
- Department of Biochemistry, Brandeis University, Waltham, MA, USA
The ability to sense mechanical, thermal, and chemical stimuli is critical to normal physiology and the perception of pain. Contact with noxious stimuli triggers a complex series of events that initiate innate protective mechanisms designed to minimize or avoid injury. Extreme temperatures, mechanical stress, and chemical irritants are detected by specific ion channels and receptors clustered on the terminals of nociceptive sensory nerve fibers and transduced into electrical information. Propagation of these signals, from distant sites in the body to the spinal cord and the higher processing centers of the brain, is also orchestrated by distinct groups of ion channels. Since their identification in 1995, evidence has emerged to support roles for K2P channels at each step along this pathway, as receptors for physiological and noxious stimuli, and as determinants of nociceptor excitability and conductivity. In addition, the many subtypes of K2P channels expressed in somatosensory neurons are also implicated in mediating the effects of volatile, general anesthetics on the central and peripheral nervous systems. Here, I offer a critical review of the existing data supporting these attributes of K2P channel function and discuss how diverse regulatory mechanisms that control the activity of K2P channels act to govern the operation of nociceptors.
The Operation of Somatosensory Neurons
The somata of the sensory neurons that innervate the limbs and torso are clustered together in dorsal root ganglia (DRG), node-like structures that flank the spinal vertebrae from the neck down. DRG neurons project axons that bifurcate into peripheral and central afferent fibers. Peripheral afferent fibers extend throughout the body, innervating skin, muscle, and other tissues while central fibers enter the spinal cord and diverge into local and ascending branches. Local branch fibers activate rapid-response reflex circuits while ascending branches project signals to the thalamus and sensory cortices of the brain. An anatomically analogous arrangement is found on each side of the cranium where sensory stimuli are detected by peripheral afferent fibers that converge on the trigeminal ganglion (TG), a structure apposed to the dura mater of the temporal bone (Kandel et al., 2000).
Noxious sensory inputs stimulate a subpopulation of peripheral fibers called nociceptors that are classified according to anatomical and functional criteria, as well as by biochemical markers (Basbaum et al., 2009). Acute, localized pain is transmitted by medium diameter (1–5 μm), lightly myelinated Aδ fibers, while diffuse pain, including itch, is mediated by unmyelinated C-fibers. Although there is functional heterogeneity in each of these groups, C-fibers are distinguished from Aδ fibers by their smaller diameter (0.2–1.5 μm) and, therefore, lower conduction velocity (∼1 m/s compared to 6–25 m/s; Fields, 1987). Proprioception and innocuous mechanical stimuli, such as light touch, are transmitted more rapidly (33–75 m/s) by large-diameter (6–12 μm), myelinated Aα, and Aβ fibers.
The termini of nociceptors are decorated by a diverse array of ion channels and other integral membrane proteins that detect and transduce sensory inputs, depolarizing nociceptive fibers, and initiating electrical signals that propagate to synaptic connections in the spinal cord and brain. Sensory receptors include transient receptor potential (TRP) channels, acid-sensing ion channels (ASICs), K2P channels, and P2X receptors. K2P channels, voltage-gated K+ (KV), Na+ (NaV), and Ca2+ (CaV) and hyperpolarization-activated cyclic nucleotide-gated (HCN) channels operate throughout somatosensory neurons to determine excitability, synaptic function, and responsiveness (Gold and Gebhart, 2010).
Nociceptors at Rest: The Importance of Being Leaky
Like all excitable cells, nociceptors are never truly at rest, even when they are electrically quiescent. Ion channels and transport proteins operate continuously to mediate ionic flux across the plasma membrane, generating, and dissipating chemical and electrostatic gradients. Transmembrane chemical gradients are established by active transport systems such as the Na+/K+ ATPase, an ATP powered enzyme pump that acts to maintain a relatively high concentration of K+ but a low concentration of Na+ in the cytoplasm. Chemical gradients are dissipated by the opening of ion channels, protein complexes that form aqueous pores in the membrane and allow the passive diffusion of select ions. When K+ channels open, K+ ions diffuse down their concentration gradient, leaving behind negative counter-charges that oppose K+ efflux. Net flux of K+ stops when the chemical and electrostatic gradients are balanced. The transmembrane voltage (Vm) where this occurs for a given ion species is called the Nernst potential and can be determined using the Nernst equation (Figure 1A). Under physiological conditions typical Nernst potentials are, approximately, −95 mV for K+, +65 mV for Na+, +120 mV for Ca2+, and −90 mV for Cl− (Hille, 2001). The resting Vm of a cell is a balance between these values, scaled to reflect the relative permeability of the membrane to each species of ion, in a manner described by the Goldman–Hodgkin–Katz equation (Figure 1B). However, permeability is dynamic because different ion channel subtypes are tuned to open and close at different potentials across the physiological voltage range. The resting Vm of sensory neurons ranges between ∼−45 and −75 mV, depending on the experimental paradigm (Baccaglini, 1978; Puil et al., 1987; Wang et al., 1994; Baumann et al., 1996). Typically, resting potentials lie closer to EK than ENa because cell membranes are populated more densely by K+ channels than any other channel type and because certain K+ channels operate across the physiological voltage range, passing background, or leak K+ currents.
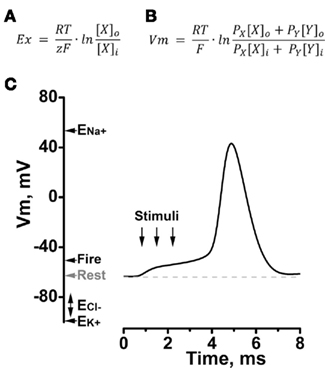
Figure 1. The neuronal membrane potential during rest and excitation. Cellular membrane potential (Vm) is determined by the internal and external ionic compositions and the relative permeability of the membrane to each ion species. Under quasi-physiological conditions, resting Vm for a neuron falls between ∼−60 and −75 mV and is determined in large part by the magnitude of the K+ leak current. (A) The Nernst equilibrium potential for cation X (Ex) is calculated using the Nernst equation, where R is the Gas constant (1.987 cal K−1 mol−1), T is the temperature in Kelvin, z is the charge of the ion and F is Faraday’s constant (9.648 × 104 C mol−1) and [X]o and [X]i are the external and internal concentrations of X, respectively. (B) The Goldman–Hodgkin–Katz equation shows that Vm is determined by the relative concentrations and permeabilities (P) of cations X and Y. (C) An exemplar action potential evoked in a neuron by depolarizing stimuli. Values for Vm corresponding to rest, the firing threshold for the action potential (Fire) and the Nernst potentials for Na+ (ENa), K+ (EK), and Cl− (ECl) are indicated.
K+ Leak Shapes Excitability
The psychophysical cognitive and innate responses to noxious stimuli begin with the opening of ion channels, such as ASICs or TRP channels at the termini of nociceptors. The resultant influx of, mainly Na+ and Ca2+ions, depolarizes the membrane. If the magnitude of depolarization is sufficient, NaV channels are activated, allowing Na+ ions to rush into the cell, further depolarizing the membrane and in-turn, activating more NaV channels. At the firing threshold, the influx of Na+is large enough to initiate a sharp depolarization toward ENa – an action potential is initiated (Figure 1C). KV channels are also activated by depolarization, usually with slower kinetics than NaV channels, and mediate an efflux of K+ that halts the action potential and contributes to repolarization of Vm. Active transport mechanisms, such as the Na+/K+ ATPase redress the displacement of ions that occurs during these periods of excitation. It is noteworthy that while neurons typically express only a few types of NaV channel they express a panoply of K+ channels. Thus, differential expression and regulation of K+ channels subtypes provides a dynamic mechanism by which excitable cells can tune many features of excitability.
The correlation between excitability and the regulation of K+ channel activity has been well established by studies elucidating the operation of the M-current, a neuronal K+ current first described in frog sympathetic neurons (Brown and Adams, 1980). M-channels are composed of Kv7 (KCNQ) subunits and have biophysical properties that stabilize the membrane potential of numerous types of peripheral and central neurons (Delmas and Brown, 2005), including DRG cells (Passmore et al., 2003). The M-current was named because it is inhibited by stimulation of muscarinic acetylcholine receptors (Brown and Adams, 1980). Activation of G-protein coupled receptors linked to Gq or G11 effector subunits by muscarine or neuroactive peptides, including angiotensin, substance P, and bradykinin increases neuronal excitability by inhibiting M-currents (Lopez and Adams, 1989; Villarroel et al., 1989). As expected, enhancement of M-channel activity hyperpolarizes neurons and decreases the frequency of action potential firing (Gamper et al., 2006). Likewise, the magnitude of the K+ leak current is a primary determinant of neuronal resting Vm. If the K+ leak current is augmented, cells become less excitable because Vm moves closer to EK, and a larger depolarization is required to initiate an action potential.
The importance of K+ leak currents to excitable membrane function has been appreciated since the 1940s (Goldman, 1943; Hodgkin et al., 1949). Leak currents stabilize cells at hyperpolarized membrane voltages, below firing threshold and, thereby, determine the magnitude of the stimulation required to initiate an action potential as well as the shape, frequency, and magnitude of each spike (Hodgkin and Huxley, 1952; Jones, 1989). Reciprocally, inhibition of K+ leak currents depolarizes Vm closer to the firing threshold, allowing smaller stimuli to initiate action potentials. Therefore, under equivalent ionic conditions, the same magnitude of excitatory stimulus may or may not elicit an action potential, depending on the magnitude of the K+ leak current. A smorgasbord of stimuli are known to regulate K+ leak currents, including neurotransmitters (Siegelbaum et al., 1982; Shen et al., 1992; Watkins and Mathie, 1996), changes in oxygen-tension (Buckler, 1997), extracellular pH (Nattel et al., 1981), and pharmaceutical agents (Nicoll and Madison, 1982). Although K+ leak pathways are fundamental to physiology, the molecules mediating background currents evaded detection until the mid-1990s.
Leak is Mediated by K2P Channels
Genome database searches predict that humans express ∼80 different types of K+ channel that fall into four distinct families based on their structure (Yu et al., 2005). Despite differences in transmembrane architecture, all K+ channels have a single aqueous pore that becomes a K+ selective permeation pathway when the channel is open. The pore is lined by four copies of a P-loop that invariably contains the signature sequence of amino acids that form the K+ selectivity filter: Gly-Tyr-Gly (Yellen et al., 1991). Thus, inward-rectifying (KIR), voltage-gated (KV), and calcium-activated (KCa) K+ channels are formed by tetramers of subunits, each containing one P-loop and two, six, or seven transmembrane domains respectively. Therefore, when TOK1 was described in 1995 it was notable as the first example of a K+ channel formed by subunits that each contain two P-loops (Ketchum et al., 1995). TOK1 was identified in the genome of Saccharomyces cerevisiae, has eight transmembrane domains and operates as an outward rectifier. Although TOK channels are not found beyond fungi, subunits with two P-loops were identified in higher organisms shortly thereafter. K2PØ (previously called dORK) was cloned from a Drosophila melanogaster neuromuscular gene library and was also found to have a novel structure with four transmembrane domains and two P-loops in each subunit (Goldstein et al., 1996; Figure 2B). K2PØ channels exhibit the functional properties expected for K+ leak channels, opening, and closing across the physiological voltage range to pass K+ selective currents that show little or no voltage or time-dependence (Goldstein et al., 1996; Zilberberg et al., 2000; Ilan and Goldstein, 2001).
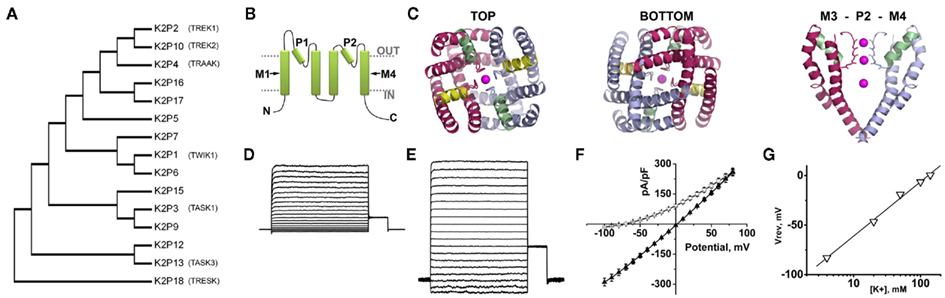
Figure 2. The structure and function of K2P channels. Fifteen K2P channel subunits have been identified in humans. Two subunits come together to form a K+ selective permeation pathway that opens and closes with little or no voltage or time-dependence. (A) A phylogenetic tree calculated from the alignment of the 15 K2P proteins expressed in humans shows the relatedness of subunits. Functional expression has not been observed for K2P7, 12, and 15. The descriptive names of the K2P channels discussed in this review are also given. (B) A cartoon showing that K2P subunits are integral membrane proteins with internal amino (N) and carboxy (C) termini, four transmembrane domains, M1–M4 and two re-entrant pore forming loops, P1 and P2. (C) A structural model of the Drosophila K2PØ channel based on experimental constrains and homology to the crystal structure of the voltage-dependent channel, Kv1.2. The top and bottom of the model are shown, as well as a side view showing the arrangement of transmembrane M3, P2, and M4. Occupation of the pore by K+ is denoted in each case. From above or below, the model shows twofold symmetry, with conservation of the fourfold symmetry required to form a K+ selective pore. Adapted from Kollewe et al. (2009). (D) A K+ leak current recorded from a Chinese Hamster Ovary cell transfected to express active human K2P1 channels. The inside of the cell is perfused with 140 mM K+ and the outside of the cell is perfused with 4 mM K+. (E) The same cell recorded in (D) with 140 mM K+ on the inside and the outside of the cell. (F) The current–voltage relationships for the cell recorded in (D) ○, and (E) (▲). (G) The same cell recorded in (D) studied in various concentrations of external K+. The voltage where zero-current was passed for each condition is plotted against the log10 of the external K+ concentration. The data are fit to a linear regression and show a shift of ∼54 mV per 10-fold change in K+. This relationship is predicted by the Nernst equation and confirms the K+ selective nature of the channel (see also Figure 1A). Elements D to F are adapted from Plant et al. (2010).
In the decade that followed, 15 KCNK genes encoding K2P subunits were identified in humans (Figure 2A; Plant et al., 2005). K2P channels share the same structure as K2PØ and because each subunit contains two P-loops the channels operate as dimers (Lopes et al., 2001; Kollewe et al., 2009; Plant et al., 2010; Figure 2B). Kollewe et al. (2009) produced a 3-dimensional homology model of K2PØ based on experimentally defined constraints and parameters determined by the crystal structure of the voltage-gated channel, KV1.2. The model predicts that, in common with other K+ channels, the K+ selectivity filter of K2PØ has fourfold symmetry, but that the channel corpus has distinct bilateral symmetry, as expected for a dimer (Figure 2C). An alternative structure–function strategy was employed to model human K2P3. Alanine-scanning mutagenesis identified residues in the pore of K2P3 channels that are required for the interaction with the specific blocker, A1899 (Streit et al., 2011). Experimentally defined constraints were then used to develop an open-state homology model of K2P3 based on the crystal structure of the arachial voltage-gated K+ channels, KvAP. Although K2PØ and K2P3 do not share a high degree of homology, common, pore-lining residues were identified in both channels (Kollewe et al., 2009; Streit et al., 2011).
In 2012, two groups reported the first crystal structures obtained for K2P channels; human K2P1 (Miller and Long, 2012) and K2P4 (Brohawn et al., 2012). Both channels appear to have crystalized in the open-state and are formed, as expected, by dimers. Corroborating the findings of Kollewe et al. (2009), both K2P1 and K2P4 channels show bilateral symmetry converging on a single, twofold symmetrical K+ conduction pathway formed by P1 and P2 loops from each subunit. Although the pore of a K2P channel is formed by pairs of distinct P-loops, both K2P1 and K2P4 share the key features associated with K+ selectivity and conduction that were first visualized in structures obtained for tetrameric K+ channels, notably KcsA (Doyle et al., 1998) and Kv1.2 (Long et al., 2005). In contrast to other K+ channels, the first extracellular loop of each K2P subunit is observed to extend ∼35 Å beyond the outer leaflet of the plasma membrane to form a “cap-domain.” The cap-domain is positioned above the outer mouth of the pore, bifurcating the K+ conduction pathway. Both K2P1 and K2P4 channels have helices that run close to the intracellular lipid interface. In K2P1, a “C-helix” follows directly from the final transmembrane domain, M4 (Miller and Long, 2012); in K2P4, a structurally analogous motif occurs at the later part of the second transmembrane domain, M2 (Brohawn et al., 2012). Both helices contain residues and motifs proposed to have roles in the gating of K2P channels.
K2P Channels in Nociceptive Neurons
Consistent with studies of native leak currents, the operation of K2P channels is tightly controlled by a plethora of regulators (Goldstein et al., 2001). Stimuli that increase the activity of K2P channels dampen excitability, while stimuli that block K2P channels, reduce the number of channels at the cell membrane, lower conductance, open probability, or selectivity all act to decrease the permeability of the membrane to K+ and, thereby, increase excitability, under physiological conditions. Since 1996, evidence has accumulated to show that the K2Ps are distinct among ion channels because they act both as receptors at sensory termini and throughout nociceptive fibers to determine intrinsic excitability and, thereby, the conduction of sensory inputs to the central nervous system (CNS). A subset of the 15 K2P channels expressed in humans has been reported to contribute to the function of DRG or TG neurons.
K2P1 Channels: Silent but Restless
K2P channels were initially cataloged by their biophysical attributes, pharmacology, or physiological regulators. K2P1 was the first K2P channel identified in humans (Lesage et al., 1996; Table 1) and was originally called TWIK1 (for tandem of P-domains in a weak inward-rectifying K+ channel) based on early evidence for weak inward rectification of single channels studied with 3 mM intracellular Mg2+ in membrane patches excised from Xenopus laevis oocytes. Subsequent studies demonstrated that K2P1 channels are electrically silent when studied by voltage-clamp (Goldstein et al., 1998; Pountney et al., 1999). Because K2P1 is expressed throughout the body, including in DRG and TG cells (Talley et al., 2001), electrical silence indicated that the channels are regulated in a physiologically relevant manner.

Table 1. The nomenclature of K2P channels.
The basis for electrical silencing of K2P1 channels was revealed to be constitutive modification of residue Lys274 by SUMO (small ubiquitin-like modifier protein; Rajan et al., 2005). Sumoylation is a reversible, enzymatic process that covalently couples SUMO to the ε-amino group of specific lysine(s) in target proteins. The discovery that K2P1 channels are sumoylated was surprising because the SUMO pathway had not previously been recognized to operate beyond the nucleus, where it modulates the activity of transcription factors and import/export signaling (Geiss-Friedlander and Melchior, 2007).
Indeed, SUMO control of K2P1 channel activity was questioned (Feliciangeli et al., 2007) before being confirmed by detailed stoichiometric analysis and the study of purified reagents in mammalian cells. Using single-molecule spectroscopy, mass-spectrometry, Förster-resonance energy transfer, and patch-clamp recording, Plant et al. (2010) showed that K2P1 channels bear one SUMO on Lys274 in each subunit but that sumoylation of only one K2P1 subunit is required to silence the channel. When Lys274 is substituted for another amino acid, or when wild type channels are desumoylated by treatment with SUMO-specific protease (SENP), K2P1 channels pass open-rectifying, K+ selective leak currents (Rajan et al., 2005; Plant et al., 2010; Figures 2D–G and 3A). The initial discrepancies reported by Feliciangeli et al., have been proposed to arise from technical difficulties that include the analysis of small currents that do not correlate with the operation of K2P1 channels, the use of confounding protein tags (HcRed–K2P1) that influence subunit trafficking and the batch and seasonal variations that influence the behavior of X. laevis oocytes (Feliciangeli et al., 2007; Plant et al., 2010). The x-ray structure of human K2P1 shows that Lys274 is exposed on the C-helix, a domain previously associated with gating of channel pore (Miller and Long, 2012).
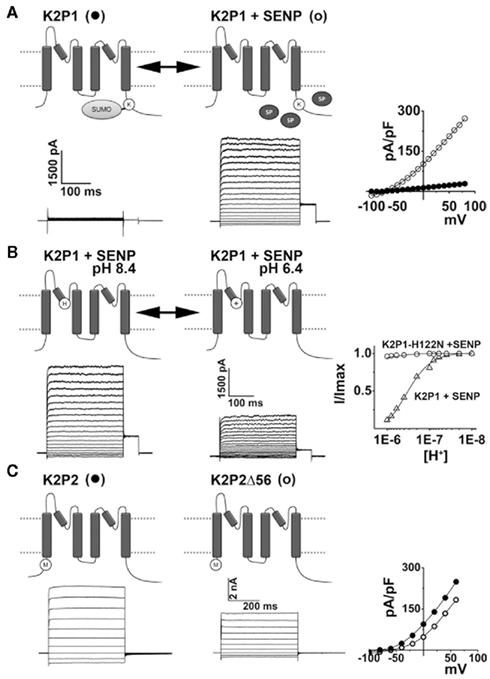
Figure 3. K2P channels are subject to regulation by diverse mechanisms. The operation of K2P channels is tightly controlled, both acutely and in the long-term, by a plethora of stimuli and regulatory pathways. (A) K2P1 channels are constitutively covalently modified at lysine residue 274 (K) by the small ubiquitin-like modifier protein, SUMO. Sumoylation silences K2P1 but is reversed by the action of SUMO-specific proteases (SENP, SP) to reveal active channels that mediate K+ selective leak currents in excitable cells such as neurons and cardiac myocytes. Example current-families and current–voltage relationships for K2P1 alone (●) and K2P1 in the presence of SENP (○) are shown; adapted from Plant et al. (2010). (B) Acid-sensitive K2P (TASK) channels have a histidine (H) adjacent to the K+ selectivity filter in the first P-loop. Thus, K2P3, K2P9, and active K2P1 channels pass currents that are reversibly blocked by protonation of this residue during acidification. Example currents-families for active K2P1 channels (in the presence of SENP) are shown at external pH 8.4 and 6.4. The proton-dependent block of active K2P1 channels is plotted (Δ) and shows that half-block occurs at pH 6.7. Active K2P1 channels in which the protonatable histidine is substituted for an asparagine residue are not sensitive to external acidification (○); adapted from Plant et al. (2010). K2P18 channels from rodents and other mammals are also acid-sensitive however, primate clones have a tyrosine rather than a histidine in the first P-loop and thus, human K2P18 channels are insensitive to acidification. (C) Full-length K2P2 channels mediate K+ selective leak currents. Alternative translation initiation of KCNK2 mRNA transcripts results in K2P2Δ, a subunits that lack the first 56 residues of the intracellular N-terminus. K2P2Δ is expressed throughout the brain and spinal cord at levels that change throughout development and pass smaller currents than full-length channels because of an increased permeability to Na+under physiological conditions. Example current-families and current–voltage relationships for full-length (●) and Δ56 variants of K2P2 (○) are shown; adapted from Thomas et al. (2008).
In addition to K2P1, SUMO modification is now recognized to impact the operation of a growing number of membrane proteins. Thus, the voltage-dependence of Kv2.1 channel activation is shifted in a graded manner, by up to 35 mV between fully sumoylated and desumoylated states to change the rate of action potential firing in hippocampal neurons (Plant et al., 2011). Sumoylation of Kv2.1 channels has also been reported to modulate the excitability of pancreatic β-cells (Dai et al., 2009). In addition, SUMO regulates the steady-state inactivation of Kv1.5 channels (Benson et al., 2007), as well as the endocytosis of neuronal GluR6 receptors (Martin et al., 2007), and a variant cardiac TRPM4 channels associated with progressive familial heart block type 1 (Kruse et al., 2009). Despite the pathophysiological importance of this post-translational modification, stimuli that alter the sumoylation status of channels, including K2P1 are yet to be identified.
Active K2P1 channels are blocked by external acidification because a histidine residue in the first P-loop is titrated by protons in the physiological pH range. When channels are studied in X. laevis oocytes or mammalian cells, half-block by protons (pKa) occurs at pH 6.7 (Rajan et al., 2005; Plant et al., 2010). This mechanism of acid-sensitivity, first recognized to operate in K2P3 channels, is shared by K2P9 and is discussed below (see also Figure 3B).
Recent evidence suggests that K2P1 channels may not be constitutively inactive in all tissues. Using shRNA to knockdown KCNK1 transcript levels, Ma et al. (2011) revealed that K2P1 channels contribute to the resting potential of cardiac myocytes. In addition to suggesting that at least some K2P1 channels are desumoylated in these cells, this work revealed that K2P1 channels might mediate the paradoxical depolarization of cardiac myocytes observed during hypokalemia (blood serum K+ concentrations below 3 mM). As the external concentration of K+ decreases relative to the internal concentration, EK will become more negative and so Vm is expected to hyperpolarize. Studies using a SUMO-insensitive K2P1 mutant (Lys274Glu) demonstrated that K2P1channels lose K+ selectivity during hypokalemia and pass a Na+ leak current of sufficient magnitude to depolarize Chinese hamster ovary (CHO) cells (Ma et al., 2011). Paradoxical depolarization is not observed in neuronal cells and dynamic changes in the selectivity of K2P1 channel, by this mechanism, are not expected to contribute to the operation of somatosensory neurons.
K2P2, K2P4, and K2P10 Channels: Thermal Rheostats?
K2P2 (previously TREK1) and K2P4 (TRAAK) were cloned from mouse brain (Fink et al., 1996, 1998) and K2P10 (TREK2) was cloned from human brain (Lesage et al., 2000) and rat cerebellar (Bang et al., 2000) cDNA libraries (Table 1). All three channels are widely expressed throughout the CNS (Medhurst et al., 2001; Talley et al., 2001; Heurteaux et al., 2006) and both K2P2 and K2P4 have been shown to traffic along peripheral nerves (Bearzatto et al., 2000). Using correlative single-channel analysis, Kang and Kim (2006) argue that of seven types of K+ channel identified, K2Ps 10 and K2P18 mediate more than ∼95% of the K+ leak current observed in cultured rat DRG cells at 37°C. However, other studies disagree on the relative expression levels of this group of channels in somatosensory neurons. Such discrepancies could reflection the detection of functionally active variants of K2P2 (Thomas et al., 2008), K2P4 (Ozaita and Vega-Saenz de Miera, 2002), and K2P10 channels (Gu et al., 2002; Simkin et al., 2008) as well as difference in expression between species and the heterogeneity of the nociceptors analyzed.
K2P2, K2P4, and K2P10 channels have been implicated in the transduction of thermal information (Maingret et al., 2000; Kang et al., 2005). From normal body temperature (37°C), the activity of this group of K2P channels increases with temperature, but falls at levels perceived as noxious heat (∼>43°C), increasing the excitability of nociceptors. The activity ratio for these channels is reported to increase by approximately sixfold for a 10°C rise in temperature (Q10). In contrast, the TRPV family of channels has a Q10 close to 20. TRPV1 opens in response to noxious heat while TRPV3 and TRPV4 channels are activated by more moderate, warm temperatures (∼>30°C; Peier et al., 2002b; Nilius et al., 2007).
The sensation of noxious cold (∼<18°C) is mediated by TRPM8 channels, an ion channel expressed by distinct groups of nociceptors (McKemy et al., 2002; Peier et al., 2002a). However, studies using mouse TG neurons show that cold depolarizes a subset of nociceptors with distinct biophysical properties by inhibiting a K+ leak current (Viana et al., 2002), mirroring similar effects observed in rat DRG cells (Reid and Flonta, 2001). While TRP channels are believed to be the primary receptors for the detection of both physiological and noxious extremes of temperature (Ramsey et al., 2006; Caterina, 2007), kcnk2, and kcnk4 knockout mice exhibit altered responses to cold and heat, suggesting that K2P2 and K2P4 channels modulate thermosensation by tuning the temperatures that elicits excitation of nociceptors (Noel et al., 2009). Consistent with this idea, immunochemical studies show that K2P2 and K2P4 channels co-localized with various TRP channels in rat TG cells (Yamamoto et al., 2009). K2P10 was also found to co-localization with TRP channels in rat TG cells (Yamamoto et al., 2009).
K2P2, K2P4, and K2P10 are proposed to respond to stimuli as diverse as free cellular lipids, including arachidonic acid, neurotransmitter activated second messengers pathways, anesthetics, and drugs (Honore, 2007; Enyedi and Czirjak, 2010). In addition, the activity of this family of channels is augmented by membrane stretch, leading to the suggestion that K2P2 and K2P4 channels act as mechanosensors (Maingret et al., 1999a,b; Alloui et al., 2006; Brohawn et al., 2012). This effect may indicate that these channels are tonically inhibited by an interaction with the cytoskeleton. In support of this notion, chemicals that disrupt cytoskeletal integrity, such as colchicine or cytochalasin D, activate the channels (Kim et al., 2001; Lauritzen et al., 2005). Given the breadth of stimuli that are reported to modulate the function of this group of channels, it is possible that K2P2, K2P4, and K2P10 act as polymodal signal integrators (Honore, 2007), important in responding to multiple stimuli including neurogenic and immune inflammatory signaling pathways, feedback control of neurotransmitter release in the CNS, and anesthesia.
The contribution of K2P2 channels to the operation of somatosensory neurons is likely to be subject to both acute and long-term regulatory mechanisms. Acute regulation was highlighted in a study that revealed one of the most surprising facets of the K2P2 channel. Phosphorylation of serine residue (Ser333) by protein kinase A converts hippocampal K2P2 channels from a K+ leak to a voltage-dependent phenotype (Bockenhauer et al., 2001). An intermediate phenotype was also observed in a number of recordings, using a mutant channel (Ser333Ala) that mimics dephosphorylation, suggesting a requirement for additional mechanistic factors to interconvert between voltage-dependent and independent behaviors. Subsequent studies have established that the voltage-dependence of K2P2 is modulated by phosphatidylinositol-4, 5-bisphosphate (PIP2). Increased cellular levels of PIP2 augment K2P2 channel currents and shift the voltage-dependence of opening toward more negative potentials (Chemin et al., 2005; Lopes et al., 2005).
Long-term changes in the biophysical properties of K2P2 channels result from alternative translation initiation (ATI) of KCNK2 mRNA transcripts (Thomas et al., 2008). Because KCNK2 has a weak Kozak sequence, translation can also be initiated at a second start-codon, located at nucleotide position 168–170, to yield an isoform of the channel that lacks the first 56 N-terminal residues of each subunit (K2P2Δ; Figure 3C). Although the truncation is intracellular, it seems to impact the operation of the K+ selectivity filter as K2P2Δ channels pass Na+ under physiological conditions with a relative permeability of 0.18 Na+ compared to K+, nearly an order or magnitude lower than that measured in full-length channels (0.02). This difference is sufficient to account for the depolarization observed in hippocampal neurons transfected to over express K2P2Δ. The ATI variant is expressed throughout the brain and spinal cord at levels that change throughout development (Thomas et al., 2008). ATI has also been reported for rat KCNK10 mRNA transcripts studied in experimental cells (Simkin et al., 2008). ATI variants of K2P10 subunits are also truncated a the N-terminus, yielding functional channels with increased single-channel conductance but no changes in selectivity for K+.
To date, the factors that regulation ATI and phosphorylation-dependent changes in the gating phenotype of K2P2 are not known. However, these pathways are expected to have profound effects on the operation of central neurons and nociceptors alike.
K2P3 and K2P9 form Acid-Sensitive Channels
K2P3 was cloned from human kidney (Duprat et al., 1997; Table 1). Originally called TASK for TWIK-related acid-sensitive K+ channel, K2P3 passes a current that is blocked by protonation of a histidine residue in the first P-loop of each subunit with a pKa of 6.4 (Lopes et al., 2000, 2001). This mechanism of acid-sensitivity is shared by K2P1 (Rajan et al., 2005; Plant et al., 2010) and K2P9 (previously TASK3), a channel cloned from rat cerebellum (Kim et al., 2000) and guinea-pig brain that has a pKa of ∼pH 6.0 (Rajan et al., 2000; Table 1). K2P3 and K2P9 channels are expressed throughout the body with particular prominence in the central and peripheral nervous systems, including in sensory neurons (Talley et al., 2000, 2001).
Both central neurons and nociceptors, including adult human DRG neurons in culture are depolarized by acidification (Baumann et al., 1996; Watkins and Mathie, 1996; Millar et al., 2000; Plant et al., 2002). A localized decrease in pH causes an intense, burning pain such as is caused by the administration of local anesthetic solutions, including lidocaine (pH 6.2–6.35) before they are buffered to a neutral pH (Moore, 1981), or by infusion of acidified saline (pH < 6.2; Steen and Reeh, 1993). Acidification also occurs as a consequence of tissue damage and is associated with the pain experienced during inflammation and ischemia (Mccarty et al., 1966; Jacobus et al., 1977).
TASK channels are proposed to mediate acidic-depolarization of nociceptive cells, including those isolated from rat DRGs (Cooper et al., 2004; Rau et al., 2006). ASICs 1–4 are also implicated in this process (Gold and Gebhart, 2010). However, whereas ASICs pass transient currents in response to acidification, prolonged depolarization is observed in many nociceptors, consistent with inhibition of K+ leak channels. In general, correlation between leak currents and cloned channels has been challenging, in part because of competing conductances but also because of the dearth of specific pharmacological probes and obfuscation of phenotypic changes in knockout animals. In addition, K2P3 and K2P9 subunits assemble to form both homo and heterodimeric TASK channels (Czirjak and Enyedi, 2002) with differential sensitivities to pH change, volatile anesthetics (Kang et al., 2004a), and pungent stimuli (Bautista et al., 2008).
Bautista et al. (2008) demonstrated that the tingling sensation experienced following the consumption of Szechuan peppercorns or other Xanthoxylum preparations result from the activation of a subsets of small-diameter, unmyelinated cells as well as large-diameter myelinated neurons. Hydroxy-α-sanshool was identified as being responsible for the anesthetic properties of these preparations and was shown to act by inhibiting K+ leak currents mediated by K2P3, K2P9, and K2P18 channels.
The contribution of TASK channels to cellular function is dependent on the number of channels at the cell membrane. Several competing signaling motifs have been identified that determine trafficking of K2P3 and K2P9 from the endoplasmic reticulum (ER) to the cell surface. Using yeast two-hybrid and biochemical analysis O’Kelly et al. (2002) demonstrated that forward trafficking of K2P3 requires phosphorylation-dependent binding of the ubiquitous, soluble adapter protein, 14-3-3β. 14-3-3 promotes forward trafficking by suppressing the interaction between K2P3 and βCOP, a COP1 vesicular transport protein that appears to hold the channel in the ER (O’Kelly et al., 2002). βCOP dependent ER retention was initially found to proceed via an N-terminal dibasic motif but was subsequently shown to be mediated by separate, basic motifs on the N- and C-termini of K2P3 subunits. Disruption of either site precluded ER retention (O’Kelly and Goldstein, 2008). In contrast, Zuzarte et al. (2009) demonstrated that βCOP interacts with a tribasic motif (KRR) on the C-terminus of K2P3 and that the activity of the N-terminal dibasic retention motif does not depend on 14-3-3 binding. The interplay between 14-3-3 and βCOP appears to regulate the forward trafficking of other membrane proteins including K2P9 (O’Kelly et al., 2002; Rajan et al., 2002; Zuzarte et al., 2009) and nicotinic acetylcholine receptors (O’Kelly et al., 2002). Forward trafficking of K2P9 was shown to require a di-acidic motif (EDE) located on the proximal C-terminus of the channel (Zuzarte et al., 2007). Mutation of the glutamate residues in this motif markedly reduced surface expression of K2P9, while mutation of the aspartate residue had no effects on surface density of the channel (Zuzarte et al., 2007).
K2P3 channels also interact with p11, an annexin II subunit (Girard et al., 2002). Several groups have explored the role of p11 in K2P3 channel trafficking with mixed results, demonstrating both that the subunits promotes (Girard et al., 2002; O’Kelly and Goldstein, 2008) and inhibits forward trafficking of K2P3 (Renigunta et al., 2006). O’Kelly and Goldstein suggested that p11 binds to K2P3 in a 14-3-3 dependent manner and, thereby, forms a ternary complex that modulates trafficking. This may occur in a tissue-dependent manner as p11 is prominently expressed in the brain and lung but not in the heart, where expression of K2P3 is high (O’Kelly and Goldstein, 2008). Of note, p11 has been shown to promote forward trafficking of other membrane proteins, including NaV1.8 (Okuse et al., 2002), TRPs V5 and V6 (van de Graaf et al., 2003), ASIC1a (Donier et al., 2005), and the serotonin receptor 5-HT1B (Svenningsson et al., 2006), all of which are expressed in various types of somatosensory neuron.
K2P18 Channels
K2P18 (also called TRESK for TWIK-related spinal cord K+ channel) was first identified in human spinal cord (Sano et al., 2003; Table 1), and subsequently in mouse cerebellum (Czirjak et al., 2004) and testis (Kang et al., 2004b) and is found in various centers of the brain (Liu et al., 2004). Expression of K2P18 channels is highest in the spinal cord, trigeminal, and DRG, where they contribute a significant component of the K+ leak current (Keshavaprasad et al., 2005; Kang and Kim, 2006; Dobler et al., 2007; Bautista et al., 2008).
In common with K2P1, K2P3, and K2P9, K2P18 channel currents are inhibited by acidification via protonation of a histidine in the first P-loop (pKa ∼ 6.8; Dobler et al., 2007; Figure 3B). However, in contrast to K2Ps 1, 3, and 9, this histidine is absent from primate isoforms of the channel. Human K2P18 channels have tyrosine residues adjacent to the K+ selectivity filter in the first P-loop and are insensitive to external acidification. Substituting the tyrosine with a histidine, to mimic the motif found in rodent isoforms, endows human K2P18 channels with almost identical proton sensitivity to the mouse clone.
K2P18 channels are reversibly activated by the calmodulin-dependent protein phosphatase, calcineurin, in response to increased levels of cytosolic Ca2+ (Czirjak et al., 2004; Czirjak and Enyedi, 2006). The kinetics by which Ca2+modulates the activity of K2P18 channels are dependent upon the phosphorylation of Ser264, a residue on the intracellular loop of K2P18 and subsequent binding of 14-3-3 proteins. Thus, function of K2P18 channels is inhibited by 14-3-3η and 14-3-3γ specifically while14-3-3β, ζ, ε, ς, and τ have no apparent impact on channel function (Czirjak et al., 2008).
Recent studies have demonstrated that the function of K2P18 channels is important for the normal operation of somatosensory neurons. Tulleuda et al. (2011) reported that axotomy of peripheral nerves leads to a ∼50% decrease in mRNA transcript levels for kcnk18 in rat DRG. Axotomy induces nociceptor hyperexcitability and an increased response to acute pain stimuli in whole animals. Lafreniere et al. (2010) studied a large, multigenerational family that presented with migraine with aura and found a frame-shift mutation (F139WfsX24) in the KCNK18 gene that was inherited dominantly and with full penetrance. Cellular and molecular studies reveal that F139WfsX24–K2P18 subunits are truncated at residue 162 residues and act, in a dominant-negative fashion to ablate the function of wild type K2P18 channels. Of note, potent immunosuppressive agents are reported to increase the frequency and intensity of headaches suffered by organ transplant patients via specific inhibition of calcineurin activity (Ferrari et al., 2005), a pathway known to decrease the activity of K2P18 channels (Czirjak et al., 2004; Czirjak and Enyedi, 2006). These studies highlight the importance of K2P channels as potential therapeutic targets for the treatment of both acute and chronic pain syndromes.
K2P Channels and General Anesthetics
Volatile anesthetics hyperpolarize neurons via effects on ligand-gated ion channels, including GABAA receptors (Franks and Lieb, 1994) and by augmenting K+ leak currents (Nicoll and Madison, 1982; Sirois et al., 1998). Several studies support important roles for K2P2, K2P10, (Patel et al., 1999), K2P3, K2P9 (Sirois et al., 2000; Talley and Bayliss, 2002; Lazarenko et al., 2010), and K2P18 channels (Liu et al., 2004) in mediating the effects of halogenated anesthetics agents such as halothane, isoflurane, and sevoflurane. K2P2 and K2P10 are also activated by chloroform and diethyl ether (Patel et al., 1999). Volatile anesthetics increase the activity of these channels, hyperpolarizing Vm and decreasing excitability. While volatile anesthetics appear to have the most pronounced effects on K2P18 in humans, marked species differences have been reported (Keshavaprasad et al., 2005). Both kcnk2 and kcnk9 knockout mice demonstrate increased tolerance for halogenated volatile anesthetics arguing for roles for K2P2 and K2P9 channels in mediating anesthesia.
Mechanistic studies have elucidated an interaction between volatile anesthetics and the proximal C-terminus of K2P2, K2P3 (Patel et al., 1999), and K2P9 (Talley and Bayliss, 2002). Disruption of this portion of the channels also prevented G-protein coupled receptor mediated inhibition by interrupting the binding of Gαq signaling molecules. These data suggest a convergent mechanism whereby volatile anesthetics augment K2P channel function via suppression of the constitutive activity of Gαq proteins (Conway and Cotten, 2012).
Conclusion
Since the revelation of 1995, evidence has accumulated to show that members of the K2P family of K+ channels determine the excitability of DRG and TG neurons and act as mechano-, thermo-, and chemoreceptors at the termini of nociceptive fibers. Accordingly, numerous, disparate stimuli, and regulatory mechanisms control the activity of K2P channels, tuning the sensitivity of nociceptors to chemical and physical changes in their microenvironment.
Despite the redoubtable importance of K2P channels to human health and disease, their utility as targets for pharmacological and therapeutic strategies has yet to be widely explored. Recent crystals structures of the human channels K2P1 and K2P4 (Brohawn et al., 2012; Miller and Long, 2012) mark a renaissance in our understanding of the structure–function relationships of these enigmatic channels. For example, both crystal structures have identified a “cap-domain” positioned directly above the outer mouth of the pore, bifurcating the K+ conduction pathway (Brohawn et al., 2012; Miller and Long, 2012). It is interesting to speculate that this large structural motif sterically precludes interactions between the pore of K2P channels and peptide toxins, to which they are notoriously insensitive. While K2P channels are, in general, sensitive to classic, K+ channel pore-blocking ions such as Ba2+, tetraethylammonium (TEA), and quinine, pharmaceuticals capable of discerning specific K+ channel subtypes are not yet commonplace. The pivotal role played by K2P channels to mediate general anesthesia argues for continued efforts to identify or engineer novel anesthetic and analgesic agents that are capable of targeting specific K2P-subtypes amongst the hordes of proteins that populate the membranes of excitable cells.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Alloui, A., Zimmermann, K., Mamet, J., Duprat, F., Noel, J., Chemin, J., Guy, N., Blondeau, N., Voilley, N., Rubat-Coudert, C., Borsotto, M., Romey, G., Heurteaux, C., Reeh, P., Eschalier, A., and Lazdunski, M. (2006). TREK-1, a K+ channel involved in polymodal pain perception. EMBO J. 25, 2368–2376.
Baccaglini, P. I. (1978). Action potentials of embryonic dorsal root ganglion neurones in Xenopus tadpoles. J. Physiol. (Lond.) 283, 585–604.
Bang, H., Kim, Y., and Kim, D. (2000). TREK-2, a new member of the mechanosensitive tandem-pore K+ channel family. J. Biol. Chem. 275, 17412–17419.
Basbaum, A. I., Bautista, D. M., Scherrer, G., and Julius, D. (2009). Cellular and molecular mechanisms of pain. Cell 139, 267–284.
Baumann, T. K., Burchiel, K. J., Ingram, S. L., and Martenson, M. E. (1996). Responses of adult human dorsal root ganglion neurons in culture to capsaicin and low pH. Pain 65, 31–38.
Bautista, D. M., Sigal, Y. M., Milstein, A. D., Garrison, J. L., Zorn, J. A., Tsuruda, P. R., Nicoll, R. A., and Julius, D. (2008). Pungent agents from Szechuan peppers excite sensory neurons by inhibiting two-pore potassium channels. Nat. Neurosci. 11, 772–779.
Bearzatto, B., Lesage, F., Reyes, R., Lazdunski, M., and Laduron, P. M. (2000). Axonal transport of TREK and TRAAK potassium channels in rat sciatic nerves. Neuroreport 11, 927–930.
Benson, M. D., Li, Q. J., Kieckhafer, K., Dudek, D., Whorton, M. R., Sunahara, R. K., Iniguez-Lluhi, J. A., and Martens, J. R. (2007). SUMO modification regulates inactivation of the voltage-gated potassium channel Kv1.5. Proc. Natl. Acad. Sci. U.S.A. 104, 1805–1810.
Bockenhauer, D., Zilberberg, N., and Goldstein, S. A. (2001). KCNK2: reversible conversion of a hippocampal potassium leak into a voltage-dependent channel. Nat. Neurosci. 4, 486–491.
Brohawn, S. G., del Marmol, J., and MacKinnon, R. (2012). Crystal structure of the human K2P TRAAK, a lipid- and mechano-sensitive K+ ion channel. Science 335, 436–441.
Brown, D. A., and Adams, P. R. (1980). Muscarinic suppression of a novel voltage-sensitive K+ current in a vertebrate neurone. Nature 283, 673–676.
Buckler, K. J. (1997). A novel oxygen-sensitive potassium current in rat carotid body type I cells. J. Physiol. (Lond.) 498(Pt 3), 649–662.
Caterina, M. J. (2007). Transient receptor potential ion channels as participants in thermosensation and thermoregulation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 292, R64–R76.
Chemin, J., Patel, A. J., Duprat, F., Lauritzen, I., Lazdunski, M., and Honore, E. (2005). A phospholipid sensor controls mechanogating of the K+ channel TREK-1. EMBO J. 24, 44–53.
Conway, K. E., and Cotten, J. F. (2012). Covalent modification of a volatile anesthetic regulatory site activates TASK-3 (KCNK9) tandem pore potassium channels. Mol. Pharmacol. 81, 393–400.
Cooper, B. Y., Johnson, R. D., and Rau, K. K. (2004). Characterization and function of twik-related acid sensing K+ channels in a rat nociceptive cell. Neuroscience 129, 209–224.
Czirjak, B., Toth, Z. E., and Enyedi, P. (2004). The two-pore domain K+ channel, TRESK, is activated by the cytoplasmic calcium signal through calcineurin. J. Biol. Chem. 279, 18550–18558.
Czirjak, G., and Enyedi, P. (2002). Formation of functional heterodimers between the TASK-1 and TASK-3 two-pore domain potassium channel subunits. J. Biol. Chem. 277, 5426–5432.
Czirjak, G., and Enyedi, P. (2006). Targeting of calcineurin to an NFAT-like docking site is required for the calcium-dependent activation of the background K(+)channel, TRESK. J. Biol. Chem. 281, 14677–14682.
Czirjak, G., Vuity, D., and Enyedi, P. (2008). Phosphorylation-dependent binding of 14-3-3 proteins controls TRESK regulation. J. Biol. Chem. 283, 15672–15680.
Dai, X. Q., Kolic, J., Marchi, P., Sipione, S., and Macdonald, P. E. (2009). SUMOylation regulates Kv2.1 and modulates pancreatic beta-cell excitability. J. Cell. Sci. 122, 775–779.
Delmas, P., and Brown, D. A. (2005). Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat. Rev. Neurosci. 6, 850–862.
Dobler, T., Springauf, A., Tovornik, S., Weber, M., Schmitt, A., Sedlmeier, R., Wischmeyer, E., and Doring, F. (2007). TRESK two-pore-domain K(+) channels constitute a significant component of background potassium currents in murine dorsal root ganglion neurones. J. Physiol. (Lond.) 585, 867–879.
Donier, E., Rugiero, F., Okuse, K., and Wood, J. N. (2005). Annexin II light chain p11 promotes functional expression of acid-sensing ion channel ASIC1a. J. Biol. Chem. 280, 38666–38672.
Doyle, D. A., Morais Cabral, J., Pfuetzner, R. A., Kuo, A., Gulbis, J. M., Cohen, S. L., Chait, B. T., and MacKinnon, R. (1998). The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science 280, 69–77.
Duprat, F., Lesage, F., Fink, M., Reyes, R., Heurteaux, C., and Lazdunski, M. (1997). TASK, a human background K+ channel to sense external pH variations near physiological pH. EMBO J. 16, 5464–5471.
Enyedi, P., and Czirjak, G. (2010). Molecular background of leak K+ currents: two-pore domain potassium channels. Physiol. Rev. 90, 559–605.
Feliciangeli, S., Bendahhou, S., Sandoz, G., Gounon, P., Reichold, M., Warth, R., Lazdunski, M., Barhanin, J., and Lesage, F. (2007). Does sumoylation control K2P1/TWIK1 background K+ channels? Cell 130, 563–569.
Ferrari, U., Empl, M., Kim, K. S., Sostak, P., Forderreuther, S., and Straube, A. (2005). Calcineurin inhibitor-induced headache: clinical characteristics and possible mechanisms. Headache 45, 211–214.
Fink, M., Duprat, F., Lesage, F., Reyes, R., Romey, G., Heurteaux, C., and Lazdunski, M. (1996). Cloning, functional expression and brain localization of a novel unconventional outward rectifier K+ channel. EMBO J. 15, 6854–6862.
Fink, M., Lesage, F., Duprat, F., Heurteaux, C., Reyes, R., Fosset, M., and Lazdunski, M. (1998). A neuronal two P domain K+ channel stimulated by arachidonic acid and polyunsaturated fatty acids. EMBO J. 17, 3297–3308.
Franks, N. P., and Lieb, W. R. (1994). Molecular and cellular mechanisms of general anaesthesia. Nature 367, 607–614.
Gamper, N., Zaika, O., Li, Y., Martin, P., Hernandez, C. C., Perez, M. R., Wang, A. Y., Jaffe, D. B., and Shapiro, M. S. (2006). Oxidative modification of M-type K(+) channels as a mechanism of cytoprotective neuronal silencing. EMBO J. 25, 4996–5004.
Geiss-Friedlander, R., and Melchior, F. (2007). Concepts in sumoylation: a decade on. Nat. Rev. Mol. Cell Biol. 8, 947–956.
Girard, C., Tinel, N., Terrenoire, C., Romey, G., Lazdunski, M., and Borsotto, M. (2002). p11, an annexin II subunit, an auxiliary protein associated with the background K+ channel, TASK-1. EMBO J. 21, 4439–4448.
Gold, M. S., and Gebhart, G. F. (2010). Nociceptor sensitization in pain pathogenesis. Nat. Med. 16, 1248–1257.
Goldman, D. E. (1943). Potential, impedance, and rectification in membranes. J. Gen. Physiol. 27, 37–60.
Goldstein, S. A., Bockenhauer, D., O’Kelly, I., and Zilberberg, N. (2001). Potassium leak channels and the KCNK family of two-P-domain subunits. Nat. Rev. Neurosci. 2, 175–184.
Goldstein, S. A., Price, L. A., Rosenthal, D. N., and Pausch, M. H. (1996). ORK1, a potassium-selective leak channel with two pore domains cloned from Drosophila melanogaster by expression in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 93, 13256–13261.
Goldstein, S. A., Wang, K. W., Ilan, N., and Pausch, M. H. (1998). Sequence and function of the two P domain potassium channels: implications of an emerging superfamily. J. Mol. Med. (Berl.) 76, 13–20.
Gu, W., Schlichthorl, G., Hirsch, J. R., Engels, H., Karschin, C., Karschin, A., Derst, C., Steinlein, O. K., and Daut, J. (2002). Expression pattern and functional characteristics of two novel splice variants of the two-pore-domain potassium channel TREK-2. J. Physiol. (Lond.) 539, 657–668.
Heurteaux, C., Lucas, G., Guy, N., El Yacoubi, M., Thummler, S., Peng, X. D., Noble, F., Blondeau, N., Widmann, C., Borsotto, M., Gobbi, G., Vaugeois, J. M., Debonnel, G., and Lazdunski, M. (2006). Deletion of the background potassium channel TREK-1 results in a depression-resistant phenotype. Nat. Neurosci. 9, 1134–1141.
Hodgkin, A. L., and Huxley, A. F. (1952). A quantitative description of membrane current and its application to conduction and excitation in nerve. J. Physiol. (Lond.) 117, 500–544.
Hodgkin, A. L., Huxley, A. F., and Katz, B. (1949). Ionic currents underlying activity in the giant axon of the squid. Arch. Sci. Physiol. (Paris) 3, 129–150.
Honore, E. (2007). The neuronal background K2P channels: focus on TREK1. Nat. Rev. Neurosci. 8, 251–261.
Ilan, N., and Goldstein, S. A. (2001). Kcnko: single, cloned potassium leak channels are multi-ion pores. Biophys. J. 80, 241–253.
Jacobus, W. E., Taylor, G. J., Hollis, D. P., and Nunnally, R. L. (1977). Phosphorus nuclear magnetic-resonance of perfused working rat hearts. Nature 265, 756–758.
Jones, S. W. (1989). On the resting potential of isolated frog sympathetic neurons. Neuron 3, 153–161.
Kandel, E. R., Schwartz, J. H., and Jessell, T. M. (2000). Principles of Neural Science. New York: McGraw-Hill, Health Professions Division, 1414.
Kang, D., Choe, C., and Kim, D. (2005). Thermosensitivity of the two-pore domain K+ channels TREK-2 and TRAAK. J. Physiol. (Lond.) 564, 103–116.
Kang, D., Han, J., Talley, E. M., Bayliss, D. A., and Kim, D. (2004a). Functional expression of TASK-1/TASK-3 heteromers in cerebellar granule cells. J. Physiol. (Lond.) 554, 64–77.
Kang, D., Mariash, E., and Kim, D. (2004b). Functional expression of TRESK-2, a new member of the tandem-pore K+ channel family. J. Biol. Chem. 279, 28063–28070.
Kang, D., and Kim, D. (2006). TREK-2 (K2P10.1) and TRESK (K2P18.1) are major background K+ channels in dorsal root ganglion neurons. Am. J. Physiol. Cell Physiol. 291, C138–C146.
Keshavaprasad, B., Liu, C., Au, J. D., Kindler, C. H., Cotten, J. F., and Yost, C. S. (2005). Species-specific differences in response to anesthetics and other modulators by the K2P channel TRESK. Anesth. Analg. 101, 1042–1049.
Ketchum, K. A., Joiner, W. J., Sellers, A. J., Kaczmarek, L. K., and Goldstein, S. A. (1995). A new family of outwardly rectifying potassium channel proteins with two pore domains in tandem. Nature 376, 690–695.
Kim, Y., Bang, H., Gnatenco, C., and Kim, D. (2001). Synergistic interaction and the role of C-terminus in the activation of TRAAK K+ channels by pressure, free fatty acids and alkali. Pflugers Arch. 442, 64–72.
Kim, Y., Bang, H., and Kim, D. (2000). TASK-3, a new member of the tandem pore K(+) channel family. J. Biol. Chem. 275, 9340–9347.
Kollewe, A., Lau, A. Y., Sullivan, A., Roux, B., and Goldstein, S. A. (2009). A structural model for K2P potassium channels based on 23 pairs of interacting sites and continuum electrostatics. J. Gen. Physiol. 134, 53–68.
Kruse, M., Schulze-Bahr, E., Corfield, V., Beckmann, A., Stallmeyer, B., Kurtbay, G., Ohmert, I., Brink, P., and Pongs, O. (2009). Impaired endocytosis of the ion channel TRPM4 is associated with human progressive familial heart block type I. J. Clin. Invest. 119, 2737–2744.
Lafreniere, R. G., Cader, M. Z., Poulin, J. F., Andres-Enguix, I., Simoneau, M., Gupta, N., Boisvert, K., Lafreniere, F., McLaughlan, S., Dube, M. P., Marcinkiewicz, M. M., Ramagopalan, S., Ansorge, O., Brais, B., Sequeiros, J., Pereira-Monteiro, J. M., Griffiths, L. R., Tucker, S. J., Ebers, G., and Rouleau, G. A. (2010). A dominant-negative mutation in the TRESK potassium channel is linked to familial migraine with aura. Nat. Med. 16, 1157–U1501.
Lauritzen, I., Chemin, J., Honore, E., Jodar, M., Guy, N., Lazdunski, M., and Jane Patel, A. (2005). Cross-talk between the mechano-gated K2P channel TREK-1 and the actin cytoskeleton. EMBO Rep. 6, 642–648.
Lazarenko, R. M., Willcox, S. C., Shu, S., Berg, A. P., Jevtovic-Todorovic, V., Talley, E. M., Chen, X., and Bayliss, D. A. (2010). Motoneuronal TASK channels contribute to immobilizing effects of inhalational general anesthetics. J. Neurosci. 30, 7691–7704.
Lesage, F., Guillemare, E., Fink, M., Duprat, F., Lazdunski, M., Romey, G., and Barhanin, J. (1996). TWIK-1, a ubiquitous human weakly inward rectifying K+ channel with a novel structure. EMBO J. 15, 1004–1011.
Lesage, F., Terrenoire, C., Romey, G., and Lazdunski, M. (2000). Human TREK2, a 2P domain mechano-sensitive K+ channel with multiple regulations by polyunsaturated fatty acids, lysophospholipids, and Gs, Gi, and Gq protein-coupled receptors. J. Biol. Chem. 275, 28398–28405.
Liu, C., Au, J. D., Zou, H. L., Cotten, J. F., and Yost, C. S. (2004). Potent activation of the human tandem pore domain K channel TRESK with clinical concentrations of volatile anesthetics. Anesth. Analg. 99, 1715–1722.
Long, S. B., Campbell, E. B., and Mackinnon, R. (2005). Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science 309, 897–903.
Lopes, C. M., Gallagher, P. G., Buck, M. E., Butler, M. H., and Goldstein, S. A. (2000). Proton block and voltage gating are potassium-dependent in the cardiac leak channel Kcnk3. J. Biol. Chem. 275, 16969–16978.
Lopes, C. M., Rohacs, T., Czirjak, G., Balla, T., Enyedi, P., and Logothetis, D. E. (2005). PIP2 hydrolysis underlies agonist-induced inhibition and regulates voltage gating of two-pore domain K+ channels. J. Physiol. (Lond.) 564, 117–129.
Lopes, C. M., Zilberberg, N., and Goldstein, S. A. (2001). Block of Kcnk3 by protons. Evidence that 2-P-domain potassium channel subunits function as homodimers. J. Biol. Chem. 276, 24449–24452.
Lopez, H. S., and Adams, P. R. (1989). A G protein mediates the inhibition of the voltage-dependent potassium M current by muscarine, LHRH, substance P and UTP in bullfrog sympathetic neurons. Eur. J. Neurosci. 1, 529–542.
Ma, L., Zhang, X., and Chen, H. (2011). TWIK-1 two-pore domain potassium channels change ion selectivity and conduct inward leak sodium currents in hypokalemia. Sci. Signal. 4, ra37.
Maingret, F., Fosset, M., Lesage, F., Lazdunski, M., and Honore, E. (1999a). TRAAK is a mammalian neuronal mechano-gated K+ channel. J. Biol. Chem. 274, 1381–1387.
Maingret, F., Patel, A. J., Lesage, F., Lazdunski, M., and Honore, E. (1999b). Mechano- or acid stimulation, two interactive modes of activation of the TREK-1 potassium channel. J. Biol. Chem. 274, 26691–26696.
Maingret, F., Lauritzen, I., Patel, A. J., Heurteaux, C., Reyes, R., Lesage, F., Lazdunski, M., and Honore, E. (2000). TREK-1 is a heat-activated background K(+) channel. EMBO J. 19, 2483–2491.
Martin, S., Nishimune, A., Mellor, J. R., and Henley, J. M. (2007). SUMOylation regulates kainate-receptor-mediated synaptic transmission. Nature 447, 321–325.
Mccarty, D. J., Phelps, P., and Pyenson, J. (1966). Crystal-induced inflammation in canine joints.1. An experimental model with quantification of host response. J. Exp. Med. 124, 99.
McKemy, D. D., Neuhausser, W. M., and Julius, D. (2002). Identification of a cold receptor reveals a general role for TRP channels in thermosensation. Nature 416, 52–58.
Medhurst, A. D., Rennie, G., Chapman, C. G., Meadows, H., Duckworth, M. D., Kelsell, R. E., Gloger, I. I., and Pangalos, M. N. (2001). Distribution analysis of human two pore domain potassium channels in tissues of the central nervous system and periphery. Brain Res. Mol. Brain Res. 86, 101–114.
Millar, J. A., Barratt, L., Southan, A. P., Page, K. M., Fyffe, R. E., Robertson, B., and Mathie, A. (2000). A functional role for the two-pore domain potassium channel TASK-1 in cerebellar granule neurons. Proc. Natl. Acad. Sci. U.S.A. 97, 3614–3618.
Miller, A. N., and Long, S. B. (2012). Crystal structure of the human two-pore domain potassium channel K2P1. Science 335, 432–436.
Nattel, S., Elharrar, V., Zipes, D. P., and Bailey, J. C. (1981). pH-dependent electrophysiological effects of quinidine and lidocaine on canine cardiac purkinje fibers. Circ. Res. 48, 55–61.
Nicoll, R. A., and Madison, D. V. (1982). General anesthetics hyperpolarize neurons in the vertebrate central nervous system. Science 217, 1055–1057.
Nilius, B., Owsianik, G., Voets, T., and Peters, J. A. (2007). Transient receptor potential cation channels in disease. Physiol. Rev. 87, 165–217.
Noel, J., Zimmermann, K., Busserolles, J., Deval, E., Alloui, A., Diochot, S., Guy, N., Borsotto, M., Reeh, P., Eschalier, A., and Lazdunski, M. (2009). The mechano-activated K+ channels TRAAK and TREK-1 control both warm and cold perception. EMBO J. 28, 1308–1318.
O’Kelly, I., Butler, M. H., Zilberberg, N., and Goldstein, S. A. (2002). Forward transport. 14-3-3 binding overcomes retention in endoplasmic reticulum by dibasic signals. Cell 111, 577–588.
O’Kelly, I., and Goldstein, S. A. (2008). Forward Transport of K2p3.1: mediation by 14-3-3 and COPI, modulation by p11. Traffic 9, 72–78.
Okuse, K., Malik-Hall, M., Baker, M. D., Poon, W. Y., Kong, H., Chao, M. V., and Wood, J. N. (2002). Annexin II light chain regulates sensory neuron-specific sodium channel expression. Nature 417, 653–656.
Ozaita, A., and Vega-Saenz de Miera, E. (2002). Cloning of two transcripts, HKT4.1a and HKT4.1b, from the human two-pore K+ channel gene KCNK4. Chromosomal localization, tissue distribution and functional expression. Brain Res. Mol. Brain Res. 102, 18–27.
Passmore, G. M., Selyanko, A. A., Mistry, M., Al-Qatari, M., Marsh, S. J., Matthews, E. A., Dickenson, A. H., Brown, T. A., Burbidge, S. A., Main, M., and Brown, D. A. (2003). KCNQ/M currents in sensory neurons: significance for pain therapy. J. Neurosci. 23, 7227–7236.
Patel, A. J., Honore, E., Lesage, F., Fink, M., Romey, G., and Lazdunski, M. (1999). Inhalational anesthetics activate two-pore-domain background K+channels. Nat. Neurosci. 2, 422–426.
Peier, A. M., Moqrich, A., Hergarden, A. C., Reeve, A. J., Andersson, D. A., Story, G. M., Earley, T. J., Dragoni, I., McIntyre, P., Bevan, S., and Patapoutian, A. (2002a). A TRP channel that senses cold stimuli and menthol. Cell 108, 705–715.
Peier, A. M., Reeve, A. J., Andersson, D. A., Moqrich, A., Earley, T. J., Hergarden, A. C., Story, G. M., Colley, S., Hogenesch, J. B., McIntyre, P., Bevan, S., and Patapoutian, A. (2002b). A heat-sensitive TRP channel expressed in keratinocytes. Science 296, 2046–2049.
Plant, L. D., Bayliss, D. A., Kim, D., Lesage, F., and Goldstein, S. A. (2005). International Union of Pharmacology. LV. Nomenclature and molecular relationships of two-P potassium channels. Pharmacol. Rev. 57, 527–540.
Plant, L. D., Dementieva, I. S., Kollewe, A., Olikara, S., Marks, J. D., and Goldstein, S. A. (2010). One SUMO is sufficient to silence the dimeric potassium channel K2P1. Proc. Natl. Acad. Sci. U.S.A. 107, 10743–10748.
Plant, L. D., Dowdell, E. J., Dementieva, I. S., Marks, J. D., and Goldstein, S. A. (2011). SUMO modification of cell surface Kv2.1 potassium channels regulates the activity of rat hippocampal neurons. J. Gen. Physiol. 137, 441–454.
Plant, L. D., Kemp, P. J., Peers, C., Henderson, Z., and Pearson, H. A. (2002). Hypoxic depolarization of cerebellar granule neurons by specific inhibition of TASK-1. Stroke 33, 2324–2328.
Pountney, D. J., Gulkarov, I., Vega-Saenz de Miera, E., Holmes, D., Saganich, M., Rudy, B., Artman, M., and Coetzee, W. A. (1999). Identification and cloning of TWIK-originated similarity sequence (TOSS): a novel human 2-pore K+ channel principal subunit. FEBS Lett. 450, 191–196.
Puil, E., Gimbarzevsky, B., and Miura, R. M. (1987). Voltage dependence of membrane properties of trigeminal root ganglion neurons. J. Neurophysiol. 58, 66–86.
Rajan, S., Plant, L. D., Rabin, M. L., Butler, M. H., and Goldstein, S. A. (2005). Sumoylation silences the plasma membrane leak K+ channel K2P1. Cell 121, 37–47.
Rajan, S., Preisig-Muller, R., Wischmeyer, E., Nehring, R., Hanley, P. J., Renigunta, V., Musset, B., Schlichthorl, G., Derst, C., Karschin, A., and Daut, J. (2002). Interaction with 14-3-3 proteins promotes functional expression of the potassium channels TASK-1 and TASK-3. J. Physiol. (Lond.) 545, 13–26.
Rajan, S., Wischmeyer, E., Xin Liu, G., Preisig-Muller, R., Daut, J., Karschin, A., and Derst, C. (2000). TASK-3, a novel tandem pore domain acid-sensitive K+ channel. An extracellular histiding as pH sensor. J. Biol. Chem. 275, 16650–16657.
Ramsey, I. S., Delling, M., and Clapham, D. E. (2006). An introduction to TRP channels. Annu. Rev. Physiol. 68, 619–647.
Rau, K. K., Cooper, B. Y., and Johnson, R. D. (2006). Expression of TWIK-related acid sensitive K+ channels in capsaicin sensitive and insensitive cells of rat dorsal root ganglia. Neuroscience 141, 955–963.
Reid, G., and Flonta, M. (2001). Cold transduction by inhibition of a background potassium conductance in rat primary sensory neurones. Neurosci. Lett. 297, 171–174.
Renigunta, V., Yuan, H., Zuzarte, M., Rinne, S., Koch, A., Wischmeyer, E., Schlichthorl, G., Gao, Y., Karschin, A., Jacob, R., Schwappach, B., Daut, J., and Preisig-Muller, R. (2006). The retention factor p11 confers an endoplasmic reticulum-localization signal to the potassium channel TASK-1. Traffic 7, 168–181.
Sano, Y., Inamura, K., Miyake, A., Mochizuki, S., Kitada, C., Yokoi, H., Nozawa, K., Okada, H., Matsushime, H., and Furuichi, K. (2003). A novel two-pore domain K(+) channel, TRESK, is localized in the spinal cord. J. Biol. Chem. 278, 27406–27412.
Shen, K. Z., North, R. A., and Surprenant, A. (1992). Potassium channels opened by noradrenaline and other transmitters in excised membrane patches of guinea-pig submucosal neurones. J. Physiol. (Lond.) 445, 581–599.
Siegelbaum, S. A., Camardo, J. S., and Kandel, E. R. (1982). Serotonin and cyclic AMP close single K+ channels in Aplysia sensory neurones. Nature 299, 413–417.
Simkin, D., Cavanaugh, E. J., and Kim, D. (2008). Control of the single channel conductance of K2P10.1 (TREK-2) by the amino-terminus: role of alternative translation initiation. J. Physiol. (Lond.) 586, 5651–5663.
Sirois, J. E., Lei, Q., Talley, E. M., Lynch, C. III, and Bayliss, D. A. (2000). The TASK-1 two-pore domain K+ channel is a molecular substrate for neuronal effects of inhalation anesthetics. J. Neurosci. 20, 6347–6354.
Sirois, J. E., Pancrazio, J. J., Iii, C. L., and Bayliss, D. A. (1998). Multiple ionic mechanisms mediate inhibition of rat motoneurones by inhalation anaesthetics. J. Physiol. (Lond.) 512(Pt 3), 851–862.
Steen, K. H., and Reeh, P. W. (1993). Sustained graded pain and hyperalgesia from harmless experimental tissue acidosis in human skin. Neurosci. Lett. 154, 113–116.
Streit, A. K., Netter, M. F., Kempf, F., Walecki, M., Rinne, S., Bollepalli, M. K., Preisig-Muller, R., Renigunta, V., Daut, J., Baukrowitz, T., Sansom, M. S., Stansfeld, P. J., and Decher, N. (2011). A specific two-pore domain potassium channel blocker defines the structure of the TASK-1 open pore. J. Biol. Chem. 286, 13977–13984.
Svenningsson, P., Chergui, K., Rachleff, I., Flajolet, M., Zhang, X., El Yacoubi, M., Vaugeois, J. M., Nomikos, G. G., and Greengard, P. (2006). Alterations in 5-HT1B receptor function by p11 in depression-like states. Science 311, 77–80.
Talley, E. M., and Bayliss, D. A. (2002). Modulation of TASK-1 (Kcnk3) and TASK-3 (Kcnk9) potassium channels: volatile anesthetics and neurotransmitters share a molecular site of action. J. Biol. Chem. 277, 17733–17742.
Talley, E. M., Lei, Q., Sirois, J. E., and Bayliss, D. A. (2000). TASK-1, a two-pore domain K+ channel, is modulated by multiple neurotransmitters in motoneurons. Neuron 25, 399–410.
Talley, E. M., Solorzano, G., Lei, Q. B., Kim, D., and Bayliss, D. A. (2001). CNS distribution of members of the two-pore-domain (KCNK) potassium channel family. J. Neurosci. 21, 7491–7505.
Thomas, D., Plant, L. D., Wilkens, C. M., McCrossan, Z. A., and Goldstein, S. A. (2008). Alternative translation initiation in rat brain yields K2P2.1 potassium channels permeable to sodium. Neuron 58, 859–870.
Tulleuda, A., Cokic, B., Callejo, G., Saiani, B., Serra, J., and Gasull, X. (2011). TRESK channel contribution to nociceptive sensory neurons excitability: modulation by nerve injury. Mol. Pain 7, 1–17.
van de Graaf, S. F., Hoenderop, J. G., Gkika, D., Lamers, D., Prenen, J., Rescher, U., Gerke, V., Staub, O., Nilius, B., and Bindels, R. J. (2003). Functional expression of the epithelial Ca(2+) channels (TRPV5 and TRPV6) requires association of the S100A10-annexin 2 complex. EMBO J. 22, 1478–1487.
Viana, F., de la Pena, E., and Belmonte, C. (2002). Specificity of cold thermotransduction is determined by differential ionic channel expression. Nat. Neurosci. 5, 254–260.
Villarroel, A., Marrion, N. V., Lopez, H., and Adams, P. R. (1989). Bradykinin inhibits a potassium M-like current in rat pheochromocytoma PC12 cells. FEBS Lett. 255, 42–46.
Wang, Z., Van den Berg, R. J., and Ypey, D. L. (1994). Resting membrane potentials and excitability at different regions of rat dorsal root ganglion neurons in culture. Neuroscience 60, 245–254.
Watkins, C. S., and Mathie, A. (1996). A non-inactivating K+ current sensitive to muscarinic receptor activation in rat cultured cerebellar granule neurons. J. Physiol. (Lond.) 491(Pt 2):401–412.
Yamamoto, Y., Hatakeyama, T., and Taniguchi, K. (2009). Immunohistochemical colocalization of TREK-1, TREK-2 and TRAAK with TRP channels in the trigeminal ganglion cells. Neurosci. Lett. 454, 129–133.
Yellen, G., Jurman, M. E., Abramson, T., and MacKinnon, R. (1991). Mutations affecting internal TEA blockade identify the probable pore-forming region of a K+ channel. Science 251, 939–942.
Yu, F. H., Yarov-Yarovoy, V., Gutman, G. A., and Catterall, W. A. (2005). Overview of molecular relationships in the voltage-gated ion channel superfamily. Pharmacol. Rev. 57, 387–395.
Zilberberg, N., Ilan, N., Gonzalez-Colaso, R., and Goldstein, S. A. (2000). Opening and closing of KCNKO potassium leak channels is tightly regulated. J. Gen. Physiol. 116, 721–734.
Zuzarte, M., Heusser, K., Renigunta, V., Schlichthorl, G., Rinne, S., Wischmeyer, E., Daut, J., Schwappach, B., and Preisig-Muller, R. (2009). Intracellular traffic of the K+ channels TASK-1 and TASK-3: role of N- and C-terminal sorting signals and interaction with 14-3-3 proteins. J. Physiol. (Lond.) 587, 929–952.
Keywords: K2P1, K2P2, K2P3, K2P4, K2P9, K2P10, K1P18, nociceptor
Citation: Plant LD (2012) A role for K2P channels in the operation of somatosensory nociceptors. Front. Mol. Neurosci. 5:21. doi: 10.3389/fnmol.2012.00021
Received: 15 December 2011;
Accepted: 09 February 2012;
Published online: 05 March 2012.
Edited by:
Nikita Gamper, University of Leeds, UKReviewed by:
Chris Peers, University of Leeds, UKNiels Decher, Philipps-University Marburg, Germany
Copyright: © 2012 Plant. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Leigh D. Plant, Department of Biochemistry, Brandeis University, 415 South Street, Waltham, MA 02454, USA. e-mail:bGRwbGFudEBicmFuZGVpcy5lZHU=