

- Laboratory for Neuroregeneration and Repair, Center for Neurology, Hertie Institute for Clinical Brain Research, University of Tuebingen, Tuebingen, Germany
Following an acute central nervous system (CNS) injury, axonal regeneration and functional recovery are extremely limited. This is due to an extrinsic inhibitory growth environment and the lack of intrinsic growth competence. Retinoic acid (RA) signaling, essential in developmental dorsoventral patterning and specification of spinal motor neurons, has been shown through its receptor, the transcription factor RA receptor β2 (RARβ2), to induce axonal regeneration following spinal cord injury (SCI). Recently, it has been shown that in dorsal root ganglion neurons (DRGs), cAMP levels were greatly increased by lentiviral RARβ2 expression and contributed to neurite outgrowth. Moreover, RARβagonists, in cerebellar granule neurons (CGN) and in the brain in vivo, induced phosphoinositide 3-kinase dependent phosphorylation of AKT that was involved in RARβ-dependent neurite outgrowth. More recently, RA-RARβpathways were shown to directly transcriptionally repress a member of the inhibitory Nogo receptor (NgR) complex, Lingo-1, under an axonal growth inhibitory environment in vitro as well as following spinal injury in vivo. This perspective focuses on these newly discovered molecular mechanisms and future directions in the field.
Introduction
The limited regenerative capacity observed following an acute central nervous system (CNS) lesion, such as stroke or brain/spinal cord traumatic injuries, is due to both the presence of an extrinsic inhibitory growth environment and the lack of intrinsic growth factors. Interestingly, the peripheral nervous system (PNS) does regenerate to a certain extent following a lesion, which has led to further research to determine the distinction between the CNS and PNS. The inhibitory environment of the CNS is vastly different than that of the PNS (Richardson et al., 1980). Following PNS injury, an inflammatory response is activated, Schwann cells infiltrate the injured area, debris is removed, neurotrophic factors are released, regeneration is initiated, and sheathing of these growing axons occurs (George and Griffin, 1994). Conversely, the glia cells in the CNS, oligodendrocytes, and astrocytes, provide an inhibitory environment for growth (McKeon et al., 1991; Qiu et al., 2000; Domeniconi et al., 2002; Niederost et al., 2002; Yiu and He, 2006).
Specifically, the neuronal insulating layer, myelin, is fragmented following a CNS lesion, releasing the extrinsic inhibitory molecules, myelin associated glycoprotein (MAG), Nogo, and Oligodendrocyte myelin glycoprotein (OMgp) (DeBellard et al., 1996; Huber and Schwab, 2000; Wang et al., 2002; He et al., 2003) that inhibit axonal outgrowth and functional recovery following injury. These myelin proteins signal through the neuronal membrane bound Nogo Receptor (NgR) complex, which includes NgR1 (Chen et al., 2000; GrandPre et al., 2000), Lingo-1 (Mi et al., 2004), and p75NTR (Domeniconi et al., 2002; Wong et al., 2002) or TROY (Park et al., 2005). Myelin protein engagement of the NgR complex activates RhoA, which induces ROCK-dependent phosphorylation of cofilin, thus actin depolymerization and growth cone collapse (He and Koprivica, 2004). When the RhoA pathway has been blocked following a CNS lesion, regeneration has been observed (Lehmann et al., 1999; Dergham et al., 2002; Fournier et al., 2003). The Paired immunoglobulin-like receptor B (PirB) is another receptor with high affinity for myelin inhibitory molecules that mediates outgrowth inhibition through dephosphorylation of tropomyosin receptor kinase (Trk) neurotrophin receptors (Atwal et al., 2008; Fujita et al., 2011).
Although following a severe CNS lesion a glial scar forms to repair the site of blood brain barrier disruption and limit inflammation (Rolls et al., 2009) it also represents a physical barrier to axonal growth (McKeon et al., 1991). The glial scar consists mainly of reactive astrocytes proteoglycans, and collagen, of which collagen IV provides the basement membrane scaffold for chondroitin sulphate proteoglycans (CSPGs) to bind (McKeon et al., 1991). It has been shown that CSPGs, of the glial scar, are inhibitory to axonal outgrowth in culture via binding to the recently discovered receptor PTPsigma (McKeon et al., 1991; Snow et al., 1996; Shen et al., 2009). Furthermore, removing CSPG glycosaminoglycan chains with chondroitinase ABC (ChABC) promotes functional recovery after spinal cord injury (SCI) (Bradbury et al., 2002).
More recently, the axonal regeneration field has partially shifted to elucidating the intrinsic growth capacity of CNS neurons. Not surprisingly, many well-defined embryonic developmental pathways have become validated in adult stem cell proliferation, regeneration, and differentiation (Tsokos et al., 1987; Terenghi, 1999; Esposito et al., 2005; Harel and Strittmatter, 2006; Reimer et al., 2009). This supports the theory that regeneration can be accomplished by a revival of developmental signals. It is believed that CNS neurons possess the ability, albeit limited, to regenerate. For example, the growth of CNS neurons into a PNS environment is possible (David and Aguayo, 1981; Benfey and Aguayo, 1982). It has been shown that the elongation of axons is linked to de novo transcription, and that blocking transcription immediately after injury inhibits regeneration from occurring (Smith and Skene, 1997). Furthermore, regeneration associated genes (RAGs) have been found following PNS injury that are absent following CNS injury in adults (Makwana and Raivich, 2005). These include GAP-43 and CAP-23 (growth cone proteins) (Chong et al., 1994; Mason et al., 2002), Sprr1a and SCG-10 (cytoskeletal proteins) (Mason et al., 2002; Starkey et al., 2009), Galanin (neuropeptide) (Holmes et al., 2000), brain derived neurotrophic factor (BDNF) (Tonra et al., 1998), as well as Chl1 and Lgals1 (cell adhesion molecules) (Zhang et al., 2000), and galectin-1 (McGraw et al., 2004). Furthermore, following a conditioning lesion, upregulation of cAMP activates protein kinase A (PKA), which in turn phosphorylates the transcription factor cAMP response element-binding protein (CREB), inducing transcription thus leading to neurite outgrowth (Neumann et al., 2002; Qiu et al., 2002; Gao et al., 2004). Interestingly, it has been shown that when neurons mature they lose their ability to overcome an inhibitory environment and this transition coincides with a decrease in cAMP levels (Cai et al., 2002). Another transcription factor thought to be involved in the retrograde signal following a conditioning lesion is signal transducer and activator of transcription 3 (STAT3) (Bareyre et al., 2011), which is activated via phosphorylation by tyrosine kinase Janus kinase (JAKs) associated with the receptor subunit glycoprotein 130 (gp130) initiated by the growth factor ciliary neurotrophic factor (CNTF) (Schwaiger et al., 2000). When either CREB or STAT3 pathways are blocked, regeneration in the CNS following a conditioning lesion is abolished (Gao et al., 2004; Qiu et al., 2005). Furthermore, it has also been shown in vitro that the conditioning lesion effect is completely abolished in a gp130 conditional knockout (Hyatt Sachs et al., 2010). However, Il-6 has been shown to be sufficient but not necessary for conditioning lesion-dependent axonal regeneration (Cao et al., 2006).
A number of injury responsive transcription factors have also been shown to affect neurite outgrowth and axonal regeneration, such as p53, c-Jun, activating transcription factor 3 (ATF3), nuclear factor of activated T cells (NFATs), nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB), SRY-box containing gene 11 (Sox11), SnoN, and the Krüppel-like factor (KLF) family (reviewed in (Moore and Goldberg, 2011).
In addition to gene regulation it has been recently discovered that signaling pathways involving protein translation can induce axonal regeneration both in the PNS and CNS (Park et al., 2008; Christie et al., 2010; Liu et al., 2010). Classically known as a tumor suppressor, phosphatase and tensin homolog (PTEN), has been shown to counteract phosphoinositide 3-kinases' (PI3K) conversion of the lipid second messenger phosphatidylinositol (4,5) bisphosphate (PIP2) into phosphatidylinositol (3,4,5) triphosphate (PIP3) (Song et al., 2005). When PTEN is inhibited or deleted, there is an increase in activation of AKT by phosphorylation (Song et al., 2005). This in turn has led to an increase in axonal regeneration, which has been determined to be through the downstream activation of the mTOR leading to protein synthesis and cell growth (Guertin and Sabatini, 2007; Park et al., 2008; Christie et al., 2010; Liu et al., 2010).
Retinoic Acid Signaling Pathways in Axonal Regeneration
Retinoic acid (RA), a degradation product of retinol after food ingestion, is a lipophilic vitamin A derivative that readily transverses the blood-brain barrier (Le Doze et al., 2000), and is a ligand for several nuclear receptors such as RA receptors (RAR, α, β, or γ, each of which have several isoforms due to alternative splicing) that play a role as transcription factors (Chambon, 1996). RA signaling is dependent upon its nuclear availability, controlled among others by retinol-binding protein-1 (RBP1), which is the carrier protein involved in the transport of retinol from the liver storage site to peripheral tissue and by cellular retinol binding protein (CRBP), which is the intracellular carrier involved in intracellular movement of retinol (Le Doze et al., 2000). Interestingly, the administration of RA not only activates the transcription factor RARβ by direct binding that releases co-repressors from promoters and recruits co-activators, but also increases its gene expression (Sucov et al., 1990; Leid et al., 1992). Importantly, a cAMP response element was found on RARβ2 promoter, which binds CREB in response to cAMP and induces RARβ2 expression (Kruyt et al., 1992). In the presence of ligand, RA bound RARβ typically forms a heterodimer with retinoid X receptor (RXR, α, β, or γ) at RA response elements (RAREs) in gene promoters, recruits co-activators (CBP/p300, the CBP/p300 associated factor PCAF, SRC1, p160 pCIP, CoA, SWI/SNF, and ACTR) and activates transcription (Chambon, 1996; Glass and Rosenfeld, 2000). However, in the absence of ligand, RARβ binds DNA in concert with co-repressors (nCo-R, SMRT, HDAC, and mSin3) and inhibits transcription (Glass and Rosenfeld, 2000). There have been several documented cases where RA bound RARβ was found to occupy promoters independently from RXR and to repress transcription (Glass et al., 1989; Lipkin et al., 1992; Schoorlemmer et al., 1994). RA signaling typically involves direct transcriptional regulation, even though there are some less-defined cases involving non-transcriptional dependent RA signaling (Lopez-Carballo et al., 2002; Masia et al., 2007; Ohashi et al., 2009).
RA Signaling in Neurite Outgrowth and Axonal Regeneration
Classically involved in development, neuronal differentiation, ventral neural patterning, and motor neuron specification (Maden et al., 1996; Diez del Corral et al., 2003; Novitch et al., 2003; Sockanathan et al., 2003), more recently RA also became a prime candidate to induce neurite outgrowth in neurons. The addition of exogenous RA and/or RARβ2 has been shown to induce neurite outgrowth in vitro in dorsal root ganglion neurons (DRGs), adult cortical neurons, and postnatal cerebellar granule neurons (CGN) (Corcoran and Maden, 1999; Corcoran et al., 2000; Wong et al., 2006; Yip et al., 2006; Puttagunta et al., 2011). In embryonic and adult DRGs as well as CGN, RA upregulated the expression RARβ2 in correlation with induced neurite outgrowth (Corcoran and Maden, 1999; Corcoran et al., 2000; Puttagunta et al., 2011). Neurotrophins, such as nerve growth factor (NGF), essential in the development of the PNS (Piirsoo et al., 2010), have been shown to upregulate retinaldehyde dehydrogenase-2 (RALDH-2) and RARβ2 expression to induce neurite outgrowth of adult mouse DRGs (Corcoran and Maden, 1999). Moreover, a RARβ agonist, but not a RARα or RARγ agonist, induced neurite outgrowth from embryonic DRGs (Corcoran et al., 2000). Additionally, both cortex and DRG explants from adult rats treated for 14 days with a RARβ agonist induced neurite outgrowth in contrast to vehicle treated animals (Agudo et al., 2010). As a proof of principle that RA mediates its effect on neurite outgrowth exclusively through RARβ, it was shown in postnatal CGN that RARβ null CGN fail to extend neurites in response to RA (Puttagunta et al., 2011). Whereas, embryonic spinal cord explants respond to RA by inducing neurite outgrowth, adult spinal cord explants do not, and the limited RARβ2 expression in the adult spinal cord in comparison to the embryonic spinal cord is believed to be the reason for this difference (McCaffery and Drager, 1994; Zetterstrom et al., 1999; Corcoran et al., 2002; So et al., 2006; Yip et al., 2006). Furthermore, when RARβ2, yet not RARβ4, was virally introduced into the adult spinal cord in organotypic preparations, it induced neurite outgrowth (Corcoran et al., 2002). Therefore, it can be concluded that RA acts specifically through RARβ2 in inducing neurite outgrowth in both embryonic and adult neurons.
Having established RARβ2 as the receptor responsible for RA induction of neurite outgrowth, the next logical step was to show that overexpression of RARβ2 in vivo would induce axonal regeneration. RARβ2 lentiviral infection of adult rat DRGs induced axonal growth and functional recovery of injured sensory neurons into the dorsal root entry zone (DREZ) following dorsal root lesion (Wong et al., 2006). In support of these findings, it was also shown that RARβ2 null mice have less axonal regeneration following a peripheral nerve crush versus wildtype mice (So et al., 2006). Additionally, RARβ2 lentiviral infection of the sensorimotor cortex three weeks prior to spinal cord lesion induced axonal and functional regeneration of the cortical spinal tracts (CST) in adult rats (Yip et al., 2006). Finally, RARβ specific agonist, CD2019, brain treated rats following a dorsal column lesion had increased growth of CST axons and functional recovery of the forelimb versus those administered with vehicle (Agudo et al., 2010). These in vivo studies confirmed previous in vitro studies, showing that indeed RA-RARβ signaling is involved in neurite outgrowth and axonal regeneration.
The Molecular Mechanisms of RA Signaling in Axonal Regeneration
While it is known that RA signaling does induce neurite outgrowth and axonal regeneration only limited research has been done on the precise molecular mechanisms involved. It was shown that lentiviral RARβ2 expression induces neurite outgrowth in DRGs and increases cAMP levels (Wong et al., 2006). The effect of RARβ2 on neurite outgrowth was significantly decreased when an adenylate cyclase inhibitor, 2′,5′-dideoxyadenosine (DDA) or a cell–permanent inhibitor of cAMP-dependent PKA was used (Wong et al., 2006). While this suggests that cAMP is involved in RA signaling, it is not sufficient, as use of dibutyryl cAMP in adult DRGs in the DREZ model did not promote functional recovery as robustly as RARβ2 alone (Wong et al., 2006). In fact, it was shown in postnatal CGN that the positive affect of a RARβ agonist (CD2019) on neurite outgrowth was fully attenuated by a PI3K inhibitor but not a PKA inhibitor. Furthermore, it was shown that RARβ induced phosphorylation of AKT in CGN and in vivo, exclusively in injured neurons (Agudo et al., 2010). Interestingly, it has been previously shown that RA-RARβ2/RXR directly activates AKT during the differentiation of human neuroblastoma cells (Lopez-Carballo et al., 2002; Ohashi et al., 2009).
Recently, we have discovered a direct transcriptional target of RA signaling in neurite outgrowth in an inhibitory environment. RA signaling increases neurite outgrowth, decreases RhoA activation and inhibits Lingo-1 gene and protein expression in a myelin-inhibitory environment (however, not on a CSPG-inhibitory substrate) in CGN, specifically through RARβ. By in silico analysis, we discovered a RARE in the Lingo-1 promoter, which bound RARβ but not RXR upon RA treatment in a myelin-inhibitory environment. Moreover, we showed this RARE to be functional by a luciferase assay that when mutated did not respond to RA signaling. Furthermore, addition of Lingo-1 in RA-treated CGN in a myelin-inhibitory environment abrogated RA-RARβ induced neurite outgrowth. Finally, in vivo RA treatment decreased Lingo-1 protein expression following SCI in wildtype but not in RARβ null mice, providing physiological relevance to the in vitro findings (Puttagunta et al., 2011) (Figure 1A). Importantly, it has previously been shown that Lingo-1 antagonists promoted axonal sprouting, improved functional recovery, decreased RhoA activation, and increased oligodendrocyte and neuronal survival following rubrospinal or CST transection (Ji et al., 2006). It will be important to determine if this RA-dependent decrease in Lingo-1 is found exclusively in the neurons or also in the oligodendrocytes that make up the myelin sheathing and if RA signaling, such as seen with Lingo-1 inhibtion (Mi et al., 2005, 2007, 2009), leads to an increase in remyelination. In fact, for proper functionality following SCI, axons must re-grow, reinnervate their targets, and remyelinate their axons.
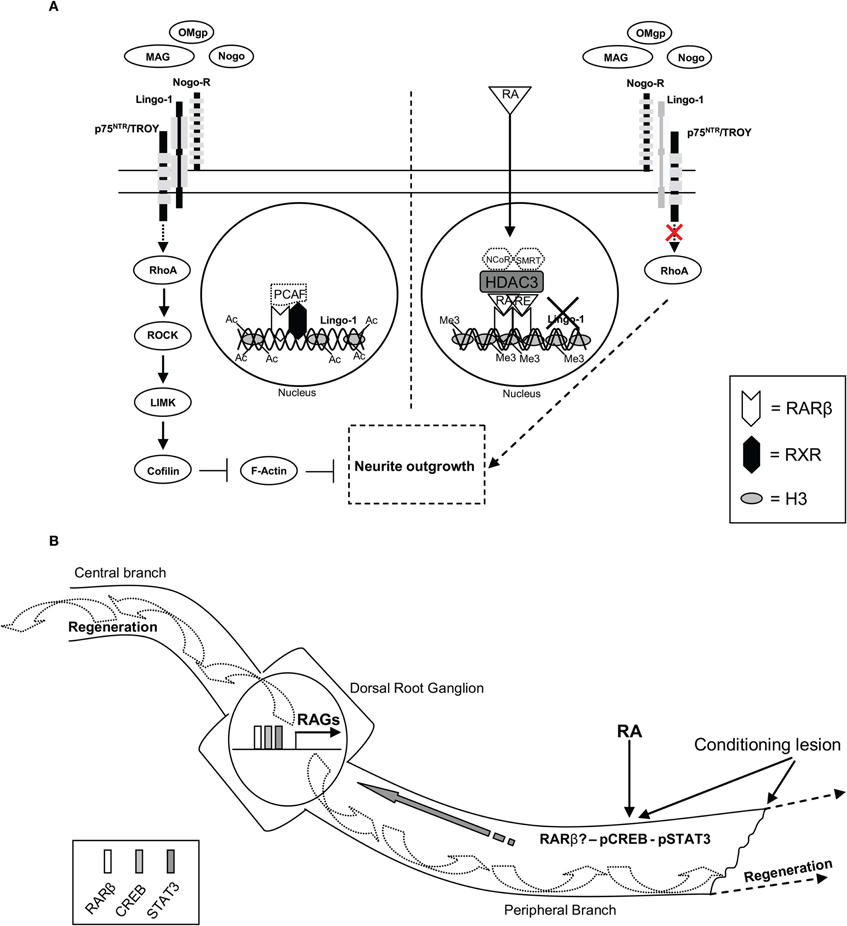
Figure 1. RA-RARβ dependent direct transcriptional repression of inhibitory myelin signaling and a hypothetical schematic of RA involvement in retrograde signaling, RAGs expression, and axonal regeneration. (A) Following RA treatment HDAC3 binding to the RARE of the Lingo-1 promoter decreases acetylation of H3 lysine 9 and possibly displaces PCAF binding. Meanwhile, an increase in methylation of H3 lysine 27 is observed, coupled with strong binding of a RARβhomodimer to the RARE of the Lingo-1 promoter repressing Lingo-1 expression. This is believed to displace the equilibrium of the NogoR complex and block the activation of RhoA, thus leading to neurite outgrowth. Other possible members of this transcriptional repressor complex may include NCoR and SMRT (indicated with dashed lines). (B) Following a conditioning peripheral lesion and RA treatment an increase in RARβ, pCREB, and pSTAT3 levels are observed. All three have been shown to be vital to PNS axonal regeneration. CREB and STAT3 have been shown to play a role in retrograde signaling involved in the induction of RAGs expression prior to axonal regeneration. It will be intriguing to examine if RA signaling activates these three transcription factors to work in concert regulating RAGs expression to induce both axonal CNS and PNS regeneration in vivo.
RA and Epigenetics
Epigenetics changes can affect gene regulation by modifying the surrounding chromatin environment. They include DNA methylation and a number of histone post-translational modifications such as phosphorylation, acetylation, and methylation. For example, methylation of promoters or histones often inhibits gene expression, yet acetylation of histones or transcription factors induces gene expression by promoting transcription factor occupancy within a more accessible chromatin environment (reviewed in (Kouzarides, 2007; Mueller and von Deimling, 2009). These epigenetic changes can be transient or more long lasting thereby allowing extrinsic and intrinsic cellular signals to immediately and strongly influence gene expression (Guasconi and Puri, 2009). The acetyltransferases, CBP/p300, are essential for mammalian cell proliferation and development (Yao et al., 1998). Importantly, p300 appears to be a critical cofactor for RAR transcriptional activity, even leading to differentiation and somitic symmetry (Kawasaki et al., 1998; Yao et al., 1998; Vilhais-Neto et al., 2010). Furthermore, in spinal motor neuron development, CBP is recruited by RA activated RAR to promoters for acetylation of histones, increasing accessibility of the promoter to transcription factors and inducing the expression of spinal motor neuron genes (Lee et al., 2009). RA signaling initiates PCAF binding to RAR/RXR heterodimers displacing co-repressor complexes and inducing gene expression (Blanco et al., 1998). Preliminary findings show (personal observations) that acetylation of histone 3 at lysine 9 (AcH3K9) at the RARE of the Lingo-1 promoter is significantly decreased upon RA treatment in a myelin-inhibitory environment. In accordance, we saw an increase in histone deacetylase 3 (HDAC3, known to form a repressor complex with NCoR/SMRT at RA regulated genes) (Hartman et al., 2005) on the RARE of the Lingo-1 promoter. In addition, we observed an increase in methylation on H3 at lysine 27 (H3K27Me3, also known to be previously linked to RA mediated differentiation) (Gillespie and Gudas, 2007). These observations further extend upon recent published work showing a decrease in Lingo-1 expression upon RA treatment in a myelin-inhibitory environment (Puttagunta et al., 2011) (Figure 1A), however, further examination is required to determine complete activator and repressor complexes involved in RA-dependent Lingo-1 regulation.
Future Perspectives
In order to achieve functional axonal regeneration and recovery following SCI several obstacles need to be overcome: (a) an inhibitory glial environment, (b) excessive inflammation, (c) lack of neuronal intrinsic capacity for axonal outgrowth, and (d) axonal demyelination. In fact, RA signaling seems to have the potential to tackle several of these limitations.
RA has long been known for its anti-inflammatory benefits (Orfanos and Bauer, 1983; Nozaki et al., 2006). Interestingly, RA has been shown to inhibit the interferon-gamma induced inflammatory response in primary rat brain cultured astrocytes by inducing suppressors of cytokine signaling (SOCS3) and inhibiting JAK and STAT3 activation (Choi et al., 2005). Thus, it will be of interest to elucidate weather RA treatment in vivo following SCI reduces the formation of a glial scar by inhibiting the inflammatory response.
As the RA-RARβ pathway seems to overcome the myelin inhibitory environment, but not necessarily proteoglycan inhibitory signaling, combining ChABC with enhancement of RA-RARβ should strongly reduce the inhibitory signaling following SCI and further promote axonal regeneration than either does alone.
Given that neuronal overexpression of RARβ promotes axonal sprouting and regeneration of the CST and dorsal columns after SCI, it will be interesting to explore whether RA-RARβ is able to enhance the intrinsic ability of CNS neurons to regenerate by driving the expression of specific RAGs. Besides a possible direct transcriptional role for RAGs, RA-RARβ signaling may be involved in the cAMP-dependent retrograde signal following a PNS lesion that induces RAGs expression (Qiu et al., 2002). In fact, RARβ null mice have significantly reduced neurite outgrowth following peripheral nerve crush compared to wildtype mice (So et al., 2006), and, as mentioned previously, cAMP is induced by RARβ overexpression and involved in its ability to induce neurite outgrowth (Wong et al., 2006). Moreover, STAT3 is thought to be a retrograde signal following a PNS lesion and its inhibition limits axonal regeneration (Qiu et al., 2002, 2005; Schweizer et al., 2002; Bareyre et al., 2011). While STAT3 is inhibited by RA signaling in astrocytes, conversely it is induced in a CNTF-dependent manner in differentiated neuroblatoma cells, retinal, and ciliary chick ganglion neurons (Malek and Halvorsen, 1997; Wang and Halvorsen, 1998). It will be of interest to inspect if RA-RARβ, CREB, and STAT3 transcriptional signaling work in concert to induce expression of RAGs and axonal regeneration (Figure 1B).
RA may also intersect with PTEN-mTOR signaling after SCI. The inhibition of PTEN results in the activation of AKT (pAKT) inducing axonal regeneration through mTOR by new protein synthesis and growth (Park et al., 2008). Accordingly, pAKT, which decreases after axonal injury (Liu et al., 2010), was shown to be only upregulated in injured CST axons following treatment with a RARβ agonist (Agudo et al., 2010). Therefore, it will be interesting to determine if AKT activation by RA signaling is through the inhibition of PTEN or the direct phosphorylation of AKT following axonal lesions and if this results in mTOR activation (Figure 2). In fact, dissection of this pro-regenerative molecular pathway may offer novel molecular targets for the effective enhancement of axonal regeneration.
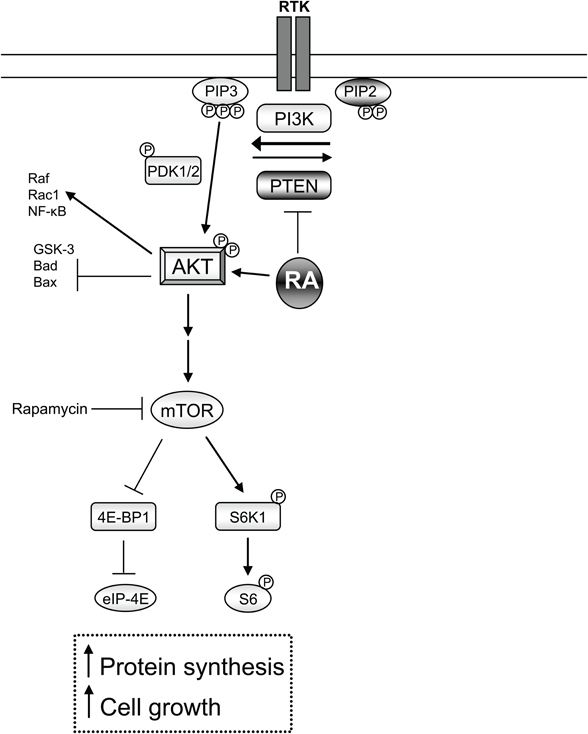
Figure 2. RA-dependent non-transcriptional involvement in neurite outgrowth. Inhibition of PTEN leads to increased levels of PIP3 via PI3K. This in turn activates AKT by phosphorylation. Active pAKT has many functions such as the inhibition of GSK-3, Bad, or Bax, conversely it can activate Raf, RacI, and NF-κB. Recently in axonal regeneration it has been shown that active pAKT leads to the activation of the mTOR pathway (which can be inhibited via rapamycin). This leads to increased protein synthesis and ribosome biogenesis as well as cell growth through the phosphorylation of the ribosomal protein S6 and the release of the eukaryotic initiation factor 4E (eIF-4E) from inhibition. RA-RARβsignaling has shown an inhibition of PTEN and a direct activation of pAKT in two separate studies. It will be of interest to examine if RA signaling induces downstream mTOR signaling in axonal regeneration.
Moreover, as RA signaling may take place via either paracrine or autocrine pathways and as Schwann cells, astrocytes, meningeal fibroblasts, and macrophages differentially express RA, RAR, or RALDH2 after PNS and CNS injury (Mey, 2006), further investigation of the relative weight of the two modes of action in axonal regeneration is warranted.
While RA pathways seem to positively affect several pathogenic aspects of SCI, further investigation is needed to clarify more direct transcriptional targets of RA-RARβ signaling following SCI and the interaction of RA pathways with other pro-regenerative signals. Only then, may we have the possibility to design RA signaling-dependent molecular therapies that may specifically enhance spinal axonal regeneration and recovery, with limited off target side effects.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank the Hertie Foundation, the DFG for financial support (funds granted to Simone Di Giovanni).
References
Agudo, M., Yip, P., Davies, M., Bradbury, E., Doherty, P., Mcmahon, S., Maden, M., and Corcoran, J. P. (2010). A retinoic acid receptor beta agonist (CD2019) overcomes inhibition of axonal outgrowth via phosphoinositide 3-kinase signalling in the injured adult spinal cord. Neurobiol. Dis. 37, 147–155.
Atwal, J. K., Pinkston-Gosse, J., Syken, J., Stawicki, S., Wu, Y., Shatz, C., and Tessier-Lavigne, M. (2008). PirB is a functional receptor for myelin inhibitors of axonal regeneration. Science 322, 967–970.
Bareyre, F. M., Garzorz, N., Lang, C., Misgeld, T., Buning, H., and Kerschensteiner, M. (2011). In vivo imaging reveals a phase-specific role of STAT3 during central and peripheral nervous system axon regeneration. Proc. Natl. Acad. Sci. U.S.A. 108, 6282–6287.
Benfey, M., and Aguayo, A. J. (1982). Extensive elongation of axons from rat brain into peripheral nerve grafts. Nature 296, 150–152.
Blanco, J. C., Minucci, S., Lu, J., Yang, X. J., Walker, K. K., Chen, H., Evans, R. M., Nakatani, Y., and Ozato, K. (1998). The histone acetylase PCAF is a nuclear receptor coactivator. Genes Dev. 12, 1638–1651.
Bradbury, E. J., Moon, L. D., Popat, R. J., King, V. R., Bennett, G. S., Patel, P. N., Fawcett, J. W., and Mcmahon, S. B. (2002). Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature 416, 636–640.
Cai, D., Deng, K., Mellado, W., Lee, J., Ratan, R. R., and Filbin, M. T. (2002). Arginase I and polyamines act downstream from cyclic AMP in overcoming inhibition of axonal growth MAG and myelin in vitro. Neuron 35, 711–719.
Cao, Z., Gao, Y., Bryson, J. B., Hou, J., Chaudhry, N., Siddiq, M., Martinez, J., Spencer, T., Carmel, J., Hart, R. B., and Filbin, M. T. (2006). The cytokine interleukin-6 is sufficient but not necessary to mimic the peripheral conditioning lesion effect on axonal growth. J. Neurosci. 26, 5565–5573.
Chen, M. S., Huber, A. B., Van Der Haar, M. E., Frank, M., Schnell, L., Spillmann, A. A., Christ, F., and Schwab, M. E. (2000). Nogo-A is a myelin-associated neurite outgrowth inhibitor and an antigen for monoclonal antibody IN-1. Nature 403, 434–439.
Choi, W. H., Ji, K. A., Jeon, S. B., Yang, M. S., Kim, H., Min, K. J., Shong, M., Jou, I., and Joe, E. H. (2005). Anti-inflammatory roles of retinoic acid in rat brain astrocytes: suppression of interferon-gamma-induced JAK/STAT phosphorylation. Biochem. Biophys. Res. Commun. 329, 125–131.
Chong, M. S., Reynolds, M. L., Irwin, N., Coggeshall, R. E., Emson, P. C., Benowitz, L. I., and Woolf, C. J. (1994). GAP-43 expression in primary sensory neurons following central axotomy. J. Neurosci. 14, 4375–4384.
Christie, K. J., Webber, C. A., Martinez, J. A., Singh, B., and Zochodne, D. W. (2010). PTEN inhibition to facilitate intrinsic regenerative outgrowth of adult peripheral axons. J. Neurosci. 30, 9306–9315.
Corcoran, J., and Maden, M. (1999). Nerve growth factor acts via retinoic acid synthesis to stimulate neurite outgrowth. Nat. Neurosci. 2, 307–308.
Corcoran, J., Shroot, B., Pizzey, J., and Maden, M. (2000). The role of retinoic acid receptors in neurite outgrowth from different populations of embryonic mouse dorsal root ganglia. J. Cell Sci. 113 (Pt. 14), 2567–2574.
Corcoran, J., So, P. L., Barber, R. D., Vincent, K. J., Mazarakis, N. D., Mitrophanous, K. A., Kingsman, S. M., and Maden, M. (2002). Retinoic acid receptor beta2 and neurite outgrowth in the adult mouse spinal cord in vitro. J. Cell Sci. 115, 3779–3786.
David, S., and Aguayo, A. J. (1981). Axonal elongation into peripheral nervous system “bridges” after central nervous system injury in adult rats. Science 214, 931–933.
DeBellard, M. E., Tang, S., Mukhopadhyay, G., Shen, Y. J., and Filbin, M. T. (1996). Myelin-associated glycoprotein inhibits axonal regeneration from a variety of neurons via interaction with a sialoglycoprotein. Mol. Cell. Neurosci. 7, 89–101.
Dergham, P., Ellezam, B., Essagian, C., Avedissian, H., Lubell, W. D., and Mckerracher, L. (2002). Rho signaling pathway targeted to promote spinal cord repair. J. Neurosci. 22, 6570–6577.
Diez del Corral, R., Olivera-Martinez, I., Goriely, A., Gale, E., Maden, M., and Storey, K. (2003). Opposing FGF and retinoid pathways control ventral neural pattern, neuronal differentiation, and segmentation during body axis extension. Neuron 40, 65–79.
Domeniconi, M., Cao, Z., Spencer, T., Sivasankaran, R., Wang, K., Nikulina, E., Kimura, N., Cai, H., Deng, K., Gao, Y., He, Z., and Filbin, M. (2002). Myelin-associated glycoprotein interacts with the Nogo66 receptor to inhibit neurite outgrowth. Neuron 35, 283–290.
Esposito, M. S., Piatti, V. C., Laplagne, D. A., Morgenstern, N. A., Ferrari, C. C., Pitossi, F. J., and Schinder, A. F. (2005). Neuronal differentiation in the adult hippocampus recapitulates embryonic development. J. Neurosci. 25, 10074–10086.
Fournier, A. E., Takizawa, B. T., and Strittmatter, S. M. (2003). Rho kinase inhibition enhances axonal regeneration in the injured CNS. J. Neurosci. 23, 1416–1423.
Fujita, Y., Endo, S., Takai, T., and Yamashita, T. (2011). Myelin suppresses axon regeneration by PIR-B/SHP-mediated inhibition of Trk activity. EMBO J. 30, 1389–1401.
Gao, Y., Deng, K., Hou, J., Bryson, J. B., Barco, A., Nikulina, E., Spencer, T., Mellado, W., Kandel, E. R., and Filbin, M. T. (2004). Activated CREB is sufficient to overcome inhibitors in myelin and promote spinal axon regeneration in vivo. Neuron 44, 609–621.
George, R., and Griffin, J. W. (1994). Delayed macrophage responses and myelin clearance during Wallerian degeneration in the central nervous system: the dorsal radiculotomy model. Exp. Neurol. 129, 225–236.
Gillespie, R. F., and Gudas, L. J. (2007). Retinoid regulated association of transcriptional co-regulators and the polycomb group protein SUZ12 with the retinoic acid response elements of Hoxa1, RARbeta(2), and Cyp26A1 in F9 embryonal carcinoma cells. J. Mol. Biol. 372, 298–316.
Glass, C. K., Lipkin, S. M., Devary, O. V., and Rosenfeld, M. G. (1989). Positive and negative regulation of gene transcription by a retinoic acid-thyroid hormone receptor heterodimer. Cell 59, 697–708.
Glass, C. K., and Rosenfeld, M. G. (2000). The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev. 14, 121–141.
GrandPre, T., Nakamura, F., Vartanian, T., and Strittmatter, S. M. (2000). Identification of the Nogo inhibitor of axon regeneration as a Reticulon protein. Nature 403, 439–444.
Guasconi, V., and Puri, P. L. (2009). Chromatin: the interface between extrinsic cues and the epigenetic regulation of muscle regeneration. Trends Cell Biol. 19, 286–294.
Guertin, D. A., and Sabatini, D. M. (2007). Defining the role of mTOR in cancer. Cancer Cell 12, 9–22.
Harel, N. Y., and Strittmatter, S. M. (2006). Can regenerating axons recapitulate developmental guidance during recovery from spinal cord injury? Nat. Rev. Neurosci. 7, 603–616.
Hartman, H. B., Yu, J., Alenghat, T., Ishizuka, T., and Lazar, M. A. (2005). The histone-binding code of nuclear receptor co-repressors matches the substrate specificity of histone deacetylase 3. EMBO Rep. 6, 445–451.
He, X. L., Bazan, J. F., Mcdermott, G., Park, J. B., Wang, K., Tessier-Lavigne, M., He, Z., and Garcia, K. C. (2003). Structure of the Nogo receptor ectodomain: a recognition module implicated in myelin inhibition. Neuron 38, 177–185.
He, Z., and Koprivica, V. (2004). The Nogo signaling pathway for regeneration block. Annu. Rev. Neurosci. 27, 341–368.
Holmes, F. E., Mahoney, S., King, V. R., Bacon, A., Kerr, N. C., Pachnis, V., Curtis, R., Priestley, J. V., and Wynick, D. (2000). Targeted disruption of the galanin gene reduces the number of sensory neurons and their regenerative capacity. Proc. Natl. Acad. Sci. U.S.A. 97, 11563–11568.
Huber, A. B., and Schwab, M. E. (2000). Nogo-A, a potent inhibitor of neurite outgrowth and regeneration. Biol. Chem. 381, 407–419.
Hyatt Sachs, H., Rohrer, H., and Zigmond, R. E. (2010). The conditioning lesion effect on sympathetic neurite outgrowth is dependent on gp130 cytokines. Exp. Neurol. 223, 516–522.
Ji, B., Li, M., Wu, W. T., Yick, L. W., Lee, X., Shao, Z., Wang, J., So, K. F., Mccoy, J. M., Pepinsky, R. B., Mi, S., and Relton, J. K. (2006). LINGO-1 antagonist promotes functional recovery and axonal sprouting after spinal cord injury. Mol. Cell. Neurosci. 33, 311–320.
Kawasaki, H., Eckner, R., Yao, T. P., Taira, K., Chiu, R., Livingston, D. M., and Yokoyama, K. K. (1998). Distinct roles of the co-activators p300 and CBP in retinoic-acid-induced F9-cell differentiation. Nature 393, 284–289.
Kruyt, F. A., Folkers, G., van Den Brink, C. E., and van Der Saag, P. T. (1992). A cyclic AMP response element is involved in retinoic acid-dependent RAR beta 2 promoter activation. Nucleic Acids Res. 20, 6393–6399.
Le Doze, F., Debruyne, D., Albessard, F., Barre, L., and Defer, G. L. (2000). Pharmacokinetics of all-trans retinoic acid, 13-cis retinoic acid, and fenretinide in plasma and brain of Rat. Drug Metab. Dispos. 28, 205–208.
Lee, S., Lee, B., Lee, J. W., and Lee, S. K. (2009). Retinoid signaling and neurogenin2 function are coupled for the specification of spinal motor neurons through a chromatin modifier CBP. Neuron 62, 641–654.
Lehmann, M., Fournier, A., Selles-Navarro, I., Dergham, P., Sebok, A., Leclerc, N., Tigyi, G., and Mckerracher, L. (1999). Inactivation of Rho signaling pathway promotes CNS axon regeneration. J. Neurosci. 19, 7537–7547.
Leid, M., Kastner, P., and Chambon, P. (1992). Multiplicity generates diversity in the retinoic acid signalling pathways. Trends Biochem. Sci. 17, 427–433.
Lipkin, S. M., Nelson, C. A., Glass, C. K., and Rosenfeld, M. G. (1992). A negative retinoic acid response element in the rat oxytocin promoter restricts transcriptional stimulation by heterologous transactivation domains. Proc. Natl. Acad. Sci. U.S.A. 89, 1209–1213.
Liu, K., Lu, Y., Lee, J. K., Samara, R., Willenberg, R., Sears-Kraxberger, I., Tedeschi, A., Park, K. K., Jin, D., Cai, B., Xu, B., Connolly, L., Steward, O., Zheng, B., and He, Z. (2010). PTEN deletion enhances the regenerative ability of adult corticospinal neurons. Nat. Neurosci. 13, 1075–1081.
Lopez-Carballo, G., Moreno, L., Masia, S., Perez, P., and Barettino, D. (2002). Activation of the phosphatidylinositol 3-kinase/Akt signaling pathway by retinoic acid is required for neural differentiation of SH-SY5Y human neuroblastoma cells. J. Biol. Chem. 277, 25297–25304.
Maden, M., Gale, E., Kostetskii, I., and Zile, M. (1996). Vitamin A-deficient quail embryos have half a hindbrain and other neural defects. Curr. Biol. 6, 417–426.
Makwana, M., and Raivich, G. (2005). Molecular mechanisms in successful peripheral regeneration. FEBS J. 272, 2628–2638.
Malek, R. L., and Halvorsen, S. W. (1997). Opposing regulation of ciliary neurotrophic factor receptors on neuroblastoma cells by distinct differentiating agents. J. Neurobiol. 32, 81–94.
Masia, S., Alvarez, S., De Lera, A. R., and Barettino, D. (2007). Rapid, nongenomic actions of retinoic acid on phosphatidylinositol-3-kinase signaling pathway mediated by the retinoic acid receptor. Mol. Endocrinol. 21, 2391–2402.
Mason, M. R., Lieberman, A. R., Grenningloh, G., and Anderson, P. N. (2002). Transcriptional upregulation of SCG10 and CAP-23 is correlated with regeneration of the axons of peripheral and central neurons in vivo. Mol. Cell. Neurosci. 20, 595–615.
McCaffery, P., and Drager, U. C. (1994). Hot spots of retinoic acid synthesis in the developing spinal cord. Proc. Natl. Acad. Sci. U.S.A. 91, 7194–7197.
McGraw, J., Oschipok, L. W., Liu, J., Hiebert, G. W., Mak, C. F., Horie, H., Kadoya, T., Steeves, J. D., Ramer, M. S., and Tetzlaff, W. (2004). Galectin-1 expression correlates with the regenerative potential of rubrospinal and spinal motoneurons. Neuroscience 128, 713–719.
McKeon, R. J., Schreiber, R. C., Rudge, J. S., and Silver, J. (1991). Reduction of neurite outgrowth in a model of glial scarring following CNS injury is correlated with the expression of inhibitory molecules on reactive astrocytes. J. Neurosci. 11, 3398–3411.
Mey, J. (2006). New therapeutic target for CNS injury? The role of retinoic acid signaling after nerve lesions. J. Neurobiol. 66, 757–779.
Mi, S., Hu, B., Hahm, K., Luo, Y., Kam Hui, E. S., Yuan, Q., Wong, W. M., Wang, L., Su, H., Chu, T. H., Guo, J., Zhang, W., So, K. F., Pepinsky, B., Shao, Z., Graff, C., Garber, E., Jung, V., Wu, E. X., and Wu, W. (2007). LINGO-1 antagonist promotes spinal cord remyelination and axonal integrity in MOG-induced experimental autoimmune encephalomyelitis. Nat. Med. 13, 1228–1233.
Mi, S., Lee, X., Shao, Z., Thill, G., Ji, B., Relton, J., Levesque, M., Allaire, N., Perrin, S., Sands, B., Crowell, T., Cate, R. L., Mccoy, J. M., and Pepinsky, R. B. (2004). LINGO-1 is a component of the Nogo-66 receptor/p75 signaling complex. Nat. Neurosci. 7, 221–228.
Mi, S., Miller, R. H., Lee, X., Scott, M. L., Shulag-Morskaya, S., Shao, Z., Chang, J., Thill, G., Levesque, M., Zhang, M., Hession, C., Sah, D., Trapp, B., He, Z., Jung, V., Mccoy, J. M., and Pepinsky, R. B. (2005). LINGO-1 negatively regulates myelination by oligodendrocytes. Nat. Neurosci. 8, 745–751.
Mi, S., Miller, R. H., Tang, W., Lee, X., Hu, B., Wu, W., Zhang, Y., Shields, C. B., Zhang, Y., Miklasz, S., Shea, D., Mason, J., Franklin, R. J., Ji, B., Shao, Z., Chedotal, A., Bernard, F., Roulois, A., Xu, J., Jung, V., and Pepinsky, B. (2009). Promotion of central nervous system remyelination by induced differentiation of oligodendrocyte precursor cells. Ann. Neurol. 65, 304–315.
Moore, D. L., and Goldberg, J. L. (2011). Multiple transcription factor families regulate axon growth and regeneration. Dev. Neurobiol. 71, 1186–1211.
Mueller, W. C., and von Deimling, A. (2009). Gene regulation by methylation. Recent Results Cancer Res. 171, 217–239.
Neumann, S., Bradke, F., Tessier-Lavigne, M., and Basbaum, A. I. (2002). Regeneration of sensory axons within the injured spinal cord induced by intraganglionic cAMP elevation. Neuron 34, 885–893.
Niederost, B., Oertle, T., Fritsche, J., Mckinney, R. A., and Bandtlow, C. E. (2002). Nogo-A and myelin-associated glycoprotein mediate neurite growth inhibition by antagonistic regulation of RhoA and Rac1. J. Neurosci. 22, 10368–10376.
Novitch, B. G., Wichterle, H., Jessell, T. M., and Sockanathan, S. (2003). A requirement for retinoic acid-mediated transcriptional activation in ventral neural patterning and motor neuron specification. Neuron 40, 81–95.
Nozaki, Y., Yamagata, T., Sugiyama, M., Ikoma, S., Kinoshita, K., and Funauchi, M. (2006). Anti-inflammatory effect of all-trans-retinoic acid in inflammatory arthritis. Clin. Immunol. 119, 272–279.
Ohashi, E., Kogai, T., Kagechika, H., and Brent, G. A. (2009). Activation of the PI3 kinase pathway by retinoic acid mediates sodium/iodide symporter induction and iodide transport in MCF-7 breast cancer cells. Cancer Res. 69, 3443–3450.
Orfanos, C. E., and Bauer, R. (1983). Evidence for anti-inflammatory activities of oral synthetic retinoids: experimental findings and clinical experience. Br. J. Dermatol. 109 (Suppl. 25), 55–60.
Park, J. B., Yiu, G., Kaneko, S., Wang, J., Chang, J., He, X. L., Garcia, K. C., and He, Z. (2005). A TNF receptor family member, TROY, is a coreceptor with Nogo receptor in mediating the inhibitory activity of myelin inhibitors. Neuron 45, 345–351.
Park, K. K., Liu, K., Hu, Y., Smith, P. D., Wang, C., Cai, B., Xu, B., Connolly, L., Kramvis, I., Sahin, M., and He, Z. (2008). Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science 322, 963–966.
Piirsoo, M., Kaljas, A., Tamm, K., and Timmusk, T. (2010). Expression of NGF and GDNF family members and their receptors during peripheral nerve development and differentiation of Schwann cells in vitro. Neurosci. Lett. 469, 135–140.
Puttagunta, R., Schmandke, A., Floriddia, E., Gaub, P., Fomin, N., Ghyselinck, N. B., and Di Giovanni, S. (2011). RA-RAR-beta counteracts myelin-dependent inhibition of neurite outgrowth via Lingo-1 repression. J. Cell Biol. 193, 1147–1156.
Qiu, J., Cafferty, W. B., Mcmahon, S. B., and Thompson, S. W. (2005). Conditioning injury-induced spinal axon regeneration requires signal transducer and activator of transcription 3 activation. J. Neurosci. 25, 1645–1653.
Qiu, J., Cai, D., Dai, H., Mcatee, M., Hoffman, P. N., Bregman, B. S., and Filbin, M. T. (2002). Spinal axon regeneration induced by elevation of cyclic AMP. Neuron 34, 895–903.
Qiu, J., Cai, D., and Filbin, M. T. (2000). Glial inhibition of nerve regeneration in the mature mammalian CNS. Glia 29, 166–174.
Reimer, M. M., Kuscha, V., Wyatt, C., Sorensen, I., Frank, R. E., Knuwer, M., Becker, T., and Becker, C. G. (2009). Sonic hedgehog is a polarized signal for motor neuron regeneration in adult zebrafish. J. Neurosci. 29, 15073–15082.
Richardson, P. M., Mcguinness, U. M., and Aguayo, A. J. (1980). Axons from CNS neurons regenerate into PNS grafts. Nature 284, 264–265.
Rolls, A., Shechter, R., and Schwartz, M. (2009). The bright side of the glial scar in CNS repair. Nat. Rev. Neurosci. 10, 235–241.
Schoorlemmer, J., Van Puijenbroek, A., van Den Eijnden, M., Jonk, L., Pals, C., and Kruijer, W. (1994). Characterization of a negative retinoic acid response element in the murine Oct4 promoter. Mol. Cell. Biol. 14, 1122–1136.
Schwaiger, F. W., Hager, G., Schmitt, A. B., Horvat, A., Hager, G., Streif, R., Spitzer, C., Gamal, S., Breuer, S., Brook, G. A., Nacimiento, W., and Kreutzberg, G. W. (2000). Peripheral but not central axotomy induces changes in Janus kinases (JAK) and signal transducers and activators of transcription (STAT). Eur. J. Neurosci. 12, 1165–1176.
Schweizer, U., Gunnersen, J., Karch, C., Wiese, S., Holtmann, B., Takeda, K., Akira, S., and Sendtner, M. (2002). Conditional gene ablation of Stat3 reveals differential signaling requirements for survival of motoneurons during development and after nerve injury in the adult. J. Cell Biol. 156, 287–297.
Shen, Y., Tenney, A. P., Busch, S. A., Horn, K. P., Cuascut, F. X., Liu, K., He, Z., Silver, J., and Flanagan, J. G. (2009). PTPsigma is a receptor for chondroitin sulfate proteoglycan, an inhibitor of neural regeneration. Science 326, 592–596.
Smith, D. S., and Skene, J. H. (1997). A transcription-dependent switch controls competence of adult neurons for distinct modes of axon growth. J. Neurosci. 17, 646–658.
Snow, D. M., Brown, E. M., and Letourneau, P. C. (1996). Growth cone behavior in the presence of soluble chondroitin sulfate proteoglycan (CSPG), compared to behavior on CSPG bound to laminin or fibronectin. Int. J. Dev. Neurosci. 14, 331–349.
So, P. L., Yip, P. K., Bunting, S., Wong, L. F., Mazarakis, N. D., Hall, S., Mcmahon, S., Maden, M., and Corcoran, J. P. (2006). Interactions between retinoic acid, nerve growth factor and sonic hedgehog signalling pathways in neurite outgrowth. Dev. Biol. 298, 167–175.
Sockanathan, S., Perlmann, T., and Jessell, T. M. (2003). Retinoid receptor signaling in postmitotic motor neurons regulates rostrocaudal positional identity and axonal projection pattern. Neuron 40, 97–111.
Song, G., Ouyang, G., and Bao, S. (2005). The activation of Akt/PKB signaling pathway and cell survival. J. Cell. Mol. Med. 9, 59–71.
Starkey, M. L., Davies, M., Yip, P. K., Carter, L. M., Wong, D. J., Mcmahon, S. B., and Bradbury, E. J. (2009). Expression of the regeneration-associated protein SPRR1A in primary sensory neurons and spinal cord of the adult mouse following peripheral and central injury. J. Comp. Neurol. 513, 51–68.
Sucov, H. M., Murakami, K. K., and Evans, R. M. (1990). Characterization of an autoregulated response element in the mouse retinoic acid receptor type beta gene. Proc. Natl. Acad. Sci. U.S.A. 87, 5392–5396.
Terenghi, G. (1999). Peripheral nerve regeneration and neurotrophic factors. J. Anat. 194 (Pt. 1), 1–14.
Tonra, J. R., Curtis, R., Wong, V., Cliffer, K. D., Park, J. S., Timmes, A., Nguyen, T., Lindsay, R. M., Acheson, A., and Distefano, P. S. (1998). Axotomy upregulates the anterograde transport and expression of brain-derived neurotrophic factor by sensory neurons. J. Neurosci. 18, 4374–4383.
Tsokos, M., Scarpa, S., Ross, R. A., and Triche, T. J. (1987). Differentiation of human neuroblastoma recapitulates neural crest development. Study of morphology, neurotransmitter enzymes, and extracellular matrix proteins. Am. J. Pathol. 128, 484–496.
Vilhais-Neto, G. C., Maruhashi, M., Smith, K. T., Vasseur-Cognet, M., Peterson, A. S., Workman, J. L., and Pourquie, O. (2010). Rere controls retinoic acid signalling and somite bilateral symmetry. Nature 463, 953–957.
Wang, K. C., Koprivica, V., Kim, J. A., Sivasankaran, R., Guo, Y., Neve, R. L., and He, Z. (2002). Oligodendrocyte-myelin glycoprotein is a Nogo receptor ligand that inhibits neurite outgrowth. Nature 417, 941–944.
Wang, X., and Halvorsen, S. W. (1998). Retinoic acid up-regulates ciliary neurotrophic factor receptors in cultured chick neurons and cardiomyocytes. Neurosci. Lett. 240, 9–12.
Wong, L. F., Yip, P. K., Battaglia, A., Grist, J., Corcoran, J., Maden, M., Azzouz, M., Kingsman, S. M., Kingsman, A. J., Mazarakis, N. D., and Mcmahon, S. B. (2006). Retinoic acid receptor beta2 promotes functional regeneration of sensory axons in the spinal cord. Nat. Neurosci. 9, 243–250.
Wong, S. T., Henley, J. R., Kanning, K. C., Huang, K. H., Bothwell, M., and Poo, M. M. (2002). A p75(NTR) and Nogo receptor complex mediates repulsive signaling by myelin-associated glycoprotein. Nat. Neurosci. 5, 1302–1308.
Yao, T. P., Oh, S. P., Fuchs, M., Zhou, N. D., Ch'ng, L. E., Newsome, D., Bronson, R. T., Li, E., Livingston, D. M., and Eckner, R. (1998). Gene dosage-dependent embryonic development and proliferation defects in mice lacking the transcriptional integrator p300. Cell 93, 361–372.
Yip, P. K., Wong, L. F., Pattinson, D., Battaglia, A., Grist, J., Bradbury, E. J., Maden, M., Mcmahon, S. B., and Mazarakis, N. D. (2006). Lentiviral vector expressing retinoic acid receptor beta2 promotes recovery of function after corticospinal tract injury in the adult rat spinal cord. Hum. Mol. Genet. 15, 3107–3118.
Yiu, G., and He, Z. (2006). Glial inhibition of CNS axon regeneration. Nat. Rev. Neurosci. 7, 617–627.
Zetterstrom, R. H., Lindqvist, E., Mata De Urquiza, A., Tomac, A., Eriksson, U., Perlmann, T., and Olson, L. (1999). Role of retinoids in the CNS: differential expression of retinoid binding proteins and receptors and evidence for presence of retinoic acid. Eur. J. Neurosci. 11, 407–416.
Keywords: RA, RARβ, axonal regeneration
Citation: Puttagunta R and Di Giovanni S (2012) Retinoic acid signaling in axonal regeneration. Front. Mol. Neurosci. 4:59. doi: 10.3389/fnmol.2011.00059
Received: 31 October 2011; Accepted: 22 December 2011;
Published online: 03 January 2012.
Edited by:
Gennadij Raivich, University College London, UKReviewed by:
Hans W. Müller, Heinrich-Heine-University Düsseldorf, GermanyRichard Zigmond, Case Western Reserve University School of Medicine, USA
Copyright: © 2012 Puttagunta and Di Giovanni. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Radhika Puttagunta and Simone Di Giovanni, Laboratory for Neuroregeneration and Repair, Hertie Institute for Clinical Brain Research, University of Tuebingen, Otfried-Mueller Strasse 27, D-72076 Tuebingen, Germany. e-mail:cmFkaGlrYS5wdXR0YWd1bnRhQG1lZGl6aW4udW5pLXR1ZWJpbmdlbi5kZQ==;c2ltb25lLmRpZ2lvdmFubmlAbWVkaXppbi51bmktdHVlYmluZ2VuLmRl