

- 1The Edmond and Lily Safra Center for Brain Sciences and the Institute of Life Sciences, The Hebrew University of Jerusalem, Jerusalem, Israel
- 2Institutes of Clinical Chemistry and Clinical Pharmacology, University Hospital Bonn, Bonn, Germany
Hematopoietic stem cells (HSCs) differentiate and generate all blood cell lineages while maintaining self-renewal ability throughout life. Systemic responses to stressful insults, either psychological or physical exert both stimulating and down-regulating effects on these dynamic members of the immune system. Stress-facilitated division and re-oriented differentiation of progenitor cells modifies hematopoietic cell type composition, while enhancing cytokine production and promoting inflammation. Inversely, stress-induced increases in the neurotransmitter acetylcholine (ACh) act to mitigate inflammatory response and regain homeostasis. This signaling process is terminated when ACh is hydrolyzed by acetylcholinesterase (AChE). Alternative splicing, which is stress-modified, changes the composition of AChE variants, modifying their terminal sequences, susceptibility for microRNA suppression, and sub-cellular localizations. Intriguingly, the effects of stress and AChE variants on hematopoietic development and inflammation in health and disease are both subject to small molecule as well as oligonucleotide-mediated manipulations in vitro and in vivo. The therapeutic agents can thus be targeted to the enzyme protein, its encoding mRNA transcripts, or the regulator microRNA-132, opening new venues for therapeutic interference with multiple nervous and immune system diseases.
Introduction
Stress can be defined as a psychological, environmental, or physiologic threat on homeostasis (Chrousos, 2009). Psychological stress begins with impulses from high cortical centers via the limbic system and results in the release of chemical mediators. Catecholamine signals, and specifically, nor-adrenaline, are then released from sympathetic nerve fibers in direct proximity to target tissues assisting the body to launch a fight-or-flight reaction, increasing heart rate, and blood flow to skeletal muscles while decreasing immune reactions (Padgett and Glaser, 2003). Another key contributor to these processes is the parasympathetic neurotransmitter acetylcholine (ACh) which is fundamental for nervous system function (Kaufer et al., 1998) and is also produced by peripheral leukocytes (Kawashima and Fujii, 2000). These two pivotal neurotransmitters are both up-regulated during stress and can activate the hypothalamic-pituitary-adrenal axis (HPA) and the sympathetic adrenal medulla (Black, 2002). Specifically, sustaining moderate ACh levels is crucial for maintaining homeostasis: abrupt stress-induced elevation of ACh signaling may be lethal whereas its gradual loss, as in Alzheimer's disease, multiple system atrophy, and other neuro-degeneration conditions, is associated with progressive deterioration of cognitive, autonomic, and neuromuscular functions (Soreq and Seidman, 2001). Also, activation of cholinergic receptors is beneficial in pathological states such as atherosclerosis, myocardial infarction, hypertensive vasculopathy and nephropathy, and heart failure, largely due to yet incompletely understood mechanisms (Chrousos, 2009; McEwen and Gianaros, 2011). In our current review, we argue that moderate ACh levels are also crucial for controlling immune and inflammatory functions, both in the brain and in peripheral tissues and that acetylcholinesterase (AChE) is a key contributor toward sustaining these levels.
The dynamic hematopoiesis process of blood cell production and differentiation is notably characterized by a continuous turnover of cells throughout life. Hematopoietic stem cells (HSCs) are characterized by their ability to generate all blood cell lineages while maintaining self-renewal. These properties are modified substantially when the host of these cells is exposed to either psychological or physical stressors (Dudakov et al., 2010), so that cells from the bone marrow and the vascular marginal pool are rapidly mobilized to the circulation (Rafii et al., 1995). Consequently, cytokines and chemokines capable of activating cells to immediately respond to various stimuli regulate the production and longevity and determine the number of circulating blood cells (Cavazzana-Calvo et al., 2011). Cytokines further regulate the division and differentiation of progenitor cells to develop into morphologically recognizable cells with distinctive phenotype features. Myelopoiesis involves the production of granulocytes, monocytes, red blood cells (RBCs) and the platelet-producing megakaryocytes (Mks). The lymphoid progenitor cells can differentiate into T, B, or natural killer (NK) lymphocytes, depending on the microenvironment. Importantly, mesenchymal stem cells carry both nicotinic and muscarinic ACh receptors (Hoogduijn et al., 2009). Stress-inducible changes in cholinergic signaling and in any of these populations can hence drastically modulate immune functions.
Stress-Affected Hematopoiesis and Neuropoiesis
There are many interactions between the nervous and hematopoietic systems. These include sequence and structural homology between neuropoietic and hematopoietic factors. Neuropoietic factors such as cholinergic differentiation factor (CDF) and ciliary neurotrophic factor (CNTF) determine the developmental fate of sympathetic neurons from noradrenergic to cholinergic function. These show homologies to the hematopoietic cytokines granulocyte colony-stimulating factor (G-CSF) and interleukin-6 (Bazan, 1991).
Hematopoietic factors may affect differentiation and regeneration of cholinergic neurons. In contrast, scopolamine, a muscarinic ACh receptor blocker, can abolish stress-induced erythropoiesis (Gol'dberg et al., 2000), and deficiencies of neuropeptides. Substance P and calcitonin gene-related peptide (CGRP) lead to a dramatic fall in neutrophil production; these neuropeptides stimulate bone marrow colony formation and affect neutrophils production via both direct and indirect effects on bone marrow colony forming units of granulocyte/monocyte progenitors (CFU-GM). The observed effects do not involve peripheral blood CFU-GM, suggesting a direct neural control over BM hematopoiesis (Broome et al., 2000). Furthermore, erythropoietin was shown to exert neurotrophic and neuroprotective activities in different in vivo and in vitro models of brain damage as well as an anti-apoptotic effect on microglia, the brain's resident immune cell (Vairano et al., 2002).
Hematopoietic factors may also affect differentiation and regeneration of cholinergic neurons. In contrast, scopolamine, a muscarinic ACh receptor blocker, can abolish stress-induced erythropoiesis, and deficiencies of neuropeptides. Substance P and CGRP lead to a dramatic fall in neutrophil production; these neuropeptides stimulate bone marrow colony formation and affect neutrophils production via both direct and indirect effects on bone marrow CFU-GM progenitors. The observed effects do not involve peripheral blood CFU-GM, suggesting a direct neural control over BM hematopoiesis.
The Parasympathetic System Acetylcholine Hydrolyzing Enzyme Acetylcholinesterase and Stress
The parasympathetic system and the ACh hydrolyzing enzyme AChE together contribute to the delicate balance of hematopoietic and immune events under stress (Figure 1). ACh produced by the vagus nerve and/or by peripheral leukocytes (Rosas-Ballina et al., 2011) can potently modulate several classical immune reactions by activating the α7 nicotinic receptor α7nAChR on the leukocytes' membrane, which in turn blocks the NF-kB-mediated production of pro-inflammatory cytokines such as IL-1 and tumor necrosis factor (TNF) α. Compatible with this mechanism of action, vagal stimulation suppresses septic shock-like response after bacterial lipopolysaccharide (LPS) injection (Gol'dberg et al., 2000; Tracey, 2002). Tissue residing mononuclear cells also receive cholinergic signaling via ACh secreted from the vagus and/or synthesized in these nucleated immune cells, which have all the components of the cholinergic signaling system: ACh, the ACh synthesizing enzyme choline acetyl-transferase (ChAT), and the co-regulated vesicular ACh transporter VAChT, transcribed from the same transcription unit, the ACh degrading enzyme AChE and functional muscarinic and nicotinic ACh receptors. Correspondingly, ACh receptors were identified on lymphocytes from the thymus, lymph nodes, spleen, and peripheral blood (Tracey, 2002). Also, vagus nerve stimulation fails to inhibit TNF α production in splenectomized animals during lethal endotoxemia, demonstrating cholinergic innervation of the spleen (Huston et al., 2006).

Figure 1. Stress modifies AChE's composition. Shown are the N-terminal and C-terminal changes representing combinatorial differences on AChE splice variants. The soluble monomeric AChE-R variant produced in all tissues expressing this gene accumulates under stress and is found in the circulation, albeit being less stable than AChE-S. Both AChE variants are associated with stress-inducible hematopoiesis. The red-labeled C-terminus of AChE-R represents the hematopoietic peptide ARP [Grisaru et al. (2006)].
In mice, peptidergic nerve fibers entering the bone marrow terminate with synapses on stromal and perivascular cells, interactions that were implicated in local inflammation and regulation of leukocyte trafficking (Gol'dberg et al., 2000). Endothelium, a key regulator of leukocyte trafficking during inflammation, is also a target of anti-inflammatory cholinergic mediators; both vagus nerve stimulation and cholinergic agonists significantly block leukocyte migration in vivo (Saeed et al., 2005). Cholinergic neurons are also present in developing thymic rudiments and their presence was associated with increased production of thymic lymphocytes. The cholinergic system also affects HSCs and their niche cells and plays an active role in T cell differentiation. Thus, CD8+ T cells from M1 receptor-deficient mice fail to differentiate into cytolytic T lymphocytes (Zimring et al., 2005). ACh release from spleen T cells can in turn attenuate TNF α production in spleen via α7nAChR expressed on innate immune cells (Rosas-Ballina et al., 2010). Furthermore, T cells show plasticity in their response to cholinergic stimuli, so that nicotine up-regulates interferon-γ (IFN-γ) and down-regulates interleukin (IL)-17 secretion, whereas muscarin enhances IL-10 and IL-17 and inhibits INF-γ secretion. As is implicated from the numerous studies stated above, ACh signaling is essential for T-cell activation and its opposite function may be required to synchronize and balance ionic and metabolic events in a single lymphocyte. Within the T-cell, the ACh regulatory axis may provide for a fine tuning of the T cells to changing environment (Qian et al., 2011).
In all peripheral tissues, ACh signals normally remain above a certain threshold sufficient to suppress the production of pro-inflammatory cytokines, yet are transiently reduced following stress due to AChE over-production which lasts several hours (Nance and Sanders, 2007). Within 24 h, elevation of the AChE-targeted microRNA-132 (Shaked et al., 2009; Soreq and Wolf, 2011) reduces AChE levels, retrieving ACh-mediated blockade of pro-inflammatory cytokines production. Therefore, stress responses also facilitate yet more IL-1 production (Gilboa-Geffen et al., 2007). Monocytes, macrophages, dendritic cells, and T lymphocytes which infiltrate peripheral organs and accelerate pathological processes are exposed to the adrenergic and cholinergic transmitters released by the autonomic innervations of these organs, which could dramatically alter cytokine release (Nance and Sanders, 2007). That serum AChE activity continuously increases with age (Sklan et al., 2004) may suggest aging-related deterioration in the efficacy of this regulation process.
Under stress, the elevated glucocorticoid cortisol interacts with two cytosine/guanosine (CpG-rich) sequence motifs in the AChE promoter to induce nuclear transcriptional and post-transcriptional events of over-production of AChE and replace the major stable AChE splice variant AChE-S by the less stable AChE-R variant (Figure 2) (Meshorer et al., 2002). This effect associates with release from AChE-R of the cleavable C-terminal peptide ARP. Both AChE-R and ARP can stimulate the proliferation of CD34+ hematopoietic progenitor cells and facilitate the proliferation and differentiation of promegakaryocytes and subsequent platelet production (Pick et al., 2006). When secreted, the extra-cellular AChE-R hydrolyzes ACh as efficiently as AChE-S, further facilitating the production of pro-inflammatory cytokines; and when retained in the intra-cellular space AChE-R interacts through the C-terminal ARP peptide with several partner signaling proteins, also promoting cell proliferation (Meshorer et al., 2005). Ex vivo, ARP was more effective than cortisol and equally effective as stem cell factor in promoting expansion and differentiation of early hematopoietic progenitor cells into myeloid and Mk lineages (Pick et al., 2006).
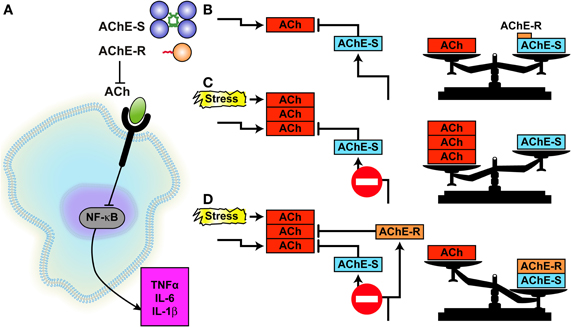
Figure 2. AChE controls the anti-inflammatory reflex.(A) At the extracellular space, both AChE-S and AChE-R hydrolyze ACh, impairing its capacity to block, through the α7 nicotinic ACh receptor, the activation of NFκB which enables pro-inflammatory cytokine secretion (the so-called “anti-inflammatory reflex”). (B) Under normal conditions, AChE-S is by far the prominent variant keeping ACh in balance, AChE-R being the soluble, minor component. (C) Under stress, excess ACh impairs the cholinergic balance. (D) Substitution of AChE-S with excess AChE-R facilitates ACh hydrolysis, shifting the imbalance to AChE excess [Ofek et al. (2007)].
Tissue macrophages are effectively deactivated when exposed to ACh, suppressing the release of the pro-inflammatory cytokines TNF-α, IL-1β, and IL-18 at the post-transcriptional level (Tracey, 2002). Among other effects, immune stimuli induce over-expression of the AChE-targeting microRNA-132 in leukocytes (Soreq and Wolf, 2011). Consequently, microRNA-132 reduces AChE levels, increasing the available ACh and finally inhibiting the secretion of pro-inflammatory cytokines (Wang et al., 2003). Inversely, mice engineered to over-express a 3′-shortened and hence microRNA-132 refractory AChE-R present excessive leukocyte recruitment into the peritoneum, higher levels of inflammatory cytokines than strain-matched controls and impaired cholinergic anti-inflammatory regulation in spite of substantial microRNA-132 up-regulation both in the bone marrow and the hypothalamus. Intravenous injection to wild-type mice of an LNA-modified anti-miR-132 oligonucleotide increases AChE activity in the bone marrow, spleen, and serum, with parallel consequences (Shaked et al., 2009). MiR-132 has been shown to be a regulator of neuronal structure and function. Transgenic mice over-expressing miR-132 in forebrain neurons display increased dendritic spine density and decreased expression of MeCP2, a protein implicated in Rett Syndrome and other disorders of mental retardation. These mice displayed significant deficits in novel object recognition (Hansen et al., 2010). However, shRNA-mediated suppression of the “synaptic” AChE-S variant prevented the increase in miR-132 following footshock stress, suggesting bidirectional regulation (Shaltiel et al., in press).
Furthermore, miR-132 has been shown to be induced during periods of active synaptogenesis and is necessary and sufficient for hippocampal spine formation. Knockdown of the miR-132 target p250GAP increases spine formation while overexpression attenuates this activity (Impey et al., 2010).
AChE and Granulocytosis
AChE-R and ARP both induce granulocytosis (Grisaru et al., 2001). The specificity of ARP is supported by the finding that ASP, the parallel yet distinct C-terminal peptide of AChE-S, fails to induce such effects (Figure 3). In the post-partum human serum, AChE-R excess was speculated to lead to reduced ACh concentrations. AChE-R accumulation might hence be perceived as an adaptive response, facilitating the production of pro-inflammatory cytokines to protect the body from post-partum conditions, such as infections. This is compatible with observations of increased production of pro-inflammatory cytokines under stress and further proliferative and cell activation signals (Broome et al., 2000). Protracted post-stress granulocytosis initiated by cortisol would then increase AChE-R production and reduce ACh's anti-inflammatory action.
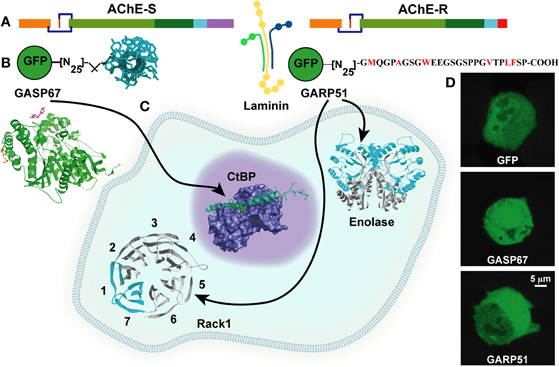
Figure 3. AChE variants exert hematopoietic effects through common and distinct protein partners.(A) The common core domain of AChE interacts with laminin. (B) To validate partner interactions, the C-termini of AChE-S and AChE-R were constructed together with GFP. (C) The C-terminus of AChE-S interacts with the anti-apoptogenic nuclear CtBp, whereas intracellular AChE-R interacts with the proliferation activating scaffold PKCβ carrier RACK1 and with Enolase. (D) Fluorescent cell images: GFP constructs direct the AChE-S and AChE-R C-termini to nucleus and cytoplasm, respectively [Perry et al. (2007)].
Subsequent secondary feedback response to the initial excess of ACh and AChE in the periphery is needed to re-balance parasympathetic activities (Erb et al., 2001), suppress the production of pro-inflammatory cytokines in macrophages (Tracey, 2002), and terminate the granulocytosis process. Such responses can potentially involve changes in miR-132 which can suppress AChE activities. In contrast, IL-1β induces ACHE gene expression in phaeochromocytoma cells (Li et al., 2000) (Figure 4). The involvement of AChE-R in immune system activities under acute stress further prolongs the beneficial (pro-inflammatory) HPA effects mediated through cortisol. While explaining the harmful aspects of stress (chronically high cytokine levels) (Grisaru et al., 2001), increased levels of AChE-R can also maintain the immune system at high alert, avoiding acute suppression of the immune system and providing protection from bacterial infection.
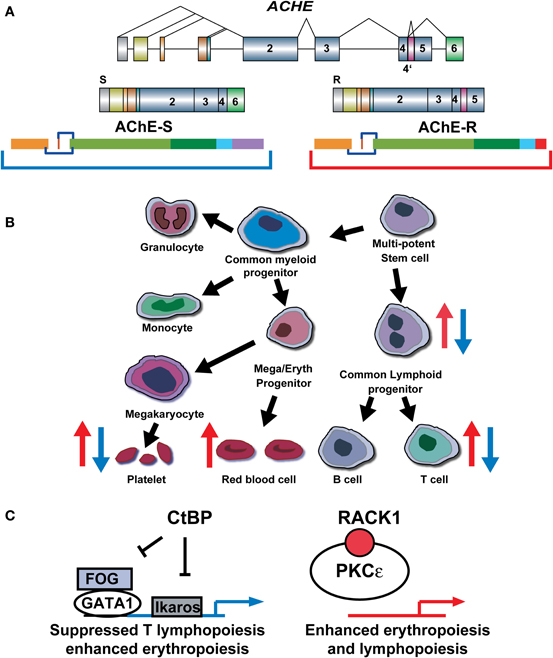
Figure 4. AChE splice variants induce inverse hematopoietic effects. (A) The AChE gene and its S and R transcripts. (B) The hematopietic pathway (black arrows). Up and down changes in cell compositions are presented as red and blue arrows for transgenic excess in AChE-R (red) and AChE-S (blue). (C) Scheme: Inverse outcome and the proteins involved [Gilboa-Geffen et al. (2007)].
The AChE-S Splice Variant Causes Leukopenia
AChE gene expression increases during apoptosis (Zhang et al., 2002), and silencing of the AChE gene by small interference (si) RNA prevents apoptosis (Pick et al., 2006). Given that AChE-S is by far the major AChE splice variant in all tissues (Soreq and Seidman, 2001), this suggests that AChE-S induces apoptosis, alternatively or additionally to its known role in the apoptosome formation. One possible mechanism through which this could occur involves the interaction of AChE-S with the anti-apoptotic C-terminal binding protein (CtBP). Supporting this notion, AChE-S excess capable of blocking the anti-apoptotic effect of CtBp reduces T lymphopoiesis and thrombopoiesis, inversely to the effect of AChE-R, which up-regulates hematopoiesis (Gilboa-Geffen et al., 2007). Correspondingly, AChE-S over-expressing mice (TgS) present elevated hemoglobin and total RBC counts accompanied by decreased platelet counts, GR1 + myeloid cells and T lymphocytes [both T helper (CD4+) and T suppressor cells (CD8+)], but unchanged content of monocytes (CD11b+) and B lymphocytes (CD19+) (Perry et al., 2007). This suggests that the CFU-GEMM progenitor cells from AChE-S over-expressing mice preferentially differentiate toward erythropoiesis rather than megakaryocytopoiesis and granulopoiesis, inverse to the stress-induced enhancement of granulopoiesis and thrombocytosis, under AChE-R over-expression (Grisaru et al., 2001). Together, under acute stress (Gilboa-Geffen et al., 2007; Perry et al., 2007), elevated AChE-R and decreased AChE-S levels can, therefore, induce a bimodal myelopoietic and thrombopoietic effect: first, by increasing AChE-R inducing proliferation, and second by enhancing the CtBP-anti-apoptotic effect due to AChE-S loss which releases CtBP from AChE/CtBP complexes (Figure 4).
Stress-Induced Thrombopoiesis
Thrombopoiesis increases in response to stress-increased production of AChE-R. This likely involves AChE-R interaction with the scaffold protein RACK1 and its cargo PKCε (Perry et al., 2007), which induces megakaryocytic differentiation in HEL and K562 cells and in primary human HSCs (Lev-Lehman et al., 1997). TPO and platelet counts also increase in the AChE-R overproducing TgR mice as compared to the strain-matched control mice. This affects the non-fatal LPS response: LPS administration (5 ug/kg) induces a rapid fall in platelet counts in both TgR and control mice, but platelet and WBC recovery is considerably faster in TgR mice, likely due to the increased capacity of TgR BM progenitors to proliferate and differentiate into pluripotent CFU-GEMM, CFU-GM, and CFU-Mk (Pick et al., 2006; Perry et al., 2007). Moreover, thrombopoietin (TPO) increases PKCε expression in mouse Mks, whereas blocking PKC activation inhibits platelet formation (Deutsch et al., 2002). Thus, the elevated levels of TPO, AChE-R, and PKCε in plasma and Mks from TgR mice support the notion of a cholinergic promotion of thrombopoietic signal transduction both through the hydrolytic and the non-enzymatic features of AChE-R. The pivotal role played by AChE-R and ARP in hematopoiesis and thrombopoiesis suggests a potential new strategy for improving post-engraftment thrombopoiesis.
Stress and Thymocyte Development
In the autoimmune disease Myasthenia gravis (MG) AChE-R translocates to the membrane-associated fraction of thymocytes, reflecting the activation state of the thymic cells. Like thymus from MG patients, the thymus from TgR mice displays an abnormally high number of immature thymocytes compared to control mice. PKCβII which plays a role in B cell receptor survival signaling (Oshevski et al., 1999) is also over-expressed in TgR mice compared to FVB/N controls. AChE-R could thus protect immature thymocytes from apoptosis, maintaining a high level of the anti-apoptotic protein Bcl-2 (Rojnuckarin and Kaushansky, 2001). The high number of thymocytes in TgR mice is compatible with production of AChE-R by cortical stromal cells exerting a protective signal from apoptosis to the neighboring thymocytes. That immature thymocytes located in the cortex require contact signals with the cortical epithelium to survive (Toiber et al., 2008), may therefore, explain why excessive AChE-R production by cortical stromal cells induces an efficient positive selection process in the TgR thymus. Also, AChE-R could be produced by the thymocytes themselves, and thymocytes cultured from TgR mice are more resistant to apoptosis than normal thymocytes, even in the absence of stromal cells. Cholinergic signals and AChE's catalytic activity thus influence thymocytic apoptosis in inverse directions, so that the pro-apoptotic effect of ACh is counterbalanced by a proliferative effect that could be predominant for the AChE-R variant. Supporting this notion, the TgR thymus presents more proliferating cells (expressing the Ki67 antigen) compared to control mice (Gilboa-Geffen et al., 2007) (Figure 5).
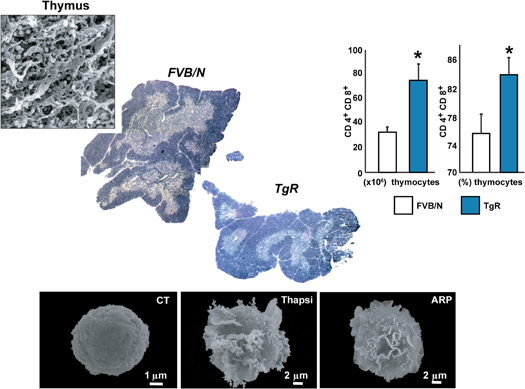
Figure 5. AChE-R potentiates lymphopoiesis and megakaryopoiesis. Excess of the stress-induced AChE-R variant induces thymocytes hyperproliferation, compatible with the clinical phenotype in myasthenic patients, as well as thrombopoiesis. This reflects on myasthenia gravis where antisense suppression of AChE-R was effective in phase I clinical trials [Argov et al. (2007)]. Below: Megakaryoblasts (Meg-01) cells scanned by electron micrographs. Control Meg-01 cells exhibit a smooth surface. Cells treated for 24 h with ARP show initial formation of flat membrane sheets (middle panels) or elongated pseudopodia reflecting proplatelet formation territories (right panels) characteristic of megakaryocytic differentiation [Guimaraes-Sternberg et al. (2006)].
Stress, AChE Splice Variants and Antisense Modulation
Antisense oligonucleotides are short synthetic strands of modified DNA or RNA designed to hybridize within the cell with target-specific mRNA by Watson–Crick base pairing. Binding activates RNAses that degrade them. RNA–antisense complexes block synthesis of the protein encoded by the mRNA sequence and release the antisense agent to bind to a further strand of mRNA, creating a recycling antisense mechanism. An antisense agent which selectively destroys AChE-R, EN101 (currently denoted BL-7040) is a 20-mer oligonucleotide, chemically modified by incorporating 2′oxymethyl groups in the last three nucleotides at its 3′ end. EN101/BL-7040 binds to a coding sequence common to all splice variants of human AChE (Evron et al., 2005). Since the AChE-R mRNA transcript includes less guanosines and cytosines than AChE-S, it is intrinsically less stable and more sensitive to this treatment. BL-7040/EN101 is an FDA approved orphan drug shown to be safe for human use and active both intravenously and orally in the suppression of the progressive muscle fatigue symptoms of rats with experimental myasthenia gravis (MG), an autoimmune neuromuscular disease involved with systemic AChE-R over-production (Brenner et al., 2003). It was also demonstrated to suppress AChE-R in primates (Evron et al., 2005) and in human myasthenic patients (Argov et al., 2007; Sussman et al., 2008).
Other short oligonucleotides, designated aptamers can be selected to tightly and specifically bind a target protein molecule (Que-Gewirth and Sullenger, 2007) and have been approved for the therapeutic use by the US FDA. A 50-mer nuclease-resistant RNA aptamer with 2′-amino-modified pyrimidines can recognize human muscarinic AChR, and cross-react with MG patient autoantibodies (Lee and Sullenger, 1997), efficiently preventing them from down-modulating AChR expression on human cells (Que-Gewirth and Sullenger, 2007). Intriguingly, BL-7040 also operates as an aptamer activator of Toll-like receptor 9 (TLR9) facilitating an alternative route of NFκB, controlling salivary function (Gilboa-Geffen et al., 2011) and avoiding post-traumatic stress in mice exposed to predator scent (Zimmerman et al., 2012).
Stress and Neuroimmune Signaling
Within the central nervous system, neuronal ACh levels rapidly increase under stress, followed by a feedback response of AChE overproduction which can tilt the balance to reduce ACh levels (Kaufer et al., 1998). Correspondingly, the key pro-inflammatory mediator IL-1 is over-expressed in the brain under stress. That IL-1 can induce neuronal AChE synthesis (Li et al., 2000) would further suppress ACh levels, inducing a feed-forward loop of stress-inducible inflammatory response in the brain. This response is pivotal for those neural systems thought to be involved in stress effects in PTSD in humans (Rauch et al., 2006) and in predator stress effects in animals (Adamec et al., 2005). Systems implicated include the amygdala, hippocampus, and medial prefrontal cortex and brain stem targets of forebrain limbic systems. It is unlikely that within such complex circuitry, changes in one system are responsible for all lasting effects of stress. Nevertheless, neuronal AChE-R has been found to be functionally involved in stress-mediated facilitation of fear learning, limbic neural plasticity, and change in affect in predator stress (Adamec and Shallow, 1993; Nijholt et al., 2004; Adamec et al., 2008). Specifically, stress-induced transcription of AChE-R facilitates neuroplasticity in the brain's limbic structures, resulting in long-lasting changes in neural systems (Meshorer et al., 2002; Nijholt et al., 2004).
Severe stress may precipitate affective disorder lasting up to a life time after traumatic stress (Yehuda, 2002). Predator stress (inescapable and unprotected exposure to a cat) likewise lastingly increases rodent anxiety (Adamec and Shallow, 1993). Increased expression of AChE-R following stress has been demonstrated in some of the same areas involved in predator stress-induced limbic neuroplasticity, including the hippocampus and amygdala. Intriguingly, BL-7040/EN101 administration limits these stress reactions. Predator stress abnormally increases the transcription of AChE-R, which in turn alters the neural substrate responsible for the changes in acoustic startle amplitude. BL-7040/EN101 reduces the mRNA for AChE-R, and prevents the occurrence of these changes (Adamec et al., 2008). This effect involves TLR9 activation and is preventable by the TLR9 blocking oligonucleotide ODN 2088 (Zimmerman et al., 2012). It has been proposed that following severely stressful events, AChE-R increases to a level which is no longer adaptive and results in physiological changes which contribute to changes in affect observed in post-traumatic stress disorder (PTSD) (Meshorer et al., 2002; Nijholt et al., 2004). Correspondingly, systemic administration of BL-7040/EN101 selectively lowers the level of AChE-R both peripherally (Argov et al., 2007) and in the brain (Evron et al., 2005; Pollak et al., 2005). In view of the TLR9 involvement, this effect indicates inter-related coordination between the innate immune system and the AChE gene expression. Termination of the granulocytosis response in the periphery thus resembles the termination of stress-inducible neurotransmission in the brain, highlighting the close inter-relationships between the neuronal and immune cell reactions to stress (Kiecolt-Glaser et al., 2003).
Non-Catalytic role for AChE in the CNS
The stress-related role of neural AChE was inferred from experiments in which over-expression of AChE-R mRNA was suppressed in mice subjected to closed head injury by administration of mouse/EN101 (Shohami et al., 2000). This treatment reduced the number of dead neurons and facilitated neurologic recovery, suggesting that AChE-R and/or secondary element(s) induced by overexpression of AChE-R contributed to neuronal death (Metz and Tracey, 2005). This could reflect a direct contribution of the increased hydrolytic capacity of the induced AChE to reduce the tissue level of ACh, promoting inflammation and tissue damage. Alternatively, or in addition, stress-induced AChE could selectively contribute to cellular apoptosis through its non-catalytic properties, inducing cell death in non-cholinergic neurons as well (Zhang et al., 2002; Park et al., 2004).
Photoreceptor loss is the primary cause of blindness in degenerative diseases such as retinitis pigmentosa and age-related macular degeneration. Exposure of albino rats to bright light provides an established model for photoreceptor damage and stress-induced photoreceptor injury (Stone et al., 1999). A variety of growth hormones, cytokines (LaVail et al., 1992), and antioxidants (Organisciak et al., 1992) were found to partially protect retinal photoreceptors from light damage, suggesting causal involvement of stress-induced processes. The importance of AChE gene expression for retinal maintenance was experimentally validated in a detailed neuro-anatomic survey that demonstrated impaired formation of the inner retina and degeneration of photoreceptors in an AChE knockout mouse where ALL of the AChE variants are missing (Bytyqi et al., 2004). The major excitatory neurotransmitter of the retina is L-glutamate, whereas ACh plays a limited role in visual information processing within the retina and is found mainly in amacrine and ganglion cells in the proximal part of the retina (Hutchins, 1987; Criswell and Brandon, 1993). Either AChE-S or AChE-R can appear either with a cleavable N-terminal signal peptide or with an extended N terminus, potentially making it membrane bound. AChE mRNA expression in human adult photoreceptors (Broide et al., 1999), cells that are not involved in cholinergic synaptic activity further raises the possibility that AChE in photoreceptors exerts stress-related morphogenic function(s). It was shown that the N-AChE protein plays a detrimental role, exacerbating photoreceptor injury after exposure to bright, damaging light. Notably, EN101 treatment limited light-induced damages to the retina (Kehat et al., 2007). Future experiments are needed to find out whether N-AChE-R accumulates in retinal (and other) neurons after other stressful episodes such as increased pressure (glaucoma), metabolic stress (diabetes), and ischemia, where it may play detrimental role(s) promoting neuronal and specifically retinal damage.
Concluding Remarks
To properly function, the immune system depends on a fine-tuned balance of immune cell activation, proliferation, and differentiation which is perturbed under stress. Consequently, hematopoietic and immune responses are largely modified under stress in an inter-related manner conserved throughout evolution. First, stress inducible changes in hematopoietic processes can substantially modulate both the immune and CNS functions, for example, by over-producing cytokines which can penetrate the brain and affect neuronal activities. Second, the parasympathetic system, largely through AChE modulations, contributes to the delicate balance between hematopoietic and immune events and CNS reactions under stress. The major AChE splice variant AChE-S is replaced by the stress-induced AChE-R variant. Due to modified splicing, consequently impaired AChE-S-CtBp interaction as well as enhanced AChE-R-RACK1 interactions, both enhance granulopoiesis and thrombocytosis. Increased levels of AChE-R can thus trigger beneficial resistance to acute overstimulation under exposure to microbial molecules such as LPS, but at the same time sustaining the harmful effects of chronically high cytokine levels. This balance of stress-inducible positive and negative effects can be maintained by diverse Oligonucleotide treatments, achieving better control over both nervous system and immune functions. While these therapeutic strategies await further studies to discern the corresponding molecular mechanisms, the promising findings can already point at numerous previously unforeseen interactions between the immune and the nervous system.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research is supported by Israel Science Foundation's Legacy Heritage grant 378/11 and the German Research Foundation's DFG Trilateral Germany-Israel Palestine Authority Cooperation Program (to Hermona Soreq). Adi Gilboa-Geffen was the incumbent of a FEBS post-doctoral fellowship for work in Bonn. The BL-7040 program has been licensed to BioLineRx for future development.
References
Adamec, R., Head, D., Soreq, H., and Blundell, J. (2008). The role of the read through variant of acetylcholinesterase in anxiogenic effects of predator stress in mice. Behav. Brain Res. 189, 180–190.
Adamec, R. E., Blundell, J., and Burton, P. (2005). Neural circuit changes mediating lasting brain and behavioral response to predator stress. Neurosci. Biobehav. Rev. 29, 1225–1241.
Adamec, R. E., and Shallow, T. (1993). Lasting effects on rodent anxiety of a single exposure to a cat. Physiol. Behav. 54, 101–109.
Argov, Z., Mckee, D., Agus, S., Brawer, S., Shlomowitz, N., Yoseph, O. B., Soreq, H., and Sussman, J. D. (2007). Treatment of human myasthenia gravis with oral antisense suppression of acetylcholinesterase. Neurology 69, 699–700.
Black, P. H. (2002). Stress and the inflammatory response: a review of neurogenic inflammation. Brain Behav. Immun. 16, 622–653.
Brenner, T., Hamra-Amitay, Y., Evron, T., Boneva, N., Seidman, S., and Soreq, H. (2003). The role of readthrough acetylcholinesterase in the pathophysiology of myasthenia gravis. FASEB J. 17, 214–222.
Broide, R. S., Grifman, M., Loewenstein, A., Grisaru, D., Timberg, R., Stone, J., Shani, M., Patrick, J. W., and Soreq, H. (1999). Manipulations of ACHE gene expression suggest non-catalytic involvement of acetylcholinesterase in the functioning of mammalian photoreceptors but not in retinal degeneration. Brain Res. Mol. Brain Res. 71, 137–148.
Broome, C. S., Whetton, A. D., and Miyan, J. A. (2000). Neuropeptide control of bone marrow neutrophil production is mediated by both direct and indirect effects on CFU-GM. Br. J. Haematol. 108, 140–150.
Bytyqi, A. H., Lockridge, O., Duysen, E., Wang, Y., Wolfrum, U., and Layer, P. G. (2004). Impaired formation of the inner retina in an AChE knockout mouse results in degeneration of all photoreceptors. Eur. J. Neurosci. 20, 2953–2962.
Cavazzana-Calvo, M., Fischer, A., Bushman, F. D., Payen, E., Hacein-Bey-Abina, S., and Leboulch, P. (2011). Is normal hematopoiesis maintained solely by long-term multipotent stem cells? Blood 117, 4420–4424.
Chrousos, G. P. (2009). Stress and disorders of the stress system. Nat. Rev. Endocrinol. 5, 374–381.
Criswell, M. H., and Brandon, C. (1993). Acetylcholinesterase and choline acetyltransferase localization patterns do correspond in cat and rat retinas. Vision Res. 33, 1747–1753.
Deutsch, V. R., Pick, M., Perry, C., Grisaru, D., Hemo, Y., Golan-Hadari, D., Grant, A., Eldor, A., and Soreq, H. (2002). The stress-associated acetylcholinesterase variant AChE-R is expressed in human CD34(+) hematopoietic progenitors and its C-terminal peptide ARP promotes their proliferation. Exp. Hematol. 30, 1153–1161.
Dudakov, J. A., Khong, D. M., Boyd, R. L., and Chidgey, A. P. (2010). Feeding the fire: the role of defective bone marrow function in exacerbating thymic involution. Trends Immunol. 31, 191–198.
Erb, C., Troost, J., Kopf, S., Schmitt, U., Loffelholz, K., Soreq, H., and Klein, J. (2001). Compensatory mechanisms enhance hippocampal acetylcholine release in transgenic mice expressing human acetylcholinesterase. J. Neurochem. 77, 638–646.
Evron, T., Moyal-Segal, L. B., Lamm, N., Geffen, A., and Soreq, H. (2005). RNA-targeted suppression of stress-induced allostasis in primate spinal cord neurons. Neurodegener. Dis. 2, 16–27.
Gilboa-Geffen, A., Lacoste, P. P., Soreq, L., Cizeron-Clairac, G., Le Panse, R., Truffault, F., Shaked, I., Soreq, H., and Berrih-Aknin, S. (2007). The thymic theme of acetylcholinesterase splice variants in myasthenia gravis. Blood 109, 4383–4391.
Gilboa-Geffen, A., Wolf, Y., Hanin, G., Melamed-Book, N., Pick, M., Bennett, E. R., Greenberg, D. S., Lester, S., Rischmueller, M., and Soreq, H. (2011). Activation of the alternative NFkappaB pathway improves disease symptoms in a model of Sjogren’s syndrome. PLoS One 6:e28727. doi: 10.1371/journal.pone.0028727
Gol’dberg, E. D., Dygai, A. M., Skurikhin, E. G., Provalova, N. V., and Suslov, N. I. (2000). Adrenergic and cholinergic mechanisms of hemopoiesis regulation during experimental neuroses. Bull. Exp. Biol. Med. 129, 323–326.
Grisaru, D., Deutsch, V., Shapira, M., Pick, M., Sternfeld, M., Melamed-Book, N., Kaufer, D., Galyam, N., Gait, M. J., Owen, D., Lessing, J. B., Eldor, A., and Soreq, H. (2001). ARP, a peptide derived from the stress-associated acetylcholinesterase variant, has hematopoietic growth promoting activities. Mol. Med. 7, 93–105.
Grisaru, D., Pick, M., Perry, C., Sklan, E. H., Almog, R., Goldberg, I., Naparstek, E., Lessing, J. B., Soreq, H., and Deutsch, V. (2006). Hydrolytic and nonenzymatic functions of acetylcholinesterase comodulate hemopoietic stress responses. J. Immunol. 176, 27–35.
Guimaraes-Sternberg, C., Meerson, A., Shaked, I., and Soreq, H. (2006). MicroRNA modulation of megakaryoblast fate involves cholinergic signaling. Leuk. Res. 30, 583–595.
Hansen, K. F., Sakamoto, K., Wayman, G. A., Impey, S., and Obrietan, K. (2010). Transgenic miR-132 alters neuronal spine density and impairs novel object recognition memory. PLoS One 5:e15497. doi: 10.1371/journal.pone.0015497
Hoogduijn, M. J., Cheng, A., and Genever, P. G. (2009). Functional nicotinic and muscarinic receptors on mesenchymal stem cells. Stem Cells Dev. 18, 103–112.
Huston, J. M., Ochani, M., Rosas-Ballina, M., Liao, H., Ochani, K., Pavlov, V. A., Gallowitsch-Puerta, M., Ashok, M., Czura, C. J., Foxwell, B., Tracey, K. J., and Ulloa, L. (2006). Splenectomy inactivates the cholinergic antiinflammatory pathway during lethal endotoxemia and polymicrobial sepsis. J. Exp. Med. 203, 1623–1628.
Hutchins, J. B. (1987). Acetylcholine as a neurotransmitter in the vertebrate retina. Exp. Eye Res. 45, 1–38.
Impey, S., Davare, M., Lasiek, A., Fortin, D., Ando, H., Varlamova, O., Obrietan, K., Soderling, T. R., Goodman, R. H., and Wayman, G. A. (2010). An activity-induced microRNA controls dendritic spine formation by regulating Rac1-PAK signaling. Mol. Cell. Neurosci. 43, 146–156.
Kaufer, D., Friedman, A., Seidman, S., and Soreq, H. (1998). Acute stress facilitates long-lasting changes in cholinergic gene expression. Nature 393, 373–377.
Kawashima, K., and Fujii, T. (2000). Extraneuronal cholinergic system in lymphocytes. Pharmacol. Ther. 86, 29–48.
Kehat, R., Zemel, E., Cuenca, N., Evron, T., Toiber, D., Loewenstein, A., Soreq, H., and Perlman, I. (2007). A novel isoform of acetylcholinesterase exacerbates photoreceptors death after photic stress. Invest. Ophthalmol. Vis. Sci. 48, 1290–1297.
Kiecolt-Glaser, J. K., Preacher, K. J., Maccallum, R. C., Atkinson, C., Malarkey, W. B., and Glaser, R. (2003). Chronic stress and age-related increases in the proinflammatory cytokine IL-6. Proc. Natl. Acad. Sci. U.S.A. 100, 9090–9095.
LaVail, M. M., Unoki, K., Yasumura, D., Matthes, M. T., Yancopoulos, G. D., and Steinberg, R. H. (1992). Multiple growth factors, cytokines, and neurotrophins rescue photoreceptors from the damaging effects of constant light. Proc. Natl. Acad. Sci. U.S.A. 89, 11249–11253.
Lee, S. W., and Sullenger, B. A. (1997). Isolation of a nuclease-resistant decoy RNA that can protect human acetylcholine receptors from myasthenic antibodies. Nat. Biotechnol. 15, 41–45.
Lev-Lehman, E., Deutsch, V., Eldor, A., and Soreq, H. (1997). Immature human megakaryocytes produce nuclear-associated acetylcholinesterase. Blood 89, 3644–3653.
Li, Y., Liu, L., Kang, J., Sheng, J. G., Barger, S. W., Mrak, R. E., and Griffin, W. S. (2000). Neuronal-glial interactions mediated by interleukin-1 enhance neuronal acetylcholinesterase activity and mRNA expression. J. Neurosci. 20, 149–155.
McEwen, B. S., and Gianaros, P. J. (2011). Stress- and allostasis-induced brain plasticity. Annu. Rev. Med. 62, 431–445.
Meshorer, E., Bryk, B., Toiber, D., Cohen, J., Podoly, E., Dori, A., and Soreq, H. (2005). SC35 promotes sustainable stress-induced alternative splicing of neuronal acetylcholinesterase mRNA. Mol. Psychiatry 10, 985–997.
Meshorer, E., Erb, C., Gazit, R., Pavlovsky, L., Kaufer, D., Friedman, A., Glick, D., Ben-Arie, N., and Soreq, H. (2002). Alternative splicing and neuritic mRNA translocation under long-term neuronal hypersensitivity. Science 295, 508–512.
Metz, C. N., and Tracey, K. J. (2005). It takes nerve to dampen inflammation. Nat. Immunol. 6, 756–757.
Nance, D. M., and Sanders, V. M. (2007). Autonomic innervation and regulation of the immune system (1987–2007). Brain Behav. Immun. 21, 736–745.
Nijholt, I., Farchi, N., Kye, M., Sklan, E. H., Shoham, S., Verbeure, B., Owen, D., Hochner, B., Spiess, J., Soreq, H., and Blank, T. (2004). Stress-induced alternative splicing of acetylcholinesterase results in enhanced fear memory and long-term potentiation. Mol. Psychiatry 9, 174–183.
Ofek, K., Krabbe, K. S., Evron, T., Debecco, M., Nielsen, A. R., Brunnsgaad, H., Yirmiya, R., Soreq, H., and Pedersen, B. K. (2007). Cholinergic status modulations in human volunteers under acute inflammation. J. Mol. Med. 85, 1239–1251.
Organisciak, D. T., Darrow, R. M., Jiang, Y. I., Marak, G. E., and Blanks, J. C. (1992). Protection by dimethylthiourea against retinal light damage in rats. Invest. Ophthalmol. Vis. Sci. 33, 1599–1609.
Oshevski, S., LeBousse-Kerdiles, M. C., Clay, D., Levashova, Z., Debili, N., Vitral, N., Jasmin, C., and Castagna, M. (1999). Differential expression of protein kinase C isoform transcripts in human hematopoietic progenitors undergoing differentiation. Biochem. Biophys. Res. Commun. 263, 603–609.
Padgett, D. A., and Glaser, R. (2003). How stress influences the immune response. Trends Immunol. 24, 444–448.
Park, S. E., Kim, N. D., and Yoo, Y. H. (2004). Acetylcholinesterase plays a pivotal role in apoptosome formation. Cancer Res. 64, 2652–2655.
Perry, C., Pick, M., Podoly, E., Gilboa-Geffen, A., Zimmerman, G., Sklan, E. H., Ben-Shaul, Y., Diamant, S., and Soreq, H. (2007). Acetylcholinesterase/C terminal binding protein interactions modify Ikaros functions, causing T lymphopenia. Leukemia 21, 1472–1480.
Pick, M., Perry, C., Lapidot, T., Guimaraes-Sternberg, C., Naparstek, E., Deutsch, V., and Soreq, H. (2006). Stress-induced cholinergic signaling promotes inflammation-associated thrombopoiesis. Blood 107, 3397–3406.
Pollak, Y., Gilboa, A., Ben-Menachem, O., Ben-Hur, T., Soreq, H., and Yirmiya, R. (2005). Acetylcholinesterase inhibitors reduce brain and blood interleukin-1beta production. Ann. Neurol. 57, 741–745.
Qian, J., Galitovskiy, V., Chernyavsky, A. I., Marchenko, S., and Grando, S. A. (2011). Plasticity of the murine spleen T-cell cholinergic receptors and their role in in vitro differentiation of naive CD4 T cells toward the Th1, Th2 and Th17 lineages. Genes Immun. 12, 222–230.
Que-Gewirth, N. S., and Sullenger, B. A. (2007). Gene therapy progress and prospects: RNA aptamers. Gene Ther. 14, 283–291.
Rafii, S., Shapiro, F., Pettengell, R., Ferris, B., Nachman, R. L., Moore, M. A., and Asch, A. S. (1995). Human bone marrow microvascular endothelial cells support long-term proliferation and differentiation of myeloid and megakaryocytic progenitors. Blood 86, 3353–3363.
Rauch, S. L., Shin, L. M., and Phelps, E. A. (2006). Neurocircuitry models of posttraumatic stress disorder and extinction: human neuroimaging research–past, present, and future. Biol. Psychiatry 60, 376–382.
Rojnuckarin, P., and Kaushansky, K. (2001). Actin reorganization and proplatelet formation in murine megakaryocytes: the role of protein kinase calpha. Blood 97, 154–161.
Rosas-Ballina, M., Ochani, M., Olofsson, P., Valdés-Ferrer, S., Chavan, S., and Tracey, K. J. (2010). Neural control of acetylcholine release by T cells attenuates TNF production in endotoxemia. J. Immunol. 184, 138.124.
Rosas-Ballina, M., Olofsson, P. S., Ochani, M., Valdes-Ferrer, S. I., Levine, Y. A., Reardon, C., Tusche, M. W., Pavlov, V. A., Andersson, U., Chavan, S., Mak, T. W., and Tracey, K. J. (2011). Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science 334, 98–101.
Saeed, R. W., Varma, S., Peng-Nemeroff, T., Sherry, B., Balakhaneh, D., Huston, J., Tracey, K. J., Al-Abed, Y., and Metz, C. N. (2005). Cholinergic stimulation blocks endothelial cell activation and leukocyte recruitment during inflammation. J. Exp. Med. 201, 1113–1123.
Shaked, I., Meerson, A., Wolf, Y., Avni, R., Greenberg, D., Gilboa-Geffen, A., and Soreq, H. (2009). MicroRNA-132 potentiates cholinergic anti-inflammatory signaling by targeting acetylcholinesterase. Immunity 31, 965–973.
Shaltiel, G., Hanan, M., Wolf, Y., Barbash, S., Kovalev, E., Shoham, S., and Soreq, H. (in press). Hippocampal microRNA-132 mediates stress-inducible cognitive deficits through its acetylcholinesterase target. Brain Struct. Funct.
Shohami, E., Kaufer, D., Chen, Y., Seidman, S., Cohen, O., Ginzberg, D., Melamed-Book, N., Yirmiya, R., and Soreq, H. (2000). Antisense prevention of neuronal damages following head injury in mice. J. Mol. Med. 78, 228–236.
Sklan, E. H., Lowenthal, A., Korner, M., Ritov, Y., Landers, D. M., Rankinen, T., Bouchard, C., Leon, A. S., Rice, T., Rao, D. C., Wilmore, J. H., Skinner, J. S., and Soreq, H. (2004). Acetylcholinesterase/paraoxonase genotype and expression predict anxiety scores in Health, Risk Factors, Exercise Training, and Genetics study. Proc. Natl. Acad. Sci. U.S.A. 101, 5512–5517.
Soreq, H., and Seidman, S. (2001). Acetylcholinesterase–new roles for an old actor. Nat. Rev. Neurosci. 2, 294–302.
Soreq, H., and Wolf, Y. (2011). NeurimmiRs: microRNAs in the neuroimmune interface. Trends Mol. Med. 17, 548–555.
Stone, J., Maslim, J., Valter-Kocsi, K., Mervin, K., Bowers, F., Chu, Y., Barnett, N., Provis, J., Lewis, G., Fisher, S. K., Bisti, S., Gargini, C., Cervetto, L., Merin, S., and Peer, J. (1999). Mechanisms of photoreceptor death and survival in mammalian retina. Prog. Retin. Eye Res. 18, 689–735.
Sussman, J. D., Argov, Z., Mckee, D., Hazum, E., Brawer, S., and Soreq, H. (2008). Antisense treatment for myasthenia gravis: experience with monarsen. Ann. N.Y. Acad. Sci. 1132, 283–290.
Toiber, D., Berson, A., Greenberg, D., Melamed-Book, N., Diamant, S., and Soreq, H. (2008). N-acetylcholinesterase-induced apoptosis in Alzheimer’s disease. PLoS One 3:e3108. doi: 10.1371/journal.pone.0003108
Vairano, M., Dello Russo, C., Pozzoli, G., Battaglia, A., Scambia, G., Tringali, G., Aloe-Spiriti, M. A., Preziosi, P., and Navarra, P. (2002). Erythropoietin exerts anti-apoptotic effects on rat microglial cells in vitro. Eur. J. Neurosci. 16, 584–592.
Wang, H., Yu, M., Ochani, M., Amella, C. A., Tanovic, M., Susarla, S., Li, J. H., Yang, H., Ulloa, L., Al-Abed, Y., Czura, C. J., and Tracey, K. J. (2003). Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 421, 384–388.
Zhang, X. J., Yang, L., Zhao, Q., Caen, J. P., He, H. Y., Jin, Q. H., Guo, L. H., Alemany, M., Zhang, L. Y., and Shi, Y. F. (2002). Induction of acetylcholinesterase expression during apoptosis in various cell types. Cell Death Differ. 9, 790–800.
Zimmerman, G., Shaltiel, G., Barabash, S., Cohen, J., Gasho, C., Shenhar, S., Shalev, H., Berliner, S., Shelef, I., Shoham, S., Friedman, A., Cohen, H., and Soreq, H. (2012). Post-traumatic anxiety associates with failure of the innate immune receptor TLR9 to evade the pro-inflammatory NFkB pathway. Transl. Psychiatry 2, e78.
Keywords: acetylcholinesterase, hematopoiesis, stress
Citation: Gilboa-Geffen A, Hartmann G and Soreq H (2012) Stressing hematopoiesis and immunity: an acetylcholinesterase window into nervous and immune system interactions. Front. Mol. Neurosci. 5:30. doi: 10.3389/fnmol.2012.00030
Received: 04 December 2011; Paper pending published: 16 January 2012;
Accepted: 22 February 2012; Published online: 16 March 2012.
Edited by:
Karl Tsim, The Hong Kong University of Science and Technology, ChinaReviewed by:
Miou Zhou, University of California, Los Angeles, USABernard Jasmin, University of Ottawa, Canada
Copyright: © 2012 Gilboa-Geffen, Hartmann and Soreq. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Hermona Soreq, The Edmond and Lily Safra Center of Brain Sciences, The Edmond J. Safra Campus, The Hebrew University of Jerusalem, Givat Ram, Jerusalem 91904, Israel. e-mail:c29yZXFAY2MuaHVqaS5hYy5pbA==