
 Fei-Peng Zhu1
Fei-Peng Zhu1


- 1State Key Laboratory of Biomembrane Engineering and the Center for Life Sciences, Institute of Molecular Medicine, Peking University, Beijing, China
- 2Cell Electrophysiology Lab, Wannan Medical College, Wuhu, China
- 3Department of Neurobiology and Beijing Institute for Neuroscience, Capital Medical University, Beijing, China
Norepinephrine (NE) released from the nerve terminal of locus coeruleus (LC) neurons contributes to about 70% of the total extracellular NE in primates brain. In addition, LC neurons also release NE from somatodendritic sites. Quantal NE release from soma of LC neurons has the characteristics of long latency, nerve activity-dependency, and autoinhibition by α2-adrenergic autoreceptor. The distinct kinetics of stimulus-secretion coupling in somata is regulated by action potential patterns. The physiological significance of soma and dendritic release is to produce negative-feedback and to down-regulate neuronal hyperactivity, which consequently inhibit NE release from axon terminal of LC projecting to many brain areas. Recent discoveries about the LC somatodendritic release may provide new insights into the pathogenesis of clinic disease involving LC-NE system dysfunction, and may help developing remedy targeted to the LC area.
Introduction
Locus coeruleus (LC) is located in the ventrallateral side of the fourth ventricle in the pontine, most of which are noradrenergic neurons projecting to the cortex, cingulate cortex, amygdala nucleus, thalamus, hypothalamus, olfactory tubercles, hippocampus, cerebellum, and spinal cord (Swanson and Hartman, 1975). Norepinephrine (NE) released from the nerve terminal of LC neurons contributes to about 70% of the total extracellular NE in primates brain (Svensson, 1987). It plays important roles not only in arousal, attention, emotion control, and stress (reviewed in Aston-Jones and Cohen, 2005; Berridge and Waterhouse, 2003; Bouret and Sara, 2005; Nieuwenhuis et al., 2005; Sara and Devauges, 1989; Valentino and Van Bockstaele, 2008), but also in sensory information processing (Svensson, 1987). LC directly modulates the somatosensory information from the peripheral system. Under the stress condition, LC could completely inhibit the input from painful stimuli through the descending projection to the spinal cord (Stahl and Briley, 2004). Dys-regulations of LC neurotransmission have been suggested to be involved in physical painful symptoms, attention deficit hyperactivity disorder (ADHD), sleep/arousal disorder, post-traumatic stress disorder, depression, schizophrenia, and Parkinson's disease (reviewed in Berridge and Waterhouse, 2003; Grimbergen et al., 2009; Mehler and Purpura, 2009).
Although extensive studies are carried out on functions of LC neurons in their targeting areas, accumulating evidences suggest that somatodendritic NE release within the LC nucleus also exerts important functions. The somatodendritic release of LC was first suspected by an early observation of a NE synthesis enzyme, dopamine β-hydroxylase, in both soma and dendrites of LC neurons (Swanson and Hartman, 1975). Microdialysis, push-pull superfusion and voltammetry are then used to investigate neurotransmitter release in the LC area in vivo or in brain slice (reviewed in Singewald and Philippu, 1998). Application of high potassium solution, field stimulation, or systematic drug all induce NE release in the LC nucleus but it is hard to differentiate whether NE is released from the somatodendritic sites or terminals of LC neuron. Recently, we have applied patch-clamp technique on the somata area of the LC neuron while recording its quantal NE release simultaneously by carbon fiber electrode (Huang et al., 2007). Single vesicle release of NE is detected after depolarization of soma of the LC neurons. In this review, we will discuss the physiology and characteristics of NE release from LC somatodendrite.
Physiology of NE Somatodendritic Release in LC Nucleus
Autoinhibition of NE Release in LC Somata
Adrenergic receptors belong to a family of G-protein coupling receptors (GPCRs) which are involved in signal transduction (reviewed in Gilsbach and Hein, 2008). Among them, functional α2A receptors are presented on the somatodendrite of LC neurons (Norenberg et al., 1997). Therefore, somatodendritic release of NE may be regulated by the interaction of released NE and α2A receptor. Using voltammetry analysis, systemic application of α2A receptor antagonist is shown to increase NE release both from LC nucleus and projecting brain area (Callado and Stamford, 1999, 2000). Consistently, local perfusion of the α2A receptor agonist clonidine decreases NE in the LC, whereas the opposite effect is observed with its antagonist BRL44408 (Pudovkina et al., 2001; Fernandez-Pastor et al., 2005). With combined measurements of amperometry and patch-clamp, a doubly enhanced individual quantal release events from LC somata is achieved upon inhibition of α2A receptor by yohimbine (Huang et al., 2007). Thus, NE released from somatodendrite inhibits the activity of LC neurons through the activation of local α2A receptors (Svensson et al., 1975; Cedarbaum and Aghajanian, 1977) (Figure 1). However, whereas inhibition via an autoreceptor was originally assumed to involve NE released from recurrent collaterals (Cedarbaum and Aghajanian, 1977), our findings identify that TTX blocked ∼20% of total secretion induced by depolarization of the soma, indicating ∼80% of NE detected in the somatodendrite of LC neuron comes from LC somata.
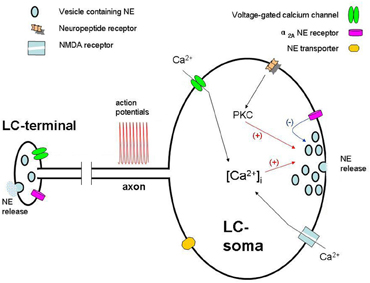
Figure 1. Schematic representation of physiology of quantal NE release from somatodendritic sites and terminal in a locus coeruleus neuron. Action potentials elevate intracellular Ca2+ ([Ca2+i]) and the release of NE from the somatodendritic sites of LC neurons. NE, through the activation of local α2A-adrenoceptors, inhibits LC neurons. Neuropeptides (i.e., hypocretin) may, via activation of their receptors and PKC, alter NMDA-induced elevation of [Ca2+]i, which in turn modulates somatodendritic secretion.
Inhibition of Neurotransmitter Release in Projecting Brain Area
In addition to autoinhibition of release, activation of α2A receptors by NE also hyperpolarizes LC neurons and reduces the firing rate of LC neurons (Williams et al., 1985). This effect on LC firing should inhibit the terminal NE release of LC neurons. It is shown that local administration of a selective NE transporter inhibitor desipramine (DMI) increases NE concentration in the LC and simultaneously decreases NE level in the cingulate cortex (Mateo et al., 1998; Fernandez-Pastor et al., 2005). Consistently, administration of α2A receptor antagonists into LC increases NE release in the cortex in a concentration-dependent manner (Fernandez-Pastor and Meana, 2002). Thus, NE-induced α2A receptor activation in the LC cell body exerts inhibitory tonic modulation on NE release in its terminal areas as well as in soma.
It is known that stress factors also activate LC neurons through neurotransmitter receptors located in LC soma, which consequently modulate NE release in projecting brain area. The emerging knowledge of how the LC-NE system is regulated by stressors and determinants of sensitivity of LC neurons to stress-related neurotransmitters is important for understanding and alleviating diverse stress-related psychiatric disorders (Valentino and Van Bockstaele, 2008).
Characteristics of NE Release from Somatodendritic Sites of Neurons in Locus Coeruleus
Action Potential Pattern Dependence
Intracellular recordings of LC neurons in slices from P8–P26 rats show that in response to stimulation, the spontaneous firing rate increases from (0.3–4 Hz) to (15–20 Hz) (Williams et al., 1987). Interestingly, quantal release events from somatodendritic sites of LC neurons are detected only when the firing rate is high (15–20 Hz) but not low (4 Hz) (Berridge and Waterhouse, 2003; Huang et al., 2007). In contrast, terminal glutamate release of LC neurons, measured by excitatory post synaptic current (EPSC), is not affected by the frequency of action potential (Huang et al., 2007). The high-frequency action potential most likely elevates the level of residual calcium in LC somata which leads to NE release, as shown in chromaffin cells (Duan et al., 2003). LC neuron firing is a premise for NE release from terminal. However, it is not consistence with NE release from soma, because we could not observe quantal release events when the firing frequency is low. Therefore, stress/stimulus-induced LC hyperactivity in firing may trigger somatodendritic NE release, which exerts auto-inhibitory function to alleviate the hyperactivity. This negative-feedback via α2A receptor enables an auto-regulatory mechanism and may play important roles in LC physiological function. If these negative-feedback mechanism is damaged, LC neurons hyperactivity induced by a wide range of stresses would not be inhibited and, therefore, contribute to many disorders such as epilepsy, ADHD, sleep/arousal disorder, post-traumatic stress disorder, main depression disease, schizophrenia, Alzheimer's disease, and Parkinson's disease et al (Svensson, 1987; Valentino and Van Bockstaele, 2008).
Timing of Quantal NE Release from Somatodendrites in LC
Combined amperometry and patch-clamp study of somatic transmitter secretion from neurons in brain slice preparation allows accurate measurement of the timing of quantal NE release following LC neuron firing or depolarization. Compared with the fast synaptic transmission in the range of milliseconds, the latency of LC somata release is in the range of seconds, which is 100–1000 times slower (Huang et al., 2007). A plausible explanation for the dramatic difference between fast glutamate release at synapse and slow NE release at the somatodendritic region is vesicle positions with respect to calcium channels. As shown in chromaffin cells, the channel-vesicle distance is about 10 nm for the fast release, but >200 nm for the slow release (Elhamdani et al., 1998). The long latency of NE release from somatodendritic area of LC neurons may contribute to the delayed autoinhibition on LC neurons.
Comparison to other Somatodendritic Exocytosis
Amperometry measurements have been used to detect quantal monoamine release in different kinds of cell body, such as chromaffin cell (Chow et al., 1992), leech neuron (Chen et al., 1995), substance nigra neuron (Jaffe et al., 1998), and dorsal raphe nucleus (Benzekhroufa et al., 2009). Quantal NE release from somatodendritic sites of neurons in LC is characterized by long latency, high sensitivity to frequency of action potential, calcium-dependence, and low sensitivity to TTX. The quantal size of NE release from soma of LC neuron is 11.9 fC, corresponding to 37,000 NE molecules (Huang et al., 2007). However, the quantal size of NE release from terminal of LC neuron could be 100 times larger. In organotypic rat brainstem slice cultures, >1 pC quantal events occurred at predominantly axonal release sites (Chiti and Teschemacher, 2007).
Other monoamines, such as dopamine and 5-HT, are also released from somatodendritic sites of neurons. Substantia nigra pars compacta (SNc) neurons release dopamine from somatodendrites with charge integrals ranging from a few to several hundred fC, corresponding to about 14,000 dopamine molecules on average (Bjorklund and Lindvall, 1975). The dopamine release in SNc is also Ca2+-dependent. A voltage-gated calcium channel blocker, Cd2+, blocks the stimulatory effects of glutamate on quantal dopamine secretion in cultured SNc neurons (Kim et al., 2008). Interestingly, somatodendritic dopamine release in the substantia nigra proceeds in a botulinum toxin B resistant pathway, unlike that of terminal release, suggesting different molecular players in somata release (Bergquist et al., 2002). In addition, central 5-HT exocytosis from cell bodies of raphe nuclei is reported with a quantal size of ∼34,000 molecules (Benzekhroufa et al., 2009), similar to what we observe for NE. Further studies on somata release from various neurons are needed to understand its physiological roles and characteristics.
Neuropeptide Regulation of NE Release in LC
LC activity is finely tuned by the convergence of afferent fibers. Many neurotransmitter receptors are expressed on the cell membrane of LC soma which may control LC functions via regulating LC firing or somatodendritic NE release (Valentino and Van Bockstaele, 2008). Two of the neuropeptides, orexin and tachykinins, are shown to regulate NE release in the LC. In general, neuropeptides play important roles in many essential physiological functions and behaviors. The success in recording NE release from LC neurons in brain slices provides a unique opportunity to investigate the modulation of NE release by neuropeptides at high temporal and spatial resolutions.
Hypocretin (Orexin)
Hypocretins are neuropeptides in the lateral hypothalamus that regulate sleep/wake states, feeding behavior, and reward processes (Ganjavi and Shapiro, 2007). Hypocretin applied in the LC area depolarizes LC neurons and increases firing rate (Hagan et al., 1999). At cellular level, high level (1 μM) of hypocretin directly produces dose-dependent somatodendritic NE release, while low level (0.1 μM) of hypocretin notably potentiates NMDA receptor-mediated somatodendritic release (Chen et al., 2008). This potentiation is blocked by SB 334867, a selective hypocretin receptor 1 antagonist, or bisindolylmaleimide, a specific protein kinase C (PKC) inhibitor, indicating the involvement of hypocretin receptor 1 and PKC. Consistently, phorbol 12-myristate 13-acetate, a PKC activator, mimics the hypocretin-induced potentiation. In addition, hypocretin enhances NMDA-induced intracellular Ca2+ elevation via activation of hypocretin receptor 1 and PKC, which mediate hypocretin-potentiated somatodendritic secretion. Thus, hypocretin not only induces NE release in LC somata at high concentrations, but also potentiates NMDA receptor-mediated NE release at low concentrations.
Tachykinins
Tachykinin neuropeptides [substance P (SP), neurokinin A, neurokinin B] and their receptors (NK1, NK2, NK3) are involved in stress-mechanisms, mood/anxiety regulation, and emotion-processing (reviewed in Ebner and Singewald, 2007). In freely moving animals, exposure to forced swimming considerably enhances the release of both SP and NE in the LC (Ebner and Singewald, 2007). Administration of a selective NK1R antagonist into the LC nucleus potentiates the NE response within LC nucleus but abolishes the stress-induced increase in terminal NE release in the medial prefrontal cortex. The NE transmission in the LC and in the medial prefrontal cortex are also affected in neurokinin-1-receptor gene knockout (NK1R−/−) mice (Fisher et al., 2007). The extracellular NE in the frontal cortex of NK1R−/− mice is two to fourfold greater than that of NK1R+/+ mice. All these data demonstrate an active interaction between SP and NE system in the LC.
Several other neuropeptides also affect the firing of LC neurons. Corticotropin-releasing factor increases both the activity of LC-noradrenergic neurons and the release of NE in a LC terminal region (Page and Abercrombie, 1999). Opioids inhibit firing by hyperpolarizing LC neurons via μ-opioid receptors (Williams et al., 1987). Neuropeptide Y and galanin also inhibit firing in LC neurons (Pieribone et al., 1995; Xu et al., 2005). It remains to be investigated whether these neuropeptides regulate NE release in LC neurons.
Future Study
The molecular mechanism of LC somatodendritic exocytosis (or any somatodendritic exocytosis in general) remains largely elusive. Compared with synaptic exocytosis, soma-exocytosis may be regulated differently in terms of calcium sensitivity, vesicle pools, and exocytosis machinery. The autoinhibition mediated by α2A receptor also begs for further investigation. As a GPCR, α2A receptor may modulate vesicle fusion through Gβγ pathway as shown in chromaffin cells (Chen et al., 2005). It is well-known that the LC-NE system is affected by stress and behavior state. Since NE release from somata inhibits LC neuron activity, it may serve as a drug target to modulate LC activity and function. A better understanding of LC somata release may prove useful for drug development in targeting to LC somatodendrite area specifically.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from the National Basic Research Program of China (2006CB500800; 2007CB512100, 2012CB518006) and the National Natural Science Foundation of China (31171026, 31100597, 30970660, 30911120491 30830043, and 30770674).
References
Aston-Jones, G., and Cohen, J. D. (2005). An integrative theory of locus coeruleus-norepinephrine function: adaptive gain and optimal performance. Annu. Rev. Neurosci. 28, 403–450.
Benzekhroufa, K., Liu, B., Tang, F., Teschemacher, A. G., and Kasparov, S. (2009). Adenoviral vectors for highly selective gene expression in central serotonergic neurons reveal quantal characteristics of serotonin release in the rat brain. BMC Biotechnol. 9, 23.
Bergquist, F., Niazi, H. S., and Nissbrandt, H. (2002). Evidence for different exocytosis pathways in dendritic and terminal dopamine release in vivo. Brain Res. 950, 245–253.
Berridge, C. W., and Waterhouse, B. D. (2003). The locus coeruleus-noradrenergic system: modulation of behavioral state and state-dependent cognitive processes. Brain Res. Brain Res. Rev. 42, 33–84.
Bjorklund, A., and Lindvall, O. (1975). Dopamine in dendrites of substantia nigra neurons: suggestions for a role in dendritic terminals. Brain Res. 83, 531–537.
Bouret, S., and Sara, S. J. (2005). Network reset: a simplified overarching theory of locus coeruleus noradrenaline function. Trends Neurosci. 28, 574–582.
Callado, L. F., and Stamford, J. A. (1999). Alpha2A- but not alpha2B/C-adrenoceptors modulate noradrenaline release in rat locus coeruleus: voltammetric data. Eur. J. Pharmacol. 366, 35–39.
Callado, L. F., and Stamford, J. A. (2000). Spatiotemporal interaction of alpha(2) autoreceptors and noradrenaline transporters in the rat locus coeruleus: implications for volume transmission. J. Neurochem. 74, 2350–2358.
Cedarbaum, J. M., and Aghajanian, G. K. (1977). Catecholamine receptors on locus coeruleus neurons: pharmacological characterization. Eur. J. Pharmacol. 44, 375–385.
Chen, G., Gavin, P. F., Luo, G., and Ewing, A. G. (1995). Observation and quantitation of exocytosis from the cell body of a fully developed neuron in Planorbis corneus. J. Neurosci. 15, 7747–7755.
Chen, X. K., Wang, L. C., Zhou, Y., Cai, Q., Prakriya, M., Duan, K. L., Sheng, Z. H., Lingle, C., and Zhou, Z. (2005). Activation of GPCRs modulates quantal size in chromaffin cells through G(betagamma) and PKC. Nat. Neurosci. 8, 1160–1168.
Chen, X. W., Mu, Y., Huang, H. P., Guo, N., Zhang, B., Fan, S. Y., Xiong, J. X., Wang, S. R., Xiong, W., Huang, W., Liu, T., Zheng, L. H., Zhang, C. X., Li, L. H., Yu, Z. P., Hu, Z. A., and Zhou, Z. (2008). Hypocretin-1 potentiates NMDA receptor-mediated somatodendritic secretion from locus ceruleus neurons. J. Neurosci. 28, 3202–3208.
Chiti, Z., and Teschemacher, A. G. (2007). Exocytosis of norepinephrine at axon varicosities and neuronal cell bodies in the rat brain. FASEB J. 21, 2540–2550.
Chow, R. H., von Ruden, L., and Neher, E. (1992). Delay in vesicle fusion revealed by electrochemical monitoring of single secretory events in adrenal chromaffin cells. Nature 356, 60–63.
Duan, K., Yu, X., Zhang, C., and Zhou, Z. (2003). Control of secretion by temporal patterns of action potentials in adrenal chromaffin cells. J. Neurosci. 23, 11235–11243.
Ebner, K., and Singewald, N. (2007). Stress-induced release of substance P in the locus coeruleus modulates cortical noradrenaline release. Naunyn Schmiedebergs Arch. Pharmacol. 376, 73–82.
Elhamdani, A., Zhou, Z., and Artalejo, C. R. (1998). Timing of dense-core vesicle exocytosis depends on the facilitation L-type Ca channel in adrenal chromaffin cells. J. Neurosci. 18, 6230–6240.
Fernandez-Pastor, B., Mateo, Y., Gomez-Urquijo, S., and Javier Meana, J. (2005). Characterization of noradrenaline release in the locus coeruleus of freely moving awake rats by in vivo microdialysis. Psychopharmacology (Berl.) 180, 570–579.
Fernandez-Pastor, B., and Meana, J. J. (2002). In vivo tonic modulation of the noradrenaline release in the rat cortex by locus coeruleus somatodendritic alpha(2)-adrenoceptors. Eur. J. Pharmacol. 442, 225–229.
Fisher, A. S., Stewart, R. J., Yan, T., Hunt, S. P., and Stanford, S. C. (2007). Disruption of noradrenergic transmission and the behavioural response to a novel environment in NK1R−/− mice. Eur. J. Neurosci. 25, 1195–1204.
Ganjavi, H., and Shapiro, C. M. (2007). Hypocretin/Orexin: a molecular link between sleep, energy regulation, and pleasure. J. Neuropsychiatry Clin. Neurosci. 19, 413–419.
Gilsbach, R., and Hein, L. (2008). Presynaptic metabotropic receptors for acetylcholine and adrenaline/noradrenaline. Handb. Exp. Pharmacol. 184, 261–288.
Grimbergen, Y. A., Langston, J. W., Roos, R. A., and Bloem, B. R. (2009). Postural instability in Parkinson's disease: the adrenergic hypothesis and the locus coeruleus. Expert Rev. Neurother. 9, 279–290.
Hagan, J. J., Leslie, R. A., Patel, S., Evans, M. L., Wattam, T. A., Holmes, S., Benham, C. D., Taylor, S. G., Routledge, C., Hemmati, P., Munton, R. P., Ashmeade, T. E., Shah, A. S., Hatcher, J. P., Hatcher, P. D., Jones, D. N., Smith, M. I., Piper, D. C., Hunter, A. J., Porter, R. A., and Upton, N. (1999). Orexin A activates locus coeruleus cell firing and increases arousal in the rat. Proc. Natl. Acad. Sci. U.S.A. 96, 10911–10916.
Huang, H. P., Wang, S. R., Yao, W., Zhang, C., Zhou, Y., Chen, X. W., Zhang, B., Xiong, W., Wang, L. Y., Zheng, L. H., Landry, M., Hokfelt, T., Xu, Z. Q., and Zhou, Z. (2007). Long latency of evoked quantal transmitter release from somata of locus coeruleus neurons in rat pontine slices. Proc. Natl. Acad. Sci. U.S.A. 104, 1401–1406.
Jaffe, E. H., Marty, A., Schulte, A., and Chow, R. H. (1998). Extrasynaptic vesicular transmitter release from the somata of substantia nigra neurons in rat midbrain slices. J. Neurosci. 18, 3548–3553.
Kim, Y., Park, M. K., and Chung, S. (2008). Voltage-operated Ca2+ channels regulate dopamine release from somata of dopamine neurons in the substantia nigra pars compacta. Biochem. Biophys. Res. Commun. 373, 665–669.
Mateo, Y., Pineda, J., and Meana, J. J. (1998). Somatodendritic alpha2-adrenoceptors in the locus coeruleus are involved in the in vivo modulation of cortical noradrenaline release by the antidepressant desipramine. J. Neurochem. 71, 790–798.
Mehler, M. F., and Purpura, D. P. (2009). Autism, fever, epigenetics and the locus coeruleus. Brain Res. Rev. 59, 388–392.
Nieuwenhuis, S., Aston-Jones, G., and Cohen, J. D. (2005). Decision making, the P3, and the locus coeruleus-norepinephrine system. Psychol. Bull. 131, 510–532.
Norenberg, W., Schoffel, E., Szabo, B., and Starke, K. (1997). Subtype determination of soma-dendritic alpha2-autoreceptors in slices of rat locus coeruleus. Naunyn Schmiedebergs Arch. Pharmacol. 356, 159–165.
Page, M. E., and Abercrombie, E. D. (1999). Discrete local application of corticotropin-releasing factor increases locus coeruleus discharge and extracellular norepinephrine in rat hippocampus. Synapse 33, 304–313.
Pieribone, V. A., Xu, Z. Q., Zhang, X., Grillner, S., Bartfai, T., and Hokfelt, T. (1995). Galanin induces a hyperpolarization of norepinephrine-containing locus coeruleus neurons in the brainstem slice. Neuroscience 64, 861–874.
Pudovkina, O. L., Kawahara, Y., de Vries, J., and Westerink, B. H. (2001). The release of noradrenaline in the locus coeruleus and prefrontal cortex studied with dual-probe microdialysis. Brain Res. 906, 38–45.
Sara, S. J., and Devauges, V. (1989). Idazoxan, an alpha-2 antagonist, facilitates memory retrieval in the rat. Behav. Neural Biol. 51, 401–411.
Singewald, N., and Philippu, A. (1998). Release of neurotransmitters in the locus coeruleus. Prog. Neurobiol. 56, 237–267.
Stahl, S., and Briley, M. (2004). Understanding pain in depression. Hum. Psychopharmacol. 19(Suppl. 1), S9–S13.
Svensson, T. H. (1987). Peripheral, autonomic regulation of locus coeruleus noradrenergic neurons in brain: putative implications for psychiatry and psychopharmacology. Psychopharmacology (Berl.) 92, 1–7.
Svensson, T. H., Bunney, B. S., and Aghajanian, G. K. (1975). Inhibition of both noradrenergic and serotonergic neurons in brain by the alpha-adrenergic agonist clonidine. Brain Res. 92, 291–306.
Swanson, L. W., and Hartman, B. K. (1975). The central adrenergic system. An immunofluorescence study of the location of cell bodies and their efferent connections in the rat utilizing dopamine-beta-hydroxylase as a marker. J. Comp. Neurol. 163, 467–505.
Valentino, R. J., and Van Bockstaele, E. (2008). Convergent regulation of locus coeruleus activity as an adaptive response to stress. Eur. J. Pharmacol. 583, 194–203.
Williams, J. T., Christie, M. J., North, R. A., and Roques, B. P. (1987). Potentiation of enkephalin action by peptidase inhibitors in rat locus ceruleus in vitro. J. Pharmacol. Exp. Ther. 243, 397–401.
Williams, J. T., Henderson, G., and North, R. A. (1985). Characterization of alpha 2-adrenoceptors which increase potassium conductance in rat locus coeruleus neurones. Neuroscience 14, 95–101.
Keywords: norepinephrine, somatodendritic, quantal release, locus coeruleus, α2A-adrenoceptor
Citation: Huang H-P, Zhu F-P, Chen X-W, Xu Z-QD, Zhang CX and Zhou Z (2012) Physiology of quantal norepinephrine release from somatodendritic sites of neurons in locus coeruleus. Front. Mol. Neurosci. 5:29. doi: 10.3389/fnmol.2012.00029
Received: 16 December 2011; Accepted: 20 February 2012;
Published online: 06 March 2012.
Edited by:
Nikita Gamper, University of Leeds, UKReviewed by:
Min Zhou, The Ohio State University, USAJiang Ye, University of Medicine and dentistry of New Jersey, USA
Copyright: © 2012 Huang, Zhu, Chen, Xu, Zhang and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Zhuan Zhou and Claire Xi Zhang, Institute of Molecular Medicine, Peking University, 5 Yiheyuan Road, Beijing 100871, China. e-mail:enpob3VAcGt1LmVkdS5jbg==;Y2xhaXJleHpAcGt1LmVkdS5jbg==;
Zhi-Qing David Xu, Department of Neurobiology and Beijing Institute for Neuroscience, Capital Medical University, Beijing 100069, China. e-mail:emhpcWluZ3hAY2NtdS5lZHUuY24=