 Jing A. Wen
Jing A. Wen Alison L. Barth*
Alison L. Barth*- Department of Biological Sciences and Center for the Neural Basis of Cognition, Carnegie Mellon University, Pittsburgh, PA, USA
Activity- or experience-dependent plasticity has been associated with the trafficking of calcium-permeable α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (CP-AMPARs) in a number of experimental systems. In some cases it has been shown that CP-AMPARs are only transiently present and can be removed in an activity-dependent manner. Here we test the hypothesis that the presence of CP-AMPARs confers instability onto recently potentiated synapses. Previously we have shown that altered sensory input (single-whisker experience; SWE) strengthens layer 4-2/3 excitatory synapses in mouse primary somatosensory cortex, in part by the trafficking of CP-AMPARs. Both in vivo and in vitro, this potentiation is labile, and can be depressed by N-Methyl-D-aspartate receptor (NMDAR)-activation. In the present study, the role of CP-AMPARs in conferring this synaptic instability after in vivo potentiation was evaluated. We develop an assay to depress the strength of individual layer 4-2/3 excitatory synapses after SWE, using a strontium (Sr++)-replaced artificial cerebrospinal fluid (ACSF) solution (Sr-depression). This method allows disambiguation of changes in quantal amplitude (a post-synaptic measure) from changes in event frequency (typically a presynaptic phenomenon). Presynaptic stimulation paired with post-synaptic depolarization in Sr++ lead to a rapid and significant reduction in EPSC amplitude with no change in event frequency. Sr-depression at recently potentiated synapses required NMDARs, but could still occur when CP-AMPARs were not present. As a further dissociation between the presence of CP-AMPARs and Sr-depression, CP-AMPARs could be detected in some cells from control, whisker-intact animals, although Sr-depression was never observed. Taken together, our findings suggest that CP-AMPARs are neither sufficient nor necessary for synaptic depression after in vivo plasticity in somatosensory cortex. This article is part of a Special Issue entitled “Calcium permeable AMPARs in synaptic plasticity and disease.”
Introduction
The activity-dependent trafficking of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR)-type glutamate receptors to enhance synaptic strength has been observed in many experimental preparations (Shi et al., 1999; Takahashi et al., 2003; Rumpel et al., 2005; Bellone and Luscher, 2006; Clem and Barth, 2006; Plant et al., 2006; Sutton et al., 2006; Clem and Huganir, 2010; Lante et al., 2011). AMPARs are tetrameric receptors, the vast majority of which contain an RNA-edited form of the GluA2 subunit that renders the complex impermeable to calcium (calcium-impermeable AMPARs; CI-AMPARS). However, under some circumstances, it has been possible to detect the presence of calcium-permeable AMPARs (CP-AMPARs) at synapses, based upon their unique electrophysiological and pharmacological properties (Man, 2011). These receptors typically lack GluA2, and may be homomers of GluA1. The novel subunit composition of these receptors, as well as their unusual calcium-permeability, has made these receptors the source of great interest in understanding the mechanisms that modulate synaptic function in health and disease (Liu and Zukin, 2007 and Isaac et al., 2007). For example, it has been hypothesized that CP-AMPARS can provide a non-contingent source of Ca++ entry that might regulate excitotoxicity or subsequent plasticity (Deng et al., 2003; Wiltgen et al., 2010).
In a number of cases, the trafficking of native CP-AMPARs has been associated with recent synaptic potentiation (Bellone and Luscher, 2006; Clem and Barth, 2006; Plant et al., 2006; Clem and Huganir, 2010), suggesting that these receptors may play an important role in the initiation or maturation of synaptic plasticity. For example, the presence of CP-AMPARs can be detected after altered sensory experience at layer 4-2/3 synapses in somatosensory cortex (Clem and Barth, 2006), as well as in the amygdala after fear conditioning (Clem and Huganir, 2010). However, other reports indicate that the trafficking of CP-AMPARs does not occur during in vitro plasticity induction (Adesnik and Nicoll, 2007; Gray et al., 2007). Thus, the requirement for CP-AMPARs in synaptic potentiation and depression is the center of debate.
A model that has some experimental support is that CP-AMPARs are specifically responsible for rapid and early, but not long-term, changes in synaptic strength. They may serve as “placeholders” for eventual substitution by CI-AMPARs (Plant et al., 2006; Clem and Huganir, 2010; Yang et al., 2010), or might provide a substrate to rapidly retune synaptic strength to maintain some dynamic range of synaptic drive (Thiagarajan et al., 2005; Sutton et al., 2006; Hou et al., 2008). Indeed, a number of reports indicate that the removal of CP-AMPARs can be triggered by synaptic stimulation or in vivo activation, leading to synaptic depression (Bellone and Luscher, 2006; Ho et al., 2007; Clem and Huganir, 2010; Yang et al., 2010; Lante et al., 2011). Taken together, these data suggest that CP-AMPARs might serve as an intermediate step in synaptic modifications, where their persistence or removal can determine how long-lasting synaptic strength may be.
The addition of CP-AMPARs to layer 4-2/3 synapses during experience-dependent plasticity in somatosensory cortex has been well-established (Clem and Barth, 2006; Clem et al., 2008; Wen and Barth, 2011). However, increasing evidence indicates that they are not required for this form of plasticity, since synaptic strengthening can be observed without CP-AMPARS, either at later developmental ages or in transgenic mice that are mutant for the GluA2 trafficking molecule PICK-1(Clem et al., 2010; Wen and Barth, 2011). Despite the fact that they are not required, their presence might nonetheless confer specific properties onto recently strengthened synapses, such as the ability to erase prior modifications. This has been proposed from previous work, and has important therapeutic implications for reducing pathological changes in synaptic strength in addiction, seizure disorders, or anxiety disorders (Bellone and Luscher, 2006; Rakhade et al., 2008; Clem and Huganir, 2010).
Here we test the hypothesis that CP-AMPARs are associated with synaptic lability, whereby recently modified synapses might be susceptible to synaptic weakening due to the removal of CP-AMPARs. Previous work from our lab has established that plasticity at excitatory layer 4-2/3 synapses undergoes an early, N-Methyl-D-aspartate (NMDAR)-dependent phase of synaptic strengthening, followed by a later, NMDAR-dependent phase of synaptic weakening (Clem et al., 2008). We show that this NMDAR-dependent depression can be triggered at individual synaptic contacts in vitro, using a novel protocol that triggers a reduction of AMPAR-EPSCs by pairing post-synaptic depolarization with presynaptic stimulation in a Sr++ based artificial cerebrospinal fluid (ACSF) solution.
This form of synaptic depression only occurs at previously potentiated synapses from animals with altered whisker input, requires NMDAR-activation, and can occur in cells where CP-AMPARs are undetectable or have been pharmacologically blocked. Further dissociating a role for CP-AMPARs in this phenomenon, Sr-depression was never observed in control, whisker-intact animals although CP-AMPARs could be detected at layer 4-2/3 synapses in some cells. Thus, we conclude that CP-AMPARs do not necessarily confer synaptic lability at layer 4-2/3 synapses, and that they are not essential for the induction or expression of synaptic depression in this assay.
Methods
Animals
Wild-type or heterozygous mice (males and females) from a fosGFP [1–3 line, C57Bl6 background (Barth et al., 2004)] transgenic line at postnatal days 13 or 14 (P13–14) were used. Bilateral whisker deprivation was performed where all but the D1 whisker on one side were removed (Glazewski et al., 2007). Animals were returned to their home cages for 24 h before recording. Control animals were whisker-intact littermates of the deprived animals. Since there was no significant difference between control wild-type C57Bl6 and fosGFP+/−, data from these animals were grouped. Recordings in control animals were not restricted to the D1 barrel column, since all columns were equivalent in whisker-intact animals. The barrel column representing the “spared” D1 whisker was identified by enhanced fosGFP expression and relative position to the hippocampus in acute brain slices.
Whole-Cell Recording
Animals were anesthetized with isoflurane and decapitated. Coronal slices (350 μ m thick) were vibratome sectioned in ACSF at 2–6°C composed of (mM): 119 NaCl, 2.5 KCl, 2.5 CaCl2, 1–1.3 MgSO4, 1 NaH2PO4, 26.2 NaHCO3, 11 glucose and equilibrated with 95/5% O2/CO2. Slices were maintained and whole-cell recordings were carried out at room temperature in ACSF. In all experiments, post-synaptic glutamatergic responses from layer 2/3 pyramidal neurons within the same barrel column were pharmacologically isolated using the GABAA antagonist picrotoxin (Ptx; 50 μ M) in the bath solution. Somata of lower layer 2/3 pyramidal neurons in barrel cortex were targeted for whole-cell recording with borosilicate glass electrodes with a resistance of 4–8 MΩ. Electrode internal solution was composed of (in mM): 130 cesium-gluconate, 10 HEPES, 0.5 EGTA, 8 NaCl, 4 Mg-ATP and 0.4 Na-GTP, 5 QX-314, at pH 7.25–7.30, 290–300 mOsm and contained trace amounts of the fluorescent dye Alexa-568. Pyramidal cell identity was confirmed after the recording session by pyramidal soma morphology and the presence of dendritic spines. Only cells with Rseries ≤ 20 MΩ and Rinput ≥ 200 MΩ, where changes in either measurement were less than 20% were included for analysis.
Stimulation of layer 4 afferents was applied at 0.1 Hz by placing glass monopolar electrodes in the center of a layer 4 barrel, and stimulation intensity was adjusted to isolate monosynaptic EPSCs without recurrent activity. Electrophysiological data was acquired by Multiclamp 700A (Axon Instruments, Foster City, CA) and a National Instruments acquisition interface. The data was filtered at 3 kHz and digitized at 10 kHz and collected by Igor Pro 6.0 (Wavemetrics, Lake Oswego, Oregon). Extracellular simulation was controlled by a Master-8 (A.M.P.I, Israel).
Depression of Evoked Sr-EPSCs
To measure the amplitude of stimulus-evoked miniature AMPAR-EPSCs, Sr++ (3 mM) was substituted for Ca++ in ACSF to drive asynchronous glutamate release. Layer 2/3 pyramidal neurons were voltage-clamped at −70 mV, where the contribution of NMDARs to the EPSC is minimal. This was experimentally verified with ACSF containing 0.5 mM Mg++, where the mean Sr-EPSC amplitude was not altered by bath application of D-APV. To induce synaptic depression of Sr-EPSCs, layer 2/3 pyramidal neurons were voltage-clamped at −70 mV for ≥5 min (baseline response), then to −20 mV for 5 min, returning to −70 mV holding potential (post-response). Stimulation frequency (0.1 Hz) and intensity were not altered during the experiment, which allowed a comparison of event frequency before and after pairing.
The evoked response has an initial synchronous component (∼50 ms post the stimulus artifact) which was excluded in the analysis. Isolated, asynchronous events that occurred from 50 to 500 ms after the stimulus were manually selected and analyzed using Minianalysis software (Synaptosoft, Inc. Decatur, GA). The detection threshold for events was set at 2× RMS noise (usually around 4–5 pA) and data were filtered with a low-pass filter at 1 kHz. Approximately 50–100 events were randomly selected from the pre- and post-response and then grouped to generate average traces. Within-cell comparisons were made between the events from the baseline and post-responses for each cell. An average trace was generated from grouping 50–100 random events from each cell. Selected events were grouped across all cells within an experimental condition and ranked ordered to generate cumulative distribution plots.
Decay time of individual Sr-EPSC was analyzed online as the difference between the time at the peak and 1/3 of the peak and the mean decay time was obtained by averaging all selected events from all cells within a group.
AMPA EPSC Measurement and Rectification Index (RI)
To isolate the multiquantal AMPAR- EPSCs, D-APV (50 μ M) and Ptx were included in the bath solution. Spermine (100 μ M) was included in the internal solution to avoid washout of endogenous polyamines. Layer 2/3 pyramidal neurons were voltage-clamped at −70 mV and stimulus intensity was adjusted until a clear monosynaptic response (2–5 ms latency, consistent across trials for a given response) was visible for every sweep. For holding potentials at −70, 0, 40 mV, 10–20 sweeps were collected and averaged. The rectification index (RI) was calculated based on the following formula:
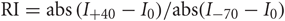
I−70, I+40, and I0 refer to the peak amplitude of the averaged responses for a given cell. If there is no rectification at positive holding potentials or the current-voltage relationship is linear, the RI in these measurements should be 4/7 (0.57).
NASPM/PhTx Application
1-Naphthylacetyl spermine trihydrochloride (NASPM, 50 μ M) or Philanthotoxin (PhTx, 10uM) were applied in the bath (containing D-APV) to assay the contribution of CP-AMPARs. Antagonist was applied for at least 10 min while the cell was being held at −70 mV, and the amplitude of the post-drug response was calculated by averaging 10–20 sweeps immediately prior to drug application versus 10 min after the onset of application. Because NASPM/PhTx is hard to wash out, data from only one cell per slice was collected. In some cases, AMPA-EPSCs increased in amplitude after NASPM application; data from these cells was included in our analysis.
To verify the reliability of using NASPM to block CP-AMPARs, the RI before and after NASPM application (10 min after wash-in) was compared. Synaptic responses from recurrent excitatory synapses onto layer 5 pyramidal cells of young postnatal C57Bl6 mice (P9–11) were obtained by stimulating layer 5. Consistent with the finding that CP-AMPARs are highly expressed around this developmental age in layer 5 (Brill and Huguenard, 2008), we observed that some AMPAR-EPSCs were rectifying. EPSC amplitude and RI before and after NASPM application was determined.
The effect of NASPM on Sr-EPSC amplitude was also evaluated. For within-cell comparisons, Sr-EPSCs were collected for ∼5 min before wash-in of NASPM-containing Sr-ACSF. Only one cell per slice was included for such NASPM wash-in experiments. For across cell comparisons of Sr-EPSC amplitude, slices were bathed in NASPM-containing ACSF for at 20–120 min with afferent stimulation before analysis of Sr-EPSC amplitude.
In experiments where the requirement of CP-AMPARs in Sr-depression was tested, slices were bathed in NASPM-containing ACSF for 20–120 min with some afferent stimulation before recordings to allow complete drug diffusion and blockade of CP-AMPARs.
Within-Cell Recording of RI and Sr-Depression
To more accurately explore the necessity of CP-AMPARs in Sr-depression, the RI was examined and then Sr-depression was induced in the same cell. In these experiments, AMPA-EPSCs were recorded at −70, 0, and +40 mV in Ca++ based ACSF, then a Sr++ based ACSF was washed in and Sr-depression was induced as described above. In a subset of cells, the Sr-based ACSF was washed out after the Sr-depression protocol, and Ca-ACSF was reapplied to obtain a second, post-depression RI measurement.
Statistics
Specific statistical tests used are indicated in the results. For Sr-EPSC amplitude comparisons before and after Sr-depression or NASPM treatment within the same cell and all other non-pair wise comparisons between two conditions (control vs. SWE), a non-parametric Mann–Whitney U-test (two-tailed) was used. For Sr-EPSC event frequency comparisons before and after Sr-depression, NASPM treatment in control and SWE conditions, and RI comparisons within the same cell before and after some manipulation, a paired t-test was used. For comparisons of distribution of Sr-EPSC amplitude before and after pairing for Sr-depression experiments, a Kolmogorov–Smirnov test was used. Summary data are presented as mean ± sem.
Results
Previous work has shown that modified whisker input, where all but a single-whisker (single-whisker experience; SWE) has been removed from one side of the mouse face, leads to the potentiation of synapses at layer 4-layer 2/3 excitatory inputs in the neocortical representation of the spared whisker. This potentiation can be accompanied by an increase in presence of CP-AMPARs, determined by their electrophysiological and pharmacological properties. The trafficking of CP-AMPARs is associated with age and input identity, where they are not implicated for plasticity induced at later developmental ages (when SWE begins at P13 or older ages) or at layer 2/3-layer 2/3 inputs (Wen and Barth, 2011).
Assays to demonstrate the experience-dependent increase in excitatory synaptic strength have relied upon a method to isolate the post-synaptic response in a pathway-specific manner, using an ACSF where Sr++ replaces Ca++. Under these conditions, neurotransmitter release is triggered by electrical stimulation to a specific input, but vesicle release is slowed such that the post-synaptic response to individual quanta can be evaluated (Goda and Stevens, 1994; Xu-Friedman and Regehr, 1999). In previous analyses (Clem and Barth, 2006; Clem et al., 2008, 2010; Wen and Barth, 2011), the AMPAR-EPSC was pharmacologically isolated from NMDAR currents by the application of D-APV. However, during the course of our investigations, we discovered that when both NMDAR- and AMPAR-mediated currents were present, there was sometimes a voltage-dependent run-down in the amplitude of average Sr-EPSC for a given cell. This run-down was only present at layer 4-2/3 synapses from SWE-treated animals, suggesting that it might be related to the recent potentiation at these synapses.
Sr-Depression is Induced by a Modest Post-Synaptic Depolarization
We formalized a method to examine this synaptic lability, named Sr-depression, by comparing mean Sr-EPSC amplitude before and after post-synaptic depolarization. We use the term Sr-depression to indicate specifically that this depression was not what has typically been considered “short-term depression” in other studies, since its onset is immediate and it is stable for many minutes following pairing. Experiments were carried out in acute brain slices from SWE-treated animals at postnatal day 13 (P13), a time when experience-dependent plasticity is pronounced (Figures 1A,B; Wen and Barth, 2011). Typically, Sr-EPSC amplitude from a given cell was calculated from the average of a 50–100 individual, well-isolated events (Figure 1C). The mean amplitude of these events was constant over the recording period when the post-synaptic cell is maintained at hyperpolarized holding potentials. Sr-EPSCs are primarily mediated by AMPARs, since application of NMDAR-antagonists does not change Sr-EPSC amplitude at hyperpolarized potentials when NMDARs exhibit a characteristic Mg-dependent voltage block.
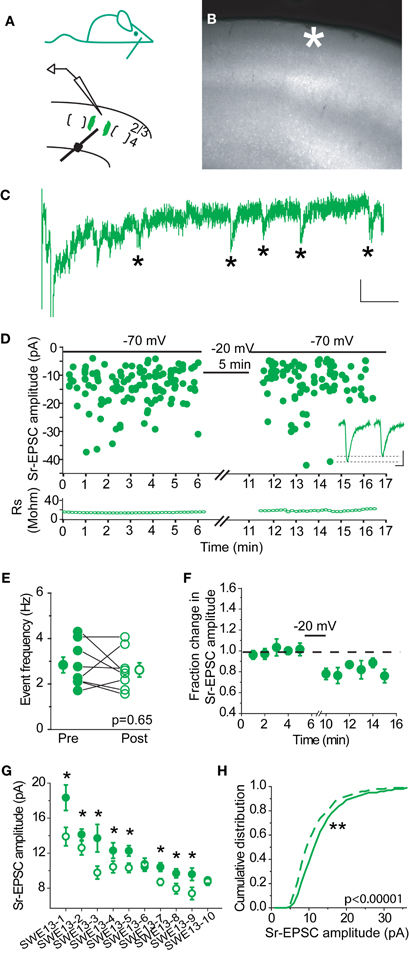
Figure 1. Sr-depression can be elicited at layer 4-2/3 synapses from SWE-treated mice. (A) Schematic of an SWE animal (top) and recording configuration in the spared barrel column (bottom). (B) Fluorescence image of a slice that contains the spared barrel column, visualized by expression of GFP fluorescence in a fosGFP transgenic mouse (*). (C) An example Sr-EPSC trace (* indicates individual Sr-EPSC event). Scale: 10 pA, 50 ms. (D) Scatter plot of individual Sr-EPSC amplitudes before and after Sr-depression induction, in a SWE-treated animal. Inset: average traces of Sr-EPSCs before (left) and after (right) Sr-depression induction. Scale bar: 5pA, 5 ms. Bottom: electrode series resistance from the same cell, which does not change between baseline vs. post-pairing window. (E) Sr-EPSC event frequency does not change after Sr-depression induction. (F) Mean change in Sr-EPSC amplitude normalized to the baseline period, for the eight cells that showed significant Sr-depression. (G) Comparison of Sr-EPSC amplitude between pre- (filled circle, green) and post-pairing window (open circle, green) for individual cells. Cells were rank ordered according to their mean amplitude of the baseline responses. Mean ± sem is plotted for each cell. *p < 0.05 by Mann–Whitney U-test. (H) Cumulative distribution histogram of Sr-EPSC amplitude before (solid line) and after (dotted line) pairing at −20 mV with presynaptic stimulation (n = 10 cells).
Depolarization of the post-synaptic cell (−20 mV, 5 min, 0.1 Hz) leads to a rapid, change in Sr-EPSC amplitude (Figure 1D), without any change in event frequency between the baseline and post-pairing period (Figure 1E, before frequency 2.85 ± 0.35, vs. after 2.63 ± 0.21, n = 8 cells, p = 0.65 by paired t-test). Since stimulation strength is not altered during the experiment, and since individual Sr-EPSCs are thought to represent individual release events at distinct synaptic contacts, these results indicate that this depression is likely to be post-synaptic in origin.
Because statistical comparisons were carried out for a large number of events before and after pairing, this method was very sensitive to small changes in Sr-EPSC amplitude. In tissue from SWE animals, 16/20 (Figures 1G and 8A) cells showed a significant reduction in Sr-EPSC amplitude, with a mean depression of ∼20% (Figures 1F,G and 7; average of 16 cells 20 ± 2%; p value range for individual cells 0.025–0.00002 for baseline vs. post-pairing window by Mann–Whitney U-test). A cumulative distribution histogram for all cells from the spared barrel column showed a highly significant reduction in event amplitude in the post-pairing window (Figure 1H; Kolmogorov–Smirnov test p < 0.00001, n = 10 cells).
In control animals, 0/7 cells showed a change in Sr-EPSC amplitude after pairing, (p value range for individual cells 0.19–0.89 for baseline vs. post-pairing window; Figures 2A–E). Depolarization did not change event frequency in the post-pairing window (Figure 2C). An absence of synaptic depression was confirmed by analysis of a cumulative distribution histogram of Sr-EPSCs from control cells, where depolarization failed to trigger any shift in event distribution (Figure 2F, Kolmogorov–Smirnov test p = 0.715, n = 7 cells).
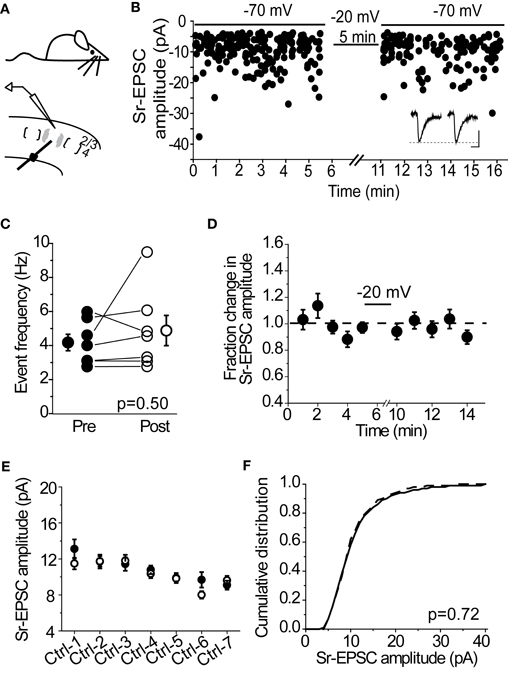
Figure 2. Layer 4-2/3 synapses from control animals did not exhibit Sr-depression. (A) Schematics of a control animal (top) and electrode configuration (bottom). (B) Example scatter plot of individual Sr-EPSC amplitudes from a control cell using the same pairing protocol. Inset: average traces, before and after pairing. Scale: 5 pA, 5 ms. (C) Sr-EPSC event frequency does not change after pairing in Sr++. (D) Mean change in Sr-EPSC amplitude normalized to the baseline period before and after pairing in control animals. (E) Within-cell comparison of Sr-EPSC amplitude between pre- (filled circle, black) and post-depression induction (open circle, black). Cells were rank ordered according to their mean amplitude of the baseline responses. (F) Cumulative distribution histogram of Sr-EPSC amplitude before (solid line) and after (dotted line) pairing at −20 mV with presynaptic stimulation (n = 7 cells).
Requirements for Sr-Depression
Many forms of synaptic depression, including depolarization-induced plasticity at layer 4-2/3 synapses in the spared barrel column (Clem et al., 2008), require NMDAR-activation. To determine whether Sr-depression requires NMDARs, we examined whether bath application of the NMDAR-antagonist D-APV was sufficient to block depression in cells from SWE-treated animals. In the presence of D-APV, 0/5 cells showed a significant reduction in Sr-EPSC amplitude after pairing (Figures 3A,B, p value range 0.11–0.65, baseline vs. post-pairing window by Mann–Whitney U-test). Analysis of a cumulative distribution histogram of Sr-EPSC events from all cells before and after pairing in Sr++ showed a small, but still significant reduction in amplitude (Figure 3B; Kolmogorov–Smirnov test p = 0.021 for baseline vs. post-pairing window; note comparison from Figure 1H where p < 0.00001).
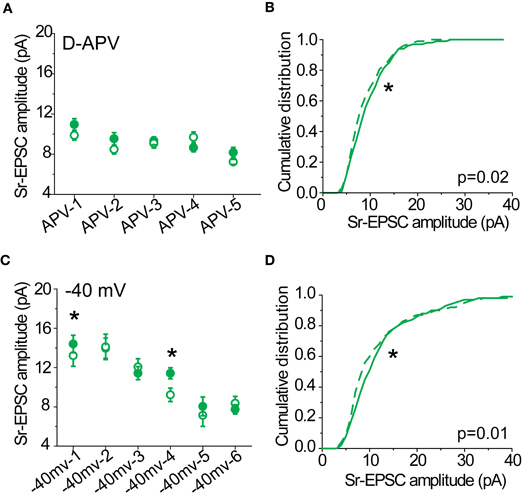
Figure 3. Sr-depression in SWE-treated animals requires NMDAR activation. (A) Within-cell comparison between pre- (filled) and post-pairing (open) windows in the presence of D-APV. (B) Cumulative distribution histogram of Sr-EPSC amplitude before (solid line) and after (dotted line) pairing at −20 mV with presynaptic stimulation in D-APV (n = 5 cells). (C) Within-cell comparison between pre- (filled) and post- responses (open) by pairing post-synaptic depolarization to −40 mV with presynaptic stimulation. *p < 0.05 by Mann–Whitney U-test. (D) Cumulative distribution histogram of Sr-EPSC amplitude before (solid line) and after (dotted line) pairing at −40 mV with presynaptic stimulation (n = 6 cells).
We also found that a more modest depolarization to −40 mV was sufficient to block Sr-depression in most cells (Figure 3C; 4/6 did not show significant depression, within-cell p value range 0.14–0.73 for baseline vs. post-pairing window by Mann–Whitney U-test), consistent with a role for NMDAR-activation which remains partially blocked at this holding potential. As above, the cumulative distribution of Sr-EPSC event amplitudes showed a small shift following pairing at −40 mV (Figure 3D; Kolmogorov–Smirnov test p = 0.01 for baseline vs. post-pairing window). Thus, Sr-depression requires post-synaptic depolarization and NMDAR activation. Based on the small but statistically significant reduction in Sr-EPSC amplitudes shown in the cumulative distribution histograms, there may be additional pathways that are involved in this depression.
The Role of CP-AMPARs in Sr-Depression
Previous work from our lab has shown that CP-AMPARs are trafficked to layer 4-2/3 synapses after SWE. Because other investigations have found that these receptors can be highly labile at the synapse, we hypothesized that the rapid depression observed might be due to the NMDAR-dependent removal of CP-AMPARs. Previous Sr-depression experiments were carried out in tissue from P13 animals, since CP-AMPARs have been detected after SWE at this age (Wen and Barth, 2011). Consistent with our previous findings, we observed significant rectification of pharmacologically isolated AMPAR-EPSCs after SWE at this age (Figures 4A,B; Control RI 0.57 ± 0.06 n = 14 cells vs. SWE RI 0.42 ± 0.04 n = 10 cells, Mann–Whitney U-test p = 0.03). To calculate the RI, EPSC amplitude was recorded at −70, 0, and +40 mV (see Methods). Cells with an RI smaller than 0.57 were classified as rectifying, and those with an RI equal to or larger than 0.57 were classified as non-rectifying.
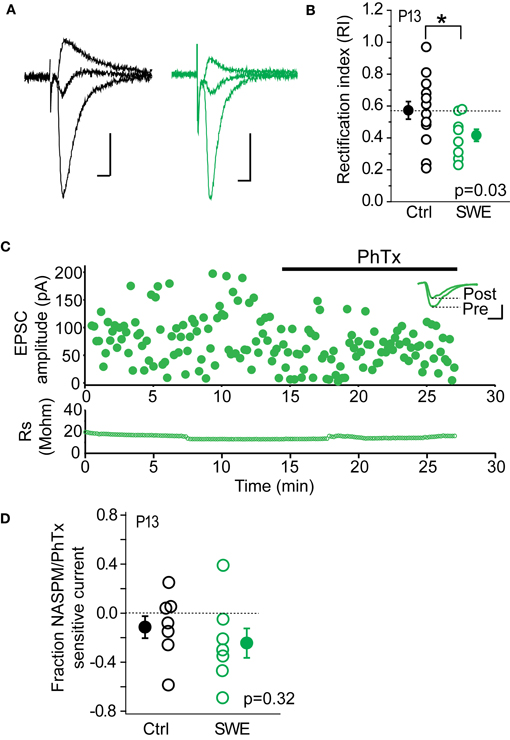
Figure 4. CP-AMPARs are present at layer 4-2/3 excitatory synapses after 24 h SWE at P13. (A) Example AMPA-EPSC traces recorded at −70, 0, and +40 mV from a control animal (left, black) and a SWE-treated animal (right, green). Scale: 20 pA, 10 ms. (B) RI in control (black) and SWE-treated (green) animals. *p < 0.05 by Mann–Whitney U-test. (C) PhTx blockade of AMPA-EPSC amplitude from a cell of P13 SWE-treated animal. Top: scatter plot of AMPA-EPSC amplitude before and after PhTx wash-in. Inset: average AMPA-EPSC traces for the pre- and post-wash in periods. Scale: 50 pA, 5 ms. Bottom: series resistance that does not change over time. (D) Fraction NASPM/PhTx sensitive current for control and SWE-treated animals at P13.
As a second method to quantify the contribution of CP-AMPARs at layer 4-2/3 inputs, we used PhTx or a synthetic analog of the CP-AMPAR antagonist Joro spider toxin, NASPM; (Koike et al., 1997) to block CP-AMPARs (Figure 4C). Although layer 4-2/3 inputs from SWE animals show significant rectification compared to age-matched controls, the PhTx/NASPM-sensitive current was not significantly different between the two groups (Figure 4D, reduction in EPSC amplitude for control 0.11 ± 0.09 n = 7 vs. SWE 0.24 ± 0.12 n = 7, p = 0.32 by Mann–Whitney U-test), likely due to large variability in magnitude of NASPM-sensitive current across cells in both control and SWE conditions. This is in contrast to previously published results showing minimal Joro spider toxin block at layer 4-2/3 synapses in control animals (Clem and Barth, 2006), which were not focused on the specific developmental age (P13) tested here. It is notable that a subset of cells in control animals showed strong rectification (5/14 cells show RI less than 0.55) and NASPM blockade (3/7 control cells showed >15% block), despite the fact that we could not induce Sr-depression in cells from this group.
CP-AMPAR Blockade Results in a Small Decrease in Sr-EPSC Amplitude
To determine whether we could detect a contribution of CP-AMPARs in Sr-EPSC amplitude, we determined the effect of NASPM on layer 4-evoked Sr-EPSCs from SWE-treated animals. We predicted that if there were a small number of CP-AMPARs at individual layer 4-2/3 synapses, NASPM blockade should reduce the mean amplitude of these events. A comparison across cells from SWE-treated animals showed that mean Sr-EPSC amplitude was 11.62 ± 0.5 pA (n = 19), compared to the amplitude of Sr-EPSCs in NASPM at 10.65 ± 0.34 pA (n = 17), a difference that was not significant (Figure 5A, p = 0.099 by Mann–Whitney U-test). Sr-EPSC amplitude before and after NASPM application within the same cell was compared for a smaller group of neurons. NASPM did not consistently decrease event amplitude (Figures 5B,C; mean EPSC amplitude before drug application, 11.94 ± 0.4 pA vs. after 10.95 ± 0.9 pA, n = 4, p = 0.43 by paired t-test).
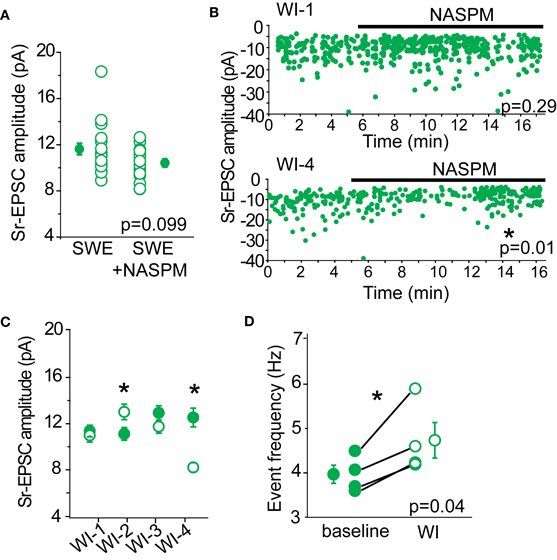
Figure 5. NASPM as a tool to block CP-AMPARs. (A) Cross-cell comparison of Sr-EPSC amplitude between SWE-treated animals (SWE) and SWE-treated animals in the presence of NASPM (SWE+NASPM). (B) Example experiments of a cell that showed no reduction in Sr-EPSC amplitude (top) and a cell that was reduced after NASPM wash-in (bottom). *p < 0.05, comparing the baseline and 5 min post-NASPM wash in window by Mann–Whitney U-test. (C) Within-cell comparison of Sr-EPSC amplitude between baseline and 5 min post-NASPM wash in (WI-1-4). (D) Comparison of Sr-EPSC event frequency between baseline and post-NASPM wash in window (WI). *p < 0.05 by paired t-test.
Because CP-AMPARs, specifically those that are homomeric for GluA1, have been shown to have moderately faster decay kinetics than GluA1-GluA2 heteromers or GluA2 homomers (Oh and Derkach, 2005), we also examined whether NASPM would slow the decay constant of the Sr-EPSC. There was no significant reduction in decay kinetics in the presence of NAPSM when compared across all cells (P13 SWE Sr-EPSC 3.27 ± 0.09 ms, n = 17 vs. in NAPSM 3.19 ± 0.08 ms, n = 15, p = 0.31 by Mann–Whitney U-test) and also when compared before and after drug application in the same cell (before 3.12 ± 0.16 ms vs. after 2.94 ± 0.19 ms, n = 4, p = 0.19 by Mann–Whitney U-test). Although this difference might become significant with a larger sample size, the lack of a pronounced effect suggests that decay kinetics of the Sr-EPSC is not a reliable indicator for CP-AMPARs.
If there were some synapses that contained primarily CP-AMPARs, NASPM application might reduce the apparent frequency of layer 4-triggered Sr-EPSCs without influencing the mean amplitude of events. Such a scenario might explain why the multi-quantal EPSC amplitude might be reduced by antagonist application, but the single-quantal response would not be markedly affected. However, a comparison of event frequency before and after NASPM application showed that event frequency was significantly increased (Figure 5D, baseline 3.96 ± 0.21 Hz vs. post-drug 4.73 ± 0.40 Hz, n = 4 cells), even when the mean amplitude of the Sr-EPSC was significantly reduced (Figure 5B, bottom panel). This increase in frequency suggests that NASPM might have some presynaptic targets that normally suppress presynaptic neurotransmitter release. Thus, we ascribe the lack of statistical significance for NASPM-blockade of multiquantal EPSCs between control and SWE layer 4-2/3 synapses (Figure 4D) to large cell-to-cell, not simply synapse-to-synapse, heterogeneity in the distribution of CP-AMPARs.
NASPM Effectively Blocks CP-AMPARs
A critical assumption behind using NASPM to block CP-AMPARs is that this compound is sufficient to fully block these receptors under our recording conditions. To verify that this was indeed the case, the RI was determined before and after drug application in the same cell. If NAPSM is sufficient to eliminate the contribution of rectifying AMPARs, the RI should become linear after drug application. This is indeed what was observed in pharmacologically isolated AMPAR-EPSCs (Figure 6). Cells were divided into two groups (rectifying vs. non-rectifying), based upon their RI values. Cells with a rectifying I–V showed a 23 ± 8% (n = 5) block in peak AMPAR-EPSC amplitude, compared to cells with a linear I–V with a 3.5 ± 5% (n = 5) block. As expected, blockade of CP-AMPARs made the I–V more linear (Figures 6C,D, pre RI 0.4 ± 0.07 vs. post RI 0.59 ± 0.03, n = 5, p < 0.05 by paired t-test). Taken together, these data indicate that NASPM application is sufficient to eliminate rectification, most likely through the selective blockade of CP-AMPARs.
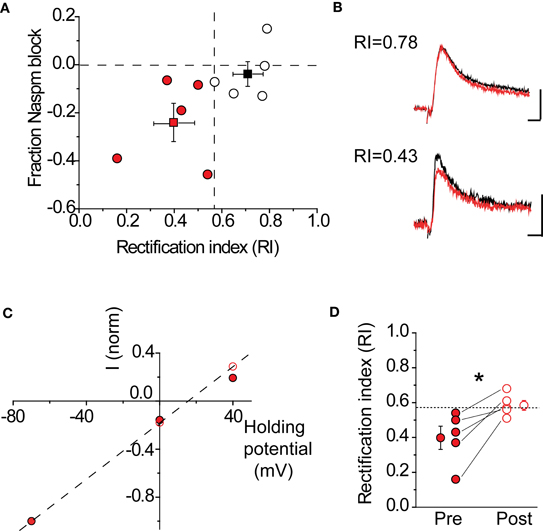
Figure 6. NASPM application blocks rectifying AMPARs. (A) Fraction AMPA-EPSC amplitude blocked by NASPM wash-in at +40 mV in rectifying cells (red) and non-rectifying cells (black). Dashed lines: horizontal, fraction NASPM = 0; vertical, RI = 0.57. Mean RI and fractional NASPM block with corresponding S.E. are shown in the same plot. (B) Example EPSC traces showing selective NASPM blockade of rectifying cells. Black: baseline EPSC before NASPM wash-in; red: 10 min after NASPM wash-in. Top: a non-rectifying cell (RI 0.78); scale bars: 20 pA, 5 ms. Bottom: a non-rectifying cell (RI 0.43); scale bars: 10 pA, 5 ms. (C) I–V relationship of AMPA-EPSC in the same cell as in (B) (bottom) became linear after NASPM application (before: filled; after: open). Current amplitudes recorded at −70, 0, and +40 mV were normalized to the amplitude at −70 mV. (D) NASPM application significantly increases the rectification index (RI) after NASPM application. *p < 0.05 by paired t-test.
Sr-Depression Does not Require CP-AMPARs
If synaptic lability at recently potentiated synapses is mediated by the removal of CP-AMPARs, we should expect that when CP-AMPARs are blocked, Sr-depression can no longer occur. To test this hypothesis, NASPM was bath applied to slices from SWE-treated animals, and Sr-depression was induced by post-synaptic depolarization. Because this antagonist is an open-channel blocker, care was taken to bath apply the drug with afferent stimulation for at least 15 min prior to pairing. In half the cells (4/9), a significant depolarization-induced reduction in Sr-EPSC amplitude was observed (Figures 7A,B), indicating that in these cells reduced current through CP-AMPARs was not required for depression. The magnitude of depression in cells that showed a significant depolarization-induced change in Sr-EPSC amplitude was identical to that which we characterized earlier, ∼20% (Figures 7B and 1G, 16 ± 3%, n = 4). The cumulative distribution of Sr-EPSC amplitude was also shifted after the pairing protocol in NASPM (Figure 7C, p < 0.001 by Kolmogorov–Smirnov test). Overall, these findings are incompatible with an obligatory role for CP-AMPARs, either for induction or expression, in Sr-depression.
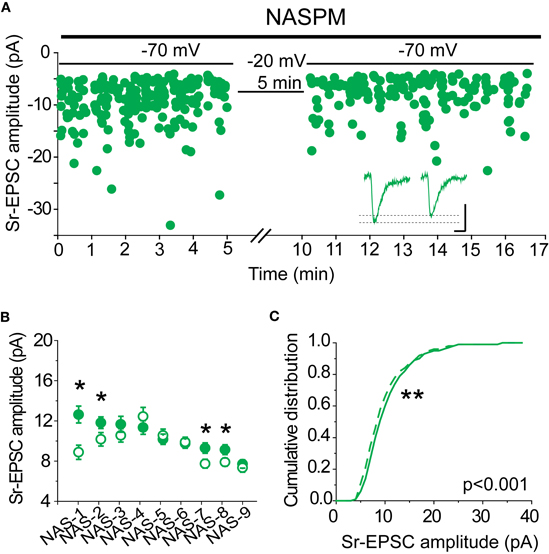
Figure 7. CP-AMPARs are not required for Sr-depression. (A) Example scatter plot of a Sr-depression experiment in the presence of NASPM that still showed depression. Inset: average traces. Scale: 5 pA, 5 ms. (B) Within-cell comparison of Sr-EPSC amplitude between pre- (filled circle) and post-depression induction (open circle) in the presence of NASPM. *p < 0.05 by Mann–Whitney U-test. (C) Cumulative distribution histogram of Sr-EPSC amplitude before (solid line) and after (dotted line) pairing at −20 mV with presynaptic stimulation in NASPM (n = 9 cells).
We also evaluated the decay kinetics of the post-pairing Sr-EPSC. If fast-decay CP-AMPARs are removed by this pairing protocol, it was reasonable to hypothesize that there might be an increase in the decay time constant. However, the lack of significant change after NAPSM application suggested we might not be able to detect a small change in decay kinetics. A comparison of the baseline and post-pairing decay time constant of the Sr-EPSC revealed that the decay time constant was not slower after Sr-depression (3.86 ± 0.09 vs. 3.27 ± 0.16 for baseline vs. post-pairing window, n = 8 cells, p = 0.52 by Mann–Whitney U-test). These data are inconsistent with the removal of fast-decay CP-AMPARs during Sr-depression.
Rectification is not Correlated with Sr-Depression
If CP-AMPARs are important for Sr-depression, either for its induction, or because they are selectively removed, then cells with more rectifying AMPAR-EPSCs should show greater depression. This was not the case (Figure 8). The amount of Sr-depression was uncorrelated with the cell's RI, when RI measurements in a Ca++ based ACSF were made before Sr++ wash-in and depolarization (Figure 8A, p = 0.53; rectifying cells, RI 0.45 ± 0.03, magnitude of depression 21 ± 4% vs. non-rectifying cells, RI 0.70 ± 0.05, magnitude of depression 23 ± 3%, n = 4 cells each). Cells that showed a linear I–V (Figure 8B, RI 0.59) or a rectifying I–V (Figure 8C, RI 0.53) showed similar depression.
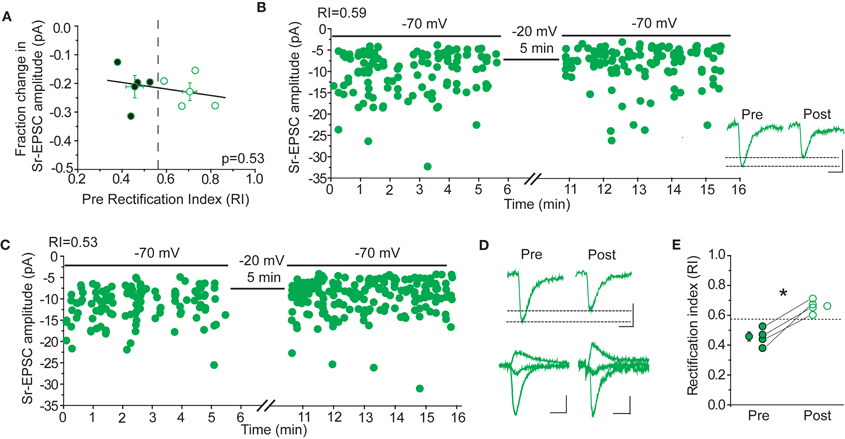
Figure 8. Sr-depression occurs irrespective of CP-AMPARs after 24 h SWE. (A) The RI of cells before the induction of Sr-depression is not correlated with the magnitude of depression in the same cells (n = 8). Cells presented here all exhibited significant Sr-depression (n = 8). Cells with rectifying (filled) and non-rectifying (open) AMPA-EPSCs all showed Sr-depression, the magnitude of which is indistinguishable between the two groups. (B) Example Sr-depression experiment at a non-rectifying cell (RI 0.59) from a SWE-treated animal. Inset: average traces of Sr-EPSCs pre- and post-Sr-depression induction. Scale bar: 5pA, 5 ms. (C) Example Sr-depression experiment at a rectifying cell (RI 0.53) from a SWE-treated animal. (D) Rectification is absent after Sr-depression. Top: average Sr-EPSC pre- and post-Sr-depression from the same cell as in (C). Scale bars: 5 pA, 5 ms. Bottom: AMPA-EPSC at −70, 0, and +40 mV before (pre) and after (post) Sr-depression from the same cell. Scale bars: 50 pA, 10 ms (pre) and 10 pA, 10 ms (post). (E) RI in rectifying cells significantly increases after Sr-depression (pre, filled; post, open). *p < 0.05 by paired t-test.
In a subset of cells, the RI was determined in a Ca++ based ACSF both before and after Sr-depression. In these cells, we noted that the RI became more linear (Figure 8E, pre RI 0.45 ± 0.03 vs. post 0.66 ± 0.02, n = 4, p < 0.05 by paired t-test), suggesting that when present, CP-AMPARs might be removed during depression. Thus, CP-AMPARs can be removed during, but their presence is not required for, Sr-depression.
Sr-Depression is Absent at Older Developmental Ages
SWE-induced increased in the strength of layer 4-2/3 synapses can be observed at least until P14, although the contribution of CP-AMPARs to SWE-induced potentiation appears minimal at this time. The RI is identical for SWE at P14 compared to control cells (control RI 0.64 ± 0.59 n = 11 vs. SWE 0.59 ± 0.13 n = 4, see also Wen and Barth, 2011), and NASPM showed a small effect on reducing the amplitude of the multiquantal EPSC (Figure 9A, control −8 ± 12% n = 6 vs. SWE −14 ± 11%, n = 4). At this age, Sr-depression in the spared whisker barrel column could not be induced in any cell (Figure 9C, 0/6 cells, p value range 0.49–0.70 baseline vs. post-pairing window by paired t-test).
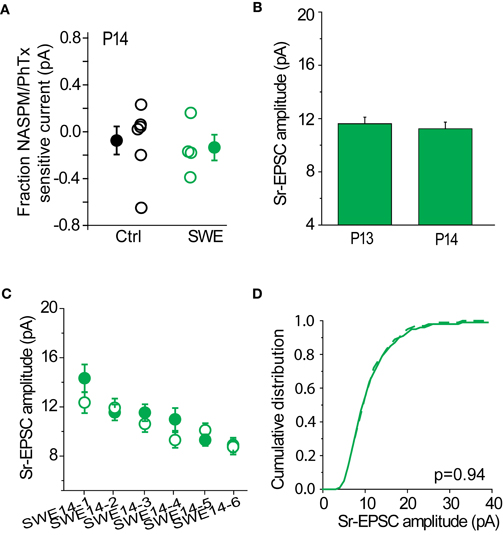
Figure 9. Sr-depression was not observed at later developmental ages. (A) Fraction NASPM/PhTx sensitive current in P14 control and SWE-treated animals. (B) Comparison of Sr-EPSC amplitude of the pre-pairing window between P13 and P14 SWE-treated animals. (C) Within-cell comparison of Sr-EPSC amplitude between pre- and post-pairing window in all P14 SWE-treated cells. (D) Cumulative distribution histogram of Sr-EPSC amplitude before (solid line) and after (dotted line) pairing at −20 mV with presynaptic stimulation (n = 6 cells).
The cumulative distribution of Sr-EPSC amplitudes were not different between pre- and post-pairing window (Figure 9D, p = 0.936). The amplitude of SWE-induced synaptic strengthening was comparable between the two ages [Figure 9B, P13 11.62 ± 0.48 pA, n = 19 cells vs. P14 11.24 ± 0.49 pA, n = 8 cells, p = 0.94 by Mann–Whitney U-test; see also (Wen and Barth, 2011)], suggesting that pre-pairing response amplitude was not a factor in the induction of Sr-depression. These data suggest the presence of a critical period for Sr-depression which concludes at the end of the second postnatal week.
Discussion
CP-AMPARs have been observed at synapses across the CNS and can be mobilized in response to activity. Despite this, it has been controversial what the role of this special AMPAR subtype might be. Because the vast majority of AMPARs are CI-AMPARs, and CP-AMPARs appear to be tightly regulated, it has been tempting to speculate that these receptors might serve some special function. For example, the calcium-permeability of these receptors might allow activation of signal transduction cascades that normally require NMDARs in mediating synaptic plasticity (Burnashev et al., 1996; Mahanty and Sah, 1998; Biou et al., 2008; Wiltgen et al., 2010).
Studies presented here were designed to evaluate the specific hypothesis that the presence of newly trafficked CP-AMPARs might confer a capacity for synaptic depression, possibly akin to the process of depotentiation that has been well-studied in other systems (O'Dell and Kandel, 1994; Kim et al., 2007). Previous work has shown that after SWE-initiated plasticity, potentiated layer 4-2/3 synapses exhibit pronounced an NMDAR-dependent depression (Clem et al., 2008). Thus, although NMDARs are required to initiate potentiation at the onset of SWE, subsequent NMDAR activation triggers synaptic depression. We have developed a novel in vitro method, Sr-depression that recapitulates essential features of this depression, and tested the hypothesis that it proceeds via the activation and/or removal of CP-AMPARs. Our findings suggest that CP-AMPARs are not essential for synaptic depression at synapses that have been recently potentiated by sensory experience.
CP-AMPARs are not Associated with Sr-Depression at Layer 4-2/3 Synapses
Our conclusions are based upon the following findings from this study and others. First, blockade of CP-AMPARs by NASPM was not sufficient to abolish Sr-depression, indicating that reduced current via CP-AMPARS, either because of their synaptic removal or decreased conductance, is not necessary for this phenomenon. Sr-depression could be induced in cells that express only CI-AMPARs, or express some CP-AMPARs, and the amount of Sr-depression was comparable between the two groups of cells. Layer 4-2/3 synapses are still labile, even when CP-AMPARs have been pharmacologically blocked. Second, although a subset of neurons from control animals showed rectifying AMPAR EPSCs (4/14 cells showed an RI < 0.5) and substantial NASPM sensitivity [4/13 showed >15% EPSC block by NASPM; see also (Kumar et al., 2002)], we never observed Sr-depression at layer 4-2/3 synapses from control animals. If the presence of CP-AMPARs was sufficient to confer a capacity for Sr-depression, we would expect to see this phenomenon at least occasionally in control tissue. Additionally, not all cells that express CP-AMPARs in SWE-treated animals exhibited Sr-depression (1/5 cells did not show Sr-depression). This dissociation between the absence of Sr-depression and the presence of CP-AMPARs was also observed at a slightly later developmental age (P14), where layer 4-2/3 inputs still display some NASPM sensitivity (in the current analysis, 3/4 cells show a > 15% EPSC block by NASPM), but no Sr-depression.
The presence of Sr-depression during NASPM blockade in a subset of cells indicates not only that depression is not mediated by the removal of CP-AMPARs, but also that CP-AMPARs are not required to initiate Sr-depression. Because CP-AMPARs can flux some Ca++ (Burnashev et al., 1996), it has been proposed that they might serve as a novel source for Ca++ entry to regulate plasticity under some conditions. However, we note that the conditions where Ca-entry via CP-AMPARs is required for the trafficking of AMPARs in excitatory neurons may be exceptional, such as in GluR2/B deficient animals (Biou et al., 2008; Wiltgen et al., 2010). Thus, at layer 4-2/3 synapses, CP-AMPARs may not be essential either for the induction or the expression of Sr-depression. Interestingly, although CP-AMPARs are not required for Sr-depression, we observed that they could be removed during Sr-depression (after Sr-depression, the AMPAR-EPSC shifted from rectifying to linear in 4/4 cells), indicating that these receptors can be mobilized during this form of depression. NAPSM application modestly reduced the fraction of cells showing Sr-depression, from 80% of the cells to 50% of cells that showed a pairing induced reduction in Sr-EPSC amplitude, suggesting that CP-AMPARs might play some role in the initiation or expression of Sr-depression at a subset of synapses.
CP-AMPARs and Potentiation
Are CP-AMPARs required for experience-dependent potentiation at neocortical synapses? Our previous studies showed that AMPARs became rectifying after LTP in vitro and also after SWE in vivo, suggesting that these receptors were acutely trafficked to and could be maintained at potentiated synapses (Clem et al., 2008). However, SWE triggers plasticity at layer 2/3–2/3 synapses where CP-AMPARs are not detectable (Wen and Barth, 2011), and in PICK-1 knock-out animals, SWE still potentiates layer 4-2/3 inputs without adding CP-AMPARS (Clem et al., 2010). Finally, we note that a capacity for further synaptic potentiation in vitro, after the onset of SWE-induced synaptic strengthening, does not require CP-AMPARs, since pharmacological blockade of CP-AMPARS does not impair LTP after the onset of SWE-induced strengthening (Clem et al., 2008). Taken together, these data indicate that CP-AMPARs are not broadly required for synaptic potentiation at neocortical synapses.
Estimating the Contribution of CP-AMPARs to the EPSC
As in other studies, both pharmacological and electrophysiological methods were used here to ascertain the presence of CP-AMPARs. Our results suggest that antagonists that have been used as specific blockers of CP-AMPARs may have some unanticipated effects on EPSCs. For example, in a number of cases we observed an increase in the multiquantal EPSC amplitude after NAPSM application (an increase of 5–40% in approximately one third of all cells), suggesting that a NASPM-sensitive receptor might normally reduce presynaptic release probability. Since these experiments were carried out in the presence of D-APV, it is unlikely that NASPM block of presynaptic NMDARS that have been hypothesized to exist at this synapses (Bender et al., 2006; Banerjee et al., 2009) are responsible for this effect. NASPM has been shown to block kainate receptors (Sun et al., 2009), and presynaptic kainate receptors have been described at thalamocortical synapses in somatosensory cortex (Kidd et al., 2002). Thus, we hypothesize that presynaptic kainate receptors may be present at layer 4-2/3 synapses. Although investigating the effects of polyamine antagonists on release probability was not a focus of the current work, further investigations into this effect may be of interest.
These NASPM effects complicate the interpretation of ours and others' results, since an increase in release probability would lead to an apparent increase in the amplitude of the post-synaptic EPSC and underestimation of the contribution of post-synaptic CP-AMPARs. However, our electrophysiological analysis showing rectification of the AMPAR-EPSC is consistent with the presence of these receptors at layer 4-2/3 synapses under some conditions.
Sr-Depression: a New Experimental Approach to Study Synaptic Plasticity in vitro
Our finding that we can induce depression of the quantal EPSC amplitude in a Sr-ACSF solution is provocative. This method offers the advantage of precisely evaluating how post-synaptic depolarization influences both the frequency and the amplitude of EPSCs at individual synaptic contacts onto a cell. Consistent with a post-synaptic locus for depression, we find EPSC amplitudes at layer 4-2/3 synapses are decreased without any change in event frequency. On average, a 20% reduction in Sr-EPSC amplitude was observed. The magnitude of the depression appears modest, but we note that a 20% reduction in Sr-EPSC amplitude normalizes SWE-induced increases back to control levels. Although a role for presynaptic NMDARs in depression of layer 4-2/3 excitatory synapses has been proposed (Bender et al., 2006; Banerjee et al., 2009), it is important to note that the effects characterized here are likely post-synaptic in origin.
The protocol developed here to elicit synaptic depression in the presence of Sr++ is novel, and we have used it to probe the mechanisms that underlie synaptic lability at recently potentiated synapses. How long does this synaptic depression persist after pairing? The post-pairing period analyzed here was admittedly short (5 min), and future experiments will be required to determine the duration of this effect. Although our data suggest that removal of CP-AMPARs can sometimes occur during Sr-depression, depression may also result from the removal of CI-AMPARs or activation of intracellular signaling cascades to reduce channel conductance. Thus, there may be several different mechanisms that underlie Sr-depression. Alternatively, Sr-depression might occur via a common mechanism involving GluR1, a subunit that could be found in both rectifying and non-rectifying AMPARs. In addition, we note that some forms of LTD might specifically target the removal of CP-AMPARs; there are likely to be diverse processes that regulate synaptic depression across the CNS. However, the relative simplicity of this assay should facilitate its use in other experimental preparations.
The mechanism by which Sr++ triggers vesicle fusion at the presynaptic terminal has been studied (Goda and Stevens, 1994; Xu-Friedman and Regehr, 1999), but a role for this ion in activating post-synaptic signaling cascades has not been evaluated. Because it is impossible to remove all Ca++ from our bath solution (even without addition of Ca++, free Ca++ may be in the low micromolar range), we cannot determine whether Sr++ is acting on normally Ca++-dependent signaling cascades, or whether residual Ca++ in the ACSF is sufficient to do this. NMDARs are permeable to Sr++ (Mayer and Westbrook, 1987), and thus, it is possible that depolarization leads to influx of this ion to activate post-synaptic signaling cascades for depression. A more detailed investigation into this phenomenon is warranted.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Adesnik, H., and Nicoll, R. A. (2007). Conservation of glutamate receptor 2-containing AMPA receptors during long-term potentiation. J. Neurosci. 27, 4598–4602.
Banerjee, A., Meredith, R. M., Rodriguez-Moreno, A., Mierau, S. B., Auberson, Y. P., and Paulsen, O. (2009). Double dissociation of spike timing-dependent potentiation and depression by subunit-preferring NMDA receptor antagonists in mouse barrel cortex. Cereb. Cortex 19, 2959–2969.
Barth, A. L., Gerkin, R. C., and Dean, K. L. (2004). Alteration of neuronal firing properties after in vivo experience in a FosGFP transgenic mouse. J. Neurosci. 24, 6466–6475.
Bellone, C., and Luscher, C. (2006). Cocaine triggered AMPA receptor redistribution is reversed in vivo by mGluR-dependent long-term depression. Nat. Neurosci. 9, 636–641.
Bender, V. A., Bender, K. J., Brasier, D. J., and Feldman, D. E. (2006). Two coincidence detectors for spike timing-dependent plasticity in somatosensory cortex. J. Neurosci. 26, 4166–4177.
Biou, V., Bhattacharyya, S., and Malenka, R. C. (2008). Endocytosis and recycling of AMPA receptors lacking GluR2/3. Proc. Natl. Acad. Sci. U.S.A. 105, 1038–1043.
Brill, J., and Huguenard, J. R. (2008). Sequential changes in AMPA receptor targeting in the developing neocortical excitatory circuit. J. Neurosci. 28, 13918–13928.
Burnashev, N., Villarroel, A., and Sakmann, B. (1996). Dimensions and ion selectivity of recombinant AMPA and kainate receptor channels and their dependence on Q/R site residues. J. Physiol. 496 (Pt 1), 165–173.
Clem, R. L., Anggono, V., and Huganir, R. L. (2010). PICK1 regulates incorporation of calcium-permeable AMPA receptors during cortical synaptic strengthening. J. Neurosci. 30, 6360–6366.
Clem, R. L., and Barth, A. (2006). Pathway-specific trafficking of native AMPARs by in vivo experience. Neuron 49, 663–670.
Clem, R. L., Celikel, T., and Barth, A. L. (2008). Ongoing in vivo experience triggers synaptic metaplasticity in the neocortex. Science 319, 101–104.
Clem, R. L., and Huganir, R. L. (2010). Calcium-permeable AMPA receptor dynamics mediate fear memory erasure. Science 330, 1108–1112.
Deng, W., Rosenberg, P. A., Volpe, J. J., and Jensen, F. E. (2003). Calcium-permeable AMPA/kainate receptors mediate toxicity and preconditioning by oxygen-glucose deprivation in oligodendrocyte precursors. Proc. Natl. Acad. Sci. U.S.A. 100, 6801–6806.
Glazewski, S., Benedetti, B. L., and Barth, A. L. (2007). Ipsilateral whiskers suppress experience-dependent plasticity in the barrel cortex. J. Neurosci. 27, 3910–3920.
Goda, Y., and Stevens, C. F. (1994). Two components of transmitter release at a central synapse. Proc. Natl. Acad. Sci. U.S.A. 91, 12942–12946.
Gray, E. E., Fink, A. E., Sarinana, J., Vissel, B., and O'Dell, T. J. (2007). Long-term potentiation in the hippocampal CA1 region does not require insertion and activation of GluR2-lacking AMPA receptors. J. Neurophysiol. 98, 2488–2492.
Ho, M. T., Pelkey, K. A., Topolnik, L., Petralia, R. S., Takamiya, K., Xia, J., Huganir, R. L., Lacaille, J. C., and McBain, C. J. (2007). Developmental expression of Ca2+-permeable AMPA receptors underlies depolarization-induced long-term depression at mossy fiber CA3 pyramid synapses. J. Neurosci. 27, 11651–11662.
Hou, Q., Zhang, D., Jarzylo, L., Huganir, R. L., and Man, H. Y. (2008). Homeostatic regulation of AMPA receptor expression at single hippocampal synapses. Proc. Natl. Acad. Sci. U.S.A. 105, 775–780.
Isaac, J. T., Ashby, M. C., and McBain, C. J. (2007). The role of the GluR2 subunit in AMPA receptor function and synaptic plasticity. Neuron 54, 859–871.
Kidd, F. L., Coumis, U., Collingridge, G. L., Crabtree, J. W., and Isaac, J. T. (2002). A presynaptic kainate receptor is involved in regulating the dynamic properties of thalamocortical synapses during development. Neuron 34, 635–646.
Kim, J., Lee, S., Park, K., Hong, I., Song, B., Son, G., Park, H., Kim, W. R., Park, E., Choe, H. K., Kim, H., Lee, C., Sun, W., Kim, K., Shin, K. S., and Choi, S. (2007). Amygdala depotentiation and fear extinction. Proc. Natl. Acad. Sci. U.S.A. 104, 20955–20960.
Koike, M., Iino, M., and Ozawa, S. (1997). Blocking effect of 1-naphthyl acetyl spermine on Ca(2+)-permeable AMPA receptors in cultured rat hippocampal neurons. Neurosci. Res. 29, 27–36.
Kumar, S. S., Bacci, A., Kharazia, V., and Huguenard, J. R. (2002). A developmental switch of AMPA receptor subunits in neocortical pyramidal neurons. J. Neurosci. 22, 3005–3015.
Lante, F., Toledo-Salas, J. C., Ondrejcak, T., Rowan, M. J., and Ulrich, D. (2011). Removal of synaptic Ca(2)+-permeable AMPA receptors during sleep. J. Neurosci. 31, 3953–3961.
Liu, S. J., and Zukin, R. S. (2007). Ca2+-permeable AMPA receptors in synaptic plasticity and neuronal death. Trends Neurosci. 30, 126–134.
Mahanty, N. K., and Sah, P. (1998). Calcium-permeable AMPA receptors mediate long-term potentiation in interneurons in the amygdala. Nature 394, 683–687.
Man, H. Y. (2011). GluA2-lacking, calcium-permeable AMPA receptors–inducers of plasticity? Curr. Opin. Neurobiol. 21, 291–298.
Mayer, M. L., and Westbrook, G. L. (1987). Permeation and block of N-methyl-D-aspartic acid receptor channels by divalent cations in mouse cultured central neurones. J. Physiol. 394, 501–527.
O'Dell, T. J., and Kandel, E. R. (1994). Low-frequency stimulation erases LTP through an NMDA receptor-mediated activation of protein phosphatases. Learn. Mem. 1, 129–139.
Oh, M. C., and Derkach, V. A. (2005). Dominant role of the GluR2 subunit in regulation of AMPA receptors by CaMKII. Nat. Neurosci. 8, 853–854.
Plant, K., Pelkey, K. A., Bortolotto, Z. A., Morita, D., Terashima, A., McBain, C. J., Collingridge, G. L., and Isaac, J. T. (2006). Transient incorporation of native GluR2-lacking AMPA receptors during hippocampal long-term potentiation. Nat. Neurosci. 9, 602–604.
Rakhade, S. N., Zhou, C., Aujla, P. K., Fishman, R., Sucher, N. J., and Jensen, F. E. (2008). Early alterations of AMPA receptors mediate synaptic potentiation induced by neonatal seizures. J. Neurosci. 28, 7979–7990.
Rumpel, S., LeDoux, J., Zador, A., and Malinow, R. (2005). Postsynaptic receptor trafficking underlying a form of associative learning. Science 308, 83–88.
Shi, S. H., Hayashi, Y., Petralia, R. S., Zaman, S. H., Wenthold, R. J., Svoboda, K., and Malinow, R. (1999). Rapid spine delivery and redistribution of AMPA receptors after synaptic NMDA receptor activation. Science 284, 1811–1816.
Sun, H. Y., Bartley, A. F., and Dobrunz, L. E. (2009). Calcium-permeable presynaptic kainate receptors involved in excitatory short-term facilitation onto somatostatin interneurons during natural stimulus patterns. J. Neurophysiol. 101, 1043–1055.
Sutton, M. A., Ito, H. T., Cressy, P., Kempf, C., Woo, J. C., and Schuman, E. M. (2006). Miniature neurotransmission stabilizes synaptic function via tonic suppression of local dendritic protein synthesis. Cell 125, 785–799.
Takahashi, T., Svoboda, K., and Malinow, R. (2003). Experience strengthening transmission by driving AMPA receptors into synapses. Science 299, 1585–1588.
Thiagarajan, T. C., Lindskog, M., and Tsien, R. W. (2005). Adaptation to synaptic inactivity in hippocampal neurons. Neuron 47, 725–737.
Wen, J. A., and Barth, A. L. (2011). Input-specific critical periods for experience-dependent plasticity in layer 2/3 pyramidal neurons. J. Neurosci. 31, 4456–4465.
Wiltgen, B. J., Royle, G. A., Gray, E. E., Abdipranoto, A., Thangthaeng, N., Jacobs, N., Saab, F., Tonegawa, S., Heinemann, S. F., O'Dell, T. J., Fanselow, M. S., and Vissel, B. (2010). A role for calcium-permeable AMPA receptors in synaptic plasticity and learning. PLoS One 5. doi: 10.1371/journal.pone.0012818
Xu-Friedman, M. A., and Regehr, W. G. (1999). Presynaptic strontium dynamics and synaptic transmission. Biophys. J. 76, 2029–2042.
Keywords: metaplasticity, NASPM, philanthotoxin, depotentiation, rectification, neocortex, development, critical period
Citation: Wen JA and Barth AL (2012) Synaptic lability after experience-dependent plasticity is not mediated by calcium-permeable AMPARs. Front. Mol. Neurosci. 5:15. doi: 10.3389/fnmol.2012.00015
Received: 13 October 2011; Accepted: 01 February 2012;
Published online: 29 February 2012.
Edited by:
R. Suzanne Zukin, Albert Einstein College of Medicine, USAReviewed by:
Hey-Kyoung Lee, Johns Hopkins University, USAYe He, University of California, San Francisco, USA
Reed Carroll, Albert Einstein College of Medicine, USA
Copyright: © 2012 Wen and Barth. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Alison L. Barth, Department of Biological Sciences and Center for the Neural Basis of Cognition, Carnegie Mellon University, 4400 Fifth Ave., Pittsburgh, PA 15213, USA. e-mail:YmFydGhAY211LmVkdQ==