.
Arber, S., Han, B., Mendelsohn, M., Smith, M., Jessell, T. M., and Sockanathan, S. (1999). Requirement for the homeobox gene Hb9 in the consolidation of motor neuron identity. Neuron 23, 659–674.
Arber, S., Ladle, D. R., Lin, J. H., Frank, E., and Jessell, T. M. (2000). ETS gene Er81 controls the formation of functional connections between group Ia sensory afferents and motor neurons. Cell 101, 485–498.
Arikkath, J., and Reichardt, L. F. (2008). Cadherins and catenins at synapses: roles in synaptogenesis and synaptic plasticity. Trends Neurosci. 31, 487–494.
Burrill, J. D., Moran, L., Goulding, M. D., and Saueressig, H. (1997). PAX2 is expressed in multiple spinal cord interneurons, including a population of EN1+ interneurons that require PAX6 for their development. Development 124, 4493–4503.
Chen, H. H., Hippenmeyer, S., Arber, S., and Frank, E. (2003). Development of the monosynaptic stretch reflex circuit. Curr. Opin. Neurobiol. 13, 96–102.
Cheng, H. J., Bagri, A., Yaron, A., Stein, E., Pleasure, S. J., and Tessier-Lavigne, M. (2001). Plexin-A3 mediates semaphorin signaling and regulates the development of hippocampal axonal projections. Neuron 32, 249–263.
Coggeshall, R. E., Pover, C. M., and Fitzgerald, M. (1994). Dorsal root ganglion cell death and surviving cell numbers in relation to the development of sensory innervation in the rat hindlimb. Brain Res. Dev. Brain Res. 82, 193–212.
Cohen, S., Funkelstein, L., Livet, J., Rougon, G., Henderson, C. E., Castellani, V., and Mann, F. (2005). A semaphorin code defines subpopulations of spinal motor neurons during mouse development. Eur. J. Neurosci. 21, 1767–1776.
da Silva, S., and Wang, F. (2011). Retrograde neural circuit specification by target-derived neurotrophins and growth factors. Curr. Opin. Neurobiol. 21, 61–67.
Dalla Torre di Sanguinetto, S. A., Dasen, J. S., and Arber, S. (2008). Transcriptional mechanisms controlling motor neuron diversity and connectivity. Curr. Opin. Neurobiol. 18, 36–43.
Dallosso, A. R., Hancock, A. L., Szemes, M., Moorwood, K., Chilukamarri, L., Tsai, H. H., Sarkar, A., Barasch, J., Vuononvirta, R., Jones, C., Pritchard-Jones, K., Royer-Pokora, B., Lee, S. B., Owen, C., Malik, S., Feng, Y., Frank, M., Ward, A., Brown, K. W., and Malik, K. (2009). Frequent long-range epigenetic silencing of protocadherin gene clusters on chromosome 5q31 in Wilms’ tumor. PLoS Genet. 5, e1000745. doi: 10.1371/journal.pgen.1000745
Danielian, P. S., Muccino, D., Rowitch, D. H., Michael, S. K., and McMahon, A. P. (1998). Modification of gene activity in mouse embryos in utero by a tamoxifen-inducible form of cre recombinase. Curr. Biol. 8, 1323–1326.
Deckwerth, T. L., Elliott, J. L., Knudson, C. M., Johnson, E. M. Jr., Snider, W. D., and Korsmeyer, S. J. (1996). BAX is required for neuronal death after trophic factor deprivation and during development. Neuron 17, 401–411.
Fitzgerald, M. (1987). Cutaneous primary afferent properties in the hind limb of the neonatal rat. J. Physiol. (Lond.) 383, 79–92.
Frank, M., Ebert, M., Shan, W., Phillips, G. R., Arndt, K., Colman, D. R., and Kemler, R. (2005). Differential expression of individual gamma-protocadherins during mouse brain development. Mol. Cell. Neurosci. 29, 603–616.
Fu, S. Y., Sharma, K., Luo, Y., Raper, J. A., and Frank, E. (2000). SEMA3A regulates developing sensory projections in the chicken spinal cord. J. Neurobiol. 45, 227–236.
Garrett, A. M., and Weiner, J. A. (2009). Control of CNS synapse development by γ-protocadherin-mediated astrocyte-neuron contact. J. Neurosci. 29, 11723–11731.
Giagtzoglou, N., Ly, C. V., and Bellen, H. J. (2009). Cell adhesion, the backbone of the synapse: “vertebrate” and “invertebrate” perspectives. Cold Spring Harb Perspect. Biol. 4, a003079.
Goulding, M. (2009). Circuits controlling vertebrate locomotion: moving in a new direction. Nat. Rev. Neurosci. 10, 507–518.
Hasegawa, S., Hamada, S., Kumode, Y., Esumi, S., Katori, S., Fukuda, E., Uchiyama, Y., Hirabayashi, T., Mombaerts, P., and Yagi, T. (2008). The protocadherin-alpha family is involved in axonal coalescence of olfactory sensory neurons into glomeruli of the olfactory bulb in mouse. Mol. Cell. Neurosci. 38, 66–79.
Honda, C. N. (1995). Differential distribution of calbindin-D28k and parvalbumin in somatic and visceral sensory neurons. Neuroscience 68, 883–892.
Kaneko, R., Kato, H., Kawamura, Y., Esumi, S., Hirayama, T., Hirabayashi, T., and Yagi, T. (2006). Allelic gene regulation of Pcdh-alpha and Pcdh-gamma clusters involving both monoallelic and biallelic expression in single Purkinje cells. J. Biol. Chem. 41, 30551–30560.
Katori, S., Hamada, S., Noguchi, Y., Fukuda, E., Yamamoto, T., Yamamoto, H., Hasegawa, S., and Yagi, T. (2009). Protocadherin-alpha family is required for serotonergic projections to appropriately innervate target brain areas. J. Neurosci. 29, 9137–9147.
Knudson, C. M., Tung, K. S., Tourtellotte, W. G., Brown, G. A., and Korsmeyer, S. J. (1995). Bax-deficient mice with lymphoid hyperplasia and male germ cell death. Science 270, 96–99.
Koerber, H. R., and Mendell, L. M. (1992). “Functional heterogeneity of dorsal root ganglion cells,” in Sensory Neurons: Diversity, Development and Plasticity, ed. S. A. Scott (New York: Oxford University Press), 77–96.
Krylova, O., Herreros, J., Cleverley, K. E., Ehler, E., Henriquez, J. P., Hughes, S. M., and Salinas, P. C. (2002). WNT-3, expressed by motoneurons, regulates terminal arborization of neurotrophin-3-responsive spinal sensory neurons. Neuron 12, 1043–1056.
Lefebvre, J. L., Zhang, Y., Meister, M., Wang, X., and Sanes, J. R. (2008). gamma-Protocadherins regulate neuronal survival but are dispensable for circuit formation in retina. Development 135, 4141–4151.
Lewandoski, M., Meyers, E. N., and Martin, G. R. (1997). Analysis of Fgf8 gene function in vertebrate development. Cold Spring Harb. Symp. Quant. Biol. 62, 159–168.
Lewis, K. E. (2006). How do genes regulate simple behaviours? Understanding how different neurons in the vertebrate spinal cord are genetically specified. Philos. Trans. R. Soc. Lond. B Biol. Sci. 361, 45–66.
Lobas, M. A., Helsper, L., Vernon, C. G., Schreiner, D., Zhang, Y., Holtzman, M. J., Thedens, D. R., and Weiner, J. A. (2011). Molecular heterogeneity in the choroid plexus epithelium: the 22-member g-protocadherin family is differentially expressed, apically localized, and implicated in CSF regulation. J. Neurochem. (in press).
Margeta, M. A., Shen, K., and Grill, B. (2008). Building a synapse: lessons on synaptic specificity and presynaptic assembly from the nematode C. elegans. Curr. Opin. Neurobiol. 18, 69–76.
Mears, S. C., and Frank, E. (1997). Formation of specific monosynaptic connections between muscle spindle afferents and motoneurons in the mouse. J. Neurosci. 17, 3128–3135.
Messersmith, E. K., Leonardo, E. D., Shatz, C. J., Tessier-Lavigne, M., Goodman, C. S., and Kolodkin, A. L. (1995). Semaphorin III can function as a selective chemorepellent to pattern sensory projections in the spinal cord. Neuron 14, 949–959.
Mirnics, K., and Koerber, H. R. (1995). Prenatal development of rat primary afferent fibers: II. Central projections. J. Comp. Neurol. 355, 601–614.
Morishita, H., and Yagi, T. (2007). Protocadherin family: diversity, structure, and function. Curr. Opin. Cell Biol. 19, 584–592.
Mu, X., Silos-Santiago, I., Carroll, S. L., and Snider, W. D. (1993). Neurotrophin receptor genes are expressed in distinct patterns in developing dorsal root ganglia. J. Neurosci. 13, 4029–4041.
Ohyama, T., and Groves, A. K. (2004). Generation of Pax2-Cre mice by modification of a Pax2 bacterial artificial chromosome. Genesis 38, 195–199.
Pecho-Vrieseling, E., Sigrist, M., Yoshida, Y., Jessell, T. M., and Arber, S. (2009). Specificity of sensory-motor connections encoded by Sema3e-Plxnd1 recognition. Nature 459, 842–846.
Perrin, F. E., Rathjen, F. G., and Stoeckli, E. T. (2001). Distinct subpopulations of sensory afferents require F11 or axonin-1 for growth to their target layers within the spinal cord of the chick. Neuron 30, 707–723.
Phillips, G. R., Tanaka, H., Frank, M., Elste, A., Fidler, L., Benson, D. L., and Colman, D. R. (2003). Gamma-protocadherins are targeted to subsets of synapses and intracellular organelles in neurons. J. Neurosci. 23, 5096–5104.
Prasad, T., Wang, X., Gray, P. A., and Weiner, J. A. (2008). A differential developmental pattern of spinal interneuron apoptosis during synaptogenesis: insights from genetic analyses of the protocadherin-gamma gene cluster. Development 135, 4153–4164.
Price, S. R., De Marco Garcia, N. V., Ranscht, B., and Jessell, T. M. (2002). Regulation of motor neuron pool sorting by differential expression of type II cadherins. Cell 109, 205–216.
Rico, B., Xu, B., and Reichardt, L. F. (2002). TrkB receptor signaling is required for establishment of GABAergic synapses in the cerebellum. Nat. Neurosci. 5, 225–233.
Sapir, T., Geiman, E. J., Wang, Z., Velasquez, T., Mitsui, S., Yoshihara, Y., Frank, E., Alvarez, F. J., and Goulding, M. (2004). Pax6 and engrailed 1 regulate two distinct aspects of renshaw cell development. J. Neurosci. 24, 1255–1264.
Schreiner, D., and Weiner, J. A. (2010). Combinatorial homophilic interaction between gamma-protocadherin multimers greatly expands the molecular diversity of cell adhesion. Proc. Natl. Acad. Sci. U.S.A. 107, 14893–14898.
Takeichi, M. (2007). The cadherin superfamily in neuronal connections and interactions. Nat. Rev. Neurosci. 8, 11–20.
Tasic, B., Nabholz, C. E., Baldwin, K. K., Kim, Y., Rueckert, E. H., Ribich, S. A., Cramer, P., Wu, Q., Axel, R., and Maniatis, T. (2002). Promoter choice determines splice site selection in protocadherin alpha and gamma pre-mRNA splicing. Mol. Cell 10, 21–33.
Umemori, H., Linhoff, M. W., Ornitz, D. M., and Sanes, J. R. (2004). FGF22 and its close relatives are presynaptic organizing molecules in the mammalian brain. Cell 118, 257–270.
Wang, X., Su, H., and Bradley, A. (2002a). Molecular mechanisms governing Pcdh-gamma gene expression: evidence for a multiple promoter and cis-alternative splicing model. Genes Dev. 16, 1890–1905.
Wang, X., Weiner, J. A., Levi, S., Craig, A. M., Bradley, A., and Sanes, J. R. (2002b). Gamma protocadherins are required for survival of spinal interneurons. Neuron 36, 843–854.
Weiner, J. A. (2006). “Protocadherins and synapse development,” in Molecular Mechanisms of Synaptogenesis, eds A. Dityatev, and A. El-Husseini (New York: Springer), 137–162.
White, F. A., Keller-Peck, C. R., Knudson, C. M., Korsmeyer, S. J., and Snider, W. D. (1998). Widespread elimination of naturally occurring neuronal death in bax-deficient mice. J. Neurosci. 18, 1428–1439.
Wu, Q., and Maniatis, T. (1999). A striking organization of a large family of human neural cadherin-like cell adhesion genes. Cell 97, 779–790.
Wu, Q., Zhang, T., Cheng, J. F., Kim, Y., Grimwood, J., Schmutz, J., Dickson, M., Noonan, J. P., Zhang, M. Q., Myers, R. M., and Maniatis, T. (2001). Comparative DNA sequence analysis of mouse and human protocadherin gene clusters. Genome Res. 11, 389–404.
Yoshida, Y., Han, B., Mendelsohn, M., and Jessell, T. M. (2006). PlexinA1 signaling directs the segregation of proprioceptive sensory axons in the developing spinal cord. Neuron 52, 775–788.
Zipursky, S. L., and Sanes, J. R. (2010). Chemoaffinity revisited: dscams, protocadherins, and neural circuit assembly. Cell 143, 343–353.
Keywords: proprioception, axons, synaptogenesis, spinal cord, cell adhesion molecule, motor neuron, interneuron, apoptosis
Citation: Prasad T and Weiner JA (2011) Direct and indirect regulation of spinal cord Ia afferent terminal formation by the γ-Protocadherins. Front. Mol. Neurosci. 4:54. doi: 10.3389/fnmol.2011.00054
Received: 16 September 2011; Paper pending published: 02 November 2011;
Accepted: 03 December 2011; Published online: 23 December 2011.
Edited by:
Alistair N. Garratt, Max Delbrück Center for Molecular Medicine, GermanyReviewed by:
John Oberdick, The Ohio State University, USAZaven Kaprielian, Albert Einstein College of Medicine of Yeshiva University, USA
Copyright: © 2011 Prasad and Weiner. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Joshua A. Weiner, Department of Biology, The University of Iowa, 143 Biology Building, Iowa City, IA 52242, USA. e-mail:am9zaHVhLXdlaW5lckB1aW93YS5lZHU=
†Present address: Tuhina Prasad, Brain Research Centre, Department of Psychiatry, University of British Columbia, Vancouver, BC, Canada V6T 2B5. e-mail:dHVoaW5hLnByYXNhZEB1YmMuY2E=