
- Department of Neurology, F. M. Kirby Neurobiology Center, Children’s Hospital Boston, Boston, MA, USA
Trauma in the adult mammalian central nervous system leads to irreversible structural and functional impairment due to failed regeneration attempts. In contrast, neurons in the peripheral nervous system exhibit a greater regenerative ability. It has been proposed that an orchestrated sequence of transcriptional events controlling the expression of specific sets of genes may be the underlying basis of an early cell-autonomous regenerative response. Understanding whether transcriptional fine tuning, in parallel with strategies aimed at counteracting extrinsic impediments promotes axon re-growth following central nervous system injuries represents an exciting challenge for future studies. Transcriptional pathways controlling axon regeneration are presented and discussed in this review.
Introduction
In addition to controlling a wide variety of cellular functions, evidence has shown that gene transcription acts as an important regulator of axon growth during development (Butler and Tear, 2007) and in response to axonal injury (Goldberg et al., 2002; Raivich et al., 2004; Moore et al., 2009). During neuronal development, transcriptional pathways regulating genes that control axon growth are particularly dynamic. Notably, this correlates with the ability of immature neurons to synthesize cytoskeletal elements and growth cone components, in addition to integrating extracellular guidance cues required during axonal elongation (Butler and Tear, 2007). Once axons reach their target, however, growth cones develop into a pre-synaptic terminal, turning off the transcriptional machinery controlling intrinsic growth programs (Baizer and Fishman, 1987; Caroni and Becker, 1992).
In contrast to immature neurons, adult CNS neurons are growth-incompetent and do not spontaneously regenerate injured axons. Is this developmental decline reversible? If so, is activation of pro-regenerative transcriptional events sufficient to regain growth abilities in adult CNS neurons? Early work done by Smith and Skene (1997) has demonstrated the presence of a transcription-dependent switch controlling growth competence in adult sensory neurons.
Primary sensory neurons with cell bodies in the DRG develop a bipolar axon that divides into two branches: one innervating peripheral targets (peripheral branch) and the other projecting into the spinal cord (central branch). Although peripheral and central axons originate from the same cell body, their respective injury-related responses differ. While the peripheral axon can regenerate and successfully re-innervate its targets, the central axon fails to attain successful regeneration within the CNS. The presence of a hostile environment encountered in the CNS partially explains this failure (Filbin, 2003; Schwab, 2004; Silver and Miller, 2004). However, neutralization of extracellular molecules that inhibit axon growth by itself has yielded limited axon regeneration (Case and Tessier-Lavigne, 2005; Harel and Strittmatter, 2006; Yiu and He, 2006; Lee et al., 2010), suggesting that removal of these inhibitory influences may not be sufficient to promote axon regeneration in the adult CNS. These studies have also highlighted the importance of overcoming the intrinsic impediments to axon re-growth as a strategy for inducing regeneration in the adult CNS (Goldberg et al., 2002; Benowitz and Yin, 2007; Di Giovanni, 2009; Liu et al., 2011).
In the last decade, the preconditioning lesion of DRG neurons has been used extensively to elucidate transcriptional pathways regulating regeneration in the adult nervous system. Importantly, this preconditioning lesion dramatically increases the intrinsic growth state, thus allowing adult sensory neurons to mount a successful regenerative response to a second lesion occurring at either the peripheral or the central branches (Richardson and Issa, 1984; Neumann and Woolf, 1999). By using a reversible inhibitor of RNA polymerase II, early work has shown that a discrete period of new transcription is essential to gain growth competence during the first hours after axotomy (Smith and Skene, 1997). Generation and shuttling of posttraumatic signals contribute to the activation of TFs through specific PTMs, and consequent nuclear translocation activates transcription of downstream targets.
In regenerating neurons, injury-induced gene transcription is the first step leading to the expression of regenerative-associated genes such as CAP-23, GAP-43, SPRR1A (Skene and Willard, 1981; Bosse et al., 2001, 2006; Schmitt et al., 2003). In addition, profound transcription-dependent changes in gene expression have been found only after severing the peripheral but not the central branch of sensory neurons (Costigan et al., 2002; Xiao et al., 2002; Bareyre and Schwab, 2003; Hoffman, 2010). Initiation of transcription is tightly controlled by a collection of TFs and co-factors that mediate the binding of RNA polymerase to specific DNA regulatory regions upstream to the transcription start site (Figure 1). Importantly, the expression of several of these TFs and co-factors changes after peripheral nerve injury, likely underlying a role in orchestrating a regenerative cell body response (Schwaiger et al., 2000; Makwana and Raivich, 2005; Qiu et al., 2005; Chen et al., 2007; Raivich and Makwana, 2007; Stam et al., 2007; Tedeschi et al., 2009a; Smith et al., 2011). It is becoming apparent that early activation of specific transcriptional pathways is likely to be one of the first steps required to mount a cell-autonomous regenerative response.
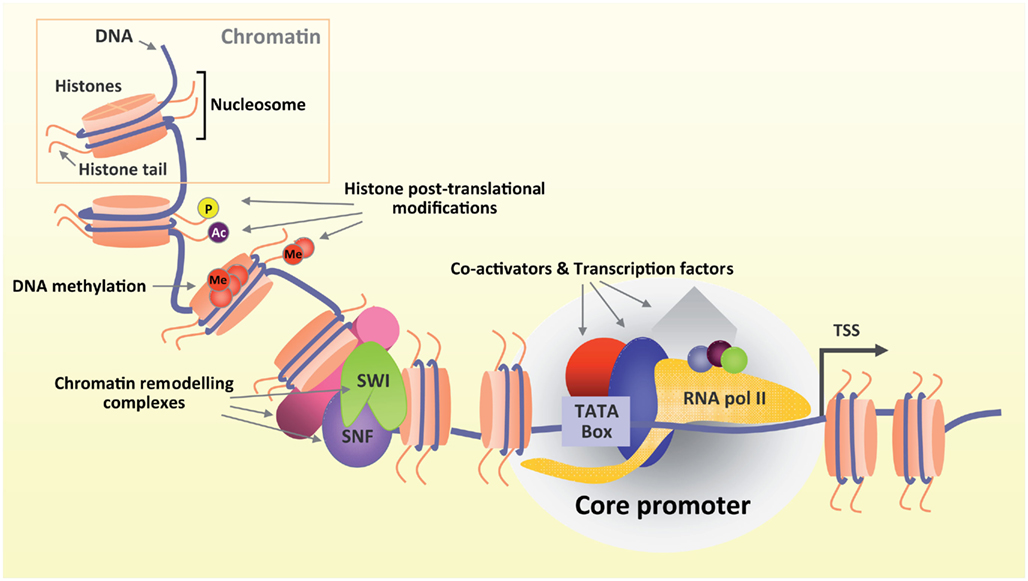
Figure 1. Gene transcription is a highly regulated process. DNA is wrapped around histones in chromatin (upper box). Several modifying complexes remodel chromatin and modify histones. Both DNA and histone tails undergo post translational modifications (Ac, acetylation; Me, methylation; P, phosphorylation) that ultimately shape chromatin architecture in a more or less favorable environment promoting or inhibiting transcription respectively. Transcription starts when TFs and co-activators bind to specific DNA regulatory elements upstream to the transcriptional start site (TSS) and recruit RNA polymerase II.
CREB-Mediated Transcriptional Pathway
In addition to controlling cellular metabolism, growth factor dependent cell survival, development and plasticity of neurons, CREB-dependent transcription has been shown to control axon regeneration in both the PNS and CNS (Cai et al., 2002; Gao et al., 2004; Figure 2).
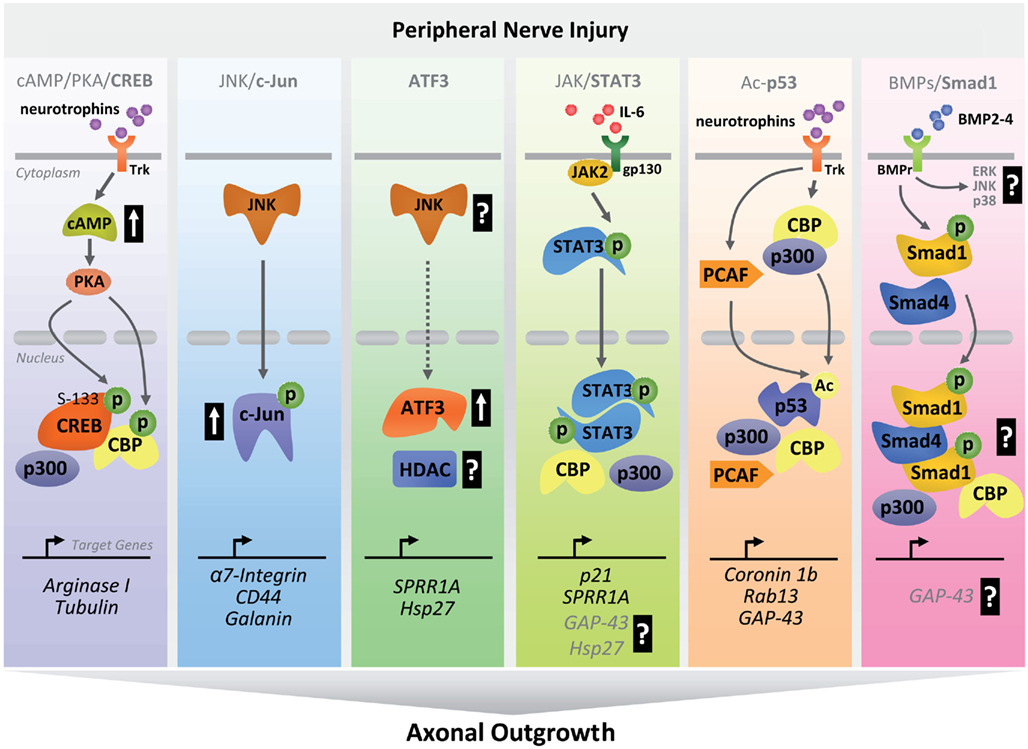
Figure 2. Schematic diagram summarizing transcriptional pathways involved in peripheral nerve regeneration. After peripheral nerve injury, activation of early clusters of TFs occurs through coordinated sequences of intracellular cascades. Upon activation, TFs and co-activators translocate to the nucleus where they bind to regulatory regions of regenerative-associated genes to drive their expression as part of an early pro-regenerative program (P, phosphorylation; Ac, acetylation; ERK, extracellular-signal-regulated kinase; p38, p38-mitogen-activated protein kinase).
Upon peripheral lesion, adult DRG neurons experience a transient increase in cAMP levels with consequent activation of PKA, which in turn phosphorylates CREB (Qiu et al., 2002; Gao et al., 2004; Teng and Tang, 2006; Hannila and Filbin, 2008). Overexpression of constitutively active CREB promotes regeneration of ascending dorsal column axons after spinal cord injury (Gao et al., 2004). CREB upregulates Arginase I, which in turn promotes polyamines synthesis. Polyamines are thought to influence axon growth by interacting with cytoskeletal elements like tubulin. A recent screening has identified daidzein as novel transcriptional activator of Arginase I capable of promoting some extent of CNS regeneration in a cAMP/CREB independent manner (Ma et al., 2010).
Overexpression of a dominant negative CREB results in decreased neurite outgrowth in vitro with consequent failure of cAMP to overcome growth inhibition in the presence of an inhibitory substrate (Redmond et al., 2002).
CREB functions in a stimulus and cellular context-dependent manner. Multiple PTMs lead to conformational changes that ultimately affect protein–protein interactions with co-factors. Nerve injury triggers calcium influx that eventually leads to phosphorylation of CREB at Ser-133, Ser-142, and Ser-143, which figures prominently in CREB-dependent transcription (Sun et al., 1994; Kornhauser et al., 2002). In addition, neurotrophin dependent phosphorylation on Ser-133 (Riccio et al., 1997, 2006; Watson et al., 1999, 2001; Lonze and Ginty, 2002; Arthur et al., 2004; Spencer et al., 2008) is required for recruitment of the co-activator CBP (Chrivia et al., 1993; Kwok et al., 1994; Figure 2). Of note, the assembly of a proper transcriptional module is necessary for recruitment of RNA polymerase II holoenzyme and dictates promoter occupancy of specific set of genes. Using a genome-wide approach, CREB-mediated promoter occupancy has been found to depend on the presence and methylation state of consensus cAMP response elements upstream to the transcription start site (Zhang et al., 2005). Acetyltransferases like CBP may be responsible for a methyl-acetylation switch, allowing CREB-transcription module to access repressed chromatin. So far, the language of the crosstalk between CREB PTMs and context-dependent histone/DNA modifications has not been deciphered yet and deserves attention for future studies.
c-Jun-Mediated Transcriptional Pathway
As a component of the heterodimeric AP-1 transcription factor (Jochum et al., 2001), c-Jun activity is strongly induced in response to numerous signals such as growth factors, cytokines and injury-related stress (Herdegen et al., 1997).
Upregulation of c-Jun is consistently accompanied by a successful regeneration response in several injury models (Herdegen et al., 1991; Jenkins and Hunt, 1991; Lindwall and Kanje, 2005). Thus far, the effects of c-Jun-mediated transcription in promoting nerve regeneration are well characterized (Raivich et al., 2004; Makwana and Raivich, 2005).
C-Jun activity is controlled by JNKs which are responsible for c-Jun N-terminal phosphorylation within its transactivation domain (Angel et al., 1988; Figure 2). Importantly, the JNK/c-Jun transcriptional pathway is thought to act as a sensor in response to nerve injury (Herdegen et al., 1997; Raivich et al., 2004; Makwana and Raivich, 2005). In this regard, JNKs can be rapidly activated and retrogradely transported following nerve injury, inducing c-Jun activation in the nucleus. In the presence of JNKs inhibitors and blockage of retrograde transport, peripheral nerve injury fails to induce c-Jun activation (Lindwall and Kanje, 2005). In addition, absence of c-Jun impairs the expression of genes associated with PNS regeneration such as CD44, Galanin, and a7b-1 integrin (Herdegen et al., 1997; Raivich et al., 2004; Lindwall and Kanje, 2005; Teng and Tang, 2006) ultimately affecting proper target reinnervation and functional recovery (Raivich et al., 2004).
After c-Jun overexpression, however, Purkinje cells fail to regenerate into a permissive graft, suggesting that c-Jun’s role in promoting regeneration is highly dependent on the cellular context (Carulli et al., 2002). Given the limitation of the cellular context, it is important to find alternatives that incorporate other TFs such as ATF3 and STAT3 to condition injured neurons to regenerate.
ATF3-Mediated Transcriptional Pathway
As a member of the ATF/CREB family of basic leucine zipper domains, ATF3 can establish functional interactions with both leucine zipper (CREB and c-Jun) and non-leucine zipper TFs (NF-κB, STAT3, and p53). Observations suggest that ATF3 and c-Jun protein–protein interaction (through heterodimerization) may synergistically regulate transcription to promote axon growth.
Similarly to c-Jun, peripheral but not central axonal injury induces early ATF3 activation (Tsujino et al., 2000; Figure 2). This strongly supports a role for ATF3 in neuronal regeneration (Pearson et al., 2003; Seijffers et al., 2007). Transgenic mice that constitutively express ATF3 in adult DRG neurons show enhanced peripheral nerve regeneration comparable to that induced by preconditioning lesion (Seijffers et al., 2007). On the other hand, constitutive ATF3 overexpression is not sufficient to overcome myelin inhibition or to promote CNS regeneration in vivo (Seijffers et al., 2007). These observations suggest that ATF3 likely contributes in promoting PNS regeneration when acting synergistically with other TFs and/or co-factors.
Several ATF3 target genes have been identified in non-neuronal cells. However, Hsp27 is the only identified ATF3 target gene in neurons so far. Peripheral nerve injury triggers robust Hsp27 expression in DRG, dorsal horn, and motor neurons in the spinal cord (Costigan et al., 1998). Through its leucine zipper DNA binding domain, ATF3 directly binds to Hsp27 promoter. In addition to increasing survival of sensory and sympathetic neurons after NGF withdrawal (Lewis et al., 1999), Hsp27 has been reported to enhance neurite outgrowth in vitro (Williams et al., 2005, 2006), and more recently to accelerate both axonal regeneration and functional recovery in vivo (Ma et al., 2011a). Whether Hsp27 also promotes CNS axon regeneration (e.g., corticospinal tract) is not known yet, and deserves further investigation.
Besides increased Hsp27 expression, ATF3 transgenic mice show enhanced SPRR1A expression in non-injured DRG neurons (Seijffers et al., 2007). Notably, SPRR1A is highly induced (>60-fold increase) by peripheral nerve injury, and its expression pattern is similar to Hsp27 (Bonilla et al., 2002; Huebner and Strittmatter, 2009). Moreover, SPRR1A is expressed at the growth cone, where it binds actin-associated proteins (Bonilla et al., 2002). Although SPRR1A overexpression enhances axonal outgrowth on permissive as well as non-permissive substrates, its functional role in promoting CNS regeneration is still lacking (Bonilla et al., 2002).
Taken together, these data suggest that ATF3 may be a point of convergence for multiple transcriptional pathways, signals, and regulators of axon growth and regeneration. Little is known about the components of the ATF3 regulatory complex in neurons.
Computational network analysis has predicted ATF3 to interact with transcriptional complexes already known to have roles in axon regeneration such as AP-1 (c-Jun, JunB, Jund1, and Fos) and NF-κB (p50; Gilchrist et al., 2006). Such interactions are tightly regulated by specific PTMs, leading to either activation or repression of transcription. The PTMs required for ATF3-dependent transcriptional changes after peripheral nerve injury are still unknown.
Nerve injury triggers ATF3 as well as c-Jun activation (Herdegen et al., 1991; Jenkins and Hunt, 1991; Tsujino et al., 2000; Pearson et al., 2003; Raivich et al., 2004; Lindwall and Kanje, 2005; Makwana and Raivich, 2005; Seijffers et al., 2007). Interestingly, both ATF3 and c-Jun promoters contain AP-1 sites (Morooka et al., 1995; Cai et al., 2000), thus supporting the idea that ATF3 and c-Jun may regulate each other’s expression. These observations suggest that coincident upregulation of ATF3 and c-Jun may act synergistically to promote axon growth after peripheral nerve injury. It is far from understood how conditional deletion of ATF3 in neurons may affect c-Jun-mediated transcription, and finally peripheral nerve regeneration. Interestingly, ATF3 can also affect gene transcription by simply sequestering repressors from specific regulatory domains, avoiding direct binding to consensus sequences (Chen et al., 1994).
In addition to TFs, gene transcription is also regulated through chromatin remodeling complexes (Figure 1) that allow or prevent access of transcription modules to DNA responsive elements. In this regard, it has been shown that ATF3 can interact “in silico” with HDAC via the NF-κB complex (Gilchrist et al., 2006). By controlling acetylation/deacetylation of histones, HDACs play a crucial role in chromatin remodeling. Histone acetylation relaxes chromatin structure (euchromatin), allowing access of transcription modules to DNA. In contrast, deacetylation limits access to DNA by condensing chromatin (heterochromatin). In neurons, the description of a functional ATF3–HDAC transcription module is still lacking.
Jak/STAT3-Mediated Transcriptional Pathway
In mammals there are seven STAT genes. In the nervous system, much attention has focused on STAT3 family member. By integrating information received from extracellular signals (growth factors and cytokines) through a transmembrane receptor, STAT3 transcriptional pathways directly target gene promoters, thereby regulating transcription without second messengers (Aaronson and Horvath, 2002). Often associated with transcriptional activation, STAT-transcription modules are also capable of repressing transcription (Ivanov et al., 2002; Ramana et al., 2002).
In the absence of stimuli, inactive STAT3 is localized in the cytoplasm. Receptor–ligand coupling rapidly activates STAT3, which is recruited to the intracellular domain of the receptor via specific binding between SH2 domains and receptor phosphotyrosine residues (Figure 2). The mammalian JAK family of proteins, consisting of JAK1, JAK2, JAK3, and TYK2, provides the tyrosine kinase activity required for STAT-activating cytokine receptors. Upon binding to intracellular cytokine receptor domains, JAKs phosphorylate themselves and tyrosine residues on receptor tails, creating STAT3 “hub stations.” Phosphorylated STATs form homo, hetero, and tetradimers (Vinkemeier et al., 1996; John et al., 1999; Zhang and Darnell, 2001) with DNA binding ability. Transcriptionally active STAT3 dimers are rapidly transported into the nucleus via importins (Levy and Darnell, 2002; Liu et al., 2005) where they bind an 8–10 bp inverted repeat DNA responsive element. Peripheral nerve injury triggers STAT3 activation within 15 min after severing (Lee et al., 2004). Studies have shown that peripheral nerve injuries result in time-dependent activation of STAT3 (Schwaiger et al., 2000; Qiu et al., 2005; Bareyre et al., 2011).
Similarly to c-Jun and ATF3, STAT3 activation appears to be restricted to peripheral and not central branch injuries, underscoring its critical role in mounting a successful peripheral regenerative response (Schwaiger et al., 2000). In fact, in the absence of STAT3, peripheral nerve regeneration is impaired in DRG neurons (Bareyre et al., 2011). Interestingly, sustained STAT3 expression promotes terminal and collateral sprouting by controlling initiation of axon growth after dorsal columns injury (Bareyre et al., 2011). However, the molecular basis that governs STAT3-mediated gene activation responsible for early axonal growth is still unknown. Future studies should aim at investigating whether these findings can be extended to other CNS axonal tracts.
TFs drive gene expression by binding to DNA responsive elements and recruiting both co-activators that remodel chromatin architecture of target promoters, and RNA polymerase II holoenzyme. Nucleosome positioning is influenced by ISWI and SWI/SNF-containing complexes (Figure 1). In addition, HATs like CBP/p300, P/CAF, and TAF250 are required for their ability to acetylate histones and other non-histone proteins such as TFs. Increased acetylation of histone and non-histone proteins facilitate access of transcription modules to core promoters, which in turn activates gene transcription. In this regard, the STAT3-mediated transcriptional pathway requires the recruitment of nuclear co-factors like CBP/p300 that are tightly associated with the RNA polymerase II holoenzyme (Paulson et al., 1999; Wang et al., 2005) and serve as connectors to the transcriptional machinery (Figure 2).
Studies in non-neuronal cells have demonstrated an unconventional nuclear function for JAK2 in phosphorylating the highly conserved tyrosine residue 41 on histone H3 (Dawson et al., 2009). Importantly, H3Y41 lies within a region known to perturb nucleosome mobility and stability (Ferreira et al., 2007). Therefore, it is likely that JAK2-mediated phosphorylation of H3Y41 regulates chromatin structure around core promoters (Dawson et al., 2009). Its involvement in disrupting heterochromatic domains further supports the JAK/STAT pathway’s role in regulating cellular epigenetic status in non-neuronal cells (Shi et al., 2006). To date, there is no evidence that peripheral and not central DRG branch injuries alter chromatin architecture to create a favorable environment driving transcription of RAGs.
In addition to SPRR1A (Wu et al., 2007) and p21/Cip1/Waf1 (Chin et al., 1996; Bellido et al., 1998), a recent high content transcriptional screen has identified several STAT3 target genes in DRG neurons that may be associated with the intrinsic ability of PNS neurons to regenerate (Smith et al., 2011). Altogether, these observations suggest that STAT3-mediated transcription is part of an early regenerative response.
CBP/p300/PCAF–p53-Mediated Transcriptional Pathway
The transcription factor and tumor suppressor p53 functions as a decision maker that contributes to directing cells toward a specific phenotype during development and following cellular damage (Jacobs et al., 2006; Helton and Chen, 2007; Tedeschi and Di Giovanni, 2009).
Following peripheral injury transcriptionally active p53 undergoes a series of acetylation events on its C-terminal domain (Di Giovanni et al., 2006; Tedeschi et al., 2009a). This acetylation leads to conformational changes that affect protein–protein interaction with transcriptional co-factors in a stimulus and cellular context-dependent manner. In this regard, overexpression of specific p53 mutants that mimic C-terminus acetylation at several lysine residues has been found to promote neurite outgrowth and neuronal maturation in vitro without affecting cell survival (Di Giovanni et al., 2006; Tedeschi et al., 2009a). Interestingly, p53’s C-terminus acetylation leads to apoptosis in cell lines (Knights et al., 2006; Tang et al., 2008; Yamaguchi et al., 2009). There has been evidence showing acetylated transcriptional modules increase the ability of p53 to both bind specific DNA elements and to activate transcription, compared with the ability shown by the total pool of p53.
After injury, active gene transcription is necessary to synthesize new proteins needed for axon growth. Acetylated-p53, together with CBP/p300 and PCAF, selectively occupies regulatory regions upstream to the TSS of pro-neurite and axon-outgrowth genes such as Coronin 1b, Rab13, and GAP-43 during an early regenerative response (Di Giovanni et al., 2006; Tedeschi et al., 2009a; Gaub et al., 2010, 2011; Floriddia et al., 2011; Figure 2). Both Coronin 1b and Rab13 are part of a gene cluster involved in neuronal plasticity, whose expression increases after traumatic spinal cord injury (Di Giovanni et al., 2005). Coronin 1b and Rab13 are also found in axonal sprouts of axotomized facial motor neurons (Di Giovanni et al., 2006). Gap-43 is highly induced after peripheral nerve injury (Skene and Willard, 1981), and when overexpressed together with CAP-23, it promotes some extent of CNS axon regeneration (Bomze et al., 2001). Similarly to Gap-43, p21/Waf1’s expression is upregulated upon peripheral axotomy (Bonilla et al., 2002). PCAF and CGN5-mediated p53 acetylation at Lys 320 increases transcriptional activation of the p21Cip1/Waf1 promoter (Liu et al., 1999). Together with p53, KLF4 transactivates the p21Cip1/Waf1 promoter (Zhang et al., 2000). P21Cip1/Waf1 is already known to influence growth cone navigation by inhibiting ROCK (Tanaka et al., 2002; Qin et al., 2009). Interestingly, p53 and NFκB compete for binding to CBP. In response to TNFα, IKKα-mediated phosphorylation of CBP results in switching CBP recruitment from p53 to NFκB target promoters (Webster and Perkins, 1999; Perkins, 2007). Absence of p53 impairs peripheral regeneration in part by affecting the pro-neurite and axon-outgrowth transcriptional program (Di Giovanni et al., 2006; Tedeschi et al., 2009a). In neurons, however, the balance between survival and axon regeneration is difficult to separate (Goldberg and Barres, 2000). Thus, given p53’s role in DNA repair and the elimination of damaged neurons, it is important to keep in mind that the impairment in axonal regeneration may also be influenced by the lack of removal of damaged cells that occurs in the absence of p53. Nevertheless, more recent observations provide further evidence that acetylated-p53 may have a critical role in modulating different transcriptional responses during axonal regeneration (Gaub et al., 2011).
KLF4-Mediated Transcriptional Pathway
The KLF family of transcription factors includes 17 members characterized by the presence of three Cys2 His2 zinc fingers located at the C-terminus. In addition to controlling cell cycle, proliferation, and cell death (Black et al., 2001), developmentally regulated KLF4 has been recently reported to affect axon growth and regeneration in vivo (Moore et al., 2009).
In the adult CNS, KLF4 restricts the intrinsic regenerative ability of certain neurons. In fact, KLF4 overexpression results in reduced axonal length in vitro and consequent KLF4 targeted deletion enhances CNS regeneration in vivo (Moore et al., 2009). The series of transcriptional events underlying KLF4-mediated regenerative response in neurons are not known (Moore et al., 2011). Depending on the promoter context and the recruitment of co-activators/co-repressors, KLFs can either activate or repress transcription. KLF4 has been reported to interact with co-factors such as CBP/p300 (Geiman et al., 2000; Evans et al., 2007) and HDAC3 (Evans et al., 2007). In cooperation with p53, KLF4 transactivates p21Cip1/Waf1 promoter (Zhang et al., 2000) which in turn influences neurite outgrowth by inhibiting ROCK (Tanaka et al., 2002). KLF4 also transactivates cGKIα, already known as p53’s transcriptional target in counteracting Semaphorin induced growth cone collapse (Tedeschi et al., 2009b). Interestingly, KLF4 directly suppresses p53, thus reflecting its anti-apoptotic properties (Rowland et al., 2005). Although speculative, suppression of p53 may be one of the underlying mechanisms for KLF4-mediated suppression of axonal growth (Subang and Richardson, 2009).
KLF4 inhibits ornithine decarboxylase activity by competing with Sp1 (Chen et al., 2002). By catalyzing the reaction from arginine to ornithine, Arginase I has been proposed as an important downstream mediator of the cAMP–PKA–CREB-dependent regenerative program (Cai et al., 2002; Gao et al., 2004; Deng et al., 2009). Finally, absence of KLF4 results in activation of genes such as SPRR1A (Bonilla et al., 2002) and ATF3 (Mason et al., 2003; Pearson et al., 2003; Campbell et al., 2005; Seijffers et al., 2007; Swamynathan et al., 2008), both of them already known to be upregulated during PNS regeneration. Nevertheless, it is unclear whether KLF4-mediated CNS regenerative response requires expression of these genes. Future work should aim to provide a better understanding of the molecular mechanisms underlying the role of KLF4 in post-axonal injury models.
BMP4/Smad1 Transcriptional Pathway
Members of the Smad family of transcription factors function as signal transducers and transcriptional modulators of the TGFβ/BMP signaling pathway, which controls a wide variety of cellular functions during development and organogenesis (Heldin et al., 1997; Massague et al., 2005).
By gene expression profile analysis, peripheral (and not central) branch axotomy has been found to increase Smad1 expression in adult DRG neurons (Zou et al., 2009). Given that Smad1 integrates signals from BMP receptors, it is conceivable that BMP signaling triggers receptor-regulated Smad1 activation after peripheral lesion. Importantly, intraganglionic delivery of BMP2-4 induces Smad1 phosphorylation and consequent nuclear translocation (Zou et al., 2009). Together with Smad4, phosphorylated Smad1 assembles a multi-subunits complex that regulates transcription (Massague and Wotton, 2000; Figure 2). In the absence of Smad1, conditioned DRG neurons show impairment in axon elongation in vitro (Parikh et al., 2011). Similarly, blockade of BMP signaling with the BMP antagonist Noggin inhibits axonal growth in both naïve and preconditioned DRG neurons (Ma et al., 2011b). Moreover, using an AAV-based approach, activation of Smad1-dependent BMP signaling has been recently reported to increase the intrinsic growth ability of adult DRG neurons in vitro, but also to promote dorsal columns axon regeneration in vivo (Parikh et al., 2011). These lines of evidence suggest that Smad1-dependent BMP signaling acts as a key player in an orchestrated transcriptional program, allowing adult sensory neurons to switch into a “growth mode” after injury.
The composition of the Smad1 transcription module as well as the identity of the DNA sequences that the module will bind in response to injury remains to be determined. In the adult nervous system, axon growth ability requires a discrete period of de novo transcription (Smith and Skene, 1997). Modules that promote transcription likely include co-activators that confer high affinity and selective interaction with DNA elements. Thus, nuclear Smad1 may serve to modulate the activity of an existing transcription module rather than assembling de novo a distinct complex. Evidence has shown that AP-1 and Smads synergistically interact to promote transcription from artificial promoters (Zhang et al., 1998; Wong et al., 1999). Members of heterodimeric AP-1 complex, including c-Jun and ATF3, play a role in peripheral nerve regeneration (Raivich et al., 2004; Seijffers et al., 2007). Notably, BMP receptor-activated Smad1 and Smad4 are able to activate transcription in part by their ability to recruit co-activators like CBP/p300 (Feng et al., 1998; Pouponnot et al., 1998; Figure 2).
HATs like CBP/p300 are required for their ability to acetylate histones and other non-histone proteins such as TFs. Increased acetylation of histone and non-histone proteins promotes chromatin remodeling, which facilitates access to core promoters. This in turn activates gene transcription. Indeed, Smad1 recruitment to DNA regulatory elements may be a crucial step in determining which set of RAGs will be activated in response to peripheral injury. Besides the fact that BMP4 overexpression drives GAP-43 expression in adult DRG neurons (Parikh et al., 2011), the downstream target genes that promote axon regeneration via Smad1-dependent transcription are still unknown.
Organization of a Pro-Regenerative Transcriptional Network
Transcriptional regulators do not operate alone. Several TFs, growth factors, chromatin remodelers, kinases, and acetyltransferases discussed above are not isolated players. In fact, they function as part of a multi-nodal transcriptional network (Figure 3). Numerous studies have reported complex pattern of transcriptional changes occurring early after axonal injury. An orchestrated regenerative program requires coordinated gene expression, as a result of the integration of a large number of connections within a functional network (Figure 3). Important nodes (also referred as “hub genes”) consistently experience waves of perturbation, and are responsible for such complex integration (van Kesteren et al., 2011). Early activated (e.g., subsequent to injury) TFs serve as “hubs,” controlling cascades of functionally related transcriptional events as well as a plethora of target genes. Within the same transcriptional network, HATs like CBP and p300 serve as scaffold for the assembly of multi-components transcriptional modules. Finally, coupling of signal transduction pathways and gene regulation is controlled by modifying enzymes. Thus far, the organization of the entire pro-regenerative transcriptional network is not completely understood. Gene co-expression network analysis (Zhang and Horvath, 2005) will eventually identify clusters of tightly co-expressed transcriptional regulators that could potentially become novel therapeutic targets to promote plasticity-related structural changes and regeneration following axonal injuries.

Figure 3. Transcriptional network. (A) List of various transcription regulators, chromatin remodelers, acetyltransferases, kinases, and growth factors implicated in an early regenerative response after nerve injury. (B) Ingenuity generated screenshot of a regenerative-associated transcriptional network. Connecting arrows and lines represent interactions between nodes.
Discussion
Although progress has been recently made in elucidating the role of transcriptional pathways in controlling axon regeneration, there is a bias toward studying gene regulation in the context of PNS regeneration (DRG and facial nerve). Indeed, it is difficult to find any data on transcriptional pathways regulating supraspinal axon (e.g., corticospinal tract) regeneration following spinal cord injuries. This apparent discrepancy can arise for a host of reasons. Due to their unique anatomical conformation, DRG neurons serve as ideal model to study the dual regenerative response of PNS and CNS axons. In contrast to the CNS, the PNS can be easily accessed, supporting the development of gene therapy applications without major surgery. The development of minimally invasive surgeries combined with the efficacy of AAV-mediated gene delivery hold great promise to study the role of transcriptional regulators in promoting CNS regeneration.
A discrete period of new transcription is crucial to gain growth competence after axonal injury (Smith and Skene, 1997). TFs drive gene expression by binding to DNA responsive elements and recruiting both co-activators and RNA polymerase II holoenzyme to core promoters. Nuclear co-activators shape chromatin architecture into a favorable environment for transcription. Interestingly, recent observations suggest that certain histone modification patterns are altered in regenerating neurons (Gaub et al., 2011). These data are intriguing and if widely represented, they suggest the worth of studying axon regeneration from an epigenetic perspective.
Histone acetyltransferases like CBP/p300, P/CAF, CGN5, and TAF250 are required for their ability to acetylate histones and other non-histone proteins such as TFs. Importantly, acetylation of lysine residues in the N-terminal tails of histones facilitates access to transcriptionally repressed chromatin. In addition, ISWI and SWI/SNF-containing complexes are known to influence nucleosome positioning. Active transcription modules do not operate alone. It is likely that several pro-regenerative regulators synergistically cluster into active transcription factories located in discrete sites within the nucleus. After peripheral injury, a successful regenerative response requires an orchestrated sequence of transcriptional events.
On the other hand, it is becoming apparent that a silent pro-regenerative transcriptional program likely restricts the regenerative ability of adult CNS neurons. It has been recently reported that unlocking such a silent transcriptional program may promote axonal regeneration in the adult CNS (Moore et al., 2009; Bareyre et al., 2011; Gaub et al., 2011; Parikh et al., 2011).
Identification of early clusters of TFs is particularly important because early activated TFs may control subsequent events of transcriptional activation. The crosstalk between early activated TFs (few important “nodes”) that sense injury signals and initiate the cascade of transcriptional events, and the actual TFs (multiple branches of the transcriptional network) that directly promote axon regeneration is not completely understood. Notably, several TFs described above share target genes, further supporting the idea that multiple transcriptional pathways are functionally related within the same network (Figure 3). Cis-regulatory analysis using bioinformatics software tools could predict representation of specific TFs binding sites in clusters of gene that are activated/repressed as part of a successful regenerative program. In addition, integration of biological networks and gene expression analysis ultimately identify potential interacting TFs based on protein–protein interaction map (Shannon et al., 2003; Figure 3). Computational analysis could further determine the density of selected TFs binding sites in relation to each other and the transcriptional start site (Shannon et al., 2003).
Future work should aim to provide a better understanding of the transcriptional outcome of TFs signaling in the context of different post-translational modification states or developmental periods behind neurite growth and regeneration. PTMs induce activation and nuclear translocation of several TFs (including c-jun, junD, ATF3, P311, Sox11, STAT3, p53, and Smad1), thus controlling gene expression in axotomized neurons (Herdegen et al., 1991; Jenkins and Hunt, 1991; Schwaiger et al., 2000; Fujitani et al., 2004; Raivich et al., 2004; Di Giovanni et al., 2006; Seijffers et al., 2007; Bareyre et al., 2011; Parikh et al., 2011). High-throughput analysis will eventually produce comprehensive epigenome mapping in regenerating neurons.
In addition to identify early cluster of TFs, future studies should aim at understanding whether altered histone modification profiles tune transcriptional pathways as part of an active regenerative program.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
I would like to thank Dr. Z. He for his support and C. Yeung for his help in editing this manuscript. I am grateful to Drs. M. Costigan, T. Omura, T. Nguyen, and C. Woolf for providing insightful comments on the manuscript. I apologize to all colleagues whose relevant work was involuntarily omitted or not included due to space limitations.
Abbreviations
AAV, adeno associated virus; AP-1, activator protein 1; ATF3, activating transcription factor 3; BMP, bone morphogenetic protein; cAMP, cyclic adenosine monophosphate; CAP-23, cytoskeleton-associated protein 23; CBP, CREB binding protein; cGKIα, cGMP-dependent protein kinase 1α; CNS, central nervous system; CREB, cyclic-AMP–response–element-binding protein; GAP-43, growth associated protein 43; HDAC, histone deacetylase; Hsp27, heat shock protein 27; JAK, janus kinase; JNKs, Jun N-terminal kinases; KLF, Krüppel-like factor; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; p21Cip1/Waf1, cyclin-dependent kinase-interacting protein 1; p300, E1A-binding protein p300; PCAF, CBP-associated factor; PKA, protein kinase A; PNS, peripheral nervous system; PTMs, post translational modifications; RAGs, regenerative-associated genes; ROCK, Rho kinase; SH2, src-homology 2; Smad1, mothers against decapentaplegic homolog 1; Sox11, SRY-box containing gene 11; Sp1, specificity protein 1; SPRR1, small proline-rich repeat protein 1A; STAT3, signal transducer and activator of transcription 3; TFs, transcription factors; TNFα, tumor necrosis factor alpha.
References
Aaronson, D. S., and Horvath, C. M. (2002). A road map for those who don’t know JAK-STAT. Science 296, 1653–1655.
Angel, P., Allegretto, E. A., Okino, S. T., Hattori, K., Boyle, W. J., Hunter, T., and Karin, M. (1988). Oncogene JUN encodes a sequence-specific trans-activator similar to AP-1. Nature 332, 166–171.
Arthur, J. S., Fong, A. L., Dwyer, J. M., Davare, M., Reese, E., Obrietan, K., and Impey, S. (2004). Mitogen- and stress-activated protein kinase 1 mediates cAMP response element-binding protein phosphorylation and activation by neurotrophins. J. Neurosci. 24, 4324–4332.
Baizer, L., and Fishman, M. C. (1987). Recognition of specific targets by cultured dorsal root ganglion neurons. J. Neurosci. 7, 2305–2311.
Bareyre, F. M., Garzorz, N., Lang, C., Misgeld, T., Buning, H., and Kerschensteiner, M. (2011). In vivo imaging reveals a phase-specific role of STAT3 during central and peripheral nervous system axon regeneration. Proc. Natl. Acad. Sci. U.S.A. 108, 6282–6287.
Bareyre, F. M., and Schwab, M. E. (2003). Inflammation, degeneration and regeneration in the injured spinal cord: insights from DNA microarrays. Trends Neurosci. 26, 555–563.
Bellido, T., O’Brien, C. A., Roberson, P. K., and Manolagas, S. C. (1998). Transcriptional activation of the p21(WAF1,CIP1,SDI1) gene by interleukin-6 type cytokines. A prerequisite for their pro-differentiating and anti-apoptotic effects on human osteoblastic cells. J. Biol. Chem. 273, 21137–21144.
Benowitz, L. I., and Yin, Y. (2007). Combinatorial treatments for promoting axon regeneration in the CNS: strategies for overcoming inhibitory signals and activating neurons’ intrinsic growth state. Dev. Neurobiol. 67, 1148–1165.
Black, A. R., Black, J. D., and Azizkhan-Clifford, J. (2001). Sp1 and kruppel-like factor family of transcription factors in cell growth regulation and cancer. J. Cell. Physiol. 188, 143–160.
Bomze, H. M., Bulsara, K. R., Iskandar, B. J., Caroni, P., and Skene, J. H. (2001). Spinal axon regeneration evoked by replacing two growth cone proteins in adult neurons. Nat. Neurosci. 4, 38–43.
Bonilla, I. E., Tanabe, K., and Strittmatter, S. M. (2002). Small proline-rich repeat protein 1A is expressed by axotomized neurons and promotes axonal outgrowth. J. Neurosci. 22, 1303–1315.
Bosse, F., Hasenpusch-Theil, K., Kury, P., and Muller, H. W. (2006). Gene expression profiling reveals that peripheral nerve regeneration is a consequence of both novel injury-dependent and reactivated developmental processes. J. Neurochem. 96, 1441–1457.
Bosse, F., Kury, P., and Muller, H. W. (2001). Gene expression profiling and molecular aspects in peripheral nerve regeneration. Restor. Neurol. Neurosci. 19, 5–18.
Butler, S. J., and Tear, G. (2007). Getting axons onto the right path: the role of transcription factors in axon guidance. Development 134, 439–448.
Cai, D., Deng, K., Mellado, W., Lee, J., Ratan, R. R., and Filbin, M. T. (2002). Arginase I and polyamines act downstream from cyclic AMP in overcoming inhibition of axonal growth MAG and myelin in vitro. Neuron 35, 711–719.
Cai, Y., Zhang, C., Nawa, T., Aso, T., Tanaka, M., Oshiro, S., Ichijo, H., and Kitajima, S. (2000). Homocysteine-responsive ATF3 gene expression in human vascular endothelial cells: activation of c-Jun NH(2)-terminal kinase and promoter response element. Blood 96, 2140–2148.
Campbell, G., Hutchins, K., Winterbottom, J., Grenningloh, G., Lieberman, A. R., and Anderson, P. N. (2005). Upregulation of activating transcription factor 3 (ATF3) by intrinsic CNS neurons regenerating axons into peripheral nerve grafts. Exp. Neurol. 192, 340–347.
Caroni, P., and Becker, M. (1992). The downregulation of growth-associated proteins in motoneurons at the onset of synapse elimination is controlled by muscle activity and IGF1. J. Neurosci. 12, 3849–3861.
Carulli, D., Buffo, A., Botta, C., Altruda, F., and Strata, P. (2002). Regenerative and survival capabilities of Purkinje cells overexpressing c-Jun. Eur. J. Neurosci. 16, 105–118.
Case, L. C., and Tessier-Lavigne, M. (2005). Regeneration of the adult central nervous system. Curr. Biol. 15, R749–R753.
Chen, B. P., Liang, G., Whelan, J., and Hai, T. (1994). ATF3 and ATF3 delta Zip. Transcriptional repression versus activation by alternatively spliced isoforms. J. Biol. Chem. 269, 15819–15826.
Chen, Z. L., Yu, W. M., and Strickland, S. (2007). Peripheral regeneration. Annu. Rev. Neurosci. 30, 209–233.
Chen, Z. Y., Shie, J. L., and Tseng, C. C. (2002). Gut-enriched Kruppel-like factor represses ornithine decarboxylase gene expression and functions as checkpoint regulator in colonic cancer cells. J. Biol. Chem. 277, 46831–46839.
Chin, Y. E., Kitagawa, M., Su, W. C., You, Z. H., Iwamoto, Y., and Fu, X. Y. (1996). Cell growth arrest and induction of cyclin-dependent kinase inhibitor p21 WAF1/CIP1 mediated by STAT1. Science 272, 719–722.
Chrivia, J. C., Kwok, R. P., Lamb, N., Hagiwara, M., Montminy, M. R., and Goodman, R. H. (1993). Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature 365, 855–859.
Costigan, M., Befort, K., Karchewski, L., Griffin, R. S., D’Urso, D., Allchorne, A., Sitarski, J., Mannion, J. W., Pratt, R. E., and Woolf, C. J. (2002). Replicate high-density rat genome oligonucleotide microarrays reveal hundreds of regulated genes in the dorsal root ganglion after peripheral nerve injury. BMC Neurosci. 3, 16. doi:10.1186/1471-2202-3-16
Costigan, M., Mannion, R. J., Kendall, G., Lewis, S. E., Campagna, J. A., Coggeshall, R. E., Meridith-Middleton, J., Tate, S., and Woolf, C. J. (1998). Heat shock protein 27: developmental regulation and expression after peripheral nerve injury. J. Neurosci. 18, 5891–5900.
Dawson, M. A., Bannister, A. J., Gottgens, B., Foster, S. D., Bartke, T., Green, A. R., and Kouzarides, T. (2009). JAK2 phosphorylates histone H3Y41 and excludes HP1alpha from chromatin. Nature 461, 819–822.
Deng, K., He, H., Qiu, J., Lorber, B., Bryson, J. B., and Filbin, M. T. (2009). Increased synthesis of spermidine as a result of upregulation of arginase I promotes axonal regeneration in culture and in vivo. J. Neurosci. 29, 9545–9552.
Di Giovanni, S. (2009). Molecular targets for axon regeneration: focus on the intrinsic pathways. Expert Opin. Ther. Targets 13, 1387–1398.
Di Giovanni, S., De Biase, A., Yakovlev, A., Finn, T., Beers, J., Hoffman, E. P., and Faden, A. I. (2005). In vivo and in vitro characterization of novel neuronal plasticity factors identified following spinal cord injury. J. Biol. Chem. 280, 2084–2091.
Di Giovanni, S., Knights, C. D., Rao, M., Yakovlev, A., Beers, J., Catania, J., Avantaggiati, M. L., and Faden, A. I. (2006). The tumor suppressor protein p53 is required for neurite outgrowth and axon regeneration. EMBO J. 25, 4084–4096.
Evans, P. M., Zhang, W., Chen, X., Yang, J., Bhakat, K. K., and Liu, C. (2007). Kruppel-like factor 4 is acetylated by p300 and regulates gene transcription via modulation of histone acetylation. J. Biol. Chem. 282, 33994–34002.
Feng, X. H., Zhang, Y., Wu, R. Y., and Derynck, R. (1998). The tumor suppressor Smad4/DPC4 and transcriptional adaptor CBP/p300 are coactivators for smad3 in TGF-beta-induced transcriptional activation. Genes Dev. 12, 2153–2163.
Ferreira, H., Somers, J., Webster, R., Flaus, A., and Owen-Hughes, T. (2007). Histone tails and the H3 alphaN helix regulate nucleosome mobility and stability. Mol. Cell. Biol. 27, 4037–4048.
Filbin, M. T. (2003). Myelin-associated inhibitors of axonal regeneration in the adult mammalian CNS. Nat. Rev. Neurosci. 4, 703–713.
Floriddia, E., Nguyen, T., and Di Giovanni, S. (2011). Chromatin immunoprecipitation from dorsal root ganglia tissue following axonal injury. J. Vis. Exp. doi: 10.3791/2803
Fujitani, M., Yamagishi, S., Che, Y. H., Hata, K., Kubo, T., Ino, H., Tohyama, M., and Yamashita, T. (2004). P311 accelerates nerve regeneration of the axotomized facial nerve. J. Neurochem. 91, 737–744.
Gao, Y., Deng, K., Hou, J., Bryson, J. B., Barco, A., Nikulina, E., Spencer, T., Mellado, W., Kandel, E. R., and Filbin, M. T. (2004). Activated CREB is sufficient to overcome inhibitors in myelin and promote spinal axon regeneration in vivo. Neuron 44, 609–621.
Gaub, P., Joshi, Y., Wuttke, A., Naumann, U., Schnichels, S., Heiduschka, P., and Di Giovanni, S. (2011). The histone acetyltransferase p300 promotes intrinsic axonal regeneration. Brain 134, 2134–2148.
Gaub, P., Tedeschi, A., Puttagunta, R., Nguyen, T., Schmandke, A., and Di Giovanni, S. (2010). HDAC inhibition promotes neuronal outgrowth and counteracts growth cone collapse through CBP/p300 and P/CAF-dependent p53 acetylation. Cell Death Differ. 17, 1392–1408.
Geiman, D. E., Ton-That, H., Johnson, J. M., and Yang, V. W. (2000). Transactivation and growth suppression by the gut-enriched Kruppel-like factor (Kruppel-like factor 4) are dependent on acidic amino acid residues and protein-protein interaction. Nucleic Acids Res. 28, 1106–1113.
Gilchrist, M., Thorsson, V., Li, B., Rust, A. G., Korb, M., Roach, J. C., Kennedy, K., Hai, T., Bolouri, H., and Aderem, A. (2006). Systems biology approaches identify ATF3 as a negative regulator of Toll-like receptor 4. Nature 441, 173–178.
Goldberg, J. L., and Barres, B. A. (2000). The relationship between neuronal survival and regeneration. Annu. Rev. Neurosci. 23, 579–612.
Goldberg, J. L., Klassen, M. P., Hua, Y., and Barres, B. A. (2002). Amacrine-signaled loss of intrinsic axon growth ability by retinal ganglion cells. Science 296, 1860–1864.
Hannila, S. S., and Filbin, M. T. (2008). The role of cyclic AMP signaling in promoting axonal regeneration after spinal cord injury. Exp. Neurol. 209, 321–332.
Harel, N. Y., and Strittmatter, S. M. (2006). Can regenerating axons recapitulate developmental guidance during recovery from spinal cord injury? Nat. Rev. Neurosci. 7, 603–616.
Heldin, C. H., Miyazono, K., and ten Dijke, P. (1997). TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature 390, 465–471.
Helton, E. S., and Chen, X. (2007). p53 modulation of the DNA damage response. J. Cell. Biochem. 100, 883–896.
Herdegen, T., Kummer, W., Fiallos, C. E., Leah, J., and Bravo, R. (1991). Expression of c-JUN, JUN B and JUN D proteins in rat nervous system following transection of vagus nerve and cervical sympathetic trunk. Neuroscience 45, 413–422.
Herdegen, T., Skene, P., and Bahr, M. (1997). The c-Jun transcription factor – bipotential mediator of neuronal death, survival and regeneration. Trends Neurosci. 20, 227–231.
Hoffman, P. N. (2010). A conditioning lesion induces changes in gene expression and axonal transport that enhance regeneration by increasing the intrinsic growth state of axons. Exp. Neurol. 223, 11–18.
Huebner, E. A., and Strittmatter, S. M. (2009). Axon regeneration in the peripheral and central nervous systems. Results Probl. Cell Differ. 48, 339–351.
Ivanov, V. N., Krasilnikov, M., and Ronai, Z. (2002). Regulation of Fas expression by STAT3 and c-Jun is mediated by phosphatidylinositol 3-kinase-AKT signaling. J. Biol. Chem. 277, 4932–4944.
Jacobs, W. B., Kaplan, D. R., and Miller, F. D. (2006). The p53 family in nervous system development and disease. J. Neurochem. 97, 1571–1584.
Jenkins, R., and Hunt, S. P. (1991). Long-term increase in the levels of c-jun mRNA and jun protein-like immunoreactivity in motor and sensory neurons following axon damage. Neurosci. Lett. 129, 107–110.
Jochum, W., Passegue, E., and Wagner, E. F. (2001). AP-1 in mouse development and tumorigenesis. Oncogene 20, 2401–2412.
John, S., Vinkemeier, U., Soldaini, E., Darnell, J. E. Jr., and Leonard, W. J. (1999). The significance of tetramerization in promoter recruitment by Stat5. Mol. Cell. Biol. 19, 1910–1918.
Knights, C. D., Catania, J., Di Giovanni, S., Muratoglu, S., Perez, R., Swartzbeck, A., Quong, A. A., Zhang, X., Beerman, T., Pestell, R. G., and Avantaggiati, M. L. (2006). Distinct p53 acetylation cassettes differentially influence gene-expression patterns and cell fate. J. Cell Biol. 173, 533–544.
Kornhauser, J. M., Cowan, C. W., Shaywitz, A. J., Dolmetsch, R. E., Griffith, E. C., Hu, L. S., Haddad, C., Xia, Z., and Greenberg, M. E. (2002). CREB transcriptional activity in neurons is regulated by multiple, calcium-specific phosphorylation events. Neuron 34, 221–233.
Kwok, R. P., Lundblad, J. R., Chrivia, J. C., Richards, J. P., Bachinger, H. P., Brennan, R. G., Roberts, S. G., Green, M. R., and Goodman, R. H. (1994). Nuclear protein CBP is a coactivator for the transcription factor CREB. Nature 370, 223–226.
Lee, J. K., Geoffroy, C. G., Chan, A. F., Tolentino, K. E., Crawford, M. J., Leal, M. A., Kang, B., and Zheng, B. (2010). Assessing spinal axon regeneration and sprouting in Nogo-, MAG-, and OMgp-deficient mice. Neuron 66, 663–670.
Lee, N., Neitzel, K. L., Devlin, B. K., and MacLennan, A. J. (2004). STAT3 phosphorylation in injured axons before sensory and motor neuron nuclei: potential role for STAT3 as a retrograde signaling transcription factor. J. Comp. Neurol. 474, 535–545.
Levy, D. E., and Darnell, J. E. Jr. (2002). Stats: transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 3, 651–662.
Lewis, S. E., Mannion, R. J., White, F. A., Coggeshall, R. E., Beggs, S., Costigan, M., Martin, J. L., Dillmann, W. H., and Woolf, C. J. (1999). A role for HSP27 in sensory neuron survival. J. Neurosci. 19, 8945–8953.
Lindwall, C., and Kanje, M. (2005). The role of p-c-Jun in survival and outgrowth of developing sensory neurons. Neuroreport 16, 1655–1659.
Liu, K., Tedeschi, A., Park, K. K., and He, Z. (2011). Neuronal intrinsic mechanisms of axon regeneration. Annu. Rev. Neurosci. 34, 131–152.
Liu, L., McBride, K. M., and Reich, N. C. (2005). STAT3 nuclear import is independent of tyrosine phosphorylation and mediated by importin-alpha3. Proc. Natl. Acad. Sci. U.S.A. 102, 8150–8155.
Liu, L., Scolnick, D. M., Trievel, R. C., Zhang, H. B., Marmorstein, R., Halazonetis, T. D., and Berger, S. L. (1999). p53 sites acetylated in vitro by PCAF and p300 are acetylated in vivo in response to DNA damage. Mol. Cell. Biol. 19, 1202–1209.
Lonze, B. E., and Ginty, D. D. (2002). Function and regulation of CREB family transcription factors in the nervous system. Neuron 35, 605–623.
Ma, C. H., Brenner, G. J., Omura, T., Samad, O. A., Costigan, M., Inquimbert, P., Niederkofler, V., Salie, R., Sun, C. C., Lin, H. Y., Arber, S., Coppola, G., Woolf, C. J., and Samad, T. A. (2011a). The BMP co-receptor RGMb promotes while the endogenous BMP antagonist Noggin reduces neurite outgrowth and peripheral nerve regeneration by modulating BMP signaling. J. Neurosci. 31, 18391–18400.
Ma, C. H., Omura, T., Cobos, E. J., Latremoliere, A., Ghasemlou, N., Brenner, G. J., van Veen, E., Barrett, L., Sawada, T., Gao, F., Coppola, G., Gertler, F., Costigan, M., Geschwind, D., and Woolf, C. J. (2011b). Accelerating axonal growth promotes motor recovery after peripheral nerve injury in mice. J. Clin. Invest. 121, 4332–4347.
Ma, T. C., Campana, A., Lange, P. S., Lee, H. H., Banerjee, K., Bryson, J. B., Mahishi, L., Alam, S., Giger, R. J., Barnes, S., Morris, S. M. Jr., Willis, D. E., Twiss, J. L., Filbin, M. T., and Ratan, R. R. (2010). A large-scale chemical screen for regulators of the arginase 1 promoter identifies the soy isoflavone daidzeinas a clinically approved small molecule that can promote neuronal protection or regeneration via a cAMP-independent pathway. J. Neurosci. 30, 739–748.
Makwana, M., and Raivich, G. (2005). Molecular mechanisms in successful peripheral regeneration. FEBS J. 272, 2628–2638.
Mason, M. R., Lieberman, A. R., and Anderson, P. N. (2003). Corticospinal neurons up-regulate a range of growth-associated genes following intracortical, but not spinal, axotomy. Eur. J. Neurosci. 18, 789–802.
Massague, J., Seoane, J., and Wotton, D. (2005). Smad transcription factors. Genes Dev. 19, 2783–2810.
Massague, J., and Wotton, D. (2000). Transcriptional control by the TGF-beta/Smad signaling system. EMBO J. 19, 1745–1754.
Moore, D. L., Apara, A., and Goldberg, J. L. (2011). Kruppel-like transcription factors in the nervous system: novel players in neurite outgrowth and axon regeneration. Mol. Cell. Neurosci. 47, 233–243.
Moore, D. L., Blackmore, M. G., Hu, Y., Kaestner, K. H., Bixby, J. L., Lemmon, V. P., and Goldberg, J. L. (2009). KLF family members regulate intrinsic axon regeneration ability. Science 326, 298–301.
Morooka, H., Bonventre, J. V., Pombo, C. M., Kyriakis, J. M., and Force, T. (1995). Ischemia and reperfusion enhance ATF-2 and c-Jun binding to cAMP response elements and to an AP-1 binding site from the c-jun promoter. J. Biol. Chem. 270, 30084–30092.
Neumann, S., and Woolf, C. J. (1999). Regeneration of dorsal column fibers into and beyond the lesion site following adult spinal cord injury. Neuron 23, 83–91.
Parikh, P., Hao, Y., Hosseinkhani, M., Patil, S. B., Huntley, G. W., Tessier-Lavigne, M., and Zou, H. (2011). Regeneration of axons in injured spinal cord by activation of bone morphogenetic protein/Smad1 signaling pathway in adult neurons. Proc. Natl. Acad. Sci. U.S.A. 108, E99–E107.
Paulson, M., Pisharody, S., Pan, L., Guadagno, S., Mui, A. L., and Levy, D. E. (1999). Stat protein transactivation domains recruit p300/CBP through widely divergent sequences. J. Biol. Chem. 274, 25343–25349.
Pearson, A. G., Gray, C. W., Pearson, J. F., Greenwood, J. M., During, M. J., and Dragunow, M. (2003). ATF3 enhances c-Jun-mediated neurite sprouting. Brain Res. Mol. Brain Res. 120, 38–45.
Perkins, N. D. (2007). Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat. Rev. Mol. Cell Biol. 8, 49–62.
Pouponnot, C., Jayaraman, L., and Massague, J. (1998). Physical and functional interaction of SMADs and p300/CBP. J. Biol. Chem. 273, 22865–22868.
Qin, Q., Baudry, M., Liao, G., Noniyev, A., Galeano, J., and Bi, X. (2009). A novel function for p53: regulation of growth cone motility through interaction with Rho kinase. J. Neurosci. 29, 5183–5192.
Qiu, J., Cafferty, W. B., McMahon, S. B., and Thompson, S. W. (2005). Conditioning injury-induced spinal axon regeneration requires signal transducer and activator of transcription 3 activation. J. Neurosci. 25, 1645–1653.
Qiu, J., Cai, D., Dai, H., McAtee, M., Hoffman, P. N., Bregman, B. S., and Filbin, M. T. (2002). Spinal axon regeneration induced by elevation of cyclic AMP. Neuron 34, 895–903.
Raivich, G., Bohatschek, M., Da Costa, C., Iwata, O., Galiano, M., Hristova, M., Nateri, A. S., Makwana, M., Riera-Sans, L., Wolfer, D. P., Lipp, H. P., Aguzzi, A., Wagner, E. F., and Behrens, A. (2004). The AP-1 transcription factor c-Jun is required for efficient axonal regeneration. Neuron 43, 57–67.
Raivich, G., and Makwana, M. (2007). The making of successful axonal regeneration: genes, molecules and signal transduction pathways. Brain Res. Rev. 53, 287–311.
Ramana, C. V., Gil, M. P., Schreiber, R. D., and Stark, G. R. (2002). Stat1-dependent and -independent pathways in IFN-gamma-dependent signaling. Trends Immunol. 23, 96–101.
Redmond, L., Kashani, A. H., and Ghosh, A. (2002). Calcium regulation of dendritic growth via CaM kinase IV and CREB-mediated transcription. Neuron 34, 999–1010.
Riccio, A., Alvania, R. S., Lonze, B. E., Ramanan, N., Kim, T., Huang, Y., Dawson, T. M., Snyder, S. H., and Ginty, D. D. (2006). A nitric oxide signaling pathway controls CREB-mediated gene expression in neurons. Mol. Cell 21, 283–294.
Riccio, A., Pierchala, B. A., Ciarallo, C. L., and Ginty, D. D. (1997). An NGF-TrkA-mediated retrograde signal to transcription factor CREB in sympathetic neurons. Science 277, 1097–1100.
Richardson, P. M., and Issa, V. M. (1984). Peripheral injury enhances central regeneration of primary sensory neurones. Nature 309, 791–793.
Rowland, B. D., Bernards, R., and Peeper, D. S. (2005). The KLF4 tumour suppressor is a transcriptional repressor of p53 that acts as a context-dependent oncogene. Nat. Cell Biol. 7, 1074–1082.
Schmitt, A. B., Breuer, S., Liman, J., Buss, A., Schlangen, C., Pech, K., Hol, E. M., Brook, G. A., Noth, J., and Schwaiger, F. W. (2003). Identification of regeneration-associated genes after central and peripheral nerve injury in the adult rat. BMC Neurosci. 4, 8. doi:10.1186/1471-2202-4-8
Schwaiger, F. W., Hager, G., Schmitt, A. B., Horvat, A., Hager, G., Streif, R., Spitzer, C., Gamal, S., Breuer, S., Brook, G. A., Nacimiento, W., and Kreutzberg, G. W. (2000). Peripheral but not central axotomy induces changes in Janus kinases (JAK) and signal transducers and activators of transcription (STAT). Eur. J. Neurosci. 12, 1165–1176.
Seijffers, R., Mills, C. D., and Woolf, C. J. (2007). ATF3 increases the intrinsic growth state of DRG neurons to enhance peripheral nerve regeneration. J. Neurosci. 27, 7911–7920.
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., Amin, N., Schwikowski, B., and Ideker, T. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504.
Shi, S., Calhoun, H. C., Xia, F., Li, J., Le, L., and Li, W. X. (2006). JAK signaling globally counteracts heterochromatic gene silencing. Nat. Genet. 38, 1071–1076.
Silver, J., and Miller, J. H. (2004). Regeneration beyond the glial scar. Nat. Rev. Neurosci. 5, 146–156.
Skene, J. H., and Willard, M. (1981). Characteristics of growth-associated polypeptides in regenerating toad retinal ganglion cell axons. J. Neurosci. 1, 419–426.
Smith, D. S., and Skene, J. H. (1997). A transcription-dependent switch controls competence of adult neurons for distinct modes of axon growth. J. Neurosci. 17, 646–658.
Smith, R. P., Lerch-Haner, J. K., Pardinas, J. R., Buchser, W. J., Bixby, J. L., and Lemmon, V. P. (2011). Transcriptional profiling of intrinsic PNS factors in the postnatal mouse. Mol. Cell. Neurosci. 46, 32–44.
Spencer, T. K., Mellado, W., and Filbin, M. T. (2008). BDNF activates CaMKIV and PKA in parallel to block MAG-mediated inhibition of neurite outgrowth. Mol. Cell. Neurosci. 38, 110–116.
Stam, F. J., MacGillavry, H. D., Armstrong, N. J., de Gunst, M. C., Zhang, Y., van Kesteren, R. E., Smit, A. B., and Verhaagen, J. (2007). Identification of candidate transcriptional modulators involved in successful regeneration after nerve injury. Eur. J. Neurosci. 25, 3629–3637.
Subang, M. C., and Richardson, P. M. (2009). Neuroscience. Nuclear power for axonal growth. Science 326, 238–239.
Sun, P., Enslen, H., Myung, P. S., and Maurer, R. A. (1994). Differential activation of CREB by Ca2+/calmodulin-dependent protein kinases type II and type IV involves phosphorylation of a site that negatively regulates activity. Genes Dev. 8, 2527–2539.
Swamynathan, S. K., Davis, J., and Piatigorsky, J. (2008). Identification of candidate Klf4 target genes reveals the molecular basis of the diverse regulatory roles of Klf4 in the mouse cornea. Invest. Ophthalmol. Vis. Sci. 49, 3360–3370.
Tanaka, H., Yamashita, T., Asada, M., Mizutani, S., Yoshikawa, H., and Tohyama, M. (2002). Cytoplasmic p21(Cip1/WAF1) regulates neurite remodeling by inhibiting Rho-kinase activity. J. Cell Biol. 158, 321–329.
Tang, Y., Zhao, W., Chen, Y., Zhao, Y., and Gu, W. (2008). Acetylation is indispensable for p53 activation. Cell 133, 612–626.
Tedeschi, A., and Di Giovanni, S. (2009). The non-apoptotic role of p53 in neuronal biology: enlightening the dark side of the moon. EMBO Rep. 10, 576–583.
Tedeschi, A., Nguyen, T., Puttagunta, R., Gaub, P., and Di Giovanni, S. (2009a). A p53-CBP/p300 transcription module is required for GAP-43 expression, axon outgrowth, and regeneration. Cell Death Differ. 16, 543–554.
Tedeschi, A., Nguyen, T., Steele, S. U., Feil, S., Naumann, U., Feil, R., and Di Giovanni, S. (2009b). The tumor suppressor p53 transcriptionally regulates cGKI expression during neuronal maturation and is required for cGMP-dependent growth cone collapse. J. Neurosci. 29, 15155–15160.
Teng, F. Y., and Tang, B. L. (2006). Axonal regeneration in adult CNS neurons – signaling molecules and pathways. J. Neurochem. 96, 1501–1508.
Tsujino, H., Kondo, E., Fukuoka, T., Dai, Y., Tokunaga, A., Miki, K., Yonenobu, K., Ochi, T., and Noguchi, K. (2000). Activating transcription factor 3 (ATF3) induction by axotomy in sensory and motoneurons: a novel neuronal marker of nerve injury. Mol. Cell. Neurosci. 15, 170–182.
van Kesteren, R. E., Mason, M. R., Macgillavry, H. D., Smit, A. B., and Verhaagen, J. (2011). A gene network perspective on axonal regeneration. Front. Mol. Neurosci. 4:46. doi:10.3389/fnmol.2011.00046
Vinkemeier, U., Cohen, S. L., Moarefi, I., Chait, B. T., Kuriyan, J., and Darnell, J. E. Jr. (1996). DNA binding of in vitro activated Stat1 alpha, Stat1 beta and truncated Stat1: interaction between NH2-terminal domains stabilizes binding of two dimers to tandem DNA sites. EMBO J. 15, 5616–5626.
Wang, R., Cherukuri, P., and Luo, J. (2005). Activation of Stat3 sequence-specific DNA binding and transcription by p300/CREB-binding protein-mediated acetylation. J. Biol. Chem. 280, 11528–11534.
Watson, F. L., Heerssen, H. M., Bhattacharyya, A., Klesse, L., Lin, M. Z., and Segal, R. A. (2001). Neurotrophins use the Erk5 pathway to mediate a retrograde survival response. Nat. Neurosci. 4, 981–988.
Watson, F. L., Heerssen, H. M., Moheban, D. B., Lin, M. Z., Sauvageot, C. M., Bhattacharyya, A., Pomeroy, S. L., and Segal, R. A. (1999). Rapid nuclear responses to target-derived neurotrophins require retrograde transport of ligand-receptor complex. J. Neurosci. 19, 7889–7900.
Webster, G. A., and Perkins, N. D. (1999). Transcriptional cross talk between NF-kappaB and p53. Mol. Cell. Biol. 19, 3485–3495.
Williams, K. L., Rahimtula, M., and Mearow, K. M. (2005). Hsp27 and axonal growth in adult sensory neurons in vitro. BMC Neurosci. 6, 24. doi:10.1186/1471-2202-6-24
Williams, K. L., Rahimtula, M., and Mearow, K. M. (2006). Heat shock protein 27 is involved in neurite extension and branching of dorsal root ganglion neurons in vitro. J. Neurosci. Res. 84, 716–723.
Wong, C., Rougier-Chapman, E. M., Frederick, J. P., Datto, M. B., Liberati, N. T., Li, J. M., and Wang, X. F. (1999). Smad3-Smad4 and AP-1 complexes synergize in transcriptional activation of the c-Jun promoter by transforming growth factor beta. Mol. Cell. Biol. 19, 1821–1830.
Wu, D., Zhang, Y., Bo, X., Huang, W., Xiao, F., Zhang, X., Miao, T., Magoulas, C., Subang, M. C., and Richardson, P. M. (2007). Actions of neuropoietic cytokines and cyclic AMP in regenerative conditioning of rat primary sensory neurons. Exp. Neurol. 204, 66–76.
Xiao, H. S., Huang, Q. H., Zhang, F. X., Bao, L., Lu, Y. J., Guo, C., Yang, L., Huang, W. J., Fu, G., Xu, S. H., Cheng, X. P., Yan, Q., Zhu, Z. D., Zhang, X., Chen, Z., Han, Z. G., and Zhang, X. (2002). Identification of gene expression profile of dorsal root ganglion in the rat peripheral axotomy model of neuropathic pain. Proc. Natl. Acad. Sci. U.S.A. 99, 8360–8365.
Yamaguchi, H., Woods, N. T., Piluso, L. G., Lee, H. H., Chen, J., Bhalla, K. N., Monteiro, A., Liu, X., Hung, M. C., and Wang, H. G. (2009). p53 acetylation is crucial for its transcription-independent proapoptotic functions. J. Biol. Chem. 284, 11171–11183.
Yiu, G., and He, Z. (2006). Glial inhibition of CNS axon regeneration. Nat. Rev. Neurosci. 7, 617–627.
Zhang, B., and Horvath, S. (2005). A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 4, Article17.
Zhang, W., Geiman, D. E., Shields, J. M., Dang, D. T., Mahatan, C. S., Kaestner, K. H., Biggs, J. R., Kraft, A. S., and Yang, V. W. (2000). The gut-enriched Kruppel-like factor (Kruppel-like factor 4) mediates the transactivating effect of p53 on the p21WAF1/Cip1 promoter. J. Biol. Chem. 275, 18391–18398.
Zhang, X., and Darnell, J. E. Jr. (2001). Functional importance of Stat3 tetramerization in activation of the alpha 2-macroglobulin gene. J. Biol. Chem. 276, 33576–33581.
Zhang, X., Odom, D. T., Koo, S. H., Conkright, M. D., Canettieri, G., Best, J., Chen, H., Jenner, R., Herbolsheimer, E., Jacobsen, E., Kadam, S., Ecker, J. R., Emerson, B., Hogenesch, J. B., Unterman, T., Young, R. A., and Montminy, M. (2005). Genome-wide analysis of cAMP-response element binding protein occupancy, phosphorylation, and target gene activation in human tissues. Proc. Natl. Acad. Sci. U.S.A. 102, 4459–4464.
Zhang, Y., Feng, X. H., and Derynck, R. (1998). Smad3 and Smad4 cooperate with c-Jun/c-Fos to mediate TGF-beta-induced transcription. Nature 394, 909–913.
Keywords: axonal regeneration, transcriptional pathways, transcription factors and co-activators, epigenetic changes, network
Citation: Tedeschi A (2012) Tuning the orchestra: transcriptional pathways controlling axon regeneration. Front. Mol. Neurosci. 4:60. doi: 10.3389/fnmol.2011.00060
Received: 31 October 2011;
Accepted: 23 December 2011;
Published online: 12 January 2012.
Edited by:
Simone Di Giovanni, University of Tuebingen, GermanyCopyright: © 2012 Tedeschi. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Andrea Tedeschi, Deutsches Zentrum für Neurodegenerative Erkrankungen, Bonn, Germany. e-mail:YW5kcmVhLnRlZGVzY2hpQGR6bmUuZGU=