

- Faculty of Medicine, Department of Cellular and Molecular Medicine, University of Ottawa, Ottawa, ON, Canada
The most characterized function of acetylcholinesterase (AChE) is to terminate cholinergic signaling at neuron-neuron and neuro-muscular synapses. In addition, AChE is causally or casually implicated in neuronal development, stress-response, cognition, and neurodegenerative diseases. Given the importance of AChE, many studies have focused on identifying the molecular mechanisms that govern its expression. Despite these efforts, post-transcriptional control of AChE mRNA expression is still relatively unclear. Here, we review the trans-acting factors and cis-acting elements that are known to control AChE pre-mRNA splicing, mature mRNA stability and translation. Moreover, since the Hu/ELAV family of RNA-binding proteins (RBPs) have emerged in recent years as “master” post-transcriptional regulators, we discuss the possibility that predominantly neuronal ELAVs (nELAVs) play multiple roles in regulating splicing, stability, localization, and translation of AChE mRNA.
Introduction
Acetylcholinesterase (AChE) is one of the most thoroughly studied enzymes and is better known for its role in hydrolyzing acetylcholine (ACh) at central and peripheral nervous system (CNS and PNS) synapses (reviewed in Legay, 2000; Soreq and Seidman, 2001; Massoulie, 2002; Rotundo, 2003; Taylor, 2010). In addition to terminating cholinergic neurotransmission, detection of AChE prior to cholinergic synapse formation and in non-cholinergic neurons led to identification of functions in several cellular processes including neurite elongation (Koenigsberger et al., 1997), synaptogenesis (Sternfeld et al., 1998), cell adherence (Sharma et al., 2001), and apoptosis (Zhang et al., 2002) (reviewed in Jiang and Zhang, 2008; Paraoanu and Layer, 2008). The functional versatility of AChE is also implicated in neocortical development (Dori et al., 2005), cognition (Beeri et al., 1995), stress-response (Kaufer et al., 1998), neuronal tumors (Karpel et al., 1994) and neurodegeneration such as in Alzheimer's and Parkinson's diseases (reviewed in Meshorer and Soreq, 2006; Greenfield et al., 2008).
AChE's tissue-specific expression pattern and diverse roles are largely attributed to the generation of multiple splice variants from a single gene (Li et al., 1991; Meshorer et al., 2004). Classically, all AChE variants are categorized depending on their different C-terminal tails into AChET (T-tailed), AChER (R-read through) and AChEH (H-hydrophobic). In neurons and skeletal muscle AChET interacts with either a collagenic tail termed ColQ, or with a proline-rich membrane anchor referred to as PRiMA. On the other hand, AChEH is primarily localized to erythrocytes and platelets, anchored to the plasma membrane via glycophospholipids. The typically low abundant variant found in several cell types, AChER, is a soluble monomer up-regulated during stress, AChE inhibition and organophosphate poisoning (reviewed in Zimmerman and Soreq, 2006). The importance of AChE for proper neuronal development and synaptic physiology, as well as its putative roles in brain pathologies, has gathered much attention over the last several decades toward delineating the molecular mechanisms that control its expression.
Neuronal transcription of AChE has first been reported during differentiation (Greene and Rukenstein, 1981, see also for example Deschenes-Furry et al., 2003), in response to stressors (Shapira et al., 2000; Meshorer et al., 2004) and in co-cultures with myotubes (Jiang et al., 2003). The murine AChE gene contains a complex 5′ regulatory region (5′RR) with alternate promoters and two enhancers (Mutero et al., 1995; Atanasova et al., 1999; Chan et al., 1999; Meshorer et al., 2004). Several transcription factors are now recognized to regulate AChE expression in neurons, including activating transcription factor-1 (ATF1) (Wan et al., 2000), glucocorticoid receptors (GR) (Meshorer et al., 2004), hepatocyte nuclear factor 3β (HNF3β) (Shapira et al., 2000) and ETS-like transcription factor (Elk-1) (Siow et al., 2010). At least 5 and 4 exon 1 (E1) variants have been identified in the mouse (mE1a–e) and human (hE1a–d) AChE 5′RR, respectively, with distinct promoters located upstream of each E1 (Figure 1A). Each leader exon generates a unique AChE 5′ untranslated region (UTR), which is presumably dependent on alternative promoter usage rather than alternative splicing. Although the purpose of these various 5′UTRs remains elusive, mE1e and hE1d encode a conserved (79%) amino acid chain that produces N-terminal extended AChE variants (N-AChET, N-AChER and in principle N-AChEH) (Meshorer et al., 2004). Interestingly, certain N-AChE variants have been shown to bind the cellular membrane, promote apoptosis in stress-induced cells and to associate with pathological markers in Alzheimer's brains (Kehat et al., 2007; Toiber et al., 2008, 2009).
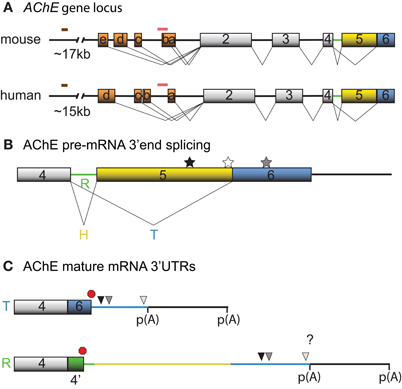
Figure 1. Schematic diagrams depicting the mouse and human AChE genes, alternative splicing options and cis-element locations. (A) Mouse and human AChE genes, including the leader exons identified in brain tissues. Noteworthy is the 5′ regulatory region that harbors alternate exons 1a–e in mouse and 1a–d in human (orange boxes), distal enhancer glucocorticoid response element (brown line) and proximal muscle-specific enhancer (pink line). Gray boxes and black lines represent constitutive exons and introns, respectively. (B) Alternative splicing at the 3′ end of AChE pre-mRNA produces tissue-specific R (green), H (yellow) and T (blue) variants. Approximate locations of the U-rich regulatory sequence, SC35 and ASF/SF2 splicing factor binding sites are denoted by black, white, and gray stars, respectively. (C) The 3′ untranslated regions (3′ UTRs) of mature T and R AChE variants and the alternative polyadenylation-dependent extended regions. The AChEH variant is not shown since it is not significantly expressed in neurons. Symbols represent the PBE (black triangle), ARE (gray triangle), miR-132 binding (white triangle), translation stop (red octagon), and polyadenylation signal (p(A)) sites. 4′ signifies a pseudo-intron that contains the AChER translation termination site. Whether E6 and the downstream region are included in AChER transcripts is unknown (?).
In addition to transcription and distinct promote usage, an increasing amount of studies are demonstrating that post-transcriptional mechanisms are equally important for AChE expression. However, our understanding of the trans-acting factors and cis-acting elements involved in these processes is still limited. Here, we review the work describing the currently identified RNA-binding proteins (RBPs), microRNAs (miRs) and RNA motifs that regulate AChE pre-mRNA splicing, mRNA stability, and translation. Given the established neuronal pleiotropic effects of one family of RBP, namely the principally neuronal ELAVs (nELAVs), we also speculate on their involvement in AChE mRNA metabolism at multiple steps.
Regulation of AChE Pre-mRNA Splicing
Recent high throughput sequencing revealed that more than 90% of human genes undergo alternative splicing (for review see Kalsotra and Cooper, 2011). In accordance with this finding, AChE pre-mRNA is subjected to alternative splicing at the 3′end, which produces AChET or AChER in neurons. The terminal sequence of the AChE gene contains a 5′ splice donor site downstream of exon 4 (E4) and two 3′ splice acceptor sites, one upstream of E5 (proximal) and the other upstream of E6 (distal). When splicing to either of the two downstream acceptor sites is inhibited, the intron (4′) and exons downstream of E4, are included into the mature transcript producing AChER. Conversely, splicing to the distal acceptor site incorporates E6 into the mature mRNA and generates AChET (reviewed in Meshorer and Soreq, 2006) (Figure 1B).
Although little is known regarding the splicing factors involved, two serine/arginine-rich (SR) auxiliary splicing factors, SC35 and ASF/SF2, have been found to inversely affect AChER versus AChET expression (Meshorer et al., 2005). In particular, in vitro experiments with an AChE minigene in COS-1 and HEK293 cells unveiled that SC35 promotes alternative splicing of AChER transcripts whereas ASF/SF2 seemed to favor expression of AChET. Furthermore, immunohistochemistry and fluorescent in situ hybridization (FISH) experiments detected a correlation between SC35 protein and AChER mRNA expression during embryonic brain development and adult stress-response. However, some discordance between SC35 protein and AChER mRNA levels in developing and post-stress cortex were apparent (Meshorer et al., 2005), indicating that other trans-acting factors may also govern splicing of precursor AChE mRNAs. In agreement with this view, Guerra et al used an AChE minigene and sequence mutagenesis to uncover various putative motifs, predominantly located in the E5 non-coding region, that influence production of the R/H/T splice variants in skeletal muscle cells (Guerra et al., 2008).
Stability-Determinants of AChE mRNA
Post-transcriptional regulation of AChE resulting in increased mRNA stability was initially documented in Neuro2a (N2a) cells under the control of the thyroid hormone T3 (Puymirat et al., 1995) and in differentiating P19 embryonal carcinoma cells (Coleman and Taylor, 1996). Moreover, increased stability of AChE mRNA through an unknown mechanism has been observed during calcium-dependent apoptosis (Zhu et al., 2007). Screening the AChET transcript sequence for cis-acting elements that might regulate mRNA stability revealed the presence of an AU-rich element (ARE) in the 3′UTR and a CU-rich sequence in E2 (Cuadrado et al., 2003; Deschenes-Furry et al., 2003). Based on the current understanding of AChE alternative splicing, it would appear that the R transcript also contains these motifs. However, whether the ARE-surrounding sequence that is encoded by E6 is also present in AChER remains controversial (Guerra et al., 2008; Camp et al., 2010; Hanin and Soreq, 2011; Marrero et al., 2011) (Figure 1C). In either case, the existence of an ARE indicates RBP-dependent regulation of AChE mRNA stability. AChE transcripts are also susceptible to alternative polyadenylation that preferentially produces a shorter 3′UTR in mature neurons with an expected altered mRNA stability (Figure 1C) (Li et al., 1991).
One of the most extensively studied families of mRNA stabilizing RBPs, termed the Hu/ELAV family, consists of the ubiquitously expressed HuR (ELAVl1) and the predominantly neuronal (known as nELAVs) HuB (ELAVl2), HuC (ELAVl3) and HuD (ELAVl4) (reviewed in Bolognani and Perrone-Bizzozero, 2008; Hinman and Lou, 2008; Pascale et al., 2008). Brain developmental and spatial expression patterns of Hu/ELAV members are partially distinct and their amino acid sequences are moderately conserved in certain regulatory regions, suggesting that these RBPs are not completely functionally redundant (Okano and Darnell, 1997; Hambardzumyan et al., 2009). Hu/ELAVs contain two RNA-Recognition Motifs (RRMs) which enable binding to AREs in the 3′UTR and a third RRM that interacts with the poly(A)-tail and other proteins (Abe et al., 1996; Ma et al., 1997; Kasashima et al., 2002; Fialcowitz-White et al., 2007). Recent studies indicate that both ARE and non-ARE sequences located in 5′UTRs, exons and introns are also targeted by Hu/ELAVs (Bolognani et al., 2010; Lebedeva et al., 2011; Mukherjee et al., 2011; Uren et al., 2011). Through these binding mechanisms, Hu/ELAVs can regulate almost all aspects of mRNA metabolism including splicing, polyadenylation, nuclear export, stabilization, localization, and translation (reviewed in Hinman and Lou, 2008; Pascale et al., 2008—and see below). Since several nELAV mRNA targets encode proteins that have essential roles ranging from neurogenesis to synaptogenesis, nELAVs act as “master” post-transcriptional regulators of neuronal development, maintenance, and function (Bolognani et al., 2010 and for review see Deschenes-Furry et al., 2006; Pascale and Govoni, 2012).
The lack of positive correlation between transcription rate and AChE mRNA levels (see for example Boudreau-Lariviere et al., 2000; Angus et al., 2001) together with the presence of an ARE motif in the AChE transcript prompted us several years ago, to initiate a series of studies to determine whether nELAVs control AChE mRNA stability. Using several complementary approaches, we were able to show that HuD binds to the ARE-containing region in the 3′UTR and enhances AChE mRNA abundance following its ectopic expression in PC12 cells (Deschenes-Furry et al., 2003). Around the same time, the group of Cuadrado et al used an in vitro RNAse protection assay to establish that HuD binds to the AChE ARE and CU-rich sequence. In a congruent experiment to ours, they were able to demonstrate that overexpression of HuD is sufficient to increase AChE mRNA expression in N2a cells (Cuadrado et al., 2003). Moreover, similar findings were obtained in a skeletal muscle differentiation model when we demonstrated that HuR also binds to the AChE 3′UTR thereby stabilizing the transcript (Deschenes-Furry et al., 2005).
In subsequent work, we ascertained that the regulation of AChE mRNA stability by HuD also occurs in vivo. To this end, we conducted experiments using two distinct models. First, we made use of transgenic mice overexpressing HuD in regions of the brain. In these animals, we detected through in situ hybridization and RT-PCR experiments, an increase in AChE levels in brain regions showing high levels of transgene expression. Furthermore, RNA immunoprecipitation experiments confirmed in vivo an interaction between the HuD transgene product and endogenous AChE transcripts. In a second approach, axotomy of neurons of the spinal cervical ganglion (SCG) produced a concomitant decrease in AChE mRNA and HuD protein levels, as well as a clear reduction in their interaction. Importantly, the decrease in AChE mRNA abundance could be rescued by localized overexpression of HuD delivered via a viral vector (Deschenes-Furry et al., 2007). Together, these in vitro and in vivo studies highlight the critical importance of HuD in controlling AChE mRNA stability (Figure 2) during neuronal development and following injury. Whether these functions extend to the other nELAVs is not presently known.
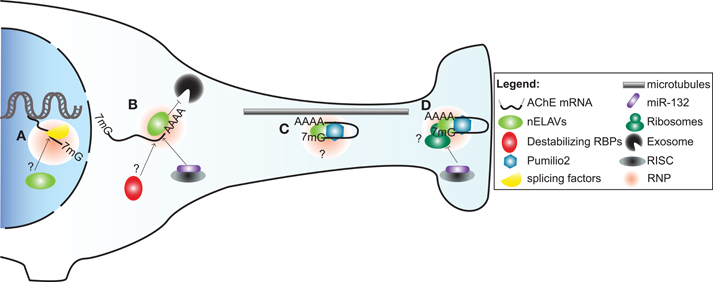
Figure 2. A model illustrating post-transcriptional regulation of AChE expression and localization in neurons by trans-acting factors. (A) Alternative splicing of AChE pre-mRNA is controlled by SC35 and ASF/SF2 general splicing factors. As part of a ribonucleoprotein particle (RNP), nELAVs might also bind to cis-elements within the AChE pre-mRNA to regulate alternative splicing. (B) AChE mRNA is stabilized by HuD and possibly other nELAVs. Stabilization of AChE mRNA could depend on nELAVs outcompeting destabilizing RBPs and/or RISC loaded miR-132, thereby preventing exosome-mediated mRNA degradation. (C) RNPs, conceivably containing nELAVs and Pumilio 2 (Pum2), transport translationally repressed AChE transcripts along microtubules into neurites. (D) At the synaptic terminal, AChE translation might be promoted by nELAVs or inhibited by Pum2 or RISC loaded miR-132.
Localization and Translation of AChE Transcripts
Transport of mRNAs into neurites necessitates a heterogeneous and dynamic assembly of proteins, known as ribonucleoprotein (RNPs) particles, onto 3′UTR cis-elements (Figure 2). RNP components stabilize the transcript, inhibit translation during transport and promote/repress protein synthesis at the synapse (reviewed in Wang et al., 2010a; Liu-Yesucevitz et al., 2011). In a potentially analogous mechanism, translocation of AChER mRNAs into neurites correlates with increased AChE activity following physiological stress, AChE inhibition or corticosterone treatment (Meshorer et al., 2002). Collectively with the discovery of augmented AChER transcript levels in cortical neurites subsequent to AChE inhibition (Kaufer et al., 1998), these findings support the notion that post-transcriptional events regulate AChER mRNA translocation and possibly local translation at the neurite terminal (Meshorer and Soreq, 2006).
In agreement with this view, a study by Marrero et al has recently identified Pumilio2 (Pum2) as a regulator of AChE translation (Marrero et al., 2011). Pum2 is a member of the PUF family of RBPs that bind to distinct Pumilio binding elements (PBEs) in 3′UTRs of mRNAs and inhibit their translation (reviewed in Quenault et al., 2011). Using protein-RNA binding assays, the authors demonstrated that Pum2 specifically binds to a PBE element in the 3′UTR and consequently represses AChE translation (Marrero et al., 2011). Although these studies were conducted in skeletal muscle, Pum2 is also found in the nervous system, where it controls synaptic function and dendrite outgrowth (Vessey et al., 2010). Furthermore, disruption of Pum2 impairs long-term memory in Drosophila while its ablation produces numerous behavioural defects in mice (Dubnau et al., 2003; Siemen et al., 2011). Together, these studies suggest that Pum2 may inhibit AChE translation during transport and at synaptic termini in neurons (Figure 2).
MicroRNA Regulation of AChE Expression
Stability and translation of mRNAs are also under the influence of miRs, which are short (∼23 nt) single-stranded non-coding RNAs. MiRs function by imperfectly binding to target sequences, usually in the 3′UTR, thereby promoting transcript degradation and/or translational silencing when loaded into the miR-induced silencing complex (miRISC) (Berezikov, 2011). Recently, Shaked et al. used a reporter assay along with binding site point mutation in cultured CHO cells to demonstrate that miR-132 targets the AChE 3′UTR. The authors then elaborated on these findings in vivo by showing that miR-132 overexpression and locked nucleic acid (LNA)-mediated reduction decreased and increased, respectively, AChE catalytic activity in bone marrow cells (Shaked et al., 2009).
MiR-132 is also abundant in the brain implying that AChE is subjected to this regulatory event in neurons (Cheng et al., 2007). Indeed, a very recent study revealed that transgenic mice overexpressing AChER transcripts lacking the 3′UTR contain elevated miR-132 levels which correlate with decreased AChET mRNA abundance in the hippocampus (Shaltiel et al., 2012). However, it remains to be tested whether miR-132 induces AChE mRNA translational inhibition, degradation or both (Figure 2). Additional bioinformatics analysis of 3′UTR sequences further identified at least 47 and 81 putative miR sites in AChET and AChER, respectively (Hanin and Soreq, 2011); though the functionality of these sites requires validation. Amongst these potential sites, at least 13 miRs, for example miR-124, -125b, -194 and -214, are of greater interest because they have been implicated in neuronal development, regeneration, and/or brain diseases (Saba and Schratt, 2010; Hanin and Soreq, 2011).
Implication of Hu/ELAVs in Regulating AChE mRNA Metabolism at Multiple Steps
It is now well established that members of the Hu/ELAV family of RBP can exert multiple effects on RNA metabolism by assuming several distinct, yet complementary functions. Thus, the multifunctional nature of Hu/ELAVs in controlling the fate of mRNAs suggests that these RBPs regulate more than stability of AChE transcripts. In support of this idea, a recent compilation of high throughput data implicates HuR in coupling pre-mRNA alternative splicing to mRNA stability. More specifically, nascent HuR targets that contain both intronic and 3′UTR binding sites were found to be more stable than those that do not (Mukherjee et al., 2011).
In view of this, a U-rich sequence in the AChE E5 non-coding region was demonstrated to control splicing to the distal 3′ splice site to favor AChET expression in skeletal muscle (Guerra et al., 2008). Interestingly, the Hu/ELAV family has been shown to control pre-mRNA splicing by binding to AU- and U-rich sequences (reviewed in Hinman and Lou, 2008). For example, one study demonstrated that nELAVs regulate splicing of the calcitonin/calcitonin gene-related peptide (CGRP) pre-mRNA in a neuron-specific manner by outcompeting TIA/TIAR splicing factors for U-rich intronic binding sites (Zhu et al., 2006). Various Hu/ELAV members were shown to similarly bind and control splicing of Fas (Izquierdo, 2010), Neurofibromatosis type 1 (NF1) (Zhu et al., 2008), Ikaros (Bellavia et al., 2007) and even HuD pre-mRNAs (Wang et al., 2010b). In most of these cases Hu/ELAVs were found to function as splicing repressors with the only exception being a splicing enhancement role on HuD pre-mRNA (Wang et al., 2010b). Based on their established nuclear functions, nELAVs could thus regulate splicing of AChE 3′ alternative exons by competing for the U-rich site within the E5 non-coding region (Figures 1B and 2).
In addition to governing pre-mRNA splicing, compelling evidence supports a role for nELAVs in regulating both localization and translation of ARE-bearing transcripts in neurites (reviewed in Pascale et al., 2008). For instance, two ARE-containing HuD target mRNAs that are both involved in axon outgrowth namely, GAP-43 and Tau, colocalize with HuD protein and polysomes in growth cones during neuronal differentiation (Smith et al., 2004; Atlas et al., 2007). Co-immunoprecipitation and in vitro binding assays revealed that HuD interacts with a motor protein, KIF3A, and directly binds to a microtubule component, MAP1B (Aronov et al., 2002; Fujiwara et al., 2011). Strengthening the notion of nELAV-mediated translation, HuD was shown to enhance cap-dependent protein synthesis and PC12 cell neurite extension by binding to eIF4A and the poly(A)-tail of transcripts (Fukao et al., 2009). Hu/ELAV members are also capable of repressing translation through a mechanism that is unclear but appears to depend on the target transcript and/or the cellular context (Kullmann et al., 2002; Meng et al., 2005). Based on these findings, it is conceivable that nELAVs regulate both AChE mRNA stability and translation in neurons, in a manner analogous to that previously observed for the mRNA targets GLUT1 and NOVA-1 (Jain et al., 1997; Ratti et al., 2008).
Concluding Remarks
Despite extensive research into the regulation of AChE expression, only a handful of studies have precisely described some of the molecular mechanisms that control AChE expression, particularly at the post-transcriptional level. Currently, our knowledge of trans-acting factors and cis-acting elements that affect AChE pre-mRNA splicing, mature mRNA stability, transport and translation is rather limited. Nevertheless, some progress has been made over the years and increasing evidence suggests that RBPs (SC35, ASF/SF2, HuD and Pum2) and at least one miR (miR-132) play integral roles in controlling AChE expression in cholinergic and non-cholinergic neurons under various physiological contexts (see above). Based on their implications in neurogenesis and synaptic plasticity, trans-acting factors may also regulate AChE expression during learning and memory (Quattrone et al., 2001; Mellios et al., 2011; Siemen et al., 2011; Tognini et al., 2011; Apostolatos et al., 2012). Under such circumstances, post-transcriptional events might differentially regulate AChET versus AChER at multiple levels, since the expression patterns and functions of these variants are often contrasting (reviewed in Meshorer and Soreq, 2006). Control of mRNA stability, transport and local translation would facilitate temporal and spatial expression of AChE variants at growth cones and mature synapses and thus contribute to the multifaceted biochemical, morphological and physiological modifications necessary for neuronal differentiation, synaptic plasticity and stress-response. Conversely, altered abundance and/or function of trans-acting factors, such as nELAVs and consequently AChE, may be directly or indirectly involved in neurodegenerative pathogenesis, namely Alzheimer's disease (Berson et al., 2008; Amadio et al., 2009). In light of the emerging role of post-transcriptional events governing AChE metabolism, it is now clear that a thorough knowledge of all mechanisms controlling AChE expression and localization is necessary if the ultimate goal is to better understand neuronal development and function including neurogenesis, stress-response, cognition, neurodegeneration, as well as other brain diseases and injuries.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to many friends and colleagues of the cholinesterase community for the scientific camaraderie and spirit of collaborations we have enjoyed over several years. We are particularly indebted to Professors Claire Legay and Nora Perrone-Bizzozero for their invaluable contributions to the original work summarized in this review. We also thank members of the Jasmin laboratory for insightful discussions. Work in the Jasmin lab has been supported over the years by grants from the Canadian Institutes of Health Research, the Muscular Dystrophy Association (USA), the Association Francaise contre les Myopathies, the Canadian Space Agency, Muscular Dystrophy Canada and the Foundation for Gene and Cell Therapy (Jesse's Journey). Finally, the authors dedicate this article to Professor Jean Massoulié whose pioneering work, scientific excellence, mentorship abilities, enthusiasm, and friendship, have inspired us all and continue to do so.
References
Abe, R., Sakashita, E., Yamamoto, K., and Sakamoto, H. (1996). Two different RNA binding activities for the AU-rich element and the poly(A) sequence of the mouse neuronal protein mHuC. Nucleic Acids Res. 24, 4895–4901.
Amadio, M., Pascale, A., Wang, J., Ho, L., Quattrone, A., Gandy, S., Haroutunian, V., Racchi, M., and Pasinetti, G. M. (2009). nELAV proteins alteration in Alzheimer's disease brain: a novel putative target for amyloid-beta reverberating on AbetaPP processing. J. Alzheimers Dis. 16, 409–419.
Angus, L. M., Chan, R. Y., and Jasmin, B. J. (2001). Role of intronic E- and N-box motifs in the transcriptional induction of the acetylcholinesterase gene during myogenic differentiation. J. Biol. Chem. 276, 17603–17609.
Apostolatos, A., Song, S., Acosta, S., Peart, M., Watson, J. E., Bickford, P., Cooper, D. R., and Patel, N. A. (2012). Insulin promotes neuronal survival via the alternatively spliced protein kinase C delta II (PKCdeltaII) isoform. J. Biol. Chem. doi: 10.1074/jbc.M111.313080. [Epub ahead of print].
Aronov, S., Aranda, G., Behar, L., and Ginzburg, I. (2002). Visualization of translated tau protein in the axons of neuronal P19 cells and characterization of tau RNP granules. J. Cell Sci. 115, 3817–3827.
Atanasova, E., Chiappa, S., Wieben, E., and Brimijoin, S. (1999). Novel messenger RNA and alternative promoter for murine acetylcholinesterase. J. Biol. Chem. 274, 21078–21084.
Atlas, R., Behar, L., Sapoznik, S., and Ginzburg, I. (2007). Dynamic association with polysomes during P19 neuronal differentiation and an untranslated-region-dependent translation regulation of the tau mRNA by the tau mRNA-associated proteins IMP1, HuD, and G3BP1. J. Neurosci. Res. 85, 173–183.
Beeri, R., Andres, C., Lev-Lehman, E., Timberg, R., Huberman, T., Shani, M., and Soreq, H. (1995). Transgenic expression of human acetylcholinesterase induces progressive cognitive deterioration in mice. Curr. Biol. 5, 1063–1071.
Bellavia, D., Mecarozzi, M., Campese, A. F., Grazioli, P., Talora, C., Frati, L., Gulino, A., and Screpanti, I. (2007). Notch3 and the Notch3-upregulated RNA-binding protein HuD regulate Ikaros alternative splicing. EMBO J. 26, 1670–1680.
Berezikov, E. (2011). Evolution of microRNA diversity and regulation in animals. Nat. Rev. Genet. 12, 846–860.
Berson, A., Knobloch, M., Hanan, M., Diamant, S., Sharoni, M., Schuppli, D., Geyer, B. C., Ravid, R., Mor, T. S., Nitsch, R. M., and Soreq, H. (2008). Changes in readthrough acetylcholinesterase expression modulate amyloid-beta pathology. Brain 131, 109–119.
Bolognani, F., Contente-Cuomo, T., and Perrone-Bizzozero, N. I. (2010). Novel recognition motifs and biological functions of the RNA-binding protein HuD revealed by genome-wide identification of its targets. Nucleic Acids Res. 38, 117–130.
Bolognani, F., and Perrone-Bizzozero, N. I. (2008). RNA-protein interactions and control of mRNA stability in neurons. J. Neurosci. Res. 86, 481–489.
Boudreau-Lariviere, C., Chan, R. Y., Wu, J., and Jasmin, B. J. (2000). Molecular mechanisms underlying the activity-linked alterations in acetylcholinesterase mRNAs in developing versus adult rat skeletal muscles. J. Neurochem. 74, 2250–2258.
Camp, S., Zhang, L., Krejci, E., Dobbertin, A., Bernard, V., Girard, E., Duysen, E. G., Lockridge, O., De Jaco, A., and Taylor, P. (2010). Contributions of selective knockout studies to understanding cholinesterase disposition and function. Chem. Biol. Interact. 187, 72–77.
Chan, R. Y., Boudreau-Lariviere, C., Angus, L. M., Mankal, F. A., and Jasmin, B. J. (1999). An intronic enhancer containing an N-box motif is required for synapse- and tissue-specific expression of the acetylcholinesterase gene in skeletal muscle fibers. Proc. Natl. Acad. Sci. U.S.A. 96, 4627–4632.
Cheng, H. Y., Papp, J. W., Varlamova, O., Dziema, H., Russell, B., Curfman, J. P., Nakazawa, T., Shimizu, K., Okamura, H., Impey, S., and Obrietan, K. (2007). microRNA modulation of circadian-clock period and entrainment. Neuron 54, 813–829.
Coleman, B. A., and Taylor, P. (1996). Regulation of acetylcholinesterase expression during neuronal differentiation. J. Biol. Chem. 271, 4410–4416.
Cuadrado, A., Navarro-Yubero, C., Furneaux, H., and Munoz, A. (2003). Neuronal HuD gene encoding a mRNA stability regulator is transcriptionally repressed by thyroid hormone. J. Neurochem. 86, 763–773.
Deschenes-Furry, J., Belanger, G., Mwanjewe, J., Lunde, J. A., Parks, R. J., Perrone-Bizzozero, N., and Jasmin, B. J. (2005). The RNA-binding protein HuR binds to acetylcholinesterase transcripts and regulates their expression in differentiating skeletal muscle cells. J. Biol. Chem. 280, 25361–25368.
Deschenes-Furry, J., Belanger, G., Perrone-Bizzozero, N., and Jasmin, B. J. (2003). Post-transcriptional regulation of acetylcholinesterase mRNAs in nerve growth factor-treated PC12 cells by the RNA-binding protein HuD. J. Biol. Chem. 278, 5710–5717.
Deschenes-Furry, J., Mousavi, K., Bolognani, F., Neve, R. L., Parks, R. J., Perrone-Bizzozero, N. I., and Jasmin, B. J. (2007). The RNA-binding protein HuD binds acetylcholinesterase mRNA in neurons and regulates its expression after axotomy. J. Neurosci. 27, 665–675.
Deschenes-Furry, J., Perrone-Bizzozero, N., and Jasmin, B. J. (2006). The RNA-binding protein HuD: a regulator of neuronal differentiation, maintenance and plasticity. Bioessays 28, 822–833.
Dori, A., Cohen, J., Silverman, W. F., Pollack, Y., and Soreq, H. (2005). Functional manipulations of acetylcholinesterase splice variants highlight alternative splicing contributions to murine neocortical development. Cereb. Cortex. 15, 419–430.
Dubnau, J., Chiang, A. S., Grady, L., Barditch, J., Gossweiler, S., McNeil, J., Smith, P., Buldoc, F., Scott, R., Certa, U., Broger, C., and Tully, T. (2003). The staufen/pumilio pathway is involved in Drosophila long-term memory. Curr. Biol. 13, 286–296.
Fialcowitz-White, E. J., Brewer, B. Y., Ballin, J. D., Willis, C. D., Toth, E. A., and Wilson, G. M. (2007). Specific protein domains mediate cooperative assembly of HuR oligomers on AU-rich mRNA-destabilizing sequences. J. Biol. Chem. 282, 20948–20959.
Fujiwara, Y., Kasashima, K., Saito, K., Fukuda, M., Fukao, A., Sasano, Y., Inoue, K., Fujiwara, T., and Sakamoto, H. (2011). Microtubule association of a neuronal RNA-binding protein HuD through its binding to the light chain of MAP1B. Biochimie 93, 817–822.
Fukao, A., Sasano, Y., Imataka, H., Inoue, K., Sakamoto, H., Sonenberg, N., Thoma, C., and Fujiwara, T. (2009). The ELAV protein HuD stimulates cap-dependent translation in a Poly(A)- and eIF4A-dependent manner. Mol. Cell. 36, 1007–1017.
Greene, L. A., and Rukenstein, A. (1981). Regulation of acetylcholinesterase activity by nerve growth factor. Role of transcription and dissociation from effects on proliferation and neurite outgrowth. J. Biol. Chem. 256, 6363–6367.
Greenfield, S. A., Zimmermann, M., and Bond, C. E. (2008). Non-hydrolytic functions of acetylcholinesterase. The significance of C-terminal peptides. FEBS J. 275, 604–611.
Guerra, M., Dobbertin, A., and Legay, C. (2008). Identification of cis-acting elements involved in acetylcholinesterase RNA alternative splicing. Mol. Cell. Neurosci. 38, 1–14.
Hambardzumyan, D., Sergent-Tanguy, S., Thinard, R., Bonnamain, V., Masip, M., Fabre, A., Boudin, H., Neveu, I., and Naveilhan, P. (2009). AUF1 and Hu proteins in the developing rat brain: implication in the proliferation and differentiation of neural progenitors. J. Neurosci. Res. 87, 1296–1309.
Hanin, G., and Soreq, H. (2011). Cholinesterase-targeting microRNAs identified in silico affect specific biological processes. Front. Mol. Neurosci. 4:28. doi: 10.3389/fnmol.2011.00028
Hinman, M. N., and Lou, H. (2008). Diverse molecular functions of Hu proteins. Cell Mol. Life Sci. 65, 3168–3181.
Izquierdo, J. M. (2010). Cell-specific regulation of Fas exon 6 splicing mediated by Hu antigen R. Biochem. Biophys. Res. Commun. 402, 324–328.
Jain, R. G., Andrews, L. G., McGowan, K. M., Pekala, P. H., and Keene, J. D. (1997). Ectopic expression of Hel-N1, an RNA-binding protein, increases glucose transporter (GLUT1) expression in 3T3-L1 adipocytes. Mol. Cell. Biol. 17, 954–962.
Jiang, H., and Zhang, X. J. (2008). Acetylcholinesterase and apoptosis. A novel perspective for an old enzyme. FEBS J. 275, 612–617.
Jiang, J. X., Choi, R. C., Siow, N. L., Lee, H. H., Wan, D. C., and Tsim, K. W. (2003). Muscle induces neuronal expression of acetylcholinesterase in neuron-muscle co-culture: transcriptional regulation mediated by cAMP-dependent signaling. J. Biol. Chem. 278, 45435–45444.
Kalsotra, A., and Cooper, T. A. (2011). Functional consequences of developmentally regulated alternative splicing. Nat. Rev. Genet. 12, 715–729.
Karpel, R., Ben Aziz-Aloya, R., Sternfeld, M., Ehrlich, G., Ginzberg, D., Tarroni, P., Clementi, F., Zakut, H., and Soreq, H. (1994). Expression of three alternative acetylcholinesterase messenger RNAs in human tumor cell lines of different tissue origins. Exp. Cell Res. 210, 268–277.
Kasashima, K., Sakashita, E., Saito, K., and Sakamoto, H. (2002). Complex formation of the neuron-specific ELAV-like Hu RNA-binding proteins. Nucleic Acids Res. 30, 4519–4526.
Kaufer, D., Friedman, A., Seidman, S., and Soreq, H. (1998). Acute stress facilitates long-lasting changes in cholinergic gene expression. Nature 393, 373–377.
Kehat, R., Zemel, E., Cuenca, N., Evron, T., Toiber, D., Loewenstein, A., Soreq, H., and Perlman, I. (2007). A novel isoform of acetylcholinesterase exacerbates photoreceptors death after photic stress. Invest. Ophthalmol. Vis. Sci. 48, 1290–1297.
Koenigsberger, C., Chiappa, S., and Brimijoin, S. (1997). Neurite differentiation is modulated in neuroblastoma cells engineered for altered acetylcholinesterase expression. J. Neurochem. 69, 1389–1397.
Kullmann, M., Gopfert, U., Siewe, B., and Hengst, L. (2002). ELAV/Hu proteins inhibit p27 translation via an IRES element in the p27 5′UTR. Genes Dev. 16, 3087–3099.
Lebedeva, S., Jens, M., Theil, K., Schwanhausser, B., Selbach, M., Landthaler, M., and Rajewsky, N. (2011). Transcriptome-wide analysis of regulatory interactions of the RNA-binding protein HuR. Mol. Cell. 43, 340–352.
Li, Y., Camp, S., Rachinsky, T. L., Getman, D., and Taylor, P. (1991). Gene structure of mammalian acetylcholinesterase. Alternative exons dictate tissue-specific expression. J. Biol. Chem. 266, 23083–23090.
Liu-Yesucevitz, L., Bassell, G. J., Gitler, A. D., Hart, A. C., Klann, E., Richter, J. D., Warren, S. T., and Wolozin, B. (2011). Local RNA translation at the synapse and in disease. J. Neurosci. 31, 16086–16093.
Ma, W. J., Chung, S., and Furneaux, H. (1997). The Elav-like proteins bind to AU-rich elements and to the poly(A) tail of mRNA. Nucleic Acids Res. 25, 3564–3569.
Marrero, E., Rossi, S. G., Darr, A., Tsoulfas, P., and Rotundo, R. L. (2011). Translational regulation of acetylcholinesterase by the RNA-binding protein Pumilio-2 at the neuromuscular synapse. J. Biol. Chem. 286, 36492–36499.
Massoulie, J. (2002). The origin of the molecular diversity and functional anchoring of cholinesterases. Neurosignals 11, 130–143.
Mellios, N., Sugihara, H., Castro, J., Banerjee, A., Le, C., Kumar, A., Crawford, B., Strathmann, J., Tropea, D., Levine, S. S., Edbauer, D., and Sur, M. (2011). miR-132, an experience-dependent microRNA, is essential for visual cortex plasticity. Nat. Neurosci. 14, 1240–1242.
Meng, Z., King, P. H., Nabors, L. B., Jackson, N. L., Chen, C. Y., Emanuel, P. D., and Blume, S. W. (2005). The ELAV RNA-stability factor HuR binds the 5′-untranslated region of the human IGF-IR transcript and differentially represses cap-dependent and IRES-mediated translation. Nucleic Acids Res. 33, 2962–2979.
Meshorer, E., Bryk, B., Toiber, D., Cohen, J., Podoly, E., Dori, A., and Soreq, H. (2005). SC35 promotes sustainable stress-induced alternative splicing of neuronal acetylcholinesterase mRNA. Mol. Psychiatry. 10, 985–997.
Meshorer, E., Erb, C., Gazit, R., Pavlovsky, L., Kaufer, D., Friedman, A., Glick, D., Ben-Arie, N., and Soreq, H. (2002). Alternative splicing and neuritic mRNA translocation under long-term neuronal hypersensitivity. Science 295, 508–512.
Meshorer, E., and Soreq, H. (2006). Virtues and woes of AChE alternative splicing in stress-related neuropathologies. Trends Neurosci. 29, 216–224.
Meshorer, E., Toiber, D., Zurel, D., Sahly, I., Dori, A., Cagnano, E., Schreiber, L., Grisaru, D., Tronche, F., and Soreq, H. (2004). Combinatorial complexity of 5′ alternative acetylcholinesterase transcripts and protein products. J. Biol. Chem. 279, 29740–29751.
Mukherjee, N., Corcoran, D. L., Nusbaum, J. D., Reid, D. W., Georgiev, S., Hafner, M., Ascano, M. Jr., Tuschl, T., Ohler, U., and Keene, J. D. (2011). Integrative regulatory mapping indicates that the RNA-binding protein HuR couples pre-mRNA processing and mRNA stability. Mol. Cell. 43, 327–339.
Mutero, A., Camp, S., and Taylor, P. (1995). Promoter elements of the mouse acetylcholinesterase gene. Transcriptional regulation during muscle differentiation. J. Biol. Chem. 270, 1866–1872.
Okano, H. J., and Darnell, R. B. (1997). A hierarchy of Hu RNA binding proteins in developing and adult neurons. J. Neurosci. 17, 3024–3037.
Paraoanu, L. E., and Layer, P. G. (2008). Acetylcholinesterase in cell adhesion, neurite growth and network formation. FEBS J. 275, 618–624.
Pascale, A., Amadio, M., and Quattrone, A. (2008). Defining a neuron: neuronal ELAV proteins. Cell. Mol. Life Sci. 65, 128–140.
Pascale, A., and Govoni, S. (2012). The complex world of post-transcriptional mechanisms: is their deregulation a common link for diseases? Focus on ELAV-like RNA-binding proteins. Cell. Mol. Life Sci. 69, 501–517.
Puymirat, J., Etongue-Mayer, P., and Dussault, J. H. (1995). Thyroid hormones stabilize acetylcholinesterase mRNA in neuro-2A cells that overexpress the beta 1 thyroid receptor. J. Biol. Chem. 270, 30651–30656.
Quattrone, A., Pascale, A., Nogues, X., Zhao, W., Gusev, P., Pacini, A., and Alkon, D. L. (2001). Posttranscriptional regulation of gene expression in learning by the neuronal ELAV-like mRNA-stabilizing proteins. Proc. Natl. Acad. Sci. U.S.A. 98, 11668–11673.
Quenault, T., Lithgow, T., and Traven, A. (2011). PUF proteins: repression, activation and mRNA localization. Trends Cell Biol. 21, 104–112.
Ratti, A., Fallini, C., Colombrita, C., Pascale, A., Laforenza, U., Quattrone, A., and Silani, V. (2008). Post-transcriptional regulation of neuro-oncological ventral antigen 1 by the neuronal RNA-binding proteins ELAV. J. Biol. Chem. 283, 7531–7541.
Rotundo, R. L. (2003). Expression and localization of acetylcholinesterase at the neuromuscular junction. J. Neurocytol. 32, 743–766.
Saba, R., and Schratt, G. M. (2010). MicroRNAs in neuronal development, function and dysfunction. Brain Res. 1338, 3–13.
Shaked, I., Meerson, A., Wolf, Y., Avni, R., Greenberg, D., Gilboa-Geffen, A., and Soreq, H. (2009). MicroRNA-132 potentiates cholinergic anti-inflammatory signaling by targeting acetylcholinesterase. Immunity 31, 965–973.
Shaltiel, G., Hanan, M., Wolf, Y., Barbash, S., Kovalev, E., Shoham, S., and Soreq, H. (2012). Hippocampal microRNA-132 mediates stress-inducible cognitive deficits through its acetylcholinesterase target. Brain Struct. Funct. doi: 10.1007/s00429-011-0376-z. [Epub ahead of print].
Shapira, M., Tur-Kaspa, I., Bosgraaf, L., Livni, N., Grant, A. D., Grisaru, D., Korner, M., Ebstein, R. P., and Soreq, H. (2000). A transcription-activating polymorphism in the ACHE promoter associated with acute sensitivity to anti-acetylcholinesterases. Hum. Mol. Genet. 9, 1273–1281.
Sharma, K. V., Koenigsberger, C., Brimijoin, S., and Bigbee, J. W. (2001). Direct evidence for an adhesive function in the noncholinergic role of acetylcholinesterase in neurite outgrowth. J. Neurosci. Res. 63, 165–175.
Siemen, H., Colas, D., Heller, H. C., Brustle, O., and Pera, R. A. (2011). Pumilio-2 function in the mouse nervous system. PLoS One 6:e25932. doi: 10.1371/journal.pone.0025932
Siow, N. L., Choi, R. C., Xie, H. Q., Kong, L. W., Chu, G. K., Chan, G. K., Simon, J., Barnard, E. A., and Tsim, K. W. (2010). ATP induces synaptic gene expressions in cortical neurons: transduction and transcription control via P2Y1 receptors. Mol. Pharmacol. 78, 1059–1071.
Smith, C. L., Afroz, R., Bassell, G. J., Furneaux, H. M., Perrone-Bizzozero, N. I., and Burry, R. W. (2004). GAP-43 mRNA in growth cones is associated with HuD and ribosomes. J. Neurobiol. 61, 222–235.
Soreq, H., and Seidman, S. (2001). Acetylcholinesterase–new roles for an old actor. Nat. Rev. Neurosci. 2, 294–302.
Sternfeld, M., Ming, G., Song, H., Sela, K., Timberg, R., Poo, M., and Soreq, H. (1998). Acetylcholinesterase enhances neurite growth and synapse development through alternative contributions of its hydrolytic capacity, core protein, and variable C termini. J. Neurosci. 18, 1240–1249.
Taylor, P. (2010). From Split to Sibenik: the tortuous pathway in the cholinesterase field. Chem. Biol. Interact. 187, 3–9.
Tognini, P., Putignano, E., Coatti, A., and Pizzorusso, T. (2011). Experience-dependent expression of miR-132 regulates ocular dominance plasticity. Nat. Neurosci. 14, 1237–1239.
Toiber, D., Berson, A., Greenberg, D., Melamed-Book, N., Diamant, S., and Soreq, H. (2008). N-acetylcholinesterase-induced apoptosis in Alzheimer's disease. PLoS One 3:e3108. doi: 10.1371/journal.pone.0003108
Toiber, D., Greenberg, D. S., and Soreq, H. (2009). Pro-apoptotic protein-protein interactions of the extended N-AChE terminus. J. Neural Transm. 116, 1435–1442.
Uren, P. J., Burns, S. C., Ruan, J., Singh, K. K., Smith, A. D., and Penalva, L. O. (2011). Genomic analyses of the RNA-binding protein Hu antigen R (HuR) identify a complex network of target genes and novel characteristics of its binding sites. J. Biol. Chem. 286, 37063–37066.
Vessey, J. P., Schoderboeck, L., Gingl, E., Luzi, E., Riefler, J., Di Leva, F., Karra, D., Thomas, S., Kiebler, M. A., and Macchi, P. (2010). Mammalian Pumilio 2 regulates dendrite morphogenesis and synaptic function. Proc. Natl. Acad. Sci. U.S.A. 107, 3222–3227.
Wan, D. C., Choi, R. C., Siow, N. L., and Tsim, K. W. (2000). The promoter of human acetylcholinesterase is activated by a cyclic adenosine 3′, 5′-monophosphate-dependent pathway in cultured NG108–15 neuroblastoma cells. Neurosci. Lett. 288, 81–85.
Wang, D. O., Martin, K. C., and Zukin, R. S. (2010a). Spatially restricting gene expression by local translation at synapses. Trends Neurosci. 33, 173–182.
Wang, H., Molfenter, J., Zhu, H., and Lou, H. (2010b). Promotion of exon 6 inclusion in HuD pre-mRNA by Hu protein family members. Nucleic Acids Res. 38, 3760–3770.
Zhang, X. J., Yang, L., Zhao, Q., Caen, J. P., He, H. Y., Jin, Q. H., Guo, L. H., Alemany, M., Zhang, L. Y., and Shi, Y. F. (2002). Induction of acetylcholinesterase expression during apoptosis in various cell types. Cell Death Differ. 9, 790–800.
Zhu, H., Gao, W., Shi, Y. F., and Zhang, X. J. (2007). The CCAAT-binding factor CBF/NF-Y regulates the human acetylcholinesterase promoter activity during calcium ionophore A23187-induced cell apoptosis. Biochim. Biophys. Acta. 1770, 1475–1482.
Zhu, H., Hasman, R. A., Barron, V. A., Luo, G., and Lou, H. (2006). A nuclear function of Hu proteins as neuron-specific alternative RNA processing regulators. Mol. Biol. Cell. 17, 5105–5114.
Zhu, H., Hinman, M. N., Hasman, R. A., Mehta, P., and Lou, H. (2008). Regulation of neuron-specific alternative splicing of neurofibromatosis type 1 pre-mRNA. Mol. Cell. Biol. 28, 1240–1251.
Keywords: Hu, nELAV, AChE, alternative splicing, mRNA stability, translation, micro-RNA
Citation: Bronicki LM and Jasmin BJ (2012) Trans-acting factors governing acetylcholinesterase mRNA metabolism in neurons. Front. Mol. Neurosci. 5:36. doi: 10.3389/fnmol.2012.00036
Received: 30 January 2012; Paper pending published: 14 February 2012;
Accepted: 06 March 2012; Published online: 22 March 2012.
Edited by:
Karl Tsim, The Hong Kong University of Science and Technology, ChinaReviewed by:
Javier SaEz-Valero, Universidad Miguel Hernandez, SpainClaire Legay, Universite Paris Descartes, France
Copyright: © 2012 Bronicki and Jasmin. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Bernard J. Jasmin, Faculty of Medicine, Department of Cellular and Molecular Medicine, University of Ottawa, 451 Smyth Rd., Ottawa, ON, K1H 8M5, Canada. e-mail:amFzbWluQHVvdHRhd2EuY2E=