

- Centre for Perinatal Brain Protection and Repair, University College London, London, UK
Following axotomy, the activation of multiple intracellular signaling cascades causes the expression of a cocktail of regeneration-associated transcription factors which interact with each other to determine the fate of the injured neurons. The nerve injury response is channeled through manifold and parallel pathways, integrating diverse inputs, and controlling a complex transcriptional output. Transcription factors form a vital link in the chain of regeneration, converting injury-induced stress signals into downstream protein expression via gene regulation. They can regulate the intrinsic ability of axons to grow, by controlling expression of whole cassettes of gene targets. In this review, we have investigated the functional roles of a number of different transcription factors – c-Jun, activating transcription factor 3, cAMP response element binding protein, signal transducer, and activator of transcription-3, CCAAT/enhancer binding proteins β and δ, Oct-6, Sox11, p53, nuclear factor kappa-light-chain-enhancer of activated B cell, and ELK3 – in peripheral nerve regeneration. Studies involving use of conditional mutants, microarrays, promoter region mapping, and different injury paradigms, have enabled us to understand their distinct as well as overlapping roles in achieving anatomical and functional regeneration after peripheral nerve injury.
Introduction
Injury to peripheral nerves elicits a sequence of molecular, cellular, and ultrastructural responses which are vital in triggering a successful regenerative program associated with neurite outgrowth, re-innervation of the denervated target and recovery of function. The rapid arrival of signals from the injured axon results in a remarkable shift of the injured neuron from a transmitting to a growth promoting phenotype. This is accomplished by the upregulation of a vast array of regeneration-associated genes (RAGs) including the rapid induction of transcription factors and enhanced synthesis of adhesion molecules, cytoskeletal elements, growth factors, cytokines, neuropeptides, and other molecules involved in regeneration.
The molecular changes are accompanied by pronounced morphological changes in the surviving neurons – the cell body undergoes a “chromatolytic” reaction characterized by swelling of the neuronal body, increase in cellular metabolism and protein synthesis, and a regional dispersion of Nissl bodies in the neuronal cytoplasm (Lieberman, 1971); and there is a rapid appearance of growth cones at the proximal tip of the lesioned axons. The distal nerve stump undergoes “Wallerian degeneration” (Waller, 1850; Dubový, 2011), leading to phagocytosis of axonal and myelin debris by Schwann cells and later also by invading macrophages.
In addition to the neuronal response, neighboring non-neuronal glial cells – in the case of brain and spinal cord motoneurons, the astrocytes, and microglia – become activated, with microglial expression of immune cell recognition molecules (ICAM1, aMb2, and aXb2 integrins, B7 system, etc.) and major histocompatibility complex (Werner et al., 1998; Bohatschek et al., 2004). There is also a rapid microglia-associated recruitment of lymphocytes within a day after injury which may assist with immune surveillance first in the absence, and later on in the presence of neuronal cell death (Raivich et al., 1998; Kalla et al., 2001). In the adult mouse facial motor nucleus model which has been studied in extensive detail, neuronal cell death is a delayed phenomenon, peaking at 2 weeks following nerve transection; the glioimmune activation is characterized by two phases – a fast but moderate response starting within 24 h after neuronal injury, and a late but much stronger response to neuronal cell death, peaking at 14 days after axotomy. Neuronal transcription factor deletions blocking cell death will suppress most of the late glioimmune response (Raivich et al., 2004; Patodia et al., 2011). However, these deletions also inhibit some of the early microglial activation – within 24 h and long before the appearance of cell death, suggesting that these transcription factors may also interfere with the synthesis of injury signals from lesioned but not dying neurons.
In contrast with the PNS, the CNS neurons normally fail to regenerate after injury, possibly due to a combination of diminished intrinsic capacity for regeneration and a heightened susceptibility to increased inhibitory factors of their extracellular environment (Maier and Schwab, 2006; Sun et al., 2011). Identifying the crucial elements responsible for successful regeneration in injured peripheral nerves, and unraveling their underlying signaling pathways and molecular components will be quintessential in improving regenerative outcomes after peripheral and central nerve injuries.
Systematic gene expression profiling using cDNA microarrays has led to a dramatic increase in the number of identified genes regulated in the injured and regenerating neurons (Bosse et al., 2001, 2006; Costigan et al., 2002; Kubo et al., 2002; Boeshore et al., 2004; Kury et al., 2004; Michaelevski et al., 2010). Interestingly, roughly half of the RAGs are part of both developmental and lesion-induced programs, suggesting that regeneration partially recapitulates development. However, this proportion of shared developmental and lesion-induced transcripts was substantially lower – only 33% – for genes encoding signal transducers or factors involved in processes such as cell death, immune response, transport, and transcriptional regulation. Most of the latter show injury-specific gene expression (Bosse et al., 2006).
Transcription factors are DNA binding proteins that can activate or repress target gene expression. Their effects are amplified by their ability to bind to multiple promoter regions, causing a large number of genes to be switched on or off by a single transcription factor. In this review, we will be discussing the key molecular events ensuing peripheral nerve injury, focusing on various transcription factors that have been implicated to play a role in neuronal regeneration.
Early Axonal Injury Sensors
Axonal injury generates three major signaling cues to the injured neurons:
A. It interferes with the retrograde flow of trophic signals, disinhibiting the normally suppressed regenerative process, within 12–24 h following injury (Raivich et al., 1991).
B. It exposes the tip of the injured axons to the intracellular content of neighboring axons and Schwann cells containing growth factors like LIF, CNTF, FGF, and NT3 (Elde et al., 1991; Sendtner et al., 1997; Kirsch et al., 2003), and later to the inflamed neural tissue environment (Lindholm et al., 1987). The above two processes can result in de novo activated molecules carrying a nuclear localization sequence (NLS) which link to importins and are retrogradely transported to the cell body by dynein motors (Schmied et al., 1993; Hanz et al., 2003).
C. It causes a disruption of the tight ionic concentration gradient between the axon and the extracellular matrix by rapid influx of extracellular ions such as calcium and sodium through the transiently open plasmalemma before it is resealed (Yoo et al., 2003). This results in depolarization and transmission of successive injury-mediated action potentials. These molecular and electrical signals cause rapid elevation of calcium and cAMP, which in turn activate multiple downstream pathways (Berdan et al., 1993).
Transcriptional Changes
The arrival of injury signals is rapidly followed by the phosphorylation and nuclear localization of a host of transcription factors. A brief summary of signaling from early sensors to mostly enzymatic cytoplasmic mediators to transcription factors and synthesis of effector molecules is shown in Figure 1. Data from phospho-proteomic and microarray studies have identified nearly 400 redundant axonal signaling networks connected to 39 transcription factors (26 transcription factor families), implicated in the sensory neuron response to axonal injury (Michaelevski et al., 2010). These include c-Jun, Jun D, activating transcription factor 3 (ATF3), cAMP response element binding protein (CREB), signal transducer and activator of transcription (STAT3), CCAAT/enhancer binding proteins (C/EBPs), p53, Oct-6, nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), nuclear factor of activated T-cells (NFATs), Kruppel-like factors (KLFs), Sox11, SnoN, ELK3, P311, and E47 among others (Schwaiger et al., 2000; Mason et al., 2003; Raivich et al., 2004; Nadeau et al., 2005; Di Giovanni et al., 2006; Jankowski et al., 2009; Ruff et al., 2009; Magoulas and Lopez-de Heredia, 2010; Moore and Goldberg, 2011; Raivich, 2011). Once in the nucleus, they bind to selective DNA promoter regions to increase or repress transcription of specific target genes. Nerve injury can also result in reduced activation of transcription factors such as islet-1, Fra-2, ATF2, and TDP43 (Doyle and Hunt, 1997; Herdegen et al., 1997a; Hol et al., 1999; Moisse et al., 2009; Sato et al., 2009), which probably contributes to the change in gene expression of the injured neuron from a fully differentiated to a growing phenotype (Raivich, 2011). The activation of transcriptional program is critical for expression of many target genes implicated in successful regeneration, and blocking of transcription at an early time point after injury changes the regenerative response of injured neurons (Smith and Skene, 1997). Recent advances in cre/loxP technology permitting cell-type and/or time specific genetic knockouts (Sauer, 1998; Akira, 2000) have begun to provide insight into the powerful roles of these transcription factors in orchestrating complex axon growth and regenerative responses. An overview of the observed phenotypes is given in Table 1.
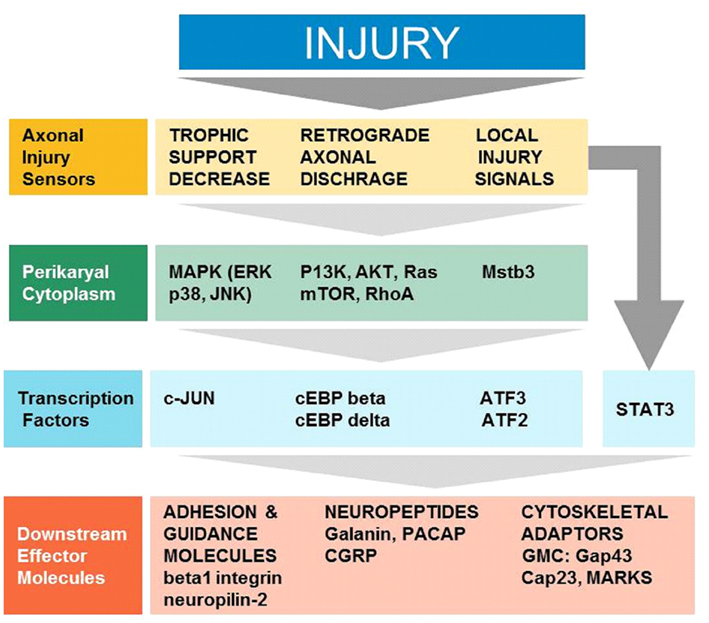
Figure 1. Cellular signaling in successful regeneration, from early sensors of injury, to cytoplasmic signals, transcription, and downstream effectors (Modified Raivich, 2011).
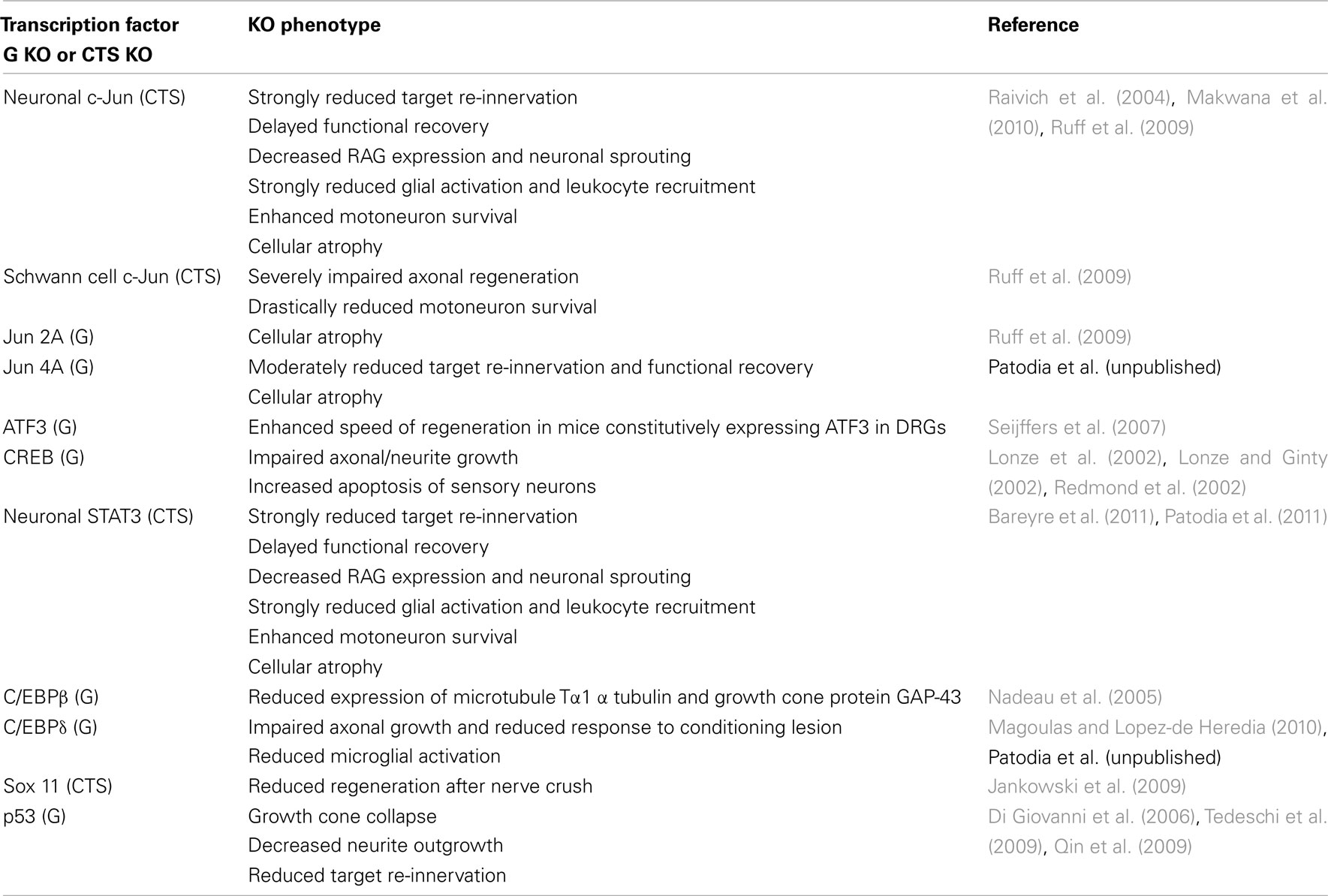
Table 1. Transcription factor deletions and peripheral nerve regeneration: effects of global (G) and cell-type specific (cts) knockouts (KO).
c-Jun
The AP-1 transcription complex is a well-characterized regulator of neural development and consists of homo- or hetero-dimeric complexes between members of the Jun, c-Fos, and ATF/CREB families. A primary component of the AP-1 complex, c-Jun, is produced as an immediate early gene (IEG) following nerve injury and persists at high levels in injured neurons during the entire peripheral regenerative process (Herdegen et al., 1991; Kenney and Kocsis, 1998; Mason et al., 2003; Raivich et al., 2004; Lindwall and Kanje, 2005; Ruff et al., 2009). Schwann cells also strongly upregulate c-Jun following axotomy, where it functions as a negative regulator of myelination and switches on local inflammation (Arthur-Farraj et al., 2007; Latouche et al., 2009; Wilton et al., 2009). Numerous studies, using targeted gene deletions or pharmacological inhibition, have examined the activation, function, and cellular basis of c-Jun and suggest a tripartite role of c-Jun action in neural degeneration, inflammation, and repair (Raivich, 2008).
Nestin-cre mediated deletion of c-Jun prevented the upregulation of axotomy-associated molecules (CD44, α7β1 integrin, galanin). It also decreased perineuronal sprouting (Makwana et al., 2010) and successful reconnection to peripheral targets by four to fivefold, and strongly reduced functional recovery (Raivich et al., 2004). This massive reduction in target re-innervation was also observed more than 3 months after facial nerve cut, suggesting that it was a persistent defect, rather than just a delay in the speed of regeneration. Incidentally, functional recovery did not appear to require re-innervation of the target by the same number of neurons. Compared with the Jun-competent littermates, functional recovery showed a longer but nevertheless transient delay, suggesting that long term, target re-innervation by a fraction of neurons could produce extensive functional recovery (Raivich et al., 2004). Importantly, though functional recovery is clinically the critical parameter, a sole reliance on this parameter could overestimate the extent of re-innervation. Non-neuronal responses like leukocyte recruitment and microglial activation were also severely compromised in the mutants. Interestingly, the facial motoneurons in the mutants showed enhanced post-axotomy survival, but were severely shrunken (Raivich et al., 2004), supporting the previously reported functional dichotomy of c-Jun in promoting post-traumatic neuronal cell death as well as axonal regeneration (Herdegen et al., 1997b; Herdegen and Waetzig, 2001).
Because this cre Recombinase was driven by a nestin-promoter expressed in early neuroepithelial cells giving rise to neurons, astrocytes, or oligodendrocytes, as well as Schwann cells it is raised the question whether the effects observed in neuronal regeneration were due to the absence of c-Jun in neurons or other nestin+ progenitor-derived cells. This issue was addressed by using transgenic mice having neuron-specific (synapsin promoter controlled cre-recombination) or Schwann cell-specific [myelin protein zero (P0) promoter controlled cre-recombination] c-Jun deletions. Neuron-specific c-Jun deletion mirrored the regeneration defect seen with nestin-cre mediated deletion of c-Jun, i.e., strongly reduced speed of axonal regeneration, significantly delayed functional recovery, strongly reduced target re-innervation, enhanced motoneuron survival but cellular atrophy and diminished astrocyte and microglial activation and T-cell influx (Ruff et al., 2009). The absence of c-Jun in Schwann cells also caused severely impaired axonal regeneration. However, it strongly increased neuronal cell death after nerve injury (Ruff et al., 2009). These results suggest a critical role of both Schwann cell and neuronal c-Jun in the axonal injury response, which led to the next challenge of understanding the mechanisms underlying its activation and signaling following axonal injury.
Activation of c-Jun mediated transcription is affected by interactions at three major sites: N-terminal phosphorylation at serines 63 and 73 and threonines 91 and 93 by the Jun N-terminal kinases (JNKs) and the ensuing ubiquitination and degradation, dephosphorylation of Thr239, and C-terminal lysine acetylation near aa 257–276 (Vries et al., 2001; Morton et al., 2003). The JNKs are rapidly activated following peripheral nerve injury, and are retrogradely transported to the cell body along with the upstream kinases MEKK1 and p-MKK4, as well as the JNK interacting protein (JIP), a scaffold protein that stabilizes the interaction between JNK and its upstream kinases (Lindwall and Kanje, 2005), reviewed in Raivich (2008). Neuronal injury increases JNK-mediated c-Jun phosphorylation (Herdegen et al., 1998; Kenney and Kocsis, 1998), and application of a JNK inhibitor to explants of injured DRGs reduces c-Jun phosphorylation, ATF3 expression, and neurite outgrowth, without affecting survival (Lindwall et al., 2004).
Removal of Ser63 and 73 phosphoacceptor sites in the JunAA mutant interfered with kainic acid excitotoxicity in hippocampal neurons (Behrens et al., 1999) at levels similar to JNK3 KO (Brecht et al., 2005), identifying c-Jun as an essential substrate of JNK signaling during kainate-induced neuronal apoptosis. While global c-Jun deletion is embryonically lethal, JunAA mice showed normal development (albeit slightly smaller than littermate wild-type mice) and were fertile (Behrens et al., 1999). Following optic nerve transection, there was a partial but significant reduction of the apoptosis of retinal ganglion cells in JunAA mice, and Fas L has been suggested to be an important target gene regulated by N-terminal Jun phosphorylation during apoptosis (Yoshida et al., 2002). JunAA and JNK3 null mutants also showed better neuronal survival after targeted dopaminergic cell death (Brecht et al., 2005; Crocker et al., 2006). Trophic factor deprivation or DNA damage-induced death was significantly delayed in JunAA neurons which correlated with delayed expression of pro-apoptotic genes (Besirli et al., 2005). In contrast, the JNK3 KO or JunAA mutations did not interfere with neuronal cell death after facial axotomy (Brecht et al., 2005; Ruff et al., 2009). This difference could be attributed to the fact that facial axotomy affects peripheral neurons, unlike previous studies which discuss effects of phosphorylation deficient c-Jun on apoptosis of CNS neurons.
Preliminary studies from our group after facial axotomy revealed that JunAA mice do show some neuronal shrinkage, but have no effect on speed of axonal regeneration for fastest axons, functional recovery, or target re-innervation (Ruff et al., 2009). Deletion of JNK1 or JNK3 single genes showed somewhat delayed functional recovery after facial nerve axotomy but the effects were quite moderate; and deletion of JNK2 had no effect on regeneration (Ruff et al., 2009). The mild effects of JunAA and global JNK deletions, and the fact that JNKs can act via c-Jun independent cytoplasmic (stathmin, MAP1b, MAP2) and nuclear (e.g., ATF2, ELK1, p53, etc.) targets (Björkblom et al., 2005; Bogoyevitch and Kobe, 2006; Barnat et al., 2010; Westerlund et al., 2011), points to a possible parting of ways: i.e., the observed JNK null effects on regeneration are c-Jun independent, and the strong Jun-dependent effects on regeneration and survival do not require N-terminal phosphorylation. Recent studies using Jun4A mice where all four JNK dependent N-terminal phosphorylation sites – Thr91 and 93 as well as Ser63 and 73 – are removed (Patodia et al., unpublished observations) partially confirm this notion. The Jun4A mutation does not interfere with the expression of injury-associated neuronal and non-neuronal markers. On the other hand, as in the JNK1 and 3 null mutants, the Jun4A strain reproduces the moderate reduction in functional recovery and a 40% lower number of reinnervating motoneurons at 28 days following facial nerve cut. However, this effect on regeneration is considerably smaller than that observed for neuronal c-Jun deletion, suggesting that the regeneration effect of c-Jun is mostly independent of its N-terminal phosphorylation. At present, no information is available with regards to the regeneration-associated Jun effects of Thr239 dephosphorylation, or for C-terminal acetylation.
Activating Transcription Factor 3
Activating transcription factor 3 is a member of the ATF/CREB family of basic leucine zipper domain (bZIP) transcription factors. It can form homo- or hetero-dimers with other bZIP transcription factors like members of the AP-1 family (ATF2, JunB, JunD, c-Jun, c-fos) and c/EBPs (Hai and Curran, 1991; Hai and Hartman, 2001). ATF3 is normally expressed at low levels, but is rapidly activated in sensory, sympathetic, and motor neurons by stress stimuli after peripheral nerve injury (Tsujino et al., 2000; Raivich and Behrens, 2006; Hyatt Sachs et al., 2007; Zigmond and Vaccariello, 2007), optic nerve injury (Takeda et al., 2000), and NGF depletion (Mayumi-Matsuda et al., 1999) but not after central injury (Tsujino et al., 2000). ATF3 expression is also induced in the disconnected Schwann cells and endoneurial fibroblasts distal to the peripheral injury site, though not on central, denervated glia (Hunt et al., 2004). Several pathways have been implicated in activating ATF3 expression such as neurotrophin deprivation (Hyatt Sachs et al., 2007), as well as JNK/SAPK and p53-dependent mechanisms (Moore and Goldberg, 2011). ATF3 is axonally transported from the periphery (Lindwall and Kanje, 2005), possibly acting as a retrograde signal.
Delivery of ATF3 to adult DRG neurons, neonatal SCG neurons and PC12 cells, enhanced both the number of neurons extending neurites and neurite length (Nakagomi et al., 2003; Pearson et al., 2003; Seijffers et al., 2006). As after conditioning peripheral injury, the pattern of growth consisted of long neurites rather than branched arborized ones (Seijffers et al., 2007). In transgenic mice constitutively expressing ATF3 in adult DRGs, the rate of peripheral nerve regeneration (measured by the nerve pinch test 2 days after sciatic nerve crush) was enhanced to an extent comparable to that produced by a preconditioning lesion (Seijffers et al., 2007). The expression of some growth-associated genes, such as Hsp27, SPRR1A, and c-Jun was increased in the non-injured neurons, but not of others like the alpha 7 integrin subunit (α7), GAP-43, CAP23, and STAT3. Unlike peripheral nerve-conditioning lesions, ATF3 overexpression also did not overcome the inhibition produced by CNS myelin in culture, suggesting the mechanisms responsible for axonal regeneration are separate from those involved in overcoming glial-associated inhibition (Seijffers et al., 2007).
Activating transcription factor 3 can bind directly to the Hsp27 promoter and activate its expression in PC12 cells (Nakagomi et al., 2003). In addition to its role as an injury-induced neuronal survival factor, Hsp27 enhances neurite outgrowth in cultured adult DRG neurons (Williams et al., 2006), possibly as a result of interactions with the cytoskeleton. The growth-associated gene SPRR1A promotes axonal outgrowth by interacting with actin structures (Bonilla et al., 2002) and ATF3 may regulate its expression either by binding directly to its promoter or by affecting other transcription factors. ATF3 also appears to have a survival role, preventing JNK-mediated neuronal death (Nakagomi et al., 2003). Thus, although ATF3 appears to contribute to nerve regeneration by increasing the intrinsic growth state of injured neurons, its upregulation is not enough to fully recapitulate the peripheral nerve regeneration program (Seijffers et al., 2007).
Activating transcription factor 3 can form hetero-dimers with c-Jun, leading to enhanced transcriptional activation of various RAGs and increased neurite outgrowth in neuronal cell lines (Hai and Curran, 1991; Hai and Hartman, 2001; Pearson et al., 2003). There has been a number of conflicting reports regarding the correlation between increased ATF3 expression and upregulation of c-Jun in surviving neurons. In 2003, Pearson et al., showed that in the absence of c-Jun, ATF3 failed to enhance neurite outgrowth in PC12 cells, suggesting that a coincident presence of c-Jun and ATF3 may act synergistically (possibly via a physical interaction) to promote nerve regeneration (Pearson et al., 2003; Lindwall et al., 2004). In contrast, Seijffers et al. (2006) showed enhanced neurite outgrowth in adult DRG neurons by ATF3 without any increase in endogenous c-Jun levels. The scope of c-Jun and ATF3 expression in DRG neurons after sciatic nerve transection also differs: ATF3 is induced in all injured neurons, phosphorylated c-Jun only in a small subset of mostly small and medium sized DRG neurons (Kenney and Kocsis, 1998; Tsujino et al., 2000; Lindwall et al., 2004). Sciatic nerve injury induces prolonged activation of JNKs in the DRGs (Kenney and Kocsis, 1998). Inhibiting JNK in this model led to decreased activation of c-Jun and ATF3 expression, and to decreased axonal growth (Lindwall et al., 2004). Recent results from our group show that deletion of neuronal c-Jun or STAT3 interferes with the nuclear transfer of ATF3 in the axotomized facial motor neurons (Patodia and Raivich, unpublished). This effect may be specific for motoneurons – in sympathetic neurons, cell-specific deletion of gp130 interferes with the appearance of phosphorylated nuclear STAT3, but does not affect the nuclear translocation of ATF3 (Habecker et al., 2009). Thus, the extent to which the growth promoting action of ATF3 is autonomous of c-Jun and STAT3 and dependent on additional injury signals needs to be explored.
cAMP Response Element Binding Protein
The CREB is a member of the ATF/CREB family of bZIP transcription factors (Hai and Hartman, 2001), and mediates cAMP signaling in the nervous system by forming homo- or hetero-dimers with other bZIP factors (Hannila and Filbin, 2008). The brains of CREB null mice show abnormal development of the corpus callosum and anterior commissure (Rudolph et al., 1998). CREB can be phosphorylated and activated by many kinases, including PKA, PKC, CAMKII, CAMKIV, AKT, MAPKAP K2, and members of RSK and MSK families (Mayr and Montminy, 2001), and it is negatively regulated by phosphatases such as PP1 and PP2A (Sun et al., 1994). Important targets of CREB include arginase I and BDNF (Mayr and Montminy, 2001; Cai et al., 2002; Deng et al., 2009).
In vitro cultured DRG and SCG neurons from CREB null mice show shorter neurites than controls. In vivo, these mutant mice exhibit impaired axonal growth and projections. Their sensory neurons undergo excess apoptosis and degeneration during the period of NGF dependency in the absence of CREB (Lonze and Ginty, 2002; Lonze et al., 2002). In addition, overexpression of a dominant negative CREB leads to decreased dendritic outgrowth in cortical neurons and loss of cAMP or neurotrophin associated neurite outgrowth on an inhibitory substrate (Redmond et al., 2002). This suggests an important role for CREB-mediated gene expression for both survival and axonal growth in PNS neurons. Recently, it has been suggested that axonal translation of CREB mRNAs may precede its phosphorylation, nuclear translocation, and transcriptional activation of pro-survival genes (Cox et al., 2008).
Signal Transducer and Activator of Transcription-3
Signal transducer and activator of transcription-3 belongs to the STAT family of seven transcription factors – STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, and STAT6 – that mediate a wide variety of biological functions in the CNS and PNS such as cell growth, regulation, inflammation, and embryological development (Dziennis and Alkayed, 2008). The STATs are comprised of six regular domains – an amino terminal (for STAT3 dimerization after activation), coiled-coil domain (for interaction with other transcription factors and regulatory proteins), DNA binding domain made up of β-sheets (for recognition of consensus binding regions on gene promoters), α-helical linker domain (for transcriptional activation and protein–protein interaction), a classical SH2 (sequence – homology) docking domain, and a transcriptional activation domain (Bromberg and Darnell, 2000; Lim and Cao, 2006). There are two highly conserved, phosphorylatable amino acids that are frequently required for STAT3 activation – a tyrosine 705 in the SH2 domain and a serine 727 in the 78-amino acid region of the transactivation domain (Bromberg and Darnell, 2000; Dziennis and Alkayed, 2008).
Injury-induced ligand binding to cell-surface receptors activates the associated receptor tyrosine kinase Janus kinases (JAKs), which phosphorylate tyrosine residues on the cytoplasmic portion of the receptor complex that acts as a binding site for STAT proteins. STATs dock onto the phospho-tyrosines via their SH2 domain, and are also phosphorylated by JAK on a tyrosine residue (e.g., Y705 in STAT3). Upon activation, they either hetero- or homodimerize and translocate to the nucleus via importins (Cimica et al., 2011), where they bind to consensus sequences (SIE/GAS/ISRE sequences) to induce gene transcription. Other mechanisms of STAT activation include phosphorylation by non-receptor tyrosine kinases such as Src, direct, or indirect phosphorylation by receptors with intrinsic tyrosine kinase activity (i.e., EGF, PDGF, and FGF receptors), and activation by G-protein coupled receptors (Ram and Iyengar, 2001). STAT3 can be deactivated by dephosphorylation or targeted degradation, and recycled back to the cytosol (reviewed by Lim and Cao, 2006; Dziennis and Alkayed, 2008; Moore and Goldberg, 2011). Further, Suppressor of Cytokine Signaling 3 (SOCS3) or protein inhibitors of activated STATS (PIAS) can also inhibit STAT3 activation (Krebs and Hilton, 2001).
In the adult brain, STATs are normally quiescent, but are activated following injury in response to multiple signaling pathways induced by several cytokines and growth factors including IL-6, LIF, CNTF, G-CSF, G-CSFR, EPO, EGF, IGF-1, NGF withdrawal, BDNF, free radicals, excitatory neurotransmitters and other inflammatory mediators that are released after injury (reviewed in Dziennis and Alkayed, 2008). Propagation of many of these signals (IL-6, LIF, and CNTF) requires a combination of various ligand binding units and a common signal-transducing unit gp130 (Taga and Kishimoto, 1995), through which they activate the JAK/STAT, Ras/MAPK, extracellular regulated-signal kinase (ERK), PI3K, and other pathways (Akira, 2000; Heinrich et al., 2003). Activation of the gp130-receptor has been implicated in neuronal response to injury and regenerative axonal growth (Habecker et al., 2009; Hyatt Sachs et al., 2010).
Signal transducer and activator of transcription-3 was identified as an acute phase response factor (Akira et al., 1994) and is expressed cytoplasmically in both neuronal and glial cells during development and in the adult (De-Fraja et al., 1998; Gautron et al., 2006). Nervous system insult leads to STAT3 activation in specific subsets of neurons with the pattern of activation dependent on the nature of the insult (Schwaiger et al., 2000; Xia et al., 2002). Global STAT3 knockout animals are embryonic lethal, dying prior to gastrulation between E6.5 and 7.5 (Takeda et al., 1997). Cell-type specific STAT3 deletion has greatly improved our knowledge of its function in different tissues (Sauer, 1998; Akira, 2000). STAT3 deletion in T-cells is associated with impaired T-cell proliferation, in macrophages with high susceptibility to endotoxin shock, in keratinocytes with compromised wound healing process, and in mammary glands with delay of involution (Akira, 2000). Nestin-promoter mediated neural-specific disruption of STAT3 resulted in mice which were hyperphagic, obese, diabetic, and infertile (Gao et al., 2004).
In the last decade, there has been growing evidence of a role of STAT3 in axonal regeneration. In vitro, STAT3 induces neurite growth in motor and sensory neurons and this growth can be inhibited by the application of SOCS3 (Liu and Snider, 2001; Miao et al., 2006). STAT3 induces GAP-43, a protein found in growth cones, and GAP-43 expression is inhibited with the JAK inhibitor AG-490, suggesting a role for the JAK/STAT3 pathway in GAP-43 expression and neurite outgrowth and regeneration (Wu and Bradshaw, 1996).
Haas et al. (1999) observed STAT3 activation at 24 h and 5 days after rat facial nerve axotomy. Following rat facial and hypoglossal nerve axotomy, a transient but significant increase in STAT3 mRNA, along with its phosphorylation and nuclear translocation within 3 h in neurons and 1 day in astrocytes was observed by Schwaiger et al. (2000). In contrast, these changes were not observed in non-regenerating neurons of Clarke’s nucleus, although both forms of injury upregulated c-Jun and GAP-43 expression (Schwaiger et al., 2000). Transient increases in STAT3 signaling were also found in sprouting neurites and astrocytes following entorhinal cortex lesions in addition to a rise in gp130-receptor cytokines, suggesting a dual role of STAT3 in both astrocytic regulation and axonal sprouting (Xia et al., 2002). Lee et al. (2004) showed that axonal STAT3, activated at the injury site, acts as both a retrograde injury signal and as a transcription factor which promotes the survival and regeneration of both sensory and motor neurons. Levels of activated STAT3 increased in the nuclei of regenerating adult cranial motor neurons, sciatic motor neurons and in sensory neuron nuclei after injury. Sciatic nerve lesion led to a very rapid activation of STAT3 in axons at the lesion site within 15 min, and by 24 h STAT3 labeling was detected in the DRG and in the spinal cord (Lee et al., 2004). These levels of STAT3 decreased when regeneration was completed, emphasizing its role in the regenerative process.
Deletion of CNTF, which is abundantly present in myelinating Schwann cells but not in or around the cell bodies of axotomized motoneurons (Dobrea et al., 1992; Rende et al., 1992) caused a delay in the appearance of phosphorylated STAT3 and its nuclear translocation in neuronal cell bodies (Kirsch et al., 2003). This delay points to a signaling cascade beginning with local release of CNTF by damaged myelinating Schwann cells, its local action on adjacent axons, intra-axonal phosphorylation of STAT3 and its retrograde transport to the cell bodies of injured neurons and finally to the nucleus. Recent studies using repetitive in vivo imaging of individual fluorescently labeled axons have shown that STAT3 selectively regulates initiation but not later perpetuation of axonal growth, thus acting as a phase-specific regulator of axonal outgrowth in both PNS and CNS (Bareyre et al., 2011). Altogether, these studies show a general trend of early and persistent expression of phosphorylated STAT3 during the regenerative process.
A number of in vitro and in vivo studies have also implicated the role of STAT3 as an intracellular survival-promoting factor. In vitro studies using cell lines have shown that STAT3 expression directly induces bcl-2 and bcl-xL gene expression, which are critical in promoting neuronal survival after injury (Dziennis and Alkayed, 2008). Mice lacking STAT3 in facial and spinal motoneurons (controlled by cre expression under NF-L promoter) showed a significant reduction in motoneuron survival after facial nerve lesion, which closely correlated with the extent of motoneuron loss seen in LIF/CNTF KO mice after nerve lesion (Sendtner et al., 1996; Schweizer et al., 2002). Moreover, upregulation of Reg-2 and Bcl-xl expression was reduced in axotomized motoneurons in STAT3 KO mice, suggesting a role of STAT3 in anti-apoptotic signaling. Interestingly, in the study by Schweizer et al. (2002) local application of CNTF strongly reduced the apparent motoneuron cell death in the absence of neuronal STAT3, but which other signaling pathways were involved is unclear.
Preliminary data from our lab suggests an important role for neuronal STAT3 in axonal regeneration (Patodia et al., 2011). Conditional mice mutants, lacking STAT3 in their neurons, had profound defects in the usual retrograde response, with a severe early and persistent reduction by 75–90% in microglial activation and recruitment of lymphocytes, and 4 days after injury in speed of axonal elongation in the distal stump. At 30 days, axotomized STAT3-deficient motoneurons appeared shrunk by 50–60% in size but displayed no cell death in contrast to results published earlier (Schweizer et al., 2002). Functional recovery was dramatically reduced and the mutants showed 80% less target muscle re-innervation. As with neuronal c-Jun deletion which interferes with regeneration (Raivich et al., 2004), expression of RAGs like CD44, β1 integrin, CGRP, galanin as well as the nuclear translocation of ATF3 was also greatly diminished or abolished in the STAT3 mutants. However, c-Jun expression itself was not affected, suggesting that deletion of either transcription factor – c-Jun or STAT3 – will produce a very similar regeneration-deficient phenotype.
CCAAT/Enhancer Binding Protein β, δ
The C/EBP family of transcription factors is comprised of six proteins (C/EBPα, β, γ, δ, ε, and ζ), each having unique properties regulating cellular proliferation and differentiation, particularly in hepatocytes, adipocytes, and hematopoietic cells (Ramji and Foka, 2002). C/EBPs also play pivotal, non-redundant roles in the development and plasticity of the nervous system, including cell fate determination, apoptosis, synthesis and response to trophic factors and responses to brain injury and ischemia (Kfoury and Kapatos, 2009). All members have a DNA binding domain and a highly homologous bZIP domain which is required for dimerization. The C/EBPs can interact with other bZIP and non-bZIP transcription factors (Huang et al., 2007).
CCAAT/enhancer binding protein-δ (also known as CELF, CRP3, and NF/IL-6) expression is typically low to undetectable in most cell-types and tissues, but it is rapidly induced by a variety of extracellular stimuli (e.g., growth hormone, insulin, IFNγ, IL-1, IL-6, LPS, TNFα, noradrenaline, and glutamate), phosphorylated, and translocated to the nucleus as an active transcription factor where is induces further IL-6 expression (Ramji and Foka, 2002). The most favorable complex responsible for the transcription of the IL-6 gene requires the interaction between p65 and C/EBPδ, with c-Jun occurring on the NF-κB site (Faggioli et al., 2004). C/EBPδ is elevated in hypoxia, glaucoma, carcinoma, traumatic brain injury and in the brains of Alzheimer’s disease patients (Huang et al., 2007; Yang et al., 2007). A recent study showed that the anti-inflammatory and neuroprotective effects of chrysin on reactive microglial cell induced neurotoxicity was partly mediated by inhibition of C/EBPδ expression at both protein and mRNA levels. C/EBPδ-deficient microglial cultures produce less NO and TNFα in response to LPS/IFNγ, re-affirming its role in microglial activation and neuroinflammation (Gresa-Arribas et al., 2010). C/EBPβδ double knockout in mouse embryonic fibroblasts showed particularly impaired production of IL-6 and TNFα in response to LPS or IL-1, suggesting complementary roles of C/EBPβ and C/EBPδ in induction of proinflammatory cytokines (Ramji and Foka, 2002).
CCAAT/enhancer binding protein β is upregulated and phosphorylated across different classes of injured neurons, including the invertebrate Aplysia model. In this model, phosphorylation of Aplysia C/EBPβ is mediated by the RISK1, a mitogen associated protein kinase homolog related to the ERK family (Sung et al., 2001). In the mouse facial nerve model, C/EBPβ is important for the expression of regeneration-associated microtubule Tα1 α tubulin and growth cone protein GAP-43, suggesting that C/EBPβ is a critical component of the regenerative response (Nadeau et al., 2005).
In the case of C/EPBδ, which is upregulated in sensory and sympathetic neurons (Boeshore et al., 2004; Magoulas and Lopez-de Heredia, 2010), initial studies using cultured primary sensory neurons from C/EPBδ null mice show impaired axonal growth and a reduced response to the conditioning lesion (Magoulas and Lopez-de Heredia, 2010). Preliminary results from our studies also confirm this finding in the motor, facial nerve injury model as well as indicate a role for C/EBPδ in early and late microglial activation (Patodia et al., unpublished).
In summary, a number of intriguing findings implicate a role of C/EBPs in injury response in neurons, i.e., they are mediators of neuro-inflammatory responses in the brain; are induced in injured neurons of invertebrates (Korneev et al., 1997; Sung et al., 2001) as well as vertebrates (Nadeau et al., 2005); have RAGs like Tα1 α tubulin and GAP-43 as direct transcriptional targets (Nadeau et al., 2005); and are upregulated by IL-6, an important post-injury molecule (Ramji et al., 1993). A number of alternative signaling pathways might also converge onto C/EBP family members following axonal injury, e.g., the JAK–STAT pathway has been shown to directly regulate C/EBPβ (Jiang and Zarnegar, 1997).
Oct-6
Transcription factors Oct-6 (SCIP/Tst-1), Krox-20 (Egr-2), and Sox 10 are major regulators of Schwann cell differentiation and myelination (Topilko et al., 1994; Bermingham et al., 1996; Jaegle and Meijer, 1998). A fragment distal to the Oct-6 gene, containing two DNase I-hypersensitive sites, acts as the Schwann cell-specific enhancer (SCE) and is responsible for the complex signaling between Schwann cells and axons driving myelination, during both normal peripheral nerve development and regeneration (Mandemakers et al., 2000). Oct-6 is present in the cytoplasm of Schwann cells associated with myelinated fibers of intact nerves. Axonal injury induces nuclear Oct-6 expression during the acute degenerative stage, reaching peak expression levels in the subsequent regenerative stage (Scherer et al., 1994; Kawasaki et al., 2003), suggesting that at the time of regeneration, Oct-6 is translocated to the nucleus to promote transcription of genes, possibly those encoding myelin proteins. Oct-6 expression is down-regulated in fully regenerated nerves and in nerves showing chronic axonal loss (Kawasaki et al., 2003). Therefore, Oct-6 may be a marker for dedifferentiation of adult Schwann cells and active nerve regeneration.
Sox11
SRY-box containing gene 11 (Sox11), a member of the Sox transcription factor family, is highly expressed in developing sensory neurons and at low levels in adult neurons (Jankowski et al., 2006; Moore and Goldberg, 2011). There has been limited study of Sox11 targets in axonal growth, but Sox11 can partner with Brn1 or -2 to regulate transcription (Kuhlbrodt et al., 1998; Tanaka et al., 2004) and drive expression of neurite growth-associated genes like β-III tubulin, MAP2 (Bergsland et al., 2006) and actin-related protein complex 3 (Arpc3; Jankowski et al., 2006). Sox11 expression in sensory and sympathetic ganglia is highly sensitive to nerve cut and crush injuries, rising steadily after injury and during subsequent regeneration, and returning to baseline levels at the end of regeneration (Boeshore et al., 2004; Jankowski et al., 2006, 2009). In Neuro2a cells, Sox11 levels dramatically increased with the number of cells extending neurites. In vitro experiments using cultured adult DRG neurons treated with Sox11 siRNAs exhibit a significant decrease in regeneration following axotomy as indicated by reduced neurite length and branching index (Jankowski et al., 2006). This suggests a correlation between Sox11 and neurite growth.
Injection of Sox11 siRNAs into mouse saphenous nerve caused a transient knockdown of Sox11 mRNA, that transiently inhibited in vivo regeneration after nerve crush. Electron microscopy analysis of Sox11 RNAi-injected nerves showed that regeneration of both myelinated and unmyelinated axons was inhibited. Nearly all neurons in the ganglia of crushed nerves that were Sox11 immunopositive showed co-labeling for ATF3 (Jankowski et al., 2009) and treatment with Sox11 siRNAs in vitro and in vivo caused a transcriptional and translational level reduction in ATF3 expression. Interestingly, ATF3 ablation using ATF3 siRNAs did not affect Sox11 levels, suggesting that Sox11 may act upstream of ATF3 and influence regulation of its gene expression (Jankowski et al., 2009). These anatomical and expression data support an intrinsic role for Sox11 in axonal regeneration that may involve interaction with ATF3.
p53
p53 is a member of a family of tumor suppressors together with p63 and p73. Like c-Jun, p53 is known to mediate both pro- and anti-apoptotic roles in the nervous system (Culmsee and Mattson, 2005; Jacobs et al., 2006). p53 can undergo multiple types of post-translational modifications including acetylation, phosphorylation, and ubiquitination, which can affect its localization and function (Lavin and Gueven, 2006; Tedeschi et al., 2009). In vitro and in vivo experiments have shown that p53 plays a role in promoting neurite growth. Overexpression of a dominant negative form of p53 in primary cortical neurons leads to growth cone collapse and decrease in neurite outgrowth, possibly due to a combination of reduced expression of its growth cone associated target genes Coronin1b, Rab13, and GAP-43 (Di Giovanni et al., 2006; Tedeschi et al., 2009), as well as its local, non-transcriptional activity at the growth cone (Qin et al., 2009). On the other hand, overexpression of wild-type p53 leads to an increase in growth cone size (Qin et al., 2009). Other axonal guidance molecules and their receptors like netrins, semaphorins, and ephrins are also regulated by p53 (Arakawa, 2005). In vivo experiments using p53 knockout mice showed a significant decrease in the number of fibers reinnervating the target muscles at 28 days after facial axotomy in the mutant mice when compared with control animals (Di Giovanni et al., 2006). Taken together, these studies suggest a role for p53 in modulating neurite growth and regeneration. Given its well described role in apoptosis of neurons and non-neuronal cells, it will be interesting to investigate its effects on neuronal survival after peripheral nerve injury.
Nuclear Factor Kappa-Light-Chain-Enhancer of Activated B Cells
Nuclear factor kappa-light-chain-enhancer of activated B cells is an inducible transcription factor dimer made up of 5 subunits, bound as homo- or hetero-dimers: RelA/p65, c-Rel, RelB, p50, and p52. Inhibitor of κB (Iκβ) family of proteins can bind to NF-κB dimer and prevent its nuclear translocation. Dissociation or proteasomal degradation of IκB reveals NF-κB’s nuclear localization sequence (NLS), allowing it to move to the nucleus and modulate gene transcription by binding to promoter and enhancer sequences (reviewed by Moore and Goldberg, 2011).
Nuclear factor kappa-light-chain-enhancer of activated B cells is significantly upregulated in DRGs and spinal cord after peripheral nerve crush (Ma and Bisby, 1998; Pollock et al., 2005). It can respond to a stimulus in neurites and retrogradely travel to the nucleus to affect gene transcription. NF-κB signaling can either enhance of inhibit neurite growth depending on the phosphorylation status of its RelA/p65 subunit. Phosphorylation of p65 by IKKβ results in reduced neurite growth, and in neurons without this IKKβ activity, there is increased neurite growth (Gutierrez et al., 2008). Identified neuronal targets of NF-κB include genes for Bcl-2, MnSOD, glutamate receptor subunits, BDNF, and calcium regulating proteins (reviewed by Mattson, 2005). In addition, NF-κB has been shown to regulate expression of cell-adhesion molecules like NCAM, tenascin C and β1 integrin in other systems (reviewed by Moore and Goldberg, 2011). NF-κB is also the downstream modulator of neurite growth for fas apoptosis inhibitory molecule (FAIM) signaling. Blocking NF-κB activation prevents the increase of neurite growth seen with FAIM overexpression (Sole et al., 2004).
ELK
The ETS-like (ELK) subfamily of transcription factors comprises of ELK1, ELK3, and ELK4 factors which can have different potential protein–protein interactions, and respond differently to MAP kinase signaling pathways (Ducret et al., 2000; Buchwalter et al., 2004). Seven days after peripheral axotomy, a 2.3-fold upregulation of ELK3 mRNA, but not of ELK1 or ELK4 mRNA was detected in adult mouse DRGs (Kerr et al., 2010). The expression of neuropeptide galanin mRNA is upregulated around 80-fold in DRGs following axotomy, and an 18 bp sequence within its promoter/enhancer region containing overlapping putative ETS, STAT, and Smad binding sites, has been shown to be critical for this response (Bacon et al., 2007), making ELK3 a potentially important player in peripheral nerve regeneration. It would be interesting to see if the injury-induced ELK3 isoforms can differently affect neuronal survival and/or regeneration after axonal injury.
Cross-Talk between Transcription Factors
Transcription factors form an intricate network in which cross-talk between different pathways leads to an integrated and input-balanced signaling outcome (Morrison and Davis, 2003; Moore and Goldberg, 2011). Some axonal regeneration still occurs in the absence of master regulators of regeneration like c-Jun, highlighting the existence of alternative pathways, and of compensatory and complementary/synergistic mechanisms, which may be shared during axonal outgrowth.
Jun N-terminal kinase signaling is interconnected with various other cellular signaling pathways, such as NFkB and the JAK/STAT system (Waetzig et al., 2006; Haeusgen et al., 2009), resulting in STAT3 phosphorylation (Levy and Lee, 2002). Besides JNK, c-Jun can also be activated by ERK1/2 (Leppa et al., 1998; Schwarz et al., 2002; Morton et al., 2003). On the other hand, STAT3 can stimulate transcription of several AP-1 members including JunB (Coffer et al., 1995) and c-fos (Yang et al., 2003; Higashi et al., 2004) which have STAT3 responsive elements in their regulatory sequence.
The interactions between AP-1 and STAT3 are multiple and complex, resulting in mutual modulation of their transcriptional activities. STAT3 and AP-1 factors can bind to independent but closely spaced DNA binding sites in the regulatory sequences of a number of genes, such as α-2 macroglobulin (Schaefer et al., 1995), VIP (Symes et al., 1997), bcl-6 (Arguni et al., 2006), and matrix metallo-proteinases and synergistically induce maximal enhancer function (Zhang et al., 1999). AP-1 complexes can also bind directly to STAT3, enhancing STAT3’s transcriptional activity (Schaefer et al., 1995; Leu et al., 2001). Mapping of the STAT3–c-Jun interactive regions by GST pull-down assays suggested that the STAT3-interactive region lies within its coiled-coiled domain, and in a portion of the DNA binding domain. The c-Jun interactive region was within its C-terminal residues 105–334. Point mutations within these regions blocked their interaction (Zhang et al., 1999). SOCS3 was discovered for its ability to inhibit tyrosine phosphorylation of STAT3 through binding to gp130 and JAKs (Krebs and Hilton, 2001). It has subsequently been shown that endogenous SOCS3 can also inhibit AP-1 activity by blocking JNK phosphorylation (Miao et al., 2008). Therefore SOCS3 could be an important molecule regulating axonal regeneration by controlling changes in JNK and JAK/STAT signaling pathways after injury.
Additional transcription factors encompassing a compensatory/complementary role include the C/EBP family members C/EBPβ and C/EBPδ. The promoter region of a c-Jun coactivator, an oncogene Jab1, contains binding sequences for C/EBP, GATA, as well as a STAT3 consensus sequence overlapping the C/EBP site. Both, mutation of the C/EBP binding site and inhibition of STAT3, significantly reduced Jab1-promoter activity (Shackleford et al., 2011). C/EBPβ is essential for appropriate induction and maintenance of GAP-43 mRNA following axonal injury (Nadeau et al., 2005). While there is no evidence of a direct binding site for C/EBP on the GAP-43 gene, it contains a functionally important AP-1 site for c-Jun and c-fos (Weber and Skene, 1998). C/EBPβ can bind to and regulate both c-fos and c-Jun (Nadeau et al., 2005), suggesting a coordinated induction of GAP-43 by C/EBPβ, c-fos, and c-Jun in injured neurons. It has already been shown that co-expression of c-Fos with c-Jun induces a greater number of cells to produce neurites than c-Jun alone (Leppa et al., 1998).
We have already discussed a number of different studies investigating the complex association between ATF3 and c-Jun expression after injury. Besides interacting with leucine zipper transcription factors, ATF3 can also interact with other transcription factors upregulated after peripheral injury, such as, STAT3. This could be via co-activators or by synergistic binding to adjacent DNA binding sites. For example, SPRR1A, a target gene for ATF3, is a gp130 pathway protein and its promoter region contains functional AP-1 binding sites (Sark et al., 1998; Pradervand et al., 2004). The levels of SPRR1A expression in DRGs of injured wild-type mice are higher than in DRGs of non-injured transgenic mice expressing constitutively active ATF3, suggesting that other downstream signals in addition to ATF3 are likely to govern SPRR1A expression. C/EBPβ, for example, is induced after peripheral nerve injury (Nadeau et al., 2005) and a functional C/EBPβ binding site is located on the SPRR1A promoter (Pradervand et al., 2004). Recently, transcription of DINE (damage-induced neuronal endopeptidase), an enzyme involved in terminal nerve branching (Nagata et al., 2010), was shown to be increased 60-fold by Sp1 mediated recruitment of STAT3, c-Jun, and ATF3 to the DINE promoter in response to LIF upregulation and NGF withdrawal following injury (Kiryu-Seo et al., 2008).
Nuclear factor kappa-light-chain-enhancer of activated B cell can also interact with Jun, ATF, CREB, and Fos transcription factors. STAT3 can induce the alternative NF-κB pathway, and bind to DNA in a complex with p52, to induce transcription. p53 can also interact with p52 to regulate expression of its target genes (reviewed by Perkins, 2007).
Mathematical models of interacting signaling pathways have been developed to predict regenerative outcomes after injury (Moya et al., 2011). Transgenic animal models with deletion of multiple signaling streams will be very useful in identification of functional complementary counterparts.
Downstream Targets of Transcription Factors
Transcription factors rapidly condition the injured nerve and within 1–4 days after injury, the neuronal perikaryon produces a plethora of RNA, protein, and glycolipid components which play a vital role in executing axonal regeneration. Downstream targets of the transcription factors include a vast number of RAGs that may be involved in cell–cell signaling, axonal growth and sprouting, and activation of the non-neuronal, cellular milieu. Successful axonal regeneration is accompanied by the appearance of functionally diverse families of molecules that regulate surface cytoskeletal interaction. Examples of regeneration-associated cytoskeletal families include the GMC family of “integral” membrane proteins – GAP-43, MARCKS, and CAP23 (Skene and Willard, 1981; Bomze et al., 2001), microtubule disassembly molecules (e.g., SCG10, stathmin, CRMP2, and RB3; Iwata et al., 2002; Mori and Morii, 2002), and the Rho GTPase family (RhoA, Rac, Cdc42, and TC10) members which act as molecular switches regulating cytoskeletal structure, dynamics, and cell-adhesion (Etienne-Manneville and Hall, 2002). Regenerating neurons also upregulate adhesion molecules like integrins α7 and β1 (Kloss et al., 1999), CD44 (Jones et al., 2000), galectin-1 (Horie and Kadoya, 2000), β2-microglobulin (Oliveira et al., 2004), ninjurin (Araki and Milbrandt, 2000), and gicerin/CD146 (Hiroi et al., 2003). Neurotrophins like BDNF and GDNF are induced in the distal stump of the injured axons, and their exogenous application promotes axonal regeneration of chronically injured neurons (Boyd and Gordon, 2003a,b). In vitro studies have shown that different neuropeptides like substance P, somatostatin, vasopressin, calcitonin, and alpha-MSH exert a moderate but significant neurite growth promoting effect on CNS and PNS neurons (Narumi and Fujita, 1978; Brinton and Gruener, 1987; Raivich, 2011). Transgenic and pharmacological studies have confirmed a similar role in vivo for peptides like galanin (Holmes et al., 2000; Wynick et al., 2001), CGRP (Toth et al., 2009), and PACAP (Pituitary Adenylate Cyclase Activating Peptide; Kimura et al., 2003; Suarez et al., 2006; Armstrong et al., 2008).
Concluding Remarks
Injury to peripheral nerves can occur due to several causes ranging from physical injury and accidental trauma to pathological conditions and degenerative diseases. Achieving successful regeneration and synchronized functional recovery following neural damage is one of the biggest challenges faced by neuroscientists and clinicians. Transcription factors are master switches controlling the expression of multiple gene programs, and orchestrating complex axon growth and regenerative responses. Blocking transcription using RNA polymerase II inhibitors at an early stage after injury inhibits the capacity of dissociated neurons to elongate neurites (Smith and Skene, 1997), highlighting their crucial role in efficient regeneration after nerve injury. In this review, we have attempted to summarize the roles of a number of different transcription factors in peripheral nerve regeneration and their complex web of interactions. An overview of the observed phenotypes following global and cell-type specific deletions is given in Table 1. Understanding the molecular mechanisms underlying nerve regeneration via various regeneration-associated molecules and their signaling components will not only help to enhance the speed and specificity of peripheral regeneration but improve the success of central axonal regeneration, as well.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Akira, S., Nishio, Y., Inoue, M., Wang, X. J., Wei, S., Matsusaka, T., Yoshida, K., Sudo, T., Naruto, M., and Kishimoto, T. (1994). Molecular cloning of Aprf, a novel Ifn-stimulated gene factor-3 p91-related transcription factor involved in the gp130-mediated signaling pathway. Cell 77, 63–71.
Araki, T., and Milbrandt, J. (2000). Ninjurin2, a novel homophilic adhesion molecule, is expressed in mature sensory and enteric neurons and promotes neurite outgrowth. J. Neurosci. 20, 187–195.
Arguni, E., Arima, M., Tsuruoka, N., Sakamoto, A., Hatano, M., and Tokuhisa, T. (2006). JunD/AP-1 and STAT3 are the major enhancer molecules for high Bcl6 expression in germinal center B cells. Int. Immunol. 18, 1079–1089.
Armstrong, B. D., Abad, C., Chhith, S., Cheung-Lau, G., Hajji, O. E., Nobuta, H., and Waschek, J. A. (2008). Impaired nerve regeneration and enhanced neuroinflammatory response in mice lacking pituitary adenylyl cyclase activating peptide. Neuroscience 151, 63–73.
Arthur-Farraj, P., Baskaran, A., Parkinson, D., Turmaine, M., Feltri, M., Wrabetz, L., Behrens, A., Mirsky, R., and Jessen, K. (2007). Schwann cells without c-Jun show retarded demyelination and dedifferentiation after peripheral nerve injury. Abstr. Soc. Neurosci. 480.
Bacon, A., Kerr, N. C. H., Holmes, F. E., Gaston, K., and Wynick, D. (2007). Characterization of an enhancer region of the galanin gene that directs expression to the dorsal root ganglion and confers responsiveness to axotomy. J. Neurosci. 27, 6573–6580.
Bareyre, F. M., Garzorz, N., Lang, C., Misgeld, T., Buning, H., and Kerschensteiner, M. (2011). In vivo imaging reveals a phase-specific role of STAT3 during central and peripheral nervous system axon regeneration. Proc. Natl. Acad. Sci. U.S.A. 108, 6282–6287.
Barnat, M., Enslen, H., Propst, F., Davis, R. J., Soares, S., and Nothias, F. (2010). Distinct roles of c-Jun N-terminal kinase isoforms in neurite initiation and elongation during axonal regeneration. J. Neurosci. 30, 7804–7816.
Behrens, A., Sibilia, M., and Wagner, E. F. (1999). Amino-terminal phosphorylation of c-Jun regulates stress-induced apoptosis and cellular proliferation. Nat. Genet. 21, 326–329.
Berdan, R. C., Easaw, J. C., and Wang, R. (1993). Alterations in membrane potential after axotomy at different distances from the soma of an identified neuron and the effect of depolarization on neurite outgrowth and calcium channel expression. J. Neurophysiol. 69, 151–164.
Bergsland, M., Werme, M., Malewicz, M., Perlmann, T., and Muhr, J. (2006). The establishment of neuronal properties is controlled by Sox4 and Sox11. Genes Dev. 20, 3475–3486.
Bermingham, J. R., Scherer, S. S., O’Connell, S., Arroyo, E., Kalla, K. A., Powell, F. L., and Rosenfeld, M. G. (1996). Tst-1/Oct-6/SCIP regulates a unique step in peripheral myelination and is required for normal respiration. Genes Dev. 10, 1751–1762.
Besirli, C. G., Wagner, E. F., and Johnson, E. M. (2005). The limited role of NH2-terminal c-Jun phosphorylation in neuronal apoptosis: identification of the nuclear pore complex as a potential target of the JNK pathway. J. Cell Biol. 170, 401–411.
Björkblom, B., Ostman, N., Hongisto, V., Komarovski, V., Filén, J. J., Nyman, T. A., Kallunki, T., Courtney, M. J., and Coffey, E. T. (2005). Constitutively active cytoplasmic c-Jun N-terminal kinase 1 is a dominant regulator of dendritic architecture: role of microtubule-associated protein 2 as an effector. J. Neurosci. 25, 6350–6361.
Boeshore, K. L., Schreiber, R. C., Vaccariello, S. A., Sachs, H. H., Salazar, R., Lee, J., Ratan, R. R., Leahy, P., and Zigmond, R. E. (2004). Novel changes in gene expression following axotomy of a sympathetic ganglion: a microarray analysis. J. Neurobiol. 59, 216–235.
Bogoyevitch, M. A., and Kobe, B. (2006). Uses for JNK: the many and varied substrates of the c-Jun N-terminal kinases. Microbiol. Mol. Biol. Rev. 70, 1061–1095.
Bohatschek, M., Kloss, C. U., Hristova, M., Pfeffer, K., and Raivich, G. (2004). Microglial major histocompatibility complex glycoprotein-1 in the axotomized facial motor nucleus: regulation and role of tumor necrosis factor receptors 1 and 2. J. Comp. Neurol. 470, 382–399.
Bomze, H. M., Bulsara, K. R., Iskandar, B. J., Caroni, P., and Skene, J. H. P. (2001). Spinal axon regeneration evoked by replacing two growth cone proteins in adult neurons. Nat. Neurosci. 4, 38–43.
Bonilla, I. E., Tanabe, K., and Strittmatter, S. M. (2002). Small proline-rich repeat protein 1A is expressed by axotomized neurons and promotes axonal outgrowth. J. Neurosci. 22, 1303–1315.
Bosse, F., Hasenpusch-Theil, K., Kury, P., and Muller, H. W. (2006). Gene expression profiling reveals that peripheral nerve regeneration is a consequence of both novel injury-dependent and reactivated developmental processes. J. Neurochem. 96, 1441–1457.
Bosse, F., Kury, P., and Muller, H. W. (2001). Gene expression profiling and molecular aspects in peripheral nerve regeneration. Restor. Neurol. Neurosci. 19, 5–18.
Boyd, J. G., and Gordon, T. (2003a). Glial cell line-derived neurotrophic factor and brain-derived neurotrophic factor sustain the axonal regeneration of chronically axotomized motoneurons in vivo. Exp. Neurol. 183, 610–619.
Boyd, J. G., and Gordon, T. (2003b). Neurotrophic factors and their receptors in axonal regeneration and functional recovery after peripheral nerve injury. Mol. Neurobiol. 27, 277–323.
Brecht, S., Kirchhof, R., Chromik, A., Willesen, M., Nicolaus, T., Raivich, G., Wessig, J., Waetzig, V., Goetz, M., Claussen, M., Pearse, D., Kuan, C. Y., Vaudano, E., Behrens, A., Wagner, E., Flavell, R. A., Davis, R. J., and Herdegen, T. (2005). Specific pathophysiological functions of JNK isoforms in the brain. Eur. J. Neurosci. 21, 363–377.
Brinton, R. E., and Gruener, R. (1987). Vasopressin promotes neurite growth in cultured embryonic neurons. Synapse 1, 329–334.
Bromberg, J., and Darnell, J. E. (2000). The role of STATs in transcriptional control and their impact on cellular function. Oncogene 19, 2468–2473.
Buchwalter, G., Gross, C., and Wasylyk, B. (2004). Ets ternary complex transcription factors. Gene 324, 1–14.
Cai, D., Deng, K., Mellado, W., Lee, J., Ratan, R. R., and Filbin, M. T. (2002). Arginase I and polyamines act downstream from cyclic AMP in overcoming inhibition of axonal growth MAG and myelin in vitro. Neuron 35, 711–719.
Cimica, V., Chen, H. C., Iyer, J. K., and Reich, N. C. (2011). Dynamics of the STAT3 transcription factor: nuclear import dependent on Ran and importin-β1. PLoS ONE 6, e20188. doi: 10.1371/journal.pone.0020188
Coffer, P., Lutticken, C., Vanpuijenbroek, A., Klopdejonge, M., Horn, F., and Kruijer, W. (1995). Transcriptional regulation of the Jun b promoter – analysis of STAT-mediated signal-transduction. Oncogene 10, 985–994.
Costigan, M., Befort, K., Karchewski, L., Griffin, R. S., D’Urso, D., Allchorne, A., Sitarski, J., Mannion, J. W., Pratt, R. E., and Woolf, C. J. (2002). Replicate high-density rat genome oligonucleotide microarrays reveal hundreds of regulated genes in the dorsal root ganglion after peripheral nerve injury. BMC Neurosci. 3, 16. doi: 10.1186/1471-2202-3-16
Cox, L. J., Hengst, U., Gurskaya, N. G., Lukyanov, K. A., and Jaffrey, S. R. (2008). Intra-axonal translation and retrograde trafficking of CREB promotes neuronal survival. Nat. Cell Biol. 10, 149–159.
Crocker, S. J., Hayley, S. P., Smith, P. D., Mount, M. P., Lamba, W. R., Callaghan, S. M., Slack, R. S., and Park, D. S. (2006). Regulation of axotomy-induced dopaminergic neuron death and c-Jun phosphorylation by targeted inhibition of cdc42 or mixed lineage kinase. J. Neurochem. 96, 489–499.
Culmsee, C., and Mattson, M. P. (2005). p53 in neuronal apoptosis. Biochem. Biophys. Res. Commun. 331, 761–777.
De-Fraja, C., Conti, L., Magrassi, L., Govoni, S., and Cattaneo, E. (1998). Members of the JAK/STAT proteins are expressed and regulated during development in the mammalian forebrain. J. Neurosci. Res. 54, 320–330.
Deng, J., Zhang, X. L., Wang, J. W., Teng, L. L., Ge, J., Takeiviori, H., Xiong, Z. Q., and Zhou, Y. (2009). Expression and regulated nuclear transport of transducers of regulated Creb 1 in retinal ganglion cells. Neuroscience 159, 1023–1031.
Di Giovanni, S., Knights, C. D., Rao, M., Yakovlev, A., Beers, J., Catania, J., Avantaggiati, M. L., and Faden, A. I. (2006). The tumor suppressor protein p53 is required for neurite outgrowth and axon regeneration. EMBO J. 25, 4084–4096.
Dobrea, G. M., Unnerstall, J. R., and Rao, M. S. (1992). The expression of CNTF message and immunoreactivity in the central and peripheral nervous system of the rat. Brain Res. Dev. Brain Res. 66, 209–219.
Doyle, C. A., and Hunt, S. P. (1997). Reduced nuclear factor kappa B (p65) expression in rat primary sensory neurons after peripheral nerve injury. Neuroreport 8, 2937–2942.
Dubový, P. (2011). Wallerian degeneration and peripheral nerve conditions for both axonal regeneration and neuropathic pain induction. Ann. Anat. 193, 267–275.
Ducret, C., Maira, S. M., Lutz, Y., and Wasylyk, B. (2000). The ternary complex factor Net contains two distinct elements that mediate different responses to MAP kinase signalling cascades. Oncogene 19, 5063–5072.
Dziennis, S., and Alkayed, N. J. (2008). Role of signal transducer and activator of transcription 3 in neuronal survival and regeneration. Rev. Neurosci. 19, 341–361.
Elde, R., Cao, Y. H., Cintra, A., Brelje, T. C., Pelto-Huikko, M., Junttila, T., Fuxe, K., Pettersson, R. F., and Hökfelt, T. (1991). Prominent expression of acidic fibroblast growth factor in motor and sensory neurons. Neuron 7, 349–364.
Faggioli, L., Costanzo, C., Donadelli, M., and Palmieri, M. (2004). Activation of the interleukin-6 promoter by a dominant negative mutant of c-Jun. Biochim. Biophys. Acta 1692, 17–24.
Gao, Q., Wolfgang, M. J., Neschen, S., Morino, K., Horvath, T. L., Shulman, G. I., and Fu, X. Y. (2004). Disruption of neural signal transducer and activator of transcription 3 causes obesity, diabetes, infertility, and thermal dysregulation. Proc. Natl. Acad. Sci. U.S.A. 101, 4661–4666.
Gautron, L., de Smedt-Peyrusse, V., and Laye, S. (2006). Characterization of STAT3-expressing cells in the postnatal rat brain. Brain Res. 1098, 26–32.
Gresa-Arribas, N., Serratosa, J., Saura, J., and Sola, C. (2010). Inhibition of CCAAT/enhancer binding protein delta expression by chrysin in microglial cells results in anti-inflammatory and neuroprotective effects. J. Neurochem. 115, 526–536.
Gutierrez, H., O’Keeffe, G. W., Gavalda, R., Gallagher, D., and Davies, A. M. (2008). Nuclear factor kappa B signaling either stimulates or inhibits neurite growth depending on the phosphorylation status of p65/RelA. J. Neurosci. 28, 8246–8256.
Haas, C. A., Hofmann, H. D., and Kirsch, M. (1999). Expression of CNTF/LIF-receptor components and activation of STAT3 signaling in axotomized facial motoneurons: evidence for a sequential postlesional function of the cytokines. J. Neurobiol. 41, 559–571.
Habecker, B. A., Sachs, H. H., Rohrer, H., and Zigmond, R. E. (2009). The dependence on gp130 cytokines of axotomy induced neuropeptide expression in adult sympathetic neurons. Dev. Neurobiol. 69, 392–400.
Haeusgen, W., Boehm, R., Zhao, Y., Herdegen, T., and Waetzig, V. (2009). Specific activities of individual c-Jun N-terminal kinases in the brain. Neuroscience 161, 951–959.
Hai, T., and Curran, T. (1991). Cross-family dimerization of transcription factors fos, Jun and atf creb alters DNA-binding specificity. Proc. Natl. Acad. Sci. U.S.A. 88, 3720–3724.
Hai, T., and Hartman, M. G. (2001). The molecular biology and nomenclature of the activating transcription factor/cAMP responsive element binding family of transcription factors: activating transcription factor proteins and homeostasis. Gene 273, 1–11.
Hannila, S. S., and Filbin, M. T. (2008). The role of cyclic AMP signaling in promoting axonal regeneration after spinal cord injury. Exp. Neurol. 209, 321–332.
Hanz, S., Perlson, E., Willis, D., Zheng, J. Q., Massarwa, R., Huerta, J. J., Koltzenburg, M., Kohler, M., van-Minnen, J., Twiss, J. L., and Fainzilber, M. (2003). Axoplasmic importins enable retrograde injury signaling in lesioned nerve. Neuron 40, 1095–1104.
Heinrich, P. C., Behrmann, I., Haan, S., Hermanns, H. M., Muller-Newen, G., and Schaper, F. (2003). Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 374, 1–20.
Herdegen, T., Blume, A., Buschmann, T., Georgakopoulos, E., Winter, C., Schmid, W., Hsieh, T. F., Zimmermann, M., and Gass, P. (1997a). Expression of activating transcription factor-2, serum response factor and cAMP/Ca response element binding protein in the adult rat brain following generalized seizures, nerve fibre lesion and ultraviolet irradiation. Neuroscience 81, 199–212.
Herdegen, T., Skene, P., and Bahr, M. (1997b). The c-Jun transcription factor – bipotential mediator of neuronal death, survival and regeneration. Trends Neurosci. 20, 227–231.
Herdegen, T., Claret, F. X., Kallunki, T., Martin-Villalba, A., Winter, C., Hunter, T., and Karin, M. (1998). Lasting N-terminal phosphorylation of c-Jun and activation of c-Jun N-terminal kinases after neuronal injury. J. Neurosci. 18, 5124–5135.
Herdegen, T., Kummer, W., Fiallos, C. E., Leah, J., and Bravo, R. (1991). Expression of C-Jun, Jun-B and Jun-D proteins in rat nervous-system following transection of vagus nerve and cervical sympathetic trunk. Neuroscience 45, 413–422.
Herdegen, T., and Waetzig, V. (2001). AP-1 proteins in the adult brain: facts and fiction about effectors of neuroprotection and neurodegeneration. Oncogene 20, 2424–2437.
Higashi, N., Kunimoto, H., Kaneko, S., Sasaki, T., Ishii, M., Kojima, H., and Nakajima, K. (2004). Cytoplasmic c-Fos induced by the YXXQ-derived STAT3 signal requires the co-operative MEK/ERK signal for its nuclear translocation. Genes Cells 9, 233–242.
Hiroi, S., Tsukamoto, Y., Sasaki, F., Miki, N., and Taira, E. (2003). Involvement of gicerin, a cell adhesion molecule, in development and regeneration of chick sciatic nerve. FEBS Lett. 554, 311–314.
Hol, E. M., Schwaiger, F. W., Werner, A., Schmitt, A., Raivich, G., and Kreutzberg, G. W. (1999). Regulation of the LIM-type homeobox gene islet-1 during neuronal regeneration. Neuroscience 88, 917–925.
Holmes, F. E., Mahoney, S., King, V. R., Bacon, A., Kerr, N. C., Pachnis, V., Curtis, R., Priestley, J. V., and Wynick, D. (2000). Targeted disruption of the galanin gene reduces the number of sensory neurons and their regenerative capacity. Proc. Natl. Acad. Sci. U.S.A. 97, 11563–11568.
Horie, H., and Kadoya, T. (2000). Identification of oxidized galectin-1 as an initial repair regulatory factor after axotomy in peripheral nerves. Neurosci. Res. 38, 131–137.
Huang, A. M., Rudelius, M., Sharan, S., McAllister, J. M., Raffeld, M., Christenson, L. K., and Sterneck, E. (2007). The Cebpd (C/EBPdelta) gene is induced by luteinizing hormones in ovarian theca and interstitial cells but is not essential for mouse ovary function. PLoS ONE 2, e1334. doi: 10.1371/journal.pone.0001334
Hunt, D., Hossain-Ibrahim, K., Mason, M. R., Coffin, R. S., Lieberman, A. R., Winterbottom, J., and Anderson, P. N. (2004). ATF3 upregulation in glia during Wallerian degeneration: differential expression in peripheral nerves and CNS white matter. BMC Neurosci. 5, 9. doi: 10.1186/1471-2202-5-9
Hyatt Sachs, H., Rohrer, H., and Zigmond, R. E. (2010). The conditioning lesion effect on sympathetic neurite outgrowth is dependent on gp130 cytokines. Exp. Neurol. 223, 516–522.
Hyatt Sachs, H., Schreiber, R. C., Shoemaker, S. E., Sabe, A., Reed, E., and Zigmond, R. E. (2007). Activating transcription factor 3 induction in sympathetic neurons after axotomy: response to decreased neurotrophin availability. Neuroscience 150, 887–897.
Iwata, T., Namikawa, K., Honma, M., Mori, N., Yachiku, S., and Kiyama, H. (2002). Increased expression of mRNAs for microtubule disassembly molecules during nerve regeneration. Brain Res. Mol. Brain Res. 102, 105–109.
Jacobs, W. B., Kaplan, D. R., and Miller, F. D. (2006). The p53 family in nervous system development and disease. J. Neurochem. 97, 1571–1584.
Jaegle, M., and Meijer, D. (1998). Role of Oct-6 in Schwann cell differentiation. Microsc. Res. Tech. 41, 372–378.
Jankowski, M. P., Cornuet, P. K., McILwrath, S., Koerber, H. R., and Albers, K. M. (2006). SRY-box containing gene 11 (Sox11) transcription factor is required for neuron survival and neurite growth. Neuroscience 143, 501–514.
Jankowski, M. P., McIlwrath, S. L., Jing, X. T., Cornuet, P. K., Salerno, K. M., Koerber, H. R., and Albers, K. M. (2009). Sox11 transcription factor modulates peripheral nerve regeneration in adult mice. Brain Res. 1256, 43–54.
Jiang, J. G., and Zarnegar, R. (1997). A novel transcriptional regulatory region within the core promoter of the hepatocyte growth factor gene is responsible for its inducibility by cytokines via the C/EBP family of transcription factors. Mol. Cell. Biol. 17, 5758–5770.
Jones, L. L., Liu, Z., Shen, J., Werner, A., Kreutzberg, G. W., and Raivich, G. (2000). Regulation of the cell adhesion molecule CD44 after nerve transection and direct trauma to the mouse brain. J. Comp. Neurol. 426, 468–492.
Kalla, R., Liu, Z., Xu, S., Koppius, A., Imai, Y., Kloss, C. U., Kohsaka, S., Gschwendtner, A., Möller, J. C., Werner, A., and Raivich, G. (2001). Microglia and the early phase of immune surveillance in the axotomized facial motor nucleus: impaired microglial activation and lymphocyte recruitment but no effect on neuronal survival or axonal regeneration in macrophage-colony stimulating factor-deficient mice. J. Comp. Neurol. 436, 182–201
Kawasaki, T., Oka, N., Tachibana, H., Akiguchi, I., and Shibasaki, H. (2003). Oct6, a transcription factor controlling myelination, is a marker for active nerve regeneration in peripheral neuropathies. Acta Neuropathol. 105, 203–208.
Kenney, A. M., and Kocsis, J. D. (1998). Peripheral axotomy induces long-term c-Jun amino-terminal kinase-1 activation and activator protein-1 binding activity by c-Jun and JunD in adult rat dorsal root ganglia in vivo. J. Neurosci. 18, 1318–1328.
Kerr, N., Pintzas, A., Holmes, F., Hobson, S. A., Pope, R., Wallace, M., Wasylyk, C., Wasylyk, B., and Wynick, D. (2010). The expression of ELK transcription factors in adult DRG: novel isoforms, antisense transcripts and upregulation by nerve damage. Mol. Cell. Neurosci. 44, 165–177.
Kfoury, N., and Kapatos, G. (2009). Identification of neuronal target genes for CCAAT/enhancer binding proteins. Mol. Cell. Neurosci. 40, 313–327.
Kimura, H., Kawatani, M., Ito, E., and Ishikawa, K. (2003). Effects of pituitary adenylate cyclase-activating polypeptide on facial nerve recovery in the guinea pig. Laryngoscope 113, 1000–1006.
Kirsch, M., Terheggen, U., and Hofmann, H. D. (2003). Ciliary neurotrophic factor is an early lesion-induced retrograde signal for axotomised facial motoneurons. Mol. Cell. Neurosci. 24, 130–138.
Kiryu-Seo, S., Kato, R., Ogawa, T., Nakagomi, S., Nagata, K., and Kiyama, H. (2008). Neuronal injury-inducible gene is synergistically regulated by ATF3, c-Jun, and STAT3 through the interaction with Sp1 in damaged neurons. J. Biol. Chem. 283, 6988–6996.
Kloss, C. U., Werner, A., Klein, M. A., Shen, J., Menuz, K., Probst, J. C., Kreutzberg, G. W., and Raivich, G. (1999). Integrin family of cell adhesion molecules in the injured brain: regulation and cellular localization in the normal and regenerating mouse facial motor nucleus. J. Comp. Neurol. 411, 162–178.
Korneev, S., Federov, A., Collins, R., Blackshaw, S., and Davies, J. (1997). A subtractive cDNA library from an identified regenerating neuron is enriched in sequences up-regulated during nerve regeneration. Invert. Neurosci. 3, 185–192.
Krebs, D. L., and Hilton, D. J. (2001). SOCS proteins: negative regulators of cytokine signalling. Stem Cells 19, 378–387.
Kubo, T., Yamashita, T., Yamaguchi, A., Hosokawa, K., and Tohyama, M. (2002). Analysis of genes induced in peripheral nerve after axotomy using cDNA microarrays. J. Neurochem. 82, 1129–1136.
Kuhlbrodt, K., Herbarth, B., Sock, E., Enderich, J., Hermans-Borgmeyer, I., and Wegner, M. (1998). Cooperative function of POU proteins and SOX proteins in glial cells. J. Biol. Chem. 273, 16050–16057.
Kury, P., Abankwa, D., Kruse, F., Greiner-Petter, R., and Muller, H. W. (2004). Gene expression profiling reveals multiple novel intrinsic and extrinsic factors associated with axonal regeneration failure. Eur. J. Neurosci. 19, 32–42.
Latouche, M., Wilton, D., Arthur-Farraj, P., DaCosta, C., Mitter, R., Turmaine, M., Raivich, G., Behrens, A., Mirsky, R., and Jessen, K. R. (2009). Schwann cell c-Jun regulates glial support for survival of PNS neurons after injury. Glia 57, S158.
Lavin, M. F., and Gueven, N. (2006). The complexity of p53 stabilization and activation. Cell Death Differ. 13, 941–950.
Lee, N., Neitzel, K. L., Devlin, B. K., and MacLennan, A. J. (2004). STAT3 phosphorylation in injured axons before sensory and motor neuron nuclei: potential role for STAT3 as a retrograde signaling transcription factor. J. Comp. Neurol. 474, 535–545.
Leppa, S., Saffrich, R., Ansorge, W., and Bohmann, D. (1998). Differential regulation of c-Jun by ERK and JNK during PC12 cell differentiation. EMBO J. 17, 4404–4413.
Leu, J. I., Crissey, M. A. S., Leu, J. P., Ciliberto, G., and Tau, R. (2001). Interleukin-6-induced STAT3 and AP-1 amplify hepatocyte nuclear factor 1-mediated transactivation of hepatic genes, an adaptive response to liver injury. Mol. Cell. Biol. 21, 414–424.
Lieberman, A. R. (1971). The axon reaction – a review of the principal features of perikaryal responses to axon injury. Int. Rev. Neurobiol. 14, 49–124.
Lim, C. P., and Cao, X. M. (2006). Structure, function, and regulation of STAT proteins. Mol. Biosyst. 2, 536–550.
Lindholm, D., Heumann, R., Meyer, M., and Thoenen, H. (1987). Interleukin-1 regulates synthesis of nerve growth-factor in non-neuronal cells of rat sciatic-nerve. Nature 330, 658–659.
Lindwall, C., Dahlin, L., Lundborg, G., and Kanje, M. (2004). Inhibition of c-Jun phosphorylation reduces axonal outgrowth of adult rat nodose ganglia and dorsal root ganglia sensory neurons. Mol. Cell. Neurosci. 27, 267–279.
Lindwall, C., and Kanje, M. (2005). Retrograde axonal transport of JNK signaling molecules influence injury induced nuclear changes in p-c-Jun and ATF3 in adult rat sensory neurons. Mol. Cell. Neurosci. 29, 269–282.
Liu, R. Y., and Snider, W. D. (2001). Different signaling pathways mediate regenerative versus developmental sensory axon growth. J. Neurosci. 21, RC164.
Lonze, B. E., and Ginty, D. D. (2002). Function and regulation of CREB family transcription factors in the nervous system. Neuron 35, 605–623.
Lonze, B. E., Riccio, A., Cohen, S., and Ginty, D. D. (2002). Apoptosis, axonal growth defects, and degeneration of peripheral neurons in mice lacking CREB. Neuron 34, 371–385.
Ma, V. Y., and Bisby, M. A. (1998). Increased activation of nuclear factor kappa B in rat lumbar dorsal root ganglion neurons following partial sciatic nerve injuries. Brain Res. 797, 243–254.
Magoulas, B., and Lopez-de Heredia, L. (2010). Functional analysis of the transcription factor C/EBPdelta in neuronal repair. FENS Abstr. 5, 040.12.
Maier, I. C., and Schwab, M. E. (2006). Sprouting, regeneration and circuit formation in the injured spinal cord: factors and activity. Philos. Trans. R. Soc. Lond. B Biol. Sci. 361, 1611–1634.
Makwana, M., Werner, A., Acosta-Saltos, A., Gonitel, R., Pararajasingham, A., Ruff, C., Rumajogee, P., Cuthill, D., Galiano, M., Bohatschek, M., Wallace, A. S., Anderson, P. N., Mayer, U., Behrens, A., and Raivich, G. (2010). Peripheral facial nerve axotomy in mice causes sprouting of motor axons into perineuronal central white matter: time course and molecular characterization. J. Comp. Neurol. 518, 699–721.
Mandemakers, W., Zwart, R., Jaegle, M., Walbeehm, E., Visser, P., Grosveld, F., and Meijer, D. (2000). A distal Schwann cell-specific enhancer mediates axonal regulation of the Oct-6 transcription factor during peripheral nerve development and regeneration. EMBO J. 19, 2992–3003.
Mason, M. R., Lieberman, A. R., and Anderson, P. N. (2003). Corticospinal neurons up-regulate a range of growth-associated genes following intracortical, but not spinal, axotomy. Eur. J. Neurosci. 18, 789–802.
Mattson, M. P. (2005). NF-kappa B in the survival and plasticity of neurons. Neurochem. Res. 30, 883–893.
Mayr, B., and Montminy, M. (2001). Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell Biol. 2, 599–609.
Mayumi-Matsuda, K., Kojima, S., Nakayama, T., Suzuki, H., and Sakata, T. (1999). Scanning gene expression during neuronal cell death evoked by nerve growth factor depletion. Biochim. Biophys. Acta 1489, 293–302.
Miao, T., Wu, D. S., Zhang, Y., Bo, X. N., Subang, M. C., Wang, P., and Richardson, P. M. (2006). Suppressor of cytokine signaling-3 suppresses the ability of activated signal transducer and activator of transcription-3 to stimulate neurite growth in rat primary sensory neurons. J. Neurosci. 26, 9512–9519.
Miao, T. Z., Wu, D. S., Zhang, Y., Bo, X. N., Xiao, F., Zhang, X. Y., Magoulas, C., Subang, M. C., Wang, P., and Richardson, P. M. (2008). SOCS3 suppresses AP-1 transcriptional activity in neuroblastoma cells through inhibition of c-Jun N-terminal kinase. Mol. Cell. Neurosci. 37, 367–375.
Michaelevski, I., Segal-Ruder, Y., Rozenbaum, M., Medzihradszky, K. F., Shalem, O., Coppola, G., Horn-Saban, S., Ben-Yaakov, K., Dagan, S. Y., Rishal, I., Geschwind, D. H., Pilpel, Y., Burlingame, A. L., and Fainzilber, M. (2010). Signalling to transcription networks in the neuronal retrograde injury response. Sci. Signal. 3, 1–11.
Moisse, K., Volkening, K., Leystra-Lantz, C., Welch, I., Hill, T., and Strong, M. J. (2009). Divergent patterns of cytosolic TDP-43 and neuronal progranulin expression following axotomy: implications for TDP-43 in the physiological response to neuronal injury. Brain Res. 1249, 202–211.
Moore, D. L., and Goldberg, J. L. (2011). Multiple transcription factor families regulate axon growth and regeneration. Dev. Neurobiol. 71, 1186–1211.
Mori, N., and Morii, H. (2002). SCG10-related neuronal growth-associated proteins in neural development, plasticity, degeneration, and aging. J. Neurosci. Res. 70, 264–273.
Morrison, D. K., and Davis, R. J. (2003). Regulation of map kinase signaling modules by scaffold proteins in mammals. Annu. Rev. Cell Dev. Biol. 19, 91–118.
Morton, S., Davis, R. J., McLaren, A., and Cohen, P. (2003). A reinvestigation of the multisite phosphorylation of the transcription factor c-Jun. EMBO J. 22, 3876–3886.
Moya, C., Huang, Z., Cheng, P., Jayaraman, A., and Hahn, J. (2011). Investigation of IL-6 and IL-10 signalling via mathematical modelling. IET Syst. Biol. 5, 15–26.
Nadeau, S., Hein, P., Fernandes, K. J., Peterson, A. C., and Miller, F. D. (2005). A transcriptional role for C/EBP beta in the neuronal response to axonal injury. Mol. Cell. Neurosci. 29, 525–535.
Nagata, K., Kiryu-Seo, S., Maeda, M., Yoshida, K., Morita, T., and Kiyama, H. (2010). Damage-induced neuronal endopeptidase is critical for presynaptic formation of neuromuscular junctions. J. Neurosci. 30, 6954–6962.
Nakagomi, S., Suzuki, Y., Namikawa, K., Kiryu-Seo, S., and Kiyama, H. (2003). Expression of the activating transcription factor 3 prevents c-Jun N-terminal kinase-induced neuronal death by promoting heat shock protein 27 expression and Akt activation. J. Neurosci. 23, 5187–5196.
Narumi, S., and Fujita, T. (1978). Stimulatory effects of substance-P and nerve growth factor (NGF) on neurite outgrowth in embryonic chick dorsal root ganglia. Neuropharmacology 17, 73–76.
Oliveira, A. L., Thams, S., Lidman, O., Piehl, F., Hokfelt, T., Karre, K., Linda, H., and Cullheim, S. (2004). A role for MHC class I molecules in synaptic plasticity and regeneration of neurons after axotomy. Proc. Natl. Acad. Sci. U.S.A. 101, 17843–17848.
Patodia, S., Hristova, M., Makwana, M., Rumajogee, P., Akira, S., and Raivich, G. (2011). Transcription factor STAT3 is required for efficient regeneration after peripheral nerve injury. Abstr. Soc. Neurosci. 784.01/BB22.
Pearson, A. G., Gray, C. W., Pearson, J. F., Greenwood, J. A., During, M. J., and Dragunow, M. (2003). ATF3 enhances c-Jun-mediated neurite sprouting. Brain Res. Mol. Brain Res. 120, 38–45.
Perkins, N. D. (2007). Integrating cell-signalling pathways with NF-kappa B and IKK function. Nat. Rev. Mol. Cell Biol. 8, 49–62.
Pollock, G., Pennypacker, K. R., Memet, S., Israel, A., and Saporta, S. (2005). Activation of NF-kappa B in the mouse spinal cord following sciatic nerve transection. Exp. Brain Res. 165, 470–477.
Pradervand, S., Yasukawa, H., Muller, O. G., Kjekshus, H., Nakamura, T., St Amand, T. R., Yajima, T., Matsumura, K., Duplain, H., Iwatate, M., Woodard, S., Pedrazzini, T., Ross, J., Firsov, D., Rossier, B. C., Hoshijima, M., and Chien, K. R. (2004). Small proline-rich protein 1A is a gp130 pathway- and stress-inducible cardioprotective protein. EMBO J. 23, 4517–4525.
Qin, Q. Y., Baudry, M., Liao, G. H., Noniyev, A., Galeano, J., and Bi, X. N. (2009). A novel function for p53: regulation of growth cone motility through Interaction with rho kinase. J. Neurosci. 29, 5183–5192.
Raivich, G. (2008). c-Jun expression, activation and function in neural cell death, inflammation and repair. J. Neurochem. 107, 898–906.
Raivich, G. (2011). Transcribing the path to neurological recovery-from early signals through transcription factors to downstream effectors of successful regeneration. Ann. Anat. 193, 248–258.
Raivich, G., and Behrens, A. (2006). Role of the AP-1 transcription factor c-Jun in developing, adult and injured brain. Prog. Neurobiol. 78, 347–363.
Raivich, G., Bohatschek, M., Da Costa, C., Iwata, O., Galiano, M., Hristova, M., Nateri, A. S., Makwana, M., Riera-Sans, L., Wolfer, D. P., Lipp, H. P., Aguzzi, A., Wagner, E. F., and Behrens, A. (2004). The AP-1 transcription factor c-Jun is required for efficient axonal regeneration. Neuron 43, 57–67.
Raivich, G., Hellweg, R., and Kreutzberg, G. W. (1991). NGF receptor-mediated reduction in axonal NGF uptake and retrograde transport following sciatic nerve injury and during regeneration. Neuron 7, 151–164.
Raivich, G., Jones, L. L., Kloss, C. U., Werner, A., Neumann, H., and Kreutzberg, G. W. (1998). Immune surveillance in the injured nervous system: T-lymphocytes invade the axotomized mouse facial motor nucleus and aggregate around sites of neuronal degeneration. J. Neurosci. 18, 5804–5816.
Ram, P. T., and Iyengar, R. (2001). G protein coupled receptor signaling through the Src and STAT3 pathway: role in proliferation and transformation. Oncogene 20, 1601–1606.
Ramji, D. P., and Foka, P. (2002). CCAAT/enhancer-binding proteins: structure, function and regulation. Biochem. J. 365, 561–575.
Ramji, D. P., Vitelli, A., Tronche, F., Cortese, R., and Ciliberto, G. (1993). The 2 c/EBP isoforms, IL-6DBP/NF-IL6 and c/EBP-delta/NF-IL6 beta, are induced by IL-6 to promote acute phase gene-transcription via different mechanisms. Nucleic Acids Res. 21, 289–294.
Redmond, L., Kashani, A. H., and Ghosh, A. (2002). Calcium regulation of dendritic growth via CaM kinase IV and CREB-mediated transcription. Neuron 34, 999–1010.
Rende, M., Muir, D., Ruoslahti, E., Hagg, T., Varon, S., and Manthorpe, M. (1992). Immunolocalization of ciliary neuronotrophic factor in adult rat sciatic nerve. Glia 5, 25–32.
Rudolph, D., Tafuri, A., Gass, P., Hammerling, G., Arnold, B., and Schutz, G. (1998). Impaired fetal T cell development and perinatal lethality in mice lacking the cAMP response element binding protein. Proc. Natl. Acad. Sci. U.S.A. 95, 4481–4486.
Ruff, C., Da Costa, C., Staak, N., Rumajogee, P., Makwana, M., Acosta-Saltos, A., Kaswich, M., Brecht, S., Hristova, M., Riera-Sans, L., Mirsky, R., Herdegen, T., Jessen, K., Behrens, A., and Raivich, G. (2009). The role of c-Jun and its N-terminal phosphorylation in facial nerve regeneration. Abstr. Soc. Neurosci. 510.9/B42.
Sark, M. W. J., Fischer, D. F., de Meijer, E., van de Putte, P., and Backendorf, C. (1998). AP-1 and ets transcription factors regulate the expression of the human SPRR1A keratinocyte terminal differentiation marker. J. Biol. Chem. 273, 24683–24692.
Sato, T., Takeuchi, S., Saito, A., Ding, W., Bamba, H., Matsuura, H., Hisa, Y., Tooyama, I., and Urushitani, M. (2009). Axonal ligation induces transient redistribution of TDP-43 in brainstem motor neurons. Neuroscience 164, 1565–1578.
Schaefer, T. S., Sanders, L. K., and Nathans, D. (1995). Cooperative transcriptional activity of Jun and STAT3-beta, a short-form of STAT3. Proc. Natl. Acad. Sci. U.S.A. 92, 9097–9101.
Scherer, S. S., Wang, D. Y., Kuhn, R., Lemke, G., Wrabetz, L., and Kamholz, J. (1994). Axons regulate Schwann-cell expression of the pou transcription factor Scip. J. Neurosci. 14, 1930–1942.
Schmied, R., Huang, C. C., Zhang, X. P., Ambron, D. A., and Ambron, R. T. (1993). Endogenous axoplasmic proteins and proteins containing nuclear localization signal sequences use the retrograde axonal transport/nuclear import pathway in Aplysia neurons. J. Neurosci. 13, 4064–4071.
Schwaiger, F. W., Hager, G., Schmitt, A. B., Horvat, A., Hager, G., Streif, R., Spitzer, C., Gamal, S., Breuer, S., Brook, G. A., Nacimiento, W., and Kreutzberg, G. W. (2000). Peripheral but not central axotomy induces changes in Janus kinases (JAK) and signal transducers and activators of transcription (STAT). Eur. J. Neurosci. 12, 1165–1176.
Schwarz, C. S., Seyfried, J., Evert, B. O., Klockgether, T., and Wullner, U. (2002). Bcl-2 up-regulates ha-ras mRNA expression and induces c-Jun phosphorylation at Ser73 via an ERK-dependent pathway in PC 12 cells. Neuroreport 13, 2439–2442.
Schweizer, U., Gunnersen, J., Karch, C., Wiese, S., Holtmann, B., Takeda, K., Akira, S., and Sendtner, M. (2002). Conditional gene ablation of STAT3 reveals differential signaling requirements for survival of motoneurons during development and after nerve injury in the adult. J. Cell Biol. 156, 287–297.
Seijffers, R., Allchorne, A. J., and Woolf, C. J. (2006). The transcription factor ATF-3 promotes neurite outgrowth. Mol. Cell. Neurosci. 32, 143–154.
Seijffers, R., Mills, C. D., and Woolf, C. J. (2007). ATF3 increases the intrinsic growth state of DRG neurons to enhance peripheral nerve regeneration. J. Neurosci. 27, 7911–7920.
Sendtner, M., Gotz, R., Holtmann, B., Escary, J. L., Masu, Y., Carroll, P., Wolf, E., Brem, G., Brulet, P., and Thoenen, H. (1996). Cryptic physiological trophic support of motoneurons by LIF revealed by double gene targeting of CNTF and LIF. Curr. Biol. 6, 686–694.
Sendtner, M., Gotz, R., Holtmann, B., and Thoenen, H. (1997). Endogenous ciliary neurotrophic factor is a lesion factor for axotomized motoneurons in adult mice. J. Neurosci. 17, 6999–7006.
Shackleford, T. J., Zhang, Q., Tian, L., Vu, T. T., Korapati, A. L., Baumgartner, A. M., Le, X. F., Liao, W. S., and Claret, F. X. (2011). STAT3 and CCAAT/enhancer binding protein beta (C/EBP-beta) regulate Jab1/CSN5 expression in mammary carcinoma cells. Breast Cancer Res. 13, R65.
Skene, J. H. P., and Willard, M. (1981). Characteristics of growth-associated polypeptides in regenerating toad retinal ganglion-cell axons. J. Neurosci. 1, 419–426.
Smith, D. S., and Skene, J. H. P. (1997). A transcription-dependent switch controls competence of adult neurons for distinct modes of axon growth. J. Neurosci. 17, 646–658.
Sole, C., Dolcet, X., Segura, M. F., Gutierrez, H., Diaz-Meco, M. T., Gozzelino, R., Sanchis, D., Bayascas, J. R., Gallego, C., Moscat, J., Davies, A. M., and Comella, J. X. (2004). The death receptor antagonist FAIM promotes neurite outgrowth by a mechanism that depends on ERK and NF-kappa B signaling. J. Cell Biol. 167, 479–492.
Suarez, V., Guntinas-Lichius, O., Streppel, M., Ingorokva, S., Grosheva, M., Neiss, W. F., Angelov, D. N., and Klimaschewski, L. (2006). The axotomy-induced neuropeptides galanin and pituitary adenylate cyclase-activating peptide promote axonal sprouting of primary afferent and cranial motor neurones. Eur. J. Neurosci. 24, 1555–1564.
Sun, F., Park, K. K., Belin, S., Wang, D., Lu, T., Chen, G., Zhang, K., Yeung, C., Feng, G., Yankner, B. A., and He, Z. (2011). Sustained axon regeneration induced by co-deletion of PTEN and SOCS3. Nature 480, 372–375.
Sun, P. Q., Enslen, H., Myung, P. S., and Maurer, R. A. (1994). Differential activation of Creb by Ca2+/calmodulin-dependent protein-kinases type-II and type-IV involves phosphorylation of a site that negatively regulates activity. Genes Dev. 8, 2527–2539.
Sung, Y. J., Povelones, M., and Ambron, R. T. (2001). RISK-1: a novel MAPK homologue in axoplasm that is activated and retrogradely transported after nerve injury. J. Neurobiol. 47, 67–79.
Symes, A., Gearan, T., Eby, J., and Fink, J. S. (1997). Integration of Jak-STAT and AP-1 signaling pathways at the vasoactive intestinal peptide cytokine response element regulates ciliary neurotrophic factor-dependent transcription. J. Biol. Chem. 272, 9648–9654.
Taga, T., and Kishimoto, T. (1995). Signaling mechanisms through cytokine receptors that share signal-transducing receptor components. Curr. Opin. Immunol. 7, 17–23.
Takeda, K., Noguchi, K., Shi, W., Tanaka, T., Matsumoto, M., Yoshida, N., Kishimoto, T., and Akira, S. (1997). Targeted disruption of the mouse STAT3 gene leads to early embryonic lethality. Proc. Natl. Acad. Sci. U.S.A. 94, 3801–3804.
Takeda, M., Kato, H., Takamiya, A., Yoshida, A., and Kiyama, H. (2000). Injury-specific expression of activating transcription factor-3 in retinal ganglion cells and its colocalized expression with phosphorylated c-Jun. Invest. Ophthalmol. Vis. Sci. 41, 2412–2421.
Tanaka, S., Kamachi, Y., Tanouchi, A., Hamada, H., Jing, N. H., and Kondoh, H. (2004). Interplay of SOX and POU factors in regulation of the Nestin gene in neural primordial cells. Mol. Cell. Biol. 24, 8834–8846.
Tedeschi, A., Nguyen, T., Puttagunta, R., Gaub, P., and Di Giovanni, S. (2009). A p53-CBP/p300 transcription module is required for GAP-43 expression, axon outgrowth, and regeneration. Cell Death Differ. 16, 543–554.
Topilko, P., Schneidermaunoury, S., Levi, G., Baronvanevercooren, A., Chennoufi, A. B., Seitanidou, T., Babinet, C., and Charnay, P. (1994). Krox-20 controls myelination in the peripheral nervous system. Nature 371, 796–799.
Toth, C. C., Willis, D., Twiss, J. L., Walsh, S., Martinez, J. A., Liu, W. Q., Midha, R., and Zochodne, D. W. (2009). Locally synthesized calcitonin gene-related peptide has a critical role in peripheral nerve regeneration. J. Neuropathol. Exp. Neurol. 68, 326–337.
Tsujino, H., Kondo, E., Fukuoka, T., Dai, Y., Tokunaga, A., Miki, K., Yonenobu, K., Ochi, T., and Noguchi, K. (2000). Activating transcription factor 3 (ATF3) induction by axotomy in sensory and motoneurons: a novel neuronal marker of nerve injury. Mol. Cell. Neurosci. 15, 170–182.
Vries, R. G. J., Prudenziati, M., Zwartjes, C., Verlaan, M., Kalkhoven, E., and Zantema, A. (2001). A specific lysine in c-Jun is required for transcriptional repression by E1A and is acetylated by p300. EMBO J. 20, 6095–6103.
Waetzig, V., Zhao, Y., and Herdegen, T. (2006). The bright side of JNKs – multitalented mediators in neuronal sprouting, brain development and nerve fiber regeneration. Prog. Neurobiol. 80, 84–97.
Waller, A. V. (1850). Experiments on the section of the glossopharyngeal and hypoglossal nerves of the frog, and observations of the alterations produced thereby in the structure of their primitive fibres. Philos. Trans. R. Soc. Lond. B Biol. Sci. 140, 423–429.
Weber, J. R. M., and Skene, J. H. P. (1998). The activity of a highly promiscuous AP-1 element can be confined to neurons by a tissue-selective repressive element. J. Neurosci. 18, 5264–5274.
Werner, A., Kloss, C. U., Walter, J., Kreutzberg, G. W., and Raivich, G. (1998). Intercellular adhesion molecule-1 (ICAM-1) in the mouse facial motor nucleus after axonal injury and during regeneration. J. Neurocytol. 27, 219–232.
Westerlund, N., Zdrojewska, J., Padzik, A., Komulainen, E., Björkblom, B., Rannikko, E., Tararuk, T., Garcia-Frigola, C., Sandholm, J., Nguyen, L., Kallunki, T., Courtney, M. J., and Coffey, E. T. (2011). Phosphorylation of SCG10/stathmin-2 determines multipolar stage exit and neuronal migration rate. Nat. Neurosci. 14, 305–313.
Williams, K. L., Rahimtula, M., and Mearow, K. M. (2006). Heat shock protein 27 is involved in neurite extension and branching of dorsal root ganglion neurons in vitro. J. Neurosci. Res. 84, 716–723.
Wilton, D., Arthur-Farraj, P., Latouche, M., Turmaine, M., Behrens, A., Mirsky, R., Raivich, G., and Jessen, K. (2009). Schwann cell C-Jun is essential for peripheral nerve regeneration. Glia 57, S158.
Wu, Y. Y., and Bradshaw, R. A. (1996). Induction of neurite outgrowth by interleukin-6 is accompanied by activation of STAT3 signaling pathway in a variant PC12 cell (E2) line. J. Biol. Chem. 271, 13023–13032.
Wynick, D., Thompson, S. W., and McMahon, S. B. (2001). The role of galanin as a multi-functional neuropeptide in the nervous system. Curr. Opin. Pharmacol. 1, 73–77.
Xia, X. G., Hofmann, H. D., Deller, T., and Kirsch, M. (2002). Induction of STAT3 signaling in activated astrocytes and sprouting septal neurons following entorhinal cortex lesion in adult rats. Mol. Cell. Neurosci. 21, 379–392.
Yang, E., Lerner, L., Besser, D., and Darnell, J. E. Jr. (2003). Independent and cooperative activation of chromosomal c-fos promoter by STAT3. J. Biol. Chem. 278, 15794–15799.
Yang, Z. Y., Quigley, H. A., Pease, M. E., Yang, Y. Q., Qian, J., Valenta, D., and Zack, D. J. (2007). Changes in gene expression in experimental glaucoma and optic nerve transection: the equilibrium between protective and detrimental mechanisms. Invest. Ophthalmol. Vis. Sci. 48, 5539–5548.
Yoo, S., Nguyen, M. P., Fukuda, M., Bittner, G. D., and Fishman, H. M. (2003). Plasmalemmal sealing of transected mammalian neurites is a gradual process mediated by Ca(2+)-regulated proteins. J. Neurosci. Res. 74, 541–551.
Yoshida, K., Behrens, A., Le-Niculescu, H., Wagner, E. F., Harada, T., Imaki, J., Ohno, S., and Karin, M. (2002). Amino-terminal phosphorylation of c-Jun regulates apoptosis in the retinal ganglion cells by optic nerve transection. Invest. Ophthalmol. Vis. Sci. 43, 1631–1635.
Zhang, X. K., Wrzeszczynska, M. H., Horvath, C. M., and Darnell, J. E. (1999). Interacting regions in STAT3 and c-Jun that participate in cooperative transcriptional activation. Mol. Cell. Biol. 19, 7138–7146.
Keywords: transcription, axotomy, facial nerve, neurite, DRG, regeneration, c-Jun, ATF3, STAT3
Citation: Patodia S and Raivich G (2012) Role of transcription factors in peripheral nerve regeneration. Front. Mol. Neurosci. 5:8. doi: 10.3389/fnmol.2012.00008
Received: 05 November 2011; Accepted: 24 January 2012;
Published online: 10 February 2012.
Edited by:
Simone Di Giovanni, University of Tuebingen, GermanyReviewed by:
Richard Zigmond, Case Western Reserve University School of Medicine, USASulayman Dib-Hajj, Yale University, USA
Copyright: © 2012 Patodia and Raivich. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Gennadij Raivich, Centre for Perinatal Brain Protection and Repair, University College London, Chenies Mews 86-96, London WC1E 6HX, UK. e-mail:Zy5yYWl2aWNoQHVjbC5hYy51aw==