 Clarrisa A. Bradley1,2
Clarrisa A. Bradley1,2 Stéphane Peineau1,5,6
Stéphane Peineau1,5,6 Changiz Taghibiglou3
Changiz Taghibiglou3 Celine S. Nicolas1
Celine S. Nicolas1 Daniel J. Whitcomb1
Daniel J. Whitcomb1 Zuner A. Bortolotto1
Zuner A. Bortolotto1 Bong-Kiun Kaang2
Bong-Kiun Kaang2 Kwangwook Cho1 Yu Tian Wang4 and Graham L. Collingridge1,2*
Kwangwook Cho1 Yu Tian Wang4 and Graham L. Collingridge1,2*- 1MRC Centre for Synaptic Plasticity, University of Bristol, Bristol, UK
- 2Department of Brain and Cognitive Sciences, Seoul National University, Seoul, Korea
- 3Department of Pharmacology, College of Medicine, University of Saskatchewan, Saskatoon, SK, Canada
- 4Brain Research Centre, University of British Columbia, Vancouver, BC, Canada
- 5Inserm, UMR U676, Paris, France
- 6Université de Médecine Denis Diderot, Paris, France
Glycogen synthase kinase-3 (GSK-3) has many cellular functions. Recent evidence suggests that it plays a key role in certain types of synaptic plasticity, in particular a form of long-term depression (LTD) that is induced by the synaptic activation of N-methyl-D-aspartate receptors (NMDARs). In the present article we summarize what is currently known concerning the roles of GSK-3 in synaptic plasticity at both glutamatergic and GABAergic synapses. We summarize its role in cognition and speculate on how alterations in the synaptic functioning of GSK-3 may be a major factor in certain neurodegenerative disorders.
Introduction
While long recognized as a critical molecular effector in the cell, and an important target for therapeutic intervention, glycogen synthase kinase-3 (GSK-3) has only recently been discovered to be a key regulator of synaptic plasticity. Here we review the studies that have illuminated our understanding of how GSK-3 is involved in long-term depression (LTD) and long-term potentiation (LTP) in the hippocampus. We describe potential signaling cascades that involve GSK-3 in neurons and how this might affect the expression of AMPA receptors at synapses. In addition, we describe the evidence that suggests that GSK-3 also regulates GABA-A receptor (GABAR) trafficking. Lastly, we relate these emerging mechanisms to cognition and propose a hypothesis of how dysregulation of GSK-3 activity at synapses could explain important aspects of neurodegenerative disease.
Role of GSK-3 in LTP and LTD
Information in the brain is stored primarily as changes in the weights of synaptic connections, via the processes of LTP and LTD. LTP, which is a long-lasting increase in the efficiency of synaptic transmission, is usually triggered by the synaptic activation of N-methyl-D-aspartate receptors (NMDARs), though NMDAR-independent forms of LTP also exist in the CNS (Bliss and Collingridge, 1993). LTD, which is the counterpart of LTP, can also be induced by the synaptic activation of NMDA receptors (NMDAR-LTD). Other forms of LTD also exist, such as those triggered by the synaptic activation of metabotropic glutamate receptors (mGluRs) and muscarinic receptors (mAChRs) (Collingridge et al., 2010). Multiple mechanisms exist to express the alteration in synaptic efficiency, including changes in the probability of neurotransmitter release and alterations in the function of postsynaptic receptors. With respect to NMDAR triggered synaptic plasticity, the most extensively investigated mechanisms involve alterations in the number of α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors (AMPARs) expressed at synapses, caused via alterations in AMPAR trafficking (Collingridge et al., 2004).
We found that inhibition of GSK-3 prevented the induction of NMDAR-LTD (Peineau et al., 2007). This effect was observed with six structurally distinct inhibitors, at the appropriate concentration range for an action on GSK-3 (Peineau et al., 2007, 2009). In contrast, inhibition of a large number of other serine/threonine (Ser/Thr) protein kinases did not affect NMDAR-LTD (Figure 1). This suggests that GSK-3 is likely to be one of a small number of Ser/Thr protein kinases involved in this form of synaptic plasticity. It is not unique, however, since we have more recently also found a role for PI3Kγ, a dual Ser/Thr protein kinase and lipid kinase, in NMDAR-LTD (Kim et al., 2011). Furthermore, a role for tyrosine phosphorylation in NMDAR-LTD has been established (Ahmadian et al., 2004; Hayashi and Huganir, 2004) and recent evidence suggests that the tyrosine kinase Janus kinase 2 (JAK2) is required for the induction of NMDAR-LTD (Nicolas et al., 2012). Therefore, GSK-3 is one component of a complex phosphorylation cascade involved in NMDAR-LTD.
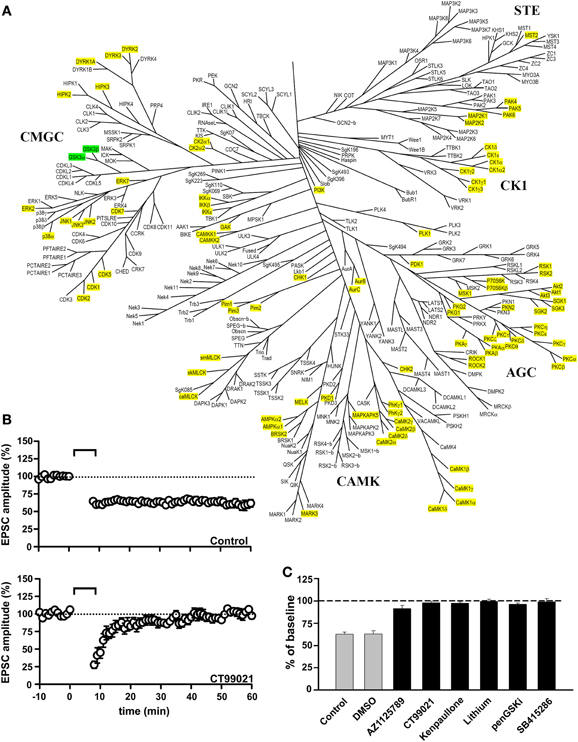
Figure 1. GSK-3 inhibitors block the induction of NMDAR-LTD. (A) Family tree of Ser/Thr protein kinases in the human genome (modified from Manning et al., 2002). The two isoforms of GSK-3 (α and β) are highlighted in green. The kinases that were excluded from having a role in the induction of NMDAR-LTD, based on inhibitor studies reported in Peineau et al. (2007, 2009), are highlighted in yellow. (B) Control NMDAR-LTD (top) and its block by the GSK-3 inhibitor CT99021 (bottom). (C) Block of the induction of NMDAR-LTD by six different GSK-3 inhibitors. See Peineau et al. (2007, 2009) for more details.
As described elsewhere, the isoform involved in NMDAR-LTD is likely to be GSK-3β, though a complementary role of GSK-3α cannot be discounted. Of the forms of synaptic plasticity that have been examined, GSK-3 is specifically involved in NMDAR-LTD (Figure 2). Thus, pharmacological inhibition of GSK-3 has no effect on NMDAR-LTP, depotentiation, or mGluR-LTD at CA3-CA1 synapses (Peineau et al., 2007). Of course, a role in one or more of the several other forms of synaptic plasticity at this or other synaptic pathways in the CNS cannot be discounted.
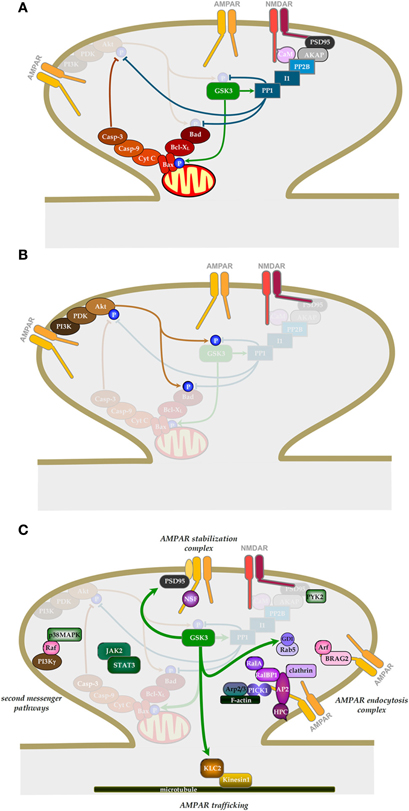
Figure 2. The role of GSK-3 in NMDAR-LTD. (A) Signaling pathways involved in the induction of NMDAR-LTD and the regulation of GSK-3β. (B) Signaling pathway by which NMDAR-LTP can inhibit NMDAR-LTD. (C) Potential downstream effectors of GSK-3β in NMDAR-LTD. (See text for details).
Although inhibition of GSK-3β has no effect on NMDAR-LTP, over-expression of GSK-3β blocks LTP (Hooper et al., 2007; Zhu et al., 2007). In addition, induction of LTP inhibits the activity of GSK-3β (Hooper et al., 2007; Peineau et al., 2007), suggesting that LTP and LTD may interact via a pathway involving GSK-3β that acts as a molecular switch.
Role of GSK-3 in Metaplasticity
Metaplasticity refers to changes in the ability of synapses to undergo plasticity after a previous synaptic or modulatory event, sometimes called “priming” (see review by Abraham and Bear, 1996). Several different forms of metaplasticity have been described so far. The prototypic form of metaplasticity is depotentiation, which is a form of LTD that can only be induced following the induction of LTP (where it reverses the potentiation to the baseline state; as reviewed by Collingridge et al., 2010). Another form of metaplasticity is where the prior activation of NMDARs can inhibit the subsequent induction of NMDAR-LTP (Coan et al., 1989; Huang et al., 1992). The findings, described above, that NMDAR-LTP can inhibit the activity of GSK-3β and that GSK-3β is required for NMDAR-LTD suggested the existence of another form of metaplasticity whereby LTP regulates LTD.
We explored this prediction experimentally and found that LTP did, indeed, potently inhibit NMDAR-LTD. The priming was induced by a protocol that could induce NMDAR-LTD but the induction of LTP per se was not a prerequisite. Indeed, many experiments were performed under conditions in which LTP was deliberately absent, due to the phenomenon of washout (Malinow and Tsien, 1990), so as to eliminate the potentially confounding situation of depotentiation. In other experiments depotentiation was blocked by an mGluR antagonist. We found that the LTP stimulus (priming stimulus) completely prevented the induction of NMDAR-LTD immediately following the priming. Maximal inhibition of NMDAR-LTD lasted for about 20 min, where after there was a slow recovery of the ability to induce NMDAR-LTD such that after approximately 1 h the ability to induce NMDAR-LTD was fully restored. The mechanism of priming involved activation of NMDARs since it was prevented if an NMDAR antagonist was present during the priming. It also involved the canonical pathway for inhibition of GSK-3, namely phosphoinositide 3-kinase (PI3K) and Akt/PKB (protein kinase B; Figure 2B; Embi et al., 1980; Peineau et al., 2007).
Upstream Regulation of GSK-3 in Synaptic Plasticity
The GSK-3 α and β isoforms are ubiquitous Ser/Thr kinases belonging to the CMGC family of protein kinases that act as key enzymes regulating various cellular signaling pathways. GSK-3 function is modulated through multiple regulatory mechanisms by protein-protein interactions, subcellular localization, priming/substrate specificity, and proteolytic cleavage, which have been recently reviewed by others (Hur and Zhou, 2010; Medina and Wandosell, 2011 and reviewed in this Special Topic by Kaidanovich-Beilin and Woodgett, 2011). Within these levels of functional regulation, phosphorylation and dephosphorylation play prominent roles.
Under certain biochemical conditions such as growth factor deprivation, the mammalian target of rapamycin complex 1(mTORC1)-S6K1 signaling can alternatively regulate and inhibit GSK-3 activity by Ser21/9 phosphorylation (Cohen and Frame, 2001; Zhang et al., 2006). There are also several other kinases such as Erk, ZAK1, MEK1/2, Pyk-2, and Fyn kinases that also have been described to interact with GSK-3 and regulate its function in other cell types (Kim et al., 1999, 2002; Lesort et al., 1999; Hartigan et al., 2001; Ding et al., 2005). However, the extent to which these regulatory processes occur in neurons and, more specifically, are involved in synaptic plasticity is largely unexplored.
We have described an upstream phosphorylation/dephosphorylation-dependent regulation of GSK-3β activity that is involved in NMDAR-LTD (Figure 2A). We cannot discount an additional role of GSK-3α, but focus on the β isoform in this review. Thus far, three sites of phosphorylation have been identified on GSK-3β: Ser9 (Sutherland et al., 1993), Tyr216 (Hughes et al., 1993), and Ser389 (Thr 390 in humans; Thornton et al., 2008) and have also been shown to be important regulatory elements in neurons. It should be noted that while regulation at Thr43 has been demonstrated in other cell types (Ding et al., 2005; Thornton et al., 2008), it remains to be explored in neurons.
The basal activity of GSK-3β is dependent on phosphorylation on Tyr216 (Hughes et al., 1991, 1993). The mechanism by which this Tyr residue becomes phosphorylated is still under debate. It is unclear whether there is an autophosphorylation mechanism (Cole et al., 2004; Lochhead et al., 2006) and/or a regulation by tyrosine kinases such as Fyn (Lesort et al., 1999) or Pyk2 (Hartigan et al., 2001). In resting neurons, Tyr216 is phosphorylated, and thus, GSK-3β is constitutively active (Hur and Zhou, 2010).
The main way neurons regulate GSK-3β activity is by controlling the phosphorylation level of Ser9 (for review see Doble and Woodgett, 2003). The phosphorylation of this site inhibits enzymatic activity. At least eight distinct signaling pathways have been identified as a regulator of the Ser9 phosphorylation state (Figure 3). Seven of them are inhibitory and mediated by kinases. These are the Akt pathway (Hong and Lee, 1997), which we have shown to be important in mediating the phosphorylation of GSK-3β during LTP (Peineau et al., 2007; Figure 2B). In addition, CaMKII has been shown to phosphorylate and inhibit GSK-3 in neurons, where it functions in a pro-survival manner (Song et al., 2010). Phosphorylation of GSK-3β (and not GSK-3α) by classical protein kinase C (PKC) isotypes (α, β1, β2, and γ) results in its inactivation (Espada et al., 2009; Ortega et al., 2010) and may protect neurons from Aβ toxicity (Garrido et al., 2002). In addition, protein kinase A (PKA; Li et al., 2000; O'Driscoll et al., 2007; Shelly et al., 2010, 2011), PrkG1 (Zhao et al., 2009), p90 ribosomal protein S6 kinase (RSK; Valerio et al., 2006) and Integrin-linked kinase (ILK; Naska et al., 2006) also regulate GSK-3 activity in neurons. Interestingly, ILK directly interacts and phosphorylates GSK-3β and also phosphorylates Akt to inhibit GSK-3β indirectly (Delcommenne et al., 1998).
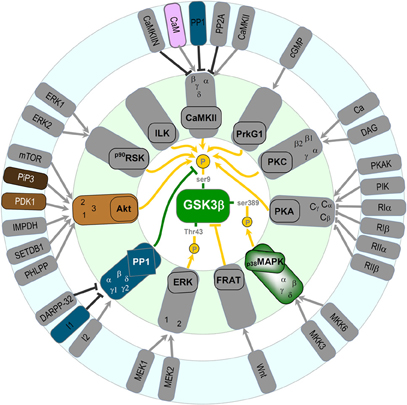
Figure 3. Potential regulators of GSK-3 β in neurons (see text for details).
A third site to regulate GSK-3β activity has been recently identified. The highly enriched neuronal enzyme p38 mitogen-activated protein kinase (p38MAPK) was found to phosphorylate GSK-3β at Ser389 (Thr390 in humans) causing inhibition of its activity similar to that of inhibition via Ser9 (Thornton et al., 2008). Inhibition of GSK-3β via p38MAPK stimulates accumulation of β-catenin.
Only one pathway has been shown to stimulate GSK-3β activity in neurons and is mediated by protein phosphatase 1 (PP1) removing the phosphate group on Ser9 (Bennecib et al., 2000; Morfini et al., 2004). Noteworthy, a positive loop between PP1 and GSK-3β has been identified (Szatmari et al., 2005). We have observed a marked activation of GSK-3β during NMDAR-LTD via dephosphorylation of Ser9 mediated by PP1 (Peineau et al., 2007). However, the involvement of additional pathways that activate GSK-3β activity during NMDAR-LTD may also exist. Indeed, in other systems there are a variety of phosphatases, which dephosphorylate and disinhibit GSK-3 activity (Wera and Hemmings, 1995). In terms of the regulation by PP1, it is likely that during NMDAR-LTD this involves Ca2+ entry through NMDARs, binding of Ca2+ to calmodulin, activation of PP2B (calcineurin) leading to dephosphorylation of Inhibitor 1 (I-1) which in turn leads to activation of PP1 (Figure 2A). This pathway has been shown to be important for NMDAR-LTD (Mulkey et al., 1994).
Downstream Effectors of GSK-3 in Synaptic Plasticity
How GSK-3β activation contributes to the expression of NMDAR-LTD remains unknown. Given that the activation of NMDARs and endocytosis of AMPARs are, respectively, involved in the induction and expression of LTD, it appears quite reasonable to investigate if GSK-3β can exert its actions by regulating either or both of the processes. Indeed, one study has reported a GSK-3 activity dependent rapid internalization of NMDARs (Chen et al., 2007). Thus, LTP might inhibit LTD by regulating the levels of NMDARs that are required for LTD induction. However, in this case, one would expect to see a rapid depression of NMDAR-mediated synaptic transmission as a result of a rapid internalization of the receptors following LTP induction. However, most studies of LTP that have monitored NMDAR-mediated synaptic transmission have reported LTP [e.g., (Bashir et al., 1991)] rather than a transient depression. Thus, further work is needed to establish the extent to which the regulation of NMDAR trafficking by GSK-3β accounts for its involvement in synaptic plasticity. Any inhibition of NMDAR activity by GSK-3 antagonists is highly unlikely to have accounted for the inhibition of NMDAR-LTD in our experiments (Peineau et al., 2007). First, the effects observed on NMDARs were typically around 20% inhibition and this may be too small to prevent NMDAR-LTD induction. Indeed, whilst we observed a complete block of NMDAR-LTD there was sufficient NMDAR activation for NMDAR-LTP to be induced (Peineau et al., 2007). GSK-3β affects not only the induction, but also the expression of LTD as lithium was able to fully block NMDAR-LTD even when applied after the induction protocol (Peineau et al., 2007). Thus, even if the inhibition of NMDAR function may contribute to the effects under certain circumstance it cannot be the sole mechanism.
Wei and colleague (2010) recently showed that constitutive GSK-3β activity enhances basal AMPAR endocytosis. They found that either pharmacological inhibition or knockdown of the protein resulted in a decrease in mEPSC amplitude in cultured hippocampal neurons (Wei et al., 2010). They went on to demonstrate that internalization of AMPARs is via enhanced activity of Rab5 and is dependent on phosphorylation of guanine nucleotide dissociation inhibitor (GDI) at Ser45. They also showed that the effects of inhibition of GSK-3 and insulin occluded, suggesting that insulin-induced endocytosis of AMPARs (Man et al., 2000) is due to inhibition of GSK-3 (Figure 2C). At first glance these observations seem at variance with the reports that inhibition of GSK-3β blocks NMDAR-LTD (Peineau et al., 2007, 2009) and Aβ-mediated inhibition of LTP (Jo et al., 2011) without affecting baseline AMPAR-mediated transmission. Although the effects on mEPSC amplitude where small (15–18%) they should have been detected as a small reduction in baseline transmission, but this was never observed. A potential explanation for these differences is the levels of activity of GSK-3β in the two preparations. As described above, GSK-3β is tightly regulated by inhibition on Ser9 by a variety of signaling pathways and is actively suppressed by LTP (via the PI3K/Akt pathway). Loss of any of these regulatory systems might result in a higher constitutive activity of GSK-3 in cultured dissociated neurons. It, therefore, remains to be determined whether endocytosis of AMPARs during LTD involves GSK-3β-dependent phosphorylation of GDI and activation of Rab5.
We have observed a complex formation between GSK-3β and AMPARs and found that the activity of GSK-3β within this complex is regulated by LTP (Peineau et al., 2007). These results strongly suggest that GSK-3β may regulate AMPAR function or trafficking, thereby affecting the expression of LTD. Consistent with this conjecture, a recent study (Du et al., 2010) has reported that GSK-3β inhibition prevented the NMDA-induced AMPAR endocytosis in a model of chemical LTD (Collingridge et al., 2010). How GSK-3β is involved in AMPAR endocytosis during LTD expression remains to be established. Given that a serine phosphorylation of the GluA2 subunit has been reported to be critical for receptor endocytosis during LTD (Kim et al., 2001a; Chung et al., 2003), GSK-3β may play an important role in mediating the expression of LTD by triggering the GluA2 phosphorylation dependent process. However, in vitro phosphorylation assays have failed to demonstrate an ability of GSK-3β to phosphorylate the carboxyl tail of GluA2 (authors' unpublished observation), suggesting that GSK-3β may phosphorylate one or more other proteins that are required for the regulated AMPAR endocytosis.
It has been proposed that one such potential protein is kinesin light chain 2 (KLC2; Du et al., 2010), a key molecule of the kinesin cargo delivery system, which may be involved in the transport of AMPAR-containing membrane-bound vesicles in neurons (Setou et al., 2002) and has previously been demonstrated as a target of neuronal GSK-3 (Morfini et al., 2002). In this study, Du et al. (2010) have shown that GSK-3β phosphorylation of KLC2 promotes its dissociation from the AMPAR/KLC2 protein complex (Figure 2C). This suggests that GSK-3 phosphorylation of KLC2 may lead to the dissociation of AMPAR-containing vesicles from the kinesin cargo system, thereby affecting the trafficking of AMPARs in neurons. To directly demonstrate that phosphorylation of KLC2 is one of the critical steps in GSK-3β promoted LTD, the authors developed a membrane permeable peptide TAT-KLCpCDK, a specific inhibitor for KLC2 phosphorylation by GSK-3β, and showed that by preventing GSK-3β phosphorylation of KLC2, it inhibited both AMPAR endocytosis and the expression of hippocampal CA1 LTD. Thus, this study has provided strong evidence that KLC2 may be a cellular target of GSK-3β capable of regulating AMPA receptor trafficking and hence synaptic plasticity. Of course, GSK-3β may also affect other components of the AMPAR endocytosis pathway. In addition to AMPAR endocytosis, GSK-3β may influence LTD expression by regulating other processes, some of which are illustrated in Figure 2C. Other potential targets include tau and presenilin-1, as well as transcription factors that may be involved in the protein synthesis dependent phase of NMDAR-LTD (Manahan-Vaughan et al., 2000).
Role in Pathological Synaptic Plasticity
A large body of evidence suggests that changes in synaptic plasticity may be responsible for the cognitive deficits that are observed in various brain disorders (Bowers et al., 2010; Goto et al., 2010; Kasai et al., 2010; Russo et al., 2010; van Spronsen and Hoogenraad, 2010; Proctor et al., 2011; Quartarone and Pisani, 2011; Zhuo et al., 2011). Increasing evidence suggests, additionally, that alterations in various molecular components of synaptic plasticity pathways may actually be the cause of many of these disorders (see review by Collingridge et al., 2010). We term this process pathological plasticity, to distinguish it from the forms of plasticity that are utilized by the CNS during the formation and stabilization of synaptic connections (developmental plasticity) and for learning and memory (cognitive plasticity; Figure 4). This basic premise is that NMDAR-LTD is a mechanism that is used by the CNS to remove AMPARs from synapses and thereby weaken the efficiency of synaptic transmission. This enables the storage of information, and indeed there is growing evidence that NMDAR-LTD is involved in forms of learning and memory (see review by Collingridge et al., 2010). NMDAR-LTD can involve the removal of all AMPARs from individual synapses (Luthi et al., 1999), via a process known as silencing. These silent synapses can then either (1) have new AMPARs inserted via the process of unsilencing (Isaac et al., 1995; Liao et al., 1995) or (2) are eliminated (Bastrikova et al., 2008; Kano et al., 2008; Egashira et al., 2010). Since NMDAR-LTD is particularly prevalent early in development (Dudek and Bear, 1993) when there is considerable pruning of superfluous synaptic connections (Rabacchi et al., 1992; Bhatt et al., 2009), it is likely that NMDAR-LTD is a physiological mechanism used to eliminate synapses during development.
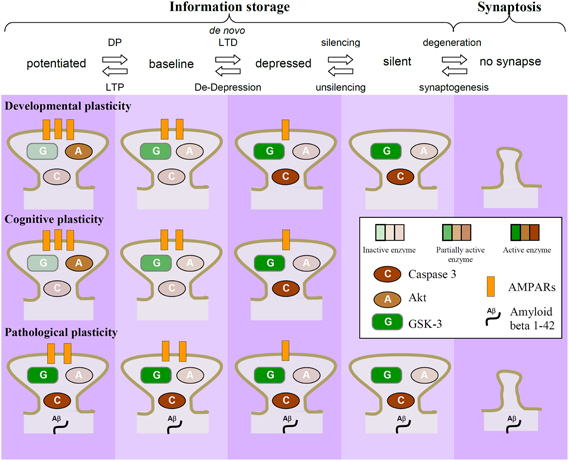
Figure 4. Dysregulation of GSK-3 as a potential causal factor in neurodegeneration. NMDAR-LTD results in a reduction in the number of AMPARs at synapses. It can lead to the removal of all AMPARs (termed “silencing”) and synapse elimination, via the process of synaptosis (localized apoptosis). This process is likely to be prominent early in development, where it may be important for the pruning of unwanted synaptic connections. During adulthood it may be more tightly regulated so that LTD involves mainly reductions in synaptic efficacy rather than the elimination of synapses. In particular, synapses that experience regular LTP will have the activity of GSK-3 dampened, by virtue of the PI3K-Akt pathway. In response to stressors (genetic, epigenetic, or environmental) the same mechanisms may be rekindled, resulting in unwanted synaptic elimination. In this example, the dysregulation of GSK-3β activity is due to activation of caspases leading to cleavage of Akt.
With respect to a molecular mechanism, we previously showed that NMDAR triggered apoptosis, and the associated increase in caspase-3 activity, requires AMPAR endocytosis (Wang et al., 2004). More recently we found that NMDAR-LTD also involves activation of a caspase cascade (Li et al., 2010). These studies suggest that the internalization of AMPARs associated with LTD triggers a local apoptosis. We will refer to this process as synaptosis, a term originally coined for the compartmentalized retraction of synapses at the neuromuscular junction (Gillingwater and Ribchester, 2001). After the period of synaptic pruning during development is over and the organization of the brain is largely complete then it makes sense that the mechanisms of synaptosis are down-regulated. We, therefore, assume that for most of the normal life-span NMDAR-LTD is used more sparingly and for mainly adjusting synaptic weights rather than for eliminating synapses because it would be an energetically more efficient way to store information. However, if these latent synaptosis mechanisms were once again awakened then synapses could be eliminated. Our hypothesis is that neurodegeneration is often triggered by just this reactivation of synaptosis, which if left unchecked leads to apoptosis of vulnerable neuronal populations. The idea that the weakening of connections between nerve cells may explain dementia has been around for over 100 years. However, the observation that GSK-3β regulates the balance between LTP and LTD provides the basis of a molecular mechanism that gives credence to this idea.
In terms of a molecular mechanism, we suggest that GSK-3β is activated to enable NMDAR-LTD and that this mechanism is especially prominent early in development for synaptic pruning. In adulthood, whilst this mechanism may still occur it is utilized less often so as to preserve synaptic connectivity. The ability of NMDAR-LTP to inhibit NMDAR-LTD via GSK-3β activity provides a potential mechanism to enable this regulation. Thus, as long as synapses are engaging LTP processes then they are protected from NMDAR-LTD by the PI3K/Akt regulatory pathway. In other words, it is a classic case of “use it or lose it” where this translates into “use LTP or lose the synapse” (because it is cognitively unimportant).
Pathological plasticity could then result from any one of a number of influences, genetic, epigenetic, or environmental, that affect the LTD process directly or its regulation by LTP so that the LTD mechanism becomes more actively re-engaged. As a formal test of this hypothesis, we used the acute application of Aβ1−42. This treatment is well known to cause a synaptic toxicity, which is characterized by an inhibition of NMDAR-LTP and facilitation of NMDAR-LTD (Cullen et al., 1997; Chen et al., 2000; Kim et al., 2001b; Li et al., 2009; Shankar et al., 2008; Witton et al., 2010). We studied the former, and tested the ability of a GSK-3β inhibitor to prevent the effect (Jo et al., 2011). What we found was that this inhibitor protected synapses from the deleterious effects of Aβ, such that LTP was no longer affected. Similar observations were made in parallel by another group using a different GSK-3 inhibitor (Shipton et al., 2011). Thus, it seems that over-activation of GSK-3β is indeed responsible for this pathological effect on plasticity.
Remarkably, in our study, the GSK-3β inhibitor was still fully effective if applied after the Aβ peptide had exerted its effects to inhibit LTP. This experiment is illustrated in Figure 5. The implications of this finding are that the Aβ-mediated inhibition of LTP is due, at least for a while, to the increased activity of GSK-3β. It is possible that in AD a chronic activation of GSK-3β (Hooper et al., 2008) is responsible for the synaptosis and consequently that GSK-3 inhibitors will reverse the cognitive decline. Of course, GSK-3 inhibitors will have many side-effects relating to the ubiquitous actions of this kinase throughout the body (see other chapters in this volume). But understanding the full cascades involved in the pathological activation of GSK-3β in synaptosis offers the possibility of finding more specific targets.
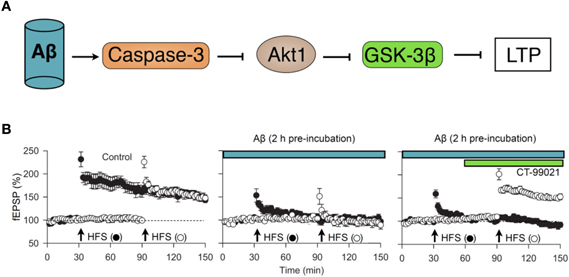
Figure 5. The CAG cascade. (A) potential mechanism by which various neuronal insults (exemplified here by Aβ1–42) may inhibit NMDAR-LTD via the activation of caspase-3 (C) which, by resulting in cleavage of Akt (A), results in persistent activation of GSK-3β (G). (B) Experimental data in support of the “CAG” cascade hypothesis. Pooled data (from left to right) showing LTP in two inputs, in response to high frequency stimulation (HFS), blockade of LTP in both inputs by pre-incubation with Aβ1–42 (500 nM, 2 h) and recovery of LTP in input two following treatment with the GSK-3 inhibitor CT-99021 (1 μM). Data re-plotted from Jo et al. (2011).
In terms of a molecular mechanism, we found that inhibition of caspase-3 and the prevention of the caspase-mediated cleavage of Akt was also able to completely prevent the Aβ-mediated inhibition of LTP. Thus, it seems that Aβ activates caspase-3, which cleaves Akt to persistently activate GSK-3β and thereby trigger synaptosis; we have termed this the CAG cascade (Figure 5). What is not known is how Aβ activates casapse-3 to initiate the process, though an action via mitochondria seems likely. Also the downstream effectors of GSK-3β pathological plasticity are not established. A role for tau is likely given that the Aβ inhibition of LTP is prevented in the tau knockout (Shipton et al., 2011) and since, more generally, tau is a likely effector of GSK-3β regulated toxicity in AD (Morris et al., 2011).
Role in Synaptic Plasticity at GABAergic Synapses
In addition to playing an important role in synaptic plasticity at glutamatergic synapses, recent evidence also suggests a role for GSK-3β in plasticity at GABAergic synapses in the CNS.
GABAergic plasticity has been demonstrated in many brain regions (for reviews see Gaiarsa et al., 2002; Mendez and Bacci, 2011). Some forms of GABAergic plasticity are dependent on Ca2+ entry via stimulation of NMDARs (Wang et al., 2003a; Marsden et al., 2007, 2010; Muir et al., 2010) and alterations in the post-synaptic GABAR distribution (for review see Luscher et al., 2011). Indeed, activation of calcineurin, which forms a complex with the intracellular loop of the γ2 subunit of GABARs, may be a critical step in the induction of GABAergic LTD (Wang et al., 2003a). Recent work has since demonstrated that activity causes rapid dispersion of GABAR clusters (Bannai et al., 2009) and is dependent on Ca2+ entry and Ser327 of the γ2 subunit (Muir et al., 2010). We have also previously shown that insulin, via the PI3 kinase-Akt pathway, causes a rapid increase of GABAergic synaptic transmission and concomitant LTD at glutamatergic synapses (Wan et al., 1997; Man et al., 2000; Wang et al., 2003b). Though GSK-3β stimulation via the insulin pathway has not yet been demonstrated in neurons, it would not be surprising to see that GSK-3 may regulate GABAergic transmission under these conditions as well.
Indeed, modulation of GABAergic function, via alteration of the scaffolding molecule gephyrin, has recently been demonstrated under conditions of chronic treatment with GSK-3β inhibitors (Tyagarajan et al., 2011). In this study the authors identified Ser270 on the inhibitory scaffolding protein gephyrin as a unique phosphorylation site targeted by GSK-3β. Mutation of this site to an alanine residue increased both the number of post-synaptic gephyrin clusters and the frequency of mIPSCs. The pharmacological inhibition of GSK-3β similarly induced greater post-synaptic clustering of gephyrin. Mechanistically, it appears that GSK-3β phosphorylation of Ser270 may cause gephyrin to be targeted for proteolytic degradation by the Ca2+-dependent protease calpain-1, and thus reduce gephyrin clusters and decrease GABAergic transmission.
However, in this study it was not clear what the implications are to GABARs on the surface of the cell as the authors did not extend their findings to look at localization of GABARs. Thus far, α1, α2, and α3 subunits of the GABARs have been demonstrated to interact with gephyrin (Mukherjee et al., 2011; Tretter et al., 2008, 2011 respectively). Gephyrin appears to have different affinities for all these receptors (Maric et al., 2011), and strongly interacts with glycine receptors (Pfeiffer et al., 1982). Thus, each receptor type may be affected differentially by GSK-3β.
Wei et al. (2010) present biochemical data that β2/3 containing GABARs were increased on the cell surface within 10 min of GSK-3β inhibition, suggesting that GABAergic transmission may be rapidly increased by the actions of this kinase. Because the previous study used long treatments with GSK-3β inhibitors more work is required to establish how rapid down-regulation or up-regulation of GSK-3β affects gephyrin clustering and GABAergic transmission.
GSK-3β, LTD, and the Implications for Learning and Memory
Since dysregulation of GSK-3 activity has been strongly implicated in a number of mood-related disorders (Lovestone et al., 2007; Du et al., 2010 and reviewed in this Special Topic by Jope, 2011) as well as AD (Giese, 2009 and reviewed in this Special Topic by Kremer et al., 2011), understanding the effects of GSK-3β's involvement in normal learning and memory, and the importance of the LTD and LTP interaction in this context, is of great interest. Spatial memory impairments on the Morris water maze task have been demonstrated in transgenic mice with a conditional over-expression of GSK-3β (Hernandez et al., 2002). In another study by Engel et al. (2006), these same mice showed significant deficits in the object recognition task, which were completely reversed when GSK-3β over-expression was restored to normal levels (Engel et al., 2006).
More recently, Kimura and colleagues sought to elucidate the role of GSK-3 by examining hypogene function on memory formation and maintenance (Kimura et al., 2008). In adult mice heterozygous for GSK-3β expression (GSK-3β+/−), the animals learned the Morris water maze task at a similar rate as wild type mice, but retrograde amnesia began to affect learning of the task in days 3–9 suggesting that long-term memory formations were adversely affected. Further testing of these heterozygous GSK-3β knock-out mice, using the contextual fear conditioning (CFC) paradigm, suggested that hypo-expression of GSK-3β affected reconsolidation of memory specifically. Inhibition of GSK-3β with the inhibitor AR-A014418 before consolidation or before reconsolidation in CFC showed severely disrupted reconsolidation. They further demonstrated with western blotting that memory reconsolidation is associated with activation of synaptosomal GSK-3β in wild type hippocampal neurons and that intermediate levels of GSK-3β activity were observed upon learning or in the consolidation state relative to naive animals. Though direct evidence is still required, because of the role of GSK-3β activity in LTD mechanisms, these experiments may indicate a greater importance of an LTD dependent cascade in reconsolidation memory.
Periods of sleep may also enhance consolidation of certain types of learning and memory (for reviews see Born et al., 2006; Ribeiro, 2011). Interestingly, in a study on sleep and awake states of plasticity, alterations in molecular, and electrophysiological LTD correlates have also been found. The sleep state (relative to awake state) was associated with low Ser9 phosphorylation of GSK-3β, and hence greater activity and low GluA1 levels as well as a long lasting depression of responses to evoked stimulation (Vyazovskiy et al., 2008). This suggests that during sleep there is an increase in LTD mechanisms relative to LTP mechanisms and that this could potentially underlie the enhancement of memory formation through improved consolidation.
Conclusions
The last few years have seen GSK-3 firmly established as a key player in synaptic plasticity. In particular, it has been shown to be involved in NMDAR-LTD and to be regulated during NMDAR-LTP. We have developed the hypothesis that the ability of LTP to inhibit LTD, via the PI3K/Akt/GSK-3β pathway, serves to stabilize synapses, which would otherwise be lost through the process of synaptosis. We argue that synapses live in a precarious state whereby if they are not protected by LTP then they die at the hands of LTD. This process may serve key physiological functions particularly during development, where synaptic pruning of unwanted connections is of paramount importance, but also during adulthood. However, we postulate that pathological alterations in this pathway, leading to excessive GSK-3β activity, may actually be a causal factor in some forms of neurodegeneration. The understanding of how GSK-3 is involved in synaptic plasticity is in its infancy. Clearly, it will be important to establish a more complete picture of how GSK-3β contributes to synaptic plasticity in both health and disease.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are supported by grants from the MRC, BBSRC, Wellcome Trust Inserm, CIHR, and the Korean Ministry of Science and Technology. In addition, Graham L. Collingridge is a WCU International Scholars and supported by the WCU program through the KOSEF funded by the MEST (R31-10089). Yu Tian Wang is a holder of the Heart and Stroke Foundation of BC and Yukon Chair of Stroke Research and a Howard Hughes Medical Institute International Scholar. Bong-Kiun Kaang is supported by the Creative Research Initiatives Program of the Korean Ministry of Science and Technology. Stéphane Peineau is also supported by Inserm, Universite Paris Diderot, PremUP, Fondation Roger de Spoelberch, Fondation Grace de Monaco, and Leducq Foundation.
References
Abraham, W. C., and Bear, M. F. (1996). Metaplasticity: the plasticity of synaptic plasticity. Trends Neurosci. 19, 126–130.
Ahmadian, G., Ju, W., Liu, L., Wyszynski, M., Lee, S. H., Dunah, A. W., Taghibiglou, C., Wang, Y., Lu, J., Wong, T. P., Sheng, M., and Wang, Y. T. (2004). Tyrosine phosphorylation of GluR2 is required for insulin-stimulated AMPA receptor endocytosis and LTD. EMBO J. 23, 1040–1050.
Bannai, H., Levi, S., Schweizer, C., Inoue, T., Launey, T., Racine, V., Sibarita, J. B., Mikoshiba, K., and Triller, A. (2009). Activity-dependent tuning of inhibitory neurotransmission based on GABAAR diffusion dynamics. Neuron 62, 670–682.
Bashir, Z. I., Alford, S., Davies, S. N., Randall, A. D., and Collingridge, G. L. (1991). Long-term potentiation of NMDA receptor-mediated synaptic transmission in the hippocampus. Nature 349, 156–158.
Bastrikova, N., Gardner, G. A., Reece, J. M., Jeromin, A., and Dudek, S. M. (2008). Synapse elimination accompanies functional plasticity in hippocampal neurons. Proc. Natl. Acad. Sci. U.S.A. 105, 3123–3127.
Bennecib, M., Gong, C. X., Grundke-Iqbal, I., and Iqbal, K. (2000). Role of protein phosphatase-2A and -1 in the regulation of GSK-3, cdk5 and cdc2 and the phosphorylation of tau in rat forebrain. FEBS Lett. 485, 87–93.
Bhatt, D. H., Zhang, S., and Gan, W. B. (2009). Dendritic spine dynamics. Annu. Rev. Physiol. 71, 261–282.
Bliss, T. V., and Collingridge, G. L. (1993). A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361, 31–39.
Bowers, M. S., Chen, B. T., and Bonci, A. (2010). AMPA receptor synaptic plasticity induced by psychostimulants: the past, present, and therapeutic future. Neuron 67, 11–24.
Chen, P., Gu, Z., Liu, W., and Yan, Z. (2007). Glycogen synthase kinase 3 regulates N-methyl-D-aspartate receptor channel trafficking and function in cortical neurons. Mol. Pharmacol. 72, 40–51.
Chen, Q. S., Kagan, B. L., Hirakura, Y., and Xie, C. W. (2000). Impairment of hippocampal long-term potentiation by Alzheimer amyloid beta-peptides. J. Neurosci. Res. 60, 65–72.
Chung, H. J., Steinberg, J. P., Huganir, R. L., and Linden, D. J. (2003). Requirement of AMPA receptor GluR2 phosphorylation for cerebellar long-term depression. Science 300, 1751–1755.
Coan, E. J., Irving, A. J., and Collingridge, G. L. (1989). Low-frequency activation of the NMDA receptor system can prevent the induction of LTP. Neurosci. Lett. 105, 205–210.
Cole, A., Frame, S., and Cohen, P. (2004). Further evidence that the tyrosine phosphorylation of glycogen synthase kinase-3 (GSK3) in mammalian cells is an autophosphorylation event. Biochem. J. 377, 249–255.
Collingridge, G. L., Isaac, J. T., and Wang, Y. T. (2004). Receptor trafficking and synaptic plasticity. Nat. Rev. Neurosci. 5, 952–962.
Collingridge, G. L., Peineau, S., Howland, J. G., and Wang, Y. T. (2010). Long-term depression in the CNS. Nat. Rev. Neurosci. 11, 459–473.
Cullen, W. K., Suh, Y. H., Anwyl, R., and Rowan, M. J. (1997). Block of LTP in rat hippocampus in vivo by beta-amyloid precursor protein fragments. Neuroreport 8, 3213–3217.
Delcommenne, M., Tan, C., Gray, V., Rue, L., Woodgett, J., and Dedhar, S. (1998). Phosphoinositide-3-OH kinase-dependent regulation of glycogen synthase kinase 3 and protein kinase B/AKT by the integrin-linked kinase. Proc. Natl. Acad. Sci. U.S.A. 95, 11211–11216.
Ding, Q., Xia, W., Liu, J. C., Yang, J. Y., Lee, D. F., Xia, J., Bartholomeusz, G., Li, Y., Pan, Y., Li, Z., Bargou, R. C., Qin, J., Lai, C. C., Tsai, F. J., Tsai, C. H., and Hung, M. C. (2005). Erk associates with and primes GSK-3beta for its inactivation resulting in upregulation of beta-catenin. Mol. Cell 19, 159–170.
Doble, B. W., and Woodgett, J. R. (2003). GSK-3: tricks of the trade for a multi-tasking kinase. J. Cell Sci. 116, 1175–1186.
Du, J., Wei, Y., Liu, L., Wang, Y., Khairova, R., Blumenthal, R., Tragon, T., Hunsberger, J. G., Machado-Vieira, R., Drevets, W., Wang, Y. T., and Manji, H. K. (2010). A kinesin signaling complex mediates the ability of GSK-3beta to affect mood-associated behaviors. Proc. Natl. Acad. Sci. U.S.A. 107, 11573–11578.
Dudek, S. M., and Bear, M. F. (1993). Bidirectional long-term modification of synaptic effectiveness in the adult and immature hippocampus. J. Neurosci. 13, 2910–2918.
Egashira, Y., Tanaka, T., Soni, P., Sakuragi, S., Tominaga-Yoshino, K., and Ogura, A. (2010). Involvement of the p75(NTR) signaling pathway in persistent synaptic suppression coupled with synapse elimination following repeated long-term depression induction. J. Neurosci. Res. 88, 3433–3446.
Embi, N., Rylatt, D. B., and Cohen, P. (1980). Glycogen synthase kinase-3 from rabbit skeletal muscle. Separation from cyclic-AMP-dependent protein kinase and phosphorylase kinase. Eur. J. Biochem. 107, 519–527.
Engel, T., Hernandez, F., Avila, J., and Lucas, J. J. (2006). Full reversal of Alzheimer's disease-like phenotype in a mouse model with conditional overexpression of glycogen synthase kinase-3. J. Neurosci. 26, 5083–5090.
Espada, S., Rojo, A. I., Salinas, M., and Cuadrado, A. (2009). The muscarinic M1 receptor activates Nrf2 through a signaling cascade that involves protein kinase C and inhibition of GSK-3beta: connecting neurotransmission with neuroprotection. J. Neurochem. 110, 1107–1119.
Gaiarsa, J. L., Caillard, O., and Ben-Ari, Y. (2002). Long-term plasticity at GABAergic and glycinergic synapses: mechanisms and functional significance. Trends Neurosci. 25, 564–570.
Garrido, J. L., Godoy, J. A., Alvarez, A., Bronfman, M., and Inestrosa, N. C. (2002). Protein kinase C inhibits amyloid beta peptide neurotoxicity by acting on members of the Wnt pathway. FASEB J. 16, 1982–1984.
Gillingwater, T. H., and Ribchester, R. R. (2001). Compartmental neurodegeneration and synaptic plasticity in the Wld(s) mutant mouse. J. Physiol. 534, 627–639.
Goto, Y., Yang, C. R., and Otani, S. (2010). Functional and dysfunctional synaptic plasticity in prefrontal cortex: roles in psychiatric disorders. Biol. Psychiatry 67, 199–207.
Hartigan, J. A., Xiong, W. C., and Johnson, G. V. (2001). Glycogen synthase kinase 3beta is tyrosine phosphorylated by PYK2. Biochem. Biophys. Res. Commun. 284, 485–489.
Hayashi, T., and Huganir, R. L. (2004). Tyrosine phosphorylation and regulation of the AMPA receptor by SRC family tyrosine kinases. J. Neurosci. 24, 6152–6160.
Hernandez, F., Borrell, J., Guaza, C., Avila, J., and Lucas, J. J. (2002). Spatial learning deficit in transgenic mice that conditionally over-express GSK-3beta in the brain but do not form tau filaments. J. Neurochem. 83, 1529–1533.
Hong, M., and Lee, V. M. (1997). Insulin and insulin-like growth factor-1 regulate tau phosphorylation in cultured human neurons. J. Biol. Chem. 272, 19547–19553.
Hooper, C., Killick, R., and Lovestone, S. (2008). The GSK3 hypothesis of Alzheimer's disease. J. Neurochem. 104, 1433–1439.
Hooper, C., Markevich, V., Plattner, F., Killick, R., Schofield, E., Engel, T., Hernandez, F., Anderton, B., Rosenblum, K., Bliss, T., Cooke, S. F., Avila, J., Lucas, J. J., Giese, K. P., Stephenson, J., and Lovestone, S. (2007). Glycogen synthase kinase-3 inhibition is integral to long-term potentiation. Eur. J. Neurosci. 25, 81–86.
Huang, Y. Y., Colino, A., Selig, D. K., and Malenka, R. C. (1992). The influence of prior synaptic activity on the induction of long-term potentiation. Science 255, 730–733.
Hughes, K., Nikolakaki, E., Plyte, S. E., Totty, N. F., and Woodgett, J. R. (1993). Modulation of the glycogen synthase kinase-3 family by tyrosine phosphorylation. EMBO J. 12, 803–808.
Hughes, K., Pulverer, B. J., Theocharous, P., and Woodgett, J. R. (1991). Baculovirus-mediated expression and characterisation of rat glycogen synthase kinase-3 beta, the mammalian homologue of the Drosophila melanogaster zeste-white 3sgg homeotic gene product. Eur. J. Biochem. 203, 305–311.
Hur, E. M., and Zhou, F. Q. (2010). GSK3 signalling in neural development. Nat. Rev. Neurosci. 11, 539–551.
Isaac, J. T., Nicoll, R. A., and Malenka, R. C. (1995). Evidence for silent synapses: implications for the expression of LTP. Neuron 15, 427–434.
Jope, R. S. (2011). Glycogen synthase kinase-3 in the etiology and treatment of mood disorders. Front. Mol. Neurosci. 4:16. doi: 10.3389/fnmol.2011.00016
Jo, J., Whitcomb, D. J., Olsen, K. M., Kerrigan, T. L., Lo, S. C., Bru-Mercier, G., Dickinson, B., Scullion, S., Sheng, M., Collingridge, G., and Cho, K. (2011). Abeta(1–42) inhibition of LTP is mediated by a signaling pathway involving caspase-3, Akt1 and GSK-3beta. Nat. Neurosci. 14, 545–547.
Kaidanovich-Beilin, O., and Woodgett, J. R. (2011). GSK-3: functional insights from cell biology and animal models. Front. Mol. Neurosci. 4:40. doi: 10.3389/fnmol.2011.00040
Kano, M., Hashimoto, K., and Tabata, T. (2008). Type-1 metabotropic glutamate receptor in cerebellar Purkinje cells: a key molecule responsible for long-term depression, endocannabinoid signalling and synapse elimination. Philos. Trans. R. Soc. Lond. B Biol. Sci. 363, 2173–2186.
Kasai, H., Fukuda, M., Watanabe, S., Hayashi-Takagi, A., and Noguchi, J. (2010). Structural dynamics of dendritic spines in memory and cognition. Trends Neurosci. 33, 121–129.
Kim, C. H., Chung, H. J., Lee, H. K., and Huganir, R. L. (2001a). Interaction of the AMPA receptor subunit GluR2/3 with PDZ domains regulates hippocampal long-term depression. Proc. Natl. Acad. Sci. U.S.A. 98, 11725–11730.
Kim, J. H., Anwyl, R., Suh, Y. H., Djamgoz, M. B., and Rowan, M. J. (2001b). Use-dependent effects of amyloidogenic fragments of (beta)-amyloid precursor protein on synaptic plasticity in rat hippocampus in vivo. J. Neurosci. 21, 1327–1333.
Kim, J. I., Lee, H. R., Sim, S. E., Beak, J., Yu, N. K., Choi, J. H., Ko, H. G., Park, S. W., Ahn, S. J., Choi, S. Y., Kim, H., Kim, K. H., Backx, P. H., Bradley, C. A., Kim, E., Jang, D. J., Lee, K., Kim, S. J., Zhuo, M., Collingridge, G. L., and Kaang, B. K. (2011). PI3Kγ is required for NMDAR-dependent long-term depression and behavioural flexibility. Nat. Neurosci. 14, 1447–1454.
Kim, L., Harwood, A., and Kimmel, A. R. (2002). Receptor-dependent and tyrosine phosphatase-mediated inhibition of GSK3 regulates cell fate choice. Dev. Cell 3, 523–532.
Kim, L., Liu, J., and Kimmel, A. R. (1999). The novel tyrosine kinase ZAK1 activates GSK3 to direct cell fate specification. Cell 99, 399–408.
Kimura, T., Yamashita, S., Nakao, S., Park, J. M., Murayama, M., Mizoroki, T., Yoshiike, Y., Sahara, N., and Takashima, A. (2008). GSK-3beta is required for memory reconsolidation in adult brain. PLoS One 3, e3540. doi: 10.1371/journal.pone.0003540
Kremer, A., Louis, J. V., Jaworski, T., and van Leuven, F. (2011). GSK3 and Alzeheimer's disease: facts and fiction. Front. Mol. Neurosci. 4:17. doi: 10.3389/fnmol.2011.00017
Lesort, M., Jope, R. S., and Johnson, G. V. (1999). Insulin transiently increases tau phosphorylation: involvement of glycogen synthase kinase-3beta and Fyn tyrosine kinase. J. Neurochem. 72, 576–584.
Li, M., Wang, X., Meintzer, M. K., Laessig, T., Birnbaum, M. J., and Heidenreich, K. A. (2000). Cyclic AMP promotes neuronal survival by phosphorylation of glycogen synthase kinase 3beta. Mol. Cell. Biol. 20, 9356–9363.
Li, S., Hong, S., Shepardson, N. E., Walsh, D. M., Shankar, G. M., and Selkoe, D. (2009). Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron 62, 788–801.
Li, Z., Jo, J., Jia, J. M., Lo, S. C., Whitcomb, D. J., Jiao, S., Cho, K., and Sheng, M. (2010). Caspase-3 activation via mitochondria is required for long-term depression and AMPA receptor internalization. Cell 141, 859–871.
Liao, D., Hessler, N. A., and Malinow, R. (1995). Activation of postsynaptically silent synapses during pairing-induced LTP in CA1 region of hippocampal slice. Nature 375, 400–404.
Lochhead, P. A., Kinstrie, R., Sibbet, G., Rawjee, T., Morrice, N., and Cleghon, V. (2006). A chaperone-dependent GSK3beta transitional intermediate mediates activation-loop autophosphorylation. Mol. Cell 24, 627–633.
Lovestone, S., Killick, R., Di Forti, M., and Murray, R. (2007). Schizophrenia as a GSK-3 dysregulation disorder. Trends Neurosci. 30, 142–149.
Luscher, B., Fuchs, T., and Kilpatrick, C. L. (2011). GABAA receptor trafficking-mediated plasticity of inhibitory synapses. Neuron 70, 385–409.
Luthi, A., Chittajallu, R., Duprat, F., Palmer, M. J., Benke, T. A., Kidd, F. L., Henley, J. M., Isaac, J. T., and Collingridge, G. L. (1999). Hippocampal LTD expression involves a pool of AMPARs regulated by the NSF-GluR2 interaction. Neuron 24, 389–399.
Malinow, R., and Tsien, R. W. (1990). Presynaptic enhancement shown by whole-cell recordings of long-term potentiation in hippocampal slices. Nature 346, 177–180.
Man, H. Y., Lin, J. W., Ju, W. H., Ahmadian, G., Liu, L., Becker, L. E., Sheng, M., and Wang, Y. T. (2000). Regulation of AMPA receptor-mediated synaptic transmission by clathrin-dependent receptor internalization. Neuron 25, 649–662.
Manahan-Vaughan, D., Kulla, A., and Frey, J. U. (2000). Requirement of translation but not transcription for the maintenance of long-term depression in the CA1 region of freely moving rats. J. Neurosci. 20, 8572–8576.
Manning, G., Whyte, D. B., Martinez, R., Hunter, T., and Sudarsanam, S. (2002). The protein kinase complement of the human genome. Science 298, 1912–1934.
Maric, H. M., Mukherjee, J., Tretter, V., Moss, S. J., and Schindelin, H. (2011). Gephyrin-mediated GABA(A) and glycine receptor clustering relies on a common binding site. J. Biol. Chem. 286, 42105–42114.
Marsden, K. C., Beattie, J. B., Friedenthal, J., and Carroll, R. C. (2007). NMDA receptor activation potentiates inhibitory transmission through GABA receptor-associated protein-dependent exocytosis of GABA(A) receptors. J. Neurosci. 27, 14326–14337.
Marsden, K. C., Shemesh, A., Bayer, K. U., and Carroll, R. C. (2010). Selective translocation of Ca2+/calmodulin protein kinase IIalpha (CaMKIIalpha) to inhibitory synapses. Proc. Natl. Acad. Sci. U.S.A. 107, 20559–20564.
Medina, M., and Wandosell, F. (2011). Deconstructing GSK-3: the fine regulation of its activity. Int. J. Alzheimers Dis. 2011, 479249.
Mendez, P., and Bacci, A. (2011). Assortment of GABAergic plasticity in the cortical interneuron melting pot. Neural Plast. 2011, 976856.
Morfini, G., Szebenyi, G., Brown, H., Pant, H. C., Pigino, G., DeBoer, S., Beffert, U., and Brady, S. T. (2004). A novel CDK5-dependent pathway for regulating GSK3 activity and kinesin-driven motility in neurons. EMBO J. 23, 2235–2245.
Morfini, G., Szebenyi, G., Elluru, R., Ratner, N., and Brady, S. T. (2002). Glycogen synthase kinase 3 phosphorylates kinesin light chains and negatively regulates kinesin-based motility. EMBO J. 21, 281–293.
Muir, J., Arancibia-Carcamo, I. L., MacAskill, A. F., Smith, K. R., Griffin, L. D., and Kittler, J. T. (2010). NMDA receptors regulate GABAA receptor lateral mobility and clustering at inhibitory synapses through serine 327 on the gamma2 subunit. Proc. Natl. Acad. Sci. U.S.A. 107, 16679–16684.
Mukherjee, J., Kretschmannova, K., Gouzer, G., Maric, H. M., Ramsden, S., Tretter, V., Harvey, K., Davies, P. A., Triller, A., Schindelin, H., and Moss, S. J. (2011). The Residence Time of GABAARs at Inhibitory Synapses Is Determined by Direct Binding of the Receptor {alpha}1 Subunit to Gephyrin. J. Neurosci. 31, 14677–14687.
Mulkey, R. M., Endo, S., Shenolikar, S., and Malenka, R. C. (1994). Involvement of a calcineurin/ inhibitor-1 phosphatase cascade in hippocampal long-term depression. Nature 369, 486–488.
Naska, S., Park, K. J., Hannigan, G. E., Dedhar, S., Miller, F. D., and Kaplan, D. R. (2006). An essential role for the integrin-linked kinase-glycogen synthase kinase-3 beta pathway during dendrite initiation and growth. J. Neurosci. 26, 13344–13356.
Nicolas, C. S., Peineau, S., Amici, M., Csaba, Z., Fafouri, A., Javalet, C., Collett, V. J., Hildebrandt, L., Seaton, G., Choi, S. L., Sim, S. E., Bradley, C., Lee, K., Zhuo, M., Kaang, B. K., Gressens, P., Dournaud, P., Fitzjohn, S. M., Bortolotto, Z. A., Cho, K., and Collingridge, G. L. (2012). The JAK/STAT pathway is involved in synaptic plasticity. Neuron 73, 374–390.
O'Driscoll, C., Wallace, D., and Cotter, T. G. (2007). bFGF promotes photoreceptor cell survival in vitro by PKA-mediated inactivation of glycogen synthase kinase 3beta and CREB-dependent Bcl-2 up-regulation. J. Neurochem. 103, 860–870.
Ortega, F., Perez-Sen, R., Morente, V., Delicado, E. G., and Miras-Portugal, M. T. (2010). P2X7, NMDA and BDNF receptors converge on GSK3 phosphorylation and cooperate to promote survival in cerebellar granule neurons. Cell. Mol. Life Sci. 67, 1723–1733.
Peineau, S., Nicolas, C. S., Bortolotto, Z. A., Bhat, R. V., Ryves, W. J., Harwood, A. J., Dournaud, P., Fitzjohn, S. M., and Collingridge, G. L. (2009). A systematic investigation of the protein kinases involved in NMDA receptor-dependent LTD: evidence for a role of GSK-3 but not other serine/threonine kinases. Mol. Brain 2, 22.
Peineau, S., Taghibiglou, C., Bradley, C., Wong, T. P., Liu, L., Lu, J., Lo, E., Wu, D., Saule, E., Bouschet, T., Matthews, P., Isaac, J. T., Bortolotto, Z. A., Wang, Y. T., and Collingridge, G. L. (2007). LTP inhibits LTD in the hippocampus via regulation of GSK3beta. Neuron 53, 703–717.
Pfeiffer, F., Graham, D., and Betz, H. (1982). Purification by affinity chromatography of the glycine receptor of rat spinal cord. J. Biol. Chem. 257, 9389–9393.
Proctor, D. T., Coulson, E. J., and Dodd, P. R. (2011). Post-synaptic scaffolding protein interactions with glutamate receptors in synaptic dysfunction and Alzheimer's disease. Prog. Neurobiol. 93, 509–521.
Quartarone, A., and Pisani, A. (2011). Abnormal plasticity in dystonia: disruption of synaptic homeostasis. Neurobiol. Dis. 42, 162–170.
Rabacchi, S., Bailly, Y., Delhaye-Bouchaud, N., and Mariani, J. (1992). Involvement of the N-methyl D-aspartate (NMDA) receptor in synapse elimination during cerebellar development. Science 256, 1823–1825.
Russo, S. J., Dietz, D. M., Dumitriu, D., Morrison, J. H., Malenka, R. C., and Nestler, E. J. (2010). The addicted synapse: mechanisms of synaptic and structural plasticity in nucleus accumbens. Trends Neurosci. 33, 267–276.
Setou, M., Seog, D. H., Tanaka, Y., Kanai, Y., Takei, Y., Kawagishi, M., and Hirokawa, N. (2002). Glutamate-receptor-interacting protein GRIP1 directly steers kinesin to dendrites. Nature 417, 83–87.
Shankar, G. M., Li, S., Mehta, T. H., Garcia-Munoz, A., Shepardson, N. E., Smith, I., Brett, F. M., Farrell, M. A., Rowan, M. J., Lemere, C. A., Regan, C. M., Walsh, D. M., Sabatini, B. L., and Selkoe, D. J. (2008). Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat. Med. 14, 837–842.
Shelly, M., Cancedda, L., Lim, B. K., Popescu, A. T., Cheng, P. L., Gao, H., and Poo, M. M. (2011). Semaphorin3A regulates neuronal polarization by suppressing axon formation and promoting dendrite growth. Neuron 71, 433–446.
Shelly, M., Lim, B. K., Cancedda, L., Heilshorn, S. C., Gao, H., and Poo, M. M. (2010). Local and long-range reciprocal regulation of cAMP and cGMP in axon/dendrite formation. Science 327, 547–552.
Shipton, O. A., Leitz, J. R., Dworzak, J., Acton, C. E., Tunbridge, E. M., Denk, F., Dawson, H. N., Vitek, M. P., Wade-Martins, R., Paulsen, O., and Vargas-Caballero, M. (2011). Tau protein is required for amyloid {beta}-induced impairment of hippocampal long-term potentiation. J. Neurosci. 31, 1688–1692.
Song, B., Lai, B., Zheng, Z., Zhang, Y., Luo, J., Wang, C., Chen, Y., Woodgett, J. R., and Li, M. (2010). Inhibitory phosphorylation of GSK-3 by CaMKII couples depolarization to neuronal survival. J. Biol. Chem. 285, 41122–41134.
Sutherland, C., Leighton, I. A., and Cohen, P. (1993). Inactivation of glycogen synthase kinase-3 beta by phosphorylation: new kinase connections in insulin and growth-factor signalling. Biochem. J. 296(Pt 1), 15–19.
Szatmari, E., Habas, A., Yang, P., Zheng, J. J., Hagg, T., and Hetman, M. (2005). A positive feedback loop between glycogen synthase kinase 3beta and protein phosphatase 1 after stimulation of NR2B NMDA receptors in forebrain neurons. J. Biol. Chem. 280, 37526–37535.
Thornton, T. M., Pedraza-Alva, G., Deng, B., Wood, C. D., Aronshtam, A., Clements, J. L., Sabio, G., Davis, R. J., Matthews, D. E., Doble, B., and Rincon, M. (2008). Phosphorylation by p38 MAPK as an alternative pathway for GSK3beta inactivation. Science 320, 667–670.
Tretter, V., Jacob, T. C., Mukherjee, J., Fritschy, J. M., Pangalos, M. N., and Moss, S. J. (2008). The clustering of GABA(A) receptor subtypes at inhibitory synapses is facilitated via the direct binding of receptor alpha 2 subunits to gephyrin. J. Neurosci. 28, 1356–1365.
Tretter, V., Kerschner, B., Milenkovic, I., Ramsden, S. L., Ramerstorfer, J., Saiepour, L., Maric, H. M., Moss, S. J., Schindelin, H., Harvey, R. J., Sieghart, W., and Harvey, K. (2011). Molecular basis of the {gamma}-aminobutyric acid A receptor {alpha}3 subunit interaction with the clustering protein gephyrin. J. Biol. Chem. 286, 37702–37711.
Tyagarajan, S. K., Ghosh, H., Yevenes, G. E., Nikonenko, I., Ebeling, C., Schwerdel, C., Sidler, C., Zeilhofer, H. U., Gerrits, B., Muller, D., and Fritschy, J. M. (2011). Regulation of GABAergic synapse formation and plasticity by GSK3beta-dependent phosphorylation of gephyrin. Proc. Natl. Acad. Sci. U.S.A. 108, 379–384.
Valerio, A., Ghisi, V., Dossena, M., Tonello, C., Giordano, A., Frontini, A., Ferrario, M., Pizzi, M., Spano, P., Carruba, M. O., and Nisoli, E. (2006). Leptin increases axonal growth cone size in developing mouse cortical neurons by convergent signals inactivating glycogen synthase kinase-3beta. J. Biol. Chem. 281, 12950–12958.
van Spronsen, M., and Hoogenraad, C. C. (2010). Synapse pathology in psychiatric and neurologic disease. Curr. Neurol. Neurosci. Rep. 10, 207–214.
Vyazovskiy, V. V., Cirelli, C., Pfister-Genskow, M., Faraguna, U., and Tononi, G. (2008). Molecular and electrophysiological evidence for net synaptic potentiation in wake and depression in sleep. Nat. Neurosci. 11, 200–208.
Wan, Q., Xiong, Z. G., Man, H. Y., Ackerley, C. A., Braunton, J., Lu, W. Y., Becker, L. E., MacDonald, J. F., and Wang, Y. T. (1997). Recruitment of functional GABA(A) receptors to postsynaptic domains by insulin. Nature 388, 686–690.
Wang, J., Liu, S., Haditsch, U., Tu, W., Cochrane, K., Ahmadian, G., Tran, L., Paw, J., Wang, Y., Mansuy, I., Salter, M. M., and Lu, Y. M. (2003a). Interaction of calcineurin and type-A GABA receptor gamma 2 subunits produces long-term depression at CA1 inhibitory synapses. J. Neurosci. 23, 826–836.
Wang, Q., Liu, L., Pei, L., Ju, W., Ahmadian, G., Lu, J., Wang, Y., Liu, F., and Wang, Y. T. (2003b). Control of synaptic strength, a novel function of Akt. Neuron 38, 915–928.
Wang, Y., Ju, W., Liu, L., Fam, S., D'Souza, S., Taghibiglou, C., Salter, M., and Wang, Y. T. (2004). alpha-Amino-3-hydroxy-5-methylisoxazole-4-propionic acid subtype glutamate receptor (AMPAR) endocytosis is essential for N-methyl-D-aspartate-induced neuronal apoptosis. J. Biol. Chem. 279, 41267–41270.
Wei, J., Liu, W., and Yan, Z. (2010). Regulation of AMPA receptor trafficking and function by glycogen synthase kinase 3. J. Biol. Chem. 285, 26369–26376.
Wera, S., and Hemmings, B. A. (1995). Serine/threonine protein phosphatases. Biochem. J. 311(Pt1), 17–29.
Witton, J., Brown, J. T., Jones, M. W., and Randall, A. D. (2010). Altered synaptic plasticity in the mossy fibre pathway of transgenic mice expressing mutant amyloid precursor protein. Mol. Brain 3, 32.
Zhang, H. H., Lipovsky, A. I., Dibble, C. C., Sahin, M., and Manning, B. D. (2006). S6K1 regulates GSK3 under conditions of mTOR-dependent feedback inhibition of Akt. Mol. Cell 24, 185–197.
Zhao, Z., Wang, Z., Gu, Y., Feil, R., Hofmann, F., and Ma, L. (2009). Regulate axon branching by the cyclic GMP pathway via inhibition of glycogen synthase kinase 3 in dorsal root ganglion sensory neurons. J. Neurosci. 29, 1350–1360.
Zhu, L. Q., Wang, S. H., Liu, D., Yin, Y. Y., Tian, Q., Wang, X. C., Wang, Q., Chen, J. G., and Wang, J. Z. (2007). Activation of glycogen synthase kinase-3 inhibits long-term potentiation with synapse-associated impairments. J. Neurosci. 27, 12211–12220.
Keywords: GSK-3 (glycogen synthase kinase-3), caspase 3, Akt (PKB, protein kinase B), synaptic plasticity, LTP (long-term potentiation), LTD (long-term depression), AMPAR (AMPA receptor), NMDAR (NMDA receptor)
Citation: Bradley CA, Peineau S, Taghibiglou C, Nicolas CS, Whitcomb DJ, Bortolotto ZA, Kaang B, Cho K, Wang Y and Collingridge GL (2012) A pivotal role of GSK-3 in synaptic plasticity. Front. Mol. Neurosci. 5:13. doi: 10.3389/fnmol.2012.00013
Received: 17 November 2011; Accepted: 31 January 2012;
Published online: 15 February 2012.
Edited by:
Jim R. Woodgett, Mount Sinai Hospital, CanadaReviewed by:
Jim R. Woodgett, Mount Sinai Hospital, CanadaKenichi Okamoto, Samuel Lunenfeld Research Institute of Mount Sinai Hospital, Canada
Copyright: © 2012 Bradley, Peineau, Taghibiglou, Nicolas, Whitcomb, Bortolotto, Kaang, Cho, Wang and Collingridge. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Graham L. Collingridge, MRC Centre for Synaptic Plasticity, University of Bristol, BS8 1TD, UK. e-mail:Zy5sLmNvbGxpbmdyaWRnZUBicmlzLmFjLnVr