

- 1 State Key Laboratory of Cell Biology, Institute of Biochemistry and Cell Biology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai, China
- 2 Edmond and Lily Safra Center of Brain Sciences, The Hebrew University of Jerusalem, Jerusalem, Israel
To date, more than 40 different types of cells from primary cultures or cell lines have shown AChE expression during apoptosis and after the induction apoptosis by different stimuli. It has been well-established that increased AChE expression or activity is detected in apoptotic cells after apoptotic stimuli in vitro and in vivo, and AChE could be therefore used as a marker of apoptosis. AChE is not an apoptosis initiator, but the cells in which AChE is overexpressed undergo apoptosis more easily than controls. Interestingly, cells with downregulated levels of AChE are not sensitive to apoptosis induction and AChE deficiency can protect against apoptosis. Some tumor cells do not express AChE, but when AChE is introduced into a tumor cell, the cells cease to proliferate and undergo apoptosis more readily. Therefore, AChE can be classified as a tumor suppressor gene. AChE plays a pivotal role in apoptosome formation, and silencing of the AChE gene prevents caspase-9 activation, with consequent decreased cell viability, nuclear condensation, and poly (adenosine diphosphate-ribose) polymerase cleavage. AChE is translocated into the nucleus, which may be an important event during apoptosis. Several questions still need to be addressed, and further studies that address the non-classical function of AChE in apoptosis are needed.
The human gene that encodes acetylcholinesterase (AChE) is located on chromosome 7q22 (Ehrlich et al., 1992; Getman et al., 1992). Acetylcholinesterase is a type B carboxylesterase that rapidly hydrolyzes the neurotransmitter acetylcholine (ACh) at cholinergic synapses, as well as at neuromuscular junctions (Grisaru et al., 1999; Sperling et al., 2008). Additional splice variants involving both the 5′ and 3′ ends of the gene have also been reported. Among these splice variants are three distinct AChE variants, each with a different carboxyterminal sequence, called the “synaptic” (S), “erythrocytic” (E), and “readthrough” (R) AChE isoforms (Soreq and Seidman, 2001; Meshorer and Soreq, 2006). “Synaptic” AChE-S constitutes the principal multimeric enzyme in brain and muscle; soluble, monomeric “readthrough” AChE-R has been reported in embryonic and tumor cells and is induced under psychological, chemical, and physical stress; and finally, glypiated dimers of erythrocytic AChE-E are associated with red blood cell membranes (Grisaru et al., 1999). In addition to its catalytic function of the hydrolysis of acetylcholine, AChE has been shown to be involved in many non-cholinergic functions, such as cell growth, stem cell differentiation (Sperling et al., 2008), neuritogenesis, cell adhesion (Paraoanu and Layer, 2008), synaptogenesis, activation of dopamine neurons, tumorigenesis, amyloid fiber assembly (Inestrosa et al., 1996; Alvarez et al., 1997), hematopoiesis, and thrombopoiesis (Greenfield, 1996; Layer, 1996; Small et al., 1996; Soreq and Seidman, 2001). Almost a decade ago, work by our group revealed that AChE-S expression can be induced during apoptosis in various cell types, including cells that did not originate from the nervous or hematopoietic system (Zhang et al., 2002). It has also been shown that AChE exerts its pivotal role in apoptosis by its participation in the formation of the apoptosome (Park et al., 2004). In this review we will summarize the functional evidence for the role of AChE during apoptosis, despite the fact that our understanding of the apoptotic role of AChE is still not complete.
AChE Levels are Increased in Apoptotic Cells after Apoptotic Stimuli Both in vitro and in vivo
AChE is expressed in cholinergic neurons, neuromuscular junctions, hematopoietic cells, including erythrocytes (Grisaru et al., 1999) and megakaryocytes (Paulus et al., 1981; Tranumjensen and Behnke, 1981), as well tissues that are not innervated by cholinergic neurons (Small et al., 1996). There is mounting evidence, however, that AChE expression is induced during apoptosis in a variety of other cell types (Zhang et al., 2002). Human lung fibroblast (HLF) cells and rat kidney (NRK) cells do not normally express AChE, but have been shown to express high levels of AChE when undergoing apoptosis or when moving from an inactive to an active state (Zhang et al., 2002; Jin et al., 2004). In general, cells that normally express low levels of AChE, such as PC-12, showed increased levels of AChE and AChE activity whilst undergoing apoptosis (Yang et al., 2002; Jing et al., 2008). Steinritz et al. (2007) reported that increased AChE activity was found in sulfur mustard (SM)-treated A549 cell culture, as determined by Ellman assay and Western blotting. Interestingly, AChE activity showed a strong correlation with the number of TUNEL-positive cells. Furthermore, AChE activity may be a potential marker of apoptosis in A549 cells after SM injury (Steinritz et al., 2007). Other diseases with a possible apoptotic role in disease pathogenesis such as Alzheimer’s disease and diabetes also show cholinesterase involvement. Allam et al. observed that butyrylcholinesterase and AChE-related proteins were found to be common to both Alzheimer’s disease and diabetes; these proteins may play an etiological role via their influence on insulin resistance and lipid metabolism (Allam et al., 2006). Toiber et al. (2008) showed that in transfected primary brain cultures, an alternative N terminal extended transcript of AChE (N-AChE-S) induced cell death, morphological impairments and caspase-3 activation. In addition in cortical tissues from AD patients, N-AChE-S overexpression coincides with Tau hyper-phosphorylation. In other pathologies, Hu et al. (2009) showed increased expression levels of AChE and caspase-3 in the brain and peripheral immune system of focal cerebral ischemic rats. Xiao et al. (2006) utilized AChE with Bax, c-fos, and p53 genes as markers of apoptosis. Huang et al. (2005) have developed a novel assay that utilizes fluorochrome-tagged physostigmine (Ph-F) in situ to detect active AChE that has been induced during apoptosis. Cells that show an increase in AChE activity during apoptosis are summarized in Table 1.
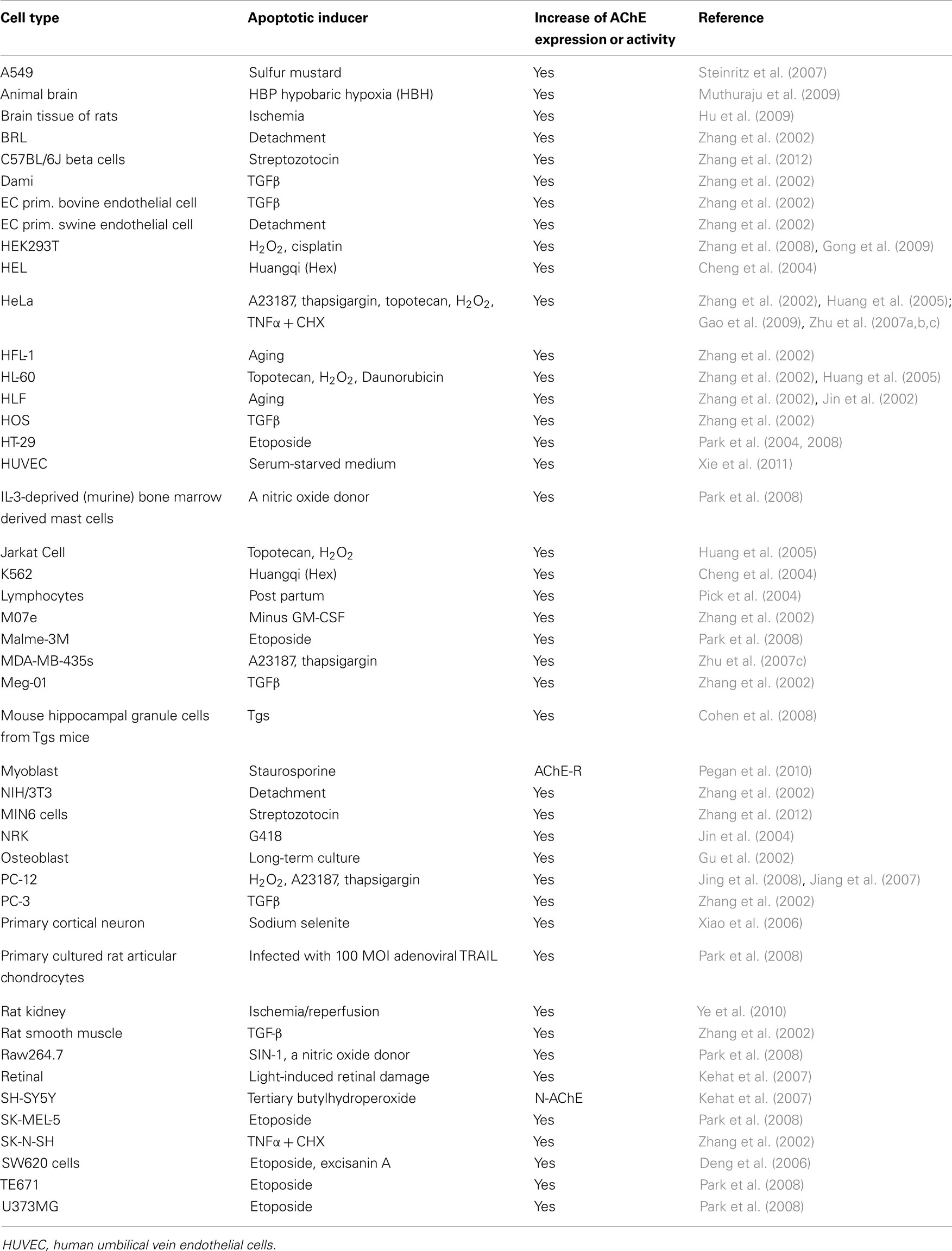
Table 1. Summarized cell lines in which AChE expression or activity were increased by the apoptotic stimuli with various apoptotic inducers.
Cell Sensitivity to Apoptosis is Related to AChE Expression Levels
AChE is not a universal activators of apoptosis as it has been well-established that cholinergic neurons and neuromuscular junctions that express AChE do not initiate apoptosis under normal conditions without an apoptotic stimulus. However, AChE-positive cells, such as PC-12 (Yang et al., 2002; Jing et al., 2008) as well as neurons have been shown to respond more sensitively to apoptotic stimuli than AChE-negative cells (Jin et al., 2002, 2004). To investigate this, a stable cell line that overexpressed AChE was established from NRK cells. Overexpression of AChE was shown to inhibit cell proliferation and promote apoptosis in these cells. Transgenic mice which overexpress catalytically active AChE-S and have a higher levels of AChE hydrolytic activity than control mice, show increased levels of cell labeling by both bromodeoxyuridine and caspase-3; which reflects increased neural apoptosis (Cohen et al., 2008) though these transgenic mice also showed increased neuronal survival through an as yet unidentified compensatory mechanism. AChE was also shown to suppress cell proliferation via catalytic hydrolysis of acetylcholine in human colorectal carcinoma (HCC) (Montenegro et al., 2006).
Zhang et al. (2002) showed that the downregulation of AChE expression with antisense inhibited apoptosis, and they suggest that AChE is potentially a marker and a regulator of apoptosis. Park et al. (2004) explored the role of AChE in apoptosis by silencing the AChE gene. Silencing of AChE abolished the expression of AChE and prevented caspase-9 activation, decreased cell viability, nuclear condensation, and poly(adenosine diphosphate-ribose) polymerase cleavage. Animal models of degenerative diseases were found to be more easily established in AChE+/+ mice than AChE+/− mice. Espallergues et al. (2010) showed that the intracerebroventricular injection of amyloid-beta (25–35) failed to induce learning deficits in AChE+/− mice, but impaired learning in AChE+/+ controls. The peptide showed reduced toxicity in the forebrain structures of AChE+/− mice, as an increase in lipid peroxidation levels was measured in the hippocampus of AChE+/+ but not AChE+/− mice. They concluded that the increase in cholinergic tonus observed in AChE+/− mice did not result in increased memory functions but allowed a significant prevention of the deleterious effects of muscarinic blockade or amyloid toxicity. Ye et al. (2010) recently reported that AChE is expressed during ischemia–reperfusion (I/R)-induced apoptosis in vivo. Their study demonstrated that AChE may be a pro-apoptotic factor and that the inhibition of AChE reduces the degree of renal I/R injury.
AChE and Tumor Suppression
Papers reporting the presence of AChE in tumors have first been reported almost 40 years ago (Gearhart and Mintz, 1974). The exact role of AChE in oncogenesis or tumor progression is still unclear and beyond the scope of this review. However a number of studies (Gearhart and Mintz, 1974; Takahashi et al., 1995; Johnson and Cotter, 1997) have shown an genetic alterations in the long arm of chromosome 7 the genetic locus of the AChE gene, in several tumors. Furthermore, the agricultural use of AChE inhibitors is known to induce several types of tumors (Dich et al., 1997; Cabello et al., 2001; Abou-Donia, 2003). Stephenson et al. (1996) investigated gene copy-number alterations at these genes in myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML). These data raise the intriguing possibility that the proaptotic role of AChE could play a role in tumor suppressor. (Stephenson et al., 1996).
Cholinesterases are downregulated in HCC (Montenegro et al., 2006), squamous cell carcinoma, and retinoblastoma (Qavi and Al-Rajhi, 2009). Zhao et al. (2011) demonstrated that AChE was significantly downregulated in the cancer tissues of 69.2% of HCC patients, and the low level of AChE expression in HCC was correlated with tumor aggressiveness, the elevated risk of postoperative recurrence, and a low survival rate. Both the recombinant AChE protein and the enhanced expression of AChE significantly inhibited HCC cell growth in vitro and tumorigenicity in vivo. The studies describe the function of AChE as a tumor growth suppressor that acts in the regulation of cell proliferation, relevant signaling pathways, and the chemosensitivity of HCC cells. AChE is a promising independent prognostic predictor for HCC recurrence and the survival of HCC patients (Montenegro et al., 2006). AChE-R was shown to increases germ cell apoptosis (Mor et al., 2008).
Localization of AChE to the Cell Nucleus and Apoptotic Bodies May Play a Role in Apoptosis Induction
Zhang et al. (2002) have shown by cytochemical staining and immunohistochemistry that the presence of AChE is not homogeneous in either the cytoplasm or in the nucleus. To establish whether this heterogeneity is due to the stage of apoptosis induction, the authors examined the time course of AChE expression in the leukemic Meg-01 cell line after treatment with transforming growth factor (TGF)-β. Cytochemical staining showed AChE initially appears in the cytosol and afterward in the nucleus. AChE-positive staining follows the nuclear morphological changes such as condensation and fragmentation. Eventually, when the cell membrane ruptures, AChE is released. They also examined the expression of AChE by transmission electron microscopy. At 2 h after TGF-β treatment, AChE was observed only in the cytosol but by 4 h had moved entirely to the nucleus. Therefore, during the induction of apoptosis, AChE is first synthesized in the cytosol and then accumulates in the nucleus. In general AChE is present in the nucleus at the mid- and late-stages of apoptosis (Yang et al., 2002; Jin et al., 2004; Xie et al., 2011). Santos et al. (2007) initially investigated the expression and subcellular localization of acetylcholinesterase (AChE) in endothelial cells and showed the expression of a 70-kDa AChE in both the cytoplasmic and nuclear compartments. The levels of this novel isoform are decreased in response to vascular endothelial growth factor via the proteasome pathway. They found that AChE was downregulated in human leukemic T cells compared with normal T cells, which suggested that the decreased expression of the 55-kDa AChE protein may contribute to an angiogenic response and be associated with tumorigenesis. Importantly, nuclear expression is not specific to endothelial cells, but is also present in non-neuronal and neuronal cells. Xie et al. (2011) showed that the 55-kDa AChE protein was induced during apoptosis and was negatively regulated by the Akt pathway. PC-12 express AChE at basal levels in normal cells, but a 55-kDa AChE protein can be found during the progression of apoptosis. This 55 kDa AChE protein was temporally expressed after the activation of caspases and was caspase-dependent. The inhibition of protein synthesis with CHX indicated that the 55-kDa AChE protein resulted from the cleavage of the 68-kDa AChE (Xie et al., 2011). There are still questions regarding AChE that need to be addressed, including its function when it enters the nucleus. Furthermore, whether AChE entry into the nucleus is an active or passive event, how is it transported and what is the signal that allows it to enter the nucleus are all questions which still need to be resolved.
AChE Could Plays a Pivotal Role in the Formation of Apoptosomes
The role of AChE in apoptosis is a key question in this field. Park et al. (2004) showed that silencing of the AChE gene inhibited the interaction between Apaf-1 and cytochrome c. They concluded that AChE plays an important role in apoptosome formation. Their data showed that an antisense AChE oligonucleotide showed a similar effect by silencing the AChE gene. Small interfering RNA (siRNA) against the cytochrome c gene blocked the interaction of AChE with Apaf-1, whereas siRNA targeting the Apaf-1 gene did not block the interaction of AChE with cytochrome c. These findings indicated that the interaction of AChE with cytochrome c is required for the interaction between cytochrome c and protease-activating factor-1. Park et al. (2008) also showed that the interactions of AChE with caveolin-1 and, subsequently with cytochrome c, appear to be indispensable for the formation of apoptosomes. To date, however, these findings have only been shown in one study and more evidence is required to confirm how AChE participates in the formation of the apoptosome.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The author is grateful to Hermona Soreq, Jerusalem, for critical evaluation of this manuscript. This work was supported in part by grants from the National Natural Science Foundation of China (Nos. 30971481 and 31071213).
References
Abou-Donia, M. B. (2003). Organophosphorus ester-induced chronic neurotoxicity. Arch. Environ. Health 58, 484–497.
Allam, A. R., Sridhar, G. R., Thota, H., Suresh Babu, C., Siva Prasad, A., and Divakar, C. (2006). Alzheimer’s disease and Type 2 diabetes mellitus: the cholinesterase connection? Lipids Health Dis. 5, 28.
Alvarez, A., Opazo, C., Alarcón, R., Garrido, J., and Inestrosa, N. C. (1997). Acetylcholinesterase promotes the aggregation of amyloid-beta-peptide fragments by forming a complex with the growing fibrils. J. Mol. Biol. 272, 348–361.
Cabello, G., Valenzuela, M., Vilaxa, A., Durán, V., Rudolph, I., Hrepic, N., and Calaf, G. (2001). A rat mammary tumor model induced by the organophosphorous pesticides parathion and malathion, possibly through acetylcholinesterase inhibition. Environ. Health Perspect. 109, 471–479.
Cheng, X. D., Hou, C. H., Zhang, X. J., Xie, H. Y., Zhou, W. Y., Yang, L., Zhang, S. B., and Qian, R. L. (2004). Effects of Huangqi (Hex) on inducing cell differentiation and cell death in K562 and HEL cells. Acta Biochim. Biophys. Sin. (Shanghai) 36, 211–217.
Cohen, J. E., Zimmerman, G., Melamed-Book, N., Friedman, A., Dori, A., and Soreq, H. (2008). Transgenic inactivation of acetylcholinesterase impairs homeostasis in mouse hippocampal granule cells. Hippocampus 18, 182–192.
Deng, R., Li, W., Guan, Z., Zhou, J. M., Wang, Y., Mei, Y. P., Li, M. T., Feng, G. K., Huang, W., Liu, Z. C., Han, Y., Zeng, Y. X., and Zhu, X. F. (2006). Acetylcholinesterase expression mediated by c-Jun-NH2-terminal kinase pathway during anticancer drug-induced apoptosis. Oncogene 25, 7070–7077.
Dich, J., Zahm, S. H., Hanberg, A., and Adami, H. O. (1997). Pesticides and cancer. Cancer Causes Control 8, 420–443.
Ehrlich, G., Viegas-Pequignot, E., Ginzberg, D., Sindel, L., Soreq, H., and Zakut, H. (1992). Mapping the human acetylcholinesterase gene to chromosome-7Q22 by fluorescent in situ hybridization coupled with selective PCR amplification from a somatic hybrid cell panel and chromosome-sorted DNA libraries. Genomics 13, 1192–1197.
Espallergues, J., Galvan, L., Sabatier, F., Rana-Poussine, V., Maurice, T., and Chatonnet, A. (2010). Behavioral phenotyping of heterozygous acetylcholinesterase knockout (AChE(+/−)) mice showed no memory enhancement but hyposensitivity to amnesic drugs. Behav. Brain Res. 206, 263–273.
Gao, W., Zhu, H., Zhang, J. Y., and Zhang, X. J. (2009). Calcium signaling-induced Smad3 nuclear accumulation induces acetylcholinesterase transcription in apoptotic HeLa cells. Cell. Mol. Life Sci. 66, 2181–2193.
Gearhart, J. D., and Mintz, B. (1974). Contact-mediated myogenesis and increased acetylcholinesterase activity in primary cultures of mouse terato-carcinoma cells. Proc. Natl. Acad. Sci. U.S.A. 71, 1734–1738.
Getman, D. K., Eubanks, J. H., Camp, S., Evans, G. A., and Taylor, P. (1992). The human gene encoding acetylcholinesterase is located on the long arm of chromosome-7. Am. J. Hum. Genet. 51, 170–177.
Gong, X. W., Ye, W., Zhou, H., Ren, X., Li, Z., Zhou, W., Wu, J., Gong, Y., Ouyang, Q., Zhao, X., and Zhang, X. (2009). RanBPM is an acetylcholinesterase-interacting protein that translocates into the nucleus during apoptosis. Acta Biochim. Biophys. Sin. (Shanghai) 41, 883–891.
Greenfield, S. (1996). Non-classical actions of cholinesterases: role in cellular differentiation, tumorigenesis and Alzheimer’s disease. Neurochem. Int. 28, 485–490.
Grisaru, D., Sternfeld, M., Eldor, A., Glick, D., and Soreq, H. (1999). Structural roles of acetylcholinesterase variants in biology and pathology. Eur. J. Biochem. 264, 672–686.
Gu, Q., Zhu, H. M., and Zhang, X. J. (2002). Apoptosis of rat osteoblasts in process of calcification in vitro. Acta Pharmacol. Sin. 23, 808–812.
Hu, T., Fu, Q., Liu, X., Zhang, H., and Dong, M. (2009). Increased acetylcholinesterase and caspase-3 expression in the brain and peripheral immune system of focal cerebral ischemic rats. J. Neuroimmunol. 211, 84–91.
Huang, X., Lee, B., Johnson, G., Naleway, J., Guzikowski, A., Dai, W., and Darzynkiewicz, Z. (2005). Novel assay utilizing fluorochrome-tagged physostigmine (Ph-F) to in situ detect active acetylcholinesterase (AChE) induced during apoptosis. Cell Cycle 4, 140–147.
Inestrosa, N. C., Alvarez, A., Pérez, C. A., Moreno, R. D., Vicente, M., Linker, C., Casanueva, O. I., Soto, C., and Garrido, J. (1996). Acetylcholinesterase accelerates assembly of amyloid-beta-peptides into Alzheimer’s fibrils: possible role of the peripheral site of the enzyme. Neuron 16, 881–891.
Jiang, H., Zhang, J., Zhu, H., Li, H., and Zhang, X. (2007). Nerve growth factor prevents the apoptosis-associated increase in acetylcholinesterase activity after hydrogen peroxide treatment by activating Akt. Acta Biochim. Biophys. Sin. (Shanghai) 39, 46–56.
Jin, Q. H., He, H. Y., Shi, Y. F., Lu, H., and Zhang, X. J. (2004). Overexpression of acetylcholinesterase inhibited cell proliferation and promoted apoptosis in NRK cells. Acta Pharmacol. Sin. 25, 1013–1021.
Jin, Q. H., Shi, Y. F., He, H. Y., Ng, K. K., Jiang, H., Yang, L., Jiang, Z. Q., and Zhang, X. J. (2002). Isolation of acetylcholinesterase from apoptotic human lung fibroblast cells by antibody affinity chromatography. Biotechniques 33, S92–S97.
Jing, P., Jin, Q., Wu, J., and Zhang, X. J. (2008). GSK3 beta mediates the induced expression of synaptic acetylcholinesterase during apoptosis. J. Neurochem. 104, 409–419.
Johnson, E., and Cotter, F. E. (1997). Monosomy 7 and 7q-associated with myeloid malignancy. Blood Rev. 11, 46–55.
Kehat, R., Zemel, E., Cuenca, N., Evron, T., Toiber, D., Loewenstein, A., Soreq, H., and Perlman, I. (2007). A novel isoform of acetylcholinesterase exacerbates photoreceptors death after photic stress. Invest. Ophthalmol. Vis. Sci. 48, 1290–1297.
Layer, P. G. (1996). Non-classical actions of cholinesterases: role in cellular differentiation, tumorigenesis and Alzheimer’s disease. Neurochem. Int. 28, 491–495.
Meshorer, E., and Soreq, H. (2006). Virtues and woes of AChE alternative splicing in stress-related neuropathologies. Trends Neurosci. 29, 216–224.
Montenegro, M. F., Ruiz-Espejo, F., Campoy, F. J., Muñoz-Delgado, E., de la Cadena, M. P., Rodríguez-Berrocal, F. J., and Vidal, C. J. (2006). Cholinesterases are down-expressed in human colorectal carcinoma. Cell. Mol. Life Sci. 63, 2175–2182.
Mor, I., Sklan, E. H., Podoly, E., Pick, M., Kirschner, M., Yogev, L., Bar-Sheshet Itach, S., Schreiber, L., Geyer, B., Mor, T., Grisaru, D., and Soreq, H. (2008). Acetylcholinesterase-R increases germ cell apoptosis but enhances sperm motility. J. Cell. Mol. Med. 12, 479–495.
Muthuraju, S., Maiti, P., Solanki, P., Sharma, A. K., Amitabh, Singh, S. B., Prasad, D., and Ilavazhagan, G. (2009). Acetylcholinesterase inhibitors enhance cognitive functions in rats following hypobaric hypoxia. Behav. Brain Res. 203, 1–14.
Paraoanu, L. E., and Layer, P. G. (2008). Acetylcholinesterase in cell adhesion, neurite growth and network formation. FEBS J. 275, 618–624.
Park, S. E., Jeong, S. H., Yee, S. B., Kim, T. H., Soung, Y. H., Ha, N. C., Kim, N. D., Park, J. Y., Bae, H. R., Park, B. S., Lee, H. J., and Yoo, Y. H. (2008). Interactions of acetylcholinesterase with caveolin-1 and subsequently with cytochrome c are required for apoptosome formation. Carcinogenesis 29, 729–737.
Park, S. E., Kim, N. D., and Yoo, Y. H. (2004). Acetylcholinesterase plays a pivotal role in apoptosome formation. Cancer Res. 64, 2652–2655.
Paulus, J. M., Maigne, J., and Keyhani, E. (1981). Mouse megakaryocytes secrete acetylcholinesterase. Blood 58, 1100–1106.
Pegan, K., Matkovic, U., Mars, T., Mis, K., Pirkmajer, S., Brecelj, J., and Grubic, Z. (2010). Acetylcholinesterase is involved in apoptosis in the precursors of human muscle regeneration. Chem. Biol. Interact. 187, 96–100.
Pick, M., Flores-Flores, C., Grisaru, D., Shochat, S., Deutsch, V., and Soreq, H. (2004). Blood-cell-specific acetylcholinesterase splice variations under changing stimuli. Int. J. Dev. Neurosci. 22, 523–531.
Qavi, H., and Al-Rajhi, A. A. (2009). Acetylcholinesterase and HHV-8 in squamous cell carcinoma and retinoblastoma. In vivo 23, 679–683.
Santos, S. C. R., Vala, I., Miguel, C., Barata, J. T., Garção, P., Agostinho, P., Mendes, M., Coelho, A. V., Calado, A., Oliveira, C. R., e Silva, J. M., and Saldanha, C. (2007). Expression and subcellular localization of a novel nuclear acetylcholinesterase protein. J. Biol. Chem. 282, 25597–25603.
Small, D. H., Michaelson, S., and Sberna, G. (1996). Non-classical actions of cholinesterases: role in cellular differentiation, tumorigenesis and Alzheimer’s disease. Neurochem. Int. 28, 453–483.
Soreq, H., and Seidman, S. (2001). Acetylcholinesterase – new roles for an old actor. Nat. Rev. Neurosci. 2, 294–302.
Sperling, L. E., Steinert, G., Boutter, J., Landgraf, D., Hescheler, J., Pollet, D., and Layer, P. G. (2008). Characterisation of cholinesterase expression during murine embryonic stem cell differentiation. Chem. Biol. Interact. 175, 156–160.
Steinritz, D., Emmler, J., Hintz, M., Worek, F., Kreppel, H., Szinicz, L., and Kehe, K. (2007). Apoptosis in sulfur mustard treated A549 cell cultures. Life Sci. 80, 2199–2201.
Stephenson, J., Czepulkowski, B., Hirst, W., and Mufti, G. J. (1996). Deletion of the acetylcholinesterase locus at 7q22 associated with myelodysplastic syndromes (MDS) and acute myeloid leukaemia (AML). Leuk. Res. 20, 235–241.
Takahashi, S., Shan, A. L., Ritland, S. R., Delacey, K. A., Bostwick, D. G., Lieber, M. M., Thibodeau, S. N., and Jenkins, R. B. (1995). Frequent loss of heterozygosity AT 7Q31.1 in primary prostate-cancer is associated with tumor aggressiveness and progression. Cancer Res. 55, 4114–4119.
Toiber, D., Berson, A., Greenberg, D., Melamed-Book, N., Diamant, S., and Soreq, H. (2008). N-acetylcholinesterase-induced apoptosis in Alzheimer’s disease. PLoS ONE 3, e3108. doi:10.1371/journal.pone.0003108
Tranumjensen, J., and Behnke, O. (1981). Acetylcholinesterase in the platelet-megakaryocyte system. 2. Structural localization in megakaryocytes of the rat, mouse, and CAT. Eur. J. Cell Biol. 24, 281–286.
Xiao, R., Qiao, J. T., Zhao, H. F., Liang, J., Yu, H. L., Liu, J., Guo, A. M., and Wang, W. (2006). Sodium selenite induces apoptosis in cultured cortical neurons with special concomitant changes in expression of the apoptosis-related genes. Neurotoxicology 27, 478–484.
Xie, J., Jiang, H., Wan, Y. H., Du, A. Y., Guo, K. J., Liu, T., Ye, W. Y., Niu, X., Wu, J., Dong, X. Q., and Zhang, X. J. (2011). Induction of a 55 kDa acetylcholinesterase protein during apoptosis and its negative regulation by the Akt pathway. J. Mol. Cell. Biol. 3, 250–259.
Yang, L., He, H. Y., and Zhang, X. J. (2002). Increased expression of intranuclear AChE involved in apoptosis of SK-N-SH cells. Neurosci. Res. 42, 261–268.
Ye, W., Gong, X., Xie, J., Wu, J., Zhang, X., Ouyang, Q., Zhao, X., Shi, Y., and Zhang, X. (2010). AChE deficiency or inhibition decreases apoptosis and p53 expression and protects renal function after ischemia/reperfusion. Apoptosis 15, 474–487.
Zhang, B., Yang, L., Yu, L., Lin, B., Hou, Y., Wu, J., Huang, Q., Han, Y., Guo, L., Ouyang, Q., Zhang, B., Lu, L., and Zhang, X. (2012). Acetylcholinesterase is associated with apoptosis in beta-cells and contributes to IDDM pathogenesis. Acta Biochim. Biophys. Sin. (Shanghai) 44, 207–216.
Zhang, J. Y., Jiang, H., Gao, W., Wu, J., Peng, K., Shi, Y. F., and Zhang, X. J. (2008). The JNK/AP1/ATF2 pathway is involved in H2O2-induced acetylcholinesterase expression during apoptosis. Cell. Mol. Life Sci. 65, 1435–1445.
Zhang, X. J., Yang, L., Zhao, Q., Caen, J. P., He, H. Y., Jin, Q. H., Guo, L. H., Alemany, M., Zhang, L. Y., and Shi, Y. F. (2002). Induction of acetylcholinesterase expression during apoptosis in various cell types. Cell Death Differ. 9, 790–800.
Zhao, Y. J., Wang, X., Wang, T., Hu, X., Hui, X., Yan, M., Gao, Q., Chen, T., Li, J, Yao, M., Wan, D., Gu, J., Fan, J., and He, X. (2011). Acetylcholinesterase, a key prognostic predictor for hepatocellular carcinoma, suppresses cell growth and induces chemosensitization. Hepatology 53, 493–503.
Zhu, H., Gao, W., Jiang, H., Wu, J., Shi, Y. F., and Zhang, X. J. (2007a). Calcineurin mediates acetylcholinesterase expression during calcium ionophore A23187-induced HeLa cell apoptosis. Biochim. Biophys. Acta 1773, 593–602.
Zhu, H., Gao, W., Shi, Y. F., and Zhang, X. J. (2007b). The CCAAT-binding factor CBF/NF-Y regulates the human acetylcholinesterase promoter activity during calcium ionophore A23187-induced cell apoptosis. Biochim. Biophys. Acta 1770, 1475–1482.
Keywords: acetylcholinesterase, apoptosis, tumor
Citation: Zhang X-J and Greenberg DS (2012) Acetylcholinesterase involvement in apoptosis. Front. Mol. Neurosci. 5:40. doi: 10.3389/fnmol.2012.00040
Received: 09 January 2012; Accepted: 18 March 2012;
Published online: 10 April 2012.
Edited by:
Hermona Soreq, The Hebrew University of Jerusalem, IsraelReviewed by:
Eran Meshorer, The Hebrew University of Jerusalem, IsraelDeborah Toiber, Harvard Medical School, USA
Adi Gilboa-Geffen, Harvard Medical School, USA
Copyright: © 2012 Zhang and Greenberg. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Xue-Jun Zhang, Institute of Biochemistry and Cell Biology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, 320 YueYang Road, Shanghai 200031, China. e-mail:eGp6aGFuZ0BzaWJzLmFjLmNu