
 Rolf Sprengel2
Rolf Sprengel2
- 1Division of Neuroscience, Medical Research Institute Ninewells Hospital and Medical School, Dundee University, Dundee, UK
- 2Department of Molecular Neuroscience, Max-Planck Institute for Medical Research, Heidelberg, Germany
The GluA2 subunit in heteromeric alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor channels restricts Ca2+ permeability and block by polyamines, rendering linear the current-voltage relationship of these glutamate-gated cation channels. Although GluA2-lacking synaptic AMPA receptors occur in GABA-ergic inhibitory neurons, hippocampal CA1 pyramidal cell synapses are widely held to feature only GluA2 containing AMPA receptors. A controversy has arisen from reports of GluA2-lacking AMPA receptors at hippocampal CA3-to-CA1 cell synapses and a study contesting these findings. Here we sought independent evidence for the presence of GluA2-lacking AMPA receptors in CA1 pyramidal cell synapses by probing the sensitivity of their gated cation channels in wild-type (WT) mice and gene-targeted mouse mutants to philanthotoxin, a specific blocker of GluA2-lacking AMPA receptors. The mutants either lacked GluA2 for maximal philanthotoxin sensitivity, or, for minimal sensitivity, expressed GluA1 solely in a Q/R site-edited version or not at all. Our comparative electrophysiological analyses provide incontrovertible evidence for the presence in wild-type CA1 pyramidal cell synapses of GluA2-less AMPA receptor channels. This article is part of a Special Issue entitled “Calcium permeable AMPARs in synaptic plasticity and disease.”
Introduction
Alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor channels mediate most of the fast synaptic excitatory transmission in the brain. Their subunit composition determines their biophysical properties, including channel gating and ion conductance. Importantly, the GluA2 subunit with its edited Q/R site (Sommer et al., 1992) ensures that AMPA receptors containing this subunit are largely Ca2+-impermeable (Hollmann et al., 1991; Burnashev et al., 1992; Jonas and Burnashev, 1995; Isaac et al., 2007). By contrast, GluA2-lacking AMPA receptor channels, i.e., any homomeric or heteromeric assembly of GluA1, A3, or A4, are Ca2+-permeable and subject to voltage-dependent block by intracellular polyamines, resulting in a characteristic doubly rectifying current-voltage (I–V) relationship. Imbalance in favor of Ca2+ permeable AMPARs can cause CNS dysfunction (Liu and Zukin, 2006). In synaptic AMPA channels, sensitivity to endogenous polyamines and the degree of rectification can be lessened by interaction with TARPs, the transmembrane AMPAR regulatory proteins (Soto et al., 2007). Thus, a low extent of rectification of synaptic AMPAR-mediated currents need not reflect a particular AMPAR composition, but may also indicate interaction with regulatory factor(s). As gleaned from the properties of AMPA receptors in different neuronal populations, the current consensus would state that principal excitatory neurons operate with GluA2-containing AMPA receptors (Adesnik and Nicoll, 2007; Lu et al., 2009), whereas GABA-ergic inhibitory neurons often additionally feature GluA2-lacking synaptic AMPA receptors (Geiger et al., 1995; Toth and McBain, 1998; Liu and Cull-Candy, 2002).
While there is firm consensus that extrasynaptic AMPARs in CA1 pyramidal cells contain GluA2 (Colquhoun et al., 1992; Lu et al., 2009), conflicting evidence surrounds the possible presence of GluA2-lacking AMPAR channels in hippocampal Schaffer collateral/commissural synapses in CA1 pyramidal cells, arguably the best-studied synapses in the brain. Several groups observed that the molecular identity of AMPARs in CA1 cell synapses is altered upon induced changes in synaptic strength. Plant et al. (2006) reported that induction of long-term potentiation (LTP) elicits a transient increase in the proportion of polyamine sensitive AMPARs at the potentiated synapses, revealed by an increased rectification index (RI). Guire et al. (2008) and Moult et al. (2010) observed a similar change in AMPAR properties. By stark contrast, Adesnik and Nicoll (2007) failed to obtain evidence for the insertion of synaptic Ca2+-permeable AMPA receptors upon LTP induction, concluding that all synaptic AMPARs in CA1 cells contain GluA2 subunits. Thus, whether GluA2-lacking AMPARs exist at glutamatergic CA1 cell synapses and contribute to fast transmission clearly needs re-evaluating.
The most common approach to detect the participation of GluA2-lacking AMPARs in synaptic transmission is to test sensitivity to the wasp polyamine toxin, philanthotoxin-433 (PhTx-433). GluA2-lacking channels can be strongly blocked by PhTx-433 in an activity dependent manner, while GluA2 containing AMPARs are insensitive to the drug (Washburn and Dingledine, 1996). However, bath-applied PhTx-433 may target a GluA2-lacking channel population already blocked by endogenous polyamines (PA), and thus, a moderate contribution of GluA2-lacking channels could be overlooked. In this context, it is important to note that virtually all GluA2-lacking AMPAR channels contain polyamine in the closed state, regardless of the membrane potential. This results in the characteristic doubly rectifying shape of their I–V curves, which reflects the speed of relief from polyamine block. This speed is dependent on the absolute transmembrane electric field, and is faster when the cell is hyperpolarized (Rozov et al., 1998). Even at physiological resting membrane potentials (∼−70 mV), a substantial portion of GluA2-lacking channels could still be blocked by polyamines during single unitary EPSCs, and repetitive high-frequency stimulation is required to relieve the block (Rozov and Burnashev, 1999). Hence, to get an accurate readout of the PhTx effect on AMPAR-mediated currents the influence of endogenous polyamines should first be minimized.
To resolve the issue of Ca2+-permeable AMPA channels in CA1 pyramidal cells of wild-type (WT), we took recourse to gene-targeted mice lacking either GluA2 (Shimshek et al., 2006) or GluA1 (Zamanillo et al., 1999) or expressing GluA1 in a Q/R site-edited version (Sanchis-Segura et al., 2006). This allowed us to analyze the artificial situations of CA1 cell synapses having AMPA receptors of maximal (GluA2−/− mutant mice) or minimal (GluA1−/− and GluA1(R) mutants) sensitivity to polyamines. The presence of Ca2+-permeable AMPA channels in WT should then be revealed by increased sensitivity to PhTx-433 relative to AMPA channels in GluA1−/− and GluA1(R) mouse mutants. No such increase in PhTx-433 sensitivity should be observed if WT CA1 pyramidal cell synapses indeed lack Ca2+-permeable AMPA channels, as posited by a prevalent view (Adesnik and Nicoll, 2007). Our data clearly demonstrate that in WT CA1 synapses, ∼8–10% of the AMPA channels are Ca2+-permeable.
Methods
Transverse hippocampal 250 μm slices were prepared from the brains of 42–56 day-old WT, GluA1−/− (Zamanillo et al., 1999), GluA1(R) (Sanchis-Segura et al., 2006) and GluA2−/− mice (Shimshek et al., 2006), killed by decapitation. All mice were backcrossed for >10 generations to C57Bl6. Genotyping was performed as detailed in http://wmn.mpimf-heidelberg.mpg.de/sprengel. The slicing chamber contained an oxygenated ice-cold solution [modified from (Dugue et al., 2005)] composed of (in mM): K-Gluconate, 140; N-(2-hydroxyethyl) piperazine-N′-ethanesulfonic acid (HEPES), 10; Na-Gluconate, 15; ethylene glycol-bis (2-aminoethyl)-N,N,N′,N′-tetraacetic acid (EGTA), 0.2; and NaCl, 4 (pH 7.2). Slices were incubated for 30 min at 35°C before being stored at room temperature in artificial CSF (ACSF) containing (in mM): NaCl, 125; NaHCO3, 25; KCl, 2.5; NaH2PO4, 1.25; MgCl2, 1; CaCl2, 2; and glucose, 25; bubbled with 95%O2 and 5%CO2.
Patch electrodes were pulled from hard borosilicate capillary glass (Sutter Instruments flaming/brown micropipette puller). These were filled with either a polyamine-free or a polyamine-containing solution. The polyamine-free solution consisted of (in mM) Cs-gluconate, 100; CsCl, 10; HEPES, 10; NaCl, 8; EGTA, 0.2; MgATP, 2; Na3GTP, 0.3; phosphocreatine, 10; and K2ATP, 20 (pH 7.3 with CsOH). The polyamine-containing solution was identical except for the addition of 0.5 mM spermine, yielding a free spermine concentration of ∼40 μM (Watanabe et al., 1991).
CA1 pyramidal cells were identified visually using IR-video microscopy. Whole-cell recordings from these neurons were made in voltage-clamp mode using a HEKA EPC-9 amplifier (List Elektronik). Cells were held at −70 mV. To evoke synaptic current, glass electrodes filled with ACSF were placed in the dendritic region of stratum radiatum, ∼50–100 μm from the cell body, to stimulate Schaffer collateral inputs at inter-stimulus intervals of 6 s. The stimulation intensity was adjusted to produce an EPSC with an amplitude of ∼50 pA at the beginning of each recording. Inhibitory synaptic transmission was blocked during recordings by adding 10 μM gabazine to the perfusion ACSF. For analysis, five subsequent responses were averaged and normalized to the mean EPSC amplitude obtained during 20–30 min of recording. PhTx-433 (10 μM) was applied 40 min after starting the experiment. The degree of blockade was calculated as a ratio of the average steady-state current amplitude before and after PhTx-433 application. Series resistance was monitored, and data from cells in which series resistance varied by >15% during recording were discarded from analysis. Glutamate (1 mM) was applied using a piezo-controlled (P 245.70, Physik Instrumente, Waldbronn, Germany) fast application system with a double-barrel application pipette onto outside-out patches of HEK293 cells constitutively expressing homomeric GluA1 receptors. Durations of the glutamate pulses were 5 and 100 ms. All fast application experiments were in the presence of 50 μM of cyclothiazide.
For statistical analysis, Student's t-test was used, and data are presented as mean ± SD.
Results
PhTx-433 Suppression of EPSCs in Absence of GluA2 Depends on Intracellular Polyamine Content
We compared the effect of PhTx-433 on EPSCs evoked in CA1 pyramidal cell synapses in hippocampal slices of GluA2−/− mice in two distinct experimental conditions, one ensuring chelation of intracellular polyamines and the other having buffered spermine added to the intracellular solution. As in this mouse mutant all AMPA channels are polyamine sensitive, we could evaluate the maximal effect of the applied concentration of PhTx-433 (10 μM) on EPSCs mediated solely by GluA2-lacking synaptic AMPA channels. This should be useful for revealing their existence and estimating their contribution in WT, in which they would co-exist with GluA2-containing AMPARs. Additionally, we evaluated the sensitivity of synaptic AMPARs to intracellular polyamines in GluA2−/− mice and compared it to that of WT. To this end, we measured the synaptic current-voltage (I–V) relationship in CA1 pyramidal neurons dialyzed with polyamine-containing intracellular solution.
Evoked EPSCs from patched CA1 pyramidal cells of GluA2−/− mice were recorded by stimulating Schaffer collaterals at interstimulus intervals of 6 s with either polyamine-containing or -free patch-pipette solution. The washout of endogenous polyamines led to a three to fourfold increase in EPSC amplitudes (0.29 ± 0.27 relative to the steady-state values; Figures 1A,B). Subsequent application of PhTx-433 (10 μM) reduced the amplitude of responses to 0.34 ± 0.15 compared to maximal amplitudes after polyamine washout (n = 7 from 4 mice; p < 0.001). Thus, 10 μM of PhTx-433 blocked ∼70% of the GluA2-less AMPA channels. However, when 0.5 mM of spermine was included in the pipette solution we typically observed a slight, though not significant, reduction in the EPSC amplitude as whole-cell dialysis was progressing. Initial amplitudes were 1.12 ± 0.28 relative to the steady-state level reached after 30 min of dialysis (p > 0.05; n = 7 from 5 mice). Reduction of postsynaptic responses by subsequently applied PhTx-433 was significantly less pronounced when compared to polyamine-free conditions (0.63 ± 0.09; p < 0.01; Figures 1A,B).
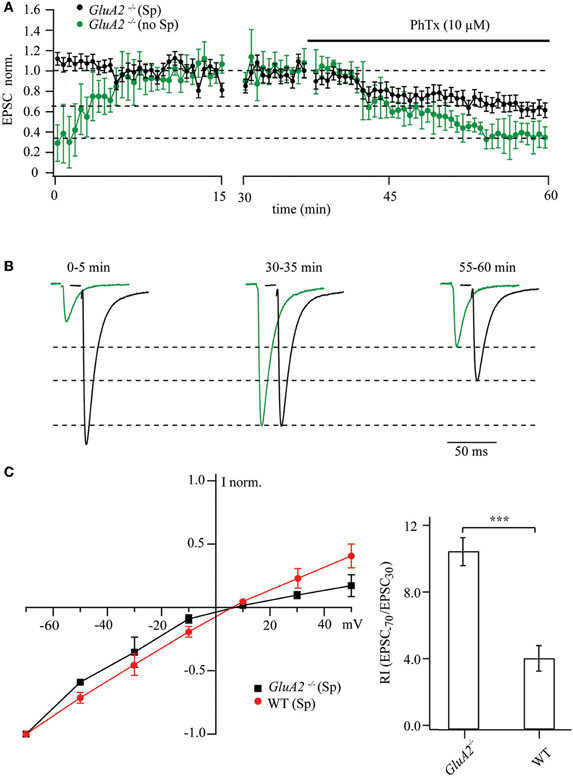
Figure 1. Maximal and polyamine-attenuated levels of PhTx-433 blockade in CA1 pyramidal cells of GluA2−/− mice. (A) The time course of EPSC amplitude changes during washout of polyamine and subsequent PhTx-433 (10 μM) application, when CA1 pyramidal neurons were dialyzed with polyamine-free intracellular solution (green circles). Also shown is the effect of exogenously added spermine on EPSC amplitudes and the masking effect of spermine on PhTx-433 suppression (black circles). (B) Representative averaged traces at the indicated times of recording in polyamine-free conditions are shown in green, in polyamine-containing condition in black. (C) Effect of intracellular polyamines on AMPAR-mediated synaptic currents in GluA2−/− and WT mice. AMPAR-mediated synaptic I–V curves measured with polyamine-containing intracellular solution in GluA2−/− and WT mice (left panel). Rectification properties of synaptic AMPAR channels in GluA2−/− and WT mice (right panel). RIs were calculated as the ratio of EPSC amplitudes measured at 30 and −70 mV (EPSC−70/EPSC30).
Collectively, the data from GluA2−/− mice suggest that endogenous or exogenously added polyamines constitutively block GluA2-lacking channels and attenuate the effect of bath-applied PhTx-433. Strong sensitivity of AMPARs to polyamines in GluA2−/− mice was further demonstrated by measuring the synaptic I–V relationship. Blockade in these mutants of the GluA2-less AMPARs by polyamines resulted in a characteristic inwardly rectifying I–V relation. Figure 1C compares the rectification properties of AMPAR channels mediating synaptic transmission at Schaffer collateral/commissural synapses in CA1 pyramidal cells of GluA2−/− and WT mice. Obviously, GluA2 ablation leads to enhanced polyamine sensitivity relative to WT, as reflected by an increased RI (GluA2−/−: RI = 10.3 ± 0.85, n = 5; WT: RI = 3.9 ± 0.77 n = 5; p < 0.01).
We next determined how the duration of agonist application affects the PhTx-433 blocking potency and to what extent the current suppression by the toxin depends on intracellular polyamine content. We employed fast agonist application on outside-out patches of HEK293 cells expressing homomeric GluA1 receptor channels in presence of 50 μM cyclothiazide to exclude an influence of channel desensitization. Application of 1 mM glutamate for 100 ms at −70 mV evoked stable inward currents, which were drastically reduced in presence of 10 μM PhTx-433, reaching a steady-state level of 3 ± 1% of the initial amplitude (Figure A1A; n = 5; p < 0.01). However, at 5 ms agonist applications, the steady-state level reached upon PhTx-433 application was significantly higher (28 ± 11% of control amplitudes; Figure A1B; n = 5; p < 0.01). Similar experiments with a polyamine-free pipette solution demonstrated a reduction of glutamate-evoked currents (5 ms; 1 mM) that was stronger (11 ± 7% relative to control values; Figure A1C; n = 5) than the 28 ± 11% observed with the PA-containing solution (p < 0.05). Thus, usage of PhTx-433 as a tool to probe the expression of GluA2-lacking AMPARs requires the washout of endogenous polyamines.
Synaptic AMPARs in CA1 Pyramidal Cells of GluA1−/− or GluA1(r) Mutants are Insensitive to PhTx-433
We performed similar experiments on GluA1−/− and GluA1(R) mouse mutants, in which the likelihood for the presence in CA1 pyramidal cells of synaptic GluA2-less, Ca2+-permeable AMPA channels is low and hence, sensitivity to polyamines and PhTx-433 should be lacking. Indeed, prolonged whole-cell dialysis with polyamine-free intracellular solution did not result in a significant increase in EPSC amplitudes in CA1 pyramidal cells in either GluA1−/− or GluA1(R) mice. We did record a moderate enhancement of AMPAR-mediated responses (0.78 ± 0.1 and 0.81 ± 0.11, n = 7 cells, for 4–5 mice of each mutant line), which is most likely due to frequency facilitation or augmentation (Thomson, 2000; Zucker and Regehr, 2002). As expected, application of PhTx-433 (10 μM) did not affect EPSC amplitudes in the GluA1−/− (0.98 ± 0.13; n = 7 from 4 mice) or GluA1(R) mutants (1.07 ± 0.16; n = 7 from 5 mice). The data obtained from these two mutant lines are depicted in Figure 2A.
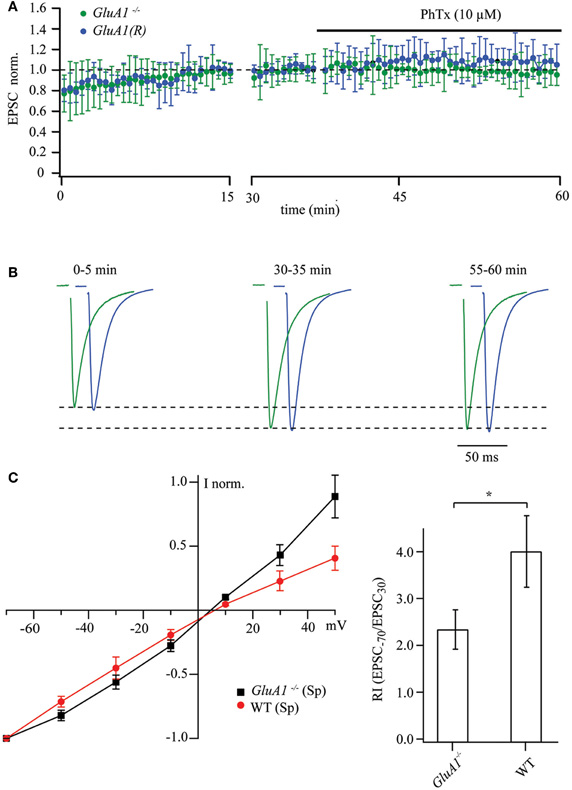
Figure 2. Synaptic AMPAR channels in CA1 pyramidal cells of GluA1−/− and GluA1(R) mouse mutants are polyamine and PhTx-433 insensitive. (A) The time course of EPSC amplitude changes recorded in CA1 pyramidal neurons of GluA1−/− (green circles) and GluA1(R) (blue circles) mutant mice during washout of polyamine and subsequent PhTx-433 (10 μM) application. (B) Representative averaged traces at the indicated times of recording from GluA1−/− mice shown in green, and for GluA1(R) mutants in blue. (C) Effect of intracellular polyamines on rectification properties of AMPAR-mediated synaptic currents in GluA1−/− and WT animals. Synaptic AMPAR-mediated I–V curves measured with polyamine-containing intracellular solution in GluA1−/− and WT mice (left panel). Rectification indices of synaptic AMPAR channels in GluA2−/− and WT mice (right panel).
Addition of 0.5 mM spermine to the intracellular solution did not cause any inward rectification of AMPAR-mediated synaptic currents in GluA1−/− mice, and the I–V curve became slightly outwardly rectifying (Figure 2C). However the shape of the I–V curve and the RI measured in GluA1−/− mice differed significantly from those in WT mice under the same experimental conditions (Figure 2C; GluA1−/−: RI = 2.32 ± 0.42, n = 5 and WT: RI = 3.9 ± 0.77 n = 5; p < 0.05). We conclude that absence of GluA1 (or expression of GluA1 in its R-form) greatly diminish the presence of synaptic Ca2+-permeable AMPA channels.
PhTx-433 Sensitive AMPA Channels in WT CA1 Pyramidal Cell Synapses
According to previous studies, EPSCs recorded from CA1 pyramidal cells are insensitive to PhTx-433 (Adesnik and Nicoll, 2007; Lu et al., 2009). This is thought to demonstrate the absence of synaptic GluA2-lacking AMPARs in these neurons. As intracellular polyamines can occlude block by PhTx-433 (Figure 1), we first determined how endogenous polyamine washout changes the potency of exogenously applied PhTx-433 on EPSC amplitudes in WT CA1 cells. When the cells were dialyzed with polyamine-free solution, the amplitudes of evoked AMPAR-mediated responses increased during the first 15–20 min of recording, finally reaching a steady-state level (Figures 3A,B). The average initial amplitude was 0.45 ± 0.21 (n = 9 from 6 mice), when normalized to the steady-state values (20–30 min of recording). Subsequent application of PhTx-433 (10 μM) caused a gradual reduction of the postsynaptic responses, reaching a plateau level (50–60 min of recording) of 0.69 ± 0.17 (p < 0.01), as compared to values prior to PhTx-433 application (Figures 3A,B).
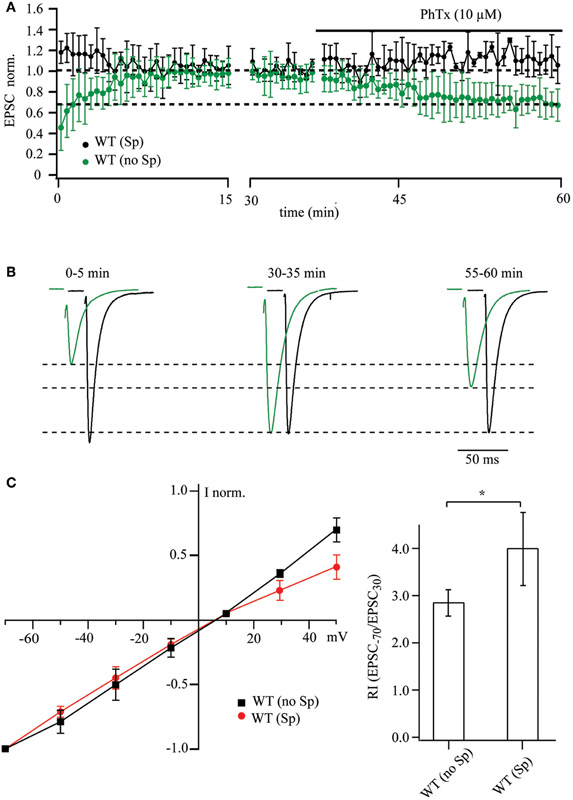
Figure 3. PhTx-433 sensitive AMPA channels in WT CA1 pyramidal cells. (A) Scatter plot shows EPSC amplitude dynamics during prolonged whole-cell dialysis and following PhTx-433 (10 μM) application, when CA1 pyramidal neurons were patched with either polyamine-free (green circles) or polyamine-containing (black circles) intracellular solution. (B) Representative averaged traces at the indicated times of recording in polyamine-free (green) or polyamine-containing (black) solution. (C) Dependence of AMPAR synaptic channel rectification properties in WT CA1 pyramidal cells on intracellular polyamine content. Synaptic AMPAR-mediated I–V curves measured with polyamine-containing and -free intracellular solutions (left panel). Dependence of rectification indices of synaptic AMPAR channels on the presence of polyamines in WT mice (right panel).
By contrast, when WT CA1 neurons were patched with polyamine-containing solution, the amplitudes of responses during the initial phase of dialysis were either stable or underwent a slight, but not significant reduction (1.18 ± 0.14, p > 0.05, n = 7 from 5 mice). Moreover, application of PhTx-433 caused no significant changes in EPSC amplitudes (1.12 ± 0.14; p > 0.05).
Thus, in WT CA1 pyramidal cells, washout of intracellular polyamines resulted in an increase in the amplitude of evoked postsynaptic responses, which was in large part counteracted by subsequent application of PhTx-433 (Figures 3A,B). This remarkable difference to the results from synaptic AMPARs in CA1 pyramidal cells of GluA1−/− and GluA1(R) mouse mutants constitutes unequivocal evidence for the presence of synaptic Ca2+-permeable AMPA receptors in WT CA1 cells. To further substantiate our finding on the existence of GluA2-lacking AMPARs at Schaffer collateral/commissural synapses in WT CA1 pyramidal cells, we evaluated the effect of the presence of intracellular spermine on the shape of AMPAR-mediated I–V curves and on the RI. The averaged I–V relation recorded with polyamine-free solution was nearly linear and deviated strongly at positive potentials from that observed with polyamine-containing solution. Also, the RI in presence of spermine was significantly higher compared to that in polyamine-free conditions [WT (Sp): 3.9 ± 0.77, n = 5 and WT (no Sp): 2.86 ± 0.28, n = 5; p < 0.05].
Discussion
The controversy concerning the presence of GluA2-less, Ca2+-permeable AMPA channels in hippocampal CA1 pyramidal cell synapses (Plant et al., 2006; Adesnik and Nicoll, 2007; Moult et al., 2010) prompted this study, for which we employed, in addition to WT mice, mouse mutants lacking either the major AMPAR subunits GluA1 or GluA2 or expressing the GluA1(R) subunit, which, like GluA2, drastically attenuates the Ca2+-permeability of AMPARs containing it (in preparation). Polyamine sensitivity of GluA2-lacking AMPARs is often used to evaluate the relative contribution of these channels to the net AMPAR-mediated synaptic current. Endogenous or exogenously added polyamines can block these receptors, resulting in strong current rectification, which can be measured as RI. Alternatively, washout of polyamines should increase EPSC amplitudes in synapses in which GluA2-lacking channels contribute to synaptic transmission (Rozov and Burnashev, 1999). The magnitude of this enhancement would depend on the endogenous polyamine concentration and the ratio between GluA2-lacking and GluA2-containing channels (Figures 4A,B). In all our experiments, we observed EPSC augmentation upon polyamine washout during the initial 20–25 min of recording, but the scale of this enhancement strongly depended on the particular mouse mutant. As expected, the increase was largest in GluA2−/− mice and smallest in GluA1−/− or GluA1(R) mutants. In the latter two mutant lines the augmentation of postsynaptic responses is unlikely to reflect the washout of polyamines, since the AMPARs in these mutants should be polyamine insensitive. A possible explanation for the EPSC amplitude rise in these mutants might be activity dependent presynaptic phenomena, such as frequency facilitation or augmentation (Thomson, 2000; Zucker and Regehr, 2002), which could also contribute to the EPSC enhancement by polyamine washout in WT and GluA2−/− mice. This assumption allows us to correct the data from WT and GluA2−/− mice and thus estimate better the putative impact of polyamine washout on fast excitatory synaptic transmission at Schaffer collateral to CA1 pyramidal cell synapses (Figure 4C).
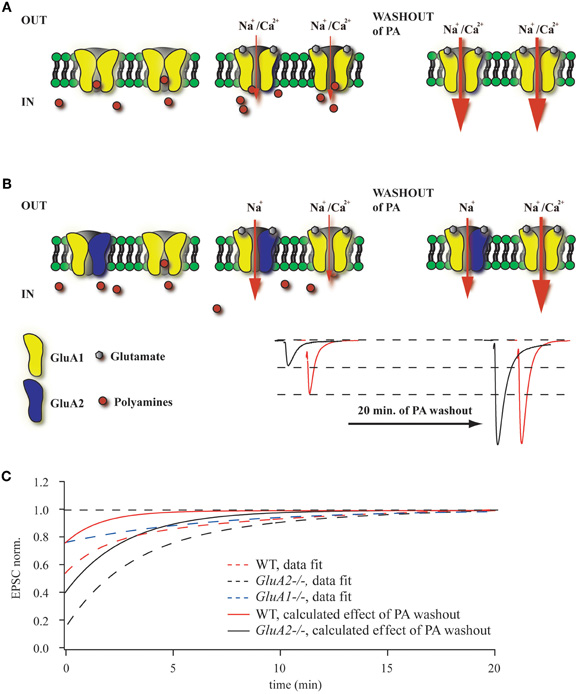
Figure 4. Washout of polyamines increases EPSC amplitudes. (A) Polyamines enter and block GluA2-lacking AMPARs in the closed state. In GluA2−/− mice almost all channels are blocked prior to glutamate release (left panel). During unitary synaptic events the channel conductance is strongly attenuated, resulting in reduced amplitudes of the net response (middle panel). In contrast, washout of polyamines results in an increased AMPAR conductance, leading to enhanced EPSC amplitudes (right panel). (B) When GluA2-lacking and GluA2-containing AMPARs are co-expressed at the same synapse, the effect of polyamine washout will depend on the ratio between the two channel types. Representative traces show enhancement of AMPAR-mediated EPSCs during polyamine washout (WT in red, GluA2−/− in black). (C) The plot summarizes data on the initial amplitude enhancement observed in WT, GluA2−/− and GluA1−/− mice. The dashed line shows the exponential fits of the time dependent amplitude augmentation during polyamine washout (WT in red, GluA2−/− in black and GluA1−/− in blue). Since the vast majority of CA1 cell AMPA channels in GluA1−/− mice should be polyamine insensitive, the slight EPSC enhancement represents another intrinsic property of Schaffer collateral/commisural to CA1 pyramidal cells synapses (e.g., frequency facilitation). Thus, by subtracting the GluA1−/− data from those obtained from WT or GluA2−/− mice, we can calculate the putative effect of endogenous polyamine washout on EPSC amplitudes in WT (red line) and GluA2−/− (black line) mice.
We also employed the sensitivity of synaptic AMPA currents to PhTx-433 to detect the presence of GluA2-lacking AMPARs. The accuracy of this PhTx-433 “probing test” is sensitive to intracellular polyamines. Indeed, as we show, the presence of ∼40 μM of free spermine reduces the PhTx-433 suppression of AMPAR-mediated EPSCs by 50% in GluA2−/− mice, in which all AMPA channels should be sensitive to the wasp toxin. The use of GluA2−/− mice allowed us to determine that after polyamine washout, 10 μM PhTx-433 blocks 70% of the EPSCs mediated by GluA2-lacking AMPA channels. The EPSCs mediated by AMPA channels in GluA1−/− or GluA1(R) mutants were insensitive to PhTx-433, consonant with the predicted lack of Ca2+-permeable AMPA channels in these mutant mice, and thus demonstrating that 10 μM PhTx-433 exerts no unspecific effects on synaptic transmission.
Notably, following polyamine washout, 10 μM PhTx-433 blocked 31% of the EPSCs in WT CA1 pyramidal cell synapses, clearly demonstrating the presence of Ca2+-permeable GluA2-less AMPA channels. While in GluA2−/− mice, where all AMPARs are polyamine sensitive, PhTx blocked 66% of the EPSC. By solving a simple rule of proportion we can calculate that in WT CA1 pyramidal cells, under artificial polyamine-free conditions, GluA2-lacking channels mediate 47% of the net EPSC. To estimate the proportion of such channels in the entire synaptic AMPAR population, one needs to consider that the conductance of GluA2-lacking AMPARs is 5–10 times higher than that of GluA2-containing channels. These data allow us to estimate the relative contribution of GluA2-lacking channels to synaptic transmission at Schaffer collateral CA1 cell synapses. In polyamine-free conditions, recombinant GluA2-lacking channels have a maximal conductance of 19–25.5 pS, whereas the conductance of GluA2-containing AMPARs is 2.5 pS (Oh and Derkach, 2005). We thus estimate the population of GluA2-lacking AMPARs to be in the range of 8–10%. Under native conditions, the contribution of GluA2-lacking channels to the synaptic response depends on the intracellular polyamine concentration and should, therefore, be <47%. However, during repetitive activity, the impact of these channels will increase due to the activity-dependent relief from polyamine block. Also, the degree of the block could be significantly attenuated by interaction of GluA2-lacking AMPARs with TARPs. Thus, the role of polyamine-sensitive AMPARs at Schaffer collateral/commissural synapses might be differentially regulated during shorter and longer activity epochs, thus providing additional ways to control the synaptic gain.
Our analyses might resolve the apparent disagreement in the field regarding GluA2-lacking AMPARs in CA1 pyramidal cells. The PhTx-433 test becomes less sensitive in the presence of intracellular polyamines, since the toxin can only block channels, which are not occupied by internal polyamines. Crucially, polyamines block GluA2-lacking channels in the closed state, and the block itself is not voltage dependent. Therefore, to maximize the measurable effect of PhTx-433, the washout of endogenous polyamines becomes essential, lest the contribution of these channels to synaptic transmission is underestimated, as demonstrated here by fast agonist application experiments on recombinant GluA1 homomeric AMPARs.
The presence of Ca2+-permeable AMPA channels in CA1 cell synapses prompts the question as to their functional role. Plant et al. (2006) observed transient incorporation of such AMPA channels following induction of hippocampal LTP and reported that the transient Ca2+-influx is essential for the subsequent maintenance phase of LTP. This postulate, still under debate, needs to be examined in future studies. Indeed, the contribution of the Ca2+-influx via gated Ca2+-permeable AMPA channels is very low relative to that of NMDA channels, considering an approximately 10-fold lower fractional Ca2+-current and a >10-fold lesser envelope of charge transfer of the AMPA channels. Notably, our data indicate no need for the transient incorporation of Ca2+-permeable AMPA channels into CA1 cell synapses upon LTP induction (Plant et al., 2006), given that such channels already constitute a small subpopulation of the synaptic AMPA receptors in WT CA1 pyramidal neurons. Rather, it appears that LTP induction may facilitate the participation in excitatory transmission of the synaptic Ca2+-permeable AMPA channels, conceivably. Via phosphorylation of intracellular subunit domains, with the additional negative charges close to the inner channel mouth accelerating the unblocking of polyamines.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Roger A. Nicoll and Georg Köhr for invaluable suggestions, and Sabine Grünewald for cell culture.
References
Adesnik, H., and Nicoll, R. A. (2007). Conservation of glutamate receptor 2-containing AMPA receptors during long-term potentiation. J. Neurosci. 27, 4598–4602.
Burnashev, N., Monyer, H., Seeburg, P. H., and Sakmann, B. (1992). Divalent ion permeability of AMPA receptor channels is dominated by the edited form of a single subunit. Neuron 8, 189–198.
Colquhoun, D., Jonas, P., and Sakmann, B. (1992). Action of brief pulses of glutamate on AMPA/kainate receptors in patches from different neurones of rat hippocampal slices. J. Physiol. 458, 261–287.
Dugue, G. P., Dumoulin, A., Triller, A., and Dieudonne, S. (2005). Target-dependent use of co-released inhibitory transmitters at central synapses. J. Neurosci. 25, 6490–6498.
Geiger, J. R., Melcher, T., Koh, D. S., Sakmann, B., Seeburg, P. H., Jonas, P., and Monyer, H. (1995). Relative abundance of subunit mRNAs determines gating and Ca2+ permeability of AMPA receptors in principal neurons and interneurons in rat CNS. Neuron 15, 193–204.
Guire, E. S., Oh, M. C., Soderling, T. R., and Derkach, V. A. (2008). Recruitment of calcium-permeable AMPA receptors during synaptic potentiation is regulated by CaM-kinase I. J. Neurosci. 28, 6000–6009.
Hollmann, M., Hartley, M., and Heinemann, S. (1991). Ca2+ permeability of KA-AMPA–gated glutamate receptor channels depends on subunit composition. Science 252, 851–853.
Isaac, J. T., Ashby, M. C., and McBain, C. (2007). The role of the GluR2 subunit in AMPA receptor function and synaptic plasticity. Neuron 54, 859–871.
Jonas, P., and Burnashev, N. (1995). Molecular mechanisms controlling calcium entry through AMPA-type glutamate receptor channels. Neuron 15, 987–990.
Liu, S. J., and Cull-Candy, S. G. (2002). Activity-dependent change in AMPA receptor properties in cerebellar stellate cells. J. Neurosci. 22, 3881–3889.
Liu, S. J., and Zukin, R. Z. (2006). Ca2+-permeable AMPA receptors in synaptic plasticity and neuronal death. Trends in Neurosci. 30, 126–134.
Lu, W., Shi, Y., Jackson, A. C., Bjorgan, K., During, M. J., Sprengel, R., Seeburg, P. H., and Nicoll, R. A. (2009). Subunit composition of synaptic AMPA receptors revealed by a single-cell genetic approach. Neuron 62, 254–268.
Moult, P. R., Cross, A., Santos, S. D., Carvalho, A. L., Lindsay, Y., Connolly, C. N., Irving, A. J., Leslie, N. R., and Harvey, J. (2010). Leptin regulates AMPA receptor trafficking via PTEN inhibition. J. Neurosci. 30, 4088–4101.
Oh, M. C., and Derkach, V. A. (2005). Dominant role of the GluR2 subunit in regulation of AMPA receptors by CaMKII. Nat. Neurosci. 8, 853–854.
Plant, K., Pelkey, K. A., Bortolotto, Z. A., Morita, D., Terashima, A., McBain, C. J., Collingridge, G. L., and Isaac, J. T. (2006). Transient incorporation of native GluR2-lacking AMPA receptors during hippocampal long-term potentiation. Nat. Neurosci. 9, 602–604.
Rozov, A., and Burnashev, N. (1999). Polyamine-dependent facilitation of postsynaptic AMPA receptors counteracts paired-pulse depression. Nature 401, 594–598.
Rozov, A., Zilberter, Y., Wollmuth, L. P., and Burnashev, N. (1998). Facilitation of currents through rat Ca2+-permeable AMPA receptor channels by activity-dependent relief from polyamine block. J. Physiol. 511(Pt 2), 361–377.
Sanchis-Segura, C., Borchardt, T., Vengeliene, V., Zghoul, T., Bachteler, D., Gass, P., Sprengel, R., and Spanagel, R. (2006). Involvement of the AMPA receptor GluR-C subunit in alcohol-seeking behavior and relapse. J. Neurosci. 26, 1231–1238.
Shimshek, D. R., Jensen, V., Celikel, T., Geng, Y., Schupp, B., Bus, T., Mack, V., Marx, V., Hvalby, O., Seeburg, P. H., and Sprengel, R. (2006). Forebrain-specific glutamate receptor B deletion impairs spatial memory but not hippocampal field long-term potentiation. J. Neurosci. 26, 8428–8440.
Sommer, B., Monyer, H., Wisden, W., Verdoorn, T. A., Burnashev, N., Sprengel, R., Sakmann, B., and Seeburg, P. H. (1992). Glutamate-gated ion channels in the brain. Genetic mechanism for generating molecular and functional diversity. Arzneimittelforschung 42, 209–210.
Soto, D., Coombs, I. D., Kelly, L., Farrant, M., and Cull-Candy, S. G. (2007). Stargazin attenuates intracellular polyamine block of calcium-permeable AMPA receptors. Nat. Neurosci. 10, 1260–1267.
Thomson, A. M. (2000). Facilitation, augmentation and potentiation at central synapses. Trends Neurosci. 23, 305–312.
Toth, K., and McBain, C. J. (1998). Afferent-specific innervation of two distinct AMPA receptor subtypes on single hippocampal interneurons. Nat. Neurosci. 1, 572–578.
Washburn, M. S., and Dingledine, R. (1996). Block of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropi onic acid (AMPA) receptors by polyamines and polyamine toxins. J. Pharmacol. Exp. Ther. 278, 669–678.
Watanabe, S., Kusama-Eguchi, K., Kobayashi, H., and Igarashi, K. (1991). Estimation of polyamine binding to macromolecules and ATP in bovine lymphocytes and rat liver. J. Biol. Chem. 266, 20803–20809.
Zamanillo, D., Sprengel, R., Hvalby, O., Jensen, V., Burnashev, N., Rozov, A., Kaiser, K. M., Koster, H. J., Borchardt, T., Worley, P., Lubke, J., Frotscher, M., Kelly, P. H., Sommer, B., Andersen, P., Seeburg, P. H., and Sakmann, B. (1999). Importance of AMPA receptors for hippocampal synaptic plasticity but not for spatial learning. Science 284, 1805–1811.
Zucker, R. S., and Regehr, W. G. (2002). Short-term synaptic plasticity. Annu. Rev. Physiol. 64, 355–405.
Appendix
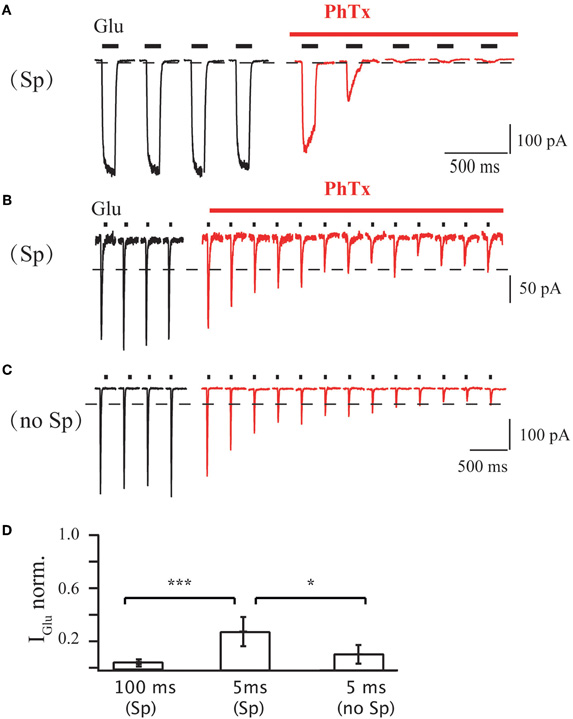
Figure A1. (A) Effect of PhTx application on GluA1-medeated currents in outside-out patches excised from HEK 293 cells. Duration of each glutamate application was 100 ms; holding potential was 70 mV; PA-containing pipette solution was used. Currents evoked before PhTx-433 application are shown in black and those recorded in the presence of toxin in red. For analysis current amplitudes at steady-state level of suppression were normalized to the control amplitude measured prior PhTx-433 application (0.03 ± 0.01; n = 5). (B) The same as in (A), but duration of each glutamate application was 5 ms. Averaged level of suppression was 0.28 ± 0.11 (n = 5). (C) The same as in (B), but PA-free pipette solution was used. Averaged level of suppression was 0.11 ± 0.07 (n = 5). (D) Bar histogram compares the effect of long (100 ms) and sort (5 ms) duration of glutamate application and effect of the presence of polyamines on the level of PhTx suppression.
Keywords: AMPA receptors, hippocampal CA1 cell synapses
Citation: Rozov A, Sprengel R and Seeburg PH (2012) GluA2-lacking AMPA receptors in hippocampal CA1 cell synapses: evidence from gene-targeted mice. Front. Mol. Neurosci. 5:22. doi: 10.3389/fnmol.2012.00022
Received: 04 January 2012; Accepted: 11 February 2012;
Published online: 24 February 2012.
Edited by:
R. Suzanne Zukin, Albert Einstein College of Medicine, USAReviewed by:
Juan Lerma, Instituto de Neurociencias, SpainMiou Zhou, University of California, Los Angeles, USA
Copyright: © 2012 Rozov, Sprengel and Seeburg. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Peter H. Seeburg, Department of Molecular Neurobiology, Max-Planck Institute for Medical Research, Jahnstrasse 29, 69120 Heidelberg, Germany. e-mail:c2VlYnVyZ0BtcGltZi1oZWlkZWxiZXJnLm1wZy5kZQ==