 Rishabh Singh1†
Rishabh Singh1†

- 1 Department of Anatomy and Neurobiology, Virginia Commonwealth University Medical Center, Richmond, VA, USA
- 2 Department of Biological Chemistry, University of Michigan Medical School, Ann Arbor, MI, USA
- 3 Molecular and Behavioral Neuroscience Institute, University of Michigan Medical School, Ann Arbor, MI, USA
At least three forms of signaling between pre- and postsynaptic partners are necessary during synapse formation. First, “targeting” signals instruct presynaptic axons to recognize and adhere to the correct portion of a postsynaptic target cell. Second, trans-synaptic “organizing” signals induce differentiation in their synaptic partner so that each side of the synapse is specialized for synaptic transmission. Finally, in many regions of the nervous system an excess of synapses are initially formed, therefore “refinement” signals must either stabilize or destabilize the synapse to reinforce or eliminate connections, respectively. Because of both their importance in processing visual information and their accessibility, retinogeniculate synapses have served as a model for studying synaptic development. Molecular signals that drive retinogeniculate “targeting” and “refinement” have been identified, however, little is known about what “organizing” cues are necessary for the differentiation of retinal axons into presynaptic terminals. To identify such “organizing” cues, we used microarray analysis to assess whether any target-derived “synaptic organizers” were enriched in the mouse dorsal lateral geniculate nucleus (dLGN) during retinogeniculate synapse formation. One candidate “organizing” molecule enriched in perinatal dLGN was FGF22, a secreted cue that induces the formation of excitatory nerve terminals in muscle, hippocampus, and cerebellum. In FGF22 knockout mice, the development of retinal terminals in dLGN was impaired. Thus, FGF22 is an important “organizing” cue for the timely development of retinogeniculate synapses.
Introduction
Proper functioning of the mammalian nervous system requires the assembly of precisely aligned pre- and postsynaptic elements between appropriate partner neurons. At least three sequential signaling mechanisms are thought to assure appropriate synaptic connectivity. First, a growing axon must recognize and adhere to an appropriate portion of a postsynaptic target cell, a process termed synaptic targeting (Sanes and Yamagata, 2009). Cell adhesion molecules, axonal guidance molecules, extracellular matrix molecules, growth factors, morphogens, and neurotrophins each have been shown to contribute to synaptic targeting in the vertebrate CNS (for examples see Yamagata et al., 2002; Yamagata and Sanes, 2008; Osterhout et al., 2011; for review see Yamagata et al., 2003; Waites et al., 2005; Sanes and Yamagata, 2009). Following axon-target recognition, signals from the presynaptic axon and postsynaptic target cell are exchanged to coordinate the transformation of these elements so that they are specialized for synaptic transmission – a process called synaptic differentiation (Waites et al., 2005; Craig et al., 2006; Fox and Umemori, 2006). Trans-synaptic cues that induce the recruitment of presynaptic release machinery (i.e., synaptic vesicles, active zone components, etc.) or the clustering of neurotransmitter receptors and intracellular scaffolds in the postsynaptic cell have been dubbed “synaptic organizers” and include many of the same families of molecules that contribute to synaptic targeting (Shen and Cowan, 2010). While synaptic targeting and organizing cues generate connections between specific neurons, an excess of these connections are initially formed, therefore, subsequent activity-dependent mechanisms lead to the stabilization and maturation of some synapses and the destabilization and pruning of others (Lichtman and Colman, 2000; Waites et al., 2005; Kano and Hashimoto, 2009).
As perturbation of synaptic structure has been implicated in contributing to a multitude of neurological diseases, identifying cellular and molecular signals responsible for synaptic targeting, differentiation, and refinement has received considerable attention. One CNS region that has served as a model for studying neural circuit formation is the dorsal lateral geniculate nucleus (dLGN), a thalamic nucleus that receives input from retinal ganglion cells (RGCs) and relays this input to visual cortex. In addition to excitatory RGC terminals, synaptic terminals in the LGN also arise from local interneurons, inhibitory projection neurons in the thalamic reticular nucleus (TRN), layer VI cortical neurons, and neurons within the midbrain and brainstem (Sherman and Guillery, 2002). While the function of this complex circuitry has been thoroughly addressed, our understanding of circuit formation in dLGN is largely limited to synapses arising from RGC axons – the retinogeniculate synapse.
In mice, RGC axons begin to invade the dLGN by the end of embryonic development (Godement et al., 1984). As these axons enter the dLGN they are targeted into topographically arranged regions, such that axons from neighboring RGCs in the retina target adjacent regions of the dLGN and thereby convey the location of stimuli in the visual field to a spatially correlated region of dLGN. Topographic targeting in mouse dLGN is driven by Eph kinase/ephrin interactions: depending upon their location in retina, RGCs (and their axons) express a variable level of Eph kinase whereas a graded expression of Eph kinase ligands, ephrins, exists in dLGN (Cheng et al., 1995; Drescher et al., 1995; Feldheim et al., 1998; Pfeiffenberger et al., 2005; Feldheim and O’Leary, 2010). Functional, albeit immature, retinogeniculate synapses form shortly after RGC axons target postsynaptic neurons in dLGN. In fact, postsynaptic responses are present in dLGN as early as the day of birth in mice, although the strength of retinogeniculate synapses during the first week of life are considerably weaker than synapses after natural eye-opening (~P14; Mooney et al., 1996; Chen and Regehr, 2000; Jaubert-Miazza et al., 2005). In additional to weaker inputs, an excess of synaptic connections exists between RGC axons and dLGN relay neurons at early ages. Before eye-opening a relay neuron may receive inputs from 10–20 RGC axons however by eye-opening only one to three retinal inputs remain on a single relay neuron (Chen and Regehr, 2000; Jaubert-Miazza et al., 2005). Although this period of synaptic refinement and remodeling occurs before natural eye-opening and visually evoked activity, it does depend upon activity in the form of waves of spontaneously generated activity in RGCs (Guido, 2008; Huberman et al., 2008; Feller, 2009). Some molecular “refinement” signals that act downstream of this spontaneous retinal activity have been elucidated in dLGN. These include components of the classical complement cascade (Stevens et al., 2007), major histocompatibility complex (MHC) class 1 (Corriveau et al., 1998; Huh et al., 2000), and neuronal pentraxins (Bjartmar et al., 2006).
Despite extensive characterization of the targeting and refinement of retinogeniculate circuits, we know little about the mechanisms regulating synaptic differentiation at this synapse. The list of organizing cues that direct synaptic differentiation at CNS synapses is considerably large (see Waites et al., 2005; Craig et al., 2006; Fox and Umemori, 2006; Eroglu and Barres, 2010; Johnson-Venkatesh and Umemori, 2010; Jones et al., 2011; Kucukdereli et al., 2011; Terauchi and Umemori, 2011), however whether any of these contribute to synapse formation in dLGN remains unclear. For this reason, we sought to identify synaptic organizers enriched in dLGN during the development of retinogeniculate synapses. Using a microarray approach we found that fibroblast growth factor 22 (FGF22) – a target-derived cue that directs excitatory nerve terminal formation in hippocampus, cerebellum, and muscle (Umemori et al., 2004; Fox et al., 2007; Umemori, 2009; Terauchi et al., 2010), is enriched in dLGN as retinal terminals form. Using targeted mouse mutants lacking FGF22, we show that the formation and maturation of retinogeniculate (but not other dLGN synapses) is impaired in the absence of this organizing cue. Thus, FGF22 contributes to the timely development of retinogeniculate synapses.
Materials and Methods
Reagents and Antibodies
All chemicals and reagents were obtained from Sigma (St. Louis, MO, USA) or Fisher (Fairlawn, NJ, USA), unless otherwise noted. A rabbit polyclonal antibody directed against melanopsin (Meln) was kindly provided by Dr. C. K. Chen [Virginia Commonwealth University, Richmond, VA, USA; diluted 1:2500 for immunohistochemistry (IHC)]. Antibodies for the following antigens were purchased: rabbit polyclonal anti-calretinin (Calr; Millipore; diluted 1:1000 for IHC), rabbit polyclonal anti-vesicular glutamate transporter 2 (VGluT2; Synaptic Systems Inc.; diluted 1:500 for IHC), mouse monoclonal anti-glutamic acid decarboxylase 67 (GAD67; Millipore; diluted 1:500 for IHC), rabbit polyclonal anti-fibroblast growth factor receptor 2 (Santa Cruz; FGFR2; diluted 1:100 for IHC), goat polyclonal anti-brain-specific homeobox/POU domain protein 3A (Brn3a; Millipore; diluted 1:125 for IHC), mouse monoclonal anti-synaptophysin 1 (Synaptic Systems Inc.; diluted 1:500 for IHC), monoclonal mouse anti-synaptotagmin 2 (previously called znp1; Fox and Sanes, 2007; Zebrafish International Resource Center, Inc.; diluted 1:250 for IHC), monoclonal mouse monoclonal anti-actin [Millipore; diluted 1:10,000 for western blots (WB)], and goat polyclonal anti-FGF22 (Santa Cruz; diluted 1:300 for WB). Fluorescent- and HRP-conjugated secondary antibodies were purchased from Invitrogen or Jackson ImmunoResearch (diluted 1:1000 for IHC and 1:5000 for WB).
Mice
Wild-type C57 and CD1 mice were from Charles River. Fgf22−/− mice (on a C57 background) were generated and described previously (Terauchi et al., 2010). All analyses conformed to National Institutes of Health guidelines and protocols approved by the Virginia Commonwealth University Institutional Animal Care and Use Committee.
Immunohistochemistry
Brains and retinas from C57 mice were fixed in 4% paraformaldehyde (PFA; in phosphate-buffered saline, PBS) for 12 h at 4°C. They were then repeatedly washed in PBS and incubated for a minimum of 24 h in 20% sucrose in PBS. Fixed tissue was frozen in Tissue Freezing Medium (Triangle Biomedical Sciences, Durham, NC, USA) and sectioned (16–20 μm) coronally on a Leica CM1850 cryostat. Sections were air-dried, incubated in blocking buffer (2.5% BSA, 5% NGS, and 0.1% Triton X-100 in PBS) for 30 min at room temperature, and subsequently incubated in primary antibodies diluted in blocking buffer for >12 h at 4°C. Following several washes in PBS, sections were treated with secondary antibodies diluted in blocking buffer for 60 min at room temperature. After four PBS washes, sections were stained with DAPI and mounted in VectaShield (Vector Laboratories, Burlingame, CA, USA). Immunostained sections were visualized on either a Leica SP2 scanning confocal microscope or a Zeiss Axiolmager A1 fluorescent microscope. dLGN could be unequivocally identified by both DAPI staining and VGluT2-immunoreactivity. When comparing different ages of tissues or between genotypes, images were acquired with identical parameters. For quantification, images were acquired from a ventromedial portion of dLGN (see Figure 1A). At least four images were acquired per animal. For quantification of the relative area (per field or per dLGN) occupied by immunoreactivity, images were thresholded in Adobe Photoshop so that no sigbal was detected in the optic tract of a P7 control animal (threshold value = 80). The same threshold value was applied to all other images regardless of age or genotype. Percent area of the thresholded signal was quantified with NIH ImageJ (Bethesda, MD, USA). All image manipulations and measurements were performed using identical procedures or parameters regardless of genotype or age. Four animals were analyzed for each genotype and age.
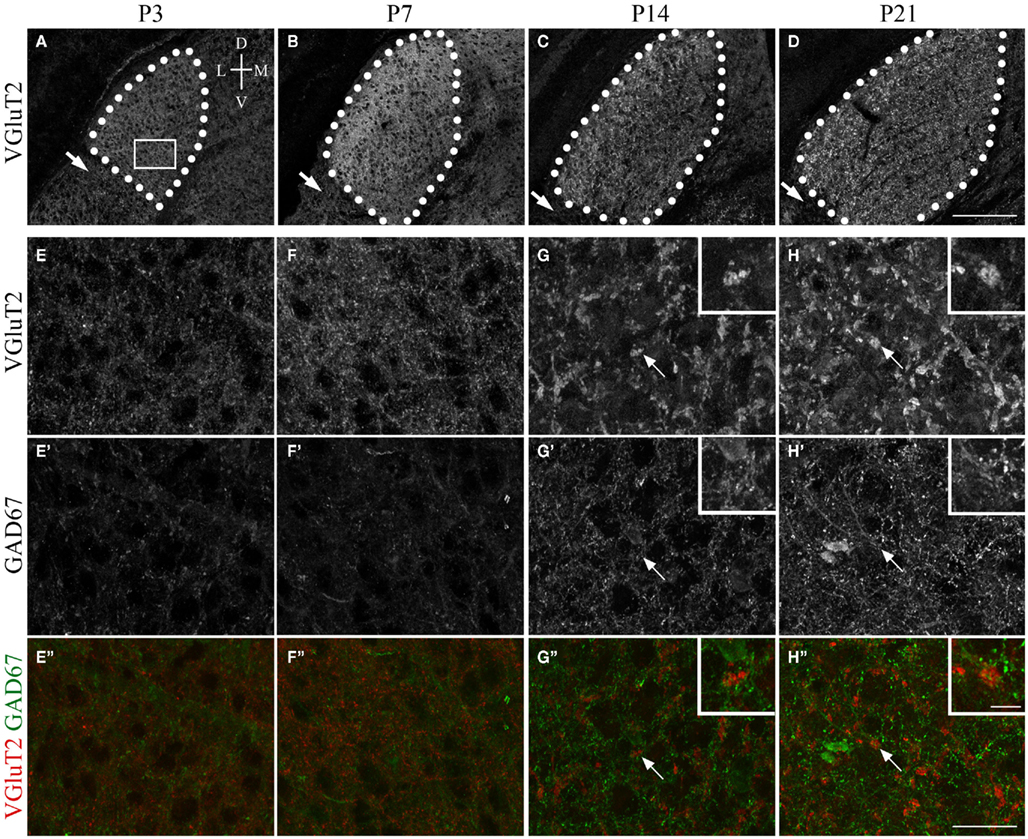
Figure 1. Synaptic development in mouse dLGN. (A–D) VGluT2-immunoreactivity in coronal sections of dLGN at P3, P7, P14, P21. dLGN are encircled by white dots. Arrows indicate the intergeniculate leaflet (IGL). Tissue orientation is indicated in (A): D, dorsal; L, lateral; M, medial; V, ventral. (E–H) High magnification of (A–D) [from areas similar to that shown by the white box in (A)] show the transformation of immature and small VGluT2-positive retinal terminals into large, morphologically distinct RLPs. (E′–H′) show the development of Gad67 inhibitory synapses in the postnatal dLGN. (E″–H″) show merged images of retinal terminals (red) and inhibitory terminals (green). Arrows in (G,H) highlight presumptive VGluT2-expressing RLPs [which are shown in higher magnification in the insets in (G,H)]. All images were acquired with identical settings on a confocal microscope. Scale bar in (D) = 200 μm for (A–D), in (H″) = 25 μm for (E–H″), and in the inset in (H″) = 5 μm for all insets.
Microarray Analysis
RNA was isolated from P3 and P8 CD1 wild-type mouse dLGN, as described previously (Su et al., 2011). Briefly, mice were decapitated, brains removed, and 300 μm coronal sections were cut in ice-cold DEPC-PBS with a vibratome. dLGN were microdissected from at least five littermates and tissues were pooled per sample. RNA was isolated using the Bio-Rad Total RNA extraction from Fibrous and Fatty Tissue kit (Bio-Rad). RNA purity assessment, first- and second-strand cDNAs preparation, cRNAs generation, hybridization to Agilent Whole Genome 44kx4 mouse arrays, and data analysis with Agilent Feature Extraction and GeneSpring GX, version 7.3.1, software packages were performed by GenUs Biosystems. To be considered differentially expressed, genes must have demonstrated a twofold difference (up- or down-regulation) in the averaged sample sets (n = 3; p < 0.05). Three pooled samples were analyzed per age.
Quantitative PCR
RNA was purified from pooled C57 dLGN samples as described above. cDNAs were generated with Superscript II Reverse Transcriptase First-Strand cDNA Synthesis kit (Invitrogen). Quantitative PCR (qPCR) was performed on a Chromo4 Four-Color Real-Time System (Bio-Rad) using iQ SYBR Green Supermix (Bio-Rad) as described previously (Su et al., 2010). The following primer pairs were used: fgf22, 5′-ACA CGG ACA GAA CGG ATC TC-3′, and 5′-CCA CTC ACT TTT TCC TGC GT-3′; actin, 5′-TTC TTT GCA GCT CCT TCG TT-3′, and 5′-ATG GAG GGG AAT ACA GCC C-3′. A minimum of three experiments (each in triplicate) was run for each age.
Western Blot
Wild-type C57 mice were perfused with PBS, brains removed and dLGN were dissected in ice-cold PBS. Tissue was pooled from at least eight littermates per age and was lysed in modified loading buffer containing 50 mmol/L Tris–HCl, pH 6.8, 2% sodium dodecyl sulfate (SDS), 10% glycerol, and protease inhibitors (1 mmol/L PMSF). Samples were homogenized, boiled for 10 min, and insoluble material was removed. Protein concentrations were determined by bicinchoninic acid (BCA) protein assay (Pierce, Rockford, IL, USA). Equal amounts of protein were loaded and separated by SDS-PAGE and transferred to PVDF membrane as described previously (Fox and Sanes, 2007). After blocking in 5% non-fat milk in PBS (containing 0.05% Tween 20), PVDF membranes were incubated with appropriate primary antibodies, followed by HRP-conjugated secondary antibodies. Immunoblotted proteins were detected with enhanced chemiluminescent detection system (ECL Plus, Amersham Pharmacia Biotech, Piscataway, NJ, USA) as previously described (Fox et al., 2003).
Results
Synaptic Development in DLGN
To begin to address the molecular mechanisms that regulate presynaptic nerve terminal development, we first documented nerve terminal formation and maturation in mouse dLGN using nerve terminal-specific markers. Retinal terminals were labeled with antibodies directed against vesicular glutamate transporter 2 (VGluT2), a synaptic vesicle associated protein present in classes of excitatory nerve terminals and expressed by RGCs (Fujiyama et al., 2003; Land et al., 2004). VGluT2 is absent from other glutamatergic, excitatory synapses within dLGN, which instead contain VGluT1 (Fujiyama et al., 2003). Thus, in dLGN VGluT2 immunolabeling specifically and selectively marks retinal terminals (Land et al., 2004; Figures 1A–D). To label inhibitory nerve terminals, which are derived from either local dLGN interneurons or inhibitory TRN neurons, antibodies directed against glutamate decarboxylase 67 (GAD67) were employed.
Using these terminal-specific markers we found that shortly after the targeting of RGC axons, the dLGN contained VGluT2-positive terminals but largely lacked GAD67-positive terminals (Figures 1A,E). By the end of the first postnatal week of development, a substantial increase in the number and density of excitatory, VGluT2-positive retinal terminals was observed (Figures 1B,F). Despite increases in retinogeniculate synapse number, little change was seen in the number or density of inhibitory synapses during this period (Figures 1E,F). The sparsity of GAD67-positive inhibitory nerve terminals support electrophysiological studies that demonstrate a general lack of inhibitory synaptic responses following optic tract stimulation in dLGN at the end of the first week of mouse development (Bickford et al., 2010). The number of GAD67-immunoreactive terminals appeared to dramatically increase by P14 (Figure 1G), which coincides with an increased incidence of inhibition following optic tract stimulation (Bickford et al., 2010).
A remarkable feature of retinal terminals in the adult dLGN is that they are morphologically distinct and significantly larger than other excitatory or inhibitory terminals in dLGN. Based upon their round shaped vesicles, large size, and pale mitochondria in ultrastructural analyses, retinogeniculate terminals have been named RLPs. IHC analysis at early perinatal ages suggested that VGluT2-immunoreactive retinal terminals were initially no larger than inhibitory terminals in dLGN (Figures 1E,F). However by P14, and at all ages thereafter, VGluT2-positive retinal terminals had transformed into larger and morphologically distinct terminals, presumably indicating the formation of RLPs (Figures 1C,D,G,H). The increase in VGluT2-positive terminal size appeared to coincide with a decrease in the number of retinal terminals in dLGN (compare Figures 1A–D). These findings confirm previous ultrastructural studies that suggested a 10-fold reduction in the number of retinogeniculate synapses from P7 to P14 (Bickford et al., 2010). Together these data demonstrate that during the first week of mouse development the numbers of retinal terminals increase dramatically but then during the second week some of these terminals are stabilized and mature while others are destabilized and pruned.
Two final observations regarding the formation and maturation of retinal terminals in LGN warrant mention. First, retinal terminal growth and maturation in dLGN appeared to occur in a dorsolateral to ventromedial gradient (compare Figures 1C,D), perhaps reflecting the fact that RGC axons innervate dorsolateral dLGN first. Second, little VGluT2-immunoreactivity was observed in the intergeniculate leaflet (IGL; arrows in Figures 1C,D), an adjacent retino-recipient nucleus (also see Fujiyama et al., 2003). This is somewhat surprising since RGC axons also innervate IGL.
Identification of Synaptic Organizing Molecules in DLGN
We next sought to identify synaptic organizing molecules whose developmental expression in dLGN coincided with the perinatal increase in synapse number. To capture the genetic profile of dLGN before the large increase in retinal terminal number, RNA was isolated from P3 mouse dLGN. Expression profiles in these pools of RNA were compared with RNA isolated from P8 dLGN, an age that corresponds to the increase in retinogeniculate terminal number and before the increase in inhibitory synaptic terminals. Differentially expressed genes in these RNA pools were identified by Agilent microarray analysis.
To assess changes in the expression of genes that might contribute to synaptic differentiation in dLGN we mined the array data, focusing on families of synaptic organizing molecules. We specifically explored the following genes: neuroligins (nlgn1–4), neurexins (nrxn1–3), cell adhesion molecules [cadm1–4 (also called SynCAM1–4)], leucine-rich repeat transmembrane molecules (lrrtm1–4), protein tyrosine phosphatase receptors (ptprf, ptprs), leucine-rich repeat and fibronectin type III domain containing molecules [lrfn1–5;also called synaptic adhesion-like molecules (SALMs1–5)], cerebellins (cbln1–4), thrombospondins (thbs1–4), secreted proteins acidic and rich in cysteine (sparc, sparcl1), neurotrophins (ngf, bdnf, ntf3, ntf4), Wnts, bone morphogenic factors (bmp1–7), ephrins (efna1–5, efnb1–3), Eph kinases (epha1–10, ephb1–6), and synaptogenic FGFs (fgf7, fgf10, fgf22). Figure 2 plots the relative change in expression in these genes in P8 dLGN compared with levels of expression at P3. Genes mentioned above that are not listed in Figure 2 were not detected by probe sets on the microarray. Of the >70 synaptic organizing genes examined, the gene most up-regulated at P8 was fgf22, which was detected as being significantly enriched by two distinct probe sets in the array (see blue bars in Figure 2). In addition to fgf22, 10 other genes encoding synaptic organizers were significantly enriched at P8: nlgn3, sparc, sparcl1, wnt5a, wnt7a, wnt7b, bmp4, lrrtm1, ephb3, and ephb4. While ~15% of the genes coding for synaptic organizers were up-regulated, the expression of another set of genes was significantly reduced at P8. Down-regulated synaptic organizers included nlgn1, nrxn1, cadm1, cbln2, ptprf, epha7, and efna2. It is unclear why these genes are down-regulated during postnatal dLGN development but may suggest roles for these cues in synaptic targeting (for examples see Osterhout et al., 2011; Su et al., 2011).
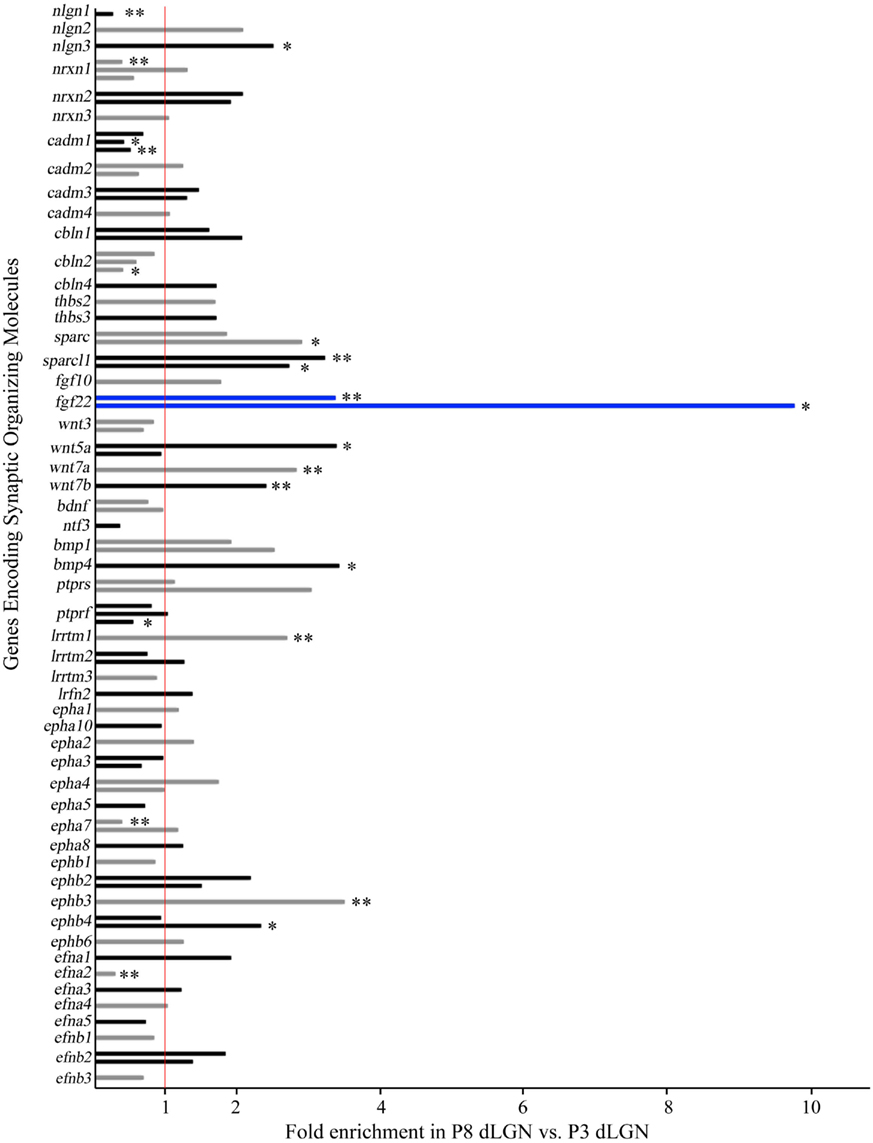
Figure 2. Changes in the expression of synaptic organizing molecules in the postnatal dLGN. Relative mRNA expression levels of known families of synaptic organizing molecules in P8 dLGN was compared to that at P3 by Agilent microarray. Bar graphs represent fold enrichment (or decrease) in P8 dLGN vs. P3 dLGN. The red line represents no change in gene expression between these ages. Bar color is alternated between gray and black for each adjacent gene for ease of viewing, with the exception of blue bars representing mRNA expressional levels for fgf22, the synaptic organizer showing the greatest enrichment. The expression of many genes is shown with multiple bars: in these cases each bar represents data from a unique probe set in the array. *Denote data that are statistically significant with p < 0.05. **Denote data that are statistically significant with p < 0.01. Expression of 11 synaptic organizers was enriched in P8 samples (nlgn3, sparc, sparcl1, fgf22, wnt5a, wnt7a, wnt7b, bmp4, lrrtm1, ephb3, ephb4) whereas seven were significantly down-regulated at P8 (nlgn1, nrxn1, cadm1, cbln2, ptprf, epha7, efna2).
FGF22 is Expressed in DLGN and Its Receptor is Expressed by RGCs
We next sought to confirm the expression of fgf22 mRNA in dLGN. RNA was isolated from wild-type dLGN at P2, P3, P8, and P14 and quantitative RT-PCR (qPCR) was performed. qPCR confirmed a considerable enrichment in fgf22 expression in P8 dLGN compared with expression levels at P2 and P3 (Figure 3A). Levels of fgf22 continued to increase postnatally until P14, an age in which retinal terminals have matured into RLPs (Figure 3A; Bickford et al., 2010).
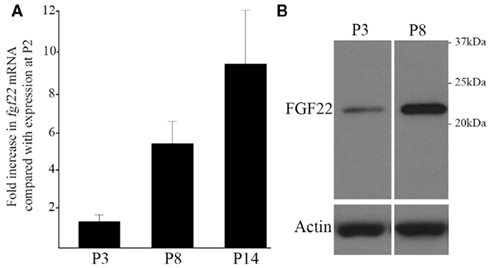
Figure 3. Increases in fgf22 mRNA and FGF22 protein levels in dLGN coincide with the maturation of retinal terminals. (A) The developmental up-regulation of fgf22 mRNA from P2 to P14 in dLGN was examined by quantitative RT-PCR (qPCR). Data from P3, P8, and P14 were compared against that at P2. All data was normalized to actin mRNA levels. Data are shown ± SEM; n > 4. (B) Western blots demonstrate an increase in FGF22 protein from P3 to P8 in dLGN protein extracts. Levels of actin were used as loading controls.
Since changes in mRNA expression levels do not always correlate with protein expression, we next used antibodies directed against FGF22 to probe protein levels in dLGN. Figure 3B demonstrates that FGF22 levels increase substantially during the first week of postnatal development. Together, these results confirm that developmental increases in FGF22 expression, at both mRNA and protein levels, coincide with the formation and maturation of retinal nerve terminals in dLGN.
Target-derived FGF22 has previously been shown to induce the formation of excitatory nerve terminals in skeletal muscle, hippocampus, and cerebellum (Umemori et al., 2004; Fox et al., 2007; Terauchi et al., 2010). Based upon these roles in synaptic differentiation and its significant enrichment in P8 dLGN, we hypothesized that dLGN-derived FGF22 induces the assembly and growth of retinal terminal. For this to be true, retinal axons must express appropriate FGF22 receptors. FGFs bind and signal through a family of alternatively spliced receptors, termed FGF receptors (FGFRs; Zhang et al., 2006). FGF22 primarily signals through FGFR2 to induce nerve terminal assembly (Umemori et al., 2004; Fox et al., 2007; Terauchi et al., 2010). Previous studies have reported postnatal increases in FGFR2 expression by mouse RGCs, which we confirm here (Figure 4A; Catalina et al., 2009). However, not all RGCs innervate dLGN. We therefore tested whether classes of RGCs known to target dLGN express FGFR2. We focused on RGCs that express either the calcium binding protein calretinin (Calr) or the transcription factor Brn3a, both of which are expressed by classes of dLGN-projecting RGCs (Luth et al., 1993; Quina et al., 2005; Badea et al., 2009; Su et al., 2011). Co-labeling of retinal cross-sections with antibodies against FGFR2 and either Calr or Brn3a revealed that dLGN-projecting classes of RGCs express the FGF22 receptor (Figures 4B,C). The expression of appropriate receptors by dLGN-projecting classes of RGCs supports our hypothesis that target-derived FGF22 regulates the formation and development of retinal terminals.
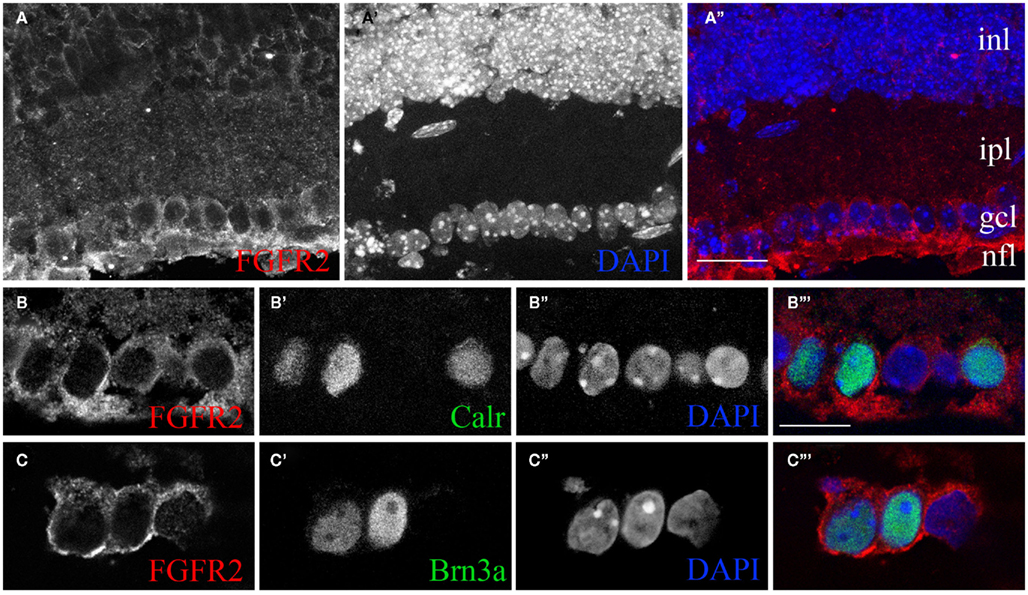
Figure 4. The FGF22 receptor FGFR2 is expressed by classes of retinal ganglion cells that innervate dLGN. (A) Confocal analysis of immunostained P14 wild-type retinal cross-sections revealed the presence of FGFR2 in the ganglion cell layer (gcl) and nerve fiber layer (nfl). Retinal layers were identified by nuclear-labeling with DAPI. (B,C) Single optical slices from confocal analyses of retinal cross-sections immunostained for FGFR2 and either Calr (B) or Brn3a (C). Nuclei were labeled with DAPI. inl, inner nuclear layer; ipl, inner plexiform layer; gcl, ganglion cell layer; nfl, nerve fiber layer. Scale bar in (A) = 50 μm and in (B) = 20 μm for (B,C).
FGF22 Contributes to Retinal Terminal Formation and Maturation in vivo
To test the role of FGF22 in the development of retinogeniculate nerve terminals we used a previously generated targeted mouse mutant that lacked FGF22 (fgf22−/−; Terauchi et al., 2010). Mutants lacking FGF22 are born in expected numbers and are phenotypically indistinguishable from littermate controls during the first weeks of postnatal development.
The development of retinal and inhibitory nerve terminals in dLGN in the absence of FGF22 was assessed by immunostaining for VGluT2 and GAD67 as described above. Terminals were examined in coronal sections of mutant and littermate control dLGN at P7, P14, and P21. While we detected no difference in GAD67-immunoreactivity in P7 dLGN, fewer VGluT2-containing nerve terminals were observed in dLGN lacking FGF22 (Figures 5A,B). The relative area occupied by VGluT2-positive terminals in mutant and control dLGN was quantified and was statistically significant (Figure 5C). We interpret these results to indicate fewer retinogeniculate synapses are present in dLGN in the absence of FGF22. Similar analyses revealed no change in the area occupied by GAD67-immunoreactive terminals in P7 mutant dLGN (Figures 5A–C).
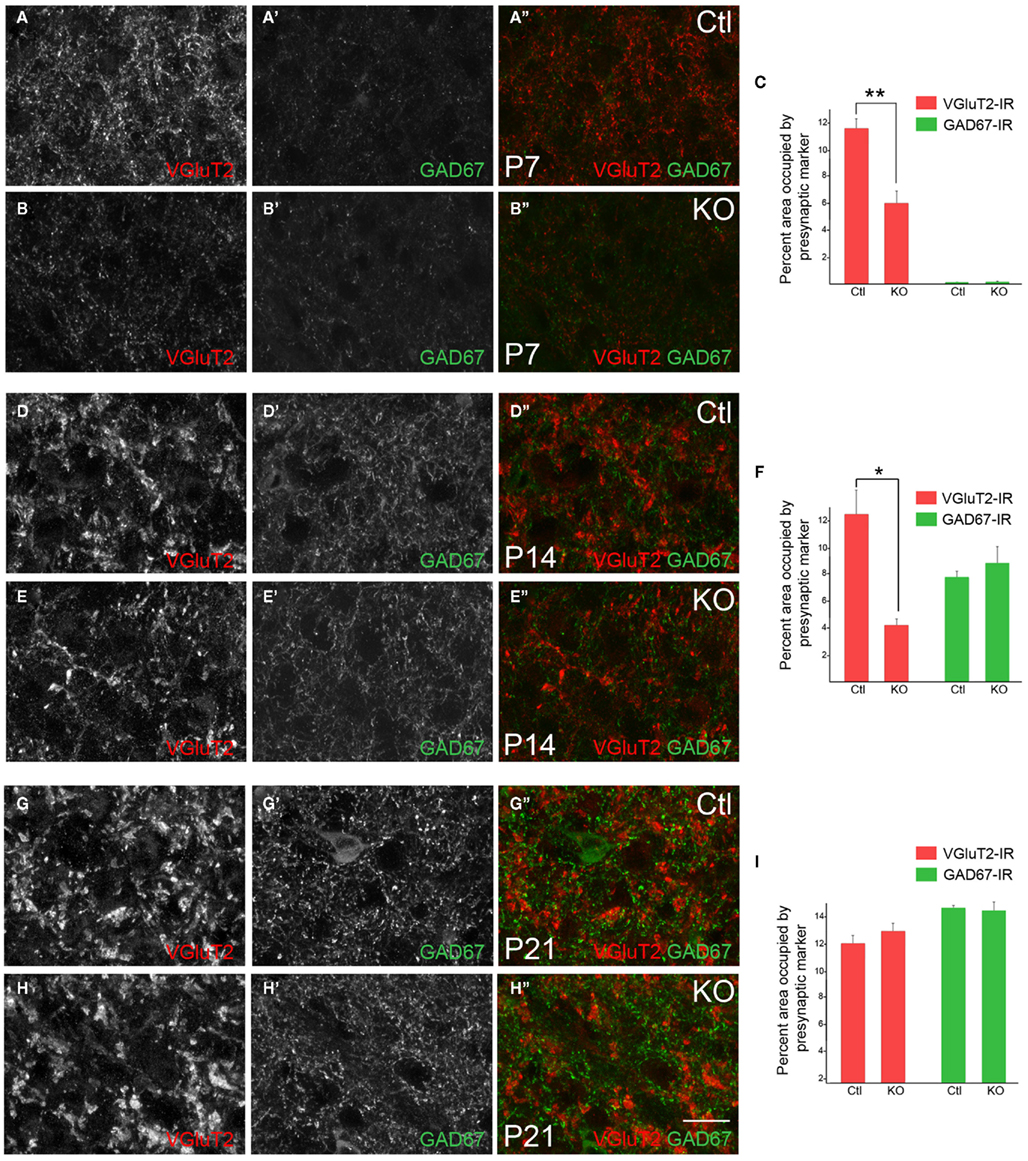
Figure 5. Deletion of FGF22 impairs the formation and maturation of retinal terminals in the dLGN. VGluT2- and GAD67-immunoreactivity in coronal sections of control (Ctl) and fgf22−/− mutant (KO) dLGN at P7 (A,B), P14 (D,E), and P21 (G,H). Differences in the percent area of immunoreactivity in each image were quantified for each age and genotype (C,F,I). At P7 fewer VGluT2-immunoreactive puncta were observed in mutant dLGN, leading to a statistically significant reduction in the area of immunoreactivity (A–C). At P14, VGluT2-immunoreactive (VGluT2-IR) terminals appeared less mature and the area that mutant terminals occupied in each field of view was significantly lower than in controls (D–F). By P21 no differences were observed in VGluT2–IHC in dLGN of mutants and controls (G–I). At all ages GAD67-immunoreactivity (GAD67-IR) appeared similar in fgf22−/− and control dLGN. For (C,F,I) data shown are ±SEM; n = 4 mice. *Differs from age-matched controls at p < 0.05 by Student’s t-test. **Differs from age-matched controls at p < 0.01 by Student’s t-test. Scale bar in (H) = 15 μm.
We next examined nerve terminal number and morphology at P14, an age in which immature terminals have differentiated into large, morphologically distinct presynaptic terminals (i.e., RLPs). As in P7 mutant dLGN, fewer VGluT2-containing nerve terminals appeared in P14 fgf22−/− dLGN (Figures 5D,E) and the area occupied by these terminals was significantly reduced (Figure 5F). In addition to fewer terminals it also appeared as if retinogeniculate terminals were smaller in P14 fgf22−/− dLGN (Figures 5D,E), a finding that suggests the normal timing of retinal terminal maturation may be delayed in the absence of FGF22. Again, in contrast to defects observed at retinal terminals, the development and distribution of inhibitory nerve terminals appeared unaffected by the loss of FGF22 at this age (Figures 5D–F).
Finally we examined retinal and inhibitory terminals at P21, an age in which synaptic development is largely complete in mouse dLGN (Chen and Regehr, 2000; Jaubert-Miazza et al., 2005). We found no appreciable differences in the number or morphology of retinal or inhibitory nerve terminals in mutants or controls at this age (Figures 5G–I). Moreover, the dorsolateral to ventromedial gradient of retinogeniculate synaptic development remained intact in mutant dLGN (data not shown). Taken together these results suggest that FGF22 contributes to retinal (but not inhibitory) nerve terminal development in dLGN, but that in its absence compensatory mechanisms eventually promote retinogeniculate circuit formation.
Normal Retinal Development in fgf22−/− Mutants
We interpret the above results to suggest a direct role for FGF22 in retinogeniculate circuit development. An alternative possibility is that these defects are secondary to intra-retinal defects or reflect a delay in the arrival of retinal axons into dLGN. To address these issues we analyzed retinal development in fgf22−/− mutants. The retina is a layered structure that contains five main cell types: photoreceptors reside in the outer most layer of the retina – the outer nuclear layer (ONL); bipolar cells, horizontal cells, amacrine cells reside in the inner nuclear layer (INL); RGCs reside in the inner most cell layer – the ganglion cell layer (Masland, 2001; Sanes and Zipursky, 2010). Two synaptic layers exist in the retina: the outer plexiform layer (OPL) where photoreceptors synapse onto dendrites of bipolar cells, and the inner plexiform layer (IPL) where bipolar and amacrine cells synapse onto RGC dendrites. Immunostaining retinal cross-sections from P14 control and fgf22−/− mutant mice showed no appreciable difference in the structure of the retina, the thickness of retinal layers, or the density of neurons in the absence of FGF22 (Figures 6A–D). Moreover, removal of FGF22 had no discernable affect on the generation of Brn3a- or Calr-expressing classes of RGCs (Figures 6A–D), or the dendritic stratification of RGCs in the IPL (Figures 6C–F). Not only did the morphology of retinal neurons and their circuitry appear unaltered in fgf22−/− mutants but the timing of retinal development also appeared unaltered. For example, in wild-type adult retina the synaptic vesicle associated protein synaptotagmin 2 (Syt2) is restricted to horizontal cells and classes of OFF-bipolar cells whose axons arborize in the outer portion of the IPL (Fox and Sanes, 2007). However during the first ~11 postnatal days of retinal development it is also transiently expressed in starburst amacrine cell processes in the IPL (Fox and Sanes, 2007). The absence of Syt2 from starburst amacrine cell processes in P14 fgf22−/− mutants (Figures 6E,F) suggests that the timing of its development is unaltered in the absence of FGF22. Thus, delayed synaptogenesis in fgf22−/− dLGN is not caused by delayed or aberrant retinal development.
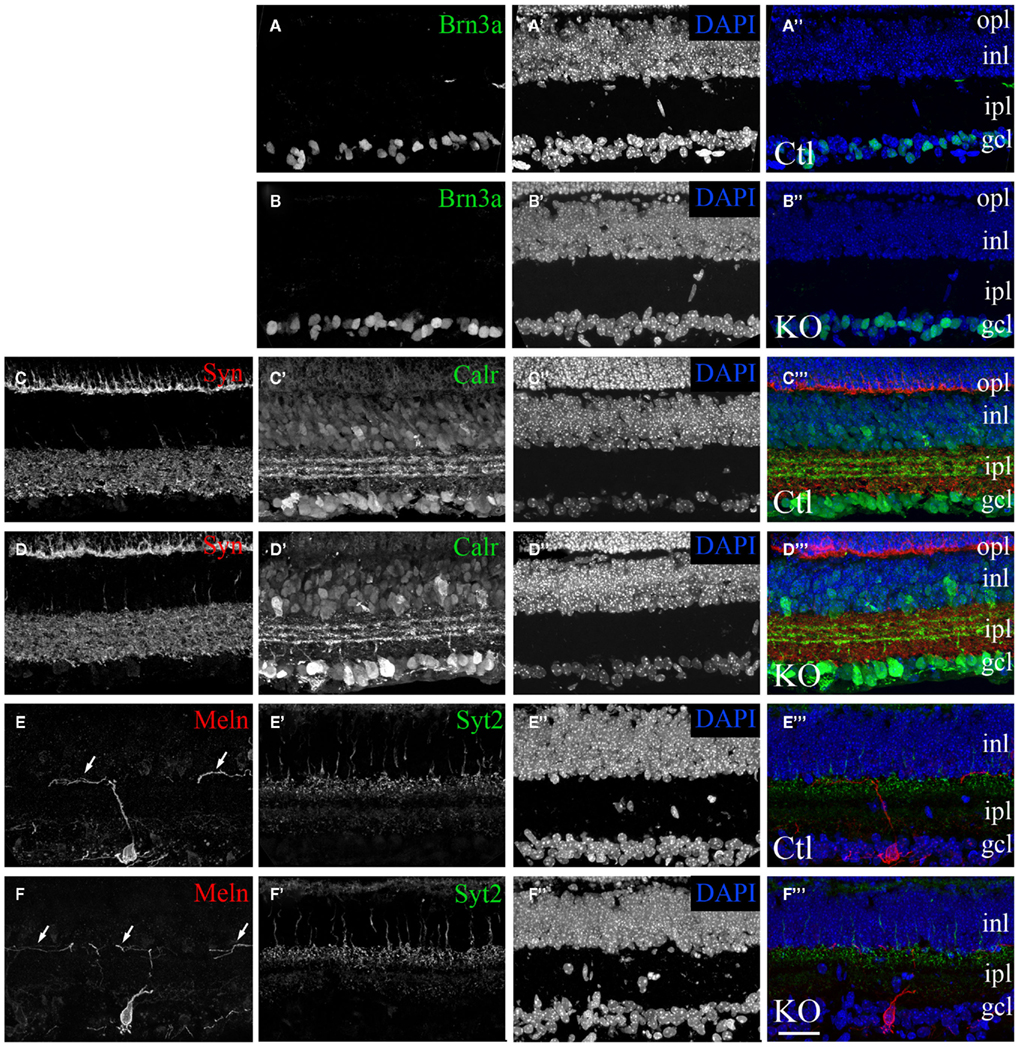
Figure 6. Normal morphological development of the retina in mice lacking FGF22. Confocal images of retinal cross-sections from P14 fgf22−/− mutant mice (B,D,F) and littermate controls (A,C,E). (A–D) Classes of dLGN-projecting RGCs were labeled by Brn3a–IHC (A,B) and Calr–IHC (C,D). The number and distribution of Brn3a- and Calr-expressing RGCs appeared similar in control and fgf22−/−retina. (C–F) Labeling of synaptic layers with anti-synaptophysin (Syn) (C,D) and synaptotagmin2 (Syt2) (E,F) revealed no remarkable differences in the density of synapses or laminar arrangement of the retina between mutants and controls. Likewise labeling with anti-Syt2, anti-Calr, and anti-melanopsin (Meln) revealed that sublaminar specificity was indistinguishable in controls and mice lacking FGF22 (D,F) and controls (C,E). Arrows in (E,F) show that dendrites from Meln-expressing RGCs correctly target the inner most region of the IPL in the absence of FGF22. In all sections nuclei were labeled with DAPI. inl, inner nuclear layer; ipl, inner plexiform layer; gcl, ganglion cell layer. Scale bar = 100 μm.
We next addressed whether the targeting of retinal axons was delayed in mutants lacking FGF22. For this, we examined the presence of Calr-expressing retinal axons in dLGN at P7 – an age in which we have previously demonstrated the presence of these axons in mouse dLGN (Su et al., 2011). The arborization of Calr-expressing RGC axons appeared similar in fgf22−/− mutants and littermate controls (Figure 7). Thus the delay in synaptogenesis in dLGN does not appear to arise from a delay in retinogeniculate targeting.
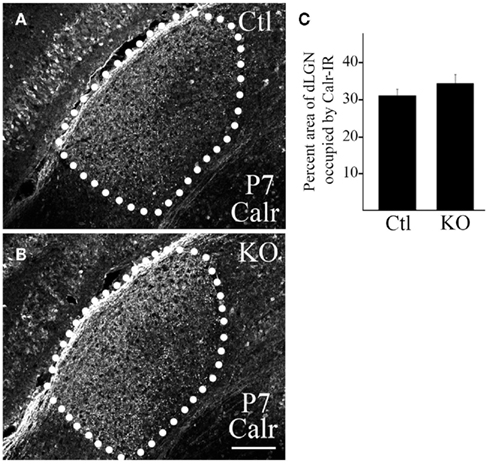
Figure 7. Deletion of FGF22 does not delay targeting of dLGN-projecting RGC axons. Calr–IHC was used to assess the development of dLGN-projecting RGC axons in P7 control [Ctl; (A)] and fgf22−/− mutant [KO; (B)] dLGN. dLGN are encircled by white dots. (C) The area of Calr-IR in control and mutant P7 dLGN was measured in ImageJ. No significant differences were detected in the percent area of dLGN innervated by Calr-expressing RGCs in P7 controls and mutants. Data are shown ± SEM; n = 4 mice. Scale bar = 125 μm.
Discussion
Identifying the mechanisms that underlie synapse formation is critical for our understanding of how neural circuits – and their associated functions – are established. Although retinogeniculate circuits have been used as a model for the mechanisms regulating synaptic targeting and refinement, the molecular mechanisms driving the differentiation of retinal axons and dendrites of thalamic relay neurons into precisely aligned pre- and postsynaptic elements remains unclear. In other brain regions synaptic differentiation is orchestrated by membrane-bound and extracellular “organizing” cues. In the present study we focused on identifying candidate synaptic organizers that regulate the timely assembly of retinal nerve terminals in dLGN. To accomplish this we explored the temporal progression of nerve terminal development in dLGN and then identified genes whose expression correlated with the formation of retinal nerve terminals. Detailed analysis of one of these genes, fgf22, revealed that target-derived FGFs contribute to the timely development of retinogeniculate synapses.
Retinal and Inhibitory Nerve Terminal Development in dLGN
Development of retinal nerve terminals in dLGN is a protracted, multistep process that shares many similarities with the pattern of nerve terminal development at the mouse neuromuscular junction (NMJ), a peripheral synapse between lower motor neurons and skeletal muscle fibers. At the NMJ, motor axons are targeted to postsynaptic sites in the central end-plate region of muscle embryonically, several weeks before pre- and postsynaptic elements are structurally and functionally mature (Fox, 2009). Motor axon–muscle contacts differentiate into immature synapses during late embryonic development in rodents (Kelly and Zacks, 1969; Lupa and Hall, 1989; Sanes and Lichtman, 1999; Fox, 2009). At these early ages, an excess of motor nerve terminals are present on each postsynaptic site, such that a single muscle fiber may receive input from 2–12 different motor neurons (Wyatt and Balice-Gordon, 2003). By P14, however, activity-dependent refinement has led to the elimination of all supernumerary nerve terminals, so that each postsynaptic site (and thus each muscle fiber) is innervated by a single motor nerve terminal (Wyatt and Balice-Gordon, 2003). As excess terminals are pruned, the remaining nerve terminal and postsynaptic apparatus mature and grow to resemble their adult-like form by P21 (Balice-Gordon and Lichtman, 1993).
Like motor axons, retinal axons begin to target appropriate regions of dLGN embryonically, weeks before the retinogeniculate synapses are adult-like in their ultrastructure or physiology (Godement et al., 1984; Chen and Regehr, 2000; Jaubert-Miazza et al., 2005). Here we show that the number of VGluT2-containing retinal nerve terminals increase dramatically in dLGN during the first postnatal week of development (Figure 1), more than a week after their initial arrival. By P7 the density of retinal terminals is so high it is not surprising that dLGN relay neurons may be innervated by up to 20 different RGCs at this age (Chen and Regehr, 2000; Jaubert-Miazza et al., 2005). Despite the increased density of presynaptic profiles at these early ages, there is much evidence that these synapses are still immature: at P7 retinogeniculate synapses are weak and lack their adult-like ultrastructural morphology (Bickford et al., 2010). Here we further show that at P7 retinal terminals are small compared to adult terminals, and in fact are similar in size to GABAergic inhibitory nerve terminals. During the second and third week of mouse development (P7–P21), spontaneous retinal activity drives the elimination of excess retinal nerve terminals (pruning the number of inputs that each dLGN relay neuron receives) and the remaining terminals mature into adult-like RLPs (Guido, 2008; Bickford et al., 2010). Like the NMJ, by P21 the morphology and function of retinogeniculate synapses reach an adult-like state (Chen and Regehr, 2000; Bickford et al., 2010).
While the timing and sequential development of excitatory retinal nerve terminals resembles that described for motor axons at the NMJ, inhibitory synapse development in dLGN follows a different pattern. We observed few inhibitory presynaptic terminals during the first postnatal week of development, supporting studies showing few inhibitory postsynaptic responses in P7 dLGN relay neurons (Bickford et al., 2010). Despite the lack of inhibitory activity or nerve terminals, interneuron-specific genes are present in mouse dLGN as early as P6, confirming that interneurons are indeed present at these early ages (Yuge et al., 2011). By eye-opening, and at all ages thereafter, we observed GAD67-containing GABAergic terminals densely populating dLGN. The late development of inhibitory circuits may reflect that interneurons in dLGN are born later than RGCs and presumably after retinal axons have begun to target the dLGN (Hayes et al., 2003; Yuge et al., 2011). However, since interneurons are indeed present at P6 (an age in which we observed few inhibitory nerve terminals) it suggests that cues necessary for inducing inhibitory synaptogenesis are not present until after retinogeniculate synapses form. Besides forming later, our data also suggest that cellular mechanisms of inhibitory nerve terminal formation differ from that of retinal terminals. For example, the size or shape of inhibitory terminals did not appear to change after their initial formation, nor did we detect a period of inhibitory synapse refinement, in which the density of inhibitory terminals decreased with age (as seen from P7 to P14 for retinal terminals).
The maturation/refinement of retinogeniculate synapses and the dramatic increase in inhibitory synapses both coincide with natural eye-opening. Moreover, retinal terminals are “multisynaptic boutons” that not only synapse onto relay neuron dendrites but also synapse onto dendritic terminals of inhibitory interneurons (which themselves synapse onto the relay neuron dendrite; Famiglietti, 1970; Sherman and Guillery, 2002; Sherman, 2004). These local circuits (or synaptic “triads”) generate feed-forward inhibition to temporally sharpen visual input to thalamus (Sherman and Guillery, 2002; Sherman, 2004). The coincident emergence of adult-like retinogeniculate terminals and inhibitory synapses, as well as there interconnection at “triads,” suggest a coordinated pattern of development. With this in mind, it was unexpected to discover that the development of inhibitory synapses was not delayed in fgf22−/− mutant mice despite delayed development of retinogeniculate synapses. This suggests that retinal and inhibitory synapses develop independently in dLGN. An alternative possibility is that retinal terminals are required for inhibitory synapse formation, but the state of their maturation does not matter.
FGF signaling is required for visual system circuit assembly
Fibroblast growth factor family members contribute to many aspects of neural development, from neural induction to the formation of neural circuits (Dono, 2003; Umemori, 2009). FGF22 was originally purified from developing mouse brain extracts for its ability to cluster synaptic vesicles into presumptive nerve terminals in cultured neurons (Umemori et al., 2004). Expression of FGF22 by postsynaptic neurons (or muscle) and its receptor FGFR2 by presynaptic neurons support in vitro studies suggesting that FGFs act as target-derived presynaptic organizers (Umemori et al., 2004; Fox et al., 2007). Moreover, genetic disruption of FGF22–FGFR2 signaling results in defects in excitatory nerve terminal assembly at mossy fiber-pyramidal neuron synapses in hippocampus, mossy fiber–granule cell synapses in cerebellum, and at the NMJ (Umemori et al., 2004; Fox et al., 2007; Terauchi et al., 2010). Together, these studies demonstrate that FGF22 is necessary for the differentiation of nerve terminals in the mammalian brain. FGF7 and FGF10, which are closely related to FGF22, share similar abilities to induce presynaptic differentiation (Umemori et al., 2004). While FGF7 and FGF22 share similar (and presumably interchangeable) roles in the assembly of nerve terminals at the NMJ, they exhibit distinct synaptogenic functions in brain. In hippocampus, FGF7 and FGF22 are both generated by CA3 pyramidal neurons but one is required at excitatory synapses and the other at inhibitory synapses (Terauchi et al., 2010).
Our studies show a novel role for this family of synaptogenic FGFs in visual system circuit assembly. Based upon results showing that FGF22 expression in dLGN coincides with synaptogenesis (Figure 3), FGFR2 expression by classes of dLGN-projecting RGCs (Figure 4), and an impairment in the development of retinal nerve terminals in the absence of FGF (Figure 5) all suggest that FGF22 acts as a target-derived presynaptic organizer in the mouse visual system. It is unclear whether other synaptogenic FGFs (namely FGF7 and FGF10) also contribute to retinogeniculate synapse formation, or are perhaps able to compensate for the absence of FGF22 in fgf22−/− mutant mice. It is also unclear whether roles for FGFs in visual system circuit assembly are confined to the dLGN or whether other retino-recipient nuclei [such as the superior colliculus (SC)] utilize these mechanisms for circuit assembly.
Multiple presynaptic organizers in dLGN
Our results demonstrate that ~15% of synaptic organizing molecules are enriched in dLGN during retinogeniculate circuit assembly (Figure 2). Why are so many target-derived organizing molecules enriched in dLGN? There are many possible answers to this question (see Fox et al., 2007), two of which we will discuss here. First, different organizers may be required for different types of synapses in dLGN. Like most regions of brain, dLGN contains a wide array of synaptic inputs. In addition to receiving input from RGCs, relay neurons also receive inputs from cortex, brainstem, midbrain, other thalamic regions, and local interneurons. In fact, retinal terminals represent only 5–10% of the inputs onto a given relay neuron in dLGN (Van Horn et al., 2000; Sherman and Guillery, 2002). A variety of synapses are also present on dLGN interneurons (Sherman and Guillery, 2002). As excitatory and inhibitory synapses generally require different synaptic organizing cues it is likely that the variety of synaptic types accounts, at least in part, for the multitude of synaptic organizers enriched during postnatal dLGN development. However it is noteworthy that we analyzed the expression of these organizing molecules at a stage in development when few non-retinal synapses are forming in dLGN. And, at least two synaptic organizers (in addition to FGF22) were identified that are known to contribute to the formation of synapses between RGC axons and target neurons. SPARC and SPARC-like 1, two glial-derived ECM molecules, contribute to retino-collicular synapse formation (Kucukdereli et al., 2011). Based upon their up-regulation in dLGN during synaptogenesis it is tempting to speculate that they play similar roles in dLGN and may be capable of compensating for the absence of FGF22 in mutants studied here.
Alternatively, different organizers may contribute to different aspects of synaptic development. At the NMJ, different muscle-derived presynaptic organizing molecules are required for sequential aspects of motor nerve terminal development: FGF7/10/22 induce the initial formation of motor nerve terminals, laminins containing the β2 subunit are required for postnatal maturation of nerve terminals, and synaptic collagens IV are required for motor nerve terminal maintenance (Fox et al., 2007). Genetic disruption of FGF signaling at the NMJ leads to a delay in nerve terminal formation, much like we observe here for FGF22 at retinogeniculate synapses. Several findings suggest that other organizers, such as muscle-derived laminin β2 and synaptic collagens, are able to compensate in the absence of FGF–FGFR2 signaling at the NMJ. First, defects in nerve terminal development are temporary, as described above. Second, genetic removal of both FGFR2 (in motor axons) and laminin β2 prolongs defects associated with FGF signaling (Fox et al., 2007). As the development of retinogeniculate synapses share similarities with NMJ development (including their dependence of FGF signaling), it may be that multiple target-derived organizing molecules are required for different aspects of retinal nerve terminal development and these additional cues are capable of partly compensating in the absence of FGF22.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by The Thomas F. Jeffress and Kate Miller Jeffress Memorial Trust (Michael A. Fox), the VCU Presidential Research Initiative Program (PRIP) Award (Michael A. Fox), and the National Institutes of Health [NIH; EY021222 (Michael A. Fox); NS070005 (Hisashi Umemori)]. Microscopy was performed at the Virginia Commonwealth University Department of Anatomy and Neurobiology Microscopy Facility supported, in part, with funding from NIH–National Institute of Neurological Disorders and Stroke Center Core Grant 5P30 NS047463-02. We thank Dr. C. K. Chen for providing anti-melanopsin antibodies.
References
Badea, T. C., Cahill, H., Ecker, J., Hattar, S., and Nathans, J. (2009). Distinct roles of transcription factors brn3a and brn3b in controlling the development, morphology, and function of retinal ganglion cells. Neuron 61, 852–864.
Balice-Gordon, R. J., and Lichtman, J. W. (1993). In vivo observations of pre- and postsynaptic changes during the transition from multiple to single innervation at developing neuromuscular junctions. J. Neurosci. 13, 834–855.
Bickford, M. E., Slusarczyk, A., Dilger, E. K., Krahe, T. E., Kucuk, C., and Guido, W. (2010). Synaptic development of the mouse dorsal lateral geniculate nucleus. J. Comp. Neurol. 518, 622–635.
Bjartmar, L., Huberman, A. D., Ullian, E. M., Renteria, R. C., Liu, X., Xu, W., Prezioso, J., Susman, M. W., Stellwagen, D., Stokes, C. C., Cho, R., Worley, P., Malenka, R. C., Ball, S., Peachey, N. S., Copenhagen, D., Chapman, B., Nakamoto, M., Barres, B. A., and Perin, M. S. (2006). Neuronal pentraxins mediate synaptic refinement in the developing visual system. J. Neurosci. 26, 6269–6281.
Catalina, E., Tomassini, S., Dal Monte, M., Bosco, L., and Casini, G. (2009). Localization patterns of fibroblast growth factor 1 and its receptors FGFR1 and FGFR2 in postnatal mouse retina. Cell Tissue Res. 336, 423–438.
Chen, C., and Regehr, W. G. (2000). Developmental remodeling of the retinogeniculate synapse. Neuron 28, 955–966.
Cheng, H. J., Nakamoto, M., Bergemann, A. D., and Flanagan, J. G. (1995). Complementary gradients in expression and binding of ELF-1 and Mek4 in development of the topographic retinotectal projection map. Cell 82, 371–381.
Corriveau, R. A., Huh, G. S., and Shatz, C. J. (1998). Regulation of class I MHC gene expression in the developing and mature CNS by neural activity. Neuron 21, 505–520.
Craig, A. M., Graf, E. R., and Linhoff, M. W. (2006). How to build a central synapse: clues from cell culture. Trends Neurosci. 29, 8–20.
Dono, R. (2003). Fibroblast growth factors as regulators of central nervous system development and function. Am. J. Physiol. Regul. Integr. Comp. Physiol. 284, R867–R881.
Drescher, U., Kremoser, C., Handwerker, C., Loschinger, J., Noda, M., and Bonhoeffer, F. (1995). In vitro guidance of retinal ganglion cell axons by RAGS, a 25 kDa tectal protein related to ligands for Eph receptor tyrosine kinases. Cell 82, 359–370.
Eroglu, C., and Barres, B. A. (2010). Regulation of synaptic connectivity by glia. Nature 468, 223–231.
Famiglietti, E. V. Jr. (1970). Dendro-dendritic synapses in the lateral geniculate nucleus of the cat. Brain Res. 20, 181–191.
Feldheim, D. A., and O’Leary, D. D. (2010) , Visual map development: bidirectional signaling, bifunctional guidance molecules, and competition. Cold Spring Harb. Perspect. Biol. 2, a001768.
Feldheim, D. A., Vanderhaeghen, P., Hansen, M. J., Frisen, J., Lu, Q., Barbacid, M., and Flanagan, J. G. (1998). Topographic guidance labels in a sensory projection to the forebrain. Neuron 21, 1303–1313.
Feller, M. B. (2009). Retinal waves are likely to instruct the formation of eye-specific retinogeniculate projections. Neural Dev. 4, 24.
Fox, M. A. (2009). “Development of the vertebrate neuromuscular junction,” in The Sticky Synapse: Cell Adhesion Molecules and Their Role in Synapse Formation and Maintenance, ed. M. Hortsch, and H. Umemori (Ney York, NY: Spinger Press), 39–84.
Fox, M. A., Colello, R. J., Macklin, W. B., and Fuss, B. (2003). Phosphodiesterase-Ialpha/autotaxin: a counteradhesive protein expressed by oligodendrocytes during onset of myelination. Mol. Cell. Neurosci. 23, 507–519.
Fox, M. A., and Sanes, J. R. (2007). Synaptotagmin I and II are present in distinct subsets of central synapses. J. Comp. Neurol. 503, 280–296.
Fox, M. A., Sanes, J. R., Borza, D. B., Eswarakumar, V. P., Fassler, R., Hudson, B. G., John, S. W., Ninomiya, Y., Pedchenko, V., Pfaff, S. L., Rheault, M. N., Sado, Y., Segal, Y., Werle, M. J., and Umemori, H. (2007). Distinct target-derived signals organize formation, maturation, and maintenance of motor nerve terminals. Cell 129, 179–193.
Fox, M. A., and Umemori, H. (2006). Seeking long-term relationship: axon and target communicate to organize synaptic differentiation. J. Neurochem. 97, 1215–1231.
Fujiyama, F., Hioki, H., Tomioka, R., Taki, K., Tamamaki, N., Nomura, S., Okamoto, K., and Kaneko, T. (2003). Changes of immunocytochemical localization of vesicular glutamate transporters in the rat visual system after the retinofugal denervation. J. Comp. Neurol. 465, 234–249.
Godement, P., Salaun, J., and Imbert, M. (1984). Prenatal and postnatal development of retinogeniculate and retinocollicular projections in the mouse. J. Comp. Neurol. 230, 552–575.
Hayes, S. G., Murray, K. D., and Jones, E. G. (2003). Two epochs in the development of gamma-aminobutyric acidergic neurons in the ferret thalamus. J. Comp. Neurol. 463, 45–65.
Huberman, A. D., Feller, M. B., and Chapman, B. (2008). Mechanisms underlying development of visual maps and receptive fields. Annu. Rev. Neurosci. 31, 479–509.
Huh, G. S., Boulanger, L. M., Du, H., Riquelme, P. A., Brotz, T. M., and Shatz, C. J. (2000). Functional requirement for class I MHC in CNS development and plasticity. Science 290, 2155–2159.
Jaubert-Miazza, L., Green, E., Lo, F. S., Bui, K., Mills, J., and Guido, W. (2005). Structural and functional composition of the developing retinogeniculate pathway in the mouse. Vis. Neurosci. 22, 661–676.
Johnson-Venkatesh, E. M., and Umemori, H. (2010). Secreted factors as synaptic organizers. Eur. J. Neurosci. 32, 181–190.
Jones, E. V., Bernardinelli, Y., Tse, Y. C., Chierzi, S., Wong, T. P., and Murai, K. K. (2011). Astrocytes control glutamate receptor levels at developing synapses through SPARC-beta-integrin interactions. J. Neurosci. 31, 4154–4165.
Kano, M., and Hashimoto, K. (2009). Synapse elimination in the central nervous system. Curr. Opin. Neurobiol. 19, 154–161.
Kelly, A. M., and Zacks, S. I. (1969). The fine structure of motor endplate morphogenesis. J. Cell Biol. 42, 154–169.
Kucukdereli, H., Allen, N. J., Lee, A. T., Feng, A., Ozlu, M. I., Conatser, L. M., Chakraborty, C., Workman, G., Weaver, M., Sage, E. H., Barres, B. A., and Eroglu, C. (2011). Control of excitatory CNS synaptogenesis by astrocyte-secreted proteins Hevin and SPARC. Proc. Natl. Acad. Sci. U.S.A. 108, E440–E449.
Land, P. W., Kyonka, E., and Shamalla-Hannah, L. (2004). Vesicular glutamate transporters in the lateral geniculate nucleus: expression of VGLUT2 by retinal terminals. Brain Res. 996, 251–254.
Lichtman, J. W., and Colman, H. (2000). Synapse elimination and indelible memory. Neuron 25, 269–278.
Lupa, M. T., and Hall, Z. W. (1989). Progressive restriction of synaptic vesicle protein to the nerve terminal during development of the neuromuscular junction. J. Neurosci. 9, 3937–3945.
Luth, H. J., Winkelmann, E., and Celio, M. R. (1993). Light- and electron microscopic localization of parvalbumin, calbindin D-28k and calretinin in the dorsal lateral geniculate nucleus of the rat. J. Hirnforsch. 34, 47–56.
Mooney, R., Penn, A. A., Gallego, R., and Shatz, C. J. (1996). Thalamic relay of spontaneous retinal activity prior to vision. Neuron 17, 863–874.
Osterhout, J. A., Josten, N., Yamada, J., Pan, F., Wu, S. W., Nguyen, P. L., Panagiotakos, G., Inoue, Y. U., Egusa, S. F., Volgyi, B., Inoue, T., Bloomfield, S. A., Barres, B. A., Berson, D. M., Feldheim, D. A., and Huberman, A. D. (2011). Cadherin-6 mediates axon-target matching in a non-image-forming visual circuit. Neuron 71, 632–639.
Pfeiffenberger, C., Cutforth, T., Woods, G., Yamada, J., Renteria, R. C., Copenhagen, D. R., Flanagan, J. G., and Feldheim, D. A. (2005). Ephrin-As and neural activity are required for eye-specific patterning during retinogeniculate mapping. Nat. Neurosci. 8, 1022–1027.
Quina, L. A., Pak, W., Lanier, J., Banwait, P., Gratwick, K., Liu, Y., Velasquez, T., O’Leary, D. D., Goulding, M., and Turner, E. E. (2005). Brn3a-expressing retinal ganglion cells project specifically to thalamocortical and collicular visual pathways. J. Neurosci. 25, 11595–11604.
Sanes, J. R., and Lichtman, J. W. (1999). Development of the vertebrate neuromuscular junction. Annu. Rev. Neurosci. 22, 389–442.
Sanes, J. R., and Yamagata, M. (2009). Many paths to synaptic specificity. Annu. Rev. Cell Dev. Biol. 25, 161–195.
Sanes, J. R., and Zipursky, S. L. (2010). Design principles of insect and vertebrate visual systems. Neuron 66, 15–36.
Shen, K., and Cowan, C. W. (2010). Guidance molecules in synapse formation and plasticity. Cold Spring Harb. Perspect. Biol. 2, a001842.
Sherman, S. M. (2004). Interneurons and triadic circuitry of the thalamus. Trends Neurosci. 27, 670–675.
Sherman, S. M., and Guillery, R. W. (2002). The role of the thalamus in the flow of information to the cortex. Philos. Trans. R. Soc. Lond. B Biol. Sci. 357, 1695–1708.
Stevens, B., Allen, N. J., Vazquez, L. E., Howell, G. R., Christopherson, K. S., Nouri, N., Micheva, K. D., Mehalow, A. K., Huberman, A. D., Stafford, B., Sher, A., Litke, A. M., Lambris, J. D., Smith, S. J., John, S. W., and Barres, B. A. (2007). The classical complement cascade mediates CNS synapse elimination. Cell 131, 1164–1178.
Su, J., Gorse, K., Ramirez, F., and Fox, M. A. (2010). Collagen XIX is expressed by interneurons and contributes to the formation of hippocampal synapses. J. Comp. Neurol. 518, 229–253.
Su, J., Haner, C. V., Imbery, T. E., Brooks, J. M., Morhardt, D. R., Gorse, K., Guido, W., and Fox, M. A. (2011). Reelin is required for class-specific retinogeniculate targeting. J. Neurosci. 31, 575–586.
Terauchi, A., Johnson-Venkatesh, E. M., Toth, A. B., Javed, D., Sutton, M. A., and Umemori, H. (2010). Distinct FGFs promote differentiation of excitatory and inhibitory synapses. Nature 465, 783–787.
Terauchi, A., and Umemori, H. (2011). Specific sets of intrinsic and extrinsic factors drive excitatory and inhibitory circuit formation. Neuroscientist. PMID: 21652588. [Epub ahead of print].
Umemori, H. (2009). Weaving the neuronal net with target-derived fibroblast growth factors. Dev. Growth Differ. 51, 263–270.
Umemori, H., Linhoff, M. W., Ornitz, D. M., and Sanes, J. R. (2004). FGF22 and its close relatives are presynaptic organizing molecules in the mammalian brain. Cell 118, 257–270.
Van Horn, S. C., Erisir, A., and Sherman, S. M. (2000). Relative distribution of synapses in the A-laminae of the lateral geniculate nucleus of the cat. J. Comp. Neurol. 416, 509–520.
Waites, C. L., Craig, A. M., and Garner, C. C. (2005). Mechanisms of vertebrate synaptogenesis. Annu. Rev. Neurosci. 28, 251–274.
Wyatt, R. M., and Balice-Gordon, R. J. (2003). Activity-dependent elimination of neuromuscular synapses. J. Neurocytol. 32, 777–794.
Yamagata, M., and Sanes, J. R. (2008). Dscam and Sidekick proteins direct lamina-specific synaptic connections in vertebrate retina. Nature 451, 465–469.
Yamagata, M., Sanes, J. R., and Weiner, J. A. (2003). Synaptic adhesion molecules. Curr. Opin. Cell Biol. 15, 621–632.
Yamagata, M., Weiner, J. A., and Sanes, J. R. (2002). Sidekicks: synaptic adhesion molecules that promote lamina-specific connectivity in the retina. Cell 110, 649–660.
Yuge, K., Kataoka, A., Yoshida, A. C., Itoh, D., Aggarwal, M., Mori, S., Blackshaw, S., and Shimogori, T. (2011). Region-specific gene expression in early postnatal mouse thalamus. J. Comp. Neurol. 519, 544–561.
Keywords: synaptogenesis, presynaptic differentiation, retina, retinal ganglion cell, lateral geniculate nucleus
Citation: Singh R, Su J, Brooks J, Terauchi A, Umemori H and Fox MA (2012) Fibroblast growth factor 22 contributes to the development of retinal nerve terminals in the dorsal lateral geniculate nucleus. Front. Mol. Neurosci. 4:61. doi: 10.3389/fnmol.2011.00061
Received: 04 November 2011; Accepted: 23 December 2011;
Published online: 10 January 2012.
Edited by:
Joshua A. Weiner, The University of Iowa, USAReviewed by:
Erik Ullian, University of California San Francisco, USAAndrew David Huberman, University of California San Diego, USA
Copyright: © 2012 Singh, Su, Brooks, Terauchi, Umemori and Fox. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Michael A. Fox, Department of Anatomy and Neurobiology, Virginia Commonwealth University Medical Center, 1101 East Marshall Street, PO Box 980709, Richmond, VA 23298-0709, USA. e-mail:bWFmb3hAdmN1LmVkdQ==
†Rishabh Singh and Jianmin Su have contributed equally to this work.